
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydrophobized poly(heptazine imide) for highly effective photocatalytic hydrogen peroxide production in a biphasic fatty alcohol–water system†‡
Igor
Krivtsov
 *ab,
Ashish
Vazirani
a,
Dariusz
Mitoraj
a,
Mohamed M.
Elnagar
a,
Christof
Neumann
c,
Andrey
Turchanin
c,
Yolanda
Patiño
b,
Salvador
Ordóñez
b,
Robert
Leiter
d,
Mika
Lindén
e,
Ute
Kaiser
d and
Radim
Beranek
*a
*ab,
Ashish
Vazirani
a,
Dariusz
Mitoraj
a,
Mohamed M.
Elnagar
a,
Christof
Neumann
c,
Andrey
Turchanin
c,
Yolanda
Patiño
b,
Salvador
Ordóñez
b,
Robert
Leiter
d,
Mika
Lindén
e,
Ute
Kaiser
d and
Radim
Beranek
*a
aInstitute of Electrochemistry Ulm University, Albert-Einstein-Allee 47, 89081 Ulm, Germany. E-mail: krivtsovigor@uniovi.es; radim.beranek@uni-ulm.de
bDepartment of Chemical and Environmental Engineering, University of Oviedo, 33006 Oviedo, Spain
cInstitute of Physical Chemistry and Abbe Center of Photonics, Friedrich Schiller University Jena, Lessingstr. 10, 07743 Jena, Germany
dElectron Microscopy of Materials Science, Central Facility for Electron Microscopy, Ulm University, Albert-Einstein-Allee 11, 89081, Ulm, Germany
eInstitute of Inorganic Chemistry II, Ulm University, Albert-Einstein-Allee 11, 89081, Ulm, Germany
First published on 6th January 2023
Abstract
Light-driven production of hydrogen peroxide via selective dioxygen reduction is an attractive “green” alternative to the conventionally used anthraquinone process. One of the most promising classes of photocatalytic materials for this conversion that excels by high selectivity, chemical stability and low cost is represented by polymeric carbon nitrides, and particularly by various types of poly(heptazine imide) (PHI), i.e., ionic variants of carbon nitride. A crucial challenge highlighted by recent studies is the problem of separation of formed H2O2 from the reaction medium and especially from the suspended photocatalyst particles since photocatalytically generated H2O2 can undergo light-driven reductive or oxidative decomposition, as well as disproportionation on the surface of the photocatalyst in the dark. Herein, we report an elegant solution to this problem by implementing a biphasic reaction system in which the hydrophobic photocatalyst oxidizes a lipophilic electron donor in the organic phase, while the produced H2O2 is instantaneously extracted into the aqueous layer. To this end, we have achieved an effective hydrophobization of ionic carbon nitride (PHI) nanoparticles in the form of a composite with alkylated silica. The hydrophobized composite effectively photocatalyzes the reduction of dioxygen to H2O2 with concurrent oxidation of a model fatty alcohol (1-octanol) in the organic phase under visible light irradiation (406 nm LED), and enables, at the same time, facile separation of the high-value H2O2/water mixture from the reaction medium at H2O2 concentrations (∼0.12 mol L−1) that are unprecedentedly high for light-driven systems reported in the literature. Notably, fatty alcohols are readily available from vegetable waxes and as pulping sub-products, and the products of their partial oxidation represent a valuable feedstock for the synthesis of pharmaceuticals and cosmetic products. This work thus showcases a rational design of a high-performance photocatalytic system for H2O2 production that enables easy separation of the product from electron donors and its recovery at high concentrations, and paves the way for sustainable and economically viable light-driven H2O2 production from easily available feedstocks.
1. Introduction
The field of photocatalysis is currently witnessing an increasing interest in the development of various selective chemical conversions beyond hydrogen production via water splitting, in particular due to the growing concerns regarding the economic viability of photo(electro)catalytic water splitting,1 and especially since the photocatalytic production of high-value compounds from low-cost chemical feedstocks holds promise for commercially viable applications on a more immediate time scale as compared to the photocatalytic production of hydrogen.2,3 One of the most attractive compounds that can be photocatalytically obtained by a simple two-electron reduction of dioxygen is hydrogen peroxide, a highly versatile and environmentally benign oxidizing agent used in a number of important industrial processes. Although photocatalytic H2O2 peroxide production from pure water and oxygen has been reported, the reaction rates were usually too low to accumulate high concentrations of the product,4–8 making the presence of additional electron donors like aliphatic or aromatic alcohols practically indispensable to achieve reasonable reaction rates.9,10 Notably, this approach appears even more attractive if also the oxidation product of this reaction has a higher value than the substrate, as it is the case for the conversion of biomass-derived feedstock.11–13Polymeric carbon nitrides (PCNs) whose photocatalytic activity was first reported by Wang et al.14 are known for their excellent performance in light-driven H2O2 production from oxygen.15 Notably, PCNs also exist in various ionic forms as poly(heptazine imide) (PHI), i.e. materials that comprise stacks of 2D heptazine-based networks and alkali metal cations and/or protons.16–20 Ionic carbon nitrides are typically not fully crystalline, thus making it challenging to determine their exact structure. That is why ionic carbon nitrides prepared by the molten salt method are often denoted by different authors as modified version of covalent melon polymer, such as CNx-NCN, KSCN- or alkali metal-modified carbon nitrides rather than PHI.13,17 The ionic variants of carbon nitride typically show improved photogenerated charge separation and effective accumulation and storage of photogenerated electrons,21–24 and exhibit an improved activity in photocatalytic H2O2 evolution compared to the conventional, non-ionic, melon polymers.12,13,25 Although, the superior activity of PHI can also be explained, among other factors, by partial substitution of the terminal NH2-groups for –N−–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N species in PHI, resulting in fewer hydrogen bond interactions and better dispersibility of the material in polar solvents.26 A major challenge in carrying out such reactions in suspension is the effective separation of the dissolved reduction and oxidation products for their subsequent use, in order to avoid the decomposition of H2O2 on the surface of the photocatalyst by photoredox decomposition or by its disproportionation in the dark. The H2O2 decomposition would lead to the loss of the overall reaction yield, a problem that typically becomes even more prominent at higher H2O2 concentrations in the reaction medium.
N species in PHI, resulting in fewer hydrogen bond interactions and better dispersibility of the material in polar solvents.26 A major challenge in carrying out such reactions in suspension is the effective separation of the dissolved reduction and oxidation products for their subsequent use, in order to avoid the decomposition of H2O2 on the surface of the photocatalyst by photoredox decomposition or by its disproportionation in the dark. The H2O2 decomposition would lead to the loss of the overall reaction yield, a problem that typically becomes even more prominent at higher H2O2 concentrations in the reaction medium.
In thermal liquid-phase catalysis the problems associated with the separation of meta-stable products are often solved by the use of biphasic systems comprising two immiscible solvents and thus enabling the separation of the catalyst and the product right after the formation of the latter.27,28 Such an approach, though highly relevant for photocatalytic production of valuable chemicals, has only recently been successfully applied in molecular photocatalysis,29 but very rarely used in the field of heterogeneous photocatalysis so far,30–34 mainly due to the lack of photocatalysts suitable for operation under biphasic conditions. To this end, Yamashita et al. recently devised a hydrophobic Ti-based MOF for the biphasic selective benzyl alcohol oxidation to benzaldehyde in the organic phase (benzyl alcohol) and dioxygen reduction to H2O2, which was continuously extracted into the aqueous layer.30–32 However, we have recently demonstrated that benzaldehyde formed under such reaction conditions acts itself as a visible light-active photocatalyst that drives the (auto)photocatalytic H2O2 production and makes the assessment of the true performance of the photocatalyst highly unreliable.35 An unambiguous demonstration of effective photocatalytic H2O2 production in a biphasic system therefore (i) requires the design of efficient hydrophobic photocatalysts, and (ii) makes the use of electron donors different from benzyl alcohol absolutely mandatory.
In order to tackle this challenge using PCN-based photocatalysts that are known to be good photocatalyst for H2O2 production, the crucial question to be addressed is: Are we able to synthesize a hydrophobic PCN-based photocatalyst for use in a well-designed biphasic system? To the best of our knowledge, no synthetic protocols for hydrophobic PCN particles that could be selectively suspended in the organic phase of a biphasic system have been available so far. Kumru et al.36–38 and Dong et al.39 developed various procedures for grafting hydrophobic functions onto carbon nitrides, resulting, however, only in amphiphilic materials. Although conventional melon polymers possessing amphiphilic properties could be further modified with alkyl- and fluoroalkyl moieties, it only resulted in the improved dispersibility of PCN in organic solvents while retaining their amphiphilic nature.36 We hypothesized that the lower degree of hydrogen bonding, better dispersibility and smaller particle size of PHI-based ionic carbon nitrides that can be achieved upon sonication40 can make them more suitable for hydrophobization than conventional melon-like PCNs. Herein, we achieve, for the first time, effective hydrophobization of ionic carbon nitride (PHI) nanoparticles in the form of a composite with hydrophobic silica bearing methyl- or alkylsilane functions. The obtained PHI-based photocatalyst is hydrophobic and can be selectively suspended in the organic layer of a biphasic water/organic solvent system. This allows us to demonstrate unambiguously the expected advantages of selective photocatalysis in a biphasic system that enables immediate extraction of the H2O2 product into the aqueous phase, thus avoiding the decomposition of H2O2 on the surface of the hydrophobized photocatalyst. Specifically, we demonstrate highly effective visible light-driven reduction of dioxygen to H2O2 with concomitant oxidation of a model fatty alcohol (1-octanol) in the organic phase, resulting in H2O2 concentrations in the aqueous phase that are unprecedentedly high for photocatalytic H2O2 production systems reported so far (Fig. 1). Since fatty alcohols are readily available as sub-products of the forestry and pulping industry, the implications of our findings for the development of sustainable and economically viable photocatalytic H2O2 production from low-cost feedstock are discussed.
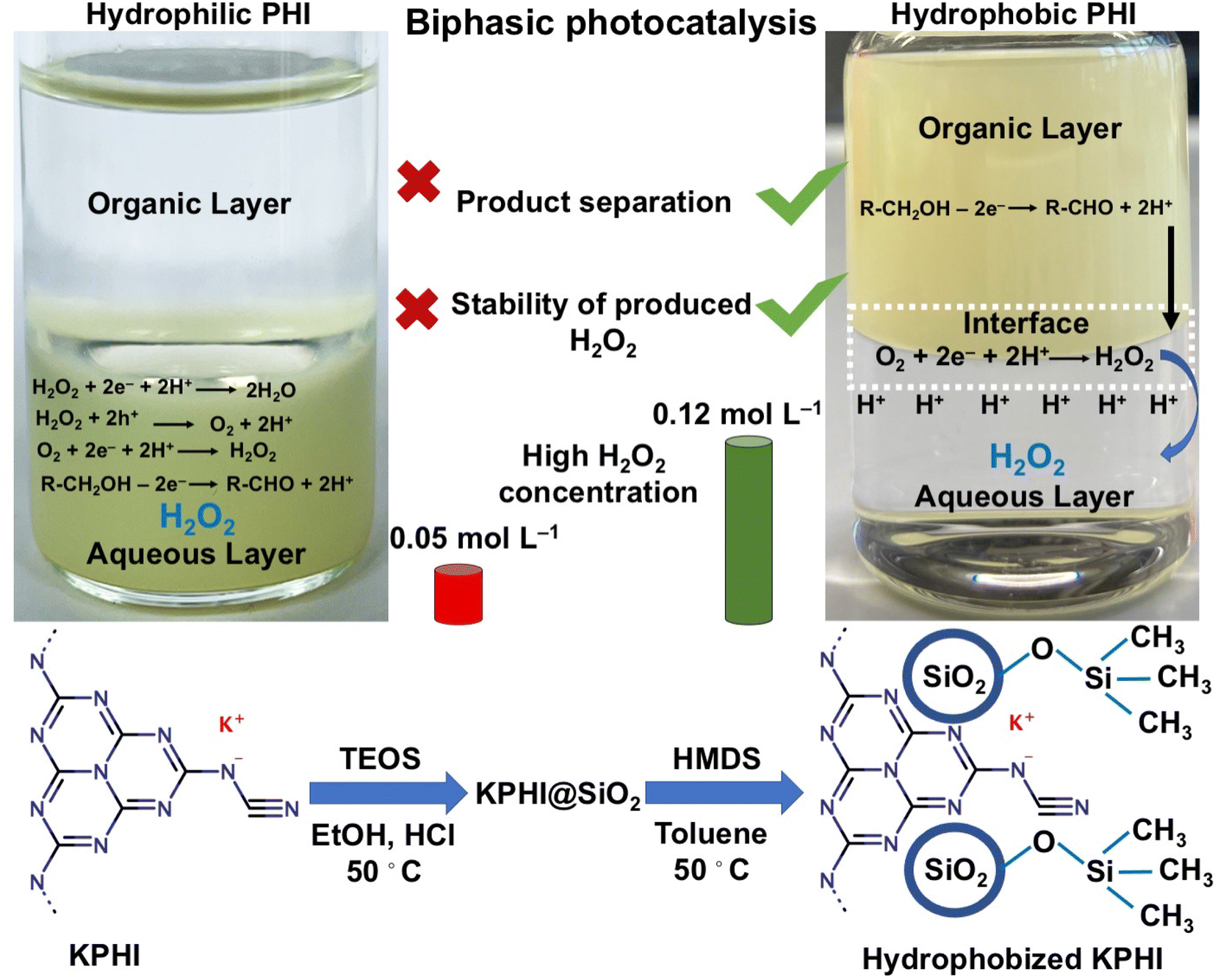 | ||
Fig. 1 Concept of biphasic photocatalysis using hydrophobized PHI. KPHI is synthesized by ionothermal treatment of melon in KSCN melt. The treatment leads to a partial exchange of surface NH2-groups for ionic ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CN− functions, thus reducing H-bonding in the polymer and allowing for the preparation of colloidally stable dispersions of small KPHI particles. The surface of suspended particles is modified with dispersed silica species via a sol–gel process and further functionalized by silylation, yielding a hydrophobic composite. Under visible light (∼406 nm) irradiation of a vigorously stirred biphasic system, the produced composite efficiently photooxidizes fatty alcohols in the organic phase (top layer in the image), while H2O2 produced by O2 photoreduction is immediately extracted into the aqueous phase (bottom layer in the image). Such separation of the product and the photocatalyst enables accumulating high concentrations of H2O2 in the water layer, which is not achievable in case of hydrophilic photocatalysts that can effectively decompose the formed H2O2. CN− functions, thus reducing H-bonding in the polymer and allowing for the preparation of colloidally stable dispersions of small KPHI particles. The surface of suspended particles is modified with dispersed silica species via a sol–gel process and further functionalized by silylation, yielding a hydrophobic composite. Under visible light (∼406 nm) irradiation of a vigorously stirred biphasic system, the produced composite efficiently photooxidizes fatty alcohols in the organic phase (top layer in the image), while H2O2 produced by O2 photoreduction is immediately extracted into the aqueous phase (bottom layer in the image). Such separation of the product and the photocatalyst enables accumulating high concentrations of H2O2 in the water layer, which is not achievable in case of hydrophilic photocatalysts that can effectively decompose the formed H2O2. | ||
2. Experimental
For the synthesis of potassium poly(heptazine imide) (KPHI), a modified procedure reported by Lotsch et al.41 based on the treatment of melon polymer with KSCN was used (for details see ESI‡). Silica-modified KPHI material (KPHI-Si) was prepared by adopting the procedure for the sol–gel fabrication of silica fibers reported by Sakka.42 To 1 g of finely ground KPHI in a round bottom flask, 65 mL of ethanol was added and the suspension was sonicated for 1 h. After sonication, 37 mL of TEOS, 3 mL of H2O and 0.15 mL of hydrochloric acid (37 wt%) were added to the mixture under vigorous stirring in this sequence. The sample was continuously stirred for 24 h, after which it was kept in a water bath at 50 °C for the next 24 h to induce silica polycondensation. In the next step, the sample was thoroughly washed by centrifugation at 9000 rpm for 10 min first with water and then with ethanol. Then, the sample was dried in an oven at 70 °C for a few hours, ground in a mortar and dried at 110 °C overnight before undergoing hydrophobization.The hydrophobization of KPHI-Si was performed according to the procedures developed by Resasco et al. and Erazo for the hydrophobization of zeolites and oxides, respectively.33,43 For this, 1 g of KPHI-Si was taken into a 20 mL vial and to that 18.5 mL of toluene was added. After adding toluene, 1.5 mL of a hydrophobic agent being (trichloro(hexyl)silane (HTS) (KPHI-Si-HTS), trichloro(octadecyl)silane (OTS) (KPHI-Si-OTS) or 1,1,1,3,3,3-hexamethyldisilazane (HMDS) (KPHI-Si-HMDS)) was introduced to the same mixture and the vial was closed and stirred overnight at 50 °C. The hydrophobized samples were washed three times with toluene (about 10 mL of solution each time) by centrifugation at 9000 rpm for 10 min. After the washing, the solid was dried at 70 °C overnight to evaporate the excess toluene and then the temperature was increased to 110 °C for the complete drying of the material (for further details on synthesis and hydrophobization see ESI‡).
The characterization techniques used for the prepared materials are described in ESI.‡
Photocatalytic H2O2 production using ethanol, 1-butanol and 1-octanol as electron donors was studied both in monophasic and biphasic conditions under vigorous stirring. The light source was a 406 nm LED (bandwidth of 16 nm) with a power density of 4.2 mW cm−2 incident on the irradiated wall of the reaction vessel. The concentration of produced H2O2 was estimated photometrically using titanyl oxysulfate method.
3. Results and discussion
3.1 Preparation and characterization of hydrophobized PHI materials
The direct reaction of KPHI with alkylchlorosilanes or HMDS did not result in formation of hydrophobic materials since polymeric carbon nitrides are known to be notoriously unreactive and difficult to modify.44 Therefore, KPHI was first modified by sol–gel deposited silica that served as a platform for the attachment of hydrophobic moieties. The success of this hydrophobization procedure is demonstrated in Fig. 2, where a water droplet in contact with the hydrophobized materials exhibits large contact angles, whereby hydrophilic KPHI and KPHI-Si films show complete wetting (Table 1). Accordingly, all hydrophobized KPHIs are also completely selectively dispersed in the organic top layer of the biphasic H2O/1-octanol system (Fig. 2). | ||
| Fig. 2 Materials characterization. Photographs of the doctor-bladed films of the prepared materials on FTO glasses with a droplet of water cast on their surface with the respective photographs of the KPHI samples suspended in a biphasic H2O (bottom phase)/1-octanol (top phase) mixture. | ||
| Sample | Absorption edge/eV | BET specific surface area/m2 g−1 | d-Spacinga/Å | Crystallite sizeb/nm | Silica contentc/wt% | Contact angled/° |
|---|---|---|---|---|---|---|
| a Calculated from XRD data. b Calculated by Scherrer equation from XRD data. c Estimated from TGA data. d Estimated by processing of photographic images of water droplets cast on the PHI films using ImageJ software with the contact angle determination plugin. | ||||||
| KPHI | 2.71 | 60 | 3.14 | 15 | — | — |
| KPHI-Si | 2.87 | 135 | 3.18 | 11 | 26.6 | — |
| KPHI-HTS | 2.98 | 136 | 3.24 | 8 | 24.3 | 137 ± 3 |
| KPHI-OTS | 2.96 | 65 | 3.24 | 8 | 26.8 | 138 ± 2 |
| KPHI-HMDS | 2.79 | 117 | 3.19 | 10 | 24.3 | 138 ± 4 |
| After 24 h of irradiation: 131 ± 4 | ||||||
The synthesized KPHI shows a typical X-ray diffraction (XRD) pattern with the maxima at ca. 28.3° (3.14 Å) and 8.1° corresponding to the (001) and (−110) crystal planes of KPHI, respectively (Fig. 3A).41 The sol–gel modification of the parent material with SiO2 does not significantly alter the diffraction pattern, with an exception that the peak intensities become lower, owing to the dilution of KPHI with silica, and a slight shift of the (001) reflection can be observed (Table 1). The latter effect is ascribed to the partial ion exchange of K+ for H+ and to the protonation of the polyheptazine structure leading to the changes in π–π stacking. This shift of the peak towards lower 2Θ values is even more pronounced for the KPHI-HTS and KPHI-OTS samples, resulting in the (001) peak position of ca. 27.4 (3.24 Å), while the XRD pattern of KPHI-HMDS does not manifest such a displacement (Fig. 3A, Table 1). This further peak displacement observed for KPHI-HTS and KPHI-OTS is possibly due to a higher degree of protonation and of the extent of exchange of K+ for H+ in the chloroalkylsilanes-modified materials, which can be explained by the protonation effect of HCl evolved during the hydrophobization as the result of the silylation agent reaction with silanols.
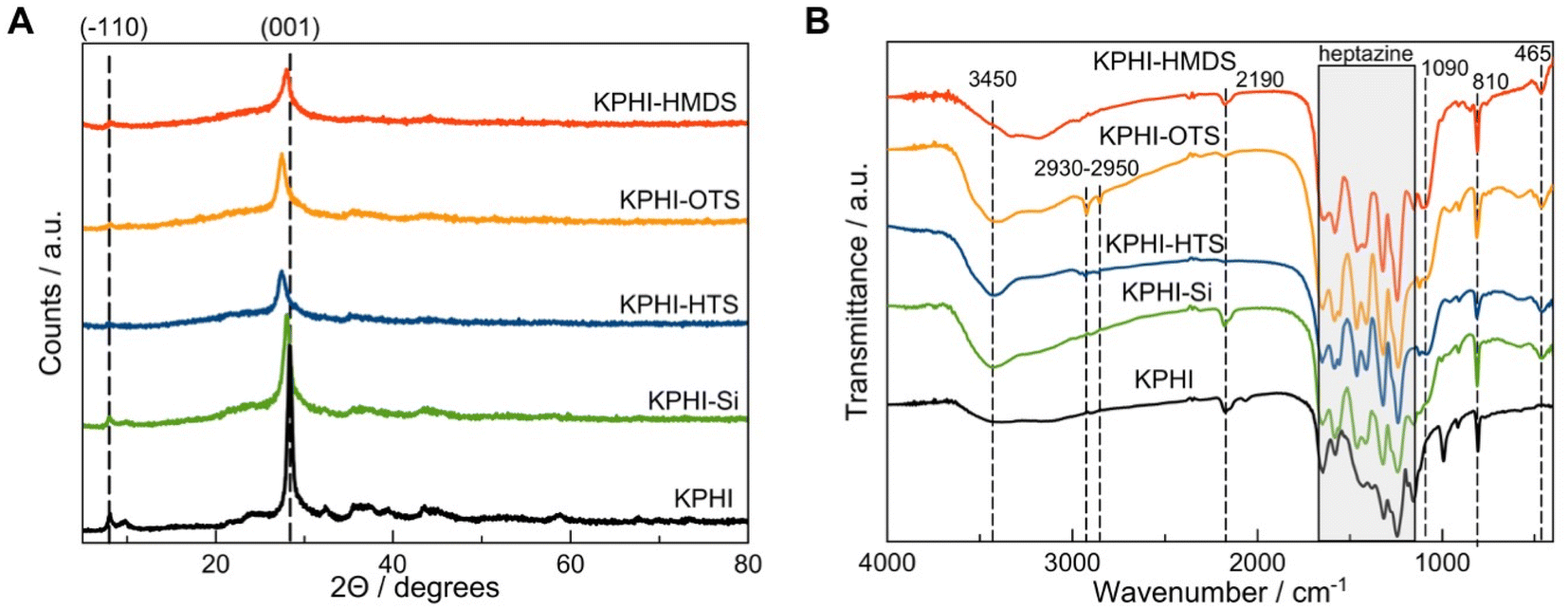 | ||
| Fig. 3 Materials characterization. (A) XRD patterns and (B) FTIR spectra of the prepared materials. | ||
The Fourier-transform infrared (FTIR) spectra confirm that the fingerprint region of PHI within 1700–1140 cm−1 remains nearly unchanged after each modification step (Fig. 3B). The typical breathing mode of triazines is also clearly visible at about 808–810 cm−1. The modification of KPHI with silica produces a broad peak on FTIR spectra centred at 1090 cm−1 assigned to asymmetric Si–O–Si stretching and a maximum at 465 cm−1 corresponding to the Si–O–Si bending modes.45 The protonation of the PHI structure is manifested in the FTIR spectra of KPHI-HTS and KPHI-OTS by the reduction of the intensity of the maximum at 2190 cm−1, which is a consequence of partial hydrolysis of the cyanamide functions induced by HCl (Fig. 3B). The incorporation of alkyl groups is evidenced on the spectra of KPHI-HTS and KPHI-OTS by the peaks in the range of 2930-2950 cm−1 ascribed to C–H stretching vibrations. These peaks do not appear in the spectra of another hydrophobized sample KPHI-HMDS, due to a shorter alkyl chain and, possibly, a smaller number of the grafted functions.
The X-ray photoelectron spectroscopy (XPS) data confirm the presence of Si in the modified PHI samples (see ESI, Fig. S1, S2 and Table S1‡). Notably, the presence of surface Si cannot be corroborated by XPS for the KPHI-OTS sample, most likely due to the long octadecyl chain of the hydrophobic agent covering the surface of the KPHI particles (see ESI, Fig. S1 and S2d‡). This is also supported by the C 1s XP spectrum of KPHI-OTS, where the peak ascribed to C–C and C–H bonds in the octadecyl chain which dominates over other carbon species contribution (see ESI, Fig. S3 and Table S1‡). In combination with the low relative sensitivity factor of Si 2p (0.82), the amount of Si on the surface is below the detection limit.
XPS data also show that the protonation of PHI causes a significant change of the peak position of different nitrogen species in the materials (see ESI, Fig. S4‡). The N 1s spectrum of KPHI-HMDS demonstrates an increase of the contribution at 399.9 eV compared to other samples, which might be attributed to the non-hydrolyzed Si–NH bonds of HMDS (see ESI, Fig. S4‡). The modified materials show a much higher surface concentration of oxygen compared to that of the parent KPHI, its presence can be linked to the deposition of SiO2 and to the hydrolysis of silane groups of the hydrophobic agents (see ESI, Fig. S5 and Table S1‡).
A detailed thermogravimetric analysis (TGA) enabled us to estimate the content of SiO2 in the modified PHI materials. The parent KPHI loses ca. 93.4% of its mass by 1000 °C with the mass loss in the high-temperature range of 850–1000 °C being assigned to the potassium carbonates or oxides produced during the decomposition of KPHI (Fig. 4A). The sol–gel modification of KPHI with silica was carried out in acidic conditions leading to K+ for H+ exchange, hence no mass loss corresponding to potassium-containing compounds is observed in the high-temperature range on the TGA profile of KPHI-Si (Fig. 4A). The residue of 26.6 wt% formed upon the KPHI-Si thermal decomposition at 1000 °C most likely corresponds to the SiO2 content in the material. The TGA data obtained for the hydrophobically modified KPHI-HTS (Fig. 4B), KPHI-OTS (Fig. 4C) and KPHI-HMDS (Fig. 4D) allow the estimation of silica content in these samples as 24.3, 26.8 and 24.3 wt%, respectively. It is worth noting that the thus determined silica concentration in the hydrophobized KPHI samples corresponds both to the SiO2 incorporated during the sol–gel modification step and to the silica formed as the result of thermal decomposition of grafted alkylsilane or methylsilazane moieties. Another important feature that can be observed in the TGA graphs is the appearance of the mass loss step in the 250-270 °C range for the KPHI-HTS (Fig. 4B) and KPHI-OTS (Fig. 4C) samples that corresponds to the oxidation of alkyl functions. This effect is not visible for KPHI-HMDS as the grafted HMDS groups tend to be more thermally stable.46
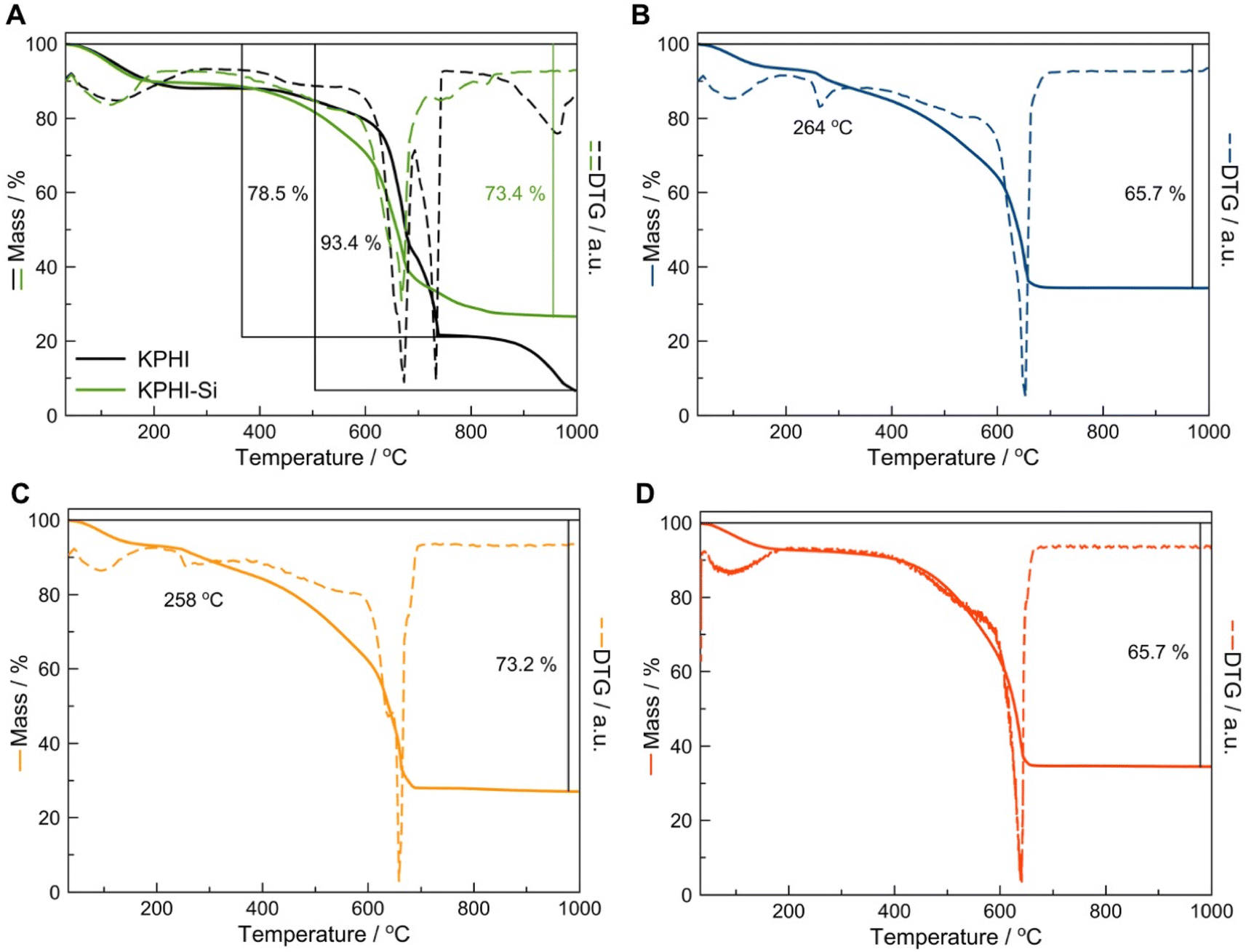 | ||
| Fig. 4 Materials characterization. Thermogravimetric (TGA) and differential thermogravimetric (DTG) analysis of the (A) hydrophilic KPHI and KPHI-Si, (B) KPHI-HTS, (C) KPHI-OTS, (D) KPHI-HMDS materials. | ||
The optical properties of the prepared materials are barely affected by the grafted hydrophobic functions and are mostly conditioned by the protonation degree of PHI (ESI, Fig. S6‡). The sol–gel silica modification of KPHI is carried out in acidic conditions, which causes the absorption edge to blue-shift from 2.7 eV to 2.9 eV (Fig. S6‡) due to increased protonation of KPHI.47 The protonation of KPHI by HCl released during the hydrophobization makes this value to increase up to 3.0 eV for the KPHI-HTS and KPHI-OTS samples (Fig. S6‡), while for KPHI-HMDS the absorption onset is 2.8 eV close to that of the parent KPHI (Fig. S6‡).
The scanning electron microscopy (SEM) images of the prepared materials show that the modification with silica and subsequent hydrophobization lead to the formation of smaller-sized PHI particles, which is possibly due to the formation of SiO2 around PHI, favouring its dispersibility (for detailed analysis of SEM images see ESI, Fig. S7‡). The SEM-EDX elemental analysis shows the presence of Si in all modified samples and evidences the reduction of potassium concentration upon the sol–gel modification and hydrophobization procedures (see ESI, Fig. S8 and Table S1‡). A comparative analysis of surface XPS and bulk SEM-EDX elemental analyses indicates that the surface-to-bulk ratio of K+ is below 1 for most of the samples. This is in agreement with other report on PHIs,25 and suggests that the potassium is likely to be found in the bulk of the material, supposedly being a structural part of poly(heptazine imide) network, while in case of KPHI-OTS the K+ ions are also present in form of separated KCl (Table S1‡). The higher surface content of C than that of N can be explained (i) by the formation of surface carbonate species in the parent material or the carbon atoms of the modifiers (Table S1‡), and (ii) by the fact that since the samples are prepared in air, there are definitely some adsorbed hydrocarbons on the sample, which will be visible in the surface sensitive XPS. As EDX probes a larger volume, this additional carbon signal is much smaller. Therefore, in XPS carbon seems to have a higher signal compared to N.
The elemental analysis using scanning transmission electron microscopy combined with energy-dispersive X-ray spectroscopy (TEM-EDX) confirms a homogeneous distribution of Si in the KPHI-Si sample as well as in the hydrophobized materials (See ESI, Fig. S9‡). Nevertheless, it also evidences some inhomogeneities in the distribution of K and Cl atoms within the hydrophobically modified PHIs corresponding to the formation of KCl salt during the silylation which was not completely removed by washing with organic solvents (see ESI, Fig. S9 and Table S1‡). These observations are consistent with the results of SEM-EDX elemental analysis (see ESI, Fig. S8 and Table S1‡). The comparison of high-resolution TEM images of KPHI and KPHI-Si shows that the silica is not only homogeneously distributed on the surface of the PHI particles, but is also present in the form of fibers wrapped around the PHI particles (See ESI, Fig. S10‡). Such fiber morphology is expected to be produced by the applied sol–gel technique that is known to favour the formation of rod-shaped silica particles.42
In line with the SEM results, the BET specific surface area of PHI is more than doubled after the modification of the parent material with silica (Table 1). This effect is mostly due to the deposition of high surface area silica particles over PHI. The increased specific surface area is observed for all modified samples with an exception of KPHI-OTS, where a longer alkyl chain of the grafted alkylsilane could cause the blockage of pores (Table 1).
3.2 Photocatalytic H2O2 production by hydrophilic and hydrophobized PHIs
The performance of our hydrophobic photocatalysts in light-driven H2O2 production in a biphasic system was tested using 1-octanol and 1-butanol as water-insoluble organic electron donors. The aim behind the choice of this substrate class is twofold. Firstly, we intentionally avoided the use of aromatic alcohols, such as benzyl alcohol, that are typically applied as model substrates for selective photocatalytic transformations31 since, in line with the autocatalytic oxidation mechanism under UV light reported recently,48,49 we found a conclusive and unambiguous evidence for benzaldehyde-mediated (auto)photocatalysis of benzyl alcohol oxidation and H2O2 production even under visible (>420 nm) light irradiation.35 In other words, the benzaldehyde present as an impurity or formed during the photocatalytic reaction can itself absorb light and further oxidize benzyl alcohol to benzaldehyde and simultaneously generate H2O2.35 Secondly, fatty alcohols, such as 1-octanol, are useful chemical compounds that can be readily obtained from biomass-derived chemicals, and the products of their partial oxidation, such as 1-octanal (caprylaldehyde), octanoic (caprylic) acid or octyl octanoate, represent a valuable feedstock for the synthesis of emulsifiers, pharmaceuticals and cosmetic products.50–56 Moreover, due to its non-activated nature, the oxidation of 1-octanol is very challenging even by the standards of conventional thermal catalysis,52,57,58 and has been seldom tackled by photocatalysis.59,60The photocatalytic performance was assessed in H2O2 production under violet LED illumination (406 nm). In order to establish the optimal photocatalyst loading, the preliminary experiments were carried out under monophasic conditions using 50% ethanol solution in water, later the established loadings were further confirmed also for the biphasic conditions. Expectedly, unmodified KPHI, among all other photocatalysts, required the lowest concentration (0.5 g L−1) for attaining its optimal performance (see ESI, Fig. S11a‡). Other materials, due to dilution of the photocatalytically active PHI phase by silica and grafted hydrophobic functions, need higher loadings for their optimal operation with values of 1.0 g L−1 for KPHI-Si and KPHI-HMDS and 1.5 g L−1 for KPHI-HTS and KPHI-OTS (ESI, Fig. S11‡). The parent KPHI material showed the highest reaction rate of H2O2 production using ethanol as an electron donor, while each subsequent stage of the photocatalyst modification, first with silica and then hydrophobization, led to a decreased activity (ESI, Fig. S11‡). This can be explained by a partial blockage of PHI with photocatalytically inactive species. The length of the grafted alkyl chains does not affect the performance of the photocatalyst to any significant extent. One may conclude that the diffusion of the reactants through the hydrophobic shell of the photocatalyst is not the limiting step of photooxidation reaction. However, in a biphasic (i.e., with two immiscible liquids) reaction system and using a more hydrophobic aliphatic alcohol as an electron donor, such as 1-butanol, KPHI shows the lowest activity of all the prepared photocatalysts while the higher concentrations of produced H2O2 are observed for KPHI-Si and KPHI-HMDS (see ESI, Fig. S12‡). 1-Butanol, although not completely miscible with water, still has a relatively high solubility in it (73 g L−1). In order to minimize the concentration of the electron donor in the aqueous phase and thus achieve efficient separation of oxidation and reduction products, 1-octanol was applied as a hydrophobic electron donor as its solubility in water is only 0.3 g L−1. When the pH value of the aqueous phase of the biphasic system was neutral, all the tested samples showed extremely low activity towards H2O2 production (Fig. 5). However, acidification upon the addition of HCl exerted a clearly positive effect on H2O2 formation and its stability in the reaction mixture with time (Fig. 5B–D, see also discussion below). Such pH effects are common in photocatalysis, in particular due to the pH dependence of surface (i.e., acidity/basicity of active sites) and redox (i.e., band edge positions) properties of most photocatalysts. It is noted, however, that protons are not consumed during the reaction since protons for H2O2 production are generated by the oxidation of 1-octanol. The hydrophilic KPHI and KPHI-Si samples were capable of producing 0.11 and 0.15 mmol of H2O2, respectively, within 24 h of irradiation when applying 0.5 mol L−1 HCl as an aqueous phase (Fig. 5C). The higher reaction rates demonstrated by the KPHI-Si with respect to that of KPHI might be attributed to a less hydrophilic character of the former rooting in incomplete hydrolysis and elimination of ethoxo-groups of TEOS. The impact of the acid concentration is much more significant in the case of hydrophobic materials that show a continuous enhancement of the photocatalytic H2O2 production with increased concentration of the acid in the aqueous layer of the biphasic system (Fig. 5). The highest amount (0.36 mmol) of H2O2 generated after 24 h irradiation was achieved by KPHI-OTS in a 1.0 mol L−1 HCl/1-octanol system (Fig. 5D). For ensuring that the observed H2O2 production is a photocatalytic reaction promoted by PHI, several control experiments were carried out (ESI, Fig. S13‡). No significant production of H2O2 was observed by irradiation of pure 1-octanol as well as the mixture of 1-octanol and 1-octanal. Conventional melon-type polymeric carbon nitride derived from dicyandiamide also showed only moderate activity in this reaction (see ESI, Fig. S13‡).
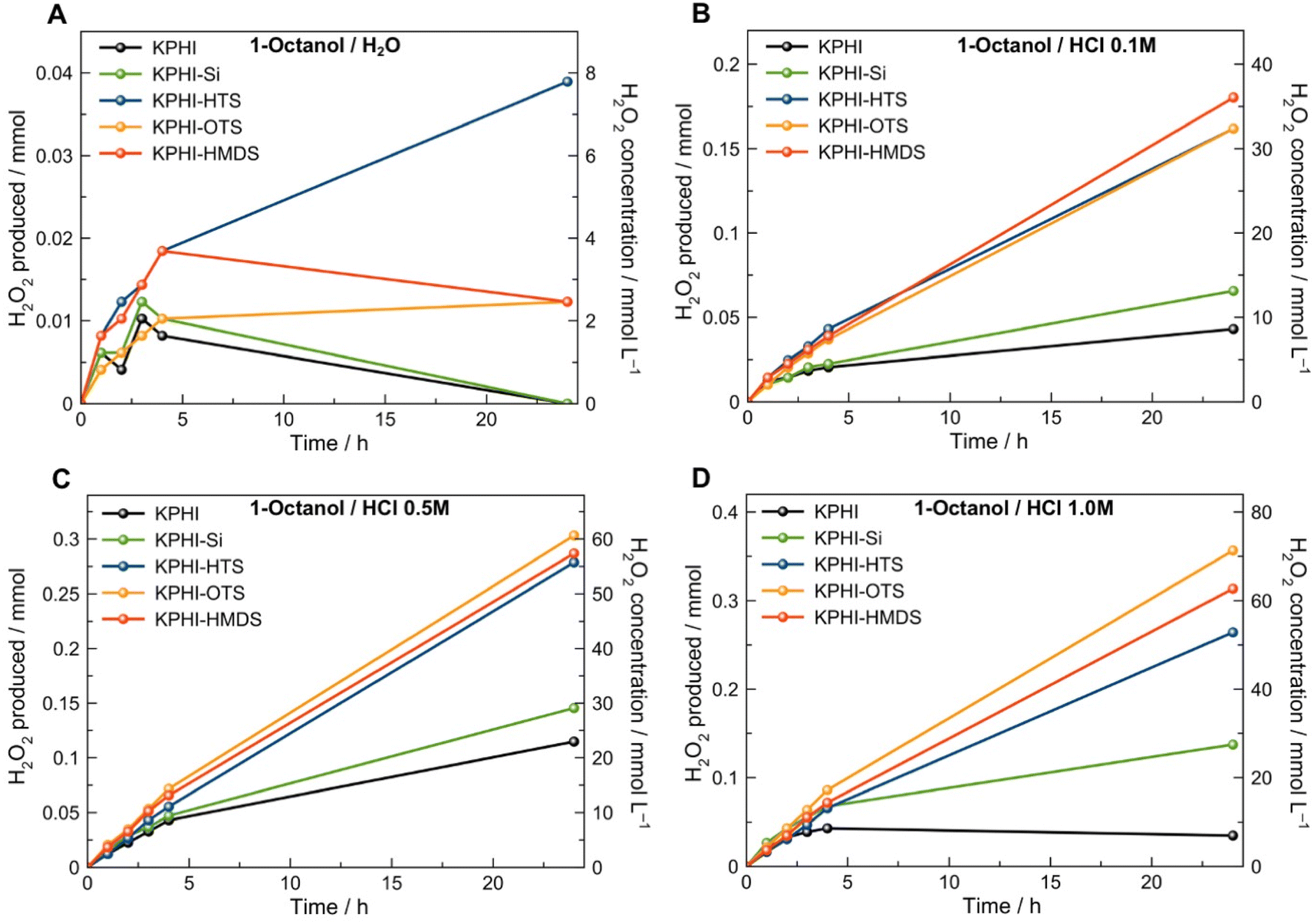 | ||
| Fig. 5 Photocatalytic tests. Photocatalytic production of H2O2 in a biphasic system comprising 1-octanol (top layer) as electron donor and an aqueous layer (bottom layer) having various HCl concentrations (A) no HCl, pH 6.5–7.0, (B) 0.1 mol L−1 HCl, (C) 0.5 mol L−1 HCl and (D) 1.0 mol L−1 HCl. Conditions: 5 mL 1-octanol, 5 mL H2O, LED 406 nm (4.2 mW cm−2), 22 °C, O2 1 atm. The indicated concentrations of HCl correspond to the 5 mL volume of the aqueous layer. The photocatalyst concentrations are 0.5 g L−1 for KPHI, 1.0 g L−1 for KPHI-Si and KPHI-HMDS and 1.5 g L−1 for KPHI-HTS and KPHI-OTS. The suspensions were saturated with pure O2 before irradiation and after each sampling. | ||
The oxidation of organic substrates by polymeric carbon nitride photocatalysts is usually a very fast process,61 while the reduction occurs on a much longer timescale and represents typically the rate-limiting step of the overall photocatalytic reaction.62,63 The obtained data show that the concentration of H3O+ ions in the aqueous phase of the biphasic system greatly affects the rate of photocatalytic H2O2 production. This is reminiscent of the electrochemical systems studied by the group of Girault64,65 and further developed by Opallo66 and Adamiak,67 where decamethyl ferrocene was used as a lipophilic electron donor in the organic layer of a biphasic mixture, however for H2O2 production it required the presence of a proton donor in the aqueous phase. As the surface activity of hydrated protons at the water/air interface is known to be very high,68 the strong dependence of the observed H2O2 production rate on HCl concentration is not surprising (Fig. 5). In other words, we speculate that the concentration of H3O+ in the aqueous phase determines the rate of the protonation of the PHI photocatalyst, which – apart from the photocatalytic function – might also act as a proton shuttle to the organic phase. Indeed, protonation/deprotonation of the hydrophobized PHI in a biphasic system takes place upon changing the pH of the aqueous phase and is accompanied by the change of the material's colour from yellow (deprotonated form) to white (protonated form) (ESI, Fig. S14‡). This reasoning is in line with the model for photocatalytic H2O2 formation at polymeric carbon nitrides proposed by Shiraishi et al.9 The model implies that the formation of the intermediate PHI surface-bound species in the form of endoperoxide and protonated heptazine nitrogen sites is a key aspect of selective H2O2 formation and its desorption from the photocatalyst surface (ESI, Fig. S15;‡ see also a simplified energy scheme in Fig. S29‡). It is also in accord with the results previously published by our group, where we found that the protonation of PHI plays a beneficial role in effective charge extraction from the photoexcited PHI.69
One of the crucial issues of photocatalytic H2O2 generation is the possibility of separating the H2O2/water mixture from the reaction medium, whereby the maximum concentration of H2O2 in the aqueous phase that can be achieved represents a key figure of merit.70 In this respect, the great advantage of the application of a biphasic system is the immediate separation of H2O2 into the water layer. In order to minimize the possibility of decomposition of the formed H2O2 at the suspended photocatalyst, it is imperative to ensure the stability of the grafted hydrophobic functions towards photogenerated charges and reactive oxygen species produced during irradiation. Long-term experiments at intermediate concentrations of HCl in the aqueous phase (0.5 mol L−1) have shown that the chloroalkylsilane-modified PHI materials reduce their activity under prolonged operation times and even the decomposition of previously produced H2O2 can take place (see ESI, Fig. S16‡). In contrast, HMDS-modified PHI remains active even after 72 h of irradiation, steadily forming hydrogen peroxide (ESI, Fig. S16‡). It is evident from the photographic images of the used photocatalysts that KPHI-HTS and KPHI-OTS partially lose their hydrophobic character after long illumination, while this is not the case for KPHI-HMDS that was found to be fully dispersible in the organic layer even after the total of 96 h of photocatalysis (ESI, Fig. S17‡). Nonetheless, at higher concentration of HCl in the aqueous phase, partial hydrophilization of KPHI-HMDS can take place after prolonged irradiation (24 h), as evident from slightly decreased H2O2 production rate (ESI, Fig. S18‡) as well as photographic images of the used material (See ESI, Fig. S19‡). One may conclude that KPHI-HMDS owes its superior photochemical stability to the absence of long alkyl groups that are present in KPHI-HTS or KPHI-OTS and can be susceptible to the reaction with photogenerated holes or reactive oxygen species.
As next we turn to a direct comparison of the photocatalytic H2O2 production performance of the hydrophilic and hydrophobic photocatalysts under biphasic conditions. During the first 4 h of irradiation all hydrophobic photocatalysts show only somewhat higher rates of H2O2 production compared to the hydrophilic ones (ESI, Fig. S20 and Table S2‡). However, this changes dramatically after 24 h of the reaction, when only a moderate increase of H2O2 concentration is observed for the systems containing the hydrophilic KPHI and KPHI-Si photocatalysts (Fig. 6). In this context it should be noted that the observed breakdown in H2O2 production rate using the hydrophilic photocatalysts is not due to the photocatalyst deactivation as the replacement of the aqueous phase with a fresh 0.5 mol L−1 HCl solution after 72 h of the reaction results in a complete regain of the initial activity. The observed behaviour of the hydrophilic photocatalysts in biphasic system is therefore suggestive of the equilibrium reached in the aqueous system between the formation of new H2O2via O2 reduction and the decomposition of the already formed H2O2, which clearly limits the maximum achievable H2O2 concentration in the aqueous phase. In contrast, the hydrophobic KPHI-HMDS material does not show any indication of such limitations for at least 96 h of total illumination (Fig. 6). Moreover, the H2O2 concentration in the separated aqueous phase achieved during photocatalysis with KPHI-HMDS after 72 h irradiation is ∼0.12 mol L−1. To the best of our knowledge, such a high value has never been reported for any other light-driven H2O2 production systems in which the obtained H2O2 would be already separated from electron donor as well as from the photocatalyst.15,71 This impressively demonstrates the advantage of performing the conversion using a hydrophobized photocatalyst in a biphasic system. It is noteworthy that after continuous 24 h irradiation of the KPHI-HMDS under the reaction conditions, it does not lose its hydrophobicity. The recovered material is clearly hydrophobic with a contact angle of 130 ± 4° (ESI, Fig. S21‡).
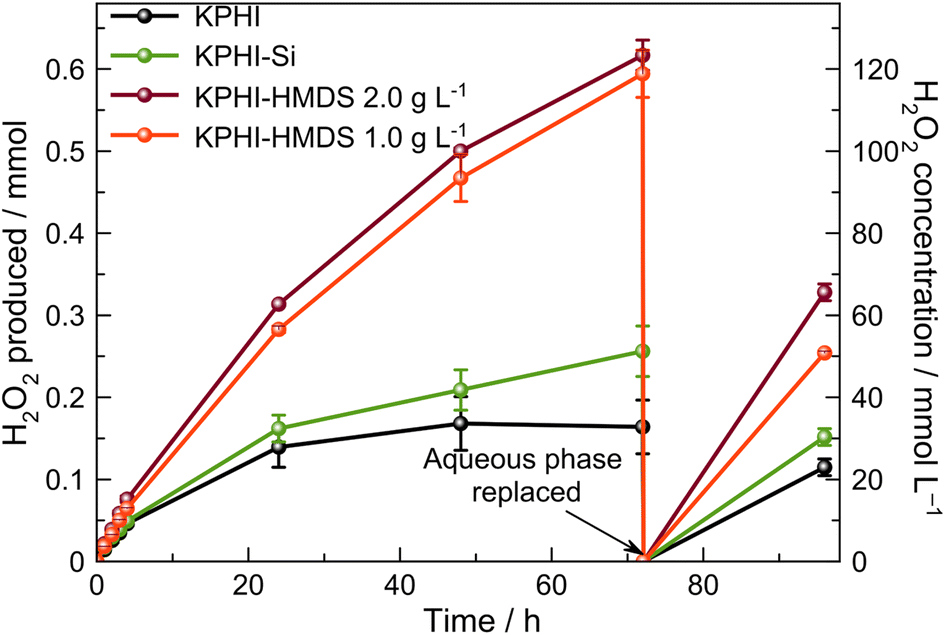 | ||
| Fig. 6 Photocatalytic tests. Photocatalytic production of H2O2 in biphasic system using 1-octanol as electron donor. Conditions: 5 mL 1-octanol, 5 mL 0.5 mol L−1 HCl, LED 406 nm (4.2 mW cm−2), 22 °C, O2 1 atm. The photocatalyst concentrations are 1.0 g L−1 and 2.0 g L−1. The suspensions were saturated with pure O2 before irradiation and after each sampling. | ||
In order to further corroborate our conclusions, we tested the tendency of the photocatalysts towards the decomposition of H2O2 under optimal biphasic conditions with H2O2 added to the reaction system from the start. The hydrophilic photocatalyst KPHI and KPHI-Si after 24 h of illumination cause the reduction of initially introduced H2O2 concentration from 91.8 mmol L−1 to 35.2 mmol L−1 and 47.6 mmol L−1, respectively (ESI, Fig. S22‡). These values are in the same range as those observed during the H2O2 evolution in biphasic system for the same photocatalysts in the interval from 24 h to 72 h of irradiation (Fig. 6). In contrast, when H2O2 is added at the start of the reaction using the hydrophobic KPHI-HMDS photocatalyst, it continues to produce H2O2 resulting in its higher concentration after 24 h of reaction compared to the initial one (ESI, Fig. S22‡). These results are in line with changes of the apparent quantum yield (AQY) of H2O2 formation (see ESI 2.2‡ for the details) in the course of the photocatalytic reaction under biphasic conditions (Fig. 7). One can see that the differences in the performance of the hydrophilic and hydrophobic photocatalysts become more pronounced under prolonged irradiation (for comparison with monophasic conditions see ESI, Fig. S23‡). The AQY of the best-performing hydrophilic photocatalyst drops drastically from 7.6% after 1 hour to 2.9% after 24 hours, i.e., by a factor of ∼2.5. In contrast, the champion hydrophobic PHI-based composite achieves AQY values of 9.4% and 5.7% after 1 h and 24 h, respectively, i.e., a drop by the factor of only ∼1.6, hence after 24 hours the AQY is nearly twice as high for the hydrophobized photocatalyst than for the hydrophilic one. All these results confirm the highly beneficial effect of using a hydrophobized photocatalyst in a biphasic system for light-driven H2O2 production. The comparison of the performance of our hydrophobic PHI photocatalysts with other materials places it among the best-performing photocatalysts for H2O2 production (ESI, Table S3‡). The fact that the produced H2O2 is already separated from the PHI and organic reaction products grants it an additional advantage over other photocatalytic systems.
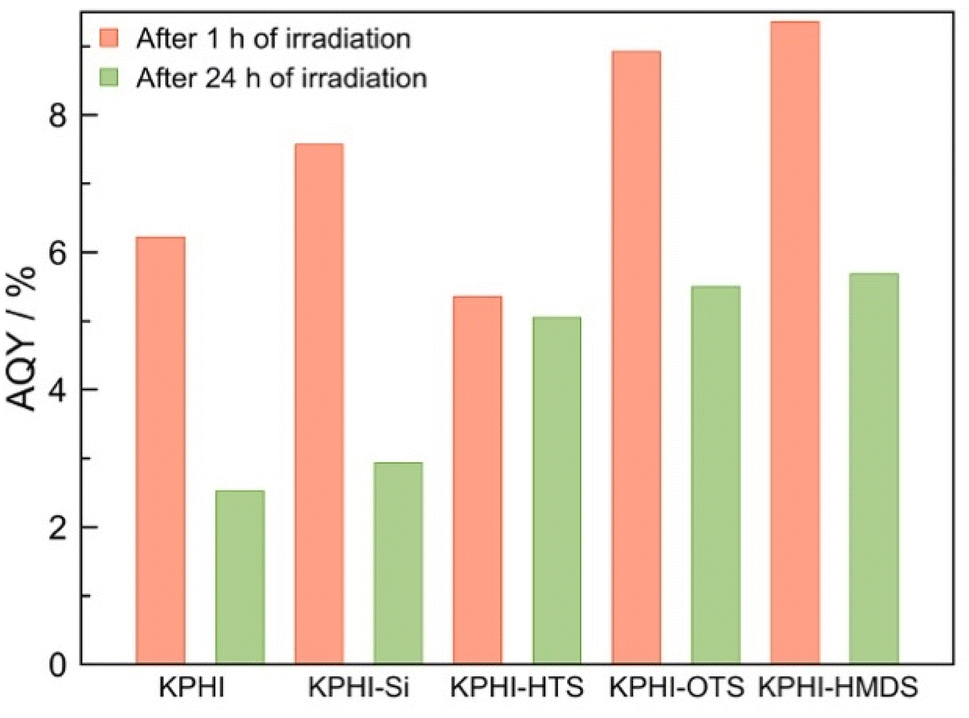 | ||
| Fig. 7 Apparent quantum yield. Apparent quantum yield of photocatalytic H2O2 production in a biphasic system using 1-octanol as electron donor. Conditions: 5 mL 1-octanol, 5 mL 0.5 mol L−1 HCl, LED 406 nm (4.2 mW cm−2), 22 °C, O2 1 atm. The photocatalyst concentrations are 0.5 g L−1 for KPHI, 1.0 g L−1 for KPHI-Si and KPHI-HMDS and 1.5 g L−1 for KPHI-HTS and KPHI-OTS. The suspensions were saturated with pure O2 before irradiation and after each sampling. | ||
In addition, we also confirmed that the optimal photocatalyst loadings established on the basis of the experiments carried out in monophasic conditions are applicable to the biphasic system as well. The increase of the KPHI-HMDS photocatalyst concentration in the biphasic system yields only moderate improvement of the H2O2 production (>0.12 mol L−1), while the doubling of the KPHI-Si loading has an opposite effect leading to worse performance (see ESI, Fig. S24‡).
Since 1-octanol represents a readily available electron donor with possibly valuable products of its oxidation, the organic phase after 96 h of irradiation of 1-octanol/0.5 mol L−1 HCl mixture in the presence of various photocatalysts was analyzed by gas chromatography with a flame ionization detector (GC-FID) (ESI, Fig. S25‡). 1-Octanal, octanoic (caprylic) acid and octyloctanoate, whose formation is promoted in the presence of water,54 were confirmed to be among the reaction products. We speculate that, similarly to the case of photooxidation of aromatic alcohols (see Krivtsov et al.35), the aldehyde is produced as the primary photocatalytic oxidation product, while octanoic acid is later formed from it by photooxidation or thermal oxidation via geminal diol formation, and finally octyl octanoate is produced by octanoic acid esterification with 1-octanol.54
The performance of the most efficient KPHI-HMDS photocatalyst was also studied under different light irradiation sources confirming its activity under simulated solar light irradiation using both AM 1.5G and 400 nm cut-off filter (ESI, Fig. S26‡).
Notably, the here developed hydrophobized photocatalysts are not only applicable for light-driven H2O2 generation, but can be also utilized for fatty alcohol reforming to molecular hydrogen using platinum as a co-catalyst (Fig. 8). Interestingly, the hydrophilic Pt–KPHI shows no activity toward H2 evolution if the pH of the aqueous solution is neutral and the hydrogen production for this material was only detected under acidic conditions. This is not the case for the hydrophobic Pt–KPHI-HMDS sample that is capable of photocatalytic H2 evolution both under neutral and acidic conditions in the aqueous phase (Fig. 8). These results suggest that the hydrophobic nature of KPHI-HMDS is highly beneficial for effective proton management that is expected to play an important role in light-driven H2 evolution in a biphasic system.
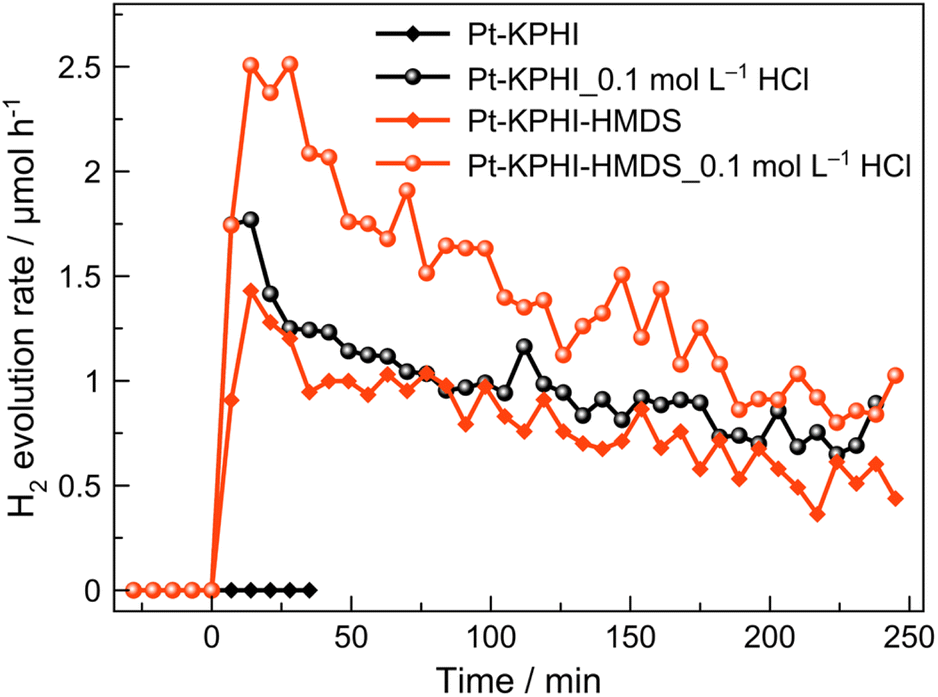 | ||
| Fig. 8 Photocatalytic H2 evolution. Photocatalytic hydrogen evolution from biphasic 1-octanol/water mixture performed by the PHI photocatalysts platinized with 3 wt% of Pt. Conditions: 5 mL 1-octanol, 5 mL H2O or 0.1 M HCl, LED 406 nm (7.1 mW cm−2), 22 °C, He flow. The photocatalyst concentrations are 0.5 g L−1 for KPHI and 1.0 g L−1 for KPHI-HMDS. | ||
4. Conclusions
In this work, we provide the first unambiguous demonstration of the advantages of carrying out photocatalytic H2O2 production in a biphasic system, yielding an aqueous H2O2 solution already separated from the photocatalyst as well as from the electron donor with unprecedentedly high H2O2 concentrations (∼0.12 mol L−1) as compared to other photocatalytic systems. This achievement was enabled by designing a hydrophobic version of poly(heptazine imide) (PHI)-based ionic polymeric carbon nitride in the form of a composite with alkylated silica. The hydrophobized PHI photocatalyst can be selectively suspended in the organic layer of a biphasic system comprising a fatty alcohol and water as two immiscible liquids. Under visible-light irradiation, the hydrophobized PHI-based photocatalyst oxidizes fatty alcohol as electron donor, while at the same time oxygen is reduced to H2O2 that is instantaneously extracted into the aqueous phase. The continuous separation of the produced H2O2 effectively prevents its decomposition at the photocatalyst surface, enabling thus achieving concentrations of H2O2 in the aqueous phase that are much higher than for conventional light-driven H2O2 production systems. We further demonstrate that effective proton management facilitates the H2O2 production in a biphasic system as evidenced by the strong effect of the proton concentration in the aqueous layer of the biphasic system on the H2O2 production rate, which is ascribed to more effective protonation of the hydrophobized PHI that is one of the key steps towards the completion of the two-electron oxygen reduction to H2O2. The long-term photocatalytic experiments confirmed the stability of the hydrophobized photocatalysts for at least up to 96 h without any loss of hydrophobicity and only a very minor decrease in activity. Finally, it should be noted that fatty alcohols are readily available from vegetable waxes and as pulping sub-products, and the products of their partial oxidation represent a valuable feedstock for the synthesis of pharmaceuticals and cosmetic products. Our work thus not only showcases a rational design of a robust photocatalyst for H2O2 production under biphasic conditions that enables easy separation of the product from electron donors and its recovery at very high concentrations, but also paves the way for sustainable and economically viable light-driven H2O2 production from easily available feedstock.Data availability
All datasets related to this work are available from the repository: DOI: doi:10.5281/zenodo.7401099.Author contributions
I. K., R. B. and D. M. conceived and supervised the project, I. K. and A. V. synthesised the materials, characterized them and performed photocatalytic experiments. M. M. E. performed SEM-EDX study and analysed the data. C. N. and A. T. performed XPS investigation of the materials and analysed the results. Y. P. and S. O. performed chromatographic studies of the reaction products and intermediates. R. L. and U. K. performed TEM-EDX analyses and processed the data. M. L. performed and analysed data of nitrogen physisorption at 77 K. I. K. and R. B. wrote the manuscript with inputs from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Projektnummer 364549901 – TRR 234 [Projects B6, B7, C2, C4 and Z2] and BE 5102/5-1. C. N. and A. T. acknowledge financial support of the DFG through the project TU 149/8-2 “Towards photo-active membranes for artificial photosynthesis” as well as the DFG through a research infrastructure grant INST 275/257-1 FUGG. I. K. acknowledges support by Spanish MCINN (PID2020-113558RB-C41) and Gobierno del Principado de Asturias (IDI-2021-000048) and the Alexander von Humboldt Foundation through the Humboldt Research Fellowship.References
- T. J. Jacobsson, Energy Environ. Sci., 2018, 11, 1977–1979 RSC.
- E. Reisner, Angew. Chem., Int. Ed., 2019, 58, 3656–3657 CrossRef CAS PubMed.
- R. Beranek, Angew. Chem., Int. Ed., 2019, 58, 16724–16729 CrossRef CAS PubMed.
- Y. Shiraishi, S. Kanazawa, Y. Kofuji, H. Sakamoto, S. Ichikawa, S. Tanaka and T. Hirai, Angew. Chem., Int. Ed., 2014, 53, 13454–13459 CrossRef CAS PubMed.
- Y. Kofuji, S. Ohkita, Y. Shiraishi, H. Sakamoto, S. Ichikawa, S. Tanaka and T. Hirai, ACS Sustainable Chem. Eng., 2017, 5, 6478–6485 CrossRef CAS.
- J. Z. Zhang, J. Y. Lang, Y. Wei, Q. Zheng, L. Y. Liu, Y. H. Hu, B. X. Zhou, C. L. Yuan and M. C. Long, Appl. Catal., B, 2021, 298, 120522 CrossRef CAS.
- Y. Shiraishi, T. Takii, T. Hagi, S. Mori, Y. Kofuji, Y. Kitagawa, S. Tanaka, S. Ichikawa and T. Hirai, Nat. Mater., 2019, 18, 985–993 CrossRef CAS PubMed.
- X. Chen, Y. Kondo, S. Li, Y. Kuwahara, K. Mori, D. Zhang, C. Louis and H. Yamashita, J. Mater. Chem. A, 2021, 9, 26371–26380 RSC.
- Y. Shiraishi, S. Kanazawa, Y. Sugano, D. Tsukamoto, H. Sakamoto, S. Ichikawa and T. Hirai, ACS Catal., 2014, 4, 774–780 CrossRef CAS.
- P. Zhang, Y. W. Tong, Y. Liu, J. J. M. Vequizo, H. W. Sun, C. Yang, A. Yamakata, F. T. Fan, W. Lin, X. C. Wang and W. Y. Choi, Angew. Chem., Int. Ed., 2020, 59, 16209–16217 CrossRef CAS PubMed.
- M. Ilkaeva, I. Krivtsov, J. Garcia, E. Diaz, S. Ordonez, E. Garcia-Lopez, G. Marci, L. Palmisano, M. Maldonado and S. Malato, Catal. Today, 2018, 315, 138–148 CrossRef CAS.
- I. Krivtsov, D. Mitoraj, C. Adler, M. Ilkaeva, M. Sardo, L. Mafra, C. Neumann, A. Turchanin, C. Y. Li, B. Dietzek, R. Leiter, J. Biskupek, U. Kaiser, C. Im, B. Kirchhoff, T. Jacob and R. Beranek, Angew. Chem., Int. Ed., 2020, 59, 487–495 CrossRef CAS PubMed.
- Y. B. Zhao, P. Zhang, Z. C. Yang, L. N. Li, J. Y. Gao, S. Chen, T. F. Xie, C. Z. Diao, S. B. Xi, B. B. Xiao, C. Hu and W. Y. Choi, Nat. Commun., 2021, 12, 3701 CrossRef CAS PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- H. L. Hou, X. K. Zeng and X. W. Zhang, Angew. Chem., Int. Ed., 2020, 59, 17356–17376 CrossRef CAS PubMed.
- A. Savateev and M. Antonietti, ChemCatChem, 2019, 11, 6166–6176 CrossRef CAS.
- C. Adler, D. Mitoraj, I. Krivtsov, L. Dos Santos-Gomez, S. Garcia-Granda, C. Neumann, J. Kund, C. Kranz, B. Mizaikoff, A. Turchanin and R. Beranek, ChemSusChem, 2021, 14, 2170–2179 CrossRef CAS PubMed.
- V. W.-h. Lau and B. V. Lotsch, Adv. Energy Mater., 2022, 12, 2101078 CrossRef CAS.
- F. K. Kessler, Y. Zheng, D. Schwarz, C. Merschjann, W. Schnick, X. Wang and M. J. Bojdys, Nat. Rev. Mater., 2017, 2, 17030 CrossRef CAS.
- D. Zheng, J. Zhou, Z. Fang, T. Heil, A. Savateev, Y. Zhang, M. Antonietti, G. Zhang and X. Wang, J. Mater. Chem. A, 2021, 9, 27370–27379 RSC.
- V. W. Lau, I. Moudrakovski, T. Botari, S. Weinberger, M. B. Mesch, V. Duppel, J. Senker, V. Blum and B. V. Lotsch, Nat. Commun., 2016, 7, 12165 CrossRef CAS PubMed.
- Y. Markushyna, P. Lamagni, C. Teutloff, J. Catalano, N. Lock, G. G. Zhang, M. Antonietti and A. Savateev, J. Mater. Chem. A, 2019, 7, 24771–24775 RSC.
- P.-S. Wu, T.-J. Lin, S.-S. Hou, C.-C. Chen, D.-L. Tsai, K.-H. Huang and J.-J. Wu, J. Mater. Chem. A, 2022, 10, 7728–7738 RSC.
- C. Li, E. Hofmeister, I. Krivtsov, D. Mitoraj, C. Adler, R. Beranek and B. Dietzek, ChemSusChem, 2021, 14, 1728–1736 CrossRef CAS PubMed.
- P. Sharma, T. J. A. Slater, M. Sharma, M. Bowker and C. R. A. Catlow, Chem. Mater., 2022, 34, 5511–5521 CrossRef CAS PubMed.
- S. Mazzanti, B. Kurpil, B. Pieber, M. Antonietti and A. Savateev, Nat. Commun., 2020, 11, 1387 CrossRef CAS PubMed.
- S. D. Bishopp, J. L. Scott and L. Torrente-Murciano, Green Chem., 2014, 16, 3281–3285 RSC.
- Y. Roman-Leshkov, J. N. Chheda and J. A. Dumesic, Science, 2006, 312, 1933–1937 CrossRef CAS PubMed.
- H. B. Vibbert, C. Bendel, J. R. Norton and A. J. Moment, ACS Sustainable Chem. Eng., 2022, 10, 11106–11116 CrossRef CAS.
- Y. Kawase, Y. Isaka, Y. Kuwahara, K. Mori and H. Yamashita, Chem. Commun., 2019, 55, 6743–6746 RSC.
- Y. Isaka, Y. Kawase, Y. Kuwahara, K. Mori and H. Yamashita, Angew. Chem., Int. Ed., 2019, 58, 5402–5406 CrossRef CAS PubMed.
- X. L. Chen, Y. Kuwahara, K. Mori, C. Louis and H. Yamashita, J. Mater. Chem. A, 2020, 8, 1904–1910 RSC.
- T. G. V. Erazo, Surface Modification of TIO2 for Photocatalytic Selective Oxidation of Organic Compounds in Biphasic and Single Phase Systems, Master thesis, University of Oklahoma, 2016, https://shareok.org/handle/11244/47041.
- X. Gong, S. Yu, M. Guan, X. Zhu and C. Xue, J. Mater. Chem. A, 2019, 7, 7373–7379 RSC.
- I. Krivtsov, A. Vazirani, D. Mitoraj and R. Beranek, ChemCatChem, 2022 DOI:10.1002/cctc.202201215.
- B. Kumru, M. Antonietti and B. V. K. J. Schmidt, Langmuir, 2017, 33, 9897–9906 CrossRef CAS PubMed.
- N. Yandrapalli, T. Robinson, M. Antonietti and B. Kumru, Small, 2020, 16, 2001180 CrossRef CAS PubMed.
- B. Kumru, D. Cruz, T. Heil, B. V. K. J. Schmidt and M. Antonietti, J. Am. Chem. Soc., 2018, 140, 17532–17537 CrossRef CAS PubMed.
- C. Yao, A. Yuan, Z. Wang, H. Lei, L. Zhang, L. Guo and X. Dong, J. Mater. Chem. A, 2019, 7, 13071–13079 RSC.
- J. Kroger, A. Jimenez-Solano, G. Savasci, V. W. H. Lau, V. Duppel, I. Moudrakovski, K. Kuster, T. Scholz, A. Gouder, M. L. Schreiber, F. Podjaski, C. Ochsenfeld and B. V. Lotsch, Adv. Funct. Mater., 2021, 31, 2102468 CrossRef.
- H. Schlomberg, J. Kroger, G. Savasci, M. W. Terban, S. Bette, I. Moudrakovski, V. Duppel, F. Podjaski, R. Siegel, J. Senker, R. E. Dinnebier, C. Ochsenfeld and B. V. Lotsch, Chem. Mater., 2019, 31, 7478–7486 CrossRef CAS PubMed.
- S. Sakka, in Better Ceramics through Chemistry, eds. C. J. Brinker, D. E. Clark and D. R. Ulrich, North-Holland, New Yourk, 1984, p. 91 Search PubMed.
- P. A. Zapata, Y. Huang, M. A. Gonzalez-Borja and D. E. Resasco, J. Catal., 2013, 308, 82–97 CrossRef CAS.
- P. Audebert, E. Kroke, C. Posern and S. H. Lee, Chem. Rev., 2021, 121, 2515–2544 CrossRef CAS PubMed.
- A. Bertoluzza, C. Fagnano, M. A. Morelli, V. Gottardi and M. Guglielmi, J. Non-Cryst. Solids, 1982, 48, 117–128 CrossRef CAS.
- S. D. Bhagat, Y. H. Kim, M. J. Moon, Y. S. Ahn and J. G. Yeo, Solid State Sci., 2007, 9, 628–635 CrossRef CAS.
- J. Kroger, A. Jimenez-Solano, G. Savasci, P. Rovo, I. Moudrakovski, K. Kuster, H. Schlomberg, H. A. Vignolo-Gonzalez, V. Duppel, L. Grunenberg, C. B. Dayan, M. Sitti, F. Podjaski, C. Ochsenfeld and B. V. Lotsch, Adv. Energy Mater., 2021, 11, 2003049 CrossRef.
- M. J. Pavan, H. Fridman, G. Segalovich, A. I. Shames, N. G. Lemcoff and T. Mokari, ChemCatChem, 2018, 10, 2541–2545 CrossRef CAS.
- R. Arcas, E. Peris, E. Mas-Marza and F. Fabregat-Santiago, Sustainable Energy Fuels, 2021, 5, 956–962 RSC.
- A. Johansson, Biomass, 1982, 2, 103–113 CrossRef CAS.
- G. Marques, J. C. del Rio and A. Gutierrez, Bioresour. Technol., 2010, 101, 260–267 CrossRef CAS PubMed.
- V. C. Corberan, M. E. Gonzalez-Perez, S. Martinez-Gonzalez and A. Gomez-Aviles, Appl. Catal., A, 2014, 474, 211–223 CrossRef CAS.
- C. Kohlpaintner, M. Schulte, J. Falbe, P. Lappe, J. Weber and G. D. Frey, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Hoboken, 2013, DOI:10.1002/14356007.a01_321.pub3.
- T. Ishida, Y. Ogihara, H. Ohashi, T. Akita, T. Honma, H. Oji and M. Haruta, ChemSusChem, 2012, 5, 2243–2248 CrossRef CAS PubMed.
- A. K. C. Schmidt and C. B. W. Stark, Org. Lett., 2011, 13, 4164–4167 CrossRef CAS PubMed.
- A. Cognigni, S. Kampichler and K. Bica, J. Colloid Interface Sci., 2017, 492, 136–145 CrossRef CAS PubMed.
- E. Pakrieva, E. Kolobova, G. Mamontov, N. Bogdanchikova, M. H. Farias, L. Pascual, V. C. Corberan, S. M. Gonzalez, S. A. C. Carabineiro and A. Pestryakov, ChemCatChem, 2019, 11, 1615–1624 CrossRef CAS.
- Y. Kotolevich, E. Kolobova, G. Mamontov, E. Khramov, J. E. C. Ortega, H. Tiznado, M. H. Farias, N. Bogdanchikova, Y. Zubavichus, J. D. Mota-Morales, V. C. Corberan, R. Zanella and A. Pestryakov, Catal. Today, 2016, 278, 104–112 CrossRef CAS.
- R. Vadakkekara, A. K. Biswas, T. Sahoo, P. Pal, B. Ganguly, S. C. Ghosh and A. B. Panda, Chem.–Asian J., 2016, 11, 3084–3089 CrossRef PubMed.
- H. Yamashita, S. Kawasaki, Y. Ichihashi, M. Harada, M. Takeuchi, M. Anpo, G. Stewart, M. A. Fox, C. Louis and M. Che, J. Phys. Chem. B, 1998, 102, 5870–5875 CrossRef CAS.
- Z. W. Chen, Q. Zhang and Y. Luo, Angew. Chem., Int. Ed., 2018, 57, 5320–5324 CrossRef CAS PubMed.
- W. Yang, R. Godin, H. Kasap, B. Moss, Y. Dong, S. A. J. Hillman, L. Steier, E. Reisner and J. R. Durrant, J. Am. Chem. Soc., 2019, 141, 11219–11229 CrossRef CAS PubMed.
- H. Kasap, C. A. Caputo, B. C. M. Martindale, R. Godin, V. W. H. Lau, B. V. Lotsch, J. R. Durrant and E. Reisner, J. Am. Chem. Soc., 2016, 138, 9183–9192 CrossRef CAS PubMed.
- B. Su, R. P. Nia, F. Li, M. Hojeij, M. Prudent, C. Corminboeuf, Z. Samec and H. H. Girault, Angew. Chem., Int. Ed., 2008, 47, 4675–4678 CrossRef CAS PubMed.
- B. Su, I. Hatay, P. Y. Ge, M. Mendez, C. Corminboeuf, Z. Samec, M. Ersoz and H. H. Girault, Chem. Commun., 2010, 46, 2918–2919 RSC.
- J. Jedraszko, W. Nogala, W. Adamiak, E. Rozniecka, I. Lubarska-Radziejewska, H. H. Girault and M. Opallo, J. Phys. Chem. C, 2013, 117, 20681–20688 CrossRef CAS.
- W. Adamiak, J. Jedraszko, W. Nogala, M. Jonsson-Niedziolka, S. Dongmo, G. Wittstock, H. H. Girault and M. Opallo, J. Phys. Chem. C, 2015, 119, 20011–20015 CrossRef CAS.
- S. Das, M. Bonn and E. H. G. Backus, Angew. Chem., Int. Ed., 2019, 58, 15636–15639 CrossRef CAS PubMed.
- C. Adler, S. Selim, I. Krivtsov, C. Y. Li, D. Mitoraj, B. Dietzek, J. R. Durrant and R. Beranek, Adv. Funct. Mater., 2021, 31, 2105369 CrossRef CAS.
- X. J. Shi, Y. R. Zhang, S. Siahrostami and X. L. Zheng, Adv. Energy Mater., 2018, 8, 1801158 CrossRef.
- Z. Xu, Y. Li, Y. Cao, R. Du, Z. Bao, S. Zhang, F. Shao, W. Ji, J. Yang, G. Zhuang, S. Deng, Z. Wei, Z. Yao, X. Zhong and J. Wang, J. Energy Chem., 2022, 64, 47–54 CrossRef CAS.
Footnotes |
| † This article is dedicated to the memory of Dr Jaromír Jirkovský (1952–2022), a dear friend and expert in photocatalysis. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta08045a |
| This journal is © The Royal Society of Chemistry 2023 |