
Highly stretchable, tough, healable and mechanoresponsive polyurethane elastomers for flexible capacitor applications†
Amir
Khan
a,
Chuan-Fu
Wang
a,
Ravinder Reddy
Kisannagar
b,
Wei-Tsung
Chuang
 c,
Pham Quoc
Nhien
ad,
Sadiq
Mahmood
ae,
Monica
Katiyar
e,
Dipti
Gupta
b,
Kung-Hwa
Wei
a and
Hong-Cheu
Lin
*af
c,
Pham Quoc
Nhien
ad,
Sadiq
Mahmood
ae,
Monica
Katiyar
e,
Dipti
Gupta
b,
Kung-Hwa
Wei
a and
Hong-Cheu
Lin
*af
aDepartment of Materials Science and Engineering, National Yang Ming Chiao Tung University, Hsinchu 300, Taiwan. E-mail: linhc@nycu.edu.tw
bDepartment of Metallurgical Engineering and Materials Science, Indian Institute of Technology Bombay, Powai, Mumbai, Maharashtra 400076, India
cNational Synchrotron Radiation Research Center, Hsinchu, Taiwan
dDepartment of Chemistry, College of Natural Sciences, Can Tho University, Can Tho City 94000, Vietnam
eDepartment of Materials Science and Engineering, Indian Institute of Technology Kanpur, Kanpur, Uttar Pradesh 208016, India
fCenter for Emergent Functional Matter Science, National Yang Ming Chiao Tung University, Hsinchu 300, Taiwan
First published on 21st November 2022
Abstract
Visualizing color changes upon mechanical force in elastomers is attractive to understand stress transfer and failure mechanisms, which is valuable for applications ranging from tamper-proof packaging to structural integrity monitoring. Herein, we synthesize mechanochromic polyurethane films with an optimum stretchability (5500% strain) and a strongest toughness (324 MJ m−3) by using the components of bis-functionalized rhodamine, Diels–Alder (DA) adduct, 2-hydroxyethyl disulfide (HEDS), polyethylene glycol, triethanolamine and hexamethylene diisocyanate via condensation polymerization. The additions of HEDS and DA moieties enhance the healing efficiency up to 87% after 24 h of curing induced by disulfide bond exchange and Diels–Alder and retro-Diels–Alder reactions. The polyurethane film exhibits a green emission (450–500 nm) before stretching, but it shows an orange-red emission (588 nm) by force stretching because of the open form of the rhodamine moieties. A high shape recovery of 90.9% and reversible mechano-fluorescence switching by heating (immersed in 60 °C hot water) after structural deformations make it useful for shape memory and soft robotic applications. Finally, a high dielectric permittivity (160 at 1 Hz) and a good capacitance value (0.28 nF cm−2 at 1 Hz) of the film exhibit its potential applications as a flexible capacitor for energy storage in modern flexible devices.
Introduction
In the past few decades, bio-inspired healable materials have gained lots of attention due to their healing ability to recover inherent properties, which is lacking in traditional non-healable materials.1–6 Healable materials can be categorized into two sectors, i.e., extrinsic and intrinsic healing.7 Extrinsic healing is dependent on external agents, such as capsules or circulatory networks,7 to initiate healing processes. These agents help to repair damage but do not interact with matrices in any way. On the other hand, there should be reversible bonds present in intrinsically healing materials that can recover by themselves after damage. Extrinsic healing processes are widely explored in thermosets and primarily epoxy resins,8–10 whereas intrinsic healing processes are intensively studied in elastomers, such as silicones,11 polyurethanes,12 and general purpose rubbers.13–15 Lately, non-covalent interactions (e.g., hydrogen bonds16 and hydrophobic interactions)17 or dynamic chemical reactions (e.g., radical-based chain transfer,18 Diels–Alder (DA) reaction,12 disulfide exchange,19 and trans-esterification)18,20 have been incorporated into polymers to prepare intrinsic self-healing polymers. Amongst them, disulfide exchange and the DA reaction have gained popularity because of their mild reaction conditions and susceptibilities to various stimuli.12,21–24Recently, various new functions have been introduced into healable materials to expand their areas of applications, such as stimuli responsiveness toward heat, light, magnetic field, pH, mechanical strain etc.25–27 Among them, mechano-responsive and thermo-responsive properties are more popular since they are easily manipulated with precision.28 These stimuli-responsive elastomers are now being studied in various fields such as shape memory devices, soft actuators, stress sensors, damage warning devices, flexible capacitors, optoelectronic devices, etc.28–33 However, mechanical stimuli such as stretching, bending and twisting for elastomers are not suitable from a lifetime perspective. Thus, the integration of both stimuli responsive and healing properties inside a single material makes it superior and stable for practical uses. A few stimuli-responsive studies have been carried out and reported by using spiropyran, rhodamine etc., as mechanophores without healing properties.28,34,35 Furthermore, rhodamine (Rh) shows an upper hand because of its excellent mechanochromic and mechanofluorescent properties.36,37 Moreover, Rh-based elastomers can be used as reusable and erasable materials for color printing/writing due to their reversibilities in color switching.28 Rhodamine, as a long time ingredient in stimuli-responsive polymers, is still in demand due to its unique isomerization-induced color switching. For example, Wang et al.28 reported a covalently bonded rhodamine (Rh)-based polyurethane (PU) film, which changed its color upon stretching as well as under light, but this film lacked healing properties. Similarly, there are a few reports on Rh-based mechanochromic elastomers, but almost all of them are non-healable and lack high stretchabilities or excellent toughnesses.28,37,38 Also, another major problem for reported mechanochromic elastomers is that most mechanically triggered chain scission reactions are irreversible, either because the reactants are spatially separated, the mechanochromic moieties react further, or the activation barriers for the reverse reactions are too high. Thus, even if the reverse reactions are priori feasible, they are not necessarily enabled upon releasing stresses but may require additional activations. In addition, it still remains challenging to fabricate PU-based flexible and stretchable films with high dielectric constants and healing properties, which are essential for flexible capacitor applications.33,39 Therefore, it is highly demanding to design healable polyurethane films that can be utilized for applications of stress-sensors, shape memory devices and flexible capacitors.
In this study, we have successfully introduced the healing units, i.e., disulfide bond-based hydroxyethyl disulfide (HEDS) and the Diels–Alder (DA) adduct, into polyurethane films containing rhodamine derivatives and explored their mechanophoric and fluorescence properties. First, we designed and synthesized healable polyurethane films with excellent mechanical and healing properties by using disulfide bond-based hydroxyethyl disulfide (HEDS), Diels–Alder (DA) adduct, rhodamine derivative (Rh-2OH), polyethylene glycol (PEG), hexamethylene diisocyanate (HDI) and triethanolamine (TEA). The components of PEG and HDI contributed to their high tensile properties due to the formation of urethane linkages. The disulfide bond exchange and Diels–Alder and retro-Diels–Alder reactions are the main contributors to their good healing behaviors. In addition, rhodamine derivatives can change their structures from close to open forms under tensile force, which can be confirmed by their orange-red fluorescence emissions (at 588 nm). The PU films are used to show shape memory applications because of the presence of reversible bonds (disulfide and DA reaction functionalities). In addition, the PU films are stretched until rhodamine deformation occurs and then immersed in 60 °C hot water, which not only helps the films recover their initial states but also brings the open-rhodamine structures back to their close forms. An optimized PU film featuring a high memory recovery of 90.9% makes this work useful for practical purposes. It also exhibits a high dielectric permittivity of 160 (at 1 Hz) along with a good capacitance value of 0.28 nF cm−2 (at 1 Hz), confirming its potential applications as a flexible capacitor for energy storage in modern flexible devices.
Result and discussion
Fig. 1a depicts the synthetic route of healable mechanochromic polymeric networks (HMPNs), which contain different amounts of the bis-functionalized rhodamine (Rh) derivative (Rh-2OH), hydroxyethyl disulfide (HEDS), Diels–Alder adduct (DA), polyethylene glycol (PEG), hexamethylene diisocyanate (HDI) and triethanolamine (TEA). The detailed stepwise synthesis of some reaction components, i.e., the Diels–Alder (DA) adduct and rhodamine derivative (Rh-2OH), for HMPNs and their related characterization studies are provided in the ESI (see Synthetic schemes and Fig. S1–S11†). The HMPN structures were synthesized by using the rhodamine derivative (Rh-2OH) as a mechanophore, polyethylene glycol (PEG) as the main chain polymer and a Diels–Alder (DA) adduct along with 2-hydroxyethyl disulfide (HEDS) as healing units in the presence of hexamethylene diisocyanate (HDI). Finally, triethanolamine (TEA) as a crosslinker was added to prepare a viscous liquid, which eventually ended up forming HMPN elastomers. All chemicals and solvents used for the synthesis are listed in Tables S1 and S2.† The formations of amide units inside elastomeric films are generated through high-yield condensation reactions between –OH and –NCO functional groups. These amide units are the main contributors toward the high stretching abilities as well as partial healabilities of HMPN elastomers, which are named according to the presence and amounts of HEDS and the DA adduct, as listed in Table S3.† Furthermore, the formations of synthesized HMPNs were confirmed by the FTIR studies illustrated in Fig. 1b, including an optimum HMPN film of PU-Rh-DA-HEDS-0.20 to demonstrate its best physical properties in our later investigations. As shown in Fig. 1b, a broad peak at around 3400 cm−1 appears because of the O–H stretching in PEG, and the band at 2252 cm−1 is attributed to the presence of –NCO moieties from HDI. However, the complete disappearance of the –NCO peak and the development of new amide bonds at 1692 cm−1 and 1530 cm−1 confirm the formation of the PU-Rh-DA-HEDS film. Also, the O–H stretching peak disappears after the polymerization, and a new broad peak of N–H stretching appears at 3328 cm−1. Moreover, the introduction of a DA adduct in the PU-Rh-HEDS unit is proved via the appearance of a weak absorption peak at 1778 cm−1. Similarly, bond formations at lower wavenumbers are revealed by using Raman spectra, as presented in Fig. 1c, since Raman vibrations are more sensitive in the lower wavenumber region. Here, the presence of vibration peaks of S–S at 513 cm−1 and C–S at 645 cm−1 verify the presence of disulfide units inside the PU-Rh-DA-HEDS structure. Additionally, we could determine the thermal properties of these healable mechanochromic polymeric networks (HMPNs) using thermogravimetric analysis (TGA) (Fig. S12†), differential scanning calorimetry (DSC) (Fig. S13†) and rheology (Fig. S14†). TGA curves showed high stabilities of the HMPN elastomers. The presence of the DA units was characterized by the DSC study, where a phase change occurred at >80 °C in the PU-Rh-DA-HEDS-0.20 film, whereas no such change could be perceived in the PU film without the DA adduct (i.e., PU-Rh-HEDS-0.20). Similarly, based on the temperature-dependent rheology curves of PU-Rh-DA-HEDS-0.20 and PU-Rh-HEDS-0.20 films shown in Fig. S14,† it is clear that the film in the presence of the DA adduct (i.e., PU-Rh-DA-HEDS-0.20) possessed stability up to 120 °C (Fig. S14a†), but the film without the DA adduct (i.e., PU-Rh-HEDS-0.20) became weak at around 80 °C (Fig. S14b†). These characterization studies display the successful incorporation of different units inside the HMPN elastomers for potential applications.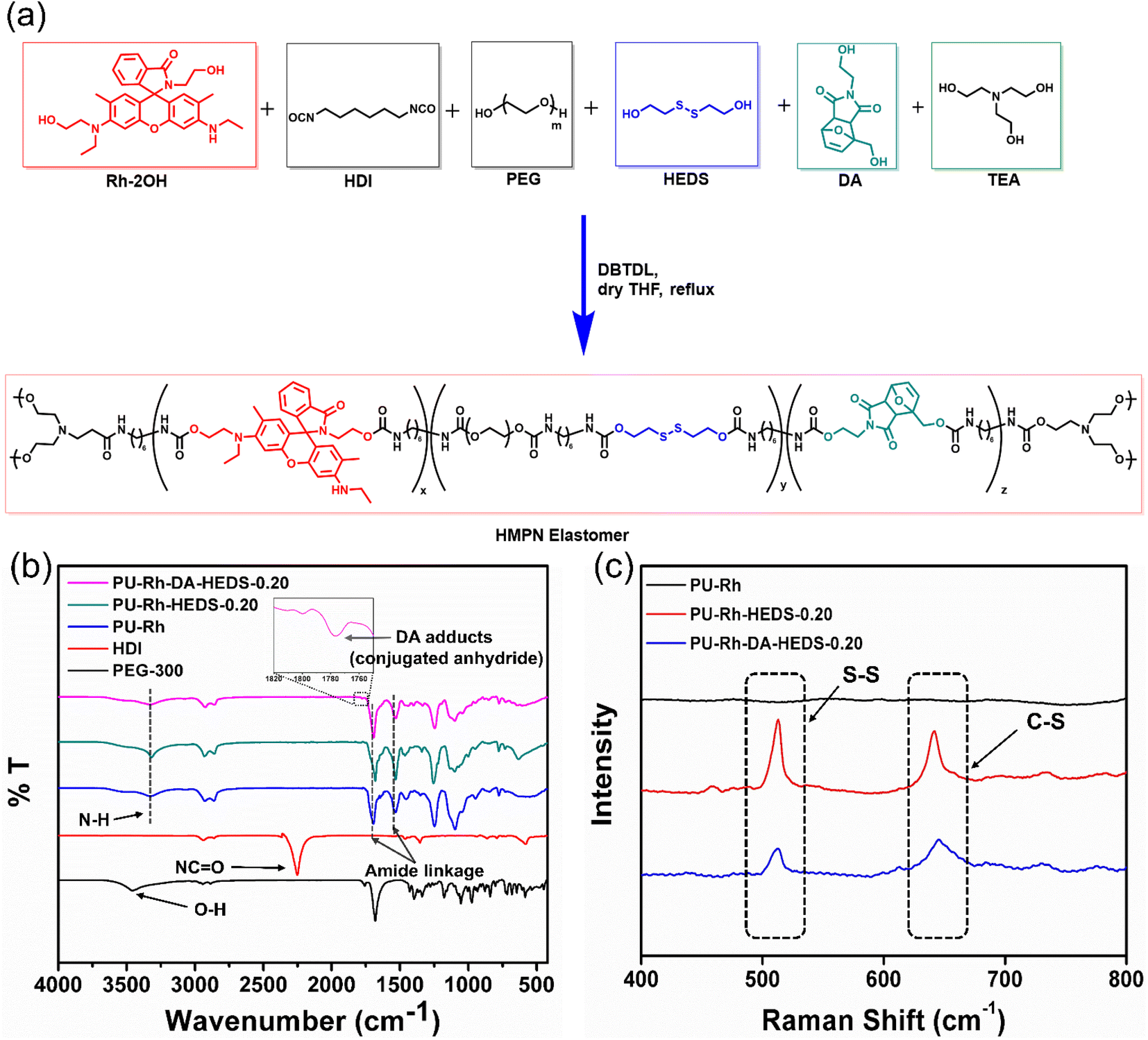 | ||
| Fig. 1 (a) Schematic synthetic route of a healable mechanochromic polymeric network (HMPN) containing a rhodamine derivative (Rh-2OH), hydroxyethyl disulfide (HEDS), Diels–Alder (DA) adduct, polyethylene glycol (PEG), hexamethylene diisocyanate (HDI) and triethanolamine (TEA). (b) FTIR and (c) Raman spectra of HMPN elastomers. | ||
Fig. 2 presents the mechanical properties of HMPN elastomers, which exhibit their outstanding mechanical strengths along with high stretchabilities and superior toughnesses. To evaluate the mechanical strengths of the HMPN elastomers, tensile tests were performed, and the tensile stresses (δ) and strains (ε) were calculated using the following equation:
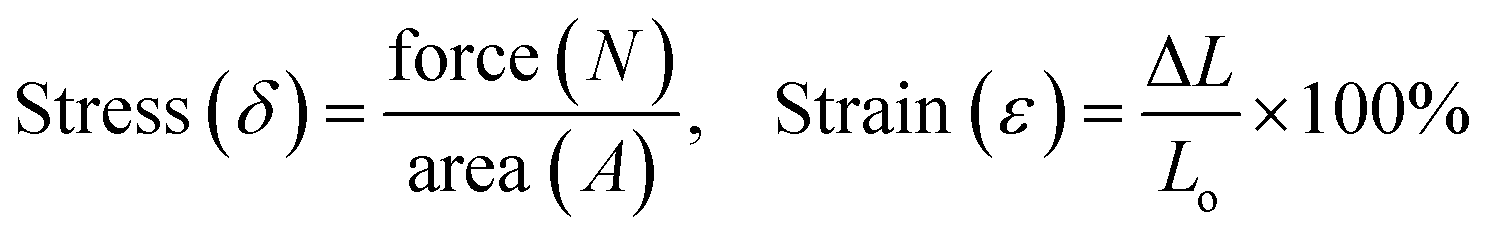
 | ||
| Fig. 2 (a) Stress–strain curves and (b) toughness and strain comparison plots of PU-Rh-HEDS-i films (i = 0.5, 0.33, 0.25 and 0.2). (c) Tensile strains of PU-Rh-HEDS-i films (i = 0.5, 0.33, 0.25, 0.2 and 0.0) with different HEDS molar ratios. (d) Stress–strain curves of PU-Rh, PU-Rh-HEDS-0.20 and PU-Rh-HEDS-DA-0.20 films. (e) Photo images of a 1600 kg car running over the PU-Rh-DA-HEDS-0.20 film 10 times. (f) Stress–strain curves of PU-Rh-HEDS-DA-0.20 films before and after car compression. | ||
The effects of disulfide bond addition inside polymer films were investigated by using different amounts of HEDS, namely PU-Rh-HEDS-i (i = HEDS/(PEG + HEDS) in a molar ratio, where i = 0.5, 0.33, 0.25 and 0.2) were measured and plotted as shown in Fig. 2a. It is worth mentioning that HMPN elastomers cannot be formed without the presence of PEG. The resulting product appeared as powder rather than a polymer film without PEG (Table S4†). From Fig. 2a, it is evident that PU-Rh-HEDS-0.5 (with the highest amount of disulfide units) showed the highest tensile strength of 12.8 MPa, but can only be stretched up to 3030% strain. However, the strain enhancements shown in Fig. 2a were observed with the reduction of disulfide units in PU-Rh-HEDS-0.33, PU-Rh-HEDS-0.25 and PU-Rh-HEDS-0.20. One possible explanation for this phenomenon is the addition of higher disulfide amounts to make the polymer films more brittle, leading to their adverse stretching properties.
Thus, PU-Rh-HEDS-0.33, PU-Rh-HEDS-0.25 and PU-Rh-HEDS-0.20 can be stretched up to tensile strains of 3525, 5100 and 5500%, respectively (Table S4†). Due to their super stretchabilities and excellent mechanical strengths, based on Fig. 2a we calculated the toughness properties of PU-Rh-HEDS-i films and plotted in Fig. 2b, where PU-Rh-HEDS-0.5, PU-Rh-HEDS-0.33, PU-Rh-HEDS-0.25 and PU-Rh-HEDS-0.20 indicate increasing toughnesses of 216, 281, 298 and 324 MJ m−3, respectively. Accordingly, it is evident that PU-Rh-HEDS-0.20 possesses the best toughness of 324 MJ m−3, which is superior to those of most of the reported healable PU films (Table S5†). Fig. 2c shows the effects of HEDS molar ratios on the film stretching properties of tensile strains, where it becomes obvious that with higher amounts of HEDS, their corresponding tensile strains eventually decrease. Due to the ultrahigh toughness and excellent stretchability of the optimum HMPN elastomer (i.e., PU-Rh-HEDS-0.20), we proceeded with further investigations of enhanced healabilities to incorporate a DA adduct inside PU-Rh-HEDS-0.20. In addition, we compared the effects of HEDS and DA additions on the mechanical properties of HMPN elastomers as plotted in Fig. 2d. Here, PU-Rh (without any additional healing units of HEDS and DA) was taken as a control film that can be stretched more than 6500%. With the addition of an HEDS unit inside PU-Rh, the previously optimized film (i.e., PU-Rh-HEDS-0.20) obtained a better mechanical strength of 8.8 MPa with an elongation of 5500%. A little enhancement in elongation of 5560% in PU-Rh-DA-HEDS-0.20 was observed when a DA unit (0.2 mmol of DA) was connected inside the structure. Besides, the resilience properties of these films were calculated from Fig. 2d and are added in Table S6.† Herein, the notably high resilience values of 0.3, 5.63 and 2.48 MJ m−3 were achieved for PU-Rh, PU-Rh-HEDS-0.20 and PU-Rh-DA-HEDS-0.20, respectively, which widen their potential applicabilities in shape memory applications. Moreover, we performed cyclic tensile tests on PU-Rh-HEDS-0.20 and PU-Rh-DA-HEDS-0.20 films to demonstrate narrow cyclic paths of tensile curves with less hysteresis of these films in their elastic region shown in Fig. S15,† which confirmed the lower energy losses because of their high resilience values owing to the existence of dynamic crosslinked networked structures. Moreover, to prove the toughness properties of the PU-Rh-DA-HEDS-0.20 film, the film was run over by a 1600 kg car 10 times, as shown in Fig. 2e. Then, the tensile test was performed after pressing by the car and curves are plotted and shown in Fig. 2f. According to this result, it is clear that the film can be stretched like the pristine film with a tensile strain ≈ 5500% even after heavy pressing by a car. However, the tensile stress at elongation break in Fig. 2f is decreased to ca. 30% after car pressing, which might be due to the partial breakage of brittle disulfide units.40
Another important function of HMPN elastomers is their healing abilities and restoration of mechanical properties after bond breakages or damage, as illustrated in Fig. 3. To introduce the healing properties inside HMPN structures, we introduced two different kinds of healing units: a disulfide bond derivative (HEDS) and Diels–Alder (DA) adduct. Fig. 3a illustrates the schematic healing mechanism of the PU-Rh-DA-HEDS network, where disulfide and the DA adduct play important roles in the healing processes. Fig. 3b(i) depicts the mechanism of the disulfide reaction, where bond exchange happens once heated to 60 °C leading to fast healing. On the other hand, the Diels–Alder reaction happens as diene and dienophile moieties of the DA adduct are heated to 60–80 °C, whereas the reverse reaction, i.e., retro Diels–Alder, occurs when continuously heated to 100–140 °C as shown in Fig. 3b(ii). Since these two healing units are the contributors to heal the PU-Rh-DA-HEDS network, we performed thorough investigations of the tensile properties of HMPN elastomers as plotted in Fig. 3c–e. For these healing tests, the HMPN films were cut first and the surfaces were brought into contact. Then, the films were heated up to 120 °C for 30 minutes to let the retro-Diels–Alder reaction happen, and cooled to 80 °C for 24 h to wait for the disulfide exchange and Diels–Alder reactions in the films to achieve faster healing effects. Regarding the PU-Rh elastomer without any additional healing units of HEDS and DA, it can be elongated up to 2473% strain after healing, which is even less than 50% of its initial stretching as shown in Fig. 3c. However, the healing efficiency of PU-Rh-HEDS-0.20 increased to more than 79% with the introduction of the HEDS unit, where this enhancement in healing efficiency occurred because of the disulfide bond exchange reaction as shown in Fig. 3d. Then, we incorporated a DA adduct inside the structure of PU-Rh-HEDS-0.20 to form PU-Rh-DA-HEDS-0.20 and checked its tensile properties after healing as shown in Fig. 3e, where the addition of HEDS and DA units enhanced the healing efficiency to 87% and the elongation of 4383% strain was observed after 24 h of healing (Table S6†).
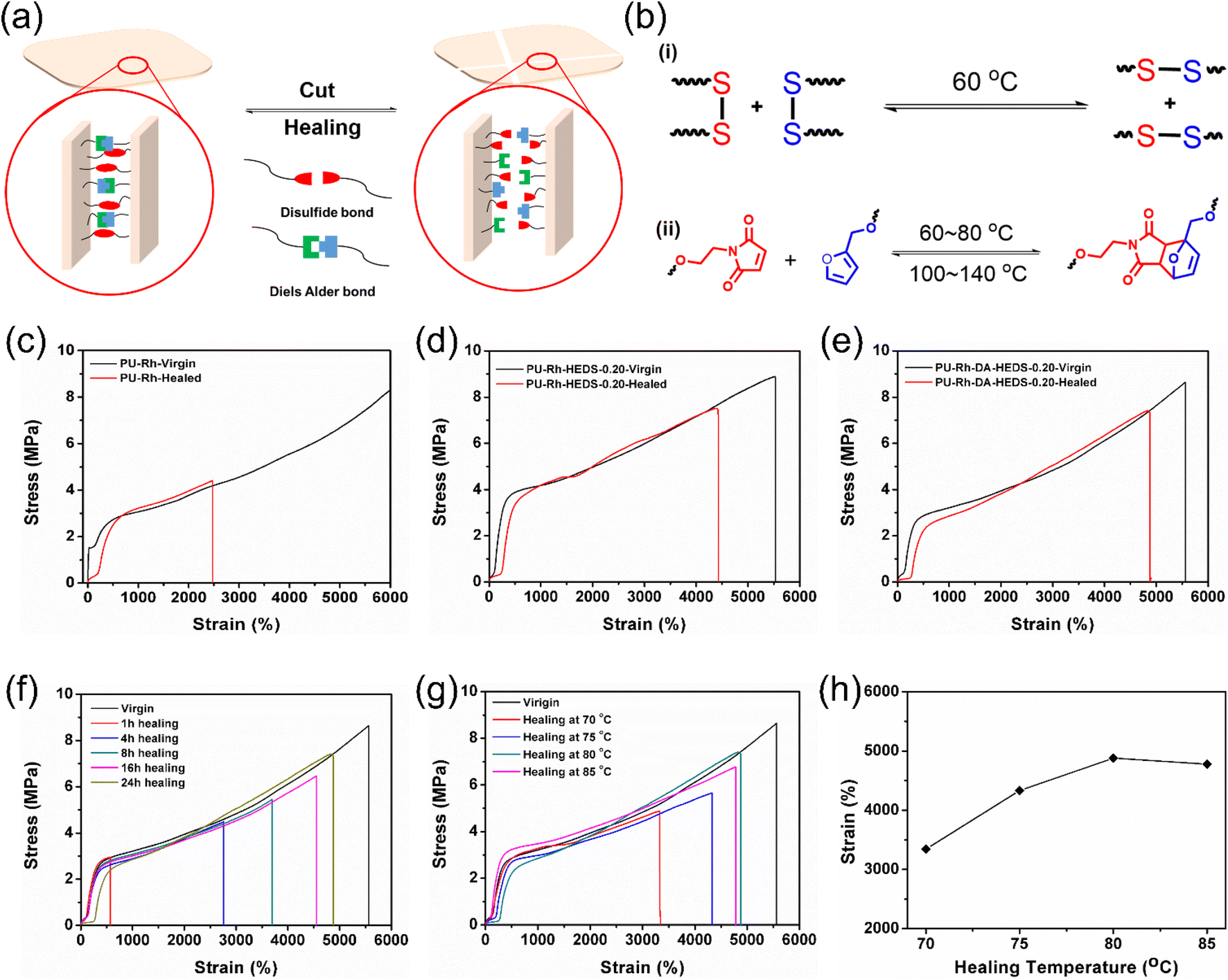 | ||
| Fig. 3 (a) Schematic illustration of the healing mechanism in healable mechanochromic polymeric networks (HMPNs). (b) Schematic diagrams of (i) disulfide bond exchange and (ii) Diels–Alder and retro-Diels–Alder reactions. Stress–strain curves of pristine and healed (c) PU-Rh, (d) PU-Rh-HEDS-0.2 and (e) PU-Rh-DA-HEDS-0.2 films. (f) Stress–strain curves of the PU-Rh-HEDS-DA-0.20 film at different healing time intervals. (g) Stress–strain curves of the PU-Rh-HEDS-DA-0.20 film healing at different temperatures. (h) Recovered strains of the PU-Rh-HEDS-DA-0.20 film after healing at different temperatures. | ||
This healing mechanism was further characterized by small-angle X-ray scattering (SAXS) for the PU-Rh-DA-HEDS-0.20 film (Fig. S16†), where the pristine and healed films showed similar diffraction patterns under the light source of TLS23A, but the healed film couldn't fully recover its initial XRD intensity. A similar trend of the healing process was also confirmed by the stress–strain curves illustrated in Fig. 3e, where under mechanical stress it was revealed that the healed film demonstrated a healing efficiency of 87% via dual covalently reversible dynamic bonds. The stress–strain curves of PU-Rh-DA-HEDS-0.20 healing at different time intervals were plotted and represented in Fig. 3f. As expected, the film can regain its better mechanical properties with more healing time, so an optimum healing efficiency of 87% was recorded after 24 h of healing (Fig. S17†). Since temperature plays an important role in the healing mechanism owing to the presence of disulfide and Diels–Alder units, a temperature-dependent healing study was carried out, and their respective tensile data are shown in Fig. 3g. In addition, the strain recoveries after healing at different temperatures are also shown in Fig. 3h, where the best strain recovery was observed at 80 °C. Once the temperature is below 80 °C, the healing efficiency is low due to the absence or partial contribution of the DA reaction. The stretchability increased from 3344 to 4880% when the healing temperature was increased from 70 to 75 °C. In contrast, the stretchability slightly decreased from 4880% to 4778% strains once the temperature exceeded 80 °C resulting in decreased recovery. This different behavior might be because the forward and reverse reactions of the DA adduct occurred at the same time when the temperature exceeded 80 °C, causing some of the DA units to de-bond, which eventually reduced the healing stretchabilities.
Mechanophores containing mechanoluminescent and mechanochromic moieties have become a hot issue recently because of their attractiveness to operate as self-reporting damage sensors. Mechanophoric responses to externally applied loads can result in color changes owing to mechanochromic and/or mechanofluorescent (mechanoluminescent) activations. The force-induced emission color changes were produced by the chemical transformations of Rh moieties from close to open forms (i.e., from the twisted spirolactam to planarized zwitterionic structures) as illustrated in Fig. 4a. Due to the presence of the rhodamine mechanophore inside PU-Rh-HEDS-0.20 and PU-Rh-DA-HEDS-0.20 structures, we performed photophysical measurements of the films to observe fluorescence emission changes during stretching. Thus, the mechanophoric characterization studies of these films were conducted and recorded by using the photoluminescence (PL) technique. As shown in Fig. 4b, the PU-Rh-HEDS-0.20 film showed a blue emission at 440 nm without stretching due to the presence of urethane linkages in the PU film. However, gradually increased orange-red emissions of open Rh moieties at 588 nm from initial close Rh moieties at 556 nm![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 28 were observed upon sequential stretching. This phenomenon of force-induced emission color changes appeared because of the transformations of Rh moieties from close to open forms under tensile stretching. The maximum PL emissions (λex = 365 nm) of the PU-Rh-HEDS-0.20 film at 588 nm upon gradual stretchings were plotted and represented as the inset image of Fig. 4b. Besides, the PU-Rh-DA-HEDS-0.2 film was prepared by adding another healable component of the DA adduct and the PL measurements were performed as reported in Fig. 4c. Here, the PU-Rh-DA-HEDS-0.2 film illustrated a mixed emission at ca. 450–500 nm before stretching because of the mixture of the blue emission of urethane groups and the green emission of DA linkages. Once the film is stretched to more than 1000%, the orange-red emission band of Rh moieties at around 588 nm was gradually enhanced due to the increased amounts of ring-opening forms in rhodamine units. The maximum PL emissions (λex = 365 nm) at 588 nm with respect to stepwise stretchings for the PU-Rh-DA-HEDS-0.2 film are shown in the inset of Fig. 4c. Additionally, the naked-eye and photoluminescence images of the PU-Rh-DA-HEDS-0.2 film during stretching are illustrated in Fig. 4d. Upon stretching, the orange-red emission of PU-Rh-DA-HEDS-0.2 was activated at ca. 3000% strain and showed blue-orange emission compared with the initial blue emission at 0% strain. This blue-orange PL emission at 3000% strain was activated by the mixed emission colors of blue urethane, green DA and red open Rh moieties. Moreover, the maximum orange-red emission at 588 nm was acquired at over 5000% strain, and rupture occurred at ca. 6000%. In addition, the reversible mechanofluorescence color changes between blue and orange-red emissions in the PU-Rh-DA-HEDS-0.2 film were also recorded with dynamic manual stretching–releasing cycles as shown in Movie S1, ESI.† Since we observed a distinct orange-red mechanoluminescent emission after fracture, a time-dependent emission decay of the fractured PU-Rh-DA-HEDS-0.20 film was surveyed. Thus, a single-recipe elastomer that is highly tough as well as super stretchable with good healing ability and reversible mechanoluminescent behavior makes this polymeric material superior to other reported polyurethane-based elastomers considering its figure of merit characteristics and wide potential applications.
28 were observed upon sequential stretching. This phenomenon of force-induced emission color changes appeared because of the transformations of Rh moieties from close to open forms under tensile stretching. The maximum PL emissions (λex = 365 nm) of the PU-Rh-HEDS-0.20 film at 588 nm upon gradual stretchings were plotted and represented as the inset image of Fig. 4b. Besides, the PU-Rh-DA-HEDS-0.2 film was prepared by adding another healable component of the DA adduct and the PL measurements were performed as reported in Fig. 4c. Here, the PU-Rh-DA-HEDS-0.2 film illustrated a mixed emission at ca. 450–500 nm before stretching because of the mixture of the blue emission of urethane groups and the green emission of DA linkages. Once the film is stretched to more than 1000%, the orange-red emission band of Rh moieties at around 588 nm was gradually enhanced due to the increased amounts of ring-opening forms in rhodamine units. The maximum PL emissions (λex = 365 nm) at 588 nm with respect to stepwise stretchings for the PU-Rh-DA-HEDS-0.2 film are shown in the inset of Fig. 4c. Additionally, the naked-eye and photoluminescence images of the PU-Rh-DA-HEDS-0.2 film during stretching are illustrated in Fig. 4d. Upon stretching, the orange-red emission of PU-Rh-DA-HEDS-0.2 was activated at ca. 3000% strain and showed blue-orange emission compared with the initial blue emission at 0% strain. This blue-orange PL emission at 3000% strain was activated by the mixed emission colors of blue urethane, green DA and red open Rh moieties. Moreover, the maximum orange-red emission at 588 nm was acquired at over 5000% strain, and rupture occurred at ca. 6000%. In addition, the reversible mechanofluorescence color changes between blue and orange-red emissions in the PU-Rh-DA-HEDS-0.2 film were also recorded with dynamic manual stretching–releasing cycles as shown in Movie S1, ESI.† Since we observed a distinct orange-red mechanoluminescent emission after fracture, a time-dependent emission decay of the fractured PU-Rh-DA-HEDS-0.20 film was surveyed. Thus, a single-recipe elastomer that is highly tough as well as super stretchable with good healing ability and reversible mechanoluminescent behavior makes this polymeric material superior to other reported polyurethane-based elastomers considering its figure of merit characteristics and wide potential applications.
 | ||
| Fig. 4 (a) Schematic illustration of force-induced ratiometric emissions for non-emissive close and red-emissive open forms (i.e., before and after stretching of elastomeric films, respectively). (b) PL spectra of the PU-Rh-HEDS-0.20 film with gradual stretchings (λex = 365 nm) (inset: relative orange-red PL intensities at 588 nm). (c) PL spectra of PU-Rh-DA-HEDS-0.20 film with gradual stretchings (λex = 365 nm) (inset: relative orange-red PL intensities at 588 nm). (d) Photographs of the PU-Rh-DA-HEDS-0.20 film under different strains, where the top and bottom images were taken under a UV lamp (λex = 365 nm) and ambient light, respectively. | ||
As revealed in Fig. 5a, the film was fractured by a uniaxial tensile strain machine and then its PL emission intensities were monitored for 24 h, where the PL emission of orange-red open Rh moieties at 588 nm recorded and shown in Fig. 5b was decreased slowly towards its final relaxed PL intensity within 24 h, which indicates the remaining 7% orange-red emission intensity of the open Rh moieties. Since the thickness of the fractured segment was thinner than its initial state, a complete emission recovery like that of the initial state was not accessible. Due to the presence of reversible disulfide, hydrogen and Diels–Alder bonds inside the elastomeric structure, it is highly expected that these bonds will try to recover their initial states after stretching. Also, the glass transition temperature of amorphous polyethylene glycol (PEG) is generally at sub-zero temperatures, and the polymeric segments of PEG possess adequate mobility in the rubbery state at room temperature. Besides, the ring-closing form of Rh moieties could be achieved by heating even after high stretching. To prove this experimentally, we stretched the PU-Rh-DA-HEDS-0.2 film to 5000% and held it for 5 minutes. Afterward, the film was directly immersed into a hot water bath at 60 °C to observe its luminescent restoration properties. The reversibility was determined by monitoring the PL intensities of the film under several heating cycles, as shown in Fig. 5c. According to Fig. 5d, it is clear that the healable polyurethane film showed good reversible stability even after three heating cycles, and its fluorescence intensity was not reduced significantly.
 | ||
| Fig. 5 (a) PL spectra (before and after fracture) and (b) relative orange-red PL intensities (λex = 365 nm) at 588 nm of the fractured PU-Rh-DA-HEDS-0.2 film at different time intervals. (c) PL spectra of the PU-Rh-DA-HEDS-0.2 film before and after stretching to 5000% strain for three cycles (with a recovery time of 24 hours). (d) The reversible abilities of two emission bands at 556 nm and 588 nm by different heating cycles. | ||
Reversible luminescent properties observed in Fig. 5 confirmed its potential applicability in the field of shape memory applications. Generally, shape memory polymers are those classes of smart materials that can restore their initial states from deformed states once triggered by external stimuli. Our previous photophysical reversible characteristics indicated the PU-Rh-DA-HEDS film to be a good candidate for shape memory applications, which can be utilized in various devices, including smart robotics, soft actuators, smart medical implants, tissue scaffolds and other medical devices. Usually, elastomers could be easily stretched in their elastic region, and the mechanical strains were stored as they were quenched below the glass transition temperature (Tg). Then, they could release the stored strains with the increased temperature and return to their original shapes. Thus, we tried to show the shape memory effect of the PU-Rh-DA-HEDS-0.20 film, where there are many driving forces (including the PEG chain and DA and disulfide linkage) to bring the elastomer back to its initial state. We performed a thermo-mechanical cycle to observe the shape memory effect of the polyurethane film, and the photo images of the film under different conditions and UV-visible light are revealed in Fig. 6. To calculate the recovery of this elastomer, an original slightly pink and green emissive PU-Rh-DA-HEDS-0.20 film with a length of 20 mm was stretched until the color changed to an obvious pink color and twisted into a spiral shape (with strong yellowish-orange emission). Then, the same film was given the shape of a twisted spiral structure and frozen below −50 °C using liquid nitrogen to maintain the temporary shape and yellowish-orange emission of open Rh moieties under this stretched condition. Later, after being immersed in 60 °C hot water for 5 minutes, the film came back to its near original shape with a length of 22 mm. Thus, a high shape recovery of 90% makes this elastomeric polymer a good candidate for industrial shape memory applications.
 | ||
| Fig. 6 Demonstration of the PU-Rh-DA-HEDS-0.20 film with a responsive shape memory effect and emission color changes upon stimuli (i.e., stretching to >3000% strain and heat treatment in a 60 °C water bath for 5 minutes). The top images were taken under a UV lamp, λex = 365 nm, and bottom images were taken under ambient light. | ||
High dielectric polymers are widely used because of their excellent energy storage abilities, dielectric properties and mechanical flexibilities with cheap costs and easy fabrication processes.33,41 To be useful for modern flexible capacitors, elastomeric films should have high dielectric permittivities along with superior capacitances.39,42,43 However, flexible polyurethane-based capacitors that possess superior dielectric constants and good healing abilities to enhance their lifetime are still attractive and demanding in the fields of flexible displays and other electronic devices. Thus, the performance of the PU-Rh-DA-HEDS-0.20 film (with 0.4 mm thickness) was measured to show its longevity and protection from damage before and after heavy car pressing as shown in Fig. 7. Most capacitors of commercially available flexible display touch panels work at higher frequencies ranging from 104 to 106 Hz,33 but a few other applications also need very low frequencies for further measurements.11 Therefore, considering the wide application varieties we performed frequency-dependent measurements in the range of 1 to 106 Hz. Fig. 7a shows frequency-dependent capacitance measurements where high capacitance values in the nF range confirmed its potential applications as a flexible and tough capacitor. Likewise, the capacitance values were measured after car compression over the film, and the reductions in capacitance values were observed in the low-frequency range (at frequencies < 100 Hz), which were dropped to ca. 30% at 1 Hz and gradually diminished to 0% reduction at 100 Hz. However, the capacitance values remained almost unchanged after car compression in the high frequency range (at frequencies > 100 Hz). A similar trend of dielectric constants was observed before and after car compression, as represented in Fig. 7b, and a similar result of ca. 30% reduction in tensile stress after car compression is also illustrated in Fig. 2f. Nonetheless, even after car compression, the film possesses an excellent dielectric constant (>12 at 106 Hz frequency), which is superior to those of commercial glass or traditional PU films. This high dielectric constant appears because of the presence of hydrogen bonding and sulfur content inside the polymeric matrix. Thus, this flexible capacitor can be used even after rough damage that enhances its lifetime after car compression. The random motions of molecules or dipoles increase in the PU-Rh-DA-HEDS-0.20 film due to thermal agitation, and the dipoles present inside get randomly aligned at high pressure and thus its dielectric constant value reduces. To check its voltage dependency, we performed a test at very low (1 Hz) and very high (1 kHz) frequencies shown in Fig. 7c and S18,† where it is evident that fluctuation appears only at low frequencies. However, voltage-independent capacitances can be obtained if we move towards higher frequencies. Then, we showed the performance of this tough and flexible capacitor under bending conditions in Fig. 7d, where it is important to note that the capacitance values in our measurements at different bending angles (0, 30, 60 and 90°) are nearly steady. As illustrated in Fig. S19,† a similar trend of steady capacitance (at 1 Hz and 100 mV) was detected even after high pressing at different bending angles (0, 30, 60 and 90°). Furthermore, the PU-Rh-DA-HEDS-0.20 film was deformed under various actions (such as bending, flattening, folding and twisting) to reconfirm its steady capacitance as shown in Fig. S20,† where no significant changes in capacitance values were observed. These consistent responses at various voltages and flexions are highly required for flexible electronics. Thus, this film appears as a potential alternative to the traditional capacitor for industrial applications.
 | ||
| Fig. 7 (a) Capacitance and (b) dielectric constant measurements at different frequencies (at 100 mV) for the PU-Rh-DA-HEDS-0.20 film before and after car compression. (c) Voltage-dependent capacitance measurements of the PU-Rh-DA-HEDS-0.20 film at low (1 Hz) and high (1 kHz) frequencies. (d) Capacitance measurements (at 1 Hz and 100 mV) of the PU-Rh-DA-HEDS-0.20 film at different bending angles (0, 30, 60 and 90°). | ||
Conclusions
In conclusion, mechanochromic, tough and healable polyurethane films were designed and successfully prepared by using various components of bis-functionalized rhodamine (Rh-2OH), Diels–Alder (DA) derivatives, 2-hydroxyethyl disulfide (HEDS), polyethylene glycol (PEG), triethanolamine (TEA) and hexamethylene diisocyanate (HDI) in the presence of a dibutyltin dilaurate (DBTDL) catalyst via condensation polymerization. The optimum elastomeric (PU-Rh-DA-HEDS-0.20) film possessed an ultrahigh stretchability of 5500% and can also retain 87% recovery of tensile strain after heat-dependent healing. This high healing recovery is because of the presence of the disulfide bonds and Diels–Alder adduct inside the polymeric matrix. Additionally, the PU-Rh-DA-HEDS-0.20 film showed a remarkable super toughness of 324 MJ m−3 that can even be maintained after healing. Furthermore, we performed PL measurements of the film under tensile conditions to study the changes of fluorescence behaviors due to the presence of rhodamine moieties. The polyurethane film (i.e., PU-Rh-DA-HEDS-0.20) exhibited a green emission (450–500 nm) before stretching, whereas an orange-red emission (588 nm) was observed by force stretching because of the open structural distortion of Rh moieties. Moreover, we found good reversibilities of the PU-Rh-DA-HEDS-0.20 film within three heating cycles, and an outstanding shape memory behavior was confirmed with a high 90% shape recovery for the polyurethane film. This shape recovery can be utilized in various applications, including smart robotics, soft actuators, smart medical implants, tissue scaffolds and medical devices. Finally, the dielectric properties of the film were measured to make its scope wider in applications. A high dielectric constant and superior capacitance value make this elastomeric polymer a useful candidate as a flexible capacitor for next-generation electronics.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors are grateful for funding from the National Science and Technology Council, Taiwan. This work was supported by the National Science and Technology Council, Taiwan (Grant No. MOST 110-2221-E-A49-003-MY3, MOST 110-2113-M-A49-018 and MOST 111-2634-F-A49-007) and this work was also supported by the Center for Emergent Functional Matter Science of National Yang Ming Chiao Tung University from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan.Notes and references
- W. Zhao, Z. Zhang, J. Hu, X. Feng, J. Xu, Y. Wu and S. Yan, Chem. Eng. J., 2022, 446, 137305 CrossRef CAS.
- G.-F. Pan, Z. Wang, X.-B. Gong, Y.-F. Wang, X. Ge and R.-G. Xing, Chem. Eng. J., 2022, 446, 137228 CrossRef CAS.
- J. Xu, T. Liu, Y. Zhang, Y. Zhang, K. Wu, C. Lei, Q. Fu and J. Fu, Matter, 2021, 4, 2474–2489 CrossRef CAS.
- Z. Gao, H. Wang and Z. Chen, Matter, 2022, 5, 387–389 CrossRef CAS.
- Y. Zhang, H. Khanbareh, J. Roscow, M. Pan, C. Bowen and C. Wan, Matter, 2020, 3, 989–1008 CrossRef.
- Y. Yang, H. Wang, S. Zhang, Y. Wei, X. He, J. Wang, Y. Zhang and Y. Ji, Matter, 2021, 4, 3354–3365 CrossRef CAS.
- S. Utrera-Barrios, R. Verdejo, M. A. López-Manchado and M. H. Santana, Mater. Horiz., 2020, 7, 2882–2902 RSC.
- X. K. D. Hillewaere, R. F. A. Teixeira, L.-T. T. Nguyen, J. A. Ramos, H. Rahier and F. E. Du Prez, Adv. Funct. Mater., 2014, 24, 5575–5583 CrossRef CAS.
- S. R. White, N. R. Sottos, P. H. Geubelle, J. S. Moore, M. R. Kessler, S. R. Sriram, E. N. Brown and S. Viswanathan, Nature, 2001, 409, 794–797 CrossRef CAS PubMed.
- S. An, M. W. Lee, A. L. Yarin and S. S. Yoon, Chem. Eng. J., 2018, 344, 206–220 CrossRef CAS.
- A. Khan, S. Ginnaram, C.-H. Wu, H.-W. Lu, Y.-F. Pu, J. I. Wu, D. Gupta, Y.-C. Lai and H.-C. Lin, Nano Energy, 2021, 90, 106525 CrossRef CAS.
- C.-M. Yeh, C.-H. Lin, T.-Y. Han, Y.-T. Xiao, Y.-A. Chen and H.-H. Chou, J. Mater. Chem. A, 2021, 9, 6109–6116 RSC.
- D. Wang, J. Guo, H. Zhang, B. Cheng, H. Shen, N. Zhao and J. Xu, J. Mater. Chem. A, 2015, 3, 12864–12872 RSC.
- C. Xu, L. Cao, B. Lin, X. Liang and Y. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 17728–17737 CrossRef CAS.
- A. Das, H. H. Le, J. Vuorinen and G. Heinrich, ACS Appl. Mater. Interfaces, 2017, 9, 14547–14551 CrossRef CAS.
- A. Khan, S. Ahmed, B.-Y. Sun, Y.-C. Chen, W.-T. Chuang, Y.-H. Chan, D. Gupta, P.-W. Wu and H.-C. Lin, Biosens. Bioelectron., 2022, 198, 113811 CrossRef CAS.
- X. Liu, X. He, B. Yang, L. Lai, N. Chen, J. Hu and Q. Lu, Adv. Funct. Mater., 2021, 31, 2008187 CrossRef CAS.
- S. Wang and M. W. Urban, Nat. Rev. Mater., 2020, 5, 562–583 CrossRef CAS.
- A. Khan, R. R. Kisannagar, C. Gouda, D. Gupta and H.-C. Lin, J. Mater. Chem. A, 2020, 8, 19954–19964 RSC.
- F. Fu, M. Huang, W. Zhang, Y. Zhao and X. Liu, Sci. Rep., 2018, 8, 10325 CrossRef.
- V. T. Tran, Md. T. I. Mredha, J. Y. Na, J.-K. Seon, J. Cui and I. Jeon, Chem. Eng. J., 2020, 394, 124941 CrossRef CAS.
- N. Tiwari, F. Ho, Ankit and N. Mathews, J. Mater. Chem. A, 2018, 6, 21428–21434 RSC.
- W. Xu, M.-C. Wong, Q. Guo, T. Jia and J. Hao, J. Mater. Chem. A, 2019, 7, 16267–16276 RSC.
- H. Wei, Y. Yang, X. Huang, Y. Zhu, H. Wang, G. Huang and J. Wu, J. Mater. Chem. A, 2020, 8, 9013–9020 RSC.
- K. Zhao, X. Cao, Y. Alsaid, J. Cheng, Y. Wang, Y. Zhao, X. He, S. Zhang and W. Niu, Chem. Eng. J., 2021, 426, 130870 CrossRef CAS.
- Z. Liu, H. K. Bisoyi, Y. Huang, M. Wang, H. Yang and Q. Li, Angew. Chem., Int. Ed., 2022, 61, e202115755 CAS.
- Y. Wang, Q. Guo, G. Su, J. Cao, J. Liu and X. Zhang, Adv. Funct. Mater., 2019, 29, 1906198 CrossRef CAS.
- Z. Wang, Z. Ma, Y. Wang, Z. Xu, Y. Luo, Y. Wei and X. Jia, Adv. Mater., 2015, 27, 6469–6474 CrossRef CAS PubMed.
- Y. Yang, L. Huang, R. Wu, Z. Niu, W. Fan, Q. Dai, L. Cui, J. He and C. Bai, ACS Appl. Mater. Interfaces, 2022, 14, 3344–3355 CrossRef CAS PubMed.
- Z. Wang, H. Jiang, G. Wu, Y. Li, T. Zhang, Y. Zhang and X. Wang, ACS Appl. Mater. Interfaces, 2022, 14, 15599–15607 CrossRef CAS.
- S. J. D. Lugger, S. J. A. Houben, Y. Foelen, M. G. Debije, A. P. H. J. Schenning and D. J. Mulder, Chem. Rev., 2022, 122, 4946–4975 CrossRef CAS.
- R. C. P. Verpaalen, T. Engels, A. P. H. J. Schenning and M. G. Debije, ACS Appl. Mater. Interfaces, 2020, 12, 38829–38844 CrossRef CAS.
- D. Li, L. Yuan, G. Liang and A. Gu, Ind. Eng. Chem. Res., 2020, 59, 6600–6608 CrossRef CAS.
- I. Jurewicz, A. A. K. King, R. Shanker, M. J. Large, R. J. Smith, R. Maspero, S. P. Ogilvie, J. Scheerder, J. Han, C. Backes, J. M. Razal, M. Florescu, J. L. Keddie, J. N. Coleman and A. B. Dalton, Adv. Funct. Mater., 2020, 30, 2002473 CrossRef CAS.
- M. H. Barbee, K. Mondal, J. Z. Deng, V. Bharambe, T. V. Neumann, J. J. Adams, N. Boechler, M. D. Dickey and S. L. Craig, ACS Appl. Mater. Interfaces, 2018, 10, 29918–29924 CrossRef CAS PubMed.
- L. Wang, W. Zhou, Q. Tang, H. Yang, Q. Zhou and X. Zhang, Polymers, 2018, 10, 994 CrossRef.
- T. Wang, N. Zhang, J. Dai, Z. Li, W. Bai and R. Bai, ACS Appl. Mater. Interfaces, 2017, 9, 11874–11881 CrossRef CAS.
- X. Chen, P. Ren, M. Li, Q. Lyu, L. Zhang and J. Zhu, Chem. Eng. J., 2021, 426, 131259 CrossRef CAS.
- W. Xu, Y. Ding, Y. Yu, S. Jiang, L. Chen and H. Hou, Mater. Lett., 2017, 192, 25–28 CrossRef CAS.
- S. Sobczak, W. Drożdż, G. I. Lampronti, A. M. Belenguer, A. Katrusiak and A. R. Stefankiewicz, Chem.–Eur. J., 2018, 24, 8769–8773 CrossRef CAS.
- L. Gao, Y. Yang, J. Xie, S. Zhang, J. Hu, R. Zeng, J. He, Q. Li and Q. Wang, Matter, 2020, 2, 451–463 CrossRef CAS.
- M. R. Benzigar, V. D. B. C. Dasireddy, X. Guan, T. Wu and G. Liu, Adv. Funct. Mater., 2020, 30, 2002993 CrossRef CAS.
- W. Tong, Y. Zhang, Q. Zhang, X. Luan, F. Lv, L. Liu and Q. An, Adv. Funct. Mater., 2015, 25, 7029–7037 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta06541j |
| This journal is © The Royal Society of Chemistry 2023 |