
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffective perspiration is essential to uphold the stability of zero-gap MEA-based cathodes used in CO2 electrolysers†
Huifang
Hu
 a,
Ying
Kong
a,
Menglong
Liu
a,
Viliam
Kolivoška
b,
Alexander V.
Rudnev
ac,
Yuhui
Hou
a,
Rolf
Erni
d,
Soma
Vesztergom
*ae and
Peter
Broekmann
*a
a,
Ying
Kong
a,
Menglong
Liu
a,
Viliam
Kolivoška
b,
Alexander V.
Rudnev
ac,
Yuhui
Hou
a,
Rolf
Erni
d,
Soma
Vesztergom
*ae and
Peter
Broekmann
*a
aNCCR Catalysis, University of Bern, Department of Chemistry, Biochemistry and Pharmaceutical Sciences, Freiestrasse 3, 3012 Bern, Switzerland. E-mail: peter.broekmann@unibe.ch; vesztergom@chem.elte.hu
bJ. Heyrovský Institute of Physical Chemistry of the Czech Academy of Sciences, Dolejškova 3, 182 23 Prague, Czechia
cA. N. Frumkin Institute of Physical Chemistry and Electrochemistry, Russian Academy of Sciences, Leninsky Prospekt 31, 119071 Moscow, Russia
dSwiss Federal Laboratories for Materials Science and Technology (EMPA), Electron Microscopy Center, Überlandstrasse 129, 8600 Dübendorf, Switzerland
eEötvös Loránd University, MTA-ELTE Momentum Interfacial Electrochemistry Research Group, Pázmány Péter Sétány 1/A, 1117 Budapest, Hungary
First published on 12th December 2022
Abstract
The application of gas diffusion electrodes (GDEs) for the electrochemical reduction of CO2 to value-added products creates the possibility of achieving current densities of a few hundred mA cm−2. To achieve stable operation at such high reaction rates remains, however, a challenging task, due to the flooding of the GDE. In order to mitigate flooding in a zero-gap membrane-electrode assembly (MEA) configuration, paths for effective electrolyte perspiration inside the GDE structure have to be kept open during the electrolysis process. Here we demonstrate that apart from the operational parameters of the electrolysis and the structural properties of the supporting gas diffusion layers, also the chemical composition of the applied catalyst inks can play a decisive role in the electrolyte management of GDEs used for CO2 electroreduction. In particular, the presence of excess amounts of polymeric capping agents (used to stabilize the catalyst nanoparticles) can lead to a blockage of micropores, which hinders perspiration and initiates the flooding of the microporous layer. Here we use a novel ICP-MS analysis-based approach to quantitatively monitor the amount of perspired electrolyte that exits a GDE-based CO2 electrolyser, and we show a direct correlation between the break-down of effective perspiration and the appearance of flooding—the latter ultimately leading to a loss of electrolyser stability. We recommend the use of an ultracentrifugation-based approach by which catalyst inks containing no excess amount of polymeric capping agents can be formulated. Using these inks, the stability of electrolyses can be ensured for much longer times.
1 Introduction
Global warming, caused by the burning of fossil fuels and the resulting rapid increase of atmospheric CO2 levels, is regarded as one of the most serious problems faced by human society. From this respect, electrochemical CO2 reduction deserves special attention. Propelled by excess renewable energy sources, this process may not only provide viable means to reduce CO2 emissions, but it can also create the possibility of converting excess energy into the form of storable fuels.1The successful design of industrial level CO2 electrolysers that can be operated for extended periods of time, converting CO2 to a set of desired products at high rate and at low energy costs, requires a well-coordinated interplay of all electrolyser constituents.2–5 From the chemistry, yet not from the overall efficiency point of view, the most essential part of a CO2 electrolyser unit is the cathode catalyst: this is the scene where the actual transition of CO2—its reduction to value-added products—takes place. In the past three decades, huge research efforts have thus been invested in the development of electrocatalysts that can achieve considerable reaction yields, preferably at low overvoltages.6,7
Today, researchers agree that apart from their chemical composition it is also the nanoscale structure of electrocatalysts that mostly affect their performance in CO2 electroreduction.8–10 Metallic catalysts of nanoparticulate (NP) form, for example, usually exhibit a much higher mass-normalised activity than planar metallic surfaces.11 This is mostly due to the large surface area of NPs that offers more active sites with low coordination numbers, enabling improved performances.12 Metallic NPs thus seem to become the most favoured catalysts used in today's CO2 electrolysers, and the reason for this is not only their low raw material cost, but also that they can easily be applied in gas diffusion electrodes (GDEs).
The main advantage of using GDEs as cathodes for CO2 electroreduction is that they circumvent the problem of limited solubility and slow transport in aqueous media, by allowing CO2 to access the catalyst layer mostly in a gas phase.13 In GDEs, the catalyst is supported by a gas diffusion layer (GDL) that is composed of a carbon fibrous layer (CFL) and a microporous layer (MPL), as shown in Fig. 1a. Besides enabling fast reactant delivery, the GDL also facilitates the release of gaseous reaction products, serves as a mechanical support and electrical contact for the catalyst, and plays important role in controlling the amount of electrolyte in the catalyst layer.14
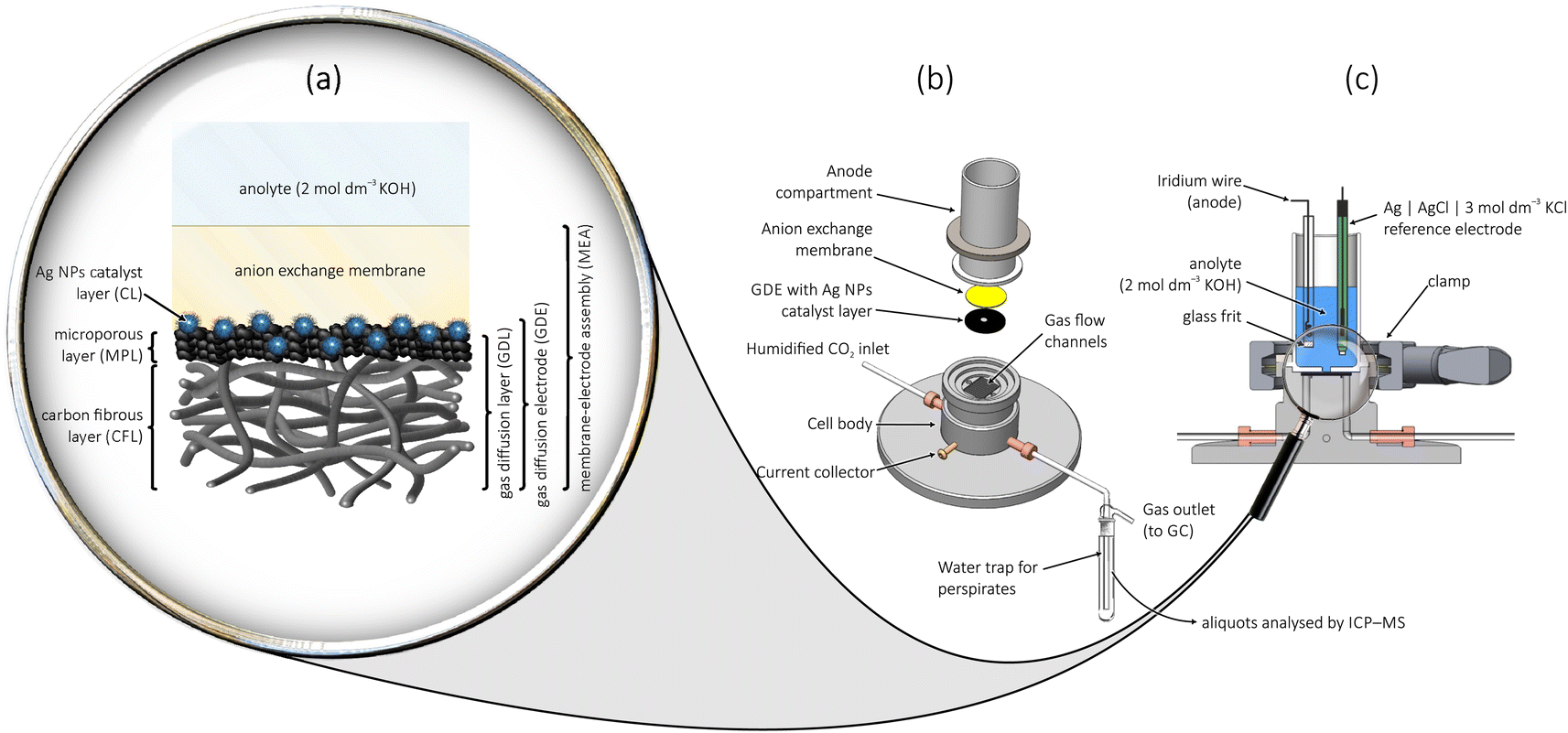 | ||
| Fig. 1 A zero-gap membrane-electrode assembly (MEA) cathode employing a gas diffusion electrode, used in a flow-cell for the electrolysis of CO2. Part (a) shows a close-up view of the MEA, parts (b) and (c) show the exploded and the cross-sectional view of the model-scale electrolyser. | ||
In so-called zero-gap membrane-electrode assemblies (MEAs, see Fig. 1a for a model-scale version), the GDE is directly interfaced to an anion exchange membrane, the primary role of which is to control the amount of water (an important reactant of cathodic CO2 reduction) reaching the catalyst layer, while anionic species (carbonate or hydroxide ions) can be transported away from it,15 in the direction of the anode. The membrane should further assure that no volatile cathode-generated products can cross over to the anode, thereby reducing the efficiency of electrolysis.16 When zero-gap MEA cathodes are employed in electrolyser set-ups similar to that shown in Fig. 1, current densities ranging a few hundred mA cm−2 can be achieved.17
Provided that at a given catalyst CO2 reduction yields only gas-phase carbon-containing products, the configuration shown in Fig. 1 indeed provides an excellent platform for the performance and stability testing of catalyst materials. We have recently shown, for example, that when Ag nanocubes are used to catalyse CO2 electroreduction (on Ag, the reaction almost exclusively yields CO), catalyst corrosion at stringent cathodic conditions occurs, and leads to truncated cube morphologies and the appearance of smaller Ag NPs.17 When using, on the other hand, Ag nanospheres protected by different ligands as a catalyst, degradation was shown to follow different pathways including corrosion, aggregation and even Ostwald ripening18—all depending on the chemical nature of the stabilizing ligand. In ref. 17 and 18 we have also found, however, that the degradation of Ag NPs, while it does occur at harshly cathodic operating conditions, is not in itself responsible for the observed, often severe, stability issues of the CO2 electrolysis process performed in zero-gap MEA cathode configurations.
In zero-gap MEA cathode-based electrolysers, stability losses rather occur due to the flooding of the MPL during prolonged electrolyses. Flooding is the excess hydration of the GDE that can severely limit or even block the access of CO2 to the catalyst layer, leading to the decrease of the observable faradaic efficiency (FE) of CO2 reduction products and the appearance of parasitic hydrogen evolution.14,19,20 The rapid flooding of GDEs, and the related stability issues are currently recognised as the biggest obstacle in front of the scale-up prospects21 of CO2 electroreduction. As a result, the focus of studies in the field of CO2 electrolysis has recently shifted from the identification of new catalyst materials to other, more engineering-related tasks that include the optimisation of electrolyser geometries, the design of GDEs and membranes, and the fine-tuning of the applied process parameters.2
Amongst these tasks, the adaptation of design strategies that can help prevent, or at least slow down flooding-related stability losses are of prime importance. It was recently shown, for example, that the presence of sub-millimeter wide cracks in the MPL can effectively mitigate the flooding of GDEs during CO2 electrolysis utilizing Ag nanowire catalysts.20 Rapid flooding, in this case, was avoided by a pathway that the surface cracks of the MPL enabled for electrolyte perspiration22,23 in addition to those provided by micropores.20
At this point it is important to emphasize the conceptual difference between electrolyte perspiration and flooding. Perspiration (a term we owe to Jeanty et al.22) refers to the cross-over of electrolyte through the GDE structure, which ultimately results in the appearance of electrolyte droplets on the back side of the GDE, and even in the gas outflow of the cathode gas compartment. The somewhat interrelated effect of flooding occurs when the electrolyte that entered the GDE structure has no means to escape it, so it remains adhered to the MPL. This trapped electrolyte, due to its large KOH concentration, reacts with CO2 and forms a large amount of carbonate (or bicarbonate) plaques that will limit and eventually block the access of CO2 to the catalyst particles.
As was recently pointed out,20,22,23 the flooding of the catalyst layer and the entrapment of electrolyte inside the GDE structure can be avoided by taking measures to uphold optimal perspiration rates, e.g., by the selection of appropriate cathode-side CO2 flow rates22,23 or the application of GDLs with cracked surfaces.20 Apart from optimizing the structure of MPLs or the operational parameters of electrolyses there is, however, more room for development, especially if we consider that essentially all parts of the MEA can influence flooding and perspiration phenomena. In this vein, the aim of the present paper will be to identify a critical point of zero-gap MEA cathode fabrication for CO2 reduction: that is, the excess use of polymeric capping agents (CAs) in the catalyst ink formulations of Ag NP-based catalysts, and its flooding promoting—perspiration blocking—effect.
CAs are commonly used in the synthesis of metallic NPs to achieve uniform and stable size distribution of the particles. By adsorbing to the surface of the NPs, CAs prevent their aggregation either by providing sterical or electro-repulsive protection.24 For the synthesis of Ag NPs, poly-(vinylpyrrolidone) (PVP) and branched polyethylenimine (BPEI) are very often used polymeric CAs that provide sterical protection to the NPs,17,25–28 while on the other hand citrate ions are often used as a non-polymeric (coulombic repulsion based) stabilizer.29
Practically regardless of the nature of the stabilizing CA, in aqueous Ag NP dispersions—either self-synthesised or purchased off-the-shelf—CAs are usually present at a rather high concentration, with not all of the ligand molecules adsorbed on the surfaces of NPs, but a majority of them being dissolved in the liquid phase.
In this study we show that the excess polymeric CA content, if not removed from the catalyst ink, blocks perspiration channels and acts as a promoter of flooding, thereby causing serious stability issues observed in zero-gap MEA cathode-based electrolysers used for CO2 electroreduction.30 In the paper we utilize a novel ICP-MS analysis-based approach to quantitatively monitor the amount of perspired electrolyte that exits the zero-gap MEA cathode and ends up in the water trap equipped to the gas outflow channel of the electrolyser (see Fig. 1b). We show a direct correlation between the break-down of effective perspiration and the appearance of flooding in the MPL—the latter ultimately leading to a loss of electrolyser stability. We also present a simple ultracentrifugation technique, by which the polymeric CA content of formulated catalyst inks can be reduced, and we show that this strategy in itself helps to provide longer time stability to CO2 electrolysers operated at high current densities. In the paper we work with PVP- and BPEI-capped Ag NPs as representatives of polymeric CA-stabilised catalysts, and we also extend our tests to Ag NPs stabilised by citrate ligands (see Fig. 2 for the applied ligand structures). We come to the conclusion that the small, monomeric ligand (citrate) has less adverse effects on the electrolysis durability.
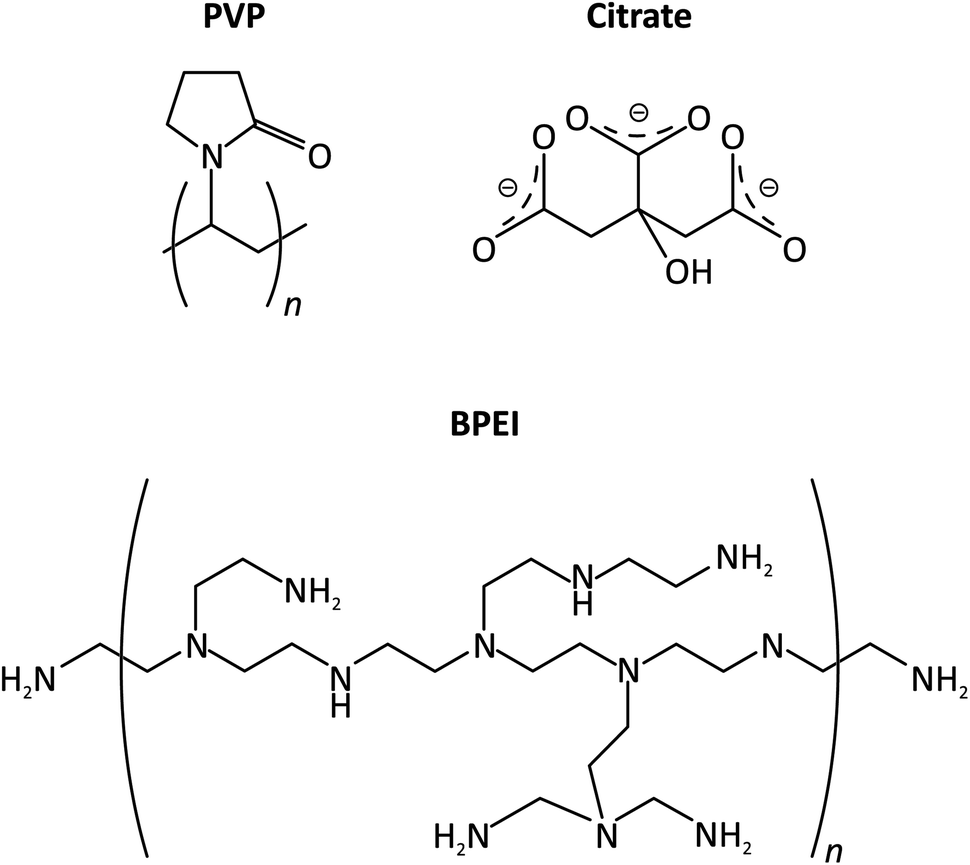 | ||
| Fig. 2 The structure of CAs studied in this paper, used for the stabilisation of Ag NP suspensions. | ||
The use of the model scale electrolyser shown in Fig. 1 for the electrochemical stress-test of GDE cathodes in a zero-gap cathode MEA configuration is particularly advantageous, since due to its small effective cross-section (that limits the geometric surface area of the cathode to 0.0707 cm2, see the Experimental section for details) the cell enables high current densities to be reached at the cost of relatively small applied currents (see Fig. S1 in ESI†). Furthermore, due to more pronounced edge effects introduced by the small cathode area,31 cell failures already manifest during relatively short (few hours long) electrolyses, making our test system a promising tool for the accelerated durability testing32 of CO2-to-CO converting GDEs.
2 Experimental
Ag NP suspensions: CA removal
PVP-capped spherical Ag NP suspensions (of ∼10 nm and ∼100 nm diameter), BPEI-capped spherical Ag NP suspensions (of ∼50 nm diameter) and citrate-capped Ag NP suspensions (also of ∼50 nm diameter) were purchased from NanoComposix (all water-based, with a concentration of 1 mg cm−3). The suspensions were either used as-purchased for the preparation of catalyst inks, or—in order to remove at least some of their CA content—were made subject to centrifugation. In this latter case, the suspensions were first diluted 8-fold, and then centrifuged in an Optima XPN-80 Ultracentrifuge (Beckman Coulter) for 40 min at 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm. (Suspensions with a nanoparticle diameter of ∼10 nm were centrifuged for an additional 40 min at 50000 rpm.) Following decantation of the supernatant, NPs in the pellet were re-dispersed in ultrapure water (Milli-Q by Merck Millipore, 18.2 MΩ cm specific resistance) to restore the original concentration of 1 mg cm−3. The 8-fold dilution/centrifugation/re-dispersion steps were then repeated once for an as thorough as possible removal of the CA content (steps are illustrated by Fig. 3). To quantify the effectiveness of CA removal, total organic carbon content (TOC) and total nitrogen content (TNC) measurements were made for the supernatant of the first centrifugation step and for the finally obtained (for better comparability, 8-fold diluted) CA-deficient suspension (see Table 1 for results). For TOC and TNC determination, 0.1 cm3 sample volumes were fed into a DIMATOC 2100 instrument (Dimatec Analysentechnik GmbH, Essen, Germany). The organic compounds were oxidized at 850 °C with Pt catalyst to CO2 and NO, the amounts of which were quantified by a non-dispersive infrared gas sensor with a reflective diffuser and a chemiluminescence detector, respectively.
000 rpm. (Suspensions with a nanoparticle diameter of ∼10 nm were centrifuged for an additional 40 min at 50000 rpm.) Following decantation of the supernatant, NPs in the pellet were re-dispersed in ultrapure water (Milli-Q by Merck Millipore, 18.2 MΩ cm specific resistance) to restore the original concentration of 1 mg cm−3. The 8-fold dilution/centrifugation/re-dispersion steps were then repeated once for an as thorough as possible removal of the CA content (steps are illustrated by Fig. 3). To quantify the effectiveness of CA removal, total organic carbon content (TOC) and total nitrogen content (TNC) measurements were made for the supernatant of the first centrifugation step and for the finally obtained (for better comparability, 8-fold diluted) CA-deficient suspension (see Table 1 for results). For TOC and TNC determination, 0.1 cm3 sample volumes were fed into a DIMATOC 2100 instrument (Dimatec Analysentechnik GmbH, Essen, Germany). The organic compounds were oxidized at 850 °C with Pt catalyst to CO2 and NO, the amounts of which were quantified by a non-dispersive infrared gas sensor with a reflective diffuser and a chemiluminescence detector, respectively.
 | ||
| Fig. 3 Scheme of the ultracentrifugation-based method used for the removal of the excess CA content of Ag NP suspensions. | ||
| Capping agent | Nominal size of Ag NPs | TOC/ppm | TNC/ppm | ||
|---|---|---|---|---|---|
| Before | After | Before | After | ||
| PVP | ∼10 nm | 43.15 | 6.12 | 7.88 | 0.69 |
| PVP | ∼100 nm | 28.51 | 4.45 | 4.24 | 0.33 |
| BPEI | ∼50 nm | 7.53 | 3.81 | 3.09 | — |
| Citrate | ∼50 nm | 15.21 | 2.88 | — | — |
Catalyst ink preparation
Ag NP suspensions with 1 mg cm−3 Ag content (either as-purchased or centrifuged) were used for the catalyst ink preparation. A 1 cm3 volume of the Ag NP suspension was mixed with 627 μl of isopropanol (VLSI Selectipur, BASF, Germany). A second suspension was then made by dispersing carbon black (Vulcan XC 72R, Cabot) in isopropanol in a 0.5 mg cm−3 concentration by 1 hour sonication. The Ag NP suspension and 353 μl of the carbon black suspension were then intermixed, 20 μl of Nafion solution (5% in a mixture of lower aliphatic alcohols and water, Sigma-Aldrich) was added, and the resulting ink was homogenised by 5 min sonication.Preparation of the GDEs
75 μl of the prepared catalyst ink was drop-cast on the microporous surface of a circular Sigracet 36BB (FuelCellStore) GDL with 4 mm of diameter to form a GDE. Electrodes were dried at ambient conditions. Drop-casting 75 μl of the catalyst ink resulted in a nominal mass loading of ∼300 μg cm−2 for silver. GDEs with lower loading were prepared by drop-casting proportionally less amount of the catalyst ink.Assembly of the electrolyser
The electrolyser shown in Fig. 1 was used to carry out electrochemical CO2 reduction. The bottom part of the electrolyser is made of stainless steel and is equipped with gas flow channels. The small circular GDEs containing the Ag NP catalyst were placed in a centrally located 4 mm diameter hole of a Freudenberg H23C8 (FuelCellStore) carbon paper with an external diameter of 2 cm, with the catalyst layer facing upwards and the bottom CFL facing the gas flow channels. The GDE was covered by a hydroxide-functionalised Sustainion anion exchange membrane (X3750 RT, Dioxide Materials, diameter of 2 cm) from above, before fixing the Teflon-made anode compartment on top. The anode compartment has a central orifice on its bottom, allowing access of the anolyte to the membrane. The orifice is of 3 mm diameter and it determines the geometric surface area (0.0707 cm2) of the GDE. The anode compartment is filled with 10 cm3 of 2 mol per dm3 KOH solution (reagent grade, 90%, Sigma-Aldrich), and contains an Ag|AgCl|3 mol per dm3 KCl reference electrode (double junction, Metrohm) and an Ir wire anode (99.9%, Goodfellow GmbH). The anode is placed inside a small chamber and is separated from the rest of the anolyte by glass frit, as shown in Fig. 1.Electrochemical measurements and product analysis
All electrochemical measurements were carried out using an ECi-200 potentiostat (Nordic Electrochemistry) at constant current. The applied currents of −20, −10 and −5 mA correspond to geometric surface area normalised current densities of −283.0, −141.5 and −70.7 mA cm−2. Cathode potentials reported in the paper are all referred to the applied Ag|AgCl|3 mol per dm3 KCl reference electrode, and are corrected for IR drop (the cell resistance was followed by high frequency impedance measurements). On the Ag NP catalysts used in our studies, CO was the only detectable gaseous product of CO2 reduction with H2 formed as the product of parasitic hydrogen evolution. Gaseous reaction products were analysed by connecting the gas outlet of the electrolyser to a gas chromatograph (SRI Instruments Multigas Analyzer). The continuous flow of the carrier CO2 gas through the electrolysis cell carried reaction products from the gas outlet of the electrolyser into the sampling loops of the gas chromatograph. The partial current Ii, corresponding to the formation of a gaseous product i, can be calculated33 as| Ii = xiniFvm, | (1) |
485.3 C mol−1 is Faraday's constant; and vm is the molar CO2 gas flow rate measured by a universal flowmeter (7000 GC flowmeter, Ellutia) at the gas outlet of the electrolyser. The faradaic efficiency (FE) of a given reaction product was determined by dividing the respective partial current, calculated from eqn (1), by the total current. A thermal conductivity detector (TCD, for the detection of H2) and a flame ionization detector (FID, for the detection of CO) were equipped to the gas chromatograph. In our experiments, the formed CO and H2 amounts accounted for an about 90% FE. Following long time electrolyses, some amounts of formate (HCOO−) were detected in the anolyte compartment, which could account for the less than 100% total FE, although an exact quantification of this product is not possible (due to the fact that formate is partially consumed by oxidation at the anode).
Scanning electron microscopy (SEM), energy-dispersive X-ray spectroscopy (EDX), and transmission electron microscopy (TEM)
A Zeiss Gemini 450 scanning electron microscope equipped with an InLens secondary electron detector and a back-scattering detector was used for the morphological analysis of GDEs, prior to and following electrolyses. An accelerating voltage of 5 kV and currents of 70 to 100 pA were applied at a working distance of 4.0 to 5.1 mm. The Smile View software was used to measure the size of particles. Particle size distribution histograms were created by determining the size of at least 500 individual particles based on micrographs taken from at least three randomly selected areas.18 EDX spectra were acquired and elemental composition maps were composed by using the AZtec 5.0 software (Oxford Instruments). The applied acceleration voltage and current were 10 kV and 1 nA, respectively, and a working distance of 8.5 mm was set for all EDX measurements. For the transmission electron microscopy (TEM) imaging, an FEI Titan Themis instrument was used with an accelerating voltage of 300 kV.Inductively coupled plasma mass spectrometry (ICP-MS)
ICP-MS (NExION 2000, PerkinElmer) was applied to determine the mass of potassium that perspired through the membrane and the GDE, and exited the electrolyser through the gas outlet. A trap containing 15 cm3 of ultrapure water was used to collect perspired potassium salts. Aliquots of the collected perspired material were diluted by an appropriate amount of 2% HNO3 solution (BASF SE, Ludwigshafen, Germany) and the resulting solution samples were injected into the ICP-MS to obtain the content of potassium in perspiration.Raw data, as well as unprocessed measurement files serving as a basis of this publication can be downloaded from Zenodo.34
3 Results and discussion
In this paper we use Ag NPs stabilised by different capping agents (PVP, BPEI and citrate, Fig. 2) for the creation of cathode GDEs that we integrate in the zero-gap MEA configuration (Fig. 1) to carry out durability tests at high current density. In order to study the effect of CAs exerted on the electrolysis stress-tests, in some cases we removed the majority of the CA content from the Ag NP suspensions before preparing the catalyst ink by using the ultracentrifugation-based method described in the Experimental section.The possibility that (especially, polymeric) CAs can have an adverse effect on the stability of electrolyses first occurred to us when we used as-purchased, PVP-stabilised Ag NPs of ∼10 nm nominal diameter in order to formulate catalyst inks with a carbon black support, and we attempted to define the optimal loading of the catalyst by one hour-long galvanostatic electrolyses.
These tests gave the rather counter-intuitive result (Fig. 4) that by increasing the nominal mass loading of Ag, the obtained GDEs became less stable, tending to lose their (initially high) CO production efficiency more quickly, and beginning to form H2 after shorter and shorter times. Concomitantly with the loss of CO faradaic efficiency, the cathode potential also shifted to more negative values as instead of CO2 reduction, hydrogen evolution became the dominant electrode reaction.
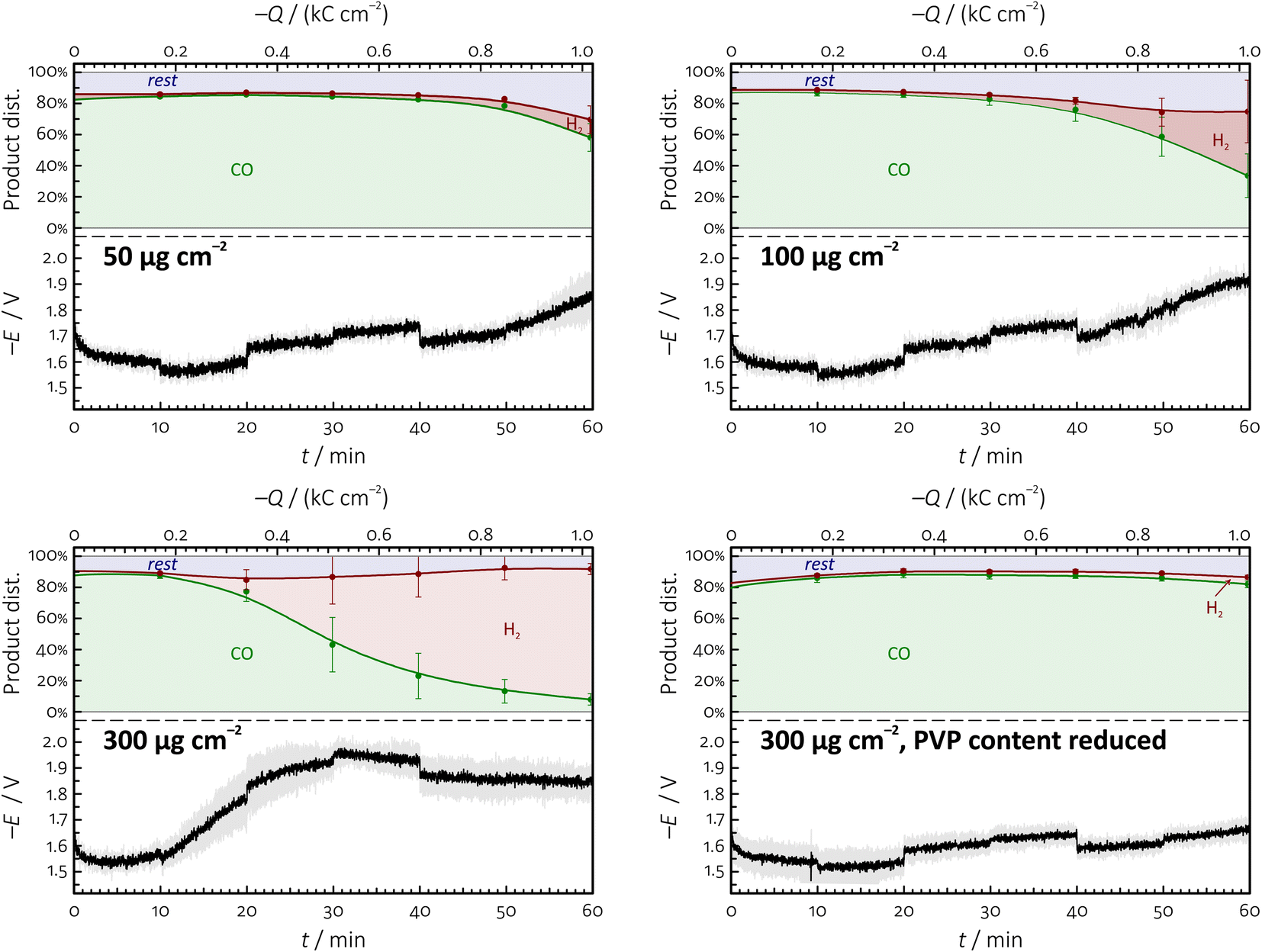 | ||
| Fig. 4 Temporal variation of the product distribution of CO2 reduction (top panels) and the measured cathode potential (bottom panels) during one hour long galvanostatic electrolyses (geometric surface area-normalised current density: −283 mA cm−2) conducted on GDEs prepared with different Ag NP loadings shown in the figure. Error bars represent 95% confidence intervals. Nominal size of the Ag NPs used as catalyst: ∼10 nm. The area labelled “rest” in the product distribution plots can mostly be attributed to the production of small amounts of formate during electrolyses, see the Experimental section for details. | ||
We found that the above equivocal result (higher loading—less stability) can be explained by another experiment, in which we reduced, by ultracentrifugation, the PVP content of the applied Ag NP dispersion before it was used for the preparation of the catalyst ink. By 2 times centrifugation, the majority of the PVP-content of the ∼10 nm Ag NP suspension was removed (see Table 1 in the Experimental section for details), and this helped to maintain the stability of electrolysis even when the catalyst was applied at high loading. Indeed, comparing the plots of the bottom row of Fig. 4 immediately makes it apparent that it was the excess amount of PVP present in the catalyst ink—and not the increase of the silver loading in itself—that exerted a detrimental effect on the stability of electrolysis.
In order to unravel the exact reasons of this effect, we made two GDEs (one prepared from an Ag NP suspension of the original PVP content, other with less PVP) subject to further investigation.
The top-down SEM images (Fig. 5) obtained from the two catalyst layers (one prepared with the original, another with reduced PVP content) reveal only slight differences. Silver NPs well-dispersed in a matrix of carbon flakes over the MPL are well distinguishable in both cases. If the catalyst ink is prepared using the as-purchased NP suspension (nominal diameter: ∼10 nm) of high PVP content, the size distribution of the NPs is fairly symmetric and is centred at about 11.5 nm. If on the other hand the majority of the PVP content is removed by centrifugation, and the catalyst ink is prepared using a suspension gained by re-dispersing the particles in pure water, slight aggregation of the particles can be observed, resulting in a broadened and tailed size distribution histogram with a slightly increased maximum located at 16.2 nm. This indicates that removing the majority of PVP from the suspension of the Ag NPs results in a detectable but minor agglomeration of the particles on the GDE surface.
 | ||
| Fig. 5 Top-down SEM images of different magnification, showing catalyst layers containing the original (to the left) and reduced amount of PVP (to the right). Reducing, by ultracentrifugation, the amount of the stabilizing agent PVP leads to a broadening of the size distribution of the NPs as shown by the histograms. The cross-sectional view of the MPLs prove that the partial removal of the PVP content from the Ag NP suspensions exerts no considerable effect on the vertical distribution of the NPs. The EDX-based Ag elemental map (with red pixels corresponding to high Ag concentration) is superimposed on the cross-sectional SEM micrographs. EDX signals averaged over some selected areas, labelled from (a)–(e) clearly exhibit peaks assigned to Ag with no particular vertical intensity variation. Nominal size of the Ag NPs used as catalyst: ∼10 nm, applied mass loading: 300 μg per cm2 Ag. For more details on the structure of PVP-capped Ag NPs (TEM images), see Fig. S2 of the ESI.† | ||
In order to determine to which extent the catalyst particles penetrate the microporous layer, we carried out cross-sectional SEM/EDX investigations, the results of which are shown in the bottom row of Fig. 5. EDX signals collected from different depth of the MPL all show strong peaks related to Ag, indicating that the NPs well penetrate the micropores of the MPL, practically regardless to whether they originate from as-purchased or centrifuged (PVP-deficient) suspensions.
Fig. 5 thus reveals no significant differences between GDEs prepared from Ag NP suspensions of the originally high PVP content and those prepared from PVP-deficient suspensions. Yet, when the two GDEs are made subject to prolonged electrolysis lasting 2.5 hours, differences between their stability immediately become apparent, as shown in Fig. 6.
 | ||
| Fig. 6 Comparison of the results of electrolysis stress-tests on two different GDEs prepared with PVP-capped Ag NPs of 10 nm nominal diameter. The PVP content of the Ag NP suspensions was either unchanged (left side) or was reduced by ultracentrifugation (right side) during the course of catalyst ink preparation; in both cases, the catalyst was applied at a mass loading of 300 μg per cm2 Ag. The plots show the variation of the product distribution of CO2 reduction, the measured electrode potential E (referenced vs. an Ag|AgCl|3 mol per dm3 KCl electrode), and the mass of K+ ions collected in the liquid trap equipped to the gas outflow of the electrolyser, as a function of both time and passed charge, for galvanostatic electrolyses carried out at −283 mA cm−2. Structural changes of the applied GDEs were monitored by recording top-down and cross-sectional SEM/EDX images of the GDEs obtained before, as well as 60 and 150 min after the electrolysis. | ||
It is clearly visible in the product distribution plots of Fig. 6 that both normal and PVP-deficient GDEs exhibit a high (>90%) faradaic efficiency for CO production at the start of the electrolysis. However, in GDEs of high excess PVP content the initially high selectivity towards CO formation is quickly lost, and in less than 30 minutes the FE of CO formation drops down to below 50%. On the other hand, FECO values measured on PVP-deficient GDEs remain rather high, with significant decrease appearing only after 1 hour, and the efficiency dropping below 50% only after about 90 minutes of electrolysis. That the shortage of CO production is due to the appearance of hydrogen evolution is also indicated by the E vs. t plots of Fig. 6, showing cathode potentials shifting to more negative values concomitantly with the observed FECO loss.
That the observed stability losses—both on GDEs with originally high and of reduced PVP content—occur due to flooding can be demonstrated by the post-electrolysis top-down and cross-sectional SEM and EDX mapping of the GDEs. The SEM micrographs and K elemental maps shown in Fig. 6, recorded after 60 and 150 minutes of electrolyses, clearly indicate the presence of a significant amount of K+ (in the form of K2CO3/KHCO3 precipitates) both on-top and inside the GDE structure. The appearance of K+ in the cathode GDE serves as a direct proof of anolyte cross-over through the membrane, and as such, the formation pattern of K+ precipitates inside the GDE acts as a tracer of electrolyte intrusion to the GDE.19,20
While the as-prepared GDEs are essentially potassium-free, the post-electrolysis determination of the K-content of GDEs by digestion in concentrated HNO3 and follow-up ICP-MS measurements indicate that already at the first 60 minutes of electrolysis, K+ ions accumulate within the GDE structure in an amount of approximately 4 mg cm−2, normalised to the geometric surface area of the GDE (see Fig. 6 for exact values).
While the total amount of precipitated K+ (determined by ICP-MS) seems not to vary much (at least, after 60 minutes) with the time of electrolysis, nor it seems to depend significantly on whether the original CA-content of the Ag NPs was reduced, a marked difference with regard to the spatial distribution of the precipitates can clearly be observed in Fig. 6, especially if we compare the top-down EDX elemental maps recorded on the two electrodes (PVP-deficient and original) after 60 min electrolysis.
These elemental maps show that for the GDE containing PVP in the original (high) concentration, precipitates first appear on top of flat parts of the catalyst layer, while in case of the GDE with reduced PVP content, precipitates tend to appear preferentially inside the cracks of the MPL, leaving the catalyst layer—at least, for some time—mostly uncovered. (In a later stage, following 2.5 hours of electrolysis, precipitates already seem to cover the entire MPL.) This observation can be explained by assuming that the excess PVP content of the catalyst ink facilitates the flooding of the GDE, and leads to a blockage of CO2 access to the catalyst NPs. On GDEs of a reduced PVP content, this effect occurs only in a later stage of electrolysis, with precipitates first appearing not on-top of the active catalyst layer but rather inside cracks of the MPL, also penetrating deeper parts of the GDE structure.
Note here that the cross-over of K+ ions through the membrane is a naturally occurring (and in fact unavoidable) phenomenon, and does not (or should not) in itself lead to a blockage of the catalyst. E.g., in case of the PVP-deficient catalyst, the electrolyte penetrating the GDE structure does not get trapped in the MPL but—through cracks and micropores—drains to deeper layers of the GDE and finally exits the cell in the form of small liquid drops through the gas flow channels of the cathode. This latter phenomenon was termed perspiration by Jeanty et al.,22 and we quantitatively monitor it here for the first time, by applying a liquid trap to collect K2CO3/KHCO3 perspirates at the gas out-flow of the electrolyser (see Fig. 1b).
If we plot the amount of K+ present in this outlet trap as a function of time (see the mK,trapvs. t plots of Fig. 6), we see that perspiration does occur (and the amount of K+ in the trap constantly rises) up to the point at which the actual flooding of the catalyst layer takes place, the access of CO2 to the catalyst layer becomes blocked, and the overall performance drops down.
Our results clearly indicate that the (at least partial) removal of PVP from the catalyst layer delays the flooding of the GDE and aids the electrolyte management of the cathode MEA by facilitating perspiration. The correlation of the mK,trapvs. t curves and the product distribution plots of Fig. 6 demonstrate that the faradaic efficiency of CO production can only be efficiently upheld until the electrolyte (and, with it, K+) can effectively perspire through the system. The distribution of precipitates on-top and within the GDE structure, visualised by the EDX maps of Fig. 6, point out that excess amounts of the capping agent PVP can easily clog the micropores of the GDL, and emphasize the role of the PVP content of Ag NP catalyst inks in creating erratic electrolysis performances.
From what was written above, the question immediately follows: is it only PVP, or other polymeric CAs as well, the presence of which in catalyst inks can have such detrimental effect on the operation of CO2-to-CO electrolysing GDEs? While we cannot give a generally valid answer to this question, we did perform additional tests on BPEI-capped Ag NPs and, similarly to the case of PVP-stabilised ones, we found that the ultracentrifugation-based removal of the excess BPEI content of the Ag NP suspensions increases the durability of GDEs in the course of high current density electrolyses (Fig. 7).
 | ||
| Fig. 7 Comparison of the results of electrolysis stress-tests on two different GDEs prepared with BPEI-capped Ag NPs of 50 nm nominal diameter. The BPEI content of the Ag NP suspensions was either unchanged (left side) or was reduced by ultracentrifugation (right side) during the course of catalyst ink preparation; in both cases, the catalyst was applied at a mass loading of 300 μg per cm2 Ag. The plots show the variation of the product distribution of CO2 reduction, the measured electrode potential E (referenced vs. an Ag|AgCl|3 mol per dm3 KCl electrode), and the mass of K+ ions collected in the liquid trap equipped to the gas outflow of the electrolyser, as a function of both time and passed charge, for galvanostatic electrolyses carried out at −283 mA cm−2. Structural changes of the applied GDEs were monitored by recording top-down and cross-sectional SEM/EDX images of the GDEs obtained before, as well as 60 and 150 min after the electrolysis. For more details on the structure of BPEI-capped Ag NPs (TEM images), see Fig. S3 of the ESI.† | ||
By comparing Fig. 6 and 7 one can immediately see that BPEI, present in catalyst inks at high amount, decreases the FE of CO formation already at the start of the electrolysis, which is in agreement with previous observations.18 As the electrolysis proceeds, CO selectivity is dropping quickly and within 25 minutes H2 already becomes the dominant product. As HER takes over the place of CO2 reduction, massive precipitate formations can be observed, especially on-top of the catalyst layer, with precipitates showing typical burst patterns around the edges of cracks.
When the catalyst ink is prepared from BPEI-deficient (Table 1) suspensions (Fig. 7, to the right), the formation of the burst-like precipitate patterns can only be observed at later stages of the electrolysis. In this case the (also initially higher) FE of CO production breaks down to below 50% only after ∼75 minutes of electrolysis. With a certain delay (that is due to the time needed for the formed precipitates to get transported out of the electrolyser) the measured mK,trapvs. t curves of Fig. 7 show the same tendency: following the drop-down of FECO the amount of K+ in the down-flow trap will also grow less rapidly. This points to that the cell seems to remain active for CO2 reduction as long as the pores of the MPL remain unclogged.
From Fig. 6 and 7 it is obvious that removing some of the (originally high) polymeric CA content of Ag NP suspensions before turning these into catalyst inks does seem to be an advisable strategy, as this will increase the durability of the prepared GDEs. We note here, however, that the effect seems observable only in the case when polymeric CAs are used for the Ag NP preparation. The operation of GDEs prepared from Ag NPs stabilised by (monomeric) citrate ligands, for example, seems less to depend on whether the citrate content of the NP suspension was reduced or not (Table 1). As shown in Fig. 8, citrate removal seems to exert no considerable effect either on the measurable product distribution, electrode potential and trapped K+ plots, or on the formed precipitate patterns.
 | ||
| Fig. 8 Comparison of the results of electrolysis stress-tests on two different GDEs prepared with citrate-capped Ag NPs of 50 nm nominal diameter. The citrate content of the Ag NP suspensions was either unchanged (left side) or was reduced by ultracentrifugation (right side) during the course of catalyst ink preparation; in both cases, the catalyst was applied at a mass loading of 300 μg per cm2 Ag. The plots show the variation of the product distribution of CO2 reduction, the measured electrode potential E (referenced vs. an Ag|AgCl|3 mol per dm3 KCl electrode), and the mass of K+ ions collected in the liquid trap equipped to the gas outflow of the electrolyser, as a function of both time and passed charge, for galvanostatic electrolyses carried out at −283 mA cm−2. Structural changes of the applied GDEs were monitored by recording top-down and cross-sectional SEM/EDX images of the GDEs obtained before, as well as 60 and 150 min after the electrolysis. | ||
In order to further demonstrate, for the case of polymeric CA-stabilised Ag NPs, the detrimental effect of the excess CA content of catalyst inks on the stability of CO2 electrolysers, we also created GDEs using PVP-capped Ag NPs of ∼100 nm (as opposed to the previously used ∼10 nm) diameter. The suspensions of larger size Ag NPs inherently contains less PVP,28 although the PVP content could again be further reduced by ultracentrifugation (see Table 1). When used for the fabrication of GDEs, the 100 nm diameter NPs seemed to penetrate the MPL lot less efficiently than the ∼10 nm particles did, and remained more concentrated in the catalyst layer on-top of the MPL, with some NPs penetrating through cracks of the MPL into the CFL, as shown by the cross-sectional EDX maps of Fig. S4 of the ESI.† Decreasing the amount of PVP in the catalyst ink helped, also in case of the ∼100 nm diameter PVP-capped Ag NPs to increase the stability time of electrolysis (see Fig. S5 of the ESI†), proving that the durability impairments reported in this paper are in-deed caused by the excess amounts of polymeric CAs clogging the pores (perspiration pathways) of the MPL (scheme shown in Fig. 9) and are not due to particle size effects.
 | ||
| Fig. 9 Schematic illustration of the clogging effect of polymeric CAs, disallowing effective perspiration through the micropores of GDEs. (Figure not to scale, micropores are depicted magnified for better visibility.) | ||
The importance of maintaining the ability of GDEs to uphold effective perspiration pathways is further emphasized by the results of sessile water drop contact angle measurements made on the as-prepared GDEs, shown in Fig. 10. These measurements all show that the ultracentrifugation-based partial removal of the excess amount of capping agents from the NP suspensions in the course of catalyst ink preparation does increase, however only a bit, the hydrophobicity of the catalyst layer. This increase of hydrophobicity can, however, not explain the durability improvements caused by the removal of polymeric capping agents from the catalyst ink, pointing to that—in contrast to prevailing views5,35,36—the perspiration properties of GDEs are probably even more important than their non-wettability in efficient, long time CO2 electrolysis.
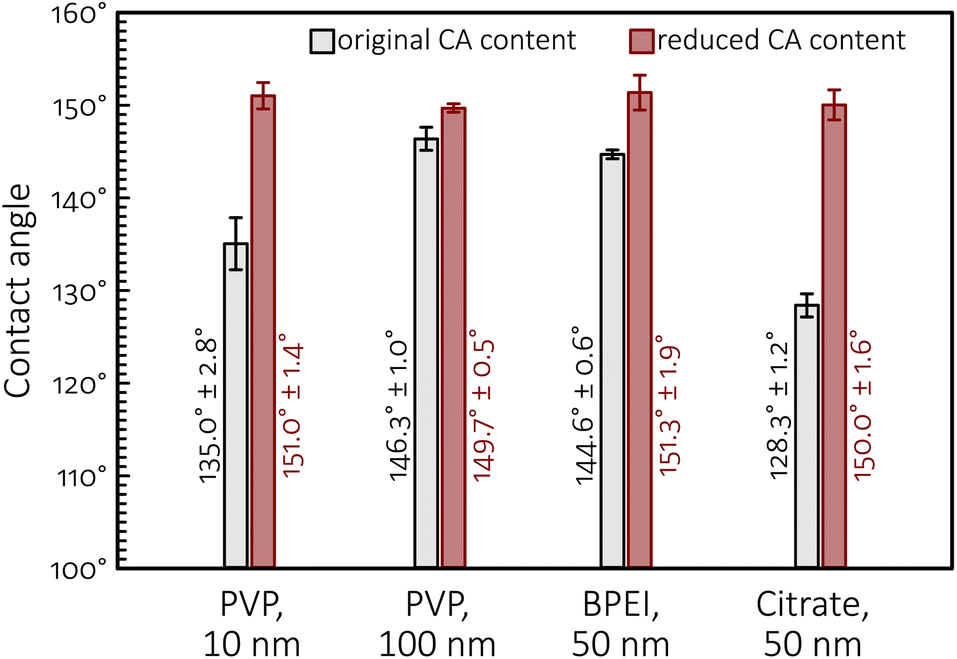 | ||
| Fig. 10 Contact angles (of sessile water drops) measured on different GDEs (as-prepared, prior to electrolyses) used in this study. | ||
4 Conclusion
Zero-gap MEA cathodes employing catalysts distributed over the surface of a gas diffusion layer allow CO2 reduction to be carried out at extreme high rate and good selectivity. E.g., by using Ag NPs with well-chosen structure and size distribution, current densities in the range of few hundreds of mA cm−2 and an almost 100% faradaic efficiency of CO production can be achieved. This efficiency increase comes, however, at the cost of severe stability issues that are mostly all related to the flooding of the GDE by the employed electrolyte. Flooding is currently recognised as the biggest obstacle in front of the scale-up prospects of CO2 electroreduction, thus the identification and possible elimination of phenomena that can lead to flooding in CO2 electrolysers is a primary goal of today's electrocatalysis studies.In the above vein, we identified in this paper one important contributor to flooding; that is, the excessive presence of polymeric capping agents in the catalyst layer of gas diffusion electrodes. CAs have an important role in stabilising Ag NPs in suspension, however when these suspensions are turned into catalyst inks and deposited on the surface of a gas diffusion layer, polymeric CAs can clog the perspiration channels of the MPL and can thus act as an initiator to flooding. The detrimental effect of polymeric CAs (PVP and BPEI were studied in this paper) present in catalyst inks can in fact be so strong that it overshadows other tendencies. E.g., it was shown in the paper that by increasing the mass loading of PVP-capped Ag NP catalysts, a counter-intuitive stability loss can often be observed. This stability loss, as we showed, is however not a result of the increased loading in itself, but of the increase of the PVP concentration in the catalyst layer.
In the paper we used a combination of electrochemical performance tests, during which we monitored the faradaic efficiency of CO production, the cathode potential, and also, using a novel ICP-MS detection-based approach, the outflow of electrolyte perspirates. We combined these measurements with ex situ SEM and EDX-based investigations of the GDEs, carried out at various stages of the electrolysis. Our investigations proved that the presence of excess amount of polymeric capping agents (PVP and BPEI) in the catalyst ink can cause a rapid flooding of the GDE cathode and can thus exert disastrous effects on the stability of zero-gap MEA cathode-based electrolysers. No such effects were observed when a monomeric CA (citrate) was used as a stabilizing ligand of Ag NPs. In order to attenuate the concentration of polymeric capping agents in the used catalyst inks, an ultracentrifugation-based method has been described in the paper, the application of which in the catalyst ink formulation immediately resulted in an improved electrolyser stability.
Author contributions
Huifang Hu: investigation, experiments, data curation, writing – first draft. Ying Kong and Menglong Liu: investigation, experiments. Viliam Kolivoška, Alexander V. Rudnev and Yuhui Hou: conceptualization, methodology. Rolf Erni: TEM measurements. Soma Vesztergom: conceptualization, methodology, visualization, data analysis, writing, funding acquisition. Peter Broekmann: conceptualization, methodology, writing – review & editing, supervision, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This publication was created as part of NCCR Catalysis (grant number 180544), a National Centre of Competence in Research funded by the Swiss National Science Foundation. V. K. acknowledges financial support from the Czech Science Foundation (project number 18-09848S and 23-07292S). A. R. acknowledges support from the Ministry of Science and Higher Education of the Russian Federation. H. H., Y. K. and M. L. acknowledge the financial support by the Chinese Scholarship Council (CSC). S. V. acknowledges support from the Lendület (Momentum) program of the Hungarian Academy of Sciences (grant LP2022-18/2022) and from the National Research, Development and Innovation Office of Hungary (NKFIH grants FK135375 and K129210).Notes and references
- D. R. Feldman, W. D. Collins, P. J. Gero, M. S. Torn, E. J. Mlawer and T. R. Shippert, Nature, 2015, 519, 339–343 CrossRef CAS PubMed.
- A. Gawel, T. Jaster, D. Siegmund, J. Holzmann, H. Lohmann, E. Klemm and U.-P. Apfel, iScience, 2022, 25, 104011 CrossRef CAS PubMed.
- J.-B. Vennekoetter, R. Sengpiel and M. Wessling, Chem. Eng. J., 2019, 364, 89–101 CrossRef CAS.
- C. M. Gabardo, A. Seifitokaldani, J. P. Edwards, C.-T. Dinh, T. Burdyny, M. G. Kibria, C. P. O'Brien, E. H. Sargent and D. Sinton, Energy Environ. Sci., 2018, 11, 2531–2539 RSC.
- U. O. Nwabara, A. D. Hernandez, D. A. Henckel, X. Chen, E. R. Cofell, M. P. de Heer, S. Verma, A. A. Gewirth and P. J. A. Kenis, ACS Appl. Energy Mater., 2021, 4, 5175–5186 CrossRef CAS.
- J. Li, S. U. Abbas, H. Wang, Z. Zhang and W. Hu, Nano-Micro Lett., 2021, 13, 216 CrossRef CAS PubMed.
- H.-R. M. Jhong, S. Ma and P. J. A. Kenis, Curr. Opin. Chem. Eng., 2013, 2, 191–199 CrossRef.
- F. Yu, P. Wei, Y. Yang, Y. Chen, L. Guo and Z. Peng, Nano Mater. Sci., 2019, 1, 60–69 CrossRef.
- D. Sun, X. Xu, Y. Qin, S. P. Jiang and Z. Shao, ChemSusChem, 2019, 13, 39–58 CrossRef PubMed.
- D. Xue, H. Xia, W. Yan, J. Zhang and S. Mu, Nano-Micro Lett., 2020, 13, 5 CrossRef PubMed.
- J. Y. Choi, W. Choi, J. W. Park, C. K. Lim and H. Song, Chem.–Asian J., 2019, 15, 253–265 CrossRef PubMed.
- Y. Pei, H. Zhong and F. Jin, Energy Sci. Eng., 2021, 9, 1012–1032 CrossRef CAS.
- D. M. Weekes, D. A. Salvatore, A. Reyes, A. Huang and C. P. Berlinguette, Acc. Chem. Res., 2018, 51, 910–918 CrossRef CAS PubMed.
- N. T. Nesbitt, T. Burdyny, H. Simonson, D. Salvatore, D. Bohra, R. Kas and W. A. Smith, ACS Catal., 2020, 10, 14093–14106 CrossRef CAS.
- A. Marcos-Madrazo, C. Casado-Coterillo, J. Iniesta and A. Irabien, Membranes, 2022, 12, 783 CrossRef CAS PubMed.
- G. Díaz-Sainz, M. Alvarez-Guerra, J. Solla-Gullón, L. García-Cruz, V. Montiel and A. Irabien, J. CO2 Util., 2019, 34, 12–19 CrossRef.
- M. de J. Gálvez-Vázquez, P. Moreno-García, H. Xu, Y. Hou, H. Hu, I. Z. Montiel, A. V. Rudnev, S. Alinejad, V. Grozovski, B. J. Wiley, M. Arenz and P. Broekmann, ACS Catal., 2020, 10, 13096–13108 CrossRef.
- M. Liu, Y. Kong, H. Hu, N. Kovács, C. Sun, I. Z. Montiel, M. de J. G. Vázquez, Y. Hou, M. Mirolo, I. Martens, J. Drnec, S. Vesztergom and P. Broekmann, J. Catal., 2021, 404, 371–382 CrossRef CAS.
- Y. Kong, H. Hu, M. Liu, Y. Hou, V. Kolivoška, S. Vesztergom and P. Broekmann, J. Catal., 2022, 408, 1–8 CrossRef CAS.
- Y. Kong, M. Liu, H. Hu, Y. Hou, S. Vesztergom, M. de J. G. Vázquez, I. Z. Montiel, V. Kolivoška and P. Broekmann, Small Methods, 2022, 6, 2200369 CrossRef CAS PubMed.
- K. Yang, R. Kas, W. A. Smith and T. Burdyny, ACS Energy Lett., 2020, 6, 33–40 CrossRef.
- P. Jeanty, C. Scherer, E. Magori, K. Wiesner-Fleischer, O. Hinrichsen and M. Fleischer, J. CO2 Util., 2018, 24, 454–462 CrossRef CAS.
- B. De Mot, J. Hereijgers, M. Duarte and T. Breugelmans, Chem. Eng. J., 2019, 378, 122224 CrossRef CAS.
- C. V. Restrepo and C. C. Villa, Environ. Nanotechnol., Monit. Manage., 2021, 15, 100428 CAS.
- S. H. Im, Y. T. Lee, B. Wiley and Y. Xia, Angew. Chem., 2005, 117, 2192–2195 CrossRef.
- J. Zhang, Q. Wang, X. Zhang, J. Wang, M. Guo, B. J. Wiley, C. Li and C. Hu, Inorg. Chem. Front., 2016, 3, 547–555 RSC.
- M. de J. Gálvez-Vázquez, S. Alinejad, H. Hu, Y. Hou, P. Moreno-García, A. Zana, G. K. H. Wiberg, P. Broekmann and M. Arenz, Chimia, 2019, 73, 922 CrossRef PubMed.
- K. M. Koczkur, S. Mourdikoudis, L. Polavarapu and S. E. Skrabalak, Dalton Trans., 2015, 44, 17883–17905 RSC.
- Q. Zhang, N. Li, J. Goebl, Z. Lu and Y. Yin, J. Am. Chem. Soc., 2011, 133, 18931–18939 CrossRef CAS PubMed.
- S. Alinejad, J. Quinson, G. K. H. Wiberg, N. Schlegel, D. Zhang, Y. Li, S. Reichenberger, S. Barcikowski and M. Arenz, ChemElectroChem, 2022, 9, e20220034 CrossRef.
- K. Ehelebe, D. Seeberger, M. T. Y. Paul, S. Thiele, K. J. J. Mayrhofer and S. Cherevko, J. Electrochem. Soc., 2019, 166, F1259–F1268 CrossRef CAS.
- U. O. Nwabara, M. P. de Heer, E. R. Cofell, S. Verma, E. Negro and P. J. A. Kenis, J. Mater. Chem. A, 2020, 8, 22557–22571 RSC.
- A. V. Rudnev, Encyclopedia of Interfacial Chemistry, Elsevier, Amsterdam, 2018, pp. 321–325 Search PubMed.
- H. Hu, Y. Kong, M. Liu, V. Kolivoška, A. V. Rudnev, Y. Hou, R. Erni, S. Vesztergom and P. Broekmann, Raw data for the article "Effective perspiration is essential to uphold the stability of zero-gap MEA-based cathodes used in CO2 electrolysers, Zenodo, 2022 Search PubMed , https://doi.org/10.5281/zenodo.7002917.
- M. Li, M. N. Idros, Y. Wu, T. Burdyny, S. Garg, X. S. Zhao, G. Wang and T. E. Rufford, J. Mater. Chem. A, 2021, 9, 19369–19409 RSC.
- M. E. Leonard, M. J. Orella, N. Aiello, Y. Román-Leshkov, A. Forner-Cuenca and F. R. Brushett, J. Electrochem. Soc., 2020, 167, 124521 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta06965b |
| This journal is © The Royal Society of Chemistry 2023 |