
Unveiling a high capacity multi-redox (Nb5+/Nb4+/Nb3+) NASICON-Nb2(PO4)3 anode for Li- and Na-ion batteries†
Biplab
Patra
 a,
Keshav
Kumar
a,
Debolina
Deb
b,
Subham
Ghosh
a,
Gopalakrishnan Sai
Gautam
b and
Premkumar
Senguttuvan
*a
a,
Keshav
Kumar
a,
Debolina
Deb
b,
Subham
Ghosh
a,
Gopalakrishnan Sai
Gautam
b and
Premkumar
Senguttuvan
*a
aNew Chemistry Unit, International Centre for Materials Science and School of Advanced Materials, Jawaharlal Nehru Centre for Advanced Scientific Research, Jakkur, Bengaluru-560064, India. E-mail: prem@jncasr.ac.in
bDepartment of Materials Engineering, Indian Institute of Science, Bengaluru-560012, India
First published on 21st February 2023
Abstract
Sodium superionic conductor (NASICON)-type materials are widely explored as Li- and Na-ion cathodes and solid-state electrolytes but are largely ignored as anodes due to their lower capacities and higher intercalation voltages, which reduce the overall energy densities of Li- and Na-ion batteries (LIBs and SIBs). Herein, we unveil high capacity multi-redox empty NASICON-Nb2(PO4)3 as a potential anode material for LIBs and SIBs, which reversibly delivers 167 and 150 mA h g−1 at the average voltages of 1.86 V vs. Li+/Li0 and 1.46 V vs. Na+/Na0, respectively. The Li and Na intercalation reactions proceed via multiple phase transitions, leading to short-range ordered Li3Nb2(PO4)3 and triclinic (P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) Na3Nb2(PO4)3, as revealed by in situ X-ray diffraction studies. Our density functional theory calculations are also in agreement with the in situ measurements in predicting a stable Na3Nb2(PO4)3 composition in the Na–Nb2(PO4)3 pseudo-binary system. X-ray absorption spectroscopy confirms the participation of multi-redox Nb5+/Nb4+/Nb3+ couples. The Nb2(PO4)3 anode delivers capacities greater than 124 and 106 mA h g−1 at 1C rate in Li and Na cells, respectively. Pairing Nb2(PO4)3 with suitable cathodes and electrolytes can lead to high energy density batteries.
) Na3Nb2(PO4)3, as revealed by in situ X-ray diffraction studies. Our density functional theory calculations are also in agreement with the in situ measurements in predicting a stable Na3Nb2(PO4)3 composition in the Na–Nb2(PO4)3 pseudo-binary system. X-ray absorption spectroscopy confirms the participation of multi-redox Nb5+/Nb4+/Nb3+ couples. The Nb2(PO4)3 anode delivers capacities greater than 124 and 106 mA h g−1 at 1C rate in Li and Na cells, respectively. Pairing Nb2(PO4)3 with suitable cathodes and electrolytes can lead to high energy density batteries.
 Premkumar Senguttuvan | Dr. Premkumar Senguttuvan is currently an Associate Professor at New Chemistry Unit and International Centre for Materials Science, Jawaharlal Nehru Centre for Advanced Scientific Research, India. He received his MS degree from Université de Picardie Jules Verne – France (2010). He pursued his PhD under the guidance of Prof. J. M. Tarascon and Prof. M. R. Palacín at UPJV-France and ICMAB-Spain (2010–2013). Thereafter, he worked as a Postdoctoral Associate in Dr C. S. Johnson's group at Argonne National Laboratory, USA (2014–2016). His research interests are solid-state chemistry, electrochemistry and rechargeable batteries. His recent honors include the DAE-BRNS Young Scientist's Research Award (2019) and DST-Early Career Research Award (2017). |
Introduction
The demand for low-cost and sustainable battery technologies is continuously increasing to achieve carbon neutrality.1,2 At present, Li-ion batteries are used extensively in portable electronics and electric vehicles, and their Na-ion analogues have emerged as the front-runner for grid storage applications.3–5 Nevertheless, there exist many technical challenges for improving the performance of such batteries, in terms of higher energy and power densities, enhanced safety, and long-term cyclability.6,7 Since these performance metrics are deeply connected with the materials used, the need for advanced Li- and Na-ion electrodes and electrolytes is continuously growing.8While graphite and hard carbon are widely used in commercial Li-ion and prototypical Na-ion cells, respectively, they pose a serious risk of lithium and sodium metal plating at higher current densities, besides their lower volumetric energy densities.9 Alternatively, intercalation-type transition metal oxides insert lithium and sodium ions at relatively higher voltages (>1.0 V), which may avoid electrolyte decomposition, and Li and Na metal electrodeposition.10 In the case of LIBs, the Li4Ti5O12 anode reversibly exchanges Li ions at 1.5 V vs. Li+/Li0 with a capacity of ∼175 mA h g−1.11 Recently, Wadsley–Roth crystallographic shear phases such as TiNb2O7,12,13 VNb9O25,14 and Ti2Nb10O29 have attracted much interest as Li-ion anodes due to their higher capacities (200–380 mA h g−1)15 and excellent rate performances (TiNb2O7 delivers capacities of 236, 219, 195 and 128 mA h g−1 at 5, 10, 20 and 50C rates, respectively). Similarly, few transition metal oxides such as Na2Ti3O7,16 TiO2,17 and Li4Ti5O12 have18 been investigated as potential Na-ion hosts with moderate capacities (100–170 mA h g−1) and limited cycle life.
Apart from oxides, transition metal-based polyanionic compounds such as sodium superionic conductor (NASICON) frameworks have been explored as possible Li- and Na-ion anodes. NASICON frameworks are appealing as electrodes19–23 and solid-electrolytes24,25 for Li- and Na-ion battery applications owing to their higher chemical, structural and thermal stabilities, and higher Na-ion conductivity. Their general chemical composition can be given as AaM2(XO4)3 (A = Li and Na; M = Sc to Fe, Zr to Mo, In and Sn; X = Si, P, S; 0 ≤ a ≤ 4) and structures are built from lantern units consisting of two MO6 octahedra and three XO4 tetrahedra, stacked along the c-axis.26,27 Due to their size differences, Li and Na ions occupy different crystallographic sites in the NASICON framework (Li ions into the 18f site of the R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) & Na ions into 6b/18e sites of the Rc).28,29 In principle, NASICON compounds can reversibly exchange a maximum of four moles of Li and Na ions, if provided with suitable redox centers.
& Na ions into 6b/18e sites of the Rc).28,29 In principle, NASICON compounds can reversibly exchange a maximum of four moles of Li and Na ions, if provided with suitable redox centers.
Experimentally, ATi2(PO4)3 (A = Li and Na) NASICONs reversibly (de)intercalate two moles of Li- and Na-ions at 2.6 and 2.1 V vs. Li+/Li0 and Na+/Na0, respectively, through the redox activity of the Ti4+/Ti3+ couple.30–32 Upon reducing the lower cut-off voltage beyond Na3Ti2(PO4)3, an additional mole of sodium can be intercalated at 0.45 V with concomitant reduction of Ti3+/Ti2+, accounting for a total capacity of 150 mA h g−1.33 Another NASICON-Na3V2(PO4)3 reversibly exchanges Na ions at a lower voltage (∼1.6 V vs. Na) but shows a limited capacity of 60 mA h g−1 (i.e., equivalent to intercalation of one mole of sodium ions).34,35 Also, a mixed NASICON compound, TiNb(PO4)3, has been preliminarily explored for Na intercalation (∼120 mA h g−1).36 Thus, NASICON cathodes and anodes explored so far operate within the limit of three moles of lithium- and/or sodium-ion exchange during cycling, thus leading to maximum storage capacities of 140–160 mA h g−1.37–39 Moreover, in terms of synthesis, NASICONs usually contain Na (or Li) ions as synthesized (e.g., Na3V2(PO4)3 is a NASICON composition that can be synthesized)40 and electrochemical/chemical oxidation routes have to be employed to remove Na (or Li).41
In this work, we report for the first time the synthesis of polycrystalline “empty” NASICON-Nb2(PO4)3 and its potential application as an anode in LIBs and SIBs. Previously Leclaire et al. reported its crystal structure using a single crystal of Nb2(PO4)3.42 This compound contains no Li or Na ions in its pristine state and is expected to exchange higher amount (∼3 moles per formula unit) of lithium and sodium ions through the activity of multi-redox-Nb5+/Nb4+/Nb3+ centers at relatively lower voltages than the Ti- or V-based NASICON anodes. In addition to our electrochemical and characterization measurements, we have also performed first-principles calculations to understand the Na-intercalation phase behavior better. We believe that pairing this “empty” NASICON anode with suitable Na-ion cathodes and/or solid electrolytes (which can also be NASICON-based) can enable building high energy density Na-ion batteries.
Experimental
Synthesis
Empty NASICON-Nb2(PO4)3 was prepared by high-temperature solid-state synthesis. Nb2O5 (0.9 mmol) (Sigma Aldrich, 99.5%), P2O5 (1.5 mmol) (Alfa Aesar, 99.0%), and Nb-powder (0.2 mmol) (Alfa Aesar, 99.8%) were mixed using a high-energy ball miller (SPEX 8000M) for 20 min. The resulting mixture was then placed in an evacuated quartz ampoule and sealed under high vacuum of 10−6 mbar. The tube was slowly heated up to 1473 K in 35 hours followed by dwelling at this temperature for 72 hours and then cooled back to room temperature in 7 hours. Finally, the tube was opened in an Ar-filled glove box and the final powder product was collected.Characterization
Synchrotron powder diffraction patterns were collected on Nb2(PO4)3 and Na3Nb2(PO4)3 sealed in a Kapton capillary (0.5 mm diameter) at the 11-BM beamline (λ = 0.458![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 969 Å) of the Advanced Photon Source, Argonne National Laboratory, and the data were analyzed using the FullProf program.43 FESEM images were taken using a Zeiss Gemini SEM 500 to observe the morphology and homogeneous distribution of the elements.
969 Å) of the Advanced Photon Source, Argonne National Laboratory, and the data were analyzed using the FullProf program.43 FESEM images were taken using a Zeiss Gemini SEM 500 to observe the morphology and homogeneous distribution of the elements.
Electrochemical testing of Nb2(PO4)3 was carried out in two-electrode Swagelok cells using either lithium or sodium (99.9% Aldrich) metal as the counter electrode in galvanostatic mode. Nb2(PO4)3 electrodes were prepared by ball milling as-synthesized samples with Super C45 (Timcal) in a 70:30 ratio for 8 min. The ball-milled mixture was collected and mixed with polyvinylidene fluoride (PVDF) (in such a way that the final electrode contains active material:carbon:PVDF in a weight ratio of 65:27:8) in N-methyl-2-pyrrolidone (NMP) solvent. The resulting slurry was uniformly coated on Cu foil, followed by drying in a vacuum oven at 90 °C for 6 hours. The dried electrodes were punched into round discs of 10 mm diameter. The active material loading of the final electrode was estimated to be 2.5–3.0 mg cm−2. Sodium cells were assembled using a 1 M solution of NaPF6 (Sigma, 98%) in diglyme (Sigma, 99.5%) as an electrolyte with sodium metal as the counter electrode. Similarly, lithium cells were fabricated using a 1 M electrolyte solution of LiPF6 in EC:DMC and lithium metal as the counter electrode. The cells were assembled in an Ar-filled glove box (O2 < 0.1 ppm and H2O < 0.1 ppm) and tested in a battery cycler (BT-lab, Biologic) using the galvanostatic protocol. For galvanostatic intermittent titration technique (GITT) experiments, first, the cells were cycled at C/10 for three cycles then a current equivalent to a C/10 rate was applied for 1 h followed by a 4 h relaxation.
For in situ XRD measurements, the Nb2(PO4)3 anode was coated on pinhole-free thin aluminum foil (Alfa, 99.99%, 10 mm thickness) and a home-made in situ cell fitted with a Be-window was used for the experiment. XRD patterns at different states-of-charge were collected using the same Bruker D8-diffractometer and Le-Bail fitting was performed using the Fullprof program.
X-ray absorption spectroscopy (XAS) measurements of pristine and cycled electrodes at different states-of-charge were carried out at PETRA-III beamline P65 at DESY in Hamburg. The measurements of the Nb–K edge at room temperature were performed in fluorescence mode as well as transmission mode using gas ionization chambers to monitor the intensities of the incident and transmitted X-ray using a PIPS diode. The energy of the Nb–K edge was calibrated by defining the inflection point (first derivative maxima) of Nb foil as 18987.5 eV. Nb2O5 and NbO2 were used as standard materials. The standard materials were thoroughly mixed with boron nitride and pressed into 12 mm pellets of 1 mm thickness and ex situ electrodes were sealed in between Kapton tapes inside an Ar-filled glove box and used directly for the data collection. All data were collected at room temperature with a Si (111) double crystal monochromator and all XAS spectra were processed using the DEMETER software package.44,45
Computations
Density functional theory (DFT) calculations were performed using the Vienna ab initio simulation package (VASP),46,47 which employs a plane wave basis (energy cut-off of 520 eV) and projector augmented wave48 potentials. To describe the electronic exchange and correlation, we used the strongly constrained and appropriately normed49 functional. We used a Γ-centred k-point mesh to sample the Brillouin zone with a density of at least 32 k-points per Å. We relaxed the lattice vectors, cell shape, and cell volume of all structures, without preserving the underlying symmetry, with structures considered converged when the total energies and atomic forces drop below 0.01 meV and |0.03| eV Å−1. We obtained the initial structure of Nb2(PO4)3 from the inorganic crystal structure database50 and we took the equivalent Na positions from the Na4V2(PO4)3 cathode NASICON structure. The Na-vacancy orderings at NaNb2(PO4)3, Na2Nb2(PO4)3, and Na3Nb2(PO4)3 compositions were enumerated using the pymatgen51 package within the primitive cell (containing two Nb2(PO4)3 formula units) and a 2 × 1 × 1 supercell of the primitive NASICON structure. The procedure to calculate average voltages, which neglect pV and entropic contributions, is detailed in our previous studies.52,53Results and discussion
Polycrystalline NASICON-Nb2(PO4)3 was prepared through a classical solid-state synthesis route using Nb2O5, Nb, and P2O5 precursors (for details, see the Experimental section). Fig. 1a shows the Rietveld refinement of the synchrotron X-ray diffraction (XRD) pattern collected on NASICON-Nb2(PO4)3 at room temperature. The XRD pattern can be completely indexed with Rc space group, thus confirming the phase purity of the sample. The scanning electron microscopy (SEM) image of NASICON-Nb2(PO4)3 (inset of Fig. 1a) shows the presence of irregular micron-sized (∼10 μm) primary particles and elemental mapping shows the homogeneous distribution of Nb, P, and O in the sample (Fig. S1†). The calculated lattice parameters (a = 8.6629(1) and c = 22.0627(6) Å) and atomic coordinates of the NASICON-Nb2(PO4)3 structure are displayed in Table S1 in the ESI.† Its crystal structure is built from lantern units,42 consisting of two (Nb5+/Nb4+)O6 and three PO4 units, stacked along the c-direction (Fig. 1b) and both Na(1) and Na(2) (i.e., 6b and 18e, respectively) sites are empty. To probe the oxidation state of Nb in NASICON-Nb2(PO4)3, X-ray absorption spectroscopy measurements were performed along with Nb2O5 and NbO2 references. Their corresponding normalized X-ray absorption near-edge structure (XANES) spectra collected at the Nb K-edge are shown in Fig. 1c. The absorption edge position of Nb2(PO4)3 is found to be between those of Nb2O5 and NbO2, thus confirming the presence of both Nb5+ and Nb4+ in the NASICON lattice.
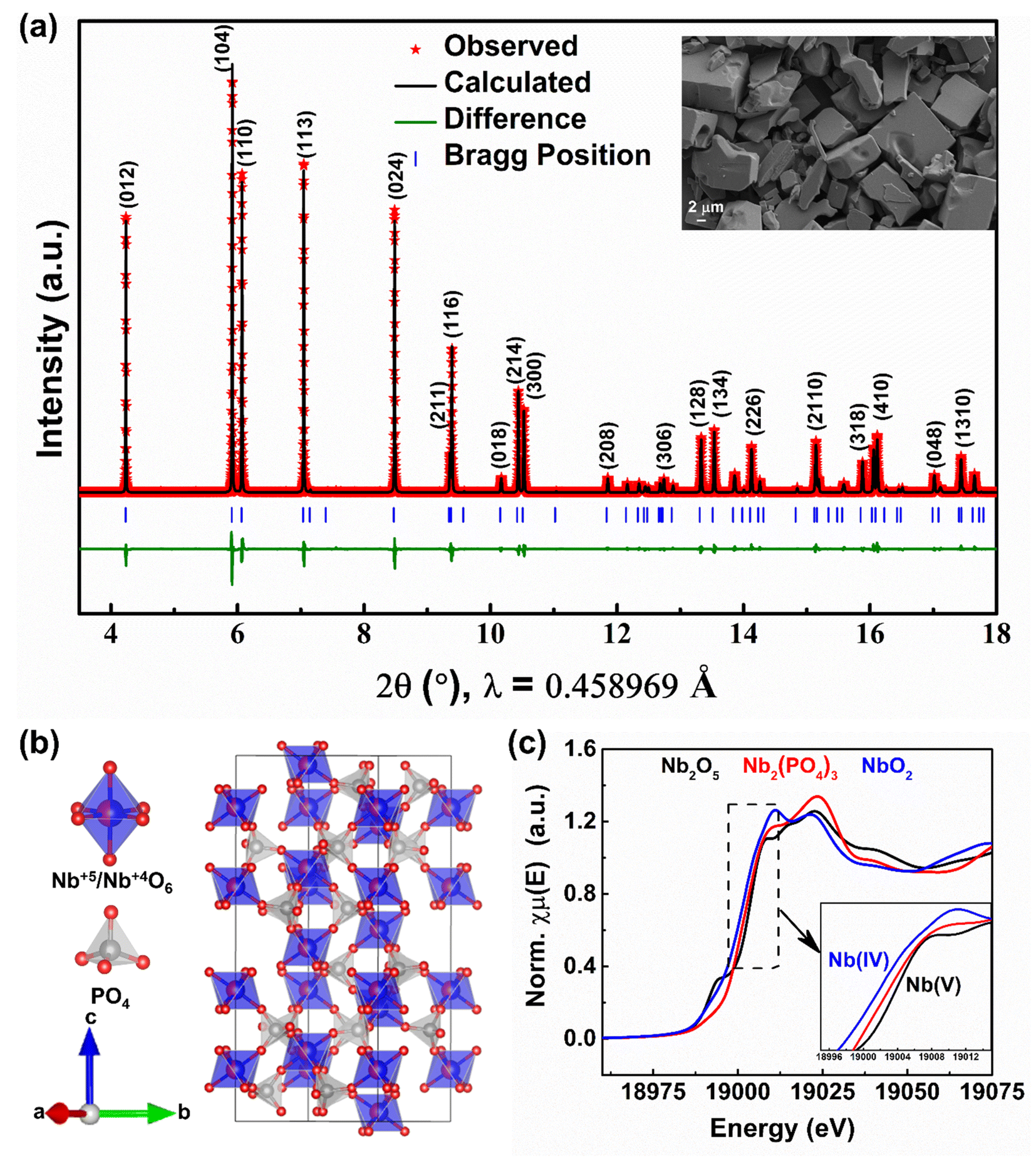 | ||
| Fig. 1 (a) Rietveld refinement of the room temperature synchrotron XRD pattern (inset: SEM image), (b) crystal structure, and (c) XANES spectra collected at the Nb K-edge of NASICON-Nb2(PO4)3. | ||
The NASICON-Nb2(PO4)3 anode is expected to reversibly exchange three moles of Li- and Na-ions through redox activities of Nb5+/Nb4+ and Nb4+/Nb3+ centers, leading to a theoretical capacity of ∼171 mA h g−1. First, we studied the electrochemical Li-ion (de)intercalation property of the NASICON-Nb2(PO4)3 anode in Li half cells. The voltage-capacity plot of the Nb2(PO4)3/Li cell cycled at C/10 rate in a voltage window of 3.0–1.2 V vs. Li+/Li0 is displayed in Fig. 2a. During the first discharge, this cell exhibits two voltage plateaus at ∼2.3 and 1.6 V vs. Li+/Li0 followed by a sloping curve until 1.2 V vs. Li+/Li0 with a total discharge capacity of 257 mA h g−1. On the subsequent charge, the two-step voltage profile is reversed with a charge capacity of 167 mA h g−1, which is equivalent to the deintercalation of ∼2.9 moles of Li-ions from the Nb2(PO4)3 framework. Nearly 1.5 moles of lithium-ions are lost during the first cycle, which could be due to electrolyte decomposition. The voltage–capacity profiles from the subsequent cycles overlap with each other with capacities of ∼167 mA h g−1. The corresponding dQ/dV curves (Fig. 2b) show two oxidation/reduction peaks located at 2.31/2.29 and 1.65/1.55 V vs. Li+/Li0, which could be tentatively assigned to the operation of Nb5+/Nb4+ and Nb4+/Nb3+ redox couples, respectively.
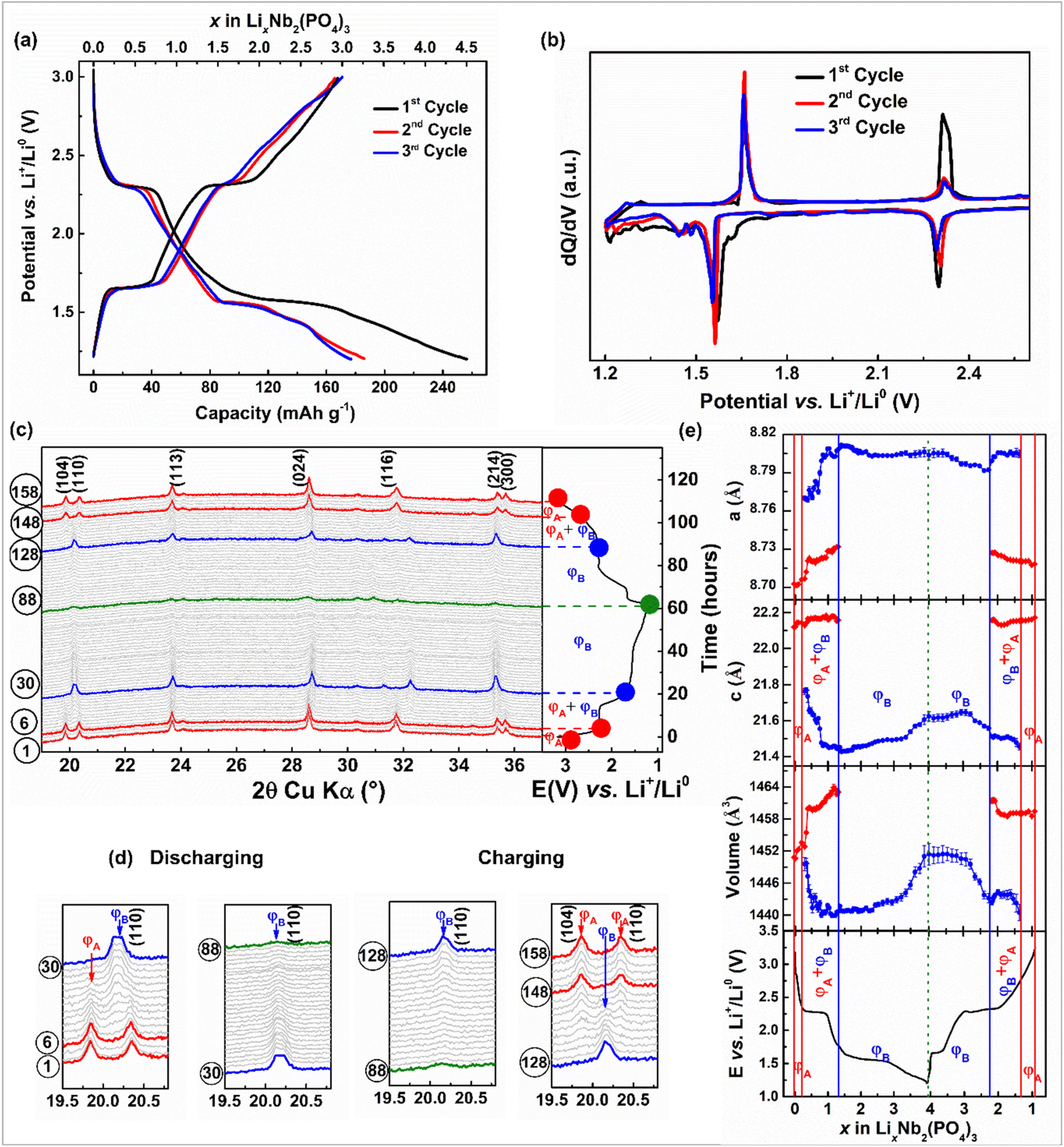 | ||
| Fig. 2 (a) Voltage vs. capacity and (b) dQ/dV profiles of the Nb2(PO4)3/Li cell. (c and d) in situ XRD patterns of the Nb2(PO4)3 anode and (e) its cell parameter evolution during the first cycle of Li-ion (de)intercalation (vertical solid and dotted lines indicate the phase boundaries during Li-ion (de)intercalation). | ||
To better understand the structural evolution of the NASICON-Nb2(PO4)3 anode upon Li (de)intercalation, we performed an in situ XRD measurement at C/15 rate during the first cycle (Fig. 2c and d) and the evolution of corresponding lattice parameters is displayed in Fig. 2e. Initially, a small solid-solution region is noticed for a Δx = 0.2 moles of Li-ion intercalation into the NASICON-Nb2(PO4)3 anode with subtle changes in the lattice parameters (ΦA: Li0–0.2Nb2(PO4)3). As the discharge proceeds through the voltage plateau at ∼2.25 V vs. Li+/Li0, we find a new set of reflections along with the parent NASICON-Nb2(PO4)3, indicating a two-phase intercalation mechanism (ΦB: Li1.3–3.0Nb2(PO4)3). However, the peak positions of both NASICON phases slightly drift as Li-ion intercalation proceeds, similar to LiVOPO4 and Na3V2(PO4)2F3 cathodes.54,55 The XRD pattern collected at the end of 1.5 V shows anisotropic lattice parameter changes, i.e., Δa/a = +1.21% and Δc/c = −3.06%, with a nominal overall unit cell volume reduction of Δv/v = −0.69%. Further, as the voltage descends to 1.2 V vs. Li+/Li0, we notice a smoother variation of peak positions, signifying solid-solution behavior. It is worth mentioning that the XRD patterns collected closer to 1.2 V present weaker reflections, implying the formation of short-range ordered “Li3Nb2(PO4)3” at the end of discharge. Note that the extra capacity (i.e., 4 moles of Li+ insertion) observed during the first discharge can be accounted for electrolyte decomposition as mentioned earlier. The overall volume change between the fully lithiated and pristine NASICON phases is estimated as Δv/v = +0.03%. Interestingly, upon subsequent charging the above-mentioned phenomena are reversed sequentially and the XRD pattern collected at the end of 1st cycle nearly superimposes on that of the pristine electrode, indicating excellent reversibility of the Li-ion (de)intercalation reaction. Further, the in situ XRD patterns collected during the second cycle quite resemble the first cycle patterns, thus confirming the same Li-ion (de)intercalation pathway in the successive cycles (Fig. S2†). It is worth mentioning that the limited quality of XRD patterns impedes us from solving the crystal structures of Li-rich NASICON phases obtained during discharge, especially given the large configurational space for Li to arrange itself in the NASICON lattice. Thus, advanced local and bulk structural studies along with DFT calculations are required to elucidate the Li-ion (de)intercalation mechanism of the Nb2(PO4)3 anode.
The voltage–capacity profiles of Nb2(PO4)3/Na cell cycled at C/10 rate in a voltage window of 3.0–1.05 V vs. Na+/Na0 are displayed in Fig. 3a. In contrast to its Li counterpart, the Nb2(PO4)3/Na cell exhibits a shorter voltage step at ∼2.35 V vs. Na+/Na0 (for Δx = 0.2 Na+), which is followed by a sloping voltage curve until 1.2 V vs. Na+/Na0 (for Δx = 2 Na+) and a voltage plateau at ∼1.14 V vs. Na+/Na0 with a total discharge capacity of 226 mA h g−1. During the subsequent charge, these voltage features are partially reversible with a de-sodiation capacity of 150 mA h g−1 (i.e., equivalent to 2.6 moles of Na-ion deintercalation from the Nb2(PO4)3 framework). A capacity of 76 mA h g−1 is lost during the first cycle, which could be due to electrolyte decomposition and/or partial entrapment of Na+ ions in the NASICON-Nb2(PO4)3 framework.56,57 The voltage–capacity profiles of the second and third cycles neatly superimpose on each other with reversible capacities and coulombic efficiencies of ∼150 mA h g−1 and 95%, respectively. The corresponding dQ/dV profiles of the Nb2(PO4)3/Na cell (Fig. 3b) show two oxidation/reduction peaks located at 2.4/2.3 and 1.22/1.12 V vs. Na+/Na0 which could be attributed to the operation of Nb5+/Nb4+ and Nb4+/Nb3+ redox couples, respectively.
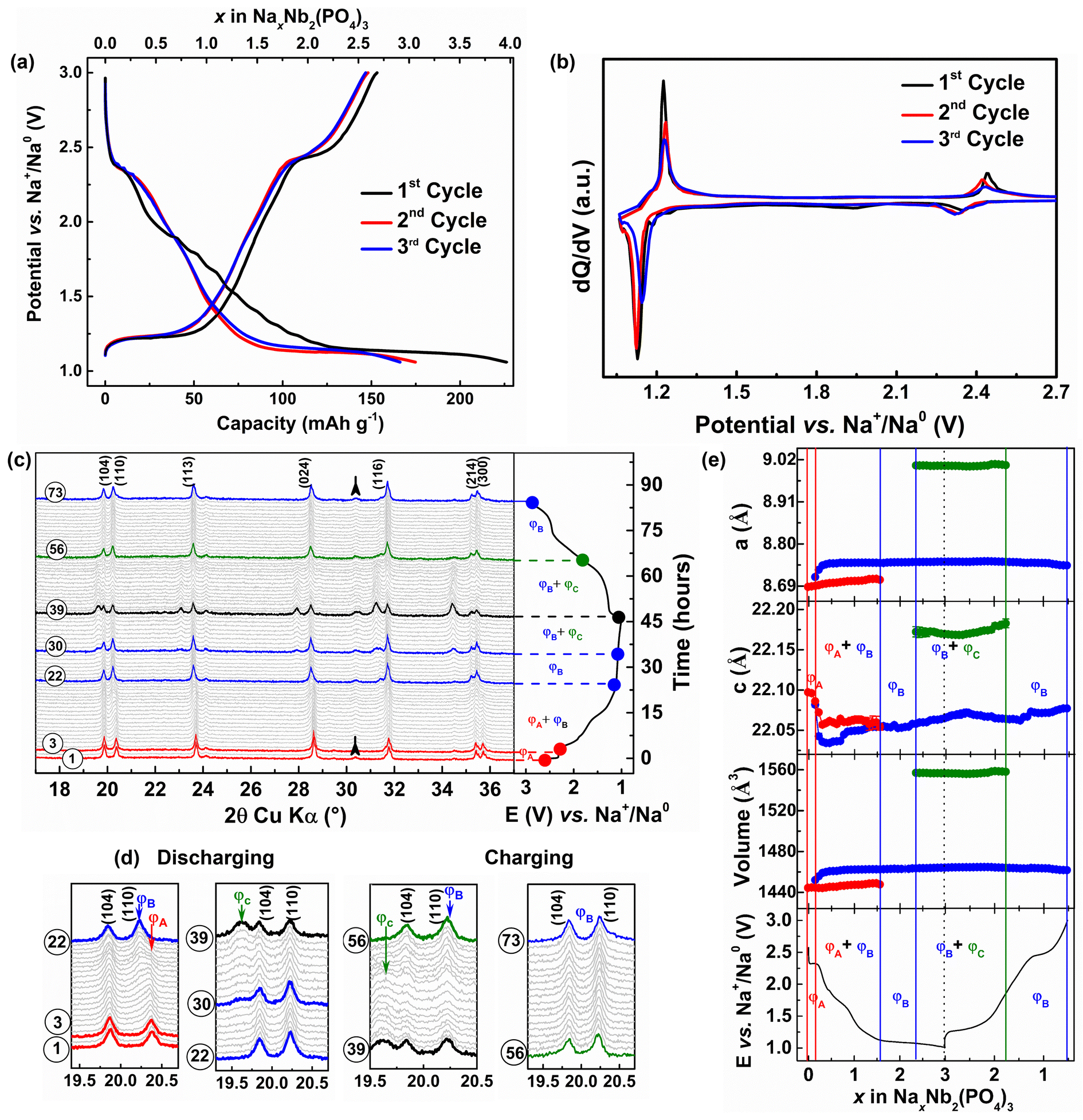 | ||
| Fig. 3 (a) Voltage vs. capacity and (b) dQ/dV profiles of the Nb2(PO4)3/Na cell. (c and d) in situ XRD patterns of the Nb2(PO4)3 anode and (e) its cell parameter evolution during the first cycle of Na-ion (de)intercalation (vertical solid and dotted lines indicate the phase boundaries during Na-ion (de)intercalation). | ||
To follow the structural changes of the Nb2(PO4)3 anode upon Na-ion (de)intercalation, we carried out an in situ XRD measurement at C/15 rate during the first cycle (Fig. 3c and d). The calculated lattice parameters of the XRD patterns are plotted against x in Fig. 3e. At the beginning of the first discharge (pattern #1–3), the formation of Na0.2Nb2(PO4)3 (denoted as ΦA phase) from the parent Nb2(PO4)3 proceeds via a solid-solution mechanism, which is self-evident from moving of XRD peaks toward lower 2θ values. During this process, the changes in lattice parameters are less pronounced (Δa/a = +0.03%) and (Δc/c = −0.05%). The decrease of the c-parameter can be attributed to the filling of Na-ions into the Na(1) site, which diminishes the electrostatic repulsion between NbO6 octahedra. As the discharge proceeds to the next Δx = 1.4 (pattern #4–22), a new set of reflections belonging to another NASICON phase (ΦB: Na1.6–2.3Nb2(PO4)3) grows at the expense of the former ΦA phase (refer Fig. 3e), which is followed by another narrow solid-solution region for Δx = 0.66 (Pattern #23–30). The c-parameters of both NASICON (ΦA and ΦB) phases initially decrease (due to the filling of the Na(1) site) and stabilize whereas their a-parameters do not change significantly. Beyond this point, the sodiation reaction associated with the low-voltage plateau (1.15 V vs. Na+/Na0) continues via a two-phase mechanism, resulting in the formation of the third NASICON phase “Na3Nb2(PO4)3” (denoted as ΦC). During this process, the cell parameters and unit cell volume of ΦC increase (Δa/a = +3.64%, Δc/c = +0.32%, and Δv/v = +7.77%) with the respect to the pristine NASICON phase. The increase of cell parameters can be correlated with the filling of sodium ions and the increase of NbO6 octahedra size (due to the reduction of Nb5+/Nb4+ to Nb4+/Nb3+).
Further, we collected the synchrotron powder XRD pattern of fully discharged Na3Nb2(PO4)3 (cycled at C/50 rate) to elucidate its crystal structure (Fig. 4a and b). The XRD pattern can be completely indexed with (P) space group with the cell parameters: a = 8.7153(2) Å, b = 9.0908(1) Å, c = 22.6495(1)Å, α = 89.112(9)°, β = 90.052(9)° and γ = 119.912(3)°, similar to NASICON-Na3Ti2(PO4)3.58,59 In this structure, sodium ions fully and partially occupy Na(1a–d) and Na(2a–f) sites which correspond to the Na(1) and Na(2) sites of the Rc structure, respectively. On the subsequent charge, the above-mentioned intercalation phenomena are mostly reversed, except the final XRD pattern (pattern #73) does not exactly match that of the pristine electrode. Indeed, its corresponding unit cell parameters and volume indicate the entrapment of Na-ions (∼0.6 moles) in Nb2(PO4)3, in agreement with our electrochemical results. Furthermore, the in situ XRD patterns collected during the second cycle display similar Na (de)intercalation phenomena as observed in the first cycle, confirming the reversibility of the (de)sodiation process (Fig. S3†).
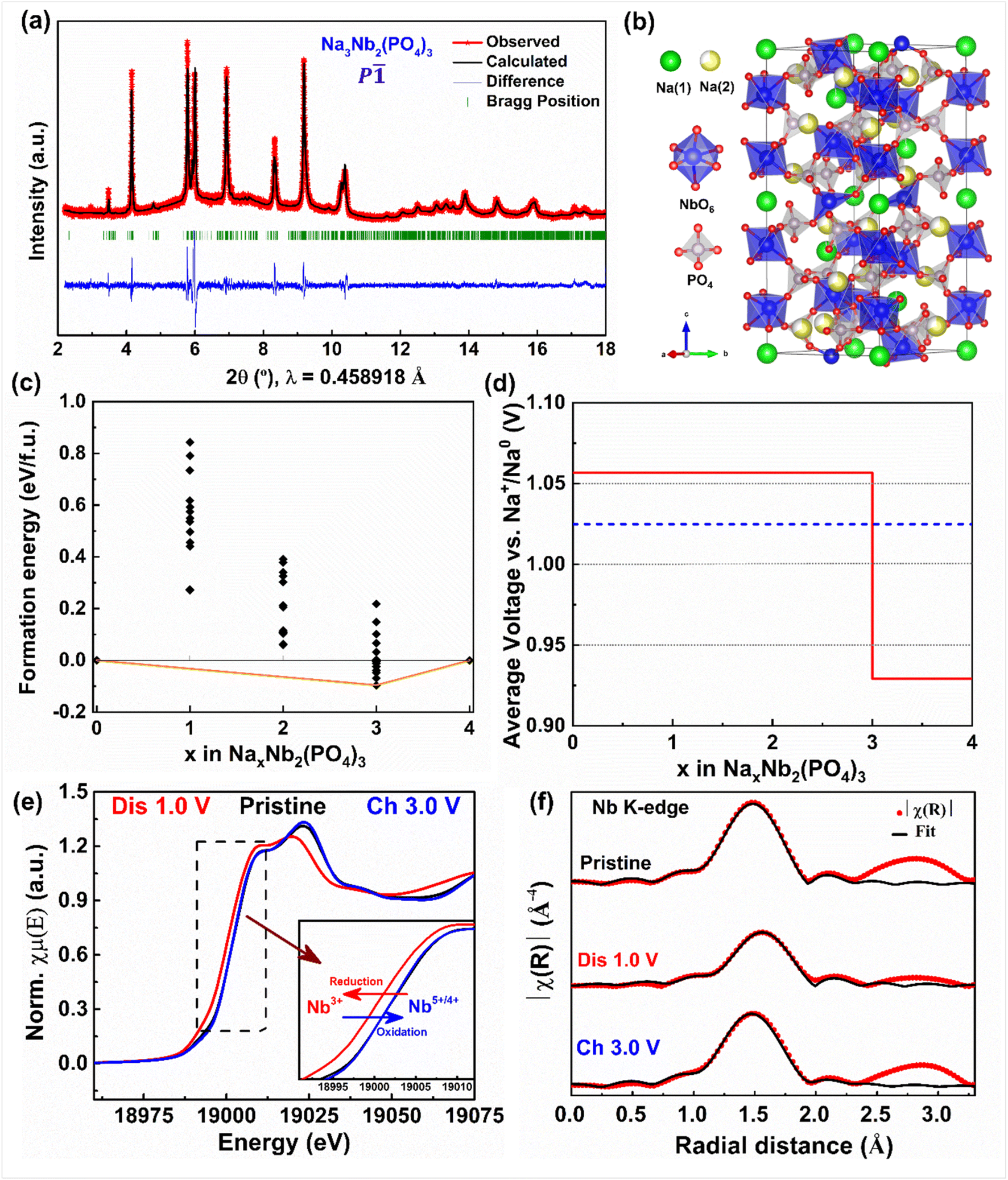 | ||
| Fig. 4 (a) Le Bail refinement of synchrotron XRD and (b) crystal structure of Na3Nb2(PO4)3. (c) 0 K formation energy and (d) voltage plots (vs. Na) as a function of Na concentration (x) in the NaxNb2(PO4)3 formula unit. The formation energy in (c) is referenced to the Nb2(PO4)3 (fully empty) and Na4Nb2(PO4)3 (fully sodiated) compositions. The orange line in (c) indicates the convex hull, i.e., the collection of lowest energy Na-vacancy configurations in NaxNb2(PO4)3, while the black diamonds indicate metastable configurations. The dashed black line in (d) indicates the overall average voltage across the entire Na concentration in Nb2(PO4)3. (e) XANES and (f) EXAFS plots of the NASICON-Nb2(PO4)3 anode collected at the Nb K-edge. | ||
To further understand the Na (de)intercalation phase behavior, we performed DFT calculations (see the Computations section), with the calculated 0 K formation energy and voltage values plotted as a function of Na concentration (x) in Nb2(PO4)3 in Fig. 4c and d, respectively. The convex hull in Fig. 4c indicates that the stable ground states of NaxNb2(PO4)3 include the fully empty (Nb2(PO4)3), fully sodiated (Na4Nb2(PO4)3), and partially sodiated (Na3Nb2(PO4)3) configurations. Importantly, the ground state at x = 3, is similar to the ground state configurations observed in other NASICON cathode chemistries.52,53 The Na3Nb2(PO4)3 ground state has Na fully occupying the Na(1) site and partially occupying the Na(2) site, which is in line with our experimental observation as well (Fig. 4b). In contrast to other cathode chemistries, however, we don't observe a ground state configuration at x = 1,52 which explains the minor solubility (x ≤ 0.2) of Na in the empty Nb2(PO4)3 structure. In terms of average voltages (Fig. 4d), we predict a voltage plateau of ∼1.06 V between x = 0 and 3, and ∼0.93 V between x = 3 and 4, partly in agreement with our observed voltage plateau of ∼1.15 V vs. Na that represents a two-phase intercalation mechanism as well. Experimentally, we observe capacity degradation to occur whenever we attempt to cycle the NaxNb2(PO4)3 system below 1 V vs. Na. This capacity degradation can be due to electrolyte degradation and/or Na-entrapment. Note that computationally, we have calculated a two-phase voltage plateau to form at voltages below 1 V vs. Na, which will result in the formation of the Na4Nb2(PO4)3 phase. Previous computational studies on NASICON systems60 showed that the Na conductivity (or diffusivity) drops significantly as Na content in NASICONs approach xNa = 4. Moreover, the redox behavior may not be fully reversible from Nb2+ till Nb4+, as reported with other transition metals as well within the NASICON framework.53 Hence, we expect partial entrapment of Na to occur at voltage ranges below 1 V vs. Na, due to the formation of the Na4Nb2(PO4)3 phase.
To monitor the changes in the Nb oxidation state and local structure, we performed X-ray absorption spectroscopy measurements. Fig. 4e displays the normalized XANES spectra of pristine, discharged, and charged electrodes collected at the Nb K-edge. As the discharge proceeds the XANES of the Nb K-edge shifts towards lower energy values, indicating the reduction of Nb5+/Nb4+ to Nb4+/Nb3+, and subsequently moves back to higher energy values at the end of the first charge. The corresponding Fourier-transformed extended X-ray absorption fine structure (EXAFS) plots are displayed in Fig. 4f. The peak located at 1.5 Å corresponds to the Nb–O shell, whereas the next three peaks located in between ∼2.0–3.25 Å represent the second and third shells of Nb–P and Nb–Na pairs. The EXAFS data collected at the end of discharge shift towards higher Å values, indicating the expansion of the NASICON structure. Note that although the EXAFS data collected on the subsequent discharge shifts towards lower Å values, it doesn't match with that of the pristine electrode due to the entrapment of Na-ions in the NASICON framework. These observations are in agreement with our electrochemical and in-situ XRD results. Furthermore, the fitting of EXAFS oscillations shows the increase of the average Nb–O bond length from 2.028 to 2.086 Å during discharge due to the reduction of niobium.
Our combined electrochemical and in situ experiments highlight the distinct intercalation behavior of the same NASICON-Nb2(PO4)3 anode against Li and Na. For instance, the Nb2(PO4)3/Li and Nb2(PO4)3/Na cells exhibit flat and sloping voltage curves for the initial one mole of Li and Na ion intercalation, respectively, even though both mostly follow a two-phase mechanism. Moreover, the volume of the Nb2(PO4)3 unit cell shrinks along with the loss of long-range crystal order during the low voltage lithiation (∼at 1.55 V vs. Li+/Li0) whereas the same anode exhibits volume expansion upon the low voltage sodiation (at ∼1.1 V vs. Na+/Na0). Hence, to better comprehend the Li and Na intercalation mechanism of the NASICON-Nb2(PO4)3 anode, we performed galvanostatic intermittent titration technique (GITT) experiments during the fourth cycle (see the Experimental section for details, Fig. 5a and b).
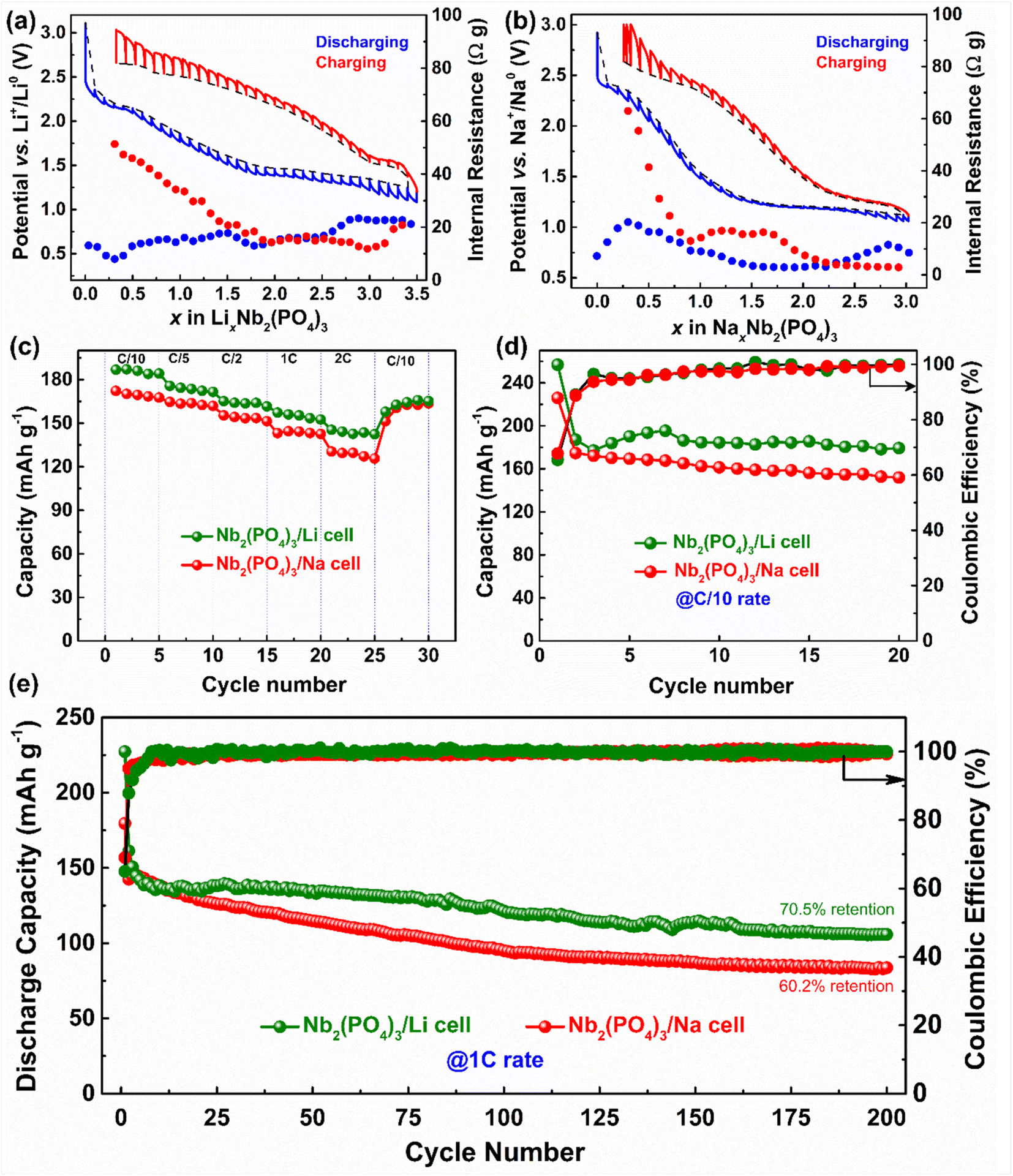 | ||
| Fig. 5 GITT curves and calculated internal resistance for (a) Nb2(PO4)3/Li and (b) Nb2(PO4)3/Na cells. Rate capability (c), capacity retention at C/10 (d) and 1C (e) plots of Nb2(PO4)3/Li and Nb2(PO4)3/Na cells. | ||
Notably, both Li and Na GITT voltage vs. composition (x in LixNb2(PO4)3 or NaxNb2(PO4)3) profiles present significant polarization between charge and discharge processes (even after 4 h relaxation). More importantly, the voltage jumps during the open circuit voltage (OCV) steps are prominent during charging compared to the discharging process, which is also reflected by the steep increase of internal resistance values and can be ascribed to slower solid-state Li and Na ion diffusion.61,62 Such a difference could arise from various thermodynamic and kinetic factors involving structural and electrochemical properties of the NASICON host. As mentioned earlier, Li and Na ions prefer to occupy different crystallographic sites, which have distinct (electro)chemical potentials for (de)intercalation.63 Moreover, it is well known that ionic conduction in the NASICON framework occurs via correlated ion migration through M(1)/M(2) pathways,24,64 thus the relative filling of these sites can tune ionic conductivities and intercalation kinetics. Besides, the different chemical characters of Li and Na ions (i.e., relative polarizability) can impart a different degree of interactions with the host lattice (such as distortion of polyhedral units),65 which can lead to stabilization of various intermediates including lower symmetry structures (C2/c and P as in the cases of Na3V2(PO4)3 and Na3Nb2(PO4)3, respectively)59,63,66 during cycling, leading to distinct electrochemical Li and Na intercalation pathways. Nevertheless, comprehensive studies involving short-range and long-range structural probes and DFT calculations are required to fully elucidate the structure–property relationship of the Nb2(PO4)3 anode.
Lastly, we have evaluated the rate performances and cycling stabilities of Nb2(PO4)3/Li and Nb2(PO4)3/Na cells at different C-rates. The Nb2(PO4)3/Li cell delivers discharge capacities of 186, 174, 164, 155, and 142 mA h g−1 at C/10, C/5, C/2, 1C, and 2C rates, respectively, whereas the Nb2(PO4)3/Na cell shows capacities of 172, 163, 153, 144 and 129 mA h g−1 under similar experimental conditions (Fig. 5c). Upon cycling at C/10 rate, the Nb2(PO4)3/Li and Nb2(PO4)3/Na cells exhibit stable discharge capacities greater than 184 and 156 mA h g−1, respectively, for 20 cycles (Fig. 5d). Further, we assessed their cycling stability at 1C rate (Fig. 5e). The Nb2(PO4)3/Li and Nb2(PO4)3/Na cells exhibit first/second cycle discharge capacities of 227/161 and 179/142 mA h g−1, respectively. The large discrepancies in discharge capacities between the first and second cycles can be attributed to the electrolyte decomposition and the entrapment of Li and Na ions in the NASICON framework. In the subsequent cycles, the discharge capacities of the Nb2(PO4)3/Li cell slowly decrease and stabilize around 122 mA h g−1 (after 100 cycles) and the cell still delivers a discharge capacity of 100 mA h g−1 after 200 cycles. The Nb2(PO4)3/Na cell retains 60.2% of its second discharge capacity after 200 cycles. The lower capacities obtained at high C-rate could be due to micron-size active particles in the as-prepared anode (Fig. S4†). It is also worth mentioning here that upon lowering the discharge voltage to 0.9 V, the capacity decay is faster in Nb2(PO4)3/Na cells due to significant electrolyte degradation (Fig. S5†). Further studies to improve the cycling performance of the Nb2(PO4)3 anode as well as to build full Li- and Na-ion cells are in progress. It is important to note that the integration of the NASICON-Nb2(PO4)3 anode and the NASICON-Na3V2(PO4)3 cathode with a suitable electrolyte is expected to produce a higher energy density Na-ion battery compared to the Ti- and V-based anodes (Fig. S6†).67
Conclusion
In conclusion, we have successfully synthesized polycrystalline Nb2(PO4)3 and demonstrated its potential application as an anode material for Li- and Na-ion batteries. The NASICON-Nb2(PO4)3 reversibly exchanged Li and Na ions at average voltages of 1.86 V and 1.46 V vs. Li+/Li0 and Na+/Na0 with intercalation capacities of ∼167 and 150 mA h g−1, respectively. Our in situ XRD measurements revealed multiple-phase transformations during Li and Na intercalation with the formation of short-range ordered Li3Nb2(PO4)3 and triclinic (P)-Na3Nb2(PO4)3 at the end of discharge. Our DFT calculations predicted the Na3Nb2(PO4)3 composition to be stable in the Na–Nb2(PO4)3 pseudo-binary system, in agreement with the in situ XRD results. The distinct electrochemical Li and Na intercalation behavior of the Nb2(PO4)3 anode can be ascribed to the relative differences in size, filling of crystallographic sites and chemical characters of Li and Na ions. Although the micron-sized anode showed moderate storage capacities (∼106 and 84 mA h g−1 for Li and Na cells, respectively) at higher C-rate (1C) after 200 cycles, further optimization of the electrode and electrolytes is expected to produce better performance, which can aid in building high energy density Na-ion batteries.
Conflicts of interest
The authors declare no conflict of interest.Author contributions
All authors have approved the final version of the manuscript.Acknowledgements
This work was supported by the Department of Science & Technology (DST), Government of India (DST/TMD/MES/2K18/188). B. P. thanks CSIR for the research fellowship. G. S. G. acknowledges support from the Science and Engineering Research Board (SERB), Government of India, under sanction number IPA/2021/000007. D. D. acknowledges financial assistance from Ministry of Human Resource Development, Government of India. The authors acknowledge DESY (Hamburg, Germany), a member of the Helmholtz Association HGF, for the provision of experimental facilities. Parts of this research were carried out at PETRA III and the authors thank Dr Edmund Welter for his assistance in using Beamline P65 to perform X-ray absorption spectroscopy measurements and DST for financial assistance for the measurement at DESY. Synchrotron X-ray diffraction data were collected at 11-BM (mail-in program; GUP-77942), at the Advanced Photon Source, Argonne National Laboratory, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. A portion of the density functional theory calculations showcased in this work were performed with the computational resources provided by the Supercomputer Education and Research Center, Indian Institute of Science.References
- Z. Yang, J. Zhang, M. C. W. Kintner-Meyer, X. Lu, D. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 CrossRef CAS PubMed.
- B. Dunn, H. Kamath and J. M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- T. Kim, W. Song, D. Y. Son, L. K. Ono and Y. Qi, J. Mater. Chem. A, 2019, 7, 2942–2964 RSC.
- Y. Tian, G. Zeng, A. Rutt, T. Shi, H. Kim, J. Wang, J. Koettgen, Y. Sun, B. Ouyang, T. Chen, Z. Lun, Z. Rong, K. Persson and G. Ceder, Chem. Rev., 2021, 121, 1623–1669 CrossRef CAS PubMed.
- N. Tapia-Ruiz, A. R. Armstrong, H. Alptekin, M. A. Amores, H. Au, J. Barker, R. Boston, W. R. Brant, J. M. Brittain, Y. Chen, M. Chhowalla, Y. S. Choi, S. I. R. Costa, M. C. Ribadeneyra, S. A. Cussen, E. J. Cussen, W. I. F. David, A. V. Desai, S. A. M. Dickson, E. I. Eweka, J. D. Forero-Saboya, C. P. Grey, J. M. Griffin, P. Gross, X. Hua, J. T. S. Irvine, P. Johansson, M. O. Jones, M. Karlsmo, E. Kendrick, E. Kim, O. V. Kolosov, Z. Li, S. F. L. Mertens, R. Mogensen, L. Monconduit, R. E. Morris, A. J. Naylor, S. Nikman, C. A. O'Keefe, D. M. C. Ould, R. G. Palgrave, P. Poizot, A. Ponrouch, S. Renault, E. M. Reynolds, A. Rudola, R. Sayers, D. O. Scanlon, S. Sen, V. R. Seymour, B. Silván, M. T. Sougrati, L. Stievano, G. S. Stone, C. I. Thomas, M. M. Titirici, J. Tong, T. J. Wood, D. S. Wright and R. Younesi, J. Phys. Energy, 2021, 3 Search PubMed 031503.
- K. Chayambuka, G. Mulder, D. L. Danilov and P. H. L. Notten, Adv. Energy Mater., 2020, 10, 2001310 CrossRef CAS.
- E. Goikolea, V. Palomares, S. Wang, I. R. de Larramendi, X. Guo, G. Wang and T. Rojo, Adv. Energy Mater., 2020, 10, 2002055 CrossRef CAS.
- J. M. Tarascon, Joule, 2020, 4, 1616–1620 CrossRef.
- E. Irisarri, A. Ponrouch and M. R. Palacin, J. Electrochem. Soc., 2015, 162, A2476–A2482 CrossRef CAS.
- G. N. Zhu, Y. G. Wang and Y. Y. Xia, Energy Environ. Sci., 2012, 5, 6652–6667 RSC.
- K. M. Colbow, J. R. Dahn and R. R. Haering, J. Power Sources, 1989, 26, 397–402 CrossRef CAS.
- J. F. Colin, V. Pralong, M. Hervieu, V. Caignaert and B. Raveau, Chem. Mater., 2008, 20, 1534–1540 CrossRef CAS.
- B. Guo, X. Yu, X. G. Sun, M. Chi, Z. A. Qiao, J. Liu, Y. S. Hu, X. Q. Yang, J. B. Goodenough and S. Dai, Energy Environ. Sci., 2014, 7, 2220–2226 RSC.
- S. Qian, H. Yu, L. Yan, H. Zhu, X. Cheng, Y. Xie, N. Long, M. Shui and J. Shu, ACS Appl. Mater. Interfaces, 2017, 9, 30608–30616 CrossRef CAS PubMed.
- Q. Cheng, J. Liang, Y. Zhu, L. Si, C. Guo and Y. Qian, J. Mater. Chem. A, 2014, 2, 17258–17262 RSC.
- P. Senguttuvan, G. Rousse, V. Seznec, J. M. Tarascon and M. R. Palacín, Chem. Mater., 2011, 23, 4109–4111 CrossRef CAS.
- W. Wang, Y. Liu, X. Wu, J. Wang, L. Fu, Y. Zhu, Y. Wu and X. Liu, Adv. Mater. Technol., 2018, 3, 1800004 CrossRef.
- Y. Sun, L. Zhao, H. Pan, X. Lu, L. Gu, Y. S. Hu, H. Li, M. Armand, Y. Ikuhara, L. Chen and X. Huang, Nat. Commun., 2013, 4, 1870 CrossRef PubMed.
- H. Gao and J. B. Goodenough, Angew. Chem., Int. Ed., 2016, 55, 12768–12772 CrossRef CAS PubMed.
- J. Wang, Y. Wang, D. H. Seo, T. Shi, S. Chen, Y. Tian, H. Kim and G. Ceder, Adv. Energy Mater., 2020, 10, 1–10 Search PubMed.
- S. Ghosh, N. Barman, M. Mazumder, S. K. Pati, G. Rousse and P. Senguttuvan, Adv. Energy Mater., 2020, 10, 1902918 CrossRef CAS.
- S. Chen, C. Wu, L. Shen, C. Zhu, Y. Huang, K. Xi, J. Maier and Y. Yu, Adv. Mater., 2017, 29, 1700431 CrossRef PubMed.
- Z. Jian, Y. S. Hu, X. Ji and W. Chen, Adv. Mater., 2017, 29, 1601925 CrossRef PubMed.
- M. Guin and F. Tietz, J. Power Sources, 2015, 273, 1056–1064 CrossRef CAS.
- Z. Zhang, Z. Zou, K. Kaup, R. Xiao, S. Shi, M. Avdeev, Y. S. Hu, D. Wang, B. He, H. Li, X. Huang, L. F. Nazar and L. Chen, Adv. Energy Mater., 2019, 9, 1–14 CrossRef.
- Z. Deng, G. Sai Gautam, S. K. Kolli, J. N. Chotard, A. K. Cheetham, C. Masquelier and P. Canepa, Chem. Mater., 2020, 32, 7908–7920 CrossRef CAS.
- R. Kahlaoui, K. Arbi, I. Sobrados, R. Jimenez, J. Sanz and R. Ternane, Inorg. Chem., 2017, 56, 1216–1224 CrossRef CAS PubMed.
- C. Masquelier, C. Wurm, J. Rodríguez-Carvajal, J. Gaubicher and L. Nazar, Chem. Mater., 2000, 12, 525–532 CrossRef CAS.
- A. Aatiq, M. Ménétrier, L. Croguennec, E. Suard and C. Delmas, J. Mater. Chem., 2002, 12, 2971–2978 RSC.
- C. Delmas, F. Cherkaoui, A. Nadiri and P. Hagenmuller, Mater. Res. Bull., 1987, 22, 631–639 CrossRef CAS.
- S. Patoux and C. Masquelier, Chem. Mater., 2002, 14, 5057–5068 CrossRef CAS.
- M. Wu, W. Ni, J. Hu and J. Ma, Nano-Micro Lett., 2019, 11, 1–36 CrossRef PubMed.
- P. Senguttuvan, G. Rousse, M. E. Arroyo Y De Dompablo, H. Vezin, J. M. Tarascon and M. R. Palacín, J. Am. Chem. Soc., 2013, 135, 3897–3903 CrossRef CAS PubMed.
- Z. Jian, L. Zhao, H. Pan, Y. S. Hu, H. Li, W. Chen and L. Chen, Electrochem. Commun., 2012, 14, 86–89 CrossRef CAS.
- Z. Jian, W. Han, X. Lu, H. Yang, Y.-S. Hu, J. Zhou, Z. Zhou, J. Li, W. Chen, D. Chen, L. Chen, Z. L. Jian, W. Z. Han, X. Lu, H. X. Yang, Y. Hu, J. Q. Li, L. Q. Chen, J. Zhou, W. Chen, D. F. Chen and Z. B. Zhou, Adv. Energy Mater., 2013, 3, 156–160 CrossRef CAS.
- O. Tillement, J. C. Couturier, J. Angenault and M. Quarton, Solid State Ionics, 1991, 48, 249–255 CrossRef CAS.
- J. Liu, K. Lin, Y. Zhao, Y. Zhou, X. Hou, X. Liu, H. Lou, K. H. Lam and F. Chen, J. Mater. Chem. A, 2021, 9, 10437–10446 RSC.
- S. Ghosh, N. Jose, B. Senthilkumar, P. Amonpattaratkit and P. Senguttuvan, J. Electrochem. Soc., 2021, 168 Search PubMed 050534.
- T. Zhu, P. Hu, X. Wang, Z. Liu, W. Luo, K. A. Owusu, W. Cao, C. Shi, J. Li, L. Zhou and L. Mai, Adv. Energy Mater., 2019, 9, 2–7 Search PubMed.
- I. V. Zatovsky, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66, i12 CrossRef CAS PubMed.
- B. Ouyang, J. Wang, T. He, C. J. Bartel, H. Huo, Y. Wang, V. Lacivita, H. Kim and G. Ceder, Nat. Commun., 2021, 12, 1–11 CrossRef PubMed.
- A. Leclaire, M.-M. Borel, A. Grandin and B. Raveau, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1989, 45, 699–701 CrossRef.
- J. Rodríguez-Carvajal, Phys. B Condens. Matter, 1993, 192, 55–69 CrossRef.
- M. Newville, J. Synchrotron Radiat., 2001, 8, 322–324 CrossRef CAS PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- J. Sun, A. Ruzsinszky and J. Perdew, Phys. Rev. Lett., 2015, 115 Search PubMed 036402.
- M. Hellenbrandt, Crystallogr. Rev., 2004, 10, 17–22 CrossRef CAS.
- S. P. Ong, W. D. Richards, A. Jain, G. Hautier, M. Kocher, S. Cholia, D. Gunter, V. L. Chevrier, K. A. Persson and G. Ceder, Comput. Mater. Sci., 2013, 68, 314–319 CrossRef CAS.
- B. Singh, Z. Wang, S. Park, G. S. Gautam, J. N. Chotard, L. Croguennec, D. Carlier, A. K. Cheetham, C. Masquelier and P. Canepa, J. Mater. Chem. A, 2021, 9, 281–292 RSC.
- Z. Wang, S. Park, Z. Deng, D. Carlier, J. N. Chotard, L. Croguennec, G. S. Gautam, A. K. Cheetham, C. Masquelier and P. Canepa, J. Mater. Chem. A, 2022, 10, 209–217 RSC.
- M. Bianchini, F. Fauth, N. Brisset, F. Weill, E. Suard, C. Masquelier and L. Croguennec, Chem. Mater., 2015, 27, 3009–3020 CrossRef CAS.
- M. Bianchini, J. M. Ateba-Mba, P. Dagault, E. Bogdan, D. Carlier, E. Suard, C. Masquelier and L. Croguennec, J. Mater. Chem. A, 2014, 2, 10182–10192 RSC.
- M. Galceran, J. Rikarte, M. Zarrabeitia, M. C. Pujol, M. Aguiló and M. Casas-Cabanas, ACS Appl. Energy Mater., 2019, 2, 1923–1931 CrossRef CAS.
- P. Senguttuvan, G. Rousse, H. Vezin, J. M. Tarascon and M. R. Palacín, Chem. Mater., 2013, 25, 2391–2393 CrossRef CAS.
- H. Kabbour, D. Coillot, M. Colmont, C. Masquelier and O. Mentré, J. Am. Chem. Soc., 2011, 133, 11900–11903 CrossRef CAS PubMed.
- P. Senguttuvan, G. Rousse, M. E. Arroyo Y De Dompablo, H. Vezin, J. M. Tarascon and M. R. Palacín, J. Am. Chem. Soc., 2013, 135, 3897–3903 CrossRef CAS PubMed.
- Z. Deng, T. P. Mishra, E. Mahayoni, Q. Ma, A. J. K. Tieu, O. Guillon, J. N. Chotard, V. Seznec, A. K. Cheetham, C. Masquelier, G. S. Gautam and P. Canepa, Nat. Commun., 2022, 13, 1–14 Search PubMed.
- G. Yan, S. Mariyappan, G. Rousse, Q. Jacquet, M. Deschamps, R. David, B. Mirvaux, J. W. Freeland and J. M. Tarascon, Nat. Commun., 2019, 10, 585 CrossRef CAS PubMed.
- J. Kim, W. Lee, J. Seok, E. Lee, W. Choi, H. Park, S. Yun, M. Kim, J. Lim and W. S. Yoon, J. Energy Chem., 2022, 66, 226–236 CrossRef CAS.
- C. Masquelier, C. Wurm, J. Rodríguez-Carvajal, J. Gaubicher and L. Nazar, Chem. Mater., 2000, 12, 525–532 CrossRef CAS.
- Z. Zhang, Z. Zou, K. Kaup, R. Xiao, S. Shi, M. Avdeev, Y. S. Hu, D. Wang, B. He, H. Li, X. Huang, L. F. Nazar and L. Chen, Adv. Energy Mater., 2019, 9, 1902373 CrossRef CAS.
- S. Zhou, G. Barim, B. J. Morgan, B. C. Melot and R. L. Brutchey, Chem. Mater., 2016, 28, 4492–4500 CrossRef CAS.
- J. N. Chotard, G. Rousse, R. David, O. Mentré, M. Courty and C. Masquelier, Chem. Mater., 2015, 27, 5982–5987 CrossRef CAS.
- M. K. Sadan, A. K. Haridas, H. Kim, C. Kim, G. B. Cho, K. K. Cho, J. H. Ahn and H. J. Ahn, Nanoscale Adv., 2020, 2, 5166–5170 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta05971a |
| This journal is © The Royal Society of Chemistry 2023 |