
A robust solvothermal-driven solid-to-solid transition route from micron SnC2O4 to tartaric acid-capped nano-SnO2 anchored on graphene for superior lithium and sodium storage†
Furong
Xie
a,
Shiqiang
Zhao
 *a,
Xiaoxu
Bo
*b,
Guanghui
Li
a,
Jiamin
Fei
a,
Ebrahim-Alkhalil M. A.
Ahmed
a,
Qingcheng
Zhang
a,
Huile
Jin
a,
Shun
Wang
*a and
Zhiqun
Lin
*c
*a,
Xiaoxu
Bo
*b,
Guanghui
Li
a,
Jiamin
Fei
a,
Ebrahim-Alkhalil M. A.
Ahmed
a,
Qingcheng
Zhang
a,
Huile
Jin
a,
Shun
Wang
*a and
Zhiqun
Lin
*c
aZhejiang Province Key Lab of Leather Engineering, Wenzhou University, Wenzhou 325035, PR China. E-mail: zhaosq@wzu.edu.cn; shunwang@wzu.edu.cn
bKey Laboratory of Crop Breeding in South Zhejiang, Department of Agriculture and Biotechnology, Wenzhou Vocational College of Science and Technology, Wenzhou 325006, PR China. E-mail: boxxsci@163.com
cDepartment of Chemical and Biomolecular Engineering, National University of Singapore, Singapore 117585, Singapore. E-mail: z.lin@nus.edu.sg
First published on 21st November 2022
Abstract
Tin dioxide (SnO2) has been widely implemented as an advanced anode material for lithium or sodium ion batteries (LIBs/SIBs) owing to its high capacity and moderate potential. However, conventional synthetic approaches often yield large-sized SnO2, which suffers from low conductivity, huge volume expansion and Sn coarsening issues, hampering its practical implementation. Herein, a unique solvothermal-driven solid-to-solid transition (SDSST) strategy is developed to craft tartaric acid (TA) capped ultrafine SnO2 nanoparticles (NPs) in situ on sacrificial SnC2O4 microrods. Ball-milling combined with solvent evaporation treatment realizes the homogeneous composition and precise mass ratio control of TA-capped SnO2 NPs and reduced graphene oxide (rGO). Remarkably, the SnO2 NPs-rGO nanocomposite manifests outstanding lithium and sodium storage capacities of 1775 and 463 mA h g−1 after 800 and 100 cycles at 1000 and 20 mA g−1, respectively, and an ultralong lifespan of 4000 cycles for LIBs. Notably, systematic electrochemical and componential characterization of the cycled electrodes reveals that SnO2 NPs-rGO manifests fully reversible three-step lithiation–delithiation reactions of SnO2 and a primary highly reversible sodiation–desodiation conversion reaction between Sn and SnO combined with a secondary partially reversible alloying–dealloying reaction between Sn and NaxSn (0 ≤ x ≤ 3.75) for lithium and sodium storage, respectively. The even encapsulation of TA-capped SnO2 NPs in the rGO matrix enables effectively suppressed volume expansion for outstanding structural stability, significantly accelerated ion/electron transport for superior reaction kinetics, greatly prohibited Sn coarsening for enhanced cycle reversibility, and dramatically increased capacitive capacity for additional energy storage. As such, the SDSST approach may represent a facile yet robust strategy for crafting a variety of nanomaterials of interest with appropriate metastable solids as the precursor under the assistance of efficient capping agents.
1. Introduction
Lithium-ion batteries (LIBs) have occupied a dominant role in a broad field of applications.1–4 However, the low theoretical capacity (TC) of the graphite anode (i.e., 372 mA h g−1) limits further improvement in the energy density.5–8 Furthermore, as a promising alternative to LIBs, sodium ion batteries (SIBs) are broadly studied in view of their similar working mechanism to LIBs, at a much lower price than LIBs due to the abundant resource of sodium in comparison to lithium.9–11 However, the larger diameter of Na+ (2.04 Å) than that of Li+ (1.52 Å) leads to the sluggish reaction kinetics of Na+ with an extremely low insertion capability in graphite.12–23 Overall, the development of high-performance and commercializable anodes has become a top priority in the development of LIBs and SIBs.16,24,25Tin dioxide (SnO2) is considered as one of the most encouraging anodes for LIBs and SIBs due to its ultrahigh mass and volume specific TCs (i.e., 1494 mA h g−1 and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 383 mA h cm−3 for LIBs and 1378 mA h g−1 and 9577 mA h cm−3 for SIBs).20,26–32 Moreover, SnO2 has a moderate lithiation and sodiation potential, affording high safety by preventing the growth of lithium and sodium dendrites.27,33 Nevertheless, conventionally synthesized SnO2 microparticles (MPs) usually exhibit poor properties, which could be principally attributed to two major issues.34–39 Firstly, the pulverization of SnO2 MPs during repeated Li+ or Na+ insertion/extraction processes would cause their stripping off from the current collector. Secondly, the continuous Sn coarsening inside the cycled SnO2 MPs would produce large-sized Sn particles with poor reactivity and reversibility. To alleviate these problems, enormous efforts have been dedicated to clarifying the in-depth deteriorative mechanisms and developing effective optimization measures. Among all the efforts, the cycling performance of SnO2 has been significantly enhanced by grafting nanoparticles (NPs) in various carbon matrices.36,40,41 Carbon matrices possess multifunctional characteristics including but not limited to enhancing the conductivity, buffering the volume changes, suppressing the coarsening of Sn particles, and stabilizing the electrode interface by inhibiting the direct contact that caused side reactions between SnO2 and the electrolyte to form a robust solid electrolyte interface layer (SEI).27,42–45
383 mA h cm−3 for LIBs and 1378 mA h g−1 and 9577 mA h cm−3 for SIBs).20,26–32 Moreover, SnO2 has a moderate lithiation and sodiation potential, affording high safety by preventing the growth of lithium and sodium dendrites.27,33 Nevertheless, conventionally synthesized SnO2 microparticles (MPs) usually exhibit poor properties, which could be principally attributed to two major issues.34–39 Firstly, the pulverization of SnO2 MPs during repeated Li+ or Na+ insertion/extraction processes would cause their stripping off from the current collector. Secondly, the continuous Sn coarsening inside the cycled SnO2 MPs would produce large-sized Sn particles with poor reactivity and reversibility. To alleviate these problems, enormous efforts have been dedicated to clarifying the in-depth deteriorative mechanisms and developing effective optimization measures. Among all the efforts, the cycling performance of SnO2 has been significantly enhanced by grafting nanoparticles (NPs) in various carbon matrices.36,40,41 Carbon matrices possess multifunctional characteristics including but not limited to enhancing the conductivity, buffering the volume changes, suppressing the coarsening of Sn particles, and stabilizing the electrode interface by inhibiting the direct contact that caused side reactions between SnO2 and the electrolyte to form a robust solid electrolyte interface layer (SEI).27,42–45
Reduced graphene oxide (rGO) is popularly considered as one of the most effective carbon substrates in view of its unique two dimensional textures, large specific surface area, robust mechanical flexibility, outstanding chemical stability and excellent electrical conductivity.46–48 The general approach of preparing SnO2/rGO nanocomposites is in situ addition of graphene oxide (GO) in the generation solution of SnO2, followed by hydrothermal or solvothermal reactions.38,49 However, GO can be uniformly dispersed in a few solvents (e.g., water and dimethyl formamide), which limits the extension of the in situ composition methods. More importantly, in the in situ composition route, the heat treatment or the ions in the reaction solution might cause the reduction and aggregation of GO nanosheets into rGO matrices prior to the generation of SnO2. As a result, the in situ composition route cannot universally realize the homogeneous combination of SnO2 with rGO and exactly control the mass ratio between SnO2 and rGO, which have critical effects on the battery performances.50,51 Therefore, it is particularly critical to explore robust strategies for the facile preparation of SnO2 nanoparticles (NPs) and their controllable composition with rGO.52
In this work, a solvothermal-driven solid-to-solid transition (SDSST) route was explored for the first time to synthesize ultrafine SnO2 NPs with an average particle size of 13 ± 10 nm with tartaric acid (TA) as a vital capping agent. Notably, the sluggish and progressive SDSST process slows down the formation rate of SnO2 NPs in situ on SnC2O4 microrods (MRs), and synergistically the TA capping agent strongly stabilizes the newly generated SnO2 NPs. In stark contrast, SnO2 submicron particles (SMPs) and SnO2 microparticles (MPs) were obtained without adding TA and TA/oxalic acid, respectively. Meanwhile, an ex situ composition method is adopted to homogeneously combine the obtained TA-capped SnO2 NPs with rGO by microthermal solvent evaporation of their ball-milling blended aqueous slurry with an artificially determined mass ratio of 4:1. The TA-capped SnO2 NPs exhibit excellent dispersibility in water, ensuring their homogeneous anchoring on rGO nanosheets and layer-by-layer assembling into a SnO2 NPs-rGO nanocomposite after ball-milling combined with solvent evaporation treatment. Contrastively, an inhomogeneous SnO2/rGO composite was obtained by in situ addition of GO in the generation solution of SnO2 NPs, which might be attributed to the poor dispersibility of GO in ethylene glycol and the solvothermal treatment induced agglomeration of GO nanosheets into rGO matrices before the generation of SnO2 NPs. The proposed SDSST route is analogous to the fact that living organisms often employ solid but poorly ordered mineral phases as precursors in the biomineralization of their functionalized body parts, which would be one of the most effective strategies to craft nanomaterials of interest by easily integrating ingredients with specific functions into the composites at the molecular level.53,54
SnO2 NPs-rGO delivered an ultrahigh lithium storage capacity of 1461 mA h g−1 after 300 cycles at 100 mA g−1, close to the TC of 1494 mA h g−1. Encouragingly, SnO2 NPs-rGO exhibited an ultrastable sodium storage capacity of 443 mA h g−1 at 50 mA g−1 in the 100th cycle. The uniform encapsulation of TA-capped SnO2 NPs in the rGO matrix effectively suppresses the volume expansion for a stable structure, improves the electrical conductivity for high reactivity, and prohibits the agglomeration of SnO2 NPs for excellent reversibility. The revealed electrochemical performance enhancement mechanisms of SnO2 NPs-rGO will shed some light on the rational structural and componential designs of advanced electrodes with outstanding reversibility and superior diffusive combined with capacitive energy storage capability.
2. Results and discussion
2.1 Solvothermal-driven solid-to-solid transition (SDSST) from SnC2O4 MRs to SnO2 NPs
The obtained SnO2 NPs (Fig. 1a, b and S1a–c†) exhibit an irregular spherical morphology and display distinct lattice fringes of the (110) facet of tetragonal SnO2 crystals. Using similar approaches, control samples of SnO2 SMPs (Fig. S2a–c†) and SnO2 MPs (Fig. S2g–i†) were prepared in the absence of TA and TA/OA, respectively, suggesting a crucial synergistic effect of SDSST and TA in the crafting of SnO2 NPs. To visually demonstrate the superiority of the proposed SDSST route in the size controlled synthesis of TA-capped SnO2 NPs in contrast to the conventional solvothermal route, representative control samples of SnO2 microspheres (MSs) and SnO2-TA MSs were synthesized by the solvothermal reaction of KSnO3·3H2O and urea. The as-prepared SnO2 MSs (Fig. S3a–c†) and SnO2-TA MSs (Fig. S3d–f†) are severely aggregated with large average particle sizes of 1.5 ± 0.9 and 1.2 ± 0.8 μm, respectively. Clearly, the traditional solvothermal route from Sn salt might probably tend to generate large-sized and aggregated SnO2 particles due to the rapid formation rate from Sn ions to SnO2 and the spontaneous agglomeration driven by the high specific surface energy of small-sized particles. | ||
| Fig. 1 TEM and SEM images of (a and b) SnO2 NPs and (c–h) SnO2 NPs-rGO. (i) XRD patterns, (k) particle size distributions measured by using a laser particle size analyzer, (m) N2 adsorption–desorption isotherms (inset shows the pore size distributions) of SnO2 NPs and counterparts. (j) Sn 3d XPS spectra of SnO2 NPs, SnO MRs and Sn MSs. (l) Particle size distributions of SnO2 NPs estimated according to TEM observation. (n) C 1s XPS spectra of SnO2 NPs-rGO and GO. (o) Raman spectra of GO, SnO2 NPs-rGO, SnO2 SMPs-rGO and SnO2 MPs-rGO. (p) TGA curves of SnO2 NPs-rGO and counterparts in an air atmosphere. | ||
The X-ray diffraction (XRD) patterns (Fig. 1i) of SnO2 samples confirm their pure crystallographic features (JCPDS card no. 41-1445). The Sn 2p XPS spectra (Fig. 1j) of SnO2 NPs, SnO microrods (MRs, Fig. S4a–d†) and Sn microspheres (MSs, Fig. S4g–j†) show Sn 3d5/2 peaks at 487.3, 486.6 and 485.8 eV and Sn 3d3/2 peaks at 495.8, 495.0 and 494.3 eV, respectively, corresponding to Sn4+, Sn2+ and Sn0. The particle size distributions of SnO2 NPs, SMPs and MPs were estimated by using a laser particle size analyzer (Fig. 1k) and are 13 ± 10 nm, 684 ± 432 nm and 4.0 ± 2.7 μm, respectively. The measured average size (Fig. 1l) of SnO2 NPs is 12 ± 10 nm based on the TEM observation (Fig. 1a–d). The N2 adsorption–desorption isotherms of the SnO2 powder (Fig. 1m) can be classified as type IV isotherms with a type H3 hysteresis loop.55,56 The calculated Brunauer–Emmett–Teller (BET) specific surface area and pore volume of SnO2 NPs (126 m2 g−1 and 0.168 cm3 g−1) are larger than those of SnO2 SMPs (93 m2 g−1 and 0.049 cm3 g−1) and SnO2 MPs (83 m2 g−1 and 0.006 cm3 g−1), agreeing with the smallest size of SnO2 NPs.
After microthermal solvent evaporation at 80 °C of the high-energy ball-milling dispersed aqueous slurries of SnO2 particles and GO nanosheets in a mass ratio of 4:1, SnO2 NPs-rGO (Fig. 1c–h and S1d–i†), SnO2 SMPs-rGO (Fig. S2d–f†), SnO2 MPs-rGO (Fig. S2j–l†), SnO2 MSs-rGO (Fig. S3g, h, k and l†) and SnO2-TA MSs-rGO (Fig. S3i–l†) composites were obtained. In the SnO2 NPs-rGO nanocomposite, the SnO2 NPs are uniformly and completely encapsulated in the rGO matrix (Fig. 1c–h). Remarkably, SnO2 NPs-rGO displays a flake-like morphology (Fig. 1e–h) similar to commercial graphite. The micron size of the SnO2 NPs-rGO nanocomposite will not only facilitate the electrode coating operation but also inhibit the intimate contact and excessive side reaction between the SnO2 NPs and electrolyte for generating a thin and stable SEI layer.20Fig. 1h clearly shows the layer-by-layer assembled structure of SnO2 NPs-rGO. It can be speculated that the hydrogen-bond interaction and electrostatic attraction among SnO2 NPs, TA molecules and oxygen-containing functional groups (i.e., hydroxyl (–OH), epoxy (–C–O–C–), and carboxyl (–COOH)) on GO might guide the uniform anchoring of TA-capped SnO2 NPs on GO nanosheets. In contrast, it is difficult for rGO nanosheets to completely encase the SnO2 SMPs (Fig. S2a–f†), SnO2 MPs (Fig. S2g–l†), SnO2 MSs (Fig. S3a, b, g and h†) and SnO2-TA MSs (Fig. S3d, e, i and j†) due to their large particle sizes and weak interactions with rGO. Notably, by in situ addition of GO in the generation solution of SnO2 NPs, a comparison sample of the SnO2/rGO composite (Fig. S5†) was also fabricated, where plenty of SnO2 particles are not encapsulated in the rGO matrix (Fig. S5b†), owing to the poor dispersibility of GO in ethylene glycol.
In the C 1s XPS spectra (Fig. 1n), SnO2 NPs-rGO presents a higher intensity of the C–C binding peak (284.6 eV); however, weaker intensities of the C–O binding peak (286.5 eV) and CO binding peak (288.7 eV) than GO confirm the reduction of GO to rGO after the ball-milling and solvent evaporation treatments. In addition, the Raman spectra (Fig. 1o) of pristine GO, SnO2 NPs-rGO, SnO2 SMPs-rGO and SnO2 MPs-rGO exhibit the characteristic G-band (1601 cm−1) and D-band (1351 cm−1) from graphene. The higher D/G intensity ratios (ID/IG) of SnO2 NPs-rGO (1.03), SnO2 SMPs-rGO (1.03) and SnO2 MPs-rGO (1.02) than pristine GO (0.87) again indicate the reduction of GO in the composites. The mass ratios of rGO in SnO2 NPs-rGO, SnO2 SMPs-rGO and SnO2 MPs-rGO composites are estimated to be 16.8, 17.9 and 18.5 wt%, respectively, in accordance with the TGA measurement curves obtained in air (Fig. 1p).
Time-dependent studies on a set of samples obtained at different solvothermal reaction times (e.g., 0, 1, 2, 5, 8 and 10 h) were performed to reveal the formation mechanism of SnO2 NPs (Fig. 2, 3, S6 and S7†). Before the solvothermal reaction (i.e., 0 h reaction time), OA acts as a precipitant and reacts with Sn2+ at room temperature to produce a white precipitate of SnC2O4 MRs with smooth surfaces and clear edges (Fig. 2a–d). Interestingly, there are abundant voids inside the SnC2O4 MRs (Fig. 2b), which might be the reason for their instability and easy pulverization in the solvothermal condition. The HRTEM image (Fig. 2c) of SnC2O4 MRs shows parallel fringes with a d-spacing of 0.32 nm attributed to the (111) planes of SnC2O4 (JCPDS card no. 51-0614). The EDX elemental mapping images (Fig. 2d) of SnC2O4 MRs display the even distribution of C, O and Sn elements.
 | ||
| Fig. 2 (a) SEM, (a–c and e–g) (HR) TEM and (d) STEM-EDX mapping images of intermediates obtained at different reaction times (i.e., (b–d) 0, (e) 2 and (f and g) 5 h) in the synthesis of SnO2 NPs. | ||
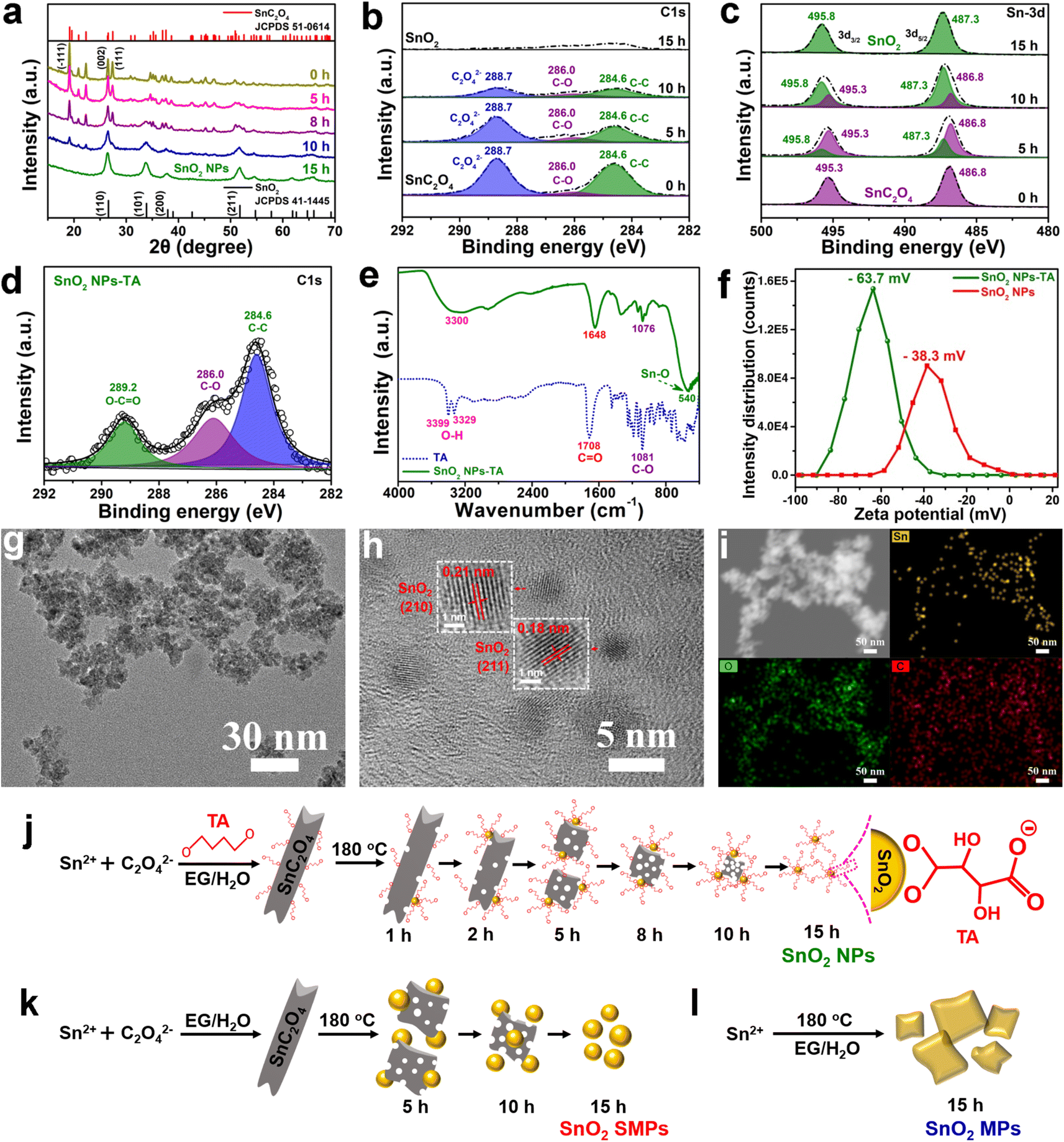 | ||
| Fig. 3 (a) XRD patterns and (b) C 1s and (c) Sn 3d XPS spectra of intermediates of SnO2 NPs. (d) C 1s XPS spectrum of SnO2 NPs-TA obtained after washing of the precipitate collected in the synthesis of SnO2 NPs with 2 mL absolute ethanol. (e) FTIR spectra of SnO2 NPs-TA and TA. (f) Zeta potential distributions of SnO2 NPs-TA and SnO2 NPs. (g and h) (HR) TEM and (i) STEM-EDX mapping images of SnO2 NPs-TA. Schematic illustrations of the formation mechanisms of (j) SnO2 NPs, (k) SnO2 SMPs and (l) SnO2 MPs. | ||
Obviously, after 1 and 2 h reactions, lots of NPs appear on the surface of SnC2O4 MRs (Fig. 2a and e). Subsequently, the SnC2O4 MRs obviously crack into scobinate irregular SnC2O4 fragments with more NPs generated on the surfaces in the 5 and 8 h products (Fig. 2a and f). The HRTEM image of 5 h products exhibits parallel fringes with d-spacings of 0.34 nm in the body area of MRs and 0.33 nm in the surface NPs, corresponding to the (002) plane of SnC2O4 and the (110) plane of SnO2 (Fig. 2g). This result confirms that the newly generated NPs on the surface of SnC2O4 MRs are SnO2 NPs, suggesting a unique SDSST process from SnC2O4 MRs to SnO2 NPs. From 8 to 10 h (Fig. 2a), the sizes of SnC2O4 fragments keep on decreasing, and meanwhile the number of scattered NPs gradually increased.
In the absence of TA, there is also a similar SDSST process from SnC2O4 MRs to SnO2 SMPs (Fig. S6a†), indicating that TA plays a critical role in the size control and agglomeration suppression of SnO2 NPs by capping on their surfaces. The XRD patterns of the intermediate products of SnO2 NPs (Fig. 3a and S7a†) and SnO2 SMPs (Fig. S7b†) clearly exhibit the gradual phase transformation from pure SnC2O4 crystals and pure SnO2 crystals during the 15 h solvothermal reaction process. In stark contrast, without addition of OA and TA, the XRD patterns (Fig. S7c†) of the intermediates display a familiar crystal growth process from the amorphous material (1 h) to less crystalline SnO2 (8 h) and then to highly crystalline SnO2 (15 h). Therein, the obtained SnO2 intermediates (Fig. S6b†) and final products (Fig. S2g–i†) exhibited large micron sizes, suggesting that the retarded SDSST process from SnC2O4 to SnO2 could effectively restrict the fast and over growth of SnO2 crystals.
The XPS spectra (Fig. 3b and c) of the intermediates of SnO2 NPs were recorded to investigate the exceptional SDSST process. The XPS spectra of SnC2O4 MRs show three C 1s peaks at 284.6, 286.0 and 288.7 eV corresponding to C–C binding, C–O binding and C2O42− groups and two Sn 3d peaks at 486.8 and 495.3 eV corresponding to Sn2+ in SnC2O4. Along with increased reaction time, the peaks at 495.3 and 486.8 eV corresponding to the C2O42− groups and C–C binding in SnC2O4 keep on shrinking and finally disappear in the 15 h product of SnO2 NPs (Fig. 3b). Simultaneously, the intensities of the two Sn 3d XPS peaks corresponding to Sn2+ in SnC2O4 (486.8 and 495.3 eV) gradually weaken and disappear in the 15 h products, and the two Sn 3d XPS peaks corresponding to Sn4+ in SnO2 (487.3 and 495.8 eV) gradually increased and pure SnO2 was generated at 15 h (Fig. 3c), confirming the progressive SDSST from SnC2O4 to SnO2.
To clearly demonstrate the capping of TA on the generated SnO2 NPs, the SnO2 NPs-TA sample was obtained by one time washing with a small amount of absolute ethanol (i.e., 2 mL) of the 15 h precipitate collected in the synthesis of SnO2 NPs. As mentioned previously for fully cleaned SnO2 NPs (Fig. 1a and b), the 15 h precipitate was washed with 9 mL absolute ethanol three times to mostly eliminate the TA molecules capped on SnO2 NPs. The C 1s XPS spectrum of SnO2 NPs-TA (Fig. 3d) exhibits three peaks at 284.6, 286.0 and 289.2 eV corresponding to the C–C, C–O and O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds in TA. The Fourier transform infrared spectroscopy (FTIR) spectrum of SnO2 NPs-TA (Fig. 3e) shows the characteristic stretching vibration peaks of O–H, CO, and C–O bonds from TA and Sn–O bonds from SnO2 at wavenumbers of 3300, 1648, 1076 and 540 cm−1, respectively.57 Furthermore, SnO2 NPs-TA possesses a higher zeta potential of −63.7 mV than the −38.3 mV of SnO2 NPs (Fig. 3f), suggesting that the capping of TA molecules improves the electronegativity of SnO2-TA NPs.58,59 The TEM images of SnO2 NPs-TA (Fig. 3g and h) exhibit a slightly better dispersion than the SnO2 NPs (Fig. 1a and b), and display clear parallel fringes with a d-spacing of 0.21 and 0.18 nm corresponding to the (210) and (211) planes of SnO2. The EDX elemental mapping images of SnO2 NPs-TA display the even distribution of O and Sn elements. Remarkably, except the effect of the carbon film on the TEM grid, the weak C signal can be reasonably ascribed to the TA molecules capped on the SnO2 NPs (Fig. 3i).
O bonds in TA. The Fourier transform infrared spectroscopy (FTIR) spectrum of SnO2 NPs-TA (Fig. 3e) shows the characteristic stretching vibration peaks of O–H, CO, and C–O bonds from TA and Sn–O bonds from SnO2 at wavenumbers of 3300, 1648, 1076 and 540 cm−1, respectively.57 Furthermore, SnO2 NPs-TA possesses a higher zeta potential of −63.7 mV than the −38.3 mV of SnO2 NPs (Fig. 3f), suggesting that the capping of TA molecules improves the electronegativity of SnO2-TA NPs.58,59 The TEM images of SnO2 NPs-TA (Fig. 3g and h) exhibit a slightly better dispersion than the SnO2 NPs (Fig. 1a and b), and display clear parallel fringes with a d-spacing of 0.21 and 0.18 nm corresponding to the (210) and (211) planes of SnO2. The EDX elemental mapping images of SnO2 NPs-TA display the even distribution of O and Sn elements. Remarkably, except the effect of the carbon film on the TEM grid, the weak C signal can be reasonably ascribed to the TA molecules capped on the SnO2 NPs (Fig. 3i).
The schematic diagram of Fig. 3j depicts the SDSST process from SnC2O4 MRs to SnO2 NPs with TA molecules as an effective capping agent to stabilize the newly generated SnO2 NPs. As a small molecule organic acid capping agent, TA has a certain stabilizing effect on the generated SnO2 NPs by reducing their surface energy via the coordination interaction between carboxyl groups on TA and Sn4+ of SnO2. Moreover, the electronegative carboxyl groups on TA will enhance the electronegativity of SnO2 NPs-TA (Fig. 3f) to prevent their agglomeration and dissolution–recrystallization in the solvothermal solution, thereby facilitating the formation of dispersed small size SnO2 NPs. In contrast, in the absence of TA, the generated SnO2 NPs via SDSST from SnC2O4 MRs spontaneously merge into submicron scale SnO2 SMPs (Fig. 3k). Additionally, in the absence of TA and OA, a conventional crystal growth procedure was revealed from Sn2+ ions to amorphous intermediates and then to crystalline large size SnO2 MPs (Fig. 3l, S6b and S7c†). Generally, the synergistic effect between distinctive SDSST and TA as a potent capping agent realizes effective size control for creating dispersed small size SnO2 NPs.
2.2 Superior lithium and sodium storage capability of SnO2 NPs-rGO
The electrochemical properties of the SnO2 samples were examined using lithium and sodium cell models within a potential window of 0.01–3.0 V vs. Li/Li+ or Na/Na+ (Fig. 4 and S8–S11†). As an anode of LIBs, in contrast to the counterparts, SnO2 NPs-rGO exhibits the best cycling stability at 100 mA g−1 (Fig. 4a, S8a, c and e†) and delivers a highest 300th discharge capacity of 1461 mA h g−1 being close to the theoretical capacity of SnO2 (1494 mA h g−1). Notably, at a high C-rate of 1000 mA g−1, the capacities of SnO2 NPs-rGO dramatically increase along with cycling and stabilized at an ultrahigh value of 1775 mA h g−1 after 800 cycles (Fig. 4b). The observations of capacity improvement during cycling in the literature are popularly recognized as the activation of electrodes due to the increased conductivity along with the continuous perfection of the solid electrolyte interphase (SEI) layer and the additional capacity contribution from capacitive lithium storage.13,27 As demonstrated in the representative charge profiles of SnO2 NPs-rGO as an anode of LIBs at 1000 mA g−1 (Fig. S8g†), the continuous extension of the charge plateaus in the voltage rang of 1–3 V along with an increased cycle number suggests the gradual activation of the delithiation reactions of SnO2 NPs-rGO in this voltage rang. Therein, the lithiation–delithiation reaction mechanisms and capacitive capacity contribution of SnO2 NPs-rGO will be discussed below in sequence. In rate performance tests (Fig. 4c), SnO2 NPs-rGO demonstrates the highest capacities at all C-rates compared to the counterparts and expresses a satisfactory capacity of 1038 mA h g−1 at 2000 mA g−1. When the current density was returned to 100 mA g−1 after 60 cycles, the capacity can be successfully recovered to 1510 mA h g−1. | ||
| Fig. 4 (a, b, and d–f) Cycling and (c and g) rate performances of SnO2 NPs-rGO and counterparts as anodes of (a–d) LIBs and (e–g) SIBs. (h) The 1st discharge–charge profiles of prelithiated/presodiated electrodes and (i) 10th discharge–charge profiles in a narrow voltage window of 0.01–2 V of SnO2 NPs-rGO as anodes of LIBs and SIBs. The capacities and cycle numbers with a log–log plot of SnO2 NPs-GO and the trends of reported experimental results on graphene-based SnO2 materials as anodes of (j) LIBs and (k) SIBs at labeled current rates as listed in Tables S1 and S2,† respectively. | ||
In stark contrast, all the SnO2 counterparts expressed significant capacity decay with lower capacities at various C-rates (Fig. 4a–c, S9 and S10†). The representative discharge/charge curves (Fig. S8a†) show that SnO2 NPs-rGO delivers almost identical charge–discharge curves after the initial 30 cycles with three stable charge plateaus at 0.48, 1.23 and 1.86 V, suggesting an excellent reaction reversibility. In stark contrast, the charge–discharge curves of SnO2 SMPs-rGO (Fig. S8c†) and SnO2 MPs-rGO (Fig. S8e†) keep on shrinking with cycling and show no obvious voltage plateaus after 70 cycles, in accord with the poor cycling reversibility (Fig. 4a). Remarkably, in a long cycling test at high current rates of 2, 5 and 10 A g−1 (Fig. 4d), SnO2 NPs-rGO delivers reversible capacities of 1680, 1353 and 753 mA h g−1 after 1500, 2500, and 4000 cycles, respectively, demonstrating an outstanding cycling lifespan.
As expected, SnO2 NPs-rGO also exhibits superior sodium storage performances. Cycling at 50 and 200 mA g−1 (Fig. 4e, f and S8–S10†), SnO2 NPs-rGO provides a stable reversible capacity of 443 and 260 mA h g−1 after 100 and 250 cycles, respectively, which are much higher than those of SnO2 SMPs-rGO (187 and 60 mA h g−1) and SnO2 MPs-rGO (140 and 38 mA h g−1). Similar to the generally reported cycling performances of SIBs, the andante capacity decline of SnO2 NPs-rGO along with cycling can be attributed to the gradual degradation of the electrochemical reaction reversibility of SnO2 NPs-rGO (Fig. S8b†) due to the sluggish reactivity of large size Na+ with most of the active materials. In the rate performance tests (Fig. 4g), SnO2 NPs-rGO demonstrates the highest capacities at all C-rates over the counterparts. The reversible capacities of 519, 443, 392, 335 and 249 mA h g−1 are achieved at the current densities ranging from 20, 50, 100, 200 to 500 mA g−1, respectively. After 60 cycles at different current densities, when the current density was returned to 20 mA g−1, the reversible capacity could be recovered to 463 mA h g−1.
To verify the practical application of SnO2 NPs-rGO, the performances of prelithiated and presodiated electrodes, cycling in a narrow voltage window and full LIB/SIB cells, were determined (Fig. 4h, i, 5a–f and S11†). Significantly, after the prelithiation and presodiation treatments (Fig. 4h, S11c and d†), the prelithiated and presodiated electrodes of SnO2 NPs-rGO yield an excellent initial Coulomb efficiency (ICE) of 98% and 94%, which are much higher than that of SnO2 SMPs-rGO (75% and 65%) and SnO2 MPs-rGO (74% and 65%), respectively. The prelithiated SnO2 NPs-rGO electrode exhibited better cycling stability with a higher 200th discharge capacity of 1625 mA h g−1 than ordinary SnO2 NPs-rGO (1396 mA h g−1) at 100 mA g−1 (Fig. S11a†). Moreover, the presodiated SnO2 NPs-rGO electrode also delivers a slightly higher 100th discharge capacity of 451 mA h g−1 than ordinary SnO2 NPs-rGO (443 mA h g−1) at 50 mA g−1 (Fig. S11b†). The above results demonstrate that the high lithium and sodium storage reactivity of SnO2 NPs can efficaciously promote the adequate prelithiation and presodiation of SnO2 NPs-rGO with superior ICE and cycling stability in contrast to SnO2 SMPs-rGO and SnO2 MPs-rGO. Operating in a narrow voltage range of 0.01–2 V,SnO2 NPs-rGO also exhibited satisfactory lithium and sodium storage capacities of 995 and 340 mA h g−1 with CEs of 98 and 98% in 10 cycles at 100 and 50 mA g−1, respectively, with excellent cycling stability (Fig. S11e–h†). Overall, benefiting from the enhanced lithium and sodium storage reactivity and reversibility, SnO2 NPs-rGO manifests outstanding performances in sharp contrast to the counterparts and the reported graphene-based SnO2 composites as anodes for LIBs and SIBs (Fig. 4j, k, Tables S1 and S2†).
Furthermore, coin full cells of LIBs or SIBs were configured with SnO2 NPs-rGO as the anode and commercial LiFePO4 (LFP) or Na3V2(PO4)3 (NVP) as the cathode with a mass ratio of 1:6, respectively. The LFP (Fig. S12a†) and NVP (Fig. S12b†) exhibit outstanding cycling stability in the half LIB and SIB cells cycling in the voltage ranges of 3.1–3.8 and 2.0–4.0 V with reversible capacities of 135 and 141 mA h g−1 after 50 and 70 cycles at 100 and 50 mA g−1, respectively. The SnO2 NPs-rGO, LFP and NVP electrodes were firstly activated after 3 cycles in the half cells of LIBs (Fig. 5a and b) and SIBs (Fig. 5d and e) at 100 and 50 mA g−1, respectively. Electrochemically activated anodes (i.e., SnO2 NPs-rGO) in the discharge state and cathodes (i.e., LFP and NVP) in the charge state were then obtained by disassembling the cycled half cells, and applied for reassembling the full cells of SnO2 NPs-rGO/LFP (Fig. 5c) and SnO2 NPs-rGO/NVP (Fig. 5f) with new separators. The SnO2 NPs-rGO/LFP full cell delivers first charge and discharge capacities of 640 and 631 mA h g−1 (ICE, 98%) in the voltage range of 2.0–3.4 V, respectively. Notably, in the charge–discharge curves of the SnO2 NPs-rGO/LFP full cell (Fig. 5c), a pair of discharge and charge plateaus at 2.92 and 3.19 V are obtained which approximate to the difference values between the discharge plateau at 3.38 of LFP and the charge plateau at 0.48 V of SnO2 NPs-rGO and between the charge plateau at 3.46 V of LFP and the discharge plateau at 0.25 V of SnO2 NPs-rGO (Fig. 5a and b), respectively.
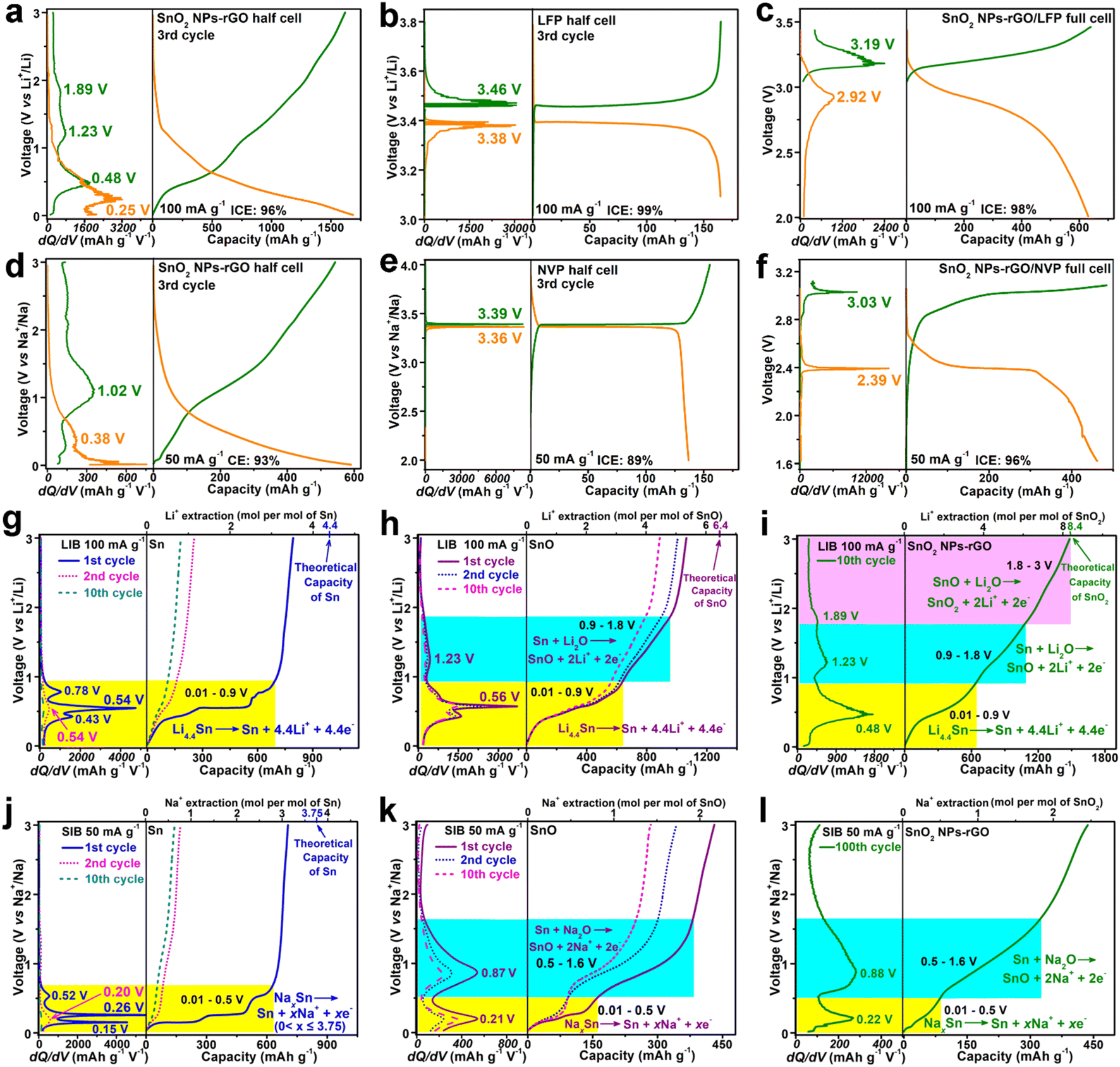 | ||
| Fig. 5 Representative discharge–charge profiles of (a and d) SnO2 NPs-rGO in the voltage range of 0.01–3.0 V, (b) LiFePO4 (LFP) in the voltage range of 3.1–3.8 V and (e) Na3V2(PO4)3 (NVP) in the voltage range of 2.0–4.0 V in the half cells of (a and b) LIBs at 100 mA g−1 and (d and e) SIBs at 50 mA g−1. First discharge–charge profiles of (c) the SnO2 NPs-rGO/LFP full LIB cell at 100 mA g−1 and (f) the SnO2 NPs-rGO/NVP full cell at 50 mA g−1. Representative charge profiles of (g and j) Sn, (h and k) SnO, and (i and l) SnO2 NPs-rGO as anodes of (g–i) LIBs at 100 mA g−1 and (j–l) SIBs at 50 mA g−1. Left panels in (a–l) show the corresponding dQ/dV curves of discharge and charge profiles. | ||
Similarly, a pair of discharge or charge plateaus at 2.39 and 3.03 V are exhibited in the first cycle of the SnO2 NPs-rGO/NVP full cell in the voltage range of 1.6–3.1 V (Fig. 5f), which approximate to the difference values between the discharge at 3.36 of NVP and the charge plateau at 1.02 V of SnO2 NPs-rGO and between the charge plateau at 3.39 V of NVP and the discharge plateau at 0.38 V of SnO2 NPs-rGO (Fig. 5d and e), respectively. The first charge and discharge capacities of 482 and 462 mA h g−1 with an ICE of 96% are expressed by the SnO2 NPs-rGO/NVP full cell, respectively. Interestingly, benefiting from the much flatter discharge and charge plateaus of NVP (Fig. 5e) than those of LFP (Fig. 5b), the SnO2 NPs-rGO/NVP full cell (Fig. 5f) exhibits obviously flatter discharge and charge plateaus than the SnO2 NPs-rGO/LFP full cell (Fig. 5c). As such, the SnO2 NPs-rGO/NVP full cell might possess a more excellent practical application prospect than the SnO2 NPs-rGO/LFP full cell due to the more stable operating voltage and the much lower price of sodium than lithium.
2.3 Lithium and sodium storage mechanisms of SnO2 NPs-rGO
To identify the reactions at each voltage plateau of SnO2 NPs-rGO for LIBs and SIBs, the charge profiles combined with differential capacity versus voltage (dQ/dV) curves of Sn MSs (Fig. S4g–l†), SnO MRs (Fig. S4a–f†) and SnO2 NPs-rGO were comparatively analyzed (Fig. 5g–l). In the first charge profiles as anodes of LIBs (Fig. 5g–i), Sn MSs, SnO MRs and SnO2 NPs-rGO delivered one (i.e., 0.54 V), two (i.e., 0.56 and 1.23 V), and three (i.e., 0.48, 1.23 and 1.89 V) plateaus, respectively. Thus, the three charge plateaus of SnO2 NPs-rGO in the voltage ranges of 0.01–0.9, 0.9–1.8 and 1.8–3.0 V could be reasonably attributed to the three-step delithiation reactions from LixSn (0 < x ≤ 4.4) to Sn, from Sn to SnO and from SnO to SnO2 (Fig. 5i), respectively. In the same way, the two charge plateaus in the voltage ranges of 0.01–0.5 and 0.5–1.6 V of SnO2 NPs-rGO for SIBs (Fig. 5j–l) could be speculatively ascribed to the desodiation reactions from NaxSn (0 < x ≤ 3.75) to Sn and from Sn to SnO (Fig. 5l), respectively.To demonstrate the above proposed reaction mechanisms of the SnO2 NPs-rGO nanocomposite as an anode for LIBs and SIBs, the CV curves, XPS spectra and HRTEM images were investigated. As anodes of LIBs, in the CV curves of SnO2 NPs-rGO (Fig. 6a), the three pairs of cathodic/anodic peaks at 1.16/1.94, 0.49/1.23 and 0.21/0.54 V correspond to the three-step reversible reactions of SnO2 + 2Li+ + 2e− ⇌ SnO + Li2O, SnO + 2Li+ + 2e− ⇌ Sn + Li2O and Sn + xLi+ + xe− ⇌ LixSn (0 < x ≤ 4.4), respectively.20,27,60 In the 30th charge–discharge profiles at 100 mA g−1 (Fig. 6b), there are three obvious charge plateaus at 0.47, 1.23 and 1.89 V for SnO2 NPs-rGO, corresponding to the delithiation reaction from LixSn to Sn, Sn to SnO, and SnO to SnO2, respectively.20,27 In contrast, the charge plateaus at 1.23 and 1.89 V almost disappear and the charge plateaus at 0.47 V obviously shrink in the 30th charge curves of SnO2 SMPs-rGO and SnO2 MPs-rGO. This suggests a severe degradation in the conversion reactions and a certain degree of decline in the alloying–dealloying reaction in the cycled electrodes of SnO2 SMPs-rGO and SnO2 MPs-rGO, in harmony with their capacity decay with continuous cycling.
 | ||
| Fig. 6 CV curves of SnO2 NPs-rGO as an anode of (a) LIBs and (d) SIBs. (b and e) Charge–discharge profiles combined with dQ/dV curves of charge profiles and (c and f) Sn 3d XPS spectra and (g–l) HRTEM images of charged electrodes of (g and j) SnO2 NPs-rGO, (h and k) SnO2 SMPs-rGO and (i and l) SnO2 MPs-rGO after 30 cycles as anodes of (b, c, and g–i) LIBs at 100 mA g−1 and (e, f, and j–l) SIBs at 50 mA g−1. | ||
To reveal the cause for the reaction reversibility degradation of SnO2 SMPs-rGO and SnO2 MPs-rGO in contrast to SnO2 NPs-rGO, the Sn 3d XPS spectra and HRTEM images of the 30th charged electrodes were comparatively analyzed. Notably, the cycled electrode of SnO2 NPs-rGO exhibits similar Sn 3d XPS spectra to pristine SnO2 NPs-rGO (Fig. 1j and 6c), confirming the complete recovery of SnO2 as depicted in the HRTEM images (Fig. 6g, S14a and d†). In contrast, the Sn 3d XPS peaks of delithiated SnO2 SMPs-rGO and SnO2 MPs-rGO can be divided into three pairs of peaks, corresponding to Sn0, Sn2+ and Sn4+, respectively, indicated that the existence of SnO and Sn (Fig. 1j, 6c, h, i, S14b, c, e and f†). The incomplete conversion of Sn to SnO2 in the charged electrodes of SnO2 SMPs-rGO and SnO2 MPs-rGO can be due to the coarsening of Sn into large size particles (Fig. 6h and i) with sluggish conversion reaction reversibility. As for SnO2 NPs-rGO, the SnO2 NPs are homogeneously and tightly encapsulated in the rGO matrix, which strongly restrains the coarsening of Sn particles among the adjacent SnO2 NPs and maintains fantastic reactivity and reversibility for superior cycling stability and rate capability (Fig. 4a–c, 6b, g, S14a and d†).
As anodes of SIBs, the CV curves (Fig. 6d, S13g and j†) of SnO2 NPs-rGO and the counterparts show great differences to those as anodes of LIBs (Fig. 6a, S13a and d†). There is an irreversible peak at 1.03 V and a large slope in the range of 0.7–0.01 V in the first cathodic scan that might be ascribed to the conversion reaction from SnO2 into Sn and the formation of a SEI layer, respectively. In the later cycles, there is one highly stable pair of cathodic/anodic peaks at 0.38/1.23 V corresponding to the reversible conversion reaction of SnO + 2Na+ + 2e− ⇌ Sn + Na2O in the CV curves of all three samples (Fig. 6d, S13g and j†).20,27 Moreover, the 30th charge profile combined with dQ/dV curve of SnO2 NPs-rGO (Fig. 6e) exhibits a primary charge plateau at 1.05 V and a slight charge plateau at 0.25 V, corresponding to the conversion reaction from Sn to SnO and the partially reversible dealloying reaction of NaxSn (0 ≤ x ≤ 3.75).20 Contrastively, the charge plateaus in the 30th charge curves of SnO2 SMPs-rGO and SnO2 MPs-rGO attenuate obviously, which indicate their lower reaction reversibility than SnO2 NPs-rGO.
In the Sn 2p XPS spectrum of the 30th charged SnO2 NPs-rGO as an anode of SIBs (Fig. 6f), the binding energies of the Sn 3d5/2 (486.6 eV) and Sn 3d3/2 (495.0 eV) peaks are the same as those of SnO (Fig. 1j), approving the generation of SnO (Fig. 6j) and substantiating that the sodium storage in SnO2 mainly relies on the conversion reaction between Sn and SnO (Fig. 6j, S15a and d†).20 However, the 30th charged SnO2 SMPs-rGO and SnO2 MPs-rGO display Sn 3d5/2 and Sn 3d3/2 XPS peaks at 494.3 and 485.8 eV, respectively, confirming the presence of metallic Sn0 (Fig. 1j, 6k, l, S15b, c, e and f†) due to the Sn coarsening induced reversibility decay. Moreover, the strong intensities of Na KLL XPS peaks at 496.6 eV in all the charged electrodes demonstrate the low reaction reversibility of the conversion reaction from SnO to SnO2, which results in the existence of unreacted Na2O in the cycled SnO2 electrodes (Fig. 6f).18 Thus, the primary reversible sodiation–desodiation reaction in SnO2 NPs-rGO can be confirmed as the conversion reaction between Sn and SnO, and the ultrafine SnO2 NPs and robustly inhibited Sn coarsening by rGO endow SnO2 NPs-rGO with superior sodium storage reaction reversibility.
2.4 Performance enhancement mechanism of SnO2 NPs-rGO
To reveal the electrochemical reaction kinetics of SnO2 NPs-rGO and the counterparts, the CV curves collected at various scan rates were systematically investigated (Fig. 7a–l, S13, S16–S18†). The CV curves of SnO2 NPs-rGO display similar shapes at varied voltage scan rates from 0.2 to 1.0 mV s−1 (Fig. 7a), indicating smaller polarization and better electrochemical reaction kinetics in contrast to SnO2 SMPs-rGO and SnO2 MPs-rGO (Fig. S13b and e†). According to the relationship between the peak current values (i) in the CV curves and voltage scan rates (v) by using the equation of i = avb, the values of slope b of the log(i) versus log(v) plots can be obtained (Fig. 7b), which serves as an indicator of the electrochemical behavior. Generally, when the b value is close to 0.5, a diffusive behavior accompanied by Faraday reaction dominates the charge storage capacity, whereas as it approaches 1.0, the capacitive behavior contributes to charge storage capacity.61 The b values are 0.57/0.54 for the CV curves of SnO2 NPs-rGO as an anode of LIBs, indicating diffusion controlled electrochemical Faraday reaction kinetics in SnO2 NPs-rGO. In contrast, the b values are higher for SnO2 SMPs-rGO (0.74/0.65) (Fig. S13c†) and SnO2 MPs-rGO (0.71/0.73) (Fig. S13f†), suggesting their diffusion and capacitance cooperatively controlled electrochemical reaction kinetics. | ||
| Fig. 7 (a and e) CV curves at various scan rates, (b and f) log(i) versus log(v) plots at representative redox peaks, (c and g) CV profiles at 0.2 mV s−1 with labeled capacitive contributions, and (d and h) ratios of diffusive capacity contributions of (a–c and e–g) SnO2 NPs-rGO and the counterparts as anodes of (a–d) LIBs and (e–h) SIBs. (i) CV curves at various scan rates, (j) log(i) versus log(v) plots at representative anodic peaks in (i), and (k) CV profile with labeled capacitive contribution at 0.2 mV s−1 of SnO2 NPs-rGO as an anode of LIBs after 800 cycles at 1 A g−1 (Fig. 4b). (l) Li+ and Na+ insertion moles in per mol of SnO2 in the 800th and 100th discharge processes of SnO2 NPs-rGO and the counterparts at 1000 and 20 mA g−1, respectively. | ||
Furthermore, the capacitive capacity ratio (CCR) and diffusive capacity ratio (DCR) can be quantitatively determined by using the equation of I = k1v + k2v0.5.62,63 Therein, the k1v and k2v0.5 correspond to the current values of capacitive and diffusive behaviors, respectively. At 0.2 mV s−1 for LIBs, SnO2 NPs-rGO shows a DCR as high as 77.9% (Fig. 7c and d), greater than the 57.4% of SnO2 SMPs-rGO (Fig. S16a†) and the 58.4% of SnO2 MPs-rGO (Fig. S16b†). Significantly, even at the highest v of 1.0 mV s−1, the DCR of SnO2 NPs-rGO (62.1%) is about 1.5 and 1.6 times that of SnO2 SMPs-rGO (42.5%) and SnO2 NPs-rGO (39.8%), verifying the highest Faraday reactivity of SnO2 NPs-rGO (Fig. 7d). Interestingly, SnO2 NPs-rGO also displays the highest DCR (Fig. 7e–h) at each v in the CV tests of all the samples as anodes of SIBs, and exhibits b values of 0.58/0.68 (Fig. 7f) resembling the 0.57/0.54 as an anode of LIBs (Fig. 7b), indicating diffusion controlled high Faraday reaction kinetics in the sodium insertion–extraction process of SnO2 NPs-rGO. In stark contrast, the b values are higher for SnO2 SMPs-rGO (0.78/0.78) (Fig. S13i†) and SnO2 MPs-rGO (0.91/0.81) (Fig. S13l†), suggesting diffusion and capacitance cooperatively controlled and capacitance primarily controlled electrochemical reaction kinetics of SnO2 SMPs-rGO and SnO2 MPs-rGO, respectively, with relatively poor Faraday reaction kinetics.
At 0.2 mV s−1, the DCR of SnO2 NPs-rGO is as high as 72.8% (Fig. 7g), which is very close to the 77.9% (Fig. 7c) as an anode of LIBs. In stark contrast, the DCRs of SnO2 SMPs-rGO (47.3%, Fig. S16c†) and SnO2 MPs-rGO (43.9%, Fig. S16d†) are lower than the values of 57.4% (Fig. S16a†) and 58.4% (Fig. S16b†) for LIBs, respectively. Remarkably, at the highest v of 0.9 mV s−1 as an anode of SIBs, the DCR of SnO2 NPs-rGO is still as high as 54.8%, which is about 1.9 times that of SnO2 SMPs-rGO (29.5%) and SnO2 MPs-rGO (28.9%) (Fig. 7h). The above CV analyses suggest the highest reactivity of SnO2 NPs-rGO in contrast to SnO2 SMPs-rGO and SnO2 MPs-rGO, benefiting from the smallest size of SnO2 NPs with the shortest ion transport distance and the most homogeneous encapsulation of SnO2 NPs in the rGO matrix with the greatest electron conductivity. It can be contemplated that the superior lithium and sodium storage capacities of SnO2 NPs-rGO can be primarily attributed to its excellent diffusion-controlled Faraday reaction activity.
However, the capacitive contribution also plays an indispensable role in the ultrahigh lithium storage capacity of SnO2 NPs-rGO, especially in the exceptional uptrend of capacities from 1168 mA h g−1 (30th cycle) to 1775 mA h g−1 (800th cycle) at 1000 mA g−1 (Fig. 4b). The CCRs of the SnO2 NPs-rGO cell after 800 cycles at 1000 mA g−1 rise to 42.1% at 0.2 mV s−1 (Fig. 7i–k), which is about 2 times higher than that of the pristine SnO2 NPs-rGO cell (22.1%). Notably, the b values are 0.94/0.96 for the CV curves of the 800th cycled SnO2 NPs-rGO electrode, indicating capacitance-controlled reaction kinetics (Fig. 7j), which in complete contrast to the b values of 0.57/0.54 for the original SnO2 NPs-rGO electrode corresponding to diffusion-controlled reaction kinetics. Namely, the extraordinary capacity rising of SnO2 NPs-rGO cycled at 1000 mA g−1 (Fig. 4b) can be partially attributed to the gradually increasing capacitive contribution (Fig. 7k). Thus, the homogeneous encapsulation of SnO2 NPs within the rGO matrix will not only effectively enhance the diffusion controlled Faraday reactivity, but also robustly strengthen the stability of abundant phase interfaces generated in the cycled electrode to contribute considerable capacitive capacity. As clearly demonstrated in the 800th and 100th discharge profiles of samples as anodes of LIBs and SIBs at 1000 and 20 mA g−1 (Fig. 4b, g and 7l), SnO2 NPs-rGO can store 10.0 mol Li+ and 2.6 mol Na+ in per mole SnO2, respectively.
To in depth unveil the structure–function relationships, the conductivities identified by electrochemical impedance spectroscopy (EIS, Fig. 8a–d) and structural stabilities revealed by morphology evolutions of cycled electrodes (Fig. 8e–p) were systematically investigated. As anodes of LIBs, the charge transfer resistance (Rct, 54.3 Ω) of SnO2 NPs-rGO is significantly lower than that of SnO2 SMPs-rGO (115.2 Ω) and SnO2 MPs-rGO (308.7 Ω) according to the Nyquist plot of the newly assembled cells (Fig. 8a). After 5 cycles, SnO2 NPs-rGO exhibited the lowest Rct and Warburg impedance (Zw) (Fig. 8b), indicating the best charge transfer capability at the electrolyte–electrode interface and the highest lithium-ion diffusion rate in the active material, respectively. Similar results were obtained in the EIS analyses of the samples as an anode of SIBs (Fig. 8c and d), demonstrating the excellent conductivity of SnO2 NPs-rGO. Notably, the cycled SnO2 NPs-rGO electrode possessed the smallest SEI layer resistances in contrast to the counterparts (Fig. 8b and d), suggesting that the rGO matrix can robustly block the direct contact between the SnO2 NPs and electrolyte to effectively promote the generation of the thinnest SEI layer with an efficient ion exchange rate.
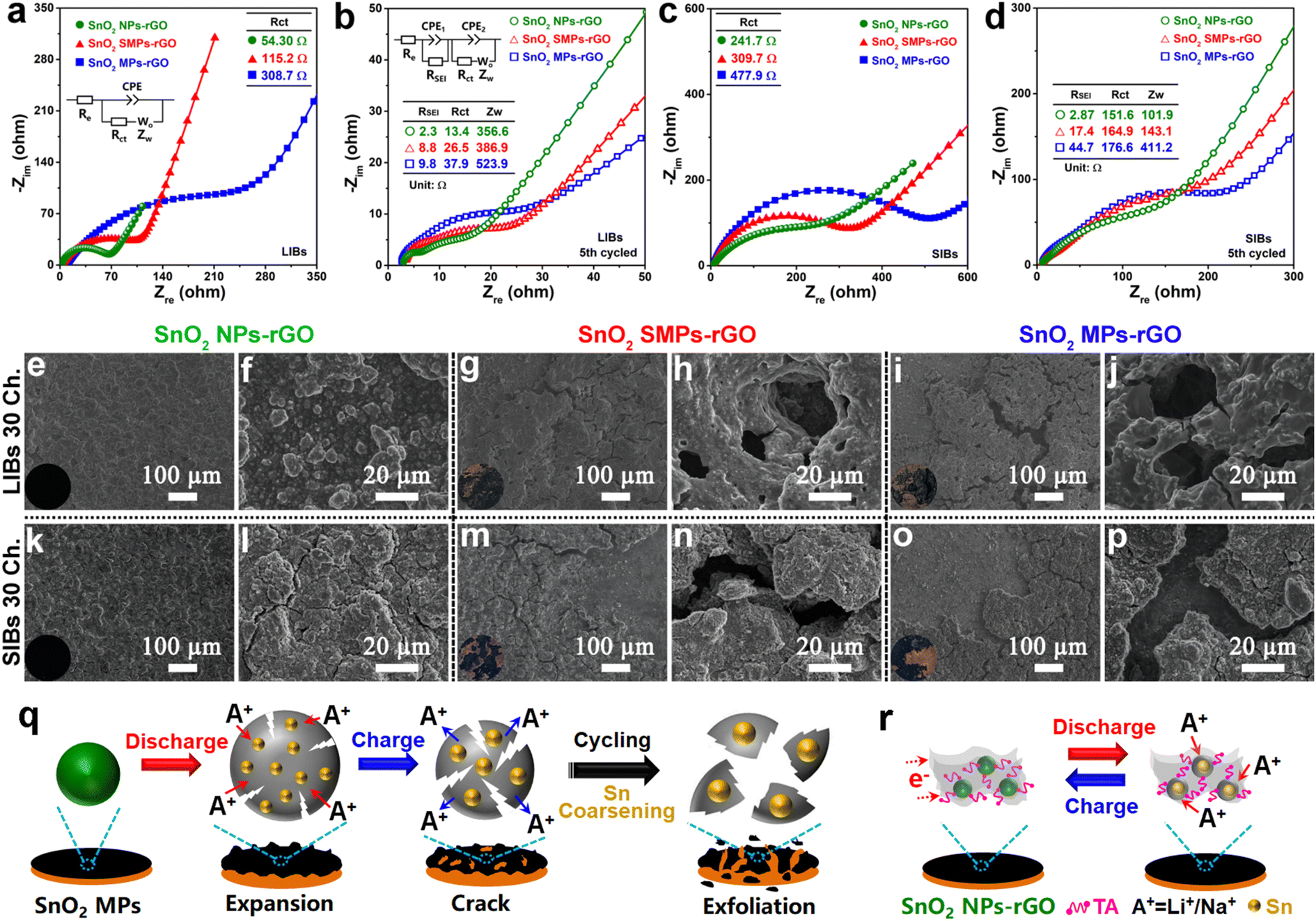 | ||
| Fig. 8 Nyquist plots and equivalent circuits of cells (a and b) before cycle and (c and d) after 5 cycles at 100 and 50 mA g−1 of the SnO2 samples as anodes of (a and c) LIBs and (b and d) SIBs, respectively. SEM images of (e–p) 30th cycled electrodes of (e, f, k, and l) SnO2 NPs-rGO, (g, h, m, and n) SnO2 SMPs-rGO and (i, j, o, and p) SnO2 MPs-rGO as anodes of (e–j) LIBs and (k–p) SIBs at 100 and 50 mA g−1, respectively. Schematic illustrations of (q) dramatic volume expansion and severe Sn coarsening in cycled SnO2 MPs and (r) robust structural stability, excellent electron conductivity and effectively inhibited Sn coarsening of SnO2 NPs-GO. | ||
The structural stability of electrodes has a decisive influence on the cycling performance. Fig. S19† and 8e–p present the SEM images and optical photographs of the pristine and 30th cycled electrodes of SnO2 NPs-rGO, SnO2 SMPs-rGO and SnO2 MPs-rGO. It is noteworthy that the surfaces of the cycled electrodes of SnO2 NPs-rGO as anodes of LIBs (Fig. 8e and f) and SIBs (Fig. 8k and l) are as flat as the pristine electrode (Fig. S19a and b†). In stark contrast, the cycled electrodes of SnO2 SMPs-rGO (Fig. 8g, h, m and n) and SnO2 MPs-rGO (Fig. 8i, j, o and p) exhibit obvious pulverization, and the active materials are seriously exfoliated from the copper foil. Namely, during repeated Li+ or Na+ insertion and extraction processes (Fig. 8q), the large volume expansion and shrinkage of SnO2 SMPs and SnO2 MPs lead to the swelling and cracking of the active material layers, respectively, which will be easily peeled off from the copper foil. Therein, the loss of electron transport channels from copper foil to active materials will cause a rapid capacity decline. Notably, the SnO2/rGO composite (Fig. S5†) obtained by the conventional in situ composition route of adding GO in the generation solution of SnO2 NPs before the solvothermal reaction exhibited worse lithium and sodium storage performances (Fig. S20†). This can be primarily attributed to the inhomogeneous composition of SnO2 particles within the rGO matrix (Fig. S5†) due to the poor dispersibility of GO in the ethylene glycol/water mixture solvent and the aggregation of rGO nanosheets into matrices prior to the formation of SnO2 particles, which results in low conductivity (Fig. S21a†) and poor structural stability (Fig. S21b–f†). Thus, after uniform encapsulation of TA-capped SnO2 NPs in the rGO matrix by the robust solvent evaporation strategy (Fig. 8r), the rGO matrices and TA molecules can effectively buffer the small volume changes of SnO2 NPs, dramatically facilitate the rapid transmission of electrons, and robustly hinder the Sn coarsening among adjacent SnO2 NPs, which give rise to the ultrahigh cycling stability, superior rate capability and excellent reaction reversibility of SnO2 NPs-rGO.
3. Conclusion
In summary, we have developed a viable solvothermal-driven solid-to-solid transition (SDSST) strategy to craft ultrafine SnO2 NPs capped with TA, which were homogeneously encapsulated in a rGO matrix by a facile ball-milling combined with solvent evaporation route to provide superior lithium and sodium storage reactivity and reversibility. The confined sluggish in situ growth of SnO2 NPs on the surface of SnC2O4 MRs limits the rapid growth and aggregation of SnO2 NPs, and synergistically the tight capping of TA on the newly produced SnO2 NPs hinders their fusion. Notably, the loading density of SnO2 NPs in rGO matrices can be precisely controlled by an ex situ composition route, that is, solvent evaporation of their ball-milling dispersed uniform aqueous slurry with a designed mass ratio. Remarkably, the SnO2 NPs-GO nanocomposite stores 10.0 mol Li+ and 2.6 mol Na+ in per mol SnO2 after 800 and 100 cycles at 1000 and 20 mA g−1 as an anode of LIBs and SIBs, respectively. The electrochemical reaction mechanisms of SnO2 NPs-GO are revealed as the fully reversible three-step reactions of SnO2 for LIBs and the highly reversible conversion reaction between SnO and Sn for SIBs. The encouraging lithium storage capacity increase of SnO2 NPs-GO is attributed to the electrochemical activation and additional capacitive contribution. The tight layer-by-layer assembled architecture of SnO2 NPs embed in the rGO matrix enables the fast ion/electron transfer, outstanding volume expansion suppression, and effective Sn coarsening inhibition capabilities of SnO2 NPs-GO. The proposed progressive SDSST procedure might stand out as a convenient and scalable strategy to fabricate nanomaterials of interest with desired structural and compositional features under the assistance of functional additives for performance enhancement in a wide diversity of applications.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (21905208) and the Natural Science Foundation of Zhejiang Province (Y23B030001).References
- Z. P. Cano, D. Banham, S. Ye, A. Hintennach, J. Lu, M. Fowler and Z. Chen, Nat. Energy, 2018, 3, 279–289 CrossRef.
- H. Xue, H. Gong, Y. Yamauchi, T. Sasaki and R. Ma, Nano Res. Energy, 2022, 1, 9120007 CrossRef.
- L. Meng and L. Li, Nano Res. Energy, 2022, 1, 9120020 CrossRef.
- Z. Zhu, W. Zhong, Y. Zhang, P. Dong, S. Sun, Y. Zhang and X. Li, Carbon Energy, 2021, 3, 541–553 CrossRef CAS.
- G. Harper, R. Sommerville, E. Kendrick, L. Driscoll, P. Slater, R. Stolkin, A. Walton, P. Christensen, O. Heidrich, S. Lambert, A. Abbott, K. Ryder, L. Gaines and P. Anderson, Nature, 2019, 575, 75–86 CrossRef CAS.
- S. Zhang, L. Sun, Q. Fan, F. Zhang, Z. Wang, J. Zou, S. Zhao, J. Mao and Z. Guo, Nano Res. Energy, 2022, 1, 9120001 CrossRef.
- T. Or, S. W. D. Gourley, K. Kaliyappan, A. Yu and Z. Chen, Carbon Energy, 2020, 2, 6–43 CrossRef CAS.
- W. Ni, X. Li, L.-Y. Shi and J. Ma, Nanoscale, 2022, 14, 9609–9635 RSC.
- W. Cai, Y. X. Yao, G. L. Zhu, C. Yan, L. L. Jiang, C. He, J. Q. Huang and Q. Zhang, Chem. Soc. Rev., 2020, 49, 3806–3833 RSC.
- Y. Liu, Y. Zhu and Y. Cui, Nat. Energy, 2019, 4, 540–550 CrossRef.
- S. Yang, C. Liu, H. Li, S. Wang, J. Choi and L. Li, Energy Storage Mater., 2021, 39, 278–286 CrossRef.
- S. Zhao, Z. Wang, Y. He, B. Jiang, Y. Harn, X. Liu, F. Yu, F. Feng, Q. Shen and Z. Lin, ACS Energy Lett., 2016, 2, 111–116 CrossRef.
- S. Zhao, Z. Wang, Y. He, H. Jiang, Y. W. Harn, X. Liu, C. Su, H. Jin, Y. Li, S. Wang, Q. Shen and Z. Lin, Adv. Energy Mater., 2019, 9, 1901093 CrossRef.
- J. Feng, C. Yang, L. Zhang, F. Lai, L. Du and X. Yang, Carbon Energy, 2020, 2, 614–623 CrossRef CAS.
- Y. Li, F. Wu, Y. Li, M. Liu, X. Feng, Y. Bai and C. Wu, Chem. Soc. Rev., 2022, 51, 4484–4536 RSC.
- C. Zhao, Q. Wang, Z. Yao, J. Wang, B. Sánchez-Lengeling, F. Ding, X. Qi, Y. Lu, X. Bai, B. Li, H. Li, A. Aspuru-Guzik, X. Huang, C. Delmas, M. Wagemaker, L. Chen and Y.-S. Hu, Science, 2020, 370, 708–711 CrossRef CAS.
- Q. Li, S. Xu, S. Guo, K. Jiang, X. Li, M. Jia, P. Wang and H. Zhou, Adv. Mater., 2020, 32, 1907936 CrossRef CAS.
- Q. Liu, Z. Hu, M. Chen, C. Zou, H. Jin, S. Wang, S.-L. Chou, Y. Liu and S.-X. Dou, Adv. Funct. Mater., 2020, 30, 1909530 CrossRef CAS.
- N. Wang, J. Ma, Z. Liu, J. Xu, D. Zhao, N. Wang, C. Yang, Y. Cao, J. Lu and J. Zhang, Chem. Eng. J., 2022, 433, 133798 CrossRef CAS.
- S. Zhao, Y. He, Z. Wang, X. Bo, S. Hao, Y. Yuan, H. Jin, S. Wang and Z. Lin, Adv. Energy Mater., 2022, 12, 2201015 CrossRef CAS.
- W. Chen, X. Zhang, L. Mi, C. Liu, J. Zhang, S. Cui, X. Feng, Y. Cao and C. Shen, Adv. Mater., 2019, 31, 1806664 CrossRef.
- Z. Zhang, R. Wang, J. Zeng, K. Shi, C. Zhu and X. Yan, Adv. Funct. Mater., 2021, 31, 2106047 CrossRef CAS.
- Z. Tian, Y. Zhang, J. Zhu, Q. Li, T. Liu and M. Antonietti, Adv. Energy Mater., 2021, 11, 2102489 CrossRef CAS.
- S. Jia, Y. Wang, X. Liu, S. Zhao, W. Zhao, Y. Huang, Z. Li and Z. Lin, Nano Energy, 2019, 59, 229–236 CrossRef CAS.
- X. Guo, C. Wang, W. Wang, Q. Zhou, W. Xu, P. Zhang, S. Wei, Y. Cao, K. Zhu, Z. Liu, X. Yang, Y. Wang, X. Wu, L. Song, S. Chen and X. Liu, Nano Res. Energy, 2022, 1, 9120026 CrossRef.
- B. Jiang, Y. He, B. Li, S. Zhao, S. Wang, Y. B. He and Z. Lin, Angew. Chem., Int. Ed., 2017, 56, 1869–1872 CrossRef CAS.
- S. Zhao, C. D. Sewell, R. Liu, S. Jia, Z. Wang, Y. He, K. Yuan, H. Jin, S. Wang, X. Liu and Z. Lin, Adv. Energy Mater., 2019, 10, 1902657 CrossRef.
- H. Qiu, L. Zhao, M. Asif, X. Huang, T. Tang, W. Li, T. Zhang, T. Shen and Y. Hou, Energy Environ. Sci., 2020, 13, 571–578 RSC.
- S. M. Jung, D. W. Kim and H. Y. Jung, J. Am. Chem. A, 2020, 8, 8244–8254 CAS.
- J. Zhu, D. Wang, L. Wang, X. Lang and W. You, Electrochim. Acta, 2013, 91, 323–329 CrossRef CAS.
- X. Wang, X. Zhou, K. Yao, J. Zhang and Z. Liu, Carbon, 2011, 49, 133–139 CrossRef CAS.
- Z. Kong, X. Liu, T. Wang, A. Fu, Y. Li, P. Guo, Y.-G. Guo, H. Li and X. S. Zhao, Appl. Surf. Sci., 2019, 479, 198–208 CrossRef CAS.
- X. Wu, X. Lan, R. Hu, Y. Yao, Y. Yu and M. Zhu, Adv. Mater., 2022, 34, 2106895 CrossRef CAS.
- L. Zhou, J. Zhang, Y. Wu, W. Wang, H. Ming, Q. Sun, L. Wang, J. Ming and H. N. Alshareef, Adv. Energy Mater., 2019, 9, 1902194 CrossRef CAS.
- X. Lan, J. Cui, X. Zhang, R. Hu, L. Tan, J. He, H. Zhang, X. Xiong, X. Yang, S. Wu and M. Zhu, Adv. Mater., 2022, 34, 2106366 CrossRef CAS PubMed.
- C. Gao, Z. Jiang, P. Wang, L. R. Jensen, Y. Zhang and Y. Yue, Nano Energy, 2020, 74, 104868 CrossRef CAS.
- B. Song, P. Loya, L. Shen, C. Sui, L. He, H. Guo, W. Guo, M.-T. F. Rodrigues, P. Dong, C. Wang, X. He, P. M. Ajayan and J. Lou, Nano Energy, 2018, 53, 277–285 CrossRef CAS.
- H.-G. Wang, Q. Wu, Y. Wang, X. Wang, L. Wu, S. Song and H. Zhang, Adv. Energy Mater., 2019, 9, 1802993 CrossRef.
- C. Zhu, X. Xia, J. Liu, Z. Fan, D. Chao, H. Zhang and H. J. Fan, Nano Energy, 2014, 4, 105–112 CrossRef CAS.
- Y. Huang, D. Wu, J. Jiang, Y. Mai, F. Zhang, H. Pan and X. Feng, Nano Energy, 2015, 12, 287–295 CrossRef CAS.
- L. Yang, T. Dai, Y. Wang, D. Xie, R. L. Narayan, J. Li and X. Ning, Nano Energy, 2016, 30, 885–891 CrossRef CAS.
- H. Wu, W. Yuan, X. Yuan and L. Cheng, Carbon Energy, 2022 DOI:10.1002/cey2.229.
- H. Xu, C. Peng, Y. Yan, F. Dong, H. Sun, J. Yang and S. Zheng, Carbon Energy, 2019, 1, 276–288 CrossRef CAS.
- W. Ni, J. Cheng, L. Shi, X. Li, B. Wang, Q. Guan, L. Huang, G. Gu and H. Li, J. Am. Chem. A, 2014, 2, 19122–19130 CAS.
- W. Ni, Y. Wang and R. Xu, Part. Part. Syst. Charact., 2013, 30, 873–880 CrossRef CAS.
- S. Choi, C. Kim, J. M. Suh and H. W. Jang, Carbon Energy, 2019, 1, 85–108 CrossRef CAS.
- R. Fang, K. Chen, L. Yin, Z. Sun, F. Li and H.-M. Cheng, Adv. Mater., 2019, 31, 1970066 CrossRef.
- Q. Zhang, X. Cheng, C. Wang, A. M. Rao and B. Lu, Energy Environ. Sci., 2021, 14, 965–974 RSC.
- J. Wang, C. Ma, X. Mu, X. Zhou, L. He, Y. Xiao, L. Song and Y. Hu, Chem. Eng. J., 2020, 402, 126221 CrossRef CAS.
- X. Liu, J. Guo, T. Liu, J. Zhang, Z. Jia and C. Zhang, Energy Storage Mater., 2021, 35, 520–529 CrossRef.
- J. Sang, K. Liu, X. Zhang, S. Zhang, G. Cao, Y. Shen and G. Shao, Energy Environ. Mater., 2022, e12431, DOI:10.1002/eem2.12431.
- S. K. Jung, I. Hwang, D. Chang, K. Y. Park, S. J. Kim, W. M. Seong, D. Eum, J. Park, B. Kim, J. Kim, J. H. Heo and K. Kang, Chem. Rev., 2020, 120, 6684–6737 CrossRef CAS PubMed.
- F. Nudelman and N. A. Sommerdijk, Angew. Chem., Int. Ed., 2012, 51, 6582–6596 CrossRef CAS.
- J. J. M. Lenders, G. Mirabello and N. Sommerdijk, Chem. Sci., 2016, 7, 5624–5634 RSC.
- A. Ramírez, L. Sierra, M. Mesa and J. Restrepo, Chem. Eng. Sci., 2005, 60, 4702–4708 CrossRef.
- Y. Zhang, D. Yan, Z. Liu, Y. Ye, F. Cheng, H. Li and A.-H. Lu, ACS Nano, 2021, 15, 16946 CrossRef CAS PubMed.
- I. Begum, S. Shamim, F. Ameen, Z. Hussain, S. A. Bhat, T. Qadri and M. Hussain, Processes, 2022, 10, 716 CrossRef CAS.
- M. T. Rahman, S. M. S. Rana, M. A. Zahed, S. Lee, E.-S. Yoon and J. Y. Park, Nano Energy, 2022, 94, 106921 CrossRef CAS.
- Z. Hu, Y. Zou, C. Xiang, L. Sun, F. Xu, M. Jiang and S. Yu, Carbon Energy, 2022, 4, 1214–1227 CrossRef CAS.
- H. Ying and W. Q. Han, Adv. Sci., 2017, 4, 1700298 CrossRef.
- V. Augustyn, J. Come, M. A. Lowe, J. W. Kim, P. L. Taberna, S. H. Tolbert, H. D. Abruna, P. Simon and B. Dunn, Nat. Mater., 2013, 12, 518–522 CrossRef CAS PubMed.
- T. Brezesinski, J. Wang, J. Polleux, B. Dunn and S. H. Tolbert, J. Am. Chem. Soc., 2009, 131, 1802–1809 CrossRef CAS PubMed.
- P. Ge, S. Li, L. Xu, K. Zou, X. Gao, X. Cao, G. Zou, H. Hou and X. Ji, Adv. Energy Mater., 2019, 9, 1803035 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, structural characterization and electrochemical properties of SnO2 NPs-rGO and control samples, HRTEM images of the cycled electrodes of SnO2 NPs-rGO and control samples, and summary of the performances of graphene-based SnO2 materials as anodes of LIBs/SIBs. See DOI: https://doi.org/10.1039/d2ta07435d |
| This journal is © The Royal Society of Chemistry 2023 |