
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBifunctional heterogeneous catalysts from biomass and waste polysaccharides for the conversion of CO2 into cyclic carbonates†
Adriano
Parodi‡
 *a,
Martina
Vagnoni‡
*a,
Lucia
Frontali
a,
Cristiano
Albonetti
b,
Francesca
De Giorgio
b,
Alessio
Mezzi
c,
Elisabetta
Petri
a,
Chiara
Samorì
a,
Francesca
Soavi
a,
Giampiero
Ruani
b and
Paola
Galletti
a
*a,
Martina
Vagnoni‡
*a,
Lucia
Frontali
a,
Cristiano
Albonetti
b,
Francesca
De Giorgio
b,
Alessio
Mezzi
c,
Elisabetta
Petri
a,
Chiara
Samorì
a,
Francesca
Soavi
a,
Giampiero
Ruani
b and
Paola
Galletti
a
aChemistry Department “Giacomo Ciamician”, University of Bologna, Via Selmi, 2, 40126, Bologna, Italy. E-mail: adriano.parodi2@unibo.it; martina.vagnoni3@unibo.it
bConsiglio Nazionale delle Ricerche, Istituto per lo Studio dei Materiali Nanostrutturati (CNR-ISMN), Via Piero Gobetti, 101, 40129 Bologna, Italy
cConsiglio Nazionale delle Ricerche, Istituto per lo Studio dei Materiali Nanostrutturati (CNR-ISMN), Via Salaria Km 29.300 – 00015 Monterotondo Scalo, Italy
First published on 5th December 2022
Abstract
A four-step methodology for the valorization of polysaccharide-based materials into bifunctional heterogeneous catalysts, active in the conversion of CO2 and epoxides into cyclic carbonates, is presented. The synthesis protocol consists of (i) pyrolysis of the starting material to produce biochar; (ii) oxidation to increase the number of –OH and –COOH functionalities; (iii) anchoring of (3-aminopropyl)triethoxysilane (APTES) on the surface of the oxidized biochar; (iv) quaternarization of the amine groups into alkylammonium iodide salts. The versatility of the method was demonstrated by applying the same protocol to six different polysaccharidic materials and wastes: six catalysts with no appreciable differences in terms of chemical composition and catalytic activity were obtained. The bifunctionality given by –OH and ammonium iodide groups was confirmed by several analyses performed on the catalysts. An extensive characterization (elemental analysis composition, FTIR, Raman, SEM, XPS and porosimetry) was done on all the functionalized biochars for every synthetic step. The catalysts were widely investigated in their activity for the conversion of CO2 and epoxides into cyclic carbonates, demonstrating to be effective under mild conditions (3 bar of CO2; 70 °C; 7 h). TONs and TOFs were calculated for each catalyst and condition. Yields up to 96%, with >99% selectivity, were obtained for terminal epoxides. The recyclability of the bifunctional heterogeneous catalysts was also confirmed over five cycles.
1. Introduction
The increasing accumulation of CO2 in the Earth's atmosphere is a clear matter of concern for all humanity.At the same time, renewable powers (e.g., wind, solar) or non-fossil fuels (e.g., hydrogen), whose use does not involve CO2 emissions, are still far from being the main sources of the planet's energy supply. A uniquely effective solution to the climate crisis is not currently available but waste recycling/valorization and CO2 reuse are among the approaches that could help in mitigating such impacts.
CO2 can be adsorbed or incorporated into molecules and materials following many reactions and protocols which were proven to be effective and useful for industries.1 In this framework, the synthesis of cyclic carbonates from CO2 and epoxides is an interesting reaction: it is a solvent-free and 100% atom economy process, and it exploits an abundant and non-toxic C1 feedstock.2 Cyclic carbonates have a potential application in the synthesis of biocompatible polymers and linear dialkyl carbonates that are used as aprotic solvents in lithium-ion battery electrolytes.3 CO2 conversion in cyclic carbonates has been the focus of many research studies. The most common and active catalytic systems for this reaction feature a Lewis acid site (a metallic center or an H-bond donor) that activates the epoxide, through coordination with the O atom, and a Lewis base (generally a halide) that is responsible for the ring-opening of the epoxide through a nucleophilic attack. After the formation of the alkoxide intermediate, the insertion of CO2 occurs, followed by the intramolecular ring closure to yield the cyclic carbonate product. The need for two different active species often requires the use of a bi-component catalytic system in which the Lewis base is the main catalyst, responsible for the epoxy-ring opening (often reported as the rate-determining step of the reaction),4 while the Lewis acid is the co-catalyst (Fig. 1).5 It is difficult to have a single catalyst bearing the two active sites, both in homogeneous and heterogeneous reactions, and finding a non-metallic, effective and recyclable catalyst that works under mild conditions is still a challenging task.6 Another interesting feature of the synthesis of cyclic carbonates is the possibility of using solvent-free conditions. Bifunctional heterogeneous catalysts are thus particularly attractive for the ease of product/catalyst separation and catalyst reuse in multiple cycles.7,8
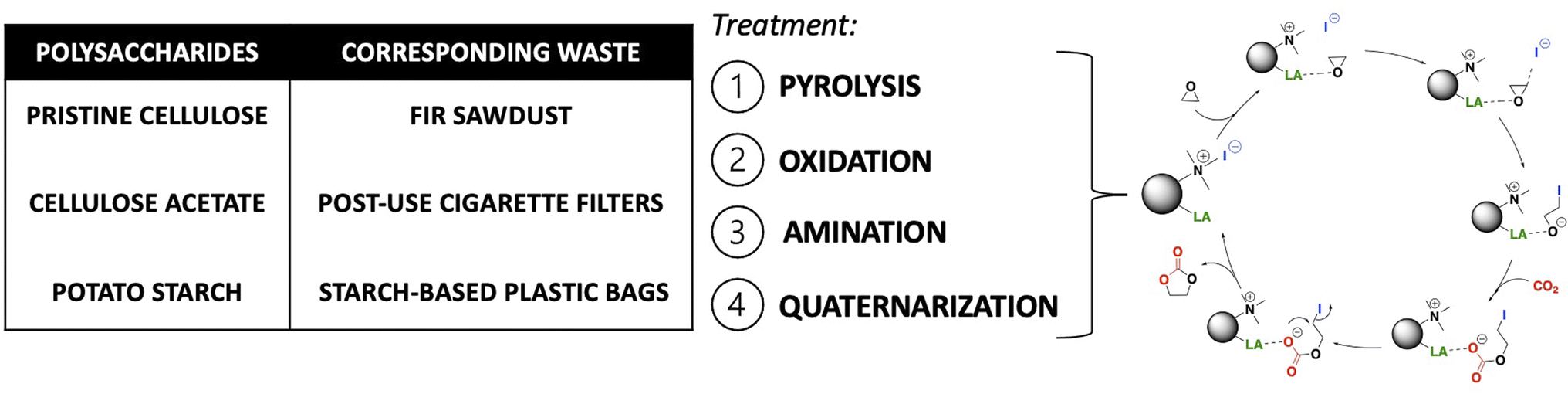 | ||
| Fig. 1 Scope of the work: four-step protocol for the synthesis of six heterogeneous catalysts starting from polysaccharide-based materials, and their use in the synthesis of cyclic carbonates form epoxides and CO2. | ||
Due to the presence of surface-OH groups, several bio-based polymeric materials (e.g., cellulose, lignin, chitosan, chitin and lignocellulose)9 have been used as Lewis acid catalysts for this kind of reaction, in combination with Lewis bases, like tetrabutylammonium halides or KI that were always needed.10–13 Bio-based polymeric materials have been also exploited as heterogeneous supports for ionic liquids or deep eutectic solvents, meanly bearing an imidazolium chain as Lewis base;14–19 or as functionalized materials (i.e. with a quaternary ammonium salt).20–22
The recycling and valorization of waste has been carried out over the years through various chemical, biological and thermochemical processes (e.g., pyrolysis, gasification or hydrothermal conversion).23,24 Specifically, various biofuels or biochemicals, such as syngas, bio-oil, biochar/char and platform chemicals can be obtained through thermochemical processes.25 Biochar is a carbonaceous material whose physicochemical properties vary significantly according to the feedstock in input, the carbonization process, and the activation or functionalization methods.5 Thanks to the porous structure, the large surface area and the high quantity of functional groups on the surface, activated or functionalized bio-chars have been widely used as activated carbons, soil amendments, carbon sequestration agents and environmental adsorbents for organic and heavy metal removal.26,27 More importantly, due to the possibility of covalently adding different functional groups on their surface, biochars can be used as versatile catalysts and/or catalyst supports in many chemical processes, such as biodiesel production.28,29
To the best of our knowledge, only one report in the literature deals with the conversion of CO2 and epoxides into cyclic carbonates using oxidized biochar from soft and hardwood with alcoholic or carboxylic groups. These moieties work as H-bond donors on the surface of biochar behaving as Lewis acid co-catalyst with the addition of the catalyst tetrabutylammonium bromide (TBAB) as Lewis base.30 See S1† for an overview of heterogeneous catalysts made from bio-based polymers (and carbonaceous materials) already reported in literature.
Here, biochars made from three different polysaccharides (cellulose, cellulose acetate, and starch) and corresponding waste/post-used materials (fir sawdust, post-use cigarette filters and starch-based plastic bags) were derivatized with a new versatile four-step protocol to obtain bifunctional heterogeneous catalysts. Such catalysts were then tested for the synthesis of cyclic carbonates by incorporating CO2 into epoxides in mild conditions (Fig. 1). An extensive characterization of the final catalysts and the materials obtained after each functionalization step was also performed.
2. Experimental section
2.1 Materials
All chemicals were purchased from Sigma-Aldrich and used without further purification. CO2 with ≥99.5% purity was purchased from Siad, Italy. Starch-based plastic bags (SBPB) were got from a local supermarket (Ravenna, Italy). Post-use cigarette filters (PUCF) were self-obtained after smoking cigarettes. Fir Sawdust (FSD) was purchased by a woodworking company (Salati e Montepietra s.r.l., Reggio Emilia, Italy).2.2 Catalysts synthesis
All the catalysts reported in this work were synthesized following an optimized experimental procedure based on four steps as reported below.1. Pyrolysis of polysaccharides and corresponding waste materials. Starting materials were pyrolyzed following a previously reported procedure.29 Hence, cellulose acetate (CA), pristine cellulose (PC), starch-based plastic bags (SBPB), post-use cigarette filters (PUCF), fir sawdust (FSD) (5 g) or potato starch (PS) (3 g) were subjected to bench-scale pyrolysis, using an apparatus consisting of a sliding sample carrier placed in a heated quartz tube connected to ice traps and a settling chamber. The quartz tube was heated by a cylindrical co-axial furnace and purged by 1.5 L per min N2 flow. Samples were moved into the heated zone of the quartz tube and heated for 15 h at 420 °C (measured temperature) under N2 flow. The resulting char (C) was collected, ground to powder in a mortar and used for the next steps without further purification. Yields of each C are reported in S2.†
2. Oxidation of chars. In a 50 mL round-bottomed flask, the selected char (500 mg) was stirred in H2O2 (30%, 25 mL) at 85 °C for 24 h. After that time, the reaction mixture was cooled to room temperature (RT) and a solution of HCl 1 M (5 mL) was added. Then the suspension was filtered and washed several times with H2O to recover oxidized chars (OC), that was dried overnight at 70 °C under reduced pressure (100 mbar). Yields of each OC are reported in S2.†
3. Anchoring of amine functionality. In a 25 mL round-bottomed flask equipped with a reflux condenser, the selected OC (80 mg) was added to a solution of (3-aminopropyl)triethoxysilane (APTES, 4.4 v/v%) in a mixture ethanol (EtOH)/H2O 96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 (5 mL). The mixture was stirred at 85 °C for 4 h, then the solution was filtered and washed three times with EtOH (10 mL). The resulting amine-functionalized char (AC) was dried overnight at 70 °C under reduced pressure (100 mbar). Yields of each AC are reported in S2.†
:4 (5 mL). The mixture was stirred at 85 °C for 4 h, then the solution was filtered and washed three times with EtOH (10 mL). The resulting amine-functionalized char (AC) was dried overnight at 70 °C under reduced pressure (100 mbar). Yields of each AC are reported in S2.†
The drying procedure for steps 2 and 3 could be avoided, with no change in activity. Here it has been performed in the case of complete characterization of catalyst intermediates.
4. Amine quaternarization. In a sealed tube the selected AC (100 mg) was put in EtOH (1 mL), then methyl iodide (MeI, 0.11 mL, 246 mg) or longer alkyl iodides were added. The reaction was stirred at 45 °C for 15 h, then the resulting mixture was filtered and washed with EtOH (2 × 5 mL) and ethyl acetate (2 × 5 mL). The resulting ammonium iodide functionalized char (HC) was dried overnight under reduced pressure (100 mbar). Yields of each HC are reported in S2.†
Main optimization phases for steps 1–4 are described in S3 (see Table S3a†).
2.3 Synthesis of cyclic carbonates
The tests with CO2 at 3–5 bar were carried out in a stainless-steel, self-made, 25 mL autoclave equipped with a heating mantle. In a typical experiment, the epoxide (0.875 mmol) and the catalyst (5–10% w/w respect to the epoxide) were weighed into a 2 mL glass vial equipped with a magnetic stirring bar and closed with a polytetrafluoroethylene (PTFE) septum pierced with a needle to let CO2 flow inside the vial. Then the vial was placed inside the steel autoclave. The air in the reactor was firstly replaced with CO2 and then the autoclave was heated and pressurized with CO2; the vial placed inside the autoclave was kept stirring (1500 rpm) for the whole reaction time. After the completion of the reaction, the autoclave was cooled to RT in 30 min and slowly depressurized.Reactions at 1 bar were conducted in a 25 mL Schlenk tube equipped with a CO2 balloon. The epoxide (0.875 mmol) and the catalyst (5–10% w/w respect to the epoxide) were weighed and put inside the Schlenk tube. The air in the tube was firstly replaced with CO2, then it was placed in an oil bath heated at 70 °C and kept stirring (1500 rpm). The CO2 in the balloon was then allowed to flow into the flask. After reaction completion, the Schlenk tube was cooled to RT.
In both cases, reaction crudes were diluted with ethyl acetate (1 mL) and the catalyst was separated from the crudes by centrifugation. The recovered catalyst was then washed twice with ethyl acetate. Crudes were weighed to check CO2 incorporation or any reagent loss, and then analyzed by GC-MS after further dilution in ethyl acetate. Selectivity was also determined by 1H NMR spectroscopy using mesitylene as the internal standard. Products have been isolated by flash column chromatography, isolated yields are reported in Tables 4–6. All obtained carbonates are known, thus they were recognized by comparison with standards or through NMR and mass spectra, matching what is reported in the NIST database. TONs have been calculated for each entry as mol of the product obtained on moles of iodide contained in the amount of catalyst (HC) used. The moles of iodide on grams of HC have been determined through XPS analysis (see S18 and S19†). TOFs have been calculated for each entry as TON values on reaction time (TON h−1). Productivities have been calculated as grams of product on grams of the catalyst, divided by reaction time: (gproduct/gcatalyst) h−1.
2.4 Catalyst recycling
The recycling of the catalysts was tested in the conversion of styrene oxide 1a (0.875 mmol) into the corresponding cyclic carbonate 2a using different HCs (10% w/w), at 3 bar of CO2, 70 °C, in 7 h. After the reaction completion, ethyl acetate (1 mL) was added to the crude and the catalyst was separated by centrifugation. The catalyst was then washed twice with ethyl acetate. The organic phase was collected to isolate the product by flash column chromatography, while the catalyst was kept under vacuum for one night to remove any traces of solvents. The recovered catalyst was weighed to check its total recovery (see S6†) and it was used for the next runs without further purification.2.5 Instrumentation
GC-MS analyses of reaction mixtures were performed using an Agilent HP 6850 gas chromatograph connected to an Agilent HP 5975 quadrupole mass spectrometer. Analytes were separated on an HP-5MS fused-silica capillary column (stationary phase 5%-phenyl-methylpolysiloxane, 30 m, 0.25 mm i.d., 0.25 μm film thickness), with helium as the carrier gas (at constant pressure, 36 cm s−1 linear velocity at 200 °C). Mass spectra were recorded under electron ionization (70 eV) at a frequency of 1 scan per s within the 12–600 m/z range. The injection port temperature was 250 °C. The temperature of the column was kept at 50 °C for 5 min, then increased from 50 to 250 °C at 10 °C min−1 and the final temperature of 250 °C was kept for 12 min. Epichlorohydrin 1c and allyl glycidyl ether 1d (more volatile than the other substrates) were analyzed through the following thermal program: 40 °C for 6 min, then from 50 to 250 °C at 10 °C min−1. 1H NMR spectra were recorded on a Varian 400 (400 MHz) spectrometer. 13C NMR spectra were recorded on a Varian 400 (100 MHz) spectrometer. Chemical shifts are reported in ppm from TMS with the solvent resonance as the internal standard (deuterochloroform: 7.26 ppm).2.6 Characterization of starting materials, chars and catalysts
The elemental composition of the starting materials, the chars at each derivatization step and the final catalysts was determined using an elemental analyzer (Thermo Scientific, Flash 2000, Organic Elemental Analyzer) through the flash combustion technique.Attenuated Total Reflectance Fourier Transform (ATR-FTIR) measurements were performed with a N2 purged Bruker Vertex 70 interferometer using a single reflection Platinum-ATR accessory (diamond crystal), a DLaTGS detector and a KBr beamsplitter. The spectra were recorded from 4000 cm−1 to 370 cm−1 with 4 cm−1 resolution at RT. The ATR-FTIR spectra correction was carried out.
The surface chemical composition of the samples was investigated by XPS. The XPS experiments were carried out by using an ESCALAB 250 Xi spectrometer, equipped with a monochromatic Al X-ray source and six channeltrons as a detection system. To avoid any charging effect, the measurements were performed by using a flood gun, neutralizing the charge induced by the photoelectrons emission. All samples were mounted fixing the powder on Au foil by mechanical pressure.
The Raman spectra were recorded with a Renishaw micro-Raman 1000 system exciting at 632.8 nm (HeNe laser) and 488 nm (diode laser) from 100.00 to 3200.00 cm−1 at RT. Results reported in S9.†
Scanning Electron Microscopy (SEM) analyses were performed with a SEM-FEG Hitachi S-4000 instrument. The images were collected with an acceleration voltage of 10–20 kV. Before the analysis, the samples were covered with an Au coating of a few nm by sputtering using a Q150R – Rotary Pumped Coater, except sawdust char. Results reported in S11.†
The porosity of the samples was evaluated by nitrogen adsorption porosimetry measurements that were carried out at 77 K with an ASAP 2020 system (Micromeritics) after a drying step for 24 h at 413 K. The N2 adsorption isotherms were analyzed by the Brunauer–Emmett–Teller (BET) and density functional (DFT) theories to obtain the specific surface area (SBET) and pore size distribution (PSD), respectively.
3. Results and discussion
3.1 Synthesis of bifunctional heterogeneous catalysts
The procedure adopted for obtaining the bifunctional catalysts from different polysaccharides and wastes is a four-step synthetic pathway consisting of (a) carbonization of the starting material to give a char (C); (b) oxidation of the char with H2O2 to give an oxidized char (OC); (c) functionalization of OC through the introduction of amine moieties by anchoring of APTES to give an amine-functionalized char (AC); (d) quaternarization of the amine functionality into ammonium iodide salt to give the final catalyst (HC) (Scheme 1). Preliminary studies for the optimization of the procedure were performed on pristine cellulose and fir sawdust and monitored through the elemental analysis (CHN) of the functionalized chars. Pyrolysis of the starting material was crucial to achieving effective oxidation of the obtained char: the higher the temperature, the less oxidizable the carbonaceous material. Higher temperatures create more aromatic and robust chars that need more energy and time to reach the same oxidation degree as a char obtained with lower pyrolysis temperatures (see S3 and Table S3a†). On the other hand, shorter times or lower temperatures gave too fragile materials that were not able to maintain their 3D-structure after the oxidation step. The best conditions corresponded to pyrolysis at 400 °C for 15 h.29,31 The oxidation was performed by screening several oxidizing agents, such as HNO3, H2O2 or KMnO4 at different times and temperatures (see S3 and Table S3a†). H2O2 was selected for the optimized methodology (85 °C for 24 h) because of its higher sustainability and safety, and also because, unlike HNO3, it cannot give side reactions such as nitration of the char. KMnO4 did not work efficiently, maintaining almost unaltered the elemental composition of the char (see S3 and Table S3a†). The oxidation significantly changed the surface of the char, increasing its oxygen content (Table 1, step 2) through alcoholic and carboxylic functionalities (see Paragraph 3.2), essential for the further amination step. After oxidation, the anchoring of APTES on the surface of the char was performed under various conditions (see S3 and Table S3a†), and it was found that a little amount of water was mandatory to produce an active catalyst, since water hydrolyzes the ethoxy groups of APTES to hydroxyl groups,32 the active Lewis acid species, together with –OH and –COOH functionalities present on its surface, in the carbonation of epoxides mechanism. The increased nitrogen content of the ACs testified to the performances of this step (Table 1, step 3). The anchoring step was also tested on the char (C) avoiding the oxidation step: no considerable amount of N was found after the reaction (see S3 and Table S3a†), testifying that the oxidation step is mandatory for successfully anchoring the amine functionality. The last step was optimized to minimize the amount of MeI necessary for the quaternarization of the amine-based chars on the effectively anchored amine group (quantified through XPS analysis, see S18†). As alternatives to MeI, longer chain alkyl iodides (butyl and octyl iodide) were successfully used even if higher reaction temperatures were needed for an effective quaternarization of the amine (see S4†).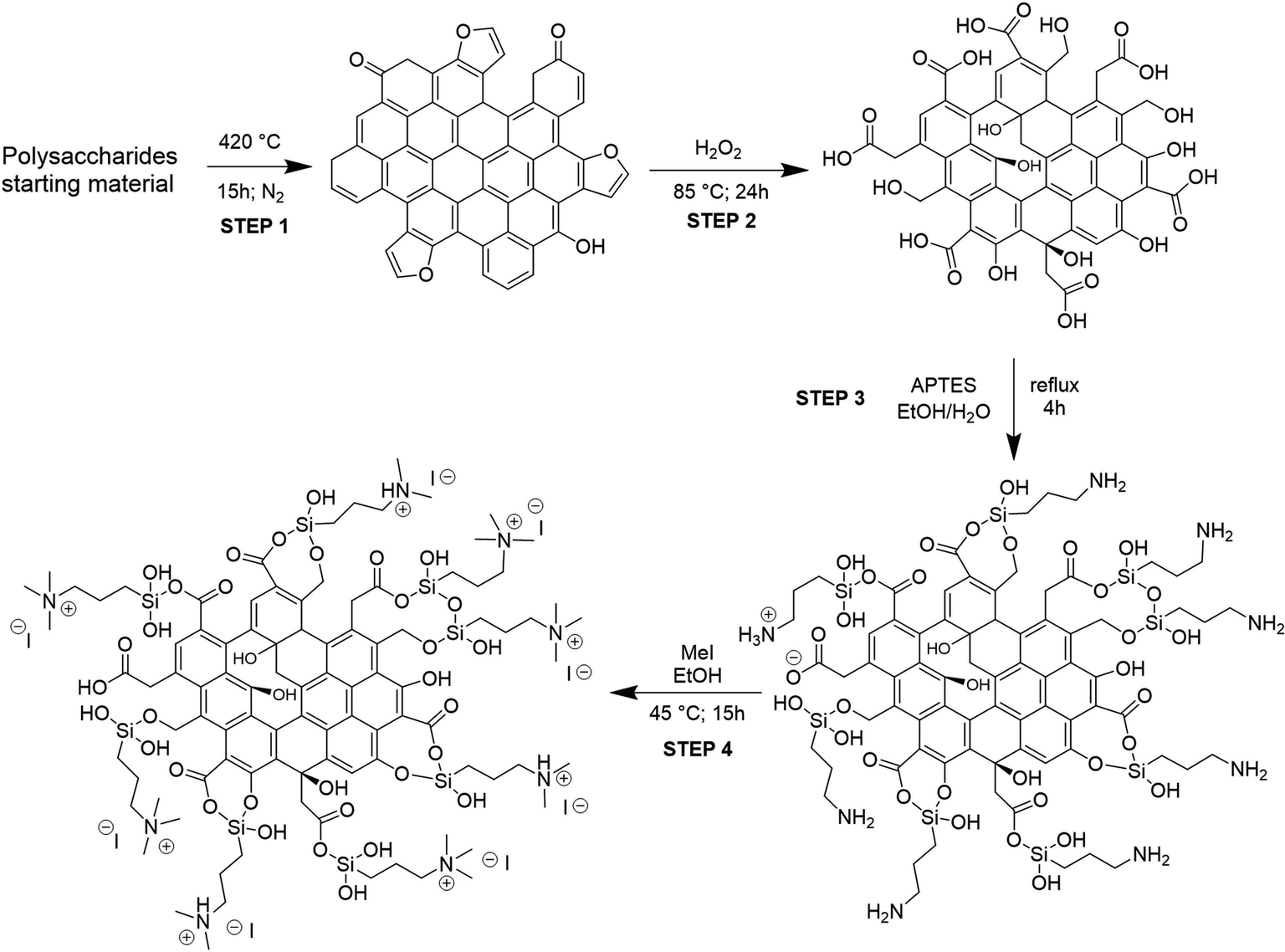 | ||
| Scheme 1 Four-step synthesis of heterogeneous catalysts. A tentative model structure of chars at each reaction step was given based on the characterization results. | ||
| Char | Pristine cellulose | Fir sawdust | ||||
|---|---|---|---|---|---|---|
| N [%] | C [%] | H [%] | N [%] | C [%] | H [%] | |
| Starting material | — | 40.5 ± 0.7 | 6.5 ± 0.1 | — | 45.7 ± 0.6 | 6.4 ± 0.1 |
| Step 1 – pyrolysis (C) | — | 85.0 ± 0.3 | 2.9 ± 0.1 | — | 79.3 ± 0.2 | 2.9 ± 0.1 |
| Step 2 – oxidation (OC) | — | 58.2 ± 0.4 | 2.8 ± 0.2 | — | 56.6 ± 0.5 | 2.9 ± 0.2 |
| Step 3 – APTES anchoring (AC) | 7.1 ± 0.1 | 42.1 ± 0.5 | 6.0 ± 0.1 | 6.7 ± 0.2 | 41.3 ± 0.4 | 5.5 ± 0.1 |
| Step 4 – quaternarization (HC) | 5.0 ± 0.1 | 29.3 ± 0.6 | 4.4 ± 0.3 | 4.9 ± 0.1 | 31.2 ± 0.3 | 4.3 ± 0.1 |
The optimized procedure was applied to six different starting materials: three polysaccharides (starch, pristine cellulose and cellulose acetate) and the three corresponding post-use wastes (starch-based plastics bags, fir sawdust and post-use cigarette filters). The elemental composition of all the catalysts resulted very close to one another, confirming the versatility of the method (Table 2).
| Starting material | Catalyst | N [%] | C [%] | H [%] |
|---|---|---|---|---|
| Potato starch | HC-PS | 4.7 ± 0.1 | 31.8 ± 0.2 | 4.5 ± 0.2 |
| Starch-based plastic bags | HC-SBPB | 4.4 ± 0.2 | 28.2 ± 0.8 | 3.8 ± 0.2 |
| Pristine cellulose | HC-PC | 5.0 ± 0.1 | 29.3 ± 0.6 | 4.4 ± 0.3 |
| Fir sawdust | HC-FSD | 4.9 ± 0.1 | 31.2 ± 0.3 | 4.3 ± 0.1 |
| Cellulose acetate | HC-CA | 5.0 ± 0.2 | 31.3 ± 0.4 | 4.4 ± 0.2 |
| Post-use cigarette filters | HC-PUCF | 4.9 ± 0.1 | 33.8 ± 0.3 | 4.6 ± 0.1 |
To demonstrate that each of the four functionalization steps were necessary to synthesize an active catalyst, the direct functionalization of cellulose with APTES, without the pyrolysis and the oxidation steps, was tested: to achieve an amine functionalization and thus an active catalyst, harsher conditions were required in the anchoring step (>130 °C and APTES to solvent ratio 1:1, Table S5a†). Even if the prepared functionalized cellulose did work well (up to 79% yield of SC see S5 and Table S5b†), it was not recyclable: after reaction the cellulose-based catalyst resulted to be partially dissolved in the reaction media and so not recoverable as an heterogenous catalyst. Presumably, this could be due to the fact that cellulose has plenty of –OH groups organized in a linear structure that can be functionalized with APTES, much more than the –OH groups available on the surface of the material obtained after the pyrolysis and oxidation steps when a condensed carbonaceous structure is formed. This allowed to keep the catalyst heterogeneous.
3.2 Characterization of the bifunctional heterogeneous catalysts
A full and comprehensive physicochemical, structural, surface, and morphological characterization via ATR-FTIR and XPS (Raman and SEM in S9 and S11†) has been carried out on all the starting materials, chars after each derivatization step (C, OC and AC) and final catalysts (HC).Since irrespective of different starting materials (i.e., PS, SBPB, PC, FSD, CA, PUCF), no significant differences are evinced in the peak positions of the ATR-FTIR spectra of the samples at each functionalization step, the obtained results by ATR-FTIR analyses are limited to the discussion of the synthesis of HC-PC and HC-FSD (Fig. 2a–d).
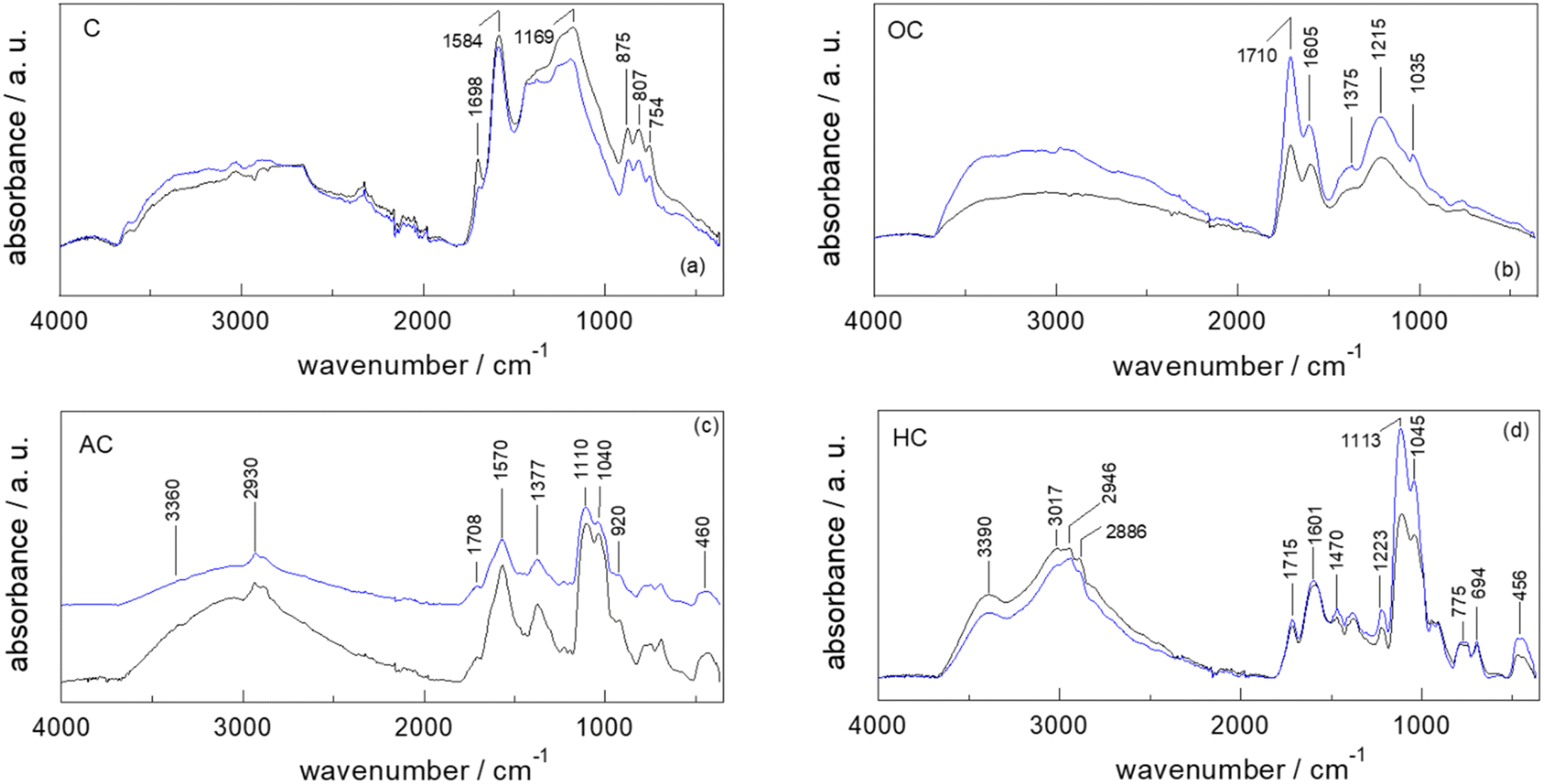 | ||
| Fig. 2 Comparison of the ATR-FTIR spectra of pristine cellulose (PC, black line)- and fir sawdust (FSD, blue line)-derived chars at different reaction steps: (a) step 1 – pyrolysis (C); (b) step 2 – oxidation (OC); (c) step 3 – APTES anchoring (AC); (d) step 4 – quaternarization (HC). | ||
The ATR-FTIR spectra of pristine cellulose (C-PC) and fir sawdust (C-FSD) chars display the peaks at 1698 cm−1 and 1584 cm−1 resulting from the stretching of conjugate C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and the skeletal vibrations involving C–C stretching within the ring of polyaromatic systems, respectively (Fig. 2a). The peak at 1169 cm−1 can be assigned to the C–O stretch of phenols, and the adsorption bands in the 875–750 cm−1 region arise from the typical out-of-plane (“oop”) bending of the C–H bond of polycyclic aromatic hydrocarbon systems.31,33–35
O and the skeletal vibrations involving C–C stretching within the ring of polyaromatic systems, respectively (Fig. 2a). The peak at 1169 cm−1 can be assigned to the C–O stretch of phenols, and the adsorption bands in the 875–750 cm−1 region arise from the typical out-of-plane (“oop”) bending of the C–H bond of polycyclic aromatic hydrocarbon systems.31,33–35
The higher degree of oxidation of both OC-PC and OC-FSD in comparison to the corresponding chars is confirmed by the peaks at 1710 (CO stretching of carboxyl groups), 1375 (in-plane O–H bending of phenol OH groups) and 1215 cm−1 (C–O stretching of phenol OH groups), as well as by the broad band in the 3500–2500 cm−1 region (O–H stretching of the phenol OH groups) (Fig. 2b).33,36,37
The spectra of AC-PC and AC-FSD display new peaks at 1110 and 1040 cm−1 that might be attributed to the C–N stretching of primary amines, Si–O–C (aliphatic), and Si–O stretching: this suggests the successful anchoring of the APTES moieties (Fig. 2c).38 The peaks at about 1110 and 460 cm−1 confirm the presence of Si–O. The broad band in the 3500–2500 cm−1 range is associated with the O–H stretching of both the phenol –OH and Si–OH groups. The characteristic Si–OH stretch signal at about 920 cm−1 is also observed.39 The signal at about 3370 cm−1 is characteristic of the N–H stretching vibrations.33,36
The new strong, broad absorption band at 3390 cm−1 in the spectra of HC-PC and HC-FSD is attributed to the N–H stretching vibrations of the ammonium salt,33 confirming that the functionalization of the amine group into ammonium iodide salt successfully occurred (Fig. 2d). As underlined in the spectra of AC-PC and AC-FSD (Fig. 2c), the broad band in the 3500–2500 cm−1 range is attributed to the O–H stretching of both the phenol –OH and Si–OH groups. The characteristic Si–OH stretch signal at 920 cm−1 is still observed.39
The peak positions of the ATR-FTIR spectra of Fig. 2a–d are summarized in S12.†
The comparison of the ATR-FTIR spectra of the six catalysts (Fig. 3) shows that all samples share the same spectral features despite the differences detected for each starting material (see Fig. S10†).
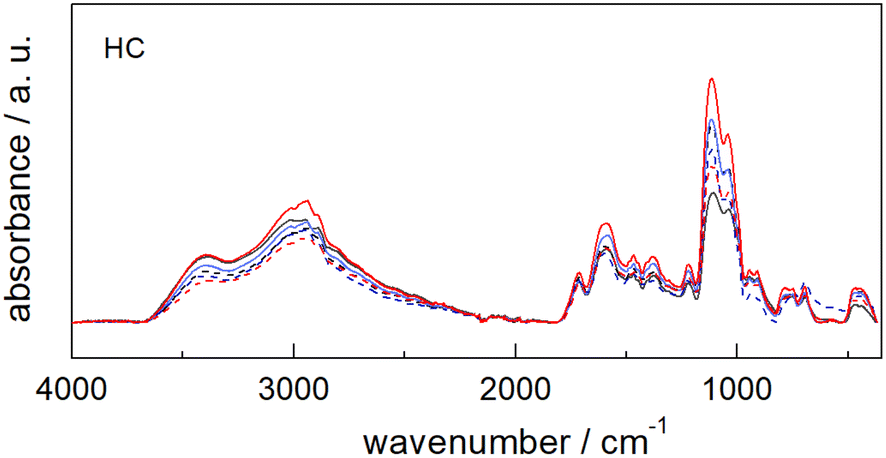 | ||
| Fig. 3 Comparison of the ATR-FTIR spectra of the six catalysts (HC) obtained from different starting materials: pristine cellulose HC-PC (black solid line); fir sawdust HC-FSD (black dash line); potato starch HC-PS (blue solid line); starch-based plastic bag HC-SBPB (blue dash line); cellulose acetate HC-CA (red solid line); post-use cigarette filters HC-PUCF (red dash line). | ||
The surface chemical composition of the samples was investigated by XPS (see S18 and Tables S18a–g† for BE values and chemical quantification). In accordance with ATR-FTIR analysis, the XPS measurements evidence that all samples have a similar composition after the amination step. The presence of Si and N, after washing with EtOH, suggests that APTES has been anchored to the char. Fig. 4 displays the comparison of the N 1s signals, acquired on the pristine cellulose and fir sawdust char samples consequently to the amination and quaternarization steps. After amination, the N 1s signal is characterized by two contributions that are assigned to the amine (BE = 399.7 eV) and the ammonium (BE = 401.6 eV) groups, respectively. By adding the MeI in the last reaction step (i.e., quaternarization), the N 1s signal was fairly modified, evidencing a remarkable increase in the amount of ammonium group (Fig. 4).
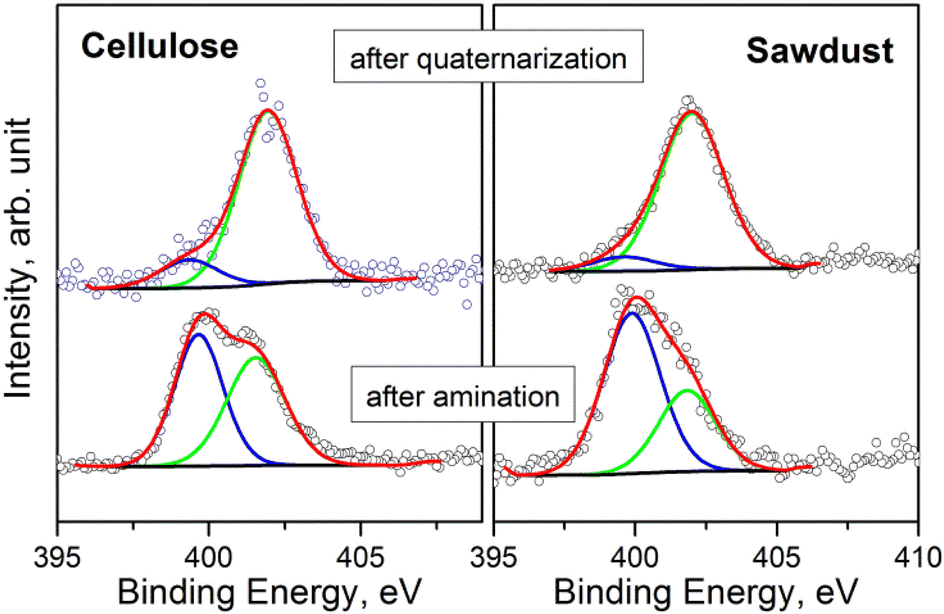 | ||
| Fig. 4 Comparison of N 1s spectra of the samples after amination and quaternarization of pristine cellulose and fir sawdust oxidized chars. | ||
Concerning the C 1s spectrum of pristine cellulose- and fir sawdust-based materials, the signal changed depending on the reaction step (Fig. 5a and b). After the pyrolysis, the char (C) was characterized by two components, positioned at BE = 285.0 eV and 286.3 eV, and assigned to C–C (1) and C–O (2) bonds, respectively. A further small contribution (∼5.5 at%) was identified at BE = 288.2 eV, due to the presence of CO groups (3).40 This component increased after the oxidation step due to –COOH formation (as demonstrated by ATR-FTIR), while it was partially reduced after amination and quaternarization, as a result of the addition of the APTES. In any case, considering the whole synthesis process, a remarkable increase of the C–O and C–N contribution (both falling around BE = 286 eV)41 was registered, higher than what registered in the char (Fig. 5a and b).
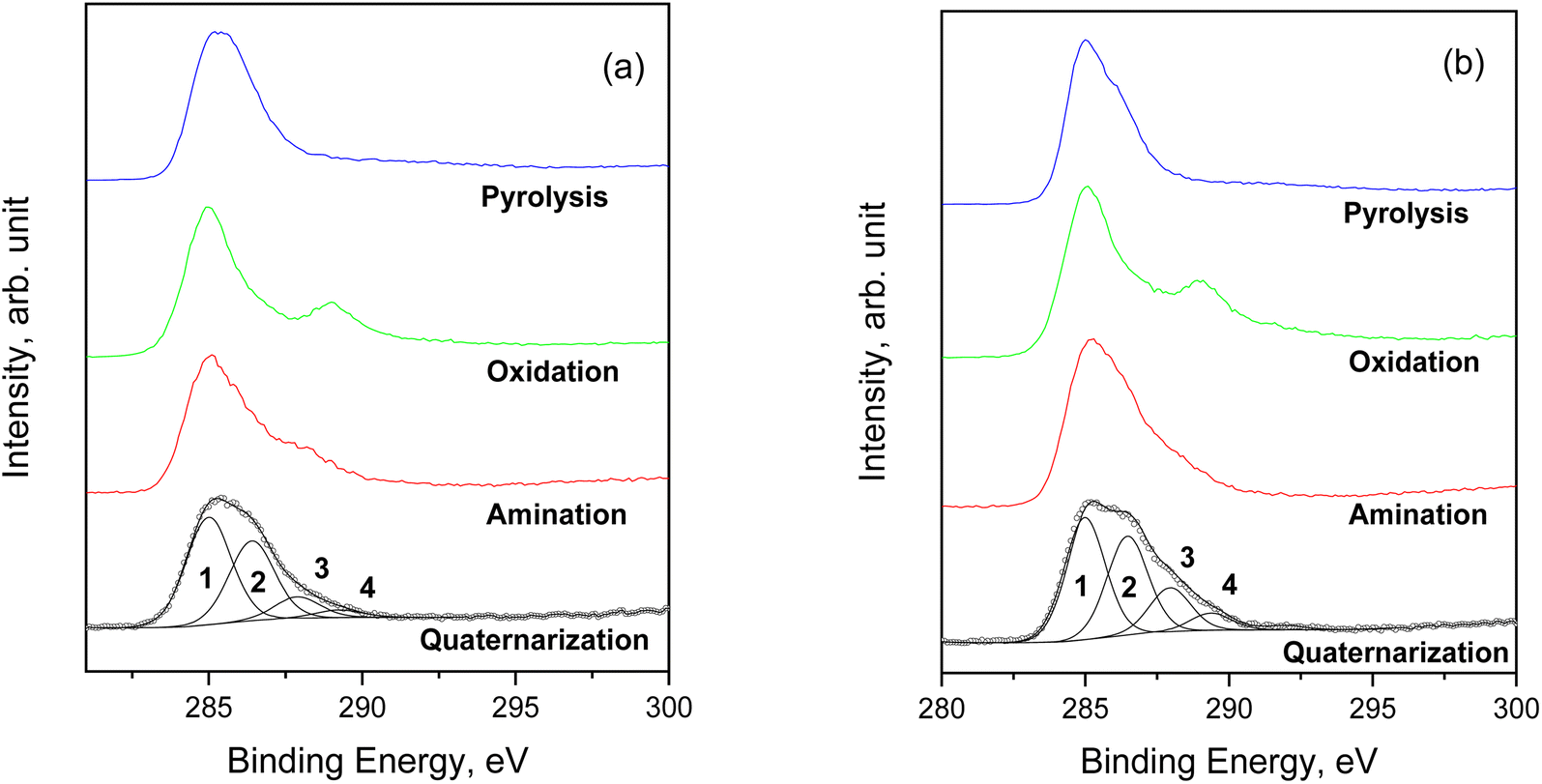 | ||
| Fig. 5 Comparison of C 1s spectra for the samples starting from (a) pristine cellulose and (b) fir sawdust. | ||
The comparison of the C 1s spectra of all samples after the quaternarization step displays that the shape of the signals is almost the same, except for the small difference in the intensity of the component of the C–O and C–N bond (Fig. 6a–f), thus demonstrating the effectiveness of each derivatization step and the versatility of the functionalization method towards different starting materials. XPS quantitative analysis and BE value of all the HC samples are reported in S18.†
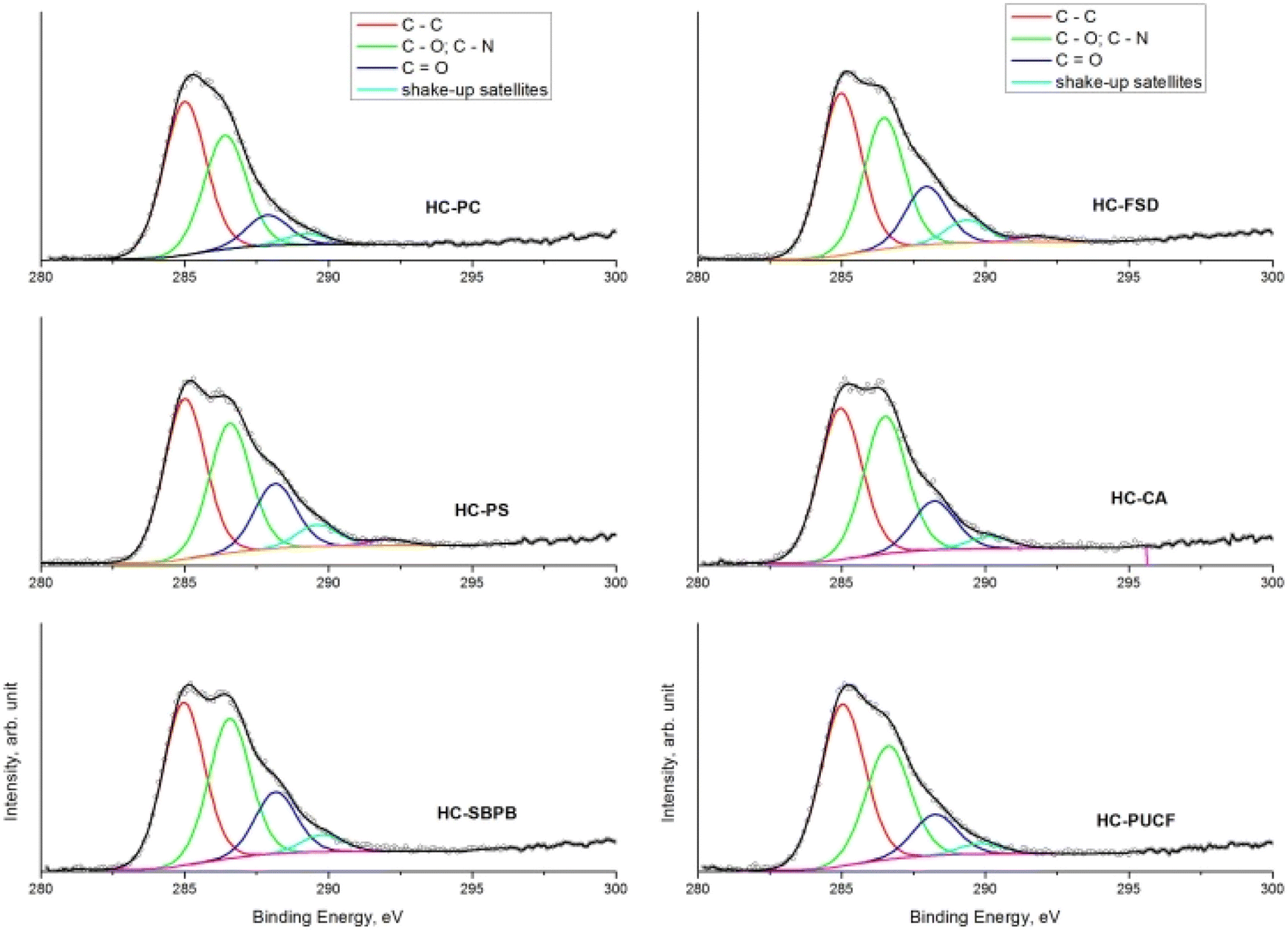 | ||
| Fig. 6 Comparison of C 1s spectra of all catalysts from different materials: pristine cellulose (HC-PC), fir sawdust (HC-FSD), potato starch (HC-PS), cellulose acetate (HC-CA), starch-based plastic bag (HC-SBPB), post-use cigarette filters (HC-PUCF). | ||
The evaluation of textural properties was carried out on three materials after the first step of pyrolysis (C-FSD, C-PUCF and C-SBPB) and on the corresponding final catalysts (HC-FSD, HC-PUCF and HC-SBPB). For each of them, the N2 adsorption/desorption isotherms at 77 K are reported in Fig. 7a. At low relative pressures, the isotherm branches of the pyrolyzed materials C-PUCF and C-FSD illustrated sharp adsorption inflections which are indicative of materials containing micropores. Adsorption at the lowest relative pressure decreases in the order C-PUCF > C-FSD > C-SBPB > HC-SBPB > HC-PUCF > HC-FSD suggesting a parallel decrease of the microporosity. The flat C-SBPB isotherm illustrates a nonporous carbon. In turn, at higher relative pressures (P/P0 > 0.2), the carbon C-PUCF displayed a type IV isotherm showing the existence of mesopores. C-FSD featured a type I isotherm that describes microporous materials, while HC-SBPB, HC-PUCF and HC-FSD displayed type II isotherms that are typical of samples with macropores.42 These observations are confirmed by the DFT pore size distribution that is reported in terms of incremental pore volume in Fig. 7b. C-PUCF pore size ranges between 2 nm and 20 nm, with the major pores at 9 nm, while in C-FSD and C-SBPB mesopores and macropores are not present and pore size is distributed below 3 nm. HC-SBPB, HC-PUCF and HC-FSD feature pores between 5 nm and 200 nm, with the highest volume provided by pores with a diameter of ca. 40 nm for HC-SBPB and HC-PUCF, and 30 nm for HC-FSD.
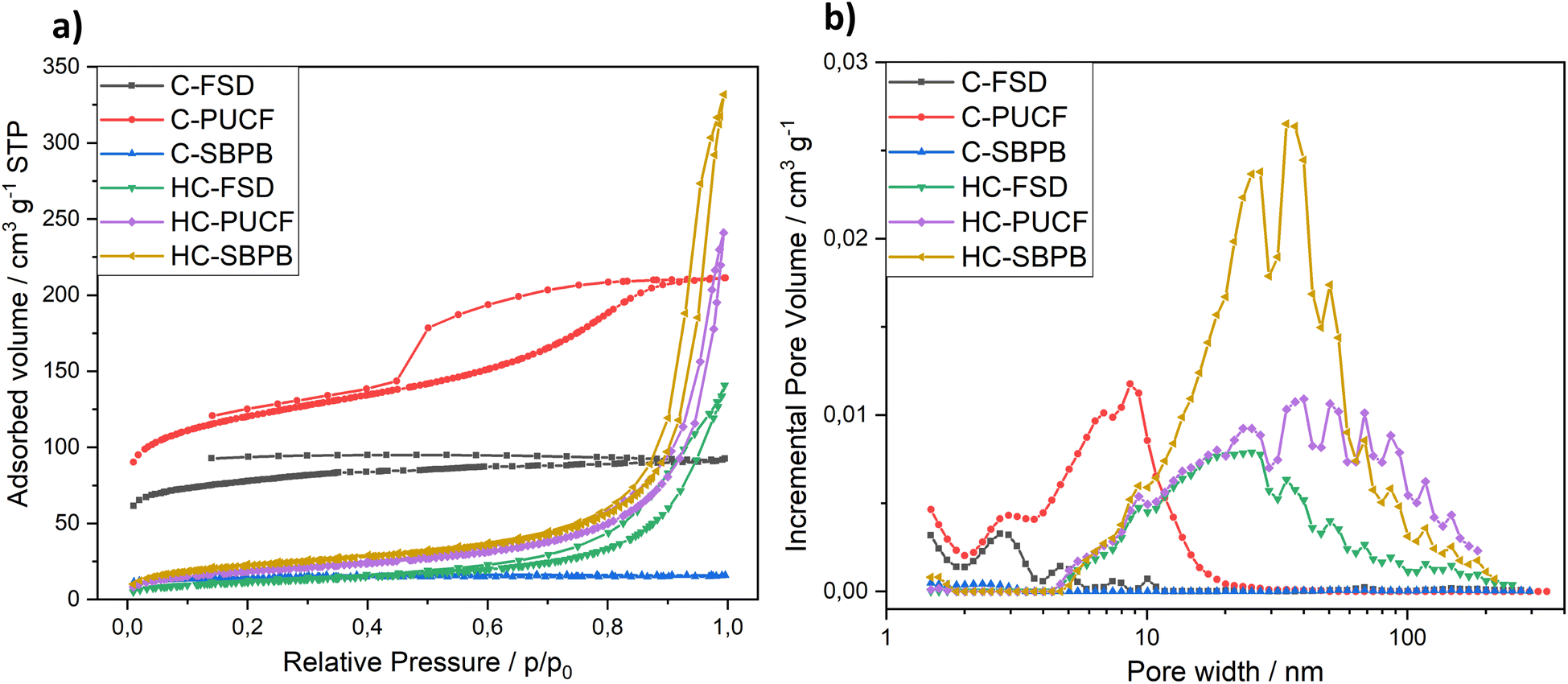 | ||
| Fig. 7 N2 adsorption–desorption isotherms (a) and pore size distribution (b) of the samples. | ||
The micropore volume (Vmicro), the mesopore volume (Vmeso) and the total pore volume (Vtotal) of the different samples are charted in Table 3 along with the BET surface area (SBET). After pyrolysis, the samples are still influenced by the nature of the starting material: with the highest Vmicro (0.123 cm3 g−1) and SBET (442 ± 1 m2 g−1) for C-PUCF and the lowest Vmicro (0.017 cm3 g−1) and SBET (54 ± 1 m2 g−1) for C-SBPB. This behavior is well known in the literature: higher amount of inorganics (i.e. ash content) in the biomass feedstock is often negatively correlated with specific surface area (SBET) in the produced biochar (where ash content is generally SBPB > FSD > PUCF).29,43–46 As expected, after treating the different materials with the same experimental procedure, similar trends with a prominent decrease in the corresponding BET surface area (82 ± 1, 66 ± 1, 42 ± 1 m2 g−1 respectively) were obtained for HC-SBPB, HC-PUCF and HC-FSD. The SBET decrease is consistent with the decrease of Vmicro in favor of the increase of Vmeso, meaning that the treatment might have caused the collapse of small pores into larger ones, in particular it is known that oxidation treatments cause an increase in pore volumes (also known as “etching”).47–49 In these samples the meso and macro-pore volume decreases in the order HC-SBPB (0.474 cm3 g−1) > HC-PUCF (0.306 cm3 g−1) > HC-FSD (0.186 cm3 g−1).
| Samples | V micro (<2 nm) cm3 g−1 | V meso (2–50 nm) cm3 g−1 | V total cm3 g−1 | S BET m2 g−1 |
|---|---|---|---|---|
| C-PUCF | 0.123 | 0.166 | 0.875 (<343 nm) | 442 ± 1 |
| C-FSD | 0.090 | 0.026 | 0.118 (<273 nm) | 290 ± 1 |
| C-SBPB | 0.017 | 0.002 | 0.021 (<294 nm) | 54 ± 1 |
| HC-SBPB | 0.003 | 0.387 | 0.474 (<217 nm) | 82 ± 1 |
| HC-PUCF | 0.0003 | 0.202 | 0.306 (<186 nm) | 66 ± 1 |
| HC-FSD | 0.0 | 0.156 | 0.186 (<253 nm) | 42 ± 1 |
3.3 Synthesis of cyclic carbonates with bifunctional heterogeneous catalysts
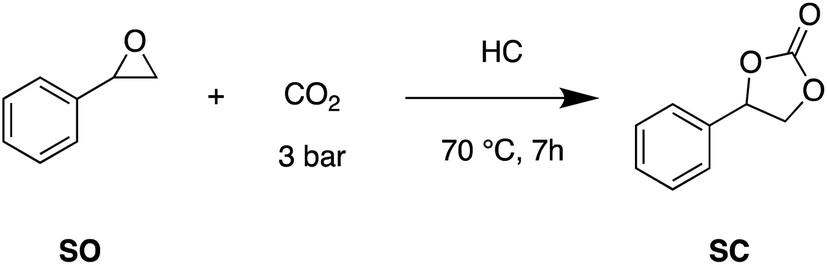
Considering the demonstrated functionalization of the different chars with both ammonium and Lewis acid groups, all the heterogeneous catalysts here prepared were tested in the conversion of CO2 and epoxides into cyclic carbonates. The cycloaddition of CO2 to styrene oxide (SO) catalyzed by HC-FSD was firstly selected as a model reaction to optimize the reaction conditions. The activity of HC-FSD was tested in a stainless-steel autoclave at maximum p (CO2) = 5 bar and T = 70 °C. As shown in Table 4, 5% of HC-FSD had an efficient catalytic activity with a yield of SC of 80%, and a TON of 48.4, at 70 °C and 3 bar of CO2 (entry 1). Increasing the amount of catalyst to 10%, the yield reached 91% (entry 2); in both cases, the selectivity towards the formation of SC (in comparison to the most common by-product 1-phenylethane-1,2-diol) was 99%. An increase of CO2 pressure to 5 bar didn't improve the yield (entry 3), while conducting the reaction at 1 bar, in a Schlenk tube using a balloon as a source of CO2 (entry 7), the yield decreased to 41%, even after a prolonged reaction time to 24 h. For what concerns the temperature, the reaction didn't proceed at room temperature even for a prolonged time (entry 4), at 50 °C there was a slight increase in yield (entry 5), and at 100 °C yields and selectivity were similar to those obtained at 70 °C (entry 6).|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entrya | Catalyst amount [% w/w] | p [bar] | T [°C] | t [h] | Yieldb [%] | Selectivityc [%] | TONe | TOFf [h−1] | Productivityg [h−1] |
| a Reaction conditions: 0.875 mmol SO (100 μL), autoclave. b Isolated yields after purification of the reaction crude by flash column chromatography. Yields are expressed as mean of two independent replicates. c Selectivity calculated by GC-MS and NMR on crude products. d Carried out using a CO2 balloon. e Turnover number, defined as molSC/molIodide. f TOF = TON h−1. g Productivity=[gsc/(gcat h−1)]. | |||||||||
| 1 | 5 | 3 | 70 | 7 | 80 | 99 | 48.4 | 6.9 | 3.2 |
| 2 | 10 | 3 | 70 | 7 | 91 | 99 | 27.5 | 3.9 | 1.8 |
| 3 | 10 | 5 | 70 | 7 | 91 | 99 | 27.5 | 3.9 | 1.8 |
| 4 | 10 | 5 | 25 | 24 | 0 | — | — | — | — |
| 5 | 10 | 3 | 50 | 7 | 13 | 98 | 3.9 | 0.6 | 0.3 |
| 6 | 10 | 3 | 100 | 7 | 92 | 99 | 27.8 | 4.0 | 1.8 |
| 7d | 10 | 1 | 70 | 24 | 41 | 99 | 12.4 | 1.8 | 0.2 |
Heterogeneous catalysts functionalized with longer alkyl iodide (butyl iodide and octyl iodide) have been successfully tested in the cycloaddition of CO2 to SO giving very good yields and selectivities (see S4 and Table S4b†).
A kinetic profile of the cycloaddition of CO2 to SO in the presence of HC-FSD at 70 °C and 3 bar of CO2 pressure is shown in Fig. 8. In the model reaction, the yield of the product increased by increasing reaction time until 7 h, after which it did not significantly increased.
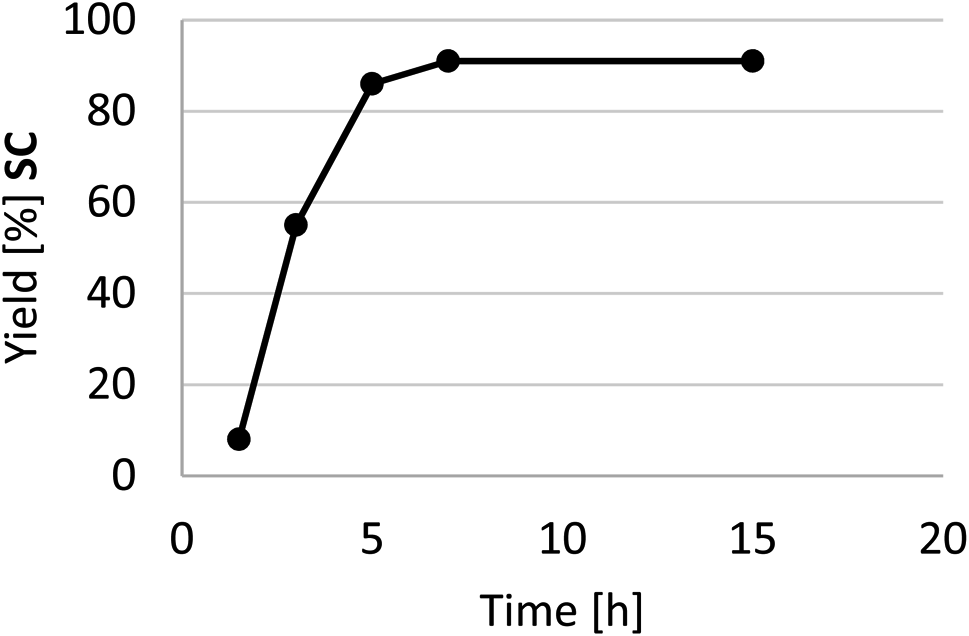 | ||
| Fig. 8 Reaction rate for the conversion of SO into SC catalyzed by HC-FSD. Reaction conditions: 0.875 mmol SO (100 μL), autoclave. Isolated yields after purification of the reaction crude by flash column chromatography. Yields are expressed as mean of two independent replicates. Selectivity was always >99%. | ||
After achieving high catalytic activity of HC-FSD for SC formation, we explored the activity of catalysts synthesized from post-use cigarette filters (HC-PUCF) and starch-based plastic bags (HC-SBPB), and from the biopolymers they are mainly composed: pristine cellulose (HC-PC), cellulose acetate (HC-CA) and potato starch (HC-PS) (Table 5). All of them have been tested at 5% and 10% w/w using the conditions optimized for HC-FSD (entry 2), 70 °C, 3 bar of CO2, 7 h. They all proved to have similar and very good activity in the cycloaddition reaction. A similar activity can be explained by the physicochemical analysis of all the HCs that do not evidence significant differences.
|
|
||||||
|---|---|---|---|---|---|---|
| Entrya | Catalyst | Catalyst amount [% w/w] | Yieldb,c [%] | TONd | TOFe [h−1] | Productivityf [h−1] |
| a Reaction conditions: 0.875 mmol SO (100 μL), autoclave, p (CO2) 3 bar, temperature 70 °C, 7 hours. b Isolated yields are given, after purification of the reaction crude by flash column chromatography. Yield of SC was the average after two runs. c Selectivity was >99% for all entries. Selectivity was calculated by GC-MS and NMR on crude products. d Turnover number, defined as molSC/molIodide. e TOF = TON h−1. f Productivity=[gsc/(gcat h−1)]. | ||||||
| 1 | HC-FSD | 5 | 80 | 48.4 | 6.9 | 3.2 |
| 2 | HC-FSD | 10 | 91 | 27.5 | 3.9 | 1.8 |
| 3 | HC-PC | 5 | 77 | 45.1 | 6.4 | 3.0 |
| 4 | HC-PC | 10 | 86 | 25.1 | 3.6 | 1.7 |
| 5 | HC-CA | 5 | 77 | 48 | 6.9 | 3.0 |
| 6 | HC-CA | 10 | 86 | 26.9 | 3.8 | 1.7 |
| 7 | HC-PUCF | 5 | 76 | 46.3 | 6.6 | 3.0 |
| 8 | HC-PUCF | 10 | 84 | 25.4 | 3.6 | 1.6 |
| 9 | HC-PS | 5 | 72 | 47.2 | 6.7 | 2.8 |
| 10 | HC-PS | 10 | 82 | 26.9 | 3.8 | 1.6 |
| 11 | HC-SBPB | 5 | 79 | 46.9 | 6.7 | 3.1 |
| 12 | HC-SBPB | 10 | 88 | 26 | 3.7 | 1.7 |
HC-PS catalyst was the less reactive one giving 82% yield at 10% w/w (entry 10), while HC-FSD the most reactive in the same conditions (entry 2). Using HC-PC (entries 3 and 4) and HC-CA (entries 4 and 5) identical yields have been obtained. As demonstrated by chemical-physical analysis, regardless of the starting material, the derivatization method used for the six chars produces similar catalysts. This shows the viability and versatility of this new method to valorize different kinds of waste. Moreover, the reaction conditions here used are definitely milder respect to those found for the use of heterogeneous catalysts made from bio-based polymers and various carbonaceous materials already present in literature (see S1†): most of the previously reported procedures for the cycloaddition of CO2 to epoxides operate at relatively high temperature, typically in the 100–150 °C range, and at a CO2 pressure > 1 MPa, mostly with the addition of a co-catalyst.
HC-FSD proved to be slightly more reactive than the other catalysts, thus it was used to explore the substrate scope under CO2 pressure in the optimized reaction conditions previously described (Table 6). Generally, all the terminal epoxides tested could be transformed into the corresponding cyclic carbonates. Very good yields have been obtained for the carbonation of epichlorohydrin 1c (entry 3) and allyl glycidyl ether 1d (entry 4). Lower but still good yields have been obtained for the reaction of benzyl glycidyl ether 1b (entry 2) and 2-ethylhexyl glycidyl ether 1e (entry 5). The selectivity toward the formation of the cyclic carbonate was 99% in all cases. As expected, for internal epoxides (entry 6), no conversion was obtained, due to the steric hindrance on both carbons.
| Entrya | Substrate | Product | Yieldb,c [%] |
|---|---|---|---|
| a Reaction conditions: 0.875 mmol substrate (100 μL), autoclave, p (CO2) 3 bar, catalyst loading 10 wt%, temperature 70 °C, 7 h. b Isolated yields after purification of the reaction crude by flash column chromatography. Yields are expressed as mean of two independent replicates. c Selectivity was > 99% for all catalytic runs. Calculated by GC-MS on crude products. | |||
| 1 |
|

|
91 |
| 2 |
|
|
76 |
| 3 |
|
|
96 |
| 4 |
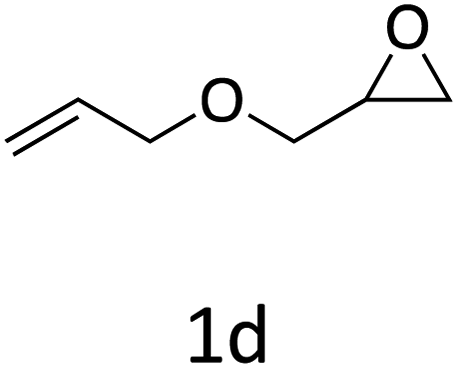
|
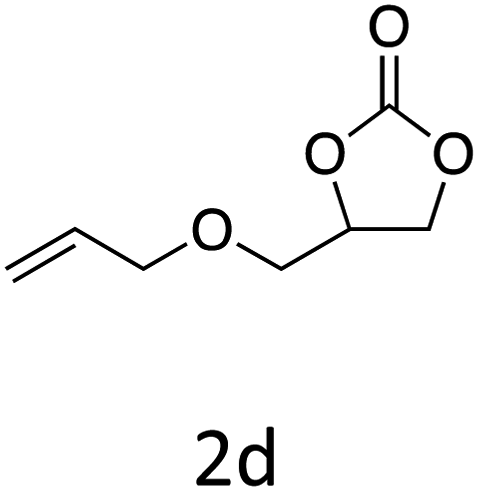
|
92 |
| 5 |
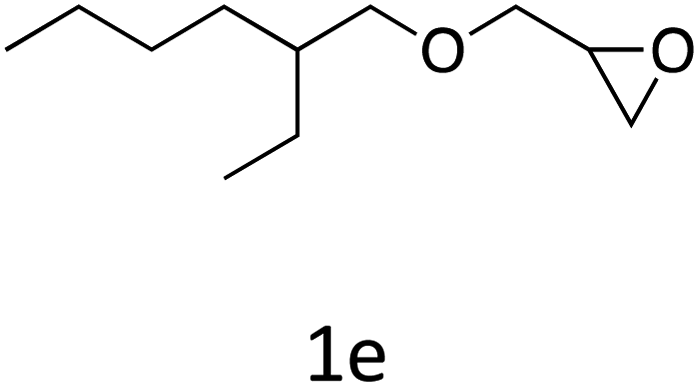
|
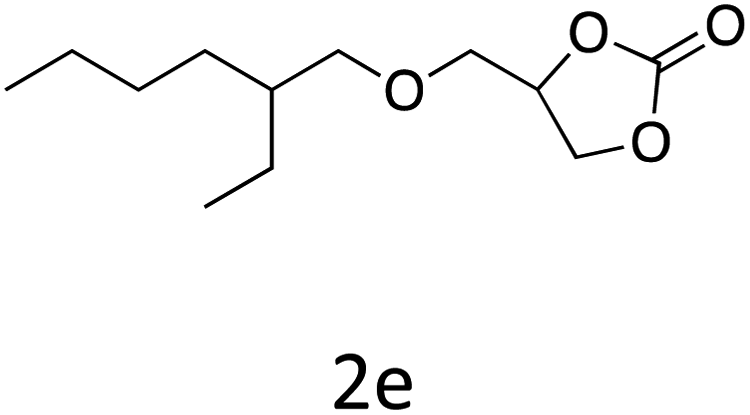
|
82 |
| 6 |
|
|
0 |
Recyclability of the catalysts HC-FSD, HC-PC and HC-CA was tested using the model reaction of SO conversion into SC, in the conditions described above (catalyst loading of 10 wt%). After the reaction completion, ethyl acetate was added, and the catalyst was separated from the crude by centrifugation as described in Section 2.4. HC-FSD could be recycled over five times (weight of the catalysts after every cycle reported in S6†) without appreciable loss of catalytic activity both in terms of conversion and selectivity (Fig. 9). HC-PC and HC-CA were also fully recyclable over five times (Fig. 9). HC-FSD was recycled also using a lower loading (5 wt%) to check if the reaction yield decreased when maximum yield was not achieved (Table S7†): the yield was constant over 5 cycles (∼80%) even at 5 wt% of catalyst loading, further demonstrating its recyclability.
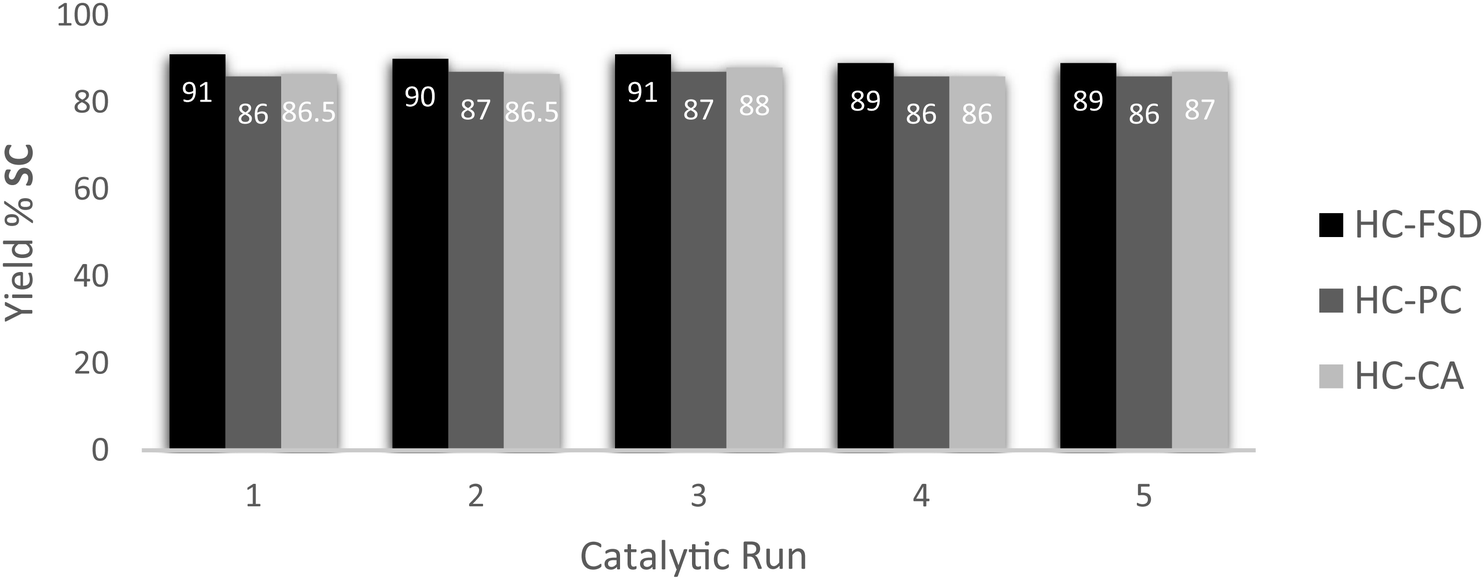 | ||
| Fig. 9 Recycling catalytic test for the coupling of SO and CO2 catalyzed by HC-FSD, HC-PC and HC-CA. Reaction conditions: 0.875 mmol SO (100 μL), autoclave, p (CO2) 3 bar, catalyst loading 10 wt%, temperature 70 °C, 7 h. Isolated yields are given, after purification of the reaction crude by flash column chromatography. Yield of SC was the average after two runs. Selectivity was > 99% for all catalytic runs. | ||
The ATR-FTIR spectrum of HC-FSD after five recycling runs shows the superposition of the spectra derived from the fir sawdust catalyst and the contamination with residues of the reaction product (as also demonstrated by the increased weight of the catalyst, see S6†). The latter component is evinced by the new peak at 1794 cm−1 as well as the shoulder at 1161 cm−1 characteristic of the ν(CO) asymmetric vibration of the carbonyl group in cyclic carbonate (Fig. 10a). The differential spectrum obtained after subtracting the spectrum of the pristine catalyst highlights the presence of cyclic carbonates (Fig. 10b).50–53
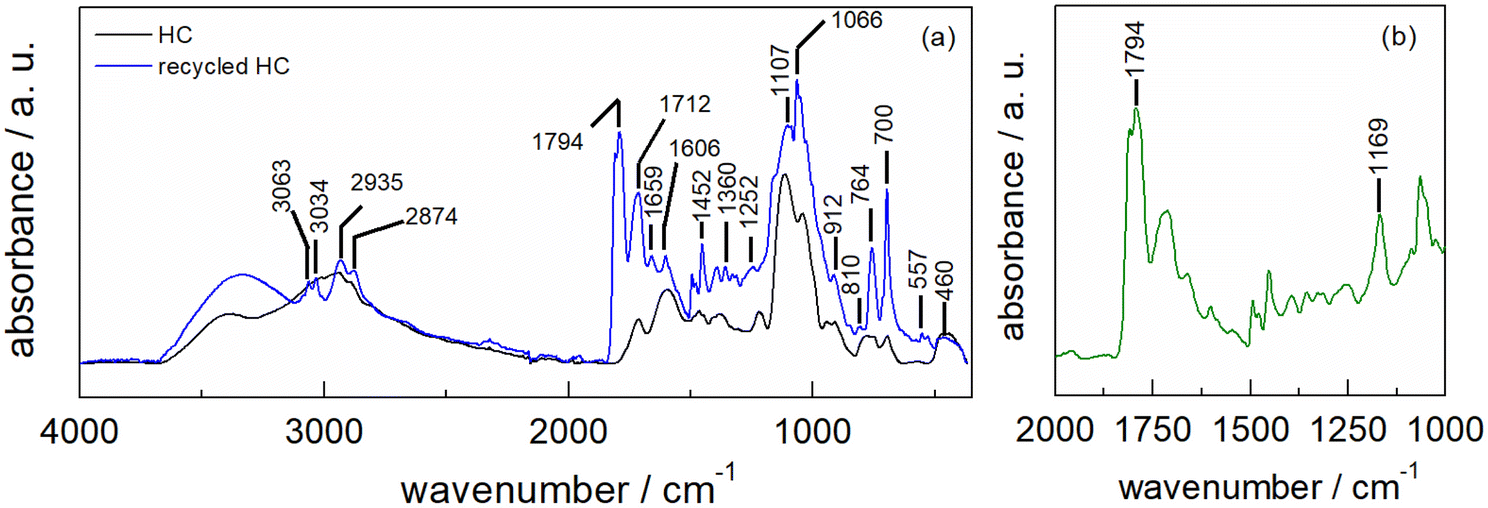 | ||
| Fig. 10 (a) ATR-FTIR spectra of fresh HC-FSD (black line) and recycled HC-FSD after five recycling runs (blue line); (b) ATR-FTIR differential spectrum of recycled HC-FSD obtained by subtracting the spectrum of fresh HC-FSD. | ||
The surface chemical composition of the recycled HC-FSD sample, characterized by XPS, was quite different compared with the fresh HC-FSD sample. The XPS quantitative analysis and the BE value are shown in Table S18b.† The obtained results evidenced that after the recycling process, an increase in the amount of C, especially in the aliphatic contribution, and a reduction of the amount of Si, I and N were registered, while the amount of O remained substantially unchanged (see Fig. S20†). Nevertheless, the catalytic activity of the three catalysts tested did not decrease after 5 cycles, even if a decrease in content of the iodide and ammonium group were observed by XPS, this could presumably be explained due the cyclic carbonate presence on the surface of the recycled catalyst as also evidenced by ATR-FTIR analysis: since XPS quantification gives information on the relative atomic concentration on the first layer of the sample (<10 nm), concentrations of Si, N and I appear to be decreased when the surface of the catalyst is covered with the conversion product.
To further investigate this behavior, elemental analysis of the recycled catalyst (HC-FSD) after 5 cycles was also performed (Table S8†): analogously to what was highlighted by the XPS characterization, a relevant increase in the C contribution from 31.2 to 48.3 wt% was observed, while a decrease from 4.9 to 4 wt% in the N contribution was observed, in line with the hypothesis of the presence of the carbonate on the catalyst surface that increases C contribution while decreasing all other atom ones (as also already observed in literature).15
4. Conclusions
This work illustrates a novel four-step methodology for the obtainment of a bifunctional heterogeneous catalyst, active in the conversion of terminal epoxides into the corresponding cyclic carbonate. The optimized methodology was versatile in the functionalization of several pristine polysaccharides (PC, PS and CA) and their corresponding waste material (FSD, SBPB and PUCF), as confirmed by FTIR, XPS and elemental analysis, that showed the presence of the same functionalities and elemental composition in all the materials produced. These functionalized chars were tested as heterogeneous catalysts in the conversion of epoxides and CO2 into cyclic carbonates using mild conditions, showing high catalytic activity: only negligible differences in yield were observed between catalysts produced starting from different sources, as further evidence of the versatility of the method presented. The reaction scope shows that as expected only terminal epoxides were successfully converted with good yields. Moreover, at least three out of six catalysts produced (HC-FSD, HC-PC and HC-CA) proved to be easily and successfully recycled over five cycles without appreciable loss in activity. The results obtained illustrate one example to valorize some of those feedstocks (CO2 and cellulose/starch-based materials) that nowadays are considered waste and whose recovery and reuse are a matter of concern.Author contributions
A. P. and M. V. conceptualization, investigation and validation of the work. Writing of original draft and visualization. L. F. preliminary investigations. A. M. Investigation and writing (concerning XPS). F. D. G. and G. R. investigation and writing (concerning AT-FTIR, SEM and Raman). C. A. writing (concerning SEM). E. P. and F. S. investigation and writing (concerning porosimetric analysis). C. S. review, funding acquisition. P. G. review and supervision, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of Bologna (RFO program), Minister of Education, University and Research (MIUR) and Regione Emilia-Romagna (Bando “Alte competenze per la ricerca e il trasferimento tecnologico” – POR FSE 2014/2020, Obiettivo tematico 10) for PhD funding. F. D. G. and G. R. would like to acknowledge funding from the Mission Innovation Program MiSE under the Grant “Italian Energy Materials Acceleration Platform—IEMAP”. The SEM images were collected in the SPM@ISMN facility.References
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS PubMed.
- P. P. Pescarmona, Curr. Opin. Green Sustainable Chem., 2021, 29, 100457 CrossRef CAS.
- P. Rollin, L. K. Soares, A. M. Barcellos, D. R. Araujo, E. J. Lenardão, R. G. Jacob and G. Perin, Appl. Sci., 2021, 11, 5024 CrossRef CAS.
- N. Kihara, N. Hara and T. Endo, J. Org. Chem., 1993, 58, 6198–6202 CrossRef CAS.
- X. Cao, S. Sun and R. Sun, RSC Adv., 2017, 7, 48793–48805 RSC.
- M. Vagnoni, C. Samorì and P. Galletti, J. CO2 Util., 2020, 42, 101302 CrossRef CAS.
- V. B. Saptal and B. M. Bhanage, Curr. Opin. Green Sustainable Chem., 2017, 3, 1–10 CrossRef.
- C. Calabrese, F. Giacalone and C. Aprile, Catalysts, 2019, 9, 1–30 CrossRef.
- C. Claver, M. Bin Yeamin, M. Reguero and A. M. Masdeu-Bultó, Green Chem., 2020, 22, 7665–7706 RSC.
- S. Liang, H. Liu, T. Jiang, J. Song, G. Yang and B. Han, Chem. Commun., 2011, 47, 2131–2133 RSC.
- Z. Wu, H. Xie, X. Yu and E. Liu, ChemCatChem, 2013, 5, 1328–1333 CrossRef CAS.
- H. Díaz Velázquez, J. Guzmán Pantoja, E. Meneses Ruiz, R. García de León and R. Martínez Palou, Catal. Lett., 2017, 147, 2260–2268 CrossRef.
- W. Chen, L. X. Zhong, X. W. Peng, R. C. Sun and F. C. Lu, ACS Sustainable Chem. Eng., 2015, 3, 147–152 CrossRef CAS.
- K. R. Roshan, G. Mathai, J. Kim, J. Tharun, G. A. Park and D. W. Park, Green Chem., 2012, 14, 2933–2940 RSC.
- X. Wu, M. Wang, Y. Xie, C. Chen, K. Li, M. Yuan, X. Zhao and Z. Hou, Appl. Catal., A, 2016, 519, 146–154 CrossRef CAS.
- X. Xiong, H. Zhang, S. L. Lai, J. Gao and L. Gao, React. Funct. Polym., 2020, 149, 104502 CrossRef.
- J. Sun, J. Wang, W. Cheng, J. Zhang, X. Li, S. Zhang and Y. She, Green Chem., 2012, 14, 654–660 RSC.
- S. Lai, J. Gao, H. Zhang, L. Cheng and X. Xiong, J. CO2 Util., 2020, 38, 148–157 CrossRef CAS.
- C. Jing-Xian, J. Bi, D. Wei-Li, D. Sen-Lin, C. Liu-Ren, C. Zong-Jie, L. Sheng-Lian, L. Xu-Biao, T. Xin-Man and A. Chak-Tong, Appl. Catal., A, 2014, 484, 26–32 CrossRef.
- K. R. Roshan, T. Jose, A. C. Kathalikkattil, D. W. Kim, B. Kim and D. W. Park, Appl. Catal., A, 2013, 467, 17–25 CrossRef CAS.
- Y. Zhao, J. S. Tian, X. H. Qi, Z. N. Han, Y. Y. Zhuang and L. N. He, J. Mol. Catal. A: Chem., 2007, 271, 284–289 CrossRef CAS.
- J. Tharun, Y. Hwang, R. Roshan, S. Ahn, A. C. Kathalikkattil and D. W. Park, Catal. Sci. Technol., 2012, 2, 1674–1680 RSC.
- D. Czajczyńska, L. Anguilano, H. Ghazal, R. Krzyżyńska, A. J. Reynolds, N. Spencer and H. Jouhara, Therm. Sci. Eng. Prog., 2017, 3, 171–197 CrossRef.
- L. Ma, T. Wang, Q. Liu, X. Zhang, W. Ma and Q. Zhang, Biotechnol. Adv., 2012, 30, 859–873 CrossRef CAS PubMed.
- C. H. Zhou, X. Xia, C. X. Lin, D. S. Tong and J. Beltramini, Chem. Soc. Rev., 2011, 40, 5588–5617 RSC.
- X. Yang, Y. Wan, Y. Zheng, F. He, Z. Yu, J. Huang, H. Wang, Y. S. Ok, Y. Jiang and B. Gao, Chem. Eng. J., 2019, 366, 608–621 CrossRef CAS.
- M. Kołtowski, B. Charmas, J. Skubiszewska-Zięba and P. Oleszczuk, Ecotoxicol. Environ. Saf., 2017, 136, 119–125 CrossRef PubMed.
- M. Toda, A. Takagaki, M. Okamura, J. N. Kondo, S. Hayashi, K. Domen and M. Hara, Nature, 2005, 438, 178 CrossRef CAS PubMed.
- C. Samorì, A. Parodi, E. Tagliavini and P. Galletti, J. Anal. Appl. Pyrolysis, 2021, 155, 105030 CrossRef.
- J. L. Vidal, V. P. Andrea, S. L. MacQuarrie and F. M. Kerton, ChemCatChem, 2019, 11, 4089–4095 CrossRef CAS.
- C. Samorí, C. Torri, D. Fabbri, G. Falini, C. Faraloni, P. Galletti, S. Spera, E. Tagliavini and G. Torzillo, ChemSusChem, 2012, 5, 1501–1512 CrossRef PubMed.
- M. C. Brochier Salon and M. N. Belgacem, Colloids Surf., A, 2010, 366, 147–154 CrossRef CAS.
- R. M. Silverstein, F. X. Webster and D. J. Kiemle, Spectrometric Identification of Organic Compounds, John Wiley Sons, Inc, 2005 Search PubMed.
- L. T. Cuba-Chiem, L. Huynh, J. Ralston and D. A. Beattie, Langmuir, 2008, 24, 8036–8044 CrossRef CAS PubMed.
- S. Pradhan, J. Hedberg, J. Rosenqvist, C. M. Jonsson, S. Wold, E. Blomberg and I. O. Wallinder, PLoS One, 2018, 13, 1–24 CrossRef PubMed.
- C. J. Pouchert, The Aldrich Library of FT-IR Spectra, Aldrich, Milwaukee, Wis, 2nd edn, 1975 Search PubMed.
- A. O. Odeh, Ranliao Huaxue Xuebao, 2015, 43, 129–137 CAS.
- W. Xiao, P. Zhao, S. Deng and N. Zhang, New J. Chem., 2015, 39, 3719–3727 RSC.
- C. Le Losq, G. D. Cody and B. O. Mysen, Am. Mineral., 2015, 100, 945–950 CrossRef.
- C. Zhang, R. Hao, H. Liao and Y. Hou, Nano Energy, 2013, 2, 88–97 CrossRef CAS.
- M. Ayiania, M. Smith, A. J. R. Hensley, L. Scudiero, J. S. McEwen and M. Garcia-Perez, Carbon, 2020, 162, 528–544 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- F. Ronsse, S. van Hecke, D. Dickinson and W. Prins, GCB Bioenergy, 2013, 5, 104–115 CrossRef CAS.
- S. Adilah, S. Nur and A. Nurhayati, J. Phys. Sci., 2014, 25, 97–112 Search PubMed.
- S. Masoudi Soltani, S. K. Yazdi and S. Hosseini, Appl. Nanosci., 2014, 4, 551–569 CrossRef CAS.
- S. Gupta, G. K. Gupta and M. K. Mondal, Energy, 2019, 181, 665–676 CrossRef CAS.
- Q. Fan, J. Sun, L. Chu, L. Cui, G. Quan, J. Yan, Q. Hussain and M. Iqbal, Chemosphere, 2018, 207, 33–40 CrossRef CAS PubMed.
- J. S. Jang, J. Lee, W. T. Koo, D. H. Kim, H. J. Cho, H. Shin and I. D. Kim, Anal. Chem., 2020, 92, 957–965 CrossRef CAS PubMed.
- W. Chen, M. S. Mirshekarloo, S. El Meragawi, G. Turpin, R. Pilkington, A. Polyzos and M. Majumder, ACS Appl. Nano Mater., 2022, 5, 3811–3823 CrossRef CAS.
- C. Moya, V. Sabater, G. Yagüe, M. Larriba and J. Palomar, J. CO2 Util., 2018, 28, 66–72 CrossRef CAS.
- Z. Guo and Q. Lin, J. Mol. Catal. A: Chem., 2014, 390, 63–68 CrossRef CAS.
- X. Li, J. Ke, J. Wang, M. Kang, F. Wang, Y. Zhao and Q. Li, RSC Adv., 2018, 8, 8615–8623 RSC.
- J. Ke, X. Li, F. Wang, S. Jiang, M. Kang, J. Wang, Q. Li and Z. Wang, RSC Adv., 2017, 7, 28841–28852 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta05906a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |