
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hybrid solid electrolyte-liquid electrolyte systems for (almost) solid-state batteries: Why, how, and where to?†
Henry M.
Woolley
ab and
Nella M.
Vargas-Barbosa
 *bc
*bc
aUniversity of Münster, MEET Battery Research Center, Institute of Physical Chemistry, Corrensstraße 46, 48149, Münster, Germany
bInternational Graduate School for Battery Chemistry, Characterization, Analysis, Recycling and Application (BACCARA), University of Münster, Corrensstraße 40, 48149 Münster, Germany
cInstitut für Energie- und Klimaforschung (IEK), IEK-12: Helmholtz Institut Münster (HI MS), Forschungszentrum Jülich, Corrensstraße 46, 48149 Münster, Germany. E-mail: n.vargas-barbosa@fz-juelich.de
First published on 16th December 2022
Abstract
All-solid-state batteries (SSBs) offer an alternative to current state of the art lithium-ion batteries, promising improved safety and higher energy densities due to the incorporation of non-flammable solid electrolytes and Li metal as an anode material. Despite this, SSBs face numerous issues, including the tendency for the solid electrolytes to decompose upon contact with anode and cathode materials as well as during cycling. In addition, poor particle on particle contact can result in sluggish transport of lithium ions to and from the solid electrolytes. One potential solution is by combining the solid electrolyte with a liquid electrolyte to form a hybrid solid–liquid electrolyte system. By using a liquid electrolyte with a wide electrochemical stability window and good wetting properties some of the problems with solid electrolytes in SSBs may be overcome. However, due to the reactive nature of solid electrolytes, a new interphase known as the solid liquid electrolyte interphase (SLEI) forms. This SLEI may be resistive and therefore increase the total impedance of the cell, thus making certain liquid/solid electrolyte combinations unsuitable for use in ASSBs. In this review we discuss the recent history of these systems, look into the ionic transport model and focus on how the chemical stability of the solid electrolyte with respect to the liquid electrolyte is a vital factor in the formation of a stable SLEI. In the case of salt-in-solvent systems the stability of the solid electrolyte is driven by the chemical nature of the solvent, therefore we also discuss what solvent properties-such as dielectric constant or donor number-may have an effect on the degree of decomposition of the solid electrolyte used.
 Henry M. Woolley | Henry Woolley received his Master of Chemistry at the University of St Andrews, United Kingdom, in 2020 working on layered transition metal oxides as cathode active materials in lithium-ion batteries. He is currently a PhD student at the MEET Battery Research Centre at the University of Münster, Germany, as well as being a member of the International Graduate School BACCARA. His research focuses on hybrid thiophosphate solid–liquid electrolytes for use in solid-state batteries. |
 Nella M. Vargas-Barbosa | Nella M. Vargas-Barbosa was born and raised in the Caribbean Island of Puerto Rico, where she grew up and completed her Bachelor's in Chemistry at the Rio Piedras Campus of the University of Puerto Rico. After completing her PhD in Chemistry at Penn State University, she moved to pursue an academic career in Germany that includes stays at the University of Marburg and the Max Planck Institute for Solid State Research. Nella is now an independent Young Investigator Group Leader at the Helmholtz Institute Münster. There she leads the iPEC lab (interfacial photoelectrochemistry lab), which focuses on studying (photo)electrochemical systems that have applications for energy generation and energy storage. The goals of the group range from elucidating the fundamentals of charge-transport at heterogeneous interfaces in model systems to more application-oriented projects characterizing solid–solid interfaces and interphases in solid-state batteries. |
Introduction
The growing threat posed by climate change alongside the increasing demand for technological devices such as laptops and mobile phones has established the lithium-ion battery (LIB) as a ubiquitous feature of modern life.1,2 In 1991, the energy density of a lithium ion cell was just 80 W h kg−1 at cell level, which has now tripled to as high as 260 W h kg−1.3 Whilst the energy density of LIBs has risen steadily over the last 30 years, the cost of a battery pack has dropped rapidly. In 2010, the standard price for an automotive battery pack was 1182.2 USD kW h−1, this dropped almost tenfold to 156 USD kW h−1 in 2019.4 The increasing drive to meet the requirements for high energy density batteries for electrical vehicles5,6 and stationary storage use means that in the coming years the theoretical energy density limit for LIBs (ca. 400 W h kg−1 in conventional LIBs) will be reached.7,8 It is clear, therefore, that unlocking the full potential of post-lithium ion batteries has now become an important research goal.9Solid state-batteries (SSBs) have been proposed as a viable alternative to the current state-of-the art LIBs.10 The main difference between a conventional LIB and a SSB is that the liquid electrolyte is replaced with a solid one. This could be in the form of an inorganic ceramic,11 a solid polymer12,13 or a combination of the two.14–16 The standard LIB suffers from safety issues, especially in high power cells, due to the flammability of organic solvents in liquid electrolytes. Thus, the use of non-flammable solids in SSBs gives them a safety advantage. Some solid electrolytes now have Li+ ionic conductivities which are equal or greater than their liquid counterparts at room temperature.11,17–20 An added benefit of solid electrolytes is the fact that they are intrinsically single-ion conductors, i.e. lithium transference and transport numbers ∼1. A single type of charge carrier minimizes the effect of dynamic ion correlations that could reduce the total ionic conductivity in the bulk.21 In addition, SSBs have revitalized the potential of using lithium metal, instead of graphite, as anode material.22
Lithium metal has a very high theoretical capacity of 3842 mA h g−1 and is widely considered the “holy grail” of batteries.23–25 The use of lithium metal in a commercial battery would allow for much higher energy and power densities. However, the incorporation of Li metal in a battery poses many challenges. The most common failure mechanism of lithium metal batteries is dendrite formation from the anode to the cathode, which eventually leads to short circuits and cell failure. As a result of their mechanical hardness, SSBs can potentially suppress dendritic formation. However, dendrite suppression is not a solved problem with electrolyte composition, and temperature and pressure control being important factors in whether dendrite suppression can occur.26 Indeed, Li dendrites can form and grow along the pores between SE crystallites, but even puncture and break crystallites.27–29
The solid–solid interface between the solid electrolyte and the electrode materials (cathode and anode) is another major bottleneck for SSBs. High internal resistance often occurs at the interface between the solid electrolyte and electrodes,30 this is especially an issue in rigid garnet electrolytes such as Li7La3Zr2O12 (LLZO).31 Poor physical contact between cathode active material and the solid electrolyte results in higher cell resistances as the lithium ions are impeded in their transport across the electrolyte-active material interface.32 Additional issues that SEs face is their (electro)chemical stability against the cathode and anode materials (their electrochemical stability window).33–35 Whilst some liquid and polymer electrolytes remain stable at high cathodic potentials and low anodic potentials,36,37 certain solid state electrolytes are readily reduced or oxidized, respectively. The solid electrolyte decomposition products are often insulators (both ionic and electronic) and therefore increase the total cell resistance.38–40Fig. 1 presents a schematic of the components in a SSB and the issues discussed above.
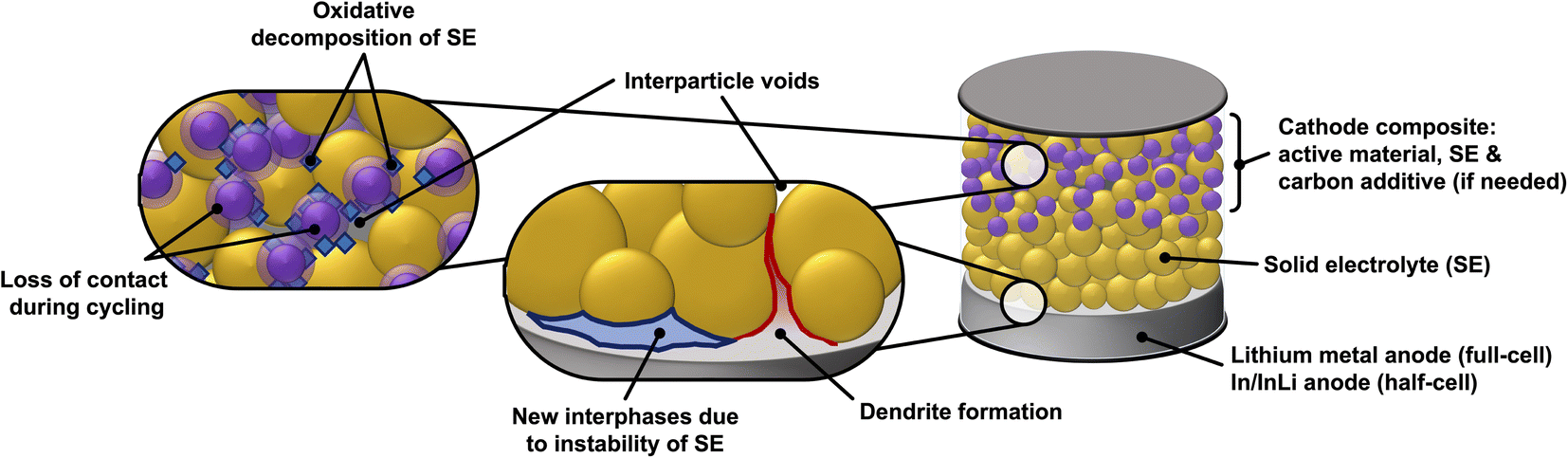 | ||
| Fig. 1 Schematic of a solid-state battery and highlights of the various issues that these systems face, some of which can be addressed by utilizing liquid electrolytes. Note that, although not shown, dendrites can also puncture the SE crystallites themselves (see text for literature references on this aspect). | ||
Despite these challenges, the commercialization of SSBs is moving forward as demonstrated by recent press-releases by QuantumScape Co. and Solid Power.41,42 A quick glance to some of the Quantumscape Co. patents demonstrates the implementation of hybrid electrolytes, as anolyte and catholyte composites in full cells.43,44 For example, a “bonding layer” consisting of a polymer, a Li ion salt and a solvent is sandwiched between a garnet oxide solid electrolyte separator and a layered cathode active material. The patented cell uses a lithium metal anode, suggesting that dendrite formation has been successfully stopped for the cells' cycle lifetime. Therefore, new hybrid electrolyte concepts are being implemented to push SSBs to commercialization.
A hybrid electrolyte could be one of the following combinations: solid electrolyte–polymer electrolyte (SE/PE), solid electrolyte–liquid electrolyte (SE/LE), or solid electrolyte–solid electrolyte (SE/SE).45 Although the combination of (solid) polymer electrolyte (membranes)–liquid electrolyte is also possible, we do not consider it a “true” hybrid because most polymer electrolytes (even commercialized ones) rely on the organic solvents to increase the mobility of lithium ions in the polymeric matrix or improve the mechanical properties of the polymer membrane itself. Notable exceptions are recently reported solid polymer electrolytes that require no solvent and show modest ionic conductivities and Li transference numbers.46,47
Since SE/PE systems have been recently and extensively reviewed by others,48–52 the present manuscript will focus on hybrid SE/LE systems, which remain comparatively unexplored. In the systems we discuss below, the purpose of the liquid electrolyte is to improve the ionic connectivity between the solid electrolyte and the electrode material. The finite and often heterogenous particle size distribution of the SE results in voids, inconsistent contact areas as well as multiple interfaces that increase the cell resistance.53 These issues could be partially mitigated with the use of a LE that can ensure ionic connectivity between the SE particles and electrode materials by filling interparticle voids. However, an often-observed side effect of the SE/LE systems is the formation of new interphases at the surface of SE particles and microstructural changes to the SE particles. The newly formed interphases are strongly dependent on the chemistry of both the SE and LE and can represent an additional transport limitation in full cells. Therefore, systematic studies and thorough characterization of the formation kinetics of such interphases is critical for the realization of hybrid SE/LE concepts in SSBs.
Hybrid SE/LE systems
In this manuscript we consider solid electrolyte–liquid electrolyte hybrid systems that consist of a ceramic solid electrolyte in contact with a liquid electrolyte that is either a lithium salt dissolved in an organic solvent or an ionic liquid. Three main factors should be considered in a hybrid SE/LE system: the composition and intrinsic properties of (i) the solid electrolyte, (ii) the liquid electrolyte, and (iii) possible chemical interactions between the components of the LE and SE phases that might lead to degradation and formation of new interphases.The first report on the transport of lithium ions across a SE/LE interface was presented by Abe and co-workers in 2005.54 For this study, the SE phases studied were crystalline Li0.35La0.55TiO3 and a Li–Al–Ti–phosphate Ohara glass. The authors' electrochemical results and their interpretation propose that (de)solvation of Li ions at the SE/LE interface has a high activation barrier and causes large interfacial resistances. This hypothesis was further supported by the fact that when the LE phase contained a pure ionic liquid as a solvent, the activation barrier of the interfacial resistance was significantly reduced. Therefore, the use of concentrated lithium liquid electrolytes (with or without ionic liquids) has garnered interest as an effective approach to mitigate interfacial ionic connectivity issues in SSBs.
One aspect that was not considered in the previous report was whether chemical interactions between components in the LE phase, specifically solvent molecules, could induce the degradation of the SE. The formation of such interphases can be driven due to the mismatch of the electrochemical potential of Li ions in between the LE and SE phases. Moreover, it is also possible that the structural units that make up the SE are susceptible to chemical attack by the solvent molecules in the LE, which also leads to dissolution/degradation of the SE. The resulting interphase, known as the solid–liquid electrolyte interphase (SLEI), will intrinsically affect the kinetics and the nature of Li+ transport in these hybrid systems.
To demonstrate the viability of SE/LE systems, a combination of fundamental and application-driven studies is needed. For the fundamental aspect, it is useful to have a theoretical model that can describe the transport of Li ions across the SE/LE interface, as well as the newly formed SLEI. For such studies, electrochemical measurements in reversible symmetric cells, i.e., Limetal|LE|SE|LE|Limetal, are often used and can provide valuable insights to the interphase formation kinetics and overall stability of the SE/LE system. From a more applied perspective, the evaluation of SE/LE hybrids in half- and full-cells is necessary, as only then can we evaluate the interaction of the SE/LE system against the target cathode active materials and anode components.
What makes up a SLEI?
At this point, it is useful to reflect on the similarities and differences between the here-presented SLEI and the more established but still not fully understood solid electrolyte interphase (SEI). The SLEI and the SEI are similar in the sense that they are (1) both made up from the decomposition products of electrolyte components, (2) must remain ionically conducting but electronically-insulating, (3) the formation kinetics and mechanism are not fully elucidated yet, and (4) ion transport across both interphases is still under study. The key difference between a SLEI and an SEI is that of the original pristine interface: whereas for a SLEI we are looking at a new interphase formed at an electrolyte–electrolyte interface, the SEI is instead formed at an electrolyte–electrode interface. In fact, in the battery community it is common to further distinguish an SEI that is formed at the anode (SEI) and a at the cathode (CEI). For the reader interested in learning more about SEIs, we suggest the following reviews: Peled and coworkers,55,56 Cheng et al.,57 Yu and Manthiram,58 Shadike et al.,59 Cekic-Laskovic & coworkers,60 Yamada & coworkers,61 Lucht & coworkers,62 Liu et al.63 and Nojabaee et al.64Since a SLEI can be considered as a more complex type of SEI, the composition, formation mechanism and microstructure of a SLEI will depend on the chosen SE and LE. Whilst there are similarities to the SEI, namely decomposition products from the liquid electrolyte, there will also be decomposition products that stem from the SE itself meaning the make up with the SLEI will contain other, particularly inorganic, compounds. A manifold of methods are necessary to fully characterize the resulting SLEIs in hybrid SE/LE systems. From an electrochemical properties point of view, a symmetrical cell as well as two additional reference electrodes on each side of the SE, i.e. 4-electrode measurement, would be necessary to try and deconvolute the impedance of the SLEI from that of the SEI at the Li electrodes. Although such cell setups and measurements can be challenging, there are reports that demonstrate their usefulness.65–67 Surface sensitive methods like X-ray photoelectron spectroscopy (XPS), or hyphenated methods like time-of-slight secondary ion mass spectrometry (ToF-SIMS) that allows depth-dependent chemical profiling in flat samples are well-suited to assess the chemical makeup of the SLEI and propose formation/decomposition mechanisms. Here we summarize recent reports that have used such methods on SE/LE hybrid systems. Note that the content will now shift more to the oxide-based hybrids because they have been studied for longer and there is more data on them.
A first example of this is in the garnet oxide Li7La3Zr2O12 (LLZO)/LiPF6 in DMC/EC hybrid system. Studies show that garnet oxides are susceptible to H+/Li+ exchange where the lithium ions on the 96 h site of the cubic garnets are replaced by H+ when in the presence of a proton source (i.e. water).68 Such an exchange reaction is shown below, followed by the subsequent reaction of lithium hydroxide with carbon dioxide to form lithium carbonate:
| Li7La3Zr2O12 + xH2O → Li7−xHxLa3La2O12 + xLiOH |
| 2LiOH + CO2 → Li2CO3 + H2O |
In this case, because of the solid electrolyte, the SLEI will contain lithium carbonate, lithium hydroxide and protonated LLZO. For the protonated LLZO, the Li+ ion mobility is reduced by the presence of H+ on the lithium positions (decreasing the number of moves that the Li+ can achieve as the O–H bond is strong enough to prevent H+ mobility). Such a scenario is supported by results from molecular dynamic simulations that show a higher activation energy for the Li+ ion transport in the protonated LLZO is larger (0.85 eV) than that of pristine LLZO (0.32 eV).69 Secondly, in this system the LiPF6 (DMC/EC) will also tend to decompose to form products common in SEIs in LIBs:70
| LiPF6 → LiF + PF5 |
| PF5 + H2O → POF3 + 2HF |
The likelihood of this reaction occurring in hybrid SE/LE electrolytes is proven by chemical analysis techniques which often show that SLEIs contain LiF and PFy species, such as PF5 (decomposition products of PF6).71 In addition, further decomposition products from the DMC/EC solvents are also included in the SLEI (such as Li2CO3 and CH3OLi). These products are also well known to form in SEIs in conventional LIBs.72
An interesting idea for limiting SLEI formation in garnet oxides was demonstrated in a tantalum-doped LLZO/LiPF6 in EC/DEC hybrid is the use of the n-BuLi superbase.73 The use n-BuLi may prevent LE decomposition, suppresses H+/Li+ exchange and lithiates the interface of the garnet solid electrolyte. Raman spectroscopy of the hybrid treated with n-BuLi shows that Li2CO3 (a decomposition product for both the SE and LE) is no longer evident at the surface of the pellet.
A recent report by Garcia-Araez and co-workers carefully studied the role of water content and other additives on the formation and resulting resistance of SLEIs for the Ohara glass LATP SE and various lithium liquid electrolytes with and without water.74 Upon analysis of XPS results of solid electrolyte samples exposed to such liquid electrolytes, the general observation is that the composition of the SLEI contains a complex mixture of inorganic and organic compounds similar to those reported before for SLEIs and even SEIs. What is most remarkable of this study is that when water is incorporated as an additive in the liquid electrolytes, it can significantly reduce the resistance for ion conduction at the solid electrolyte/liquid electrolyte interface. The authors propose various explanations to this observation, e.g. improved lithium (de)solvation kinetics, targeted interactions with the SLEI that aid ion transport across it, among others, but neither has been fully demonstrated. Nonetheless, it serves as inspiration to test and identify solvent additives that can help reduce the resistance of SLEIs in sulfide SE/liquid electrolyte hybrid systems.
Another example, in which the role of additives and salts typical in the Li-ion battery community were studied, was reported by Sakamoto and co-workers.75 Here the Ta-doped Li7La3Zr2O12 garnet oxide (LLZTO) is tested against a series of salt-in-solvent LEs. For this study, acetonitrile was used as a solvent because it has a low viscosity, a low donor number and shows high solubility for many lithium salts. The latter is particularly useful to deconvolute solvent from lithium salt effects in such hybrid systems. The salts studied were LiTFSI, LiBOB and LiPF6. The LLZTO was exposed to LEs of 0.2 M lithium salt in ACN for up to 48 h. During this time, the impedance of the SE and the SE/LE interface was monitored and showed that all hybrid systems have an initial interfacial resistance (not present in the pristine LLZTO) that increases over time. The LiBOB/ACN system shows the highest impedance with an initial resistance of ca. 500 Ω cm2, whereas the impedances of the LiTFSI/ACN and LiPF6/ACN systems are in the order of 20 Ω cm2. The final impedances are in the order of 2000, 100 and 60 Ω cm2, for the LiBOB, LiPF6 and LiTFSI systems, respectively. Ex situ X-ray photoelectron spectroscopy was then used to study the surface of the LLZTO and the formed SLEI. For the LiPF6/ACN system showed a large compositional increase in F whilst a 10% decrease in Li. It was concluded that LiPF6 reacts with the LLZTO to form LiF, LaF3 and ZrF4. For LiBOB/ACN, a decrease in Li content indicates that the high interfacial resistances could stem from a SLEI with a depleted Li charge carrier concentration. The LiTFSI sample showed the smallest compositional change when compared with the pristine LLZTO sample indicating minimal decomposition of the SE. The main takeaway from this study is that the solvent alone does not drive the SLEI formation, but lithium salts in the LE play an active role instead.
The use of LiTFSI as a lithium salt is common in many types of electrolytes, including ionic liquid and salt in solvent electrolytes. Therefore, LiTFSI is often a key component of solid–liquid hybrid electrolytes. As LiTFSI can electrochemically decompose, such products of LiTFSI will also be evident in SLEIs. Below are a series of decomposition reactions that can occur during cycling of a cell containing LiTFSI. It is therefore clear that SLEIs for LiTFSI hybrid systems with contain lithium nitrides, fluorides and sulfides.76
| LiN(SO2CF3)2 + 2e− + 2Li+ → Li2NSO2CF3 + LiSO2CF3 |
| LiN(SO2CF3)2 + ne− + nLi+ → Li3N + LiOSO2 + LiCF3 |
| LiN(SO2CF3)3 + ne− + nLi+ → Li2S2O4 + LiF + Li3N + C2FxLi |
| Li2S2O4 + 6e− + 6Li+ → 2Li2S + 4Li2O |
For example, post-mortem analysis of the SLEI of a Li2S–P2S5–LiTFSI(DME/DOL) hybrid showed to presence of –S*O2CF3, LiF and Li2S all decomposition products of LiTFSI.77 Whilst the full composition of SLEIs is not fully understood, using surface sensitive techniques such as XPS or SEM may help to investigate SLEIs further. Whilst there is an extensive amount of literature on SEIs, solid–liquid electrolytes are still in their infancy. Therefore, due to the general similarities between SLEIs and SEIs, taking influence from research work done on SEIs should be an important part of any studies done on hybrid solid–liquid electrolyte systems.
The tendency of LE decomposition products to be the dominating influence in some SLEIs is shown in a paper from Busche et al.78 In this case hybrid electrolytes consisting of either LIPON or Ohara LiCGC and LiTFSI in DME, DOL or the combination of the two were studied. It was shown that the SLEI when LiTFSI in DME is used for the electrolyte TFSI can deprotonate the DME leading to the formation of ethers and lithium alkoxides. Whilst when LiTFSI in DOL is used, deprotonation leads to ring opening and subsequently to the polymerization and formation of poly-DOLs. In the case of the DME, LiTFSI is then incorporated into the R–CH2OLi structure where it decomposes leading to the formation of LiF. However, in DOL this LiTFSI decomposition occurs less readily leading to a more stable, less resistive SLEI. In this case, the dominating factor in SLEI formation is LE decomposition however again the SE is still important. In comparison of Ohara LICGC with LIPON using the same electrolyte the SLEI for the LIPON tends to be larger than for LICGC indicating that LICGC is more stable than LIPON with respect to the solvents. This difference, however, is far less marked than the change when moving from DOL to DME.
In the case of sulfide electrolytes, the dominating case for the composition of the SLEI is the tendency of thiophosphates to decompose in the presence of hard bases. Hatz et al. demonstrated the degradation of thiophosphates when exposed to alcohol solvents (Scheme 1).
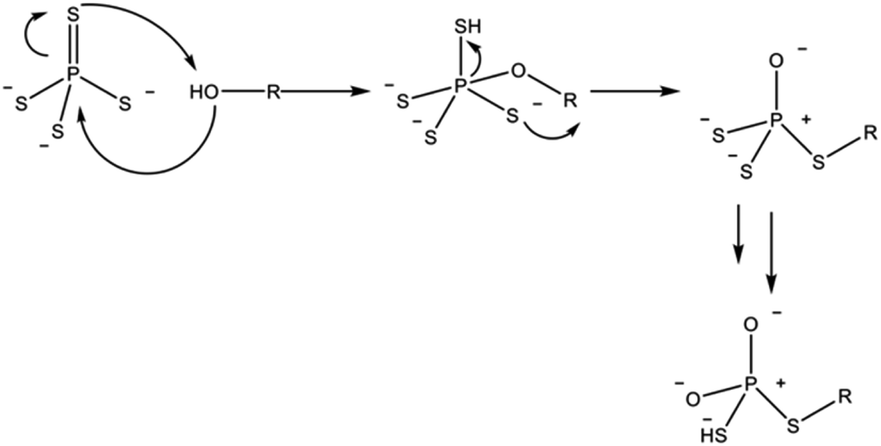 | ||
| Scheme 1 Proposed reaction pathway for the degradation of thiophosphate units by alcohol solvent molecules. Adapted from ref. 86. | ||
It is likely that a similar mechanism is at play when the solvent is replaced by an ether. Oh et al.79 systematically studied the effect of free and (under)coordinated triglyme molecules in solvent ionic liquid electrolytes. The authors show that when all glyme molecules are bound to the Li+ ions in the LE phase, a greater stability of the sulfide-based solid electrolyte/liquid electrolyte hybrid is observed. In the presence of free or undercoordinated glyme molecules, the likelihood of nucleophilic attack of the phosphorous cation in the solid electrolyte increases. The resulting composition of the formed SLEI in such systems remains elusive due to the lack of chemically specific characterization of such samples. However, the reported trends are consistent with the hard and soft acid and bases (HSAB) theory, in which hard bases (e.g. ether) will preferentially react with hard acids (the phosphorous cation in the PS43− tetrahedra of the solid electrolytes).
Ion transport model in SE/LE systems and experimental support
Ion transfer across a solid electrolyte SE/LE interface, in its simplest consideration, can be described as electron transfer kinetics at electrode–electrolyte junctions (Fig. 2). Specifically, the ion transfer across this interface must consider the difference in electrochemical potential of the ions in both the SE and LE phases, and the energetics of such process is thermally activated. A recent study by Korte and coworkers considered two current limiting scenarios for ion transfer at a SE/LE interface:67 (a) ion transfer is limited by ion diffusion, i.e. activity gradients; and (b) ion transfer is limited by thermally-activated charge-transfer. Using a sophisticated electrochemical cell setup with 2-active Li electrodes and 6 additional electrode probes for monitoring potential differences under galvanostatic conditions, the authors demonstrated that scenario (b) is at play in a system that uses a tantalum-substituted lithium garnet (Li6.6La3Zr1.6Ta0.4O12) as SE, and liquid electrolyte solutions of LiPF6 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of ethylene carbonate and dimethyl carbonate. Mathematically, the ion transfer in scenario (b) can be described as a polarization resistance (Rp) that incorporates a modified Butler–Volmer kinetics model and an additional constant series resistance to account for the SLEI (RSLEI, eqn (1)):
:1 ratio of ethylene carbonate and dimethyl carbonate. Mathematically, the ion transfer in scenario (b) can be described as a polarization resistance (Rp) that incorporates a modified Butler–Volmer kinetics model and an additional constant series resistance to account for the SLEI (RSLEI, eqn (1)):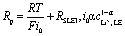 | (1) |
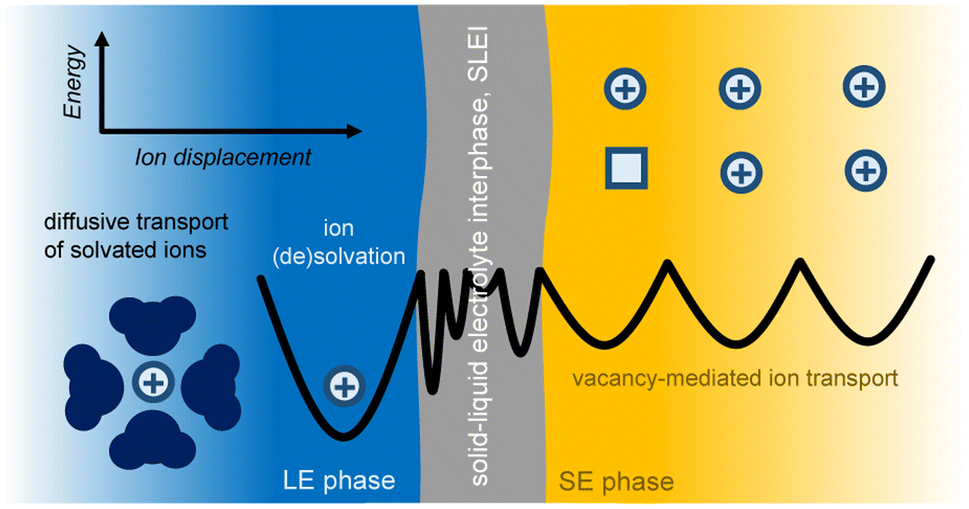 | ||
| Fig. 2 Schematic of the transport of Li ions across the interface of a LE in contact with a SE. In the LE, the transport of solvated Li is due to diffusion. In the SE phase, the ion transport is mediated via vacancies. For an ion that is moving from the LE to the SE phase, at the LE/SE interface, the ion must lose its solvation shell prior to “jumping”. The transport of ions in such a LE/SE hybrid system may be further complicated by the presence of a solid–liquid electrolyte interphase (SLEI), a phase for which the energy landscape to describe ion transport across it is not fully understood. The parabolic black lines are meant to schematically show the thermally activated energetics of ion transfer (displacement) from one phase to another. The minima of the parabolas describe local ground states for the ion, and the intersection of the parabolas show arbitrary transition states that the ion might experience while displacing from one ground state to another across the various phases. | ||
Janek and co-workers have pioneered experimental approaches to demonstrate that the formation of the SLEI is a dynamic process.65,78 The nature of the solvent used was shown to be a driving factor in SLEI formation in LiPON SE thin films. Time-dependent impedance studies showed that when dimethyl ether (DME) was used as a solvent, RSLEI increased from 539 Ω cm2 after 24 h of exposure to 1511.7 Ω cm2 after 144 h, whilst with dioxolane (DOL) the RSLEI shifted from 67.7 Ω cm2 to 129.2 Ω cm2 at the same time points. A separate study by the same group evaluated the formation of SLEIs on sputtered LiPON thin films using in situ neutron reflectometry (NR), quartz crystal microbalance (QCM) and impedance measurements.83 Here, the interaction between LIPON and LEs composed of LiTFSI salt in DME, DOL and diglyme solvents and the resulting SLEIs was reported. The NR experiments confirmed previous observations that the SLEI is primarily composed of lithium carbonate species. However, there was a mismatch in terms of the estimated thickness of the SLEIs because the QCM results suggested a continuous SLEI formation due to the perpetual mass increase observed, whereas NR results propose a fixed thickness for the formed SLEIs. Based on impedance results, it is proposed that the growth of the SLEI is not uniform (dense) and pinholes in the SLEI allow for ion transfer from the LE to the SE phase until the SLEI densifies, becomes chemically stable and a constant RSLEI is attained. The presence of pinholes that allow for the percolation and further transport of ions and molecules across SEIs in model carbon electrodes has been recently demonstrated by Roling and coworkers.84–86
Fig. 3a presents a selection of SE/LE systems recently reported in the literature. A more comprehensive survey has been compiled by Weiss et al.45 Note that, although the formation of the SLEI is a time-dependent process, it tends to a constant value in all the oxide and LiPON SE/LE systems. This is consistent with the model discussed above that allows for a constant RSLEI. Therefore, the initial assessment by Abe and co-workers regarding the (de)solvation of Li ions as the rate-limiting process is likely correct. Irrespective if the SE is oxide- or sulfide-based, we observe that stable hybrid systems show stabilized SLEI resistances that range between 100 and 1000 Ω cm2. These values translate to a gravimetric energy density loss of at least 18% in model Li–S SSB cells (assuming 100% sulfur utilization and a rate of 1C, Fig. 3b).65 As shown before,65 practical energy densities are achieved at a separator thickness below 10−5 m, irrespective of the presence of the SLEI. Replacing the separator with a faster conductor, does not mitigate the energy density losses because RSLEI is not thickness dependent. Despite these energy loses we must underscore that adding liquid electrolytes may prevent the safety issues associated with thermal runaway under applied external heat as shown in a recent paper by Bates et al.87 At a fixed separator thickness, we can estimate the effect of RSLEI on the round-trip efficiency of a model Li–S solid-state battery at various C-rates (Fig. 3c). We considered typical ionic conductivities for both oxide (0.01 S m−1) and sulfide (1 S m−1) solid electrolytes. Assuming 100% sulfur utilization, we observe that the fast-conducting sulfide-based hybrid electrolytes only make sense if we can limit the value of RSLEI to ca. 10 Ω cm2. In the case of the sulfide-based SEs, one observes that the resistances tend to increase over time, consistent with sample degradation and/or the persistent formation of a highly resistive SLEI. However, specific systems show appropriate initial RSLEI values (e.g. Li10GeP2S12|7 M LiTFSI in DOL/DME:TTE) or rapid stabilization of RSLEI (e.g. β-Li3PS4|(MeCN)2-LiTFSI:TTE) and serve as the foundation for identifying suitable sulfide SE and LE combinations that result in stable hybrid electrolyte systems. In the latter systems, the TTE was incorporated to lower the viscosity of the LE phase.
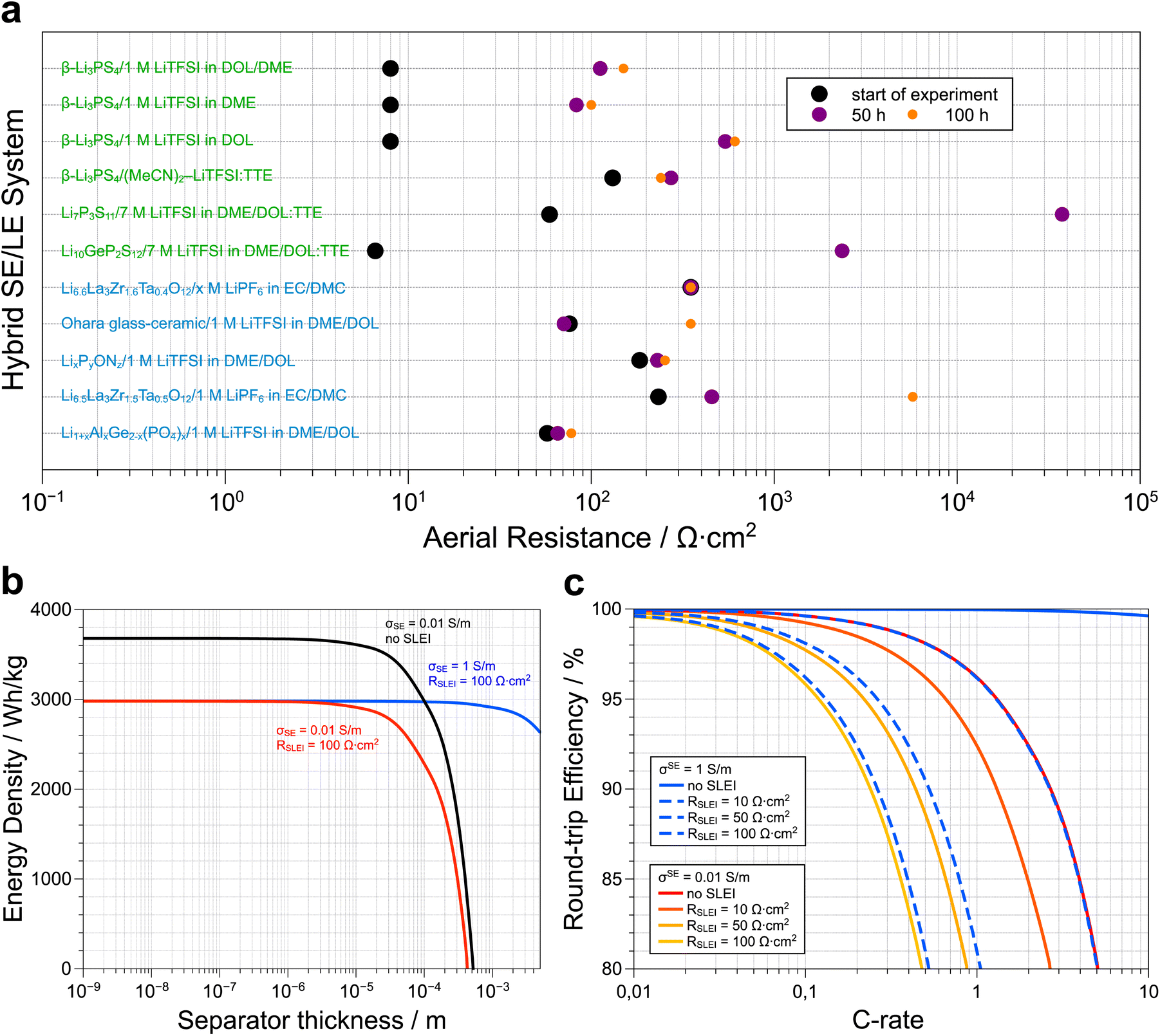 | ||
| Fig. 3 (a) The temporal evolution of the aereal resistance of the solid liquid interface (SLEI) for a series of solid electrolyte–liquid electrolyte hybrid systems reported in the literature. Although the sulfide-based systems show lower initial resistances than the oxide-based hybrids, the final resistances are in the order of 100 up to 1000 Ω cm2. Note that the system used by Korte and coworkers to develop the model that describes ion transport in these hybrid systems, i.e. Li6.6La3Zr0.6Ta0.4O12/1 M LiTFSI in EC/DMC, shows a constant resistance of 350 Ω cm2 as this value was not directly measured via impedance spectroscopy but rather extracted from polarization experiments (more details, see ref. 67). The corresponding citations as well as the exact resistance values used to generate (a) are available in the ESI.† (b) Loss in energy density of a Li–S solid-state battery due to the overpotentials of the separator and the SLEI as function of separator thickness at a fixed rate of 1C (adapted from ref. 65). (c) C-rate dependent losses in round-trip efficiency for a Li–S solid-state battery with a separator thickness of 10−5 m as a function of electrolyte conductivity (σSE) and resistance of the SLEI (RSLEI). As the value of RSLEI increases, the 80% RTE cut-off is attained at lower C-rates and the use of faster conductors is only practical when the RSLEI is limited to <10 Ω cm2. Details about how plots (b) and (c) were generated are available in the ESI.† | ||
The theoretical framework built by Korte combined with the experimental demonstrations of the Janek group can be applied to numerous combinations of liquid and solid electrolyte, for this review the focus will shift to the fast ion conducting sulfide electrolytes, which despite their high ionic conductivities, are often unstable in full cells and thus benefit from coatings or the use liquid electrolytes. However, their intrinsic reactivity means the fast formation of SLEIs and/or irreversible SE degradation and thus careful studies must be applied to hybrid sulfide-liquid electrolyte systems.
Sulfide-based hybrid systems
Sulfide electrolytes display excellent ionic conductivities above 10−3 S cm−1 at room temperature, approaching and in some cases surpassing liquid electrolytes.19,20,88,89 Despite the excellent bulk ionic transport properties of these materials issues remain, especially with regards to their chemical and electrochemical stabilities.90–94 Fast ionic conductors such as Li10GeP2S12 have a high ionic conductivity of 1.2 × 10−2 S cm−1 at room temperature,19 however are unstable against lithium metal. The electrochemical window of thiophosphate electrolytes has been studied both experimentally and computationally.33–35,38–40 Stepwise cyclic voltammetry experiments show the practical oxidative stability of LPS (Li2P–P2S5 70:30 and 75:25 glasses), LGPS and the Li6PS5Cl argyrodite are limited to a range of 2.8–3.1 V vs. Li/Li+.33 This means that thiophosphate electrolytes will oxidize upon contact with state-of-the-art commercial cathode active materials such as LiCoO2, LiNixMnyCozO2 or LiNixCoyAlzO2 all of which operate at potentials above 4 V vs. Li/Li+.95 Furthermore, studies of thiophosphate electrolytes with Ni-rich cathode materials show that the oxidation of the electrolyte material causes a resistive layer in between the cathode and electrolyte causing poor performance of the cell after 200 cycles.96 Interparticle contact between the electrolyte and cathode active materials has also proved to be an transport bottleneck for solid state batteries.91 All these issues have meant that despite their high ionic conductivities sulfide electrolytes are yet to reach their full potential in SSB systems. As with the oxide, LIPON and NASCION solid electrolytes the concept of applying a small amount of liquid electrolyte to the surface is a possible solution to these issues.
Sulfide SE against neat solvents
Due to their inherent reactivity, the study of sulfide electrolyte's sensitivity to solvents is of importance. Firstly, to achieve commercial viability sulfide electrolyte separators and cathode composites may need to be prepared in via slurry method analogous to cathode preparation in conventional electrolytes.97–99 Moreover, the typical liquid electrolytes used for lithium-ion batteries, tend to be “salt-in-solvent” type, which means that one should expect a certain amount of free solvent molecules that can react with the SE. As mentioned before, solvents are commonly used in liquid electrolytes, and therefore it is vital we understand the role of electrolyte solvents in hybrid systems. Several parameters can be used to predict the tendency of a specific solvent to cause solid electrolyte decomposition such as the dielectric constant, relative polarity, and donor number of the solvent. The donor number (DN) was defined by Viktor Gutmann as the enthalpy for the reaction of a donor and SbCl5 in a 0.0001 M solution of dichloroethane (i.e. a donor number of 0 for DCE).100 The dielectric constant and relative polarity are intrinsic properties of a solvent that define how electron density is distributed within a solvent. Lee et al. examined the stability 75Li2S–25P2S5 glass ceramic against several solvents and showed that the decomposition of the SE increases with increasing solvent polarity.101 Yamamoto et al. showed a direct relationship between a solvent's DN when in contact with LPS and the ratio of ionic conductivity of the bare and solvent treated LPS pellets.102 As seen in Fig. 4 the use of non-polar solvents such as toluene or decane results in a smaller decrease in ionic conductivity when compared with highly polar solvents such as glymes, 1,4-dioxane and PC. These solvents, which have donor numbers greater than 15 kcal per mole, cause the ionic conductivity to reduce ten-fold. These polar solvents used in liquid electrolytes therefore can be detrimental to the performance of solid-state electrolytes when used in tandem.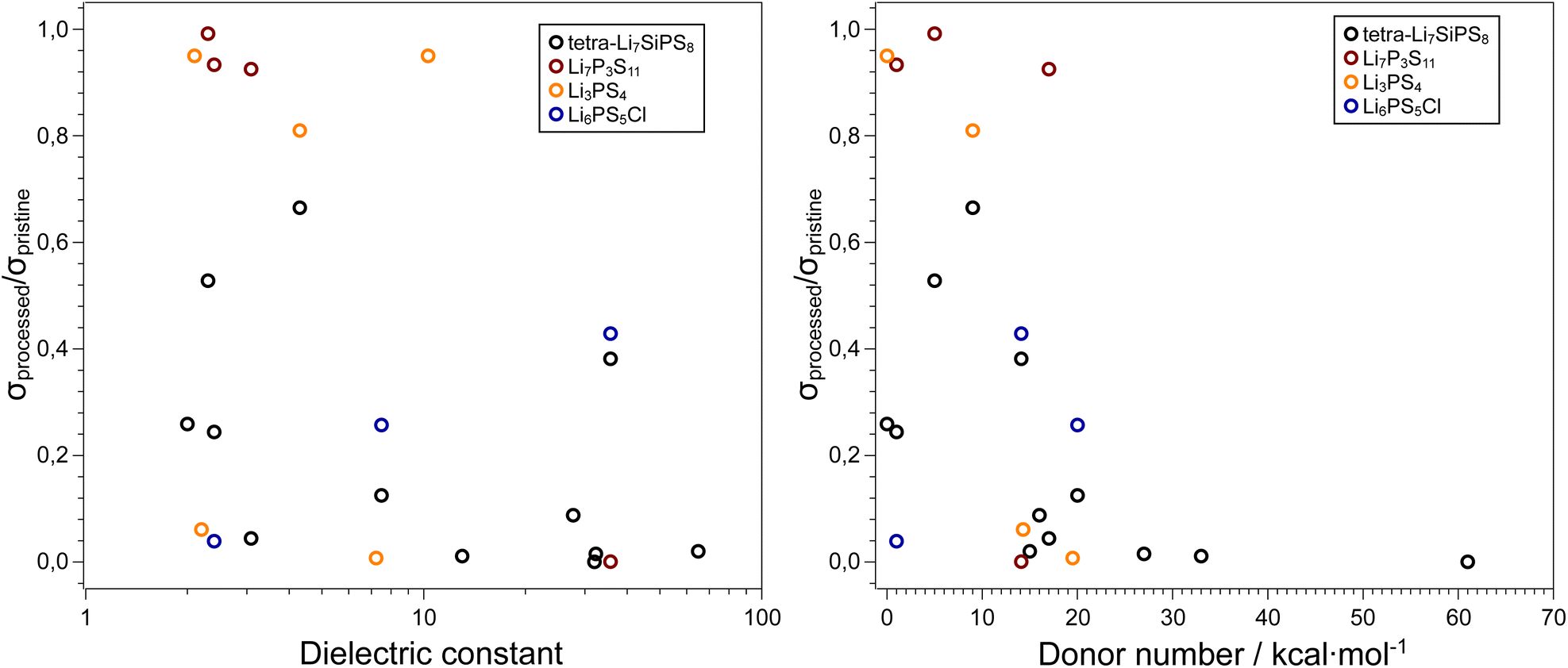 | ||
| Fig. 4 Influence of solvent properties on the ionic conductivity of thiophosphate-based SEs. Data adapted from ref. 101, 102, 103, and 104. In general, high dielectric solvents and solvents with donor numbers > 15 kcal mol−1 are unsuitable for processing these electrolytes and retaining the initial ionic conductivities. The exact values used to generate these plots, along with the respective citations are available in the ESI.† | ||
Ruhl et al. showed that the ionic conductivity of the argyrodite Li6PS5Cl decreases when solvent processed.103 This effect is less pronounced for solvents such as THF or toluene which have lower dielectric constants. XRD patterns of solvent-processed solid electrolyte show the presence of LiCl and Li2S (precursors to argyrodite synthesis) in the samples that were processed in more polar solvents.
In a recent paper Hatz et al. reported the stability of the sulfide superionic conductor tetra-Li7SiPS8 (LiSiPS) with a series of solvents.104 In a similar vein to Li6PS5Cl, the LiSiPS solid electrolyte showed chemical stability against solvents with low donor numbers such as hexane, toluene as well as acetonitrile. Based on their observations, and consistent with previous reports, a threshold DN of 14 kcal mol−1 was proposed to ensure a high enough ionic conductivity (σ > 1 mS cm−1)105 for a solid electrolyte sample after solvent processing. Note that the conductivity of pristine LiSiPS is ca. 4 mS cm−1. Moreover, the use of protic solvents, especially alcohols, should be avoided as they fully degrade the thiophosphate electrolyte. The degradation of the SE can be partially reversed by high-temperature processing, consistent with previous reports on liquid-based syntheses of thiophosphate electrolytes.106,107 Interestingly, irrespective of the solvent used, all samples showed some morphological changes after processing. Although LiSiPS is considered a glass-ceramic108 and the solvents mainly interact with the glassy phase, it is clear that particle microstructure and perhaps newly formed interphases (e.g. a SLEI) plays a crucial role in the resulting ionic conductivity of the samples.
We note that although all four thiophosphate SE presented in this section contain PS43− tetrahedra that could be prone to attack by polar solvent molecules, all materials have entirely different structures which can also affect the reactivity. However, irrespective of the SE, we observe a qualitative trend that with increasing DN, the ionic conductivity of the processed SE decreases. Conversely, the dielectric constant of the solvent shows significant scatter in the data and is not a good predictor for the effect of solvent processing on the resulting ionic conductivity of the sulfide SE. We acknowledge that the truly interesting systems to study the stability and reactivity of sulfide SEs is against LEs and not neat solvents, but the results summarized here can serve as an initial guide to the selection of compatible LE systems.
Sulfide SEs against lithium liquid electrolytes and their electrochemical performance
Gewirth and coworkers examined the effect of highly fluorinated ether cosolvents on the stability of β-Li3PS4 and Li10GeP2S12 against ether-based LEs.109 The advantage of fluorinated cosolvents has been demonstrated before and mostly impact the formation of (more) stable SEIs at Li metal anodes and result in less flammable electrolytes.110–112 Impedance spectroscopy results in reversible symmetric cells show that the β-Li3PS4–hybrid system has a higher cell resistance than the pristine β-Li3PS4 system. Conversely, the Li10GeP2S12–hybrid system shows lower cell resistances than the pristine SE. The authors attribute such differences in performance to the varying solubilities that the SEs have in the LE, namely the β-Li3PS4 has a higher solubility which produces a thicker SLEI that increases the overall impedance.An earlier study on the potential of sulfide solid electrolytes alongside liquid electrolytes was done by Oh et al.79 Here, they used Li3PS4 and Li10GeP2S12 alongside a solvate ionic liquid electrolytes Li(G3)4TFSI and Li(G3)TFSI where G3 is triethylene glycol dimethyl ether (triglyme). For their studies, pellets of the SEs were exposed to the two solvate LEs as well as pure triglyme and characterized using time-dependent impedance measurements. Note that the only difference between the Li(G3)4TFSI and the Li(G3)TFSI electrolytes is the molar ratio between Li+ and G3 molecules. Specifically, in the case of Li(G3)4TFSI, one can find free and as well as undercoordinated G3 molecules that are not fully bound to Li+. In the case of the Li10GeP2S12 hybrid, the system showed excellent stability against both liquid electrolytes, with only a small increase (in the order of 10 Ω) in total impedance compared to the pristine LGPS for up to 20 h of exposure. The latter suggests that SLEI formation in this system is quite rapid and self-limiting. Similarly, for the Li3PS4-based hybrids, the impedance after 3 h compared to the analogous LGPS system was larger (by about a factor of 5), albeit with very little change over the course of 20 h of exposure. This hints that the SLEI, despite being more resistive, is stable after its initial formation. Due to the lower impedances of the Li10GeP2S12 hybrids, the authors tested its performance in half-cell configuration using In/InLi as an anode, LiFePO4 as a cathode, and the Li10GeP2S12/Li(G3)TFSI hybrid as an electrolyte. Such solid-state batteries showed stable cycling and initial discharge capacities up to 144 mA h g−1.
Fan et al.113 tested the susceptibility of thiophosphate SEs to react with an ether-based LE. Two types of electrolytes, β-Li3PS4 and Li7P3S11 (Li2S:P2S5 glass-ceramic), were exposed for 48 h to a commercially available LE, namely 1 M LiTFSI in DOL/DME (1:1 vol%) with 1% LiNO3. Both SEs were characterized with impedance spectroscopy in ion-blocking symmetric cells to monitor how the impedance at the SE/LE interface changed during exposure. Within 48 h, the interfacial resistance of both systems increased similarly, up to ca. 88 Ω cm2. Although the β-Li3PS4-based system maintains such an interfacial resistance value after 200 h of exposure, the interfacial resistance of the Li2S:P2S5 continues to increase up to 700 Ω cm2. Based on the Raman spectra, the authors propose the formation of an ether-solvated PS43− moiety at the surface of β-Li3PS4, which presumably prevents the decomposition of the SE. Such protection layer, which can also be considered as a SLEI, is not observed in the case of the Li2S:P2S5 glass-ceramic SE, and instead the material continues to decompose after exposure to the ether-based LE. Similarly, the electrochemical stability of β-Li3PS4 hybrid system against Li metal is greater than that of the Li2S:P2S5 hybrid system. Taking all these results together, the authors design a three-component layered hybrid electrolyte system for the efficient cycling of Li–S solid-state batteries: a Li2S:P2S5 pellet (ca. 1 mm thick) is coated with a thin layer of β-Li3PS4 (ca. 1 μm thick) and is then exposed to the LE. Here, the dense glass-ceramic separator prevents the shuttling of polysulfides as well as ensures high lithium ionic conductivity across the cell, the LE ensures good interparticle connectivity at the cathode-side and the formation of a stable solid electrolyte interface at the Li metal anode, and the β-Li3PS4 protects the separator from degradation due to exposure to an ether-based solvent. Such three-component hybrid electrolyte Li–S solid-state cells show cycling stabilities that are better than reference cells that utilize a commercial polymer-based separator. Moreover, it is an example in which β-Li3PS4 is stable in an ether-based LE as well as underscores the crucial role additives (here 1% LiNO3) play in the preparation of stable and efficient sulfide-SE/LE systems.
Umeshbabu et al. report on the stability of Li10GeP2S12 against 1 M LiTFSI in N-methyl-N-propylpyrrolidinium bis(trifluoromethanesulfonyl)imide (Pyr13TFSI) and its application in a hybrid Li–S solid-state battery.114 Impedance spectroscopy measurements show that the resistance at the Li metal/electrolyte interface of the hybrid SE/LE system is much lower than with the pristine Li10GeP2S12, 142 and 2021 Ω cm2, respectively. Similar results were reported by the same group for the Li10SnP2S12 solid electrolyte when tested with 1.5 M LiTFSI in Pyr13TFSI as LE,115 namely the Li metal/electrolyte interfacial resistance of the hybrid system was in the order of 200 Ω cm2, whereas the pristine system showed a value up to 1500 Ω cm2. The Li10SnP2S12-ionic liquid hybrid systems served as good electrolytes for cycling Li|hybrid electrolyte|LiFePO4 cells.
Taken together, these reports demonstrate the potential of sulfide-based SE/LE systems for applications in solid-state batteries. Moreover, it underscores the fact that the resulting solid–liquid electrolyte interphase (SLEI) caused by the decomposition of the electrolytes is not necessarily a hindrance for applications. Similar to the solid electrolyte interface (SEI) that enabled Li-ion batteries,116 the resulting SLEIs can be exploited to design better performing and longer-life solid-state batteries.
Conclusion and outlook
Although the number of reports on sulfide-based SE/LE systems is not as extensive as for the oxide SEs, it shows that hybrid sulfide SE/LE systems may be effective in addressing some of the challenges in solid-state batteries. However, more systematic and fundamental studies are necessary to increase our understanding of the underlying chemistry as well as comparability between reported data. For example, we know that the synthesis conditions as well as the sample morphology can affect the initial ionic conductivity of the SE.117 Similarly, variability in the conditions for pellet preparation and electrochemical measurements can influence the obtained ionic conductivities.118,119 Therefore, we should expect these parameters to also play a role in the reactivity of the SE against LEs, and the resulting degradation products and SLEIs. Moreover, it is possible to tune the reactivity of the SE against a LE via chemical modification of the solid electrolyte itself, namely through substitutions. Therefore, systematic studies of the reactivity on substituted series of sulfide-based solid electrolytes that are prepared and processed under the same conditions might provide some guidelines that help us design better hybrid sulfide SE/LE systems.Shifting the focus to the use of SE/LE hybrids to address the stability of sulfide-based SEs against Li metal, the field of solid-state batteries could surely benefit from the strategies and additives already reported for liquid electrolyte cells. At this point it is important to mention that in LE systems, the solvent as well as the nature and concentration of the lithium salt and additives are parameters to explore, making this a multicomponent and very challenging optimization problem. Moreover, the analysis of electrochemical results, in particular impedance data, must be done carefully because we need to consider both the SEI formed at the Li metal as well as the SLEI formed at the SE surface, both of which could have similar time constants and be difficult to deconvolute from one another. To aid in this challenge, the combined spectro-electrochemical approach to the characterization of SE/LE hybrid systems by Janek and coworkers should be the pursued.65,78,83 From a purely electrochemical approach, the 4-electrode measurements can be described as “tricky” though. For the analysis of impedance spectra based on such measurements to work in a straightforward manner, it is necessary that the 2 reference electrodes are at the same distance from the separator as well as the 2 active (anode and cathode) electrodes. Achieving this configuration is not trivial and requires a design that can fix/seal the separator pellet on both sides without destroying it. Moreover, the cells need to be leak-proof and sealed against water and air. This means that researchers need to design home-made cells that adapt to their own independent sample requirements (regarding the mechanical and chemical properties of their SE/LE system), and it is therefore an iterative process that can produce a lot of frustration. Although there are some commercial options that could be adapted to these kinds of measurements, some trial and error will be needed to optimize the cell design and obtain high quality data. We hope however, that by emphasizing the value of such measurements for the characterization of interfacial processes and interphases, we encourage more researchers to use this approach.
From a more fundamental perspective, the formation of a double layer at the LE/SE interface should be expected because the SE will intrinsically have some sort of surface charge that will be compensated by the ions in the LE phase. The nature, structure, and dynamicity of such a double layer will naturally depend on the stability of the LE/SE interface and, should a SLEI form, then the double layer would change to accommodate the surface charge present at the SLEI. It is well-established that the double layer can influence the charge transfer kinetics of simple electron transfer reactions.120 Therefore we should expect that it could also play a role in the ion transfer kinetics in these LE/SE systems. Space-charge layers will also form at the surface of the SE phase, but these have been demonstrated to play a negligible role in causing resistive losses to ion transport, in specific SE-electrode material interfaces.121
With respect to the dynamicity of the two phases, namely the double layer and the SLEI, it is difficult to tell which one is more dynamic. An experimental method to identify and distinguish the double layer from the SLEI is quite challenging. If the resistance and dielectric properties of the SLEI are significantly different than that of the double layer formed, such that the time constant of both differ significantly, then one could perhaps use impedance spectroscopy. The challenge is of course the design of the experimental set up: one would need reference electrodes, and work under symmetrical cell conditions, likely with reversible electrodes. This would only work if one knew that the formed SLEI is dense. If the SLEI is porous, then the (dis)charging of the double layer would show a significant spread/distribution of time constants and would make it very challenging to distinguish the SLEI from the double layer. Alternatively, one could envision using spectroscopic, diffraction and/or reflection methods that are surface sensitive, and one studies model thin film solid electrolyte systems exposed to liquid electrolytes. Since the preparation of sulfide-based solid electrolytes in controlled thin film configuration with physical methods such as pulsed laser deposition, chemical vapor deposition, atomic layer deposition, among others, is an underdeveloped field of research, it is unclear if we could even prepare such model samples for characterization. We believe that the community will have to rely on theoretical/computational methods to learn more about double layers, space charge layers and SLEIs in SE/LE hybrid electrolyte systems for a while.
So far, most half- and full-cell demonstrations of sulfide-based SE/LE hybrids have focused on composite sulfur–carbon and LiFePO4 cathodes. However, incorporating these hybrid systems to high-potential cathode cells should be pursued as there is more interest from companies for commercial applications. It is likely that the ether based LE systems studied so far are not stable enough themselves and we need to look again at the state-of-the-art in LE cells. (Solvate) ionic liquids are examples of such systems that show stability with respect to both high voltage cathodes and to solid electrolytes.122–124 A recent report by Bates et al.87 challenges the generally accepted idea that SSBs are intrinsically safer than LIBs. Here, the authors considered cells that contained a Li metal anode, an oxide garnet SE, a porous LiNi0.33Co0.33Mn0.33O2 cathode and an ethyl methyl carbonate-based lithium liquid electrolyte. The authors use thermodynamic models to demonstrate that if the failure mechanism of SSBs is that of Li dendrite penetration and short-circuiting the temperature rise due to released heat can range from 100–1800 °C, irrespective if the cells incorporate a small amount of LE in the cathode compartment. For comparison, the range of temperature rise for a LIB is in the order of 1200–1400 °C. If the failure scenario is thermal runaway due to external heating, the SSBs show significantly reduced temperature rises when compared to LIBs. The latter results serve as motivation for evaluating the compatibility of ionic liquids as solvents due to their intrinsic negligible flammability, as it could allow a way forward for safer and possibly commercially viable hybrid solid–liquid electrolyte systems. Hybrid solid–liquid electrolytes are an exciting new solution to the interfacial and cell resistance problems that has prevented several solid electrolytes from becoming successful candidates to replace the conventional liquid electrolytes. Nonetheless, there are many challenges that need to be addressed to make such hybrid systems an applicable technology for next-generation energy storage devices (Fig. 5). Further exploration of them could make this seem a much more viable reality.
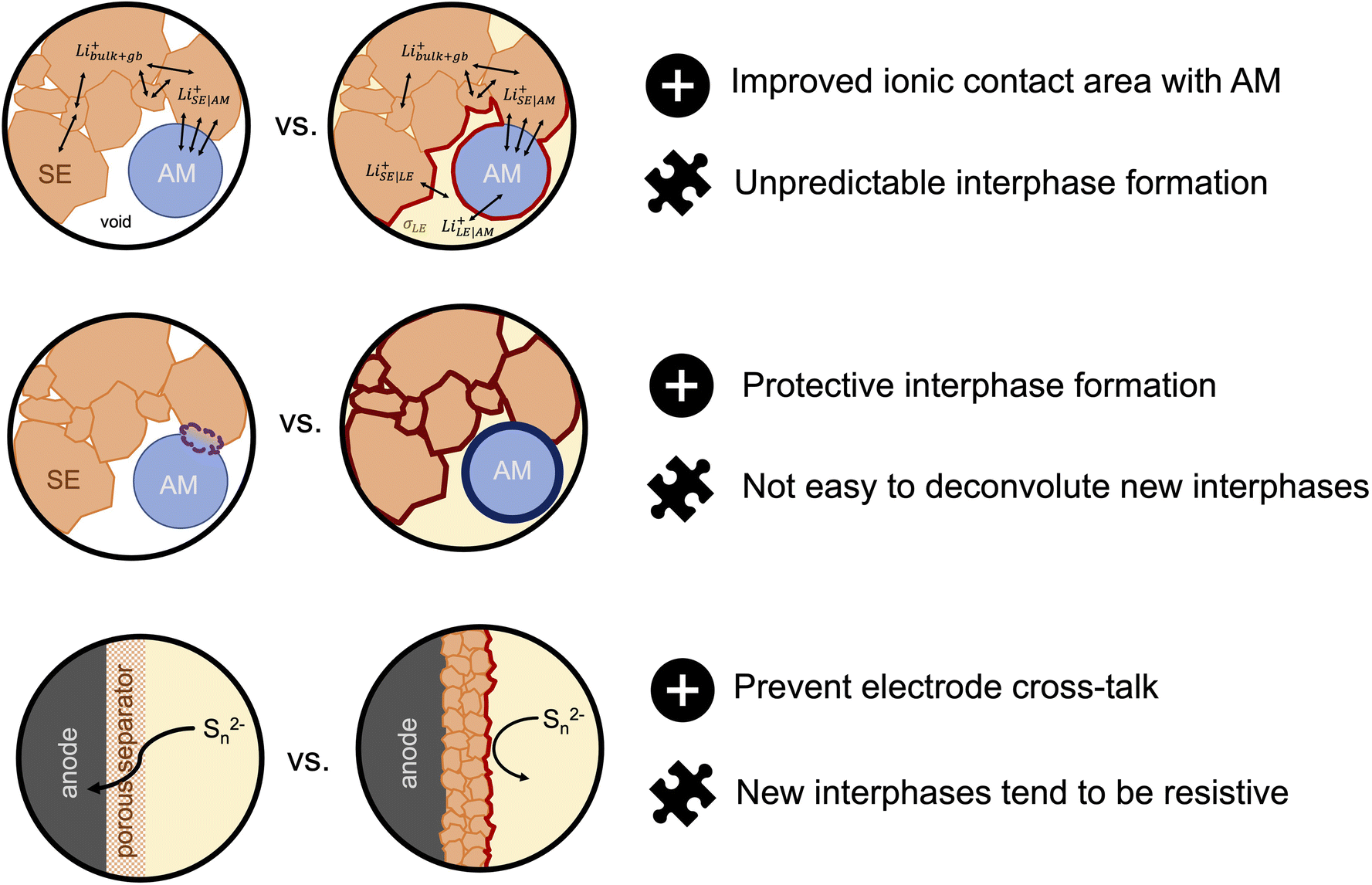 | ||
| Fig. 5 Advantages (represented with a plus sign icon) and challenges (represented with a puzzle piece icon) for hybrid solid electrolyte/liquid electrolyte systems for next-generation batteris. | ||
Author contributions
All authors have contributed to the preparation and editing of the manuscript. Moreover, all authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
N. M. V.-B. gratefully acknowledges the support of the Helmholtz Association via the Young Investigator Group Program. Moreover, we are thankful to the Center for Nanotechnology (CeNTech) at the University of Münster for providing start-up laboratory facilities. H. M. W. is a member of the International Graduate School for Battery Chemistry, Characterization, Analysis, Recycling and Application (BACCARA), which is funded by the Ministry for Culture and Science of North Rhine Westphalia, Germany. Prof. Jürgen Janek is gratefully acknowledged for coining a new definition for ASSBs, namely Almost Solid-State Batteries.References
- B. Scrosati and J. Garche, J. Power Sources, 2010, 195, 2419–2430 CrossRef.
- G. Zubi, R. Dufo-López, M. Carvalho and G. Pasaoglu, Renewable Sustainable Energy Rev., 2018, 89, 292–308 CrossRef.
- M. Armand, P. Axmann, D. Bresser, M. Copley, K. Edström, C. Ekberg, D. Guyomard, B. Lestriez, P. Novák, M. Petranikova, W. Porcher, S. Trabesinger, M. Wohlfahrt-Mehrens and H. Zhang, J. Power Sources, 2020, 479, 228708 CrossRef CAS.
- IEA, Evolution of Li-ion battery price, 1995–2019, https://www.iea.org/data-and-statistics/charts/evolution-of-li-ion-battery-price-1995-2019, (accessed March 14, 2022) Search PubMed.
- L. Lu, X. Han, J. Li, J. Hua and M. Ouyang, J. Power Sources, 2013, 226, 272–288 CrossRef CAS.
- X. Zeng, M. Li, A. E. Deia, W. Alshitari, A. Al-Bogami, J. Lu and K. Amine, Adv. Energy Mater., 2019, 9, 1900161 CrossRef.
- M. M. Thackeray, C. Wolverton and E. D. Isaacs, Energy Environ. Sci., 2012, 5, 7854–7863 RSC.
- U.-H. Kim, D.-W. Jun, K.-J. Park, Q. Zhang, P. Kaghazchi, D. Aurbach, D. T. Major, G. Goobes, M. Dixit, N. Leifer, C. M. Wang, P. Yan, D. Ahn, K.-H. Kim, C. S. Yoon and Y.-K. Sun, Energy Environ. Sci., 2018, 11, 1271–1279 RSC.
- M. Walter, M. V. Kovalenko and K. V. Kravchyk, New J. Chem., 2020, 44, 1677–1683 RSC.
- J. Janek and W. G. Zeier, Nat. Energy, 2016, 1, 16141 CrossRef.
- J. C. Bachman, S. Muy, A. Grimaud, H.-H. Chang, N. Pour, S. F. Lux, O. Paschos, F. Maglia, S. Lupart, P. Lamp, L. Giordano and Y. Shao-Horn, Chem. Rev., 2016, 116, 140–162 CrossRef CAS PubMed.
- K. S. Ngai, S. Ramesh, K. Ramesh and J. C. Juan, Ionics, 2016, 22, 1259–1279 CrossRef CAS.
- H. Zhang, C. Li, M. Piszcz, E. Coya, T. Rojo, L. M. Rodriguez-Martinez, M. Armand and Z. Zhou, Chem. Soc. Rev., 2017, 46, 797–815 RSC.
- S. Li, S. Zhang, L. Shen, Q. Liu, J. Ma, W. Lv, Y. He and Q. Yang, Adv. Sci., 2020, 7, 1903088 CrossRef CAS.
- S. Wang, X. Zhang, S. Liu, C. Xin, C. Xue, F. Richter, L. Li, L. Fan, Y. Lin, Y. Shen, J. Janek and C.-W. Nan, J Materiomics, 2020, 6, 70–76 CrossRef.
- Y. Zheng, Y. Yao, J. Ou, M. Li, D. Luo, H. Dou, Z. Li, K. Amine, A. Yu and Z. Chen, Chem. Soc. Rev., 2020, 49, 8790–8839 RSC.
- Z. Zhang, Y. Shao, B. Lotsch, Y.-S. Hu, H. Li, J. Janek, L. F. Nazar, C.-W. Nan, J. Maier, M. Armand and L. Chen, Energy Environ. Sci., 2018, 11, 1945–1976 RSC.
- S. Ohno, A. Banik, G. F. Dewald, M. A. Kraft, T. Krauskopf, N. Minafra, P. Till, M. Weiss and W. G. Zeier, Prog Energy, 2020, 2, 022001 CrossRef.
- N. Kamaya, K. Homma, Y. Yamakawa, M. Hirayama, R. Kanno, M. Yonemura, T. Kamiyama, Y. Kato, S. Hama, K. Kawamoto and A. Mitsui, Nat. Mater., 2011, 10, 682–686 CrossRef CAS.
- Y. Kato, S. Hori, T. Saito, K. Suzuki, M. Hirayama, A. Mitsui, M. Yonemura, I. Hideki and R. Kanno, Nat. Energy, 2016, 1, 16030 CrossRef CAS.
- N. M. Vargas-Barbosa and B. Roling, ChemElectroChem, 2020, 7, 367–385 CrossRef.
- S. Randau, D. A. Weber, O. Kötz, R. Koerver, P. Braun, A. Weber, E. Ivers-Tiffée, T. Adermann, J. Kulisch, W. G. Zeier, F. H. Richter and J. Janek, Nat. Energy, 2020, 5, 259–270 CrossRef.
- C. Xin-Bing, Z. Rui, Z. Chen-Zi and Z. Qiang, Chem. Rev., 2017, 117, 10403–10473 CrossRef.
- T. Placke, R. Kloepsch, S. Dühnen and M. Winter, J. Solid State Electrochem., 2017, 21, 1939–1964 CrossRef.
- B. Liu, J.-G. Zhang and W. Xu, Joule, 2018, 2, 833–845 CrossRef.
- T. Krauskopf, F. H. Richter, W. G. Zeier and J. Janek, Chem. Rev., 2020, 120, 7745–7794 CrossRef.
- M. B. Dixit, N. Singh, J. P. Horwath, P. D. Shevchenko, M. Jones, E. A. Stach, T. S. Arthur and K. B. Hatzell, Matter, 2020, 3, 2138–2159 CrossRef.
- D. K. Singh, A. Henss, B. Mogwitz, A. Gautam, J. Horn, T. Krauskopf, S. Burkhardt, J. Sann, F. H. Richter and J. Janek, Cell Rep. Phys. Sci., 2022, 101043 CrossRef CAS.
- L. Xu, T. Feng, J. Huang, Y. Hu, L. Zhang and L. Luo, ACS Appl. Energy Mater., 2022, 5, 3741–3747 CrossRef CAS.
- K. Park, B.-C. Yu, J.-W. Jung, Y. Li, W. Zhou, H. Gao, S. Son and J. B. Goodenough, Chem. Mater., 2016, 28, 8051–8059 CrossRef CAS.
- H. Fudong, Y. Jie, C. Cheng, Z. Ning, F. Xiulin, M. Zhaohui, G. Tao, W. Fei, G. Xiangxin and W. Chunsheng, Joule, 2018, 2, 497–508 CrossRef.
- S. A. Pervez, M. A. Cambaz, V. Thangadurai and M. Fichtner, ACS Appl. Mater. Interfaces, 2019, 11, 22029–22050 CrossRef CAS.
- G. F. Dewald, S. Ohno, M. A. Kraft, R. Koerver, P. Till, N. M. Vargas-Barbosa, J. Janek and W. G. Zeier, Chem. Mater., 2019, 31, 8328–8337 CrossRef CAS.
- T. Thompson, S. Yu, L. Williams, R. D. Schmidt, R. Garcia-Mendez, J. Wolfenstine, J. L. Allen, E. Kioupakis, D. J. Siegel and J. Sakamoto, ACS Energy Lett., 2017, 2, 462–468 CrossRef CAS.
- T. K. Schwietert, V. A. Arszelewska, C. Wang, C. Yu, A. Vasileiadis, N. J. J. de Klerk, J. Hageman, T. Hupfer, I. Kerkamm, Y. Xu, E. van der Maas, E. M. Kelder, S. Ganapathy and M. Wagemaker, Nat. Mater., 2020, 19, 428–435 CrossRef.
- B.-T. Yu, W.-H. Qiu, F.-S. Li and L. Cheng, J. Power Sources, 2007, 166, 499–502 CrossRef CAS.
- C. F. N. Marchiori, R. P. Carvalho, M. Ebadi, D. Brandell and C. M. Araujo, Chem. Mater., 2020, 32, 7237–7246 CrossRef CAS.
- Y. Tian, T. Shi, W. D. Richards, J. Li, J. C. Kim, S.-H. Bo and G. Ceder, Energy Environ. Sci., 2017, 10, 1150–1166 RSC.
- Y. Zhu, X. He and Y. Mo, J. Mater. Chem. A, 2016, 4, 3253–3266 RSC.
- Y. Xiao, Y. Wang, S.-H. Bo, J. C. Kim, L. J. Miara and G. Ceder, Nat. Rev. Mater., 2020, 5, 105–126 CrossRef CAS.
- Solid Power's High Energy, Automotive-Scale All Solid-State Batteries Surpass Commercial Lithium-Ion Energy Densities, https://www.prnewswire.com/news-releases/solid-powers-high-energy-automotive-scale-all-solid-state-batteries-surpass-commercial-lithium-ion-energy-densities-301190450.html?tc=eml_cleartime, (accessed March 18, 2022) Search PubMed.
- QuantumScape, QuantumScape Releases Performance Data for its Solid-State Battery Technology, https://cleantechnica.com/2020/12/08/quantumscape-releases-performance-data-on-its-solid-state-battery-technology/, (accessed March 14, 2022) Search PubMed.
- Z. Chen, N. Donnelly, T. Holme and D. Singh, US Pat., US2017/0331092A1, QuantumScape Corporation, San Jose, CA, US, 2017.
- C.-C. Chao, Z. Chen, T. Holme, M. A. Mayer and G. N. Riley Jr, US Pat., US10535878B2, QuantumScape Corporation, San Jose, CA (US), 2020.
- M. Weiss, F. J. Simon, M. R. Busche, T. Nakamura, D. Schröder, F. H. Richter and J. Janek, Electrochem. Energy Rev., 2020, 3, 221–238 CrossRef.
- A. J. Butzelaar, P. Röring, T. P. Mach, M. Hoffmann, F. Jeschull, M. Wilhelm, M. Winter, G. Brunklaus and P. Théato, ACS Appl. Mater. Interfaces, 2021, 13, 39257–39270 CrossRef CAS.
- L. Imholt, T. S. Dörr, P. Zhang, L. Ibing, I. Cekic-Laskovic, M. Winter and G. Brunklaus, J. Power Sources, 2019, 409, 148–158 CrossRef.
- J. Popovic, D. Brandell, S. Ohno, K. B. Hatzell, J. Zheng and Y.-Y. Hu, J. Mater. Chem. A, 2021, 9, 6050–6069 RSC.
- L. R. Mangani and C. Villevieille, J. Mater. Chem. A, 2020, 8, 10150–10167 RSC.
- M. Keller, A. Varzi and S. Passerini, J. Power Sources, 2018, 392, 206–225 CrossRef.
- S. Tang, W. Guo and Y. Fu, Adv. Energy Mater., 2021, 11, 2000802 CrossRef.
- T. Zhang, W. He, W. Zhang, T. Wang, P. Li, Z. Sun and X. Yu, Chem. Sci., 2020, 11, 8686–8707 RSC.
- A. Banerjee, X. Wang, C. Fang, E. A. Wu and Y. S. Meng, Chem. Rev., 2020, 120, 6878–6933 CrossRef CAS PubMed.
- T. Abe, F. Sagane, M. Ohtsuka, Y. Iriyama and Z. Ogumi, J. Electrochem. Soc., 2005, 152, A2151 CrossRef.
- E. Peled and S. Menkin, J. Electrochem. Soc., 2017, 164, A1703–A1719 CrossRef CAS.
- E. Peled, J. Electrochem. Soc., 1979, 126, 2047–2051 CrossRef CAS.
- X. Cheng, R. Zhang, C. Zhao, F. Wei, J. Zhang and Q. Zhang, Adv. Sci., 2016, 3, 1500213 CrossRef PubMed.
- X. Yu and A. Manthiram, Energy Environ. Sci., 2018, 11, 527–543 RSC.
- Z. Shadike, S. Tan, R. Lin, X. Cao, E. Hu and X.-Q. Yang, Chem. Sci., 2021, 13, 1547–1568 RSC.
- S. P. Kühn, K. Edström, M. Winter and I. Cekic-Laskovic, Adv. Mater. Interfaces, 2022, 9, 2102078 CrossRef.
- N. Takenaka, A. Bouibes, Y. Yamada, M. Nagaoka and A. Yamada, Adv. Mater., 2021, 33, 2100574 CrossRef CAS.
- S. K. Heiskanen, J. Kim and B. L. Lucht, Joule, 2019, 3, 2322–2333 CrossRef CAS.
- M. Liu, S. Zhang, E. R. H. van Eck, C. Wang, S. Ganapathy and M. Wagemaker, Nat. Nanotechnol., 2022, 1–9 Search PubMed.
- M. Nojabaee, D. Kopljar, N. Wagner and K. A. Friedrich, Batteries Supercaps, 2021, 4, 909–922 CrossRef CAS.
- M. R. Busche, T. Drossel, T. Leichtweiss, D. A. Weber, M. Falk, M. Schneider, M.-L. Reich, H. Sommer, P. Adelhelm and J. Janek, Nat. Chem., 2016, 8, 426–434 CrossRef CAS.
- L. Jingyuan, G. Xiangwen, H. O. Gareth, R. J. Gregory, G. Chen, R. H. Felix, J. Jürgen, X. Yongyao, R. W. Alex, J. R. Lee and B. G. Peter, Joule, 2020, 4, 101–108 CrossRef.
- M. Schleutker, J. Bahner, C.-L. Tsai, D. Stolten and C. Korte, Phys. Chem. Chem. Phys., 2017, 19, 26596–26605 RSC.
- R. Ye, M. Ihrig, N. Imanishi, M. Finsterbusch and E. Figgemeier, Chemsuschem, 2021, 14, 4397–4407 CrossRef CAS.
- Z. F. Yow, Y. L. Oh, W. Gu, R. P. Rao and S. Adams, Solid State Ionics, 2016, 292, 122–129 CrossRef CAS.
- T. Kawamura, S. Okada and J. Yamaki, J. Power Sources, 2006, 156, 547–554 CrossRef CAS.
- J. Liu, X. Gao, G. O. Hartley, G. J. Rees, C. Gong, F. H. Richter, J. Janek, Y. Xia, A. W. Robertson, L. R. Johnson and P. G. Bruce, Joule, 2019, 4, 101–108 CrossRef.
- M. Leißing, C. Peschel, F. Horsthemke, S. Wiemers-Meyer, M. Winter and S. Nowak, Batteries Supercaps, 2021, 4, 1731–1738 CrossRef.
- B. Xu, H. Duan, H. Liu, C. Wang and S. Zhong, ACS Appl. Mater. Interfaces, 2017, 9, 21077–21082 CrossRef.
- J. P. Vivek, N. Meddings and N. Garcia-Araez, ACS Appl. Mater. Interfaces, 2022, 14, 633–646 CrossRef.
- A. Gupta, E. Kazyak, N. P. Dasgupta and J. Sakamoto, J. Power Sources, 2020, 474, 228598 CrossRef.
- Y. Chen, W. Li, C. Sun, J. Jin, Q. Wang, X. Chen, W. Zha and Z. Wen, Adv. Energy Mater., 2021, 11, 2002545 CrossRef.
- B. Fan, Y. Xu, R. Ma, Z. Luo, F. Wang, X. Zhang, H. Ma, P. Fan, B. Xue and W. Han, ACS Appl. Mater. Interfaces, 2020, 12, 52845–52856 CrossRef CAS PubMed.
- M. R. Busche, M. Weiss, T. Leichtweiss, C. Fiedler, T. Drossel, M. Geiss, A. Kronenberger, D. A. Weber and J. Janek, Adv. Mater. Interfaces, 2020, 7, 2000380 CrossRef CAS.
- D. Y. Oh, Y. J. Nam, K. H. Park, S. H. Jung, S. Cho, Y. K. Kim, Y. Lee, S. Lee and Y. S. Jung, Adv. Energy Mater., 2015, 5, 1500865 CrossRef.
- D. T. Boyle, X. Kong, A. Pei, P. E. Rudnicki, F. Shi, W. Huang, Z. Bao, J. Qin and Y. Cui, ACS Energy Lett., 2020, 5, 701–709 CrossRef CAS.
- R. Kurchin and V. Viswanathan, J. Chem. Phys., 2020, 153, 134706 CrossRef CAS.
- S. Sripad, D. Korff, S. C. DeCaluwe and V. Viswanathan, J. Chem. Phys., 2020, 153, 194701 CrossRef CAS.
- M. Weiss, B.-K. Seidlhofer, M. Geiß, C. Geis, M. R. Busche, M. Becker, N. M. Vargas-Barbosa, L. Silvi, W. G. Zeier, D. Schröder and J. Janek, ACS Appl. Mater. Interfaces, 2019, 11, 9539–9547 CrossRef CAS.
- T. Kranz, S. Kranz, V. Miß, J. Schepp and B. Roling, J. Electrochem. Soc., 2017, 164, A3777–A3784 CrossRef CAS.
- F. T. Krauss, I. Pantenburg and B. Roling, Adv. Mater. Interfaces, 2022, 2101891 CrossRef CAS.
- S. Kranz, T. Kranz, T. Graubner, Y. Yusim, L. Hellweg and B. Roling, Batteries Supercaps, 2019, 2, 1026–1036 CrossRef CAS.
- A. M. Bates, Y. Preger, L. Torres-Castro, K. L. Harrison, S. J. Harris and J. Hewson, Joule, 2022, 6, 742–755 CrossRef CAS.
- P. Bonnick, K. Niitani, M. Nose, K. Suto, T. S. Arthur and J. Muldoon, J. Mater. Chem. A, 2019, 7, 24173–24179 RSC.
- M. A. Kraft, S. Ohno, T. Zinkevich, R. Koerver, S. P. Culver, T. Fuchs, A. Senyshyn, S. Indris, B. J. Morgan and W. G. Zeier, J. Am. Chem. Soc., 2018, 140, 16330–16339 CrossRef CAS.
- A. Neumann, S. Randau, K. Becker-Steinberger, T. Danner, S. Hein, Z. Ning, J. Marrow, F. H. Richter, J. Janek and A. Latz, ACS Appl. Mater. Interfaces, 2020, 12, 9277–9291 CrossRef CAS.
- R. Koerver, I. Aygün, T. Leichtweiß, C. Dietrich, W. Zhang, J. O. Binder, P. Hartmann, W. G. Zeier and J. Janek, Chem. Mater., 2017, 29, 5574–5582 CrossRef CAS.
- W. Zhang, F. H. Richter, S. P. Culver, T. Leichtweiss, J. G. Lozano, C. Dietrich, P. G. Bruce, W. G. Zeier and J. Janek, ACS Appl. Mater. Interfaces, 2018, 10, 22226–22236 CrossRef CAS PubMed.
- W. D. Richards, L. J. Miara, Y. Wang, J. C. Kim and G. Ceder, Chem. Mater., 2016, 28, 266–273 CrossRef CAS.
- F. Han, Y. Zhu, X. He, Y. Mo and C. Wang, Adv. Energy Mater., 2016, 6, 1501590 CrossRef.
- A. Manthiram, Nat. Commun., 2020, 11, 1550 CrossRef CAS.
- X. Li, H. Guan, Z. Ma, M. Liang, D. Song, H. Zhang, X. Shi, C. Li, L. Jiao and L. Zhang, J. Energy Chem., 2020, 48, 195–202 CrossRef.
- Y. J. Nam, S.-J. Cho, D. Y. Oh, J.-M. Lim, S. Y. Kim, J. H. Song, Y.-G. Lee, S.-Y. Lee and Y. S. Jung, Nano Lett., 2015, 15, 3317–3323 CrossRef PubMed.
- D. Cao, Y. Zhao, X. Sun, A. Natan, Y. Wang, P. Xiang, W. Wang and H. Zhu, ACS Energy Lett., 2020, 5, 3468–3489 CrossRef.
- V. Wenzel, H. Nirschl and D. Nötzel, Energy Technol., 2015, 3, 692–698 CrossRef.
- V. Gutmann and V. Gutmann, The Donor-Acceptor Approach to Molecular Interactions, Springer, 1st edn, 1978, vol. 228 Search PubMed.
- K. Lee, S. Kim, J. Park, S. H. Park, A. Coskun, D. S. Jung, W. Cho and J. W. Choi, J. Electrochem. Soc., 2017, 164, A2075–A2081 CrossRef.
- M. Yamamoto, Y. Terauchi, A. Sakuda and M. Takahashi, Sci. Rep., 2018, 8, 1212 CrossRef PubMed.
- J. Ruhl, L. M. Riegger, M. Ghidiu and W. G. Zeier, Adv. Energy Sustain. Res., 2021, 2, 2000077 CrossRef.
- A.-K. Hatz, R. Calaminus, J. Feijoo, F. Treber, J. Blahusch, T. Lenz, M. Reichel, K. Karaghiosoff, N. M. Vargas-Barbosa and B. V. Lotsch, ACS Appl. Energy Mater., 2021, 4, 9932–9943 CrossRef CAS.
- A. Bielefeld, D. A. Weber and J. Janek, ACS Appl. Mater. Interfaces, 2020, 12, 12821–12833 CrossRef CAS.
- K. Suzuki, A. Yageta, Y. Ikeda, N. Mashimo, S. Hori, M. Hirayama and R. Kanno, Chem. Lett., 2020, 49, 1379–1381 CrossRef CAS.
- S. Yubuchi, M. Uematsu, C. Hotehama, A. Sakuda, A. Hayashi and M. Tatsumisago, J. Mater. Chem. A, 2018, 7, 558–566 RSC.
- S. Harm, A.-K. Hatz, I. Moudrakovski, R. Eger, A. Kuhn, C. Hoch and B. V. Lotsch, Chem. Mater., 2019, 31, 1280–1288 CrossRef CAS.
- M. A. Philip, P. T. Sullivan, R. Zhang, G. A. Wooley, S. A. Kohn and A. A. Gewirth, ACS Appl. Mater. Interfaces, 2019, 11, 2014–2021 CrossRef CAS PubMed.
- Y. Zhao, T. Zhou, M. E. Kazzi and A. Coskun, ACS Appl. Energy Mater., 2022, 5, 7784–7790 CrossRef CAS.
- N. Azimi, Z. Xue, N. D. Rago, C. Takoudis, M. L. Gordin, J. Song, D. Wang and Z. Zhang, J. Electrochem. Soc., 2015, 162, A64–A68 CrossRef CAS.
- N. Xu, J. Shi, G. Liu, X. Yang, J. Zheng, Z. Zhang and Y. Yang, J. Power Sources Adv., 2021, 7, 100043 CrossRef CAS.
- B. Fan, W. Li, Z. Luo, X. Zhang, H. Ma, P. Fan and B. Xue, ACS Appl. Mater. Interfaces, 2022, 14, 933–942 CrossRef CAS.
- E. Umeshbabu, B. Zheng, J. Zhu, H. Wang, Y. Li and Y. Yang, ACS Appl. Mater. Interfaces, 2019, 11, 18436–18447 CrossRef CAS.
- B. Zheng, J. Zhu, H. Wang, M. Feng, E. Umeshbabu, Y. Li, Q.-H. Wu and Y. Yang, ACS Appl. Mater. Interfaces, 2018, 10, 25473–25482 CrossRef CAS.
- P. Verma, P. Maire and P. Novák, Electrochim. Acta, 2010, 55, 6332–6341 CrossRef CAS.
- A. Banik, T. Famprikis, M. Ghidiu, S. Ohno, M. A. Kraft and W. G. Zeier, Chem. Sci., 2021, 12, 6238–6263 RSC.
- S. Ohno, T. Bernges, J. Buchheim, M. Duchardt, A.-K. Hatz, M. A. Kraft, H. Kwak, A. L. Santhosha, Z. Liu, N. Minafra, F. Tsuji, A. Sakuda, R. Schlem, S. Xiong, Z. Zhang, P. Adelhelm, H. Chen, A. Hayashi, Y. S. Jung, B. V. Lotsch, B. Roling, N. M. Vargas-Barbosa and W. G. Zeier, ACS Energy Lett., 2020, 5, 910–915 CrossRef CAS.
- M. Cronau, M. Szabo, C. König, T. B. Wassermann and B. Roling, ACS Energy Lett., 2021, 6, 3072–3077 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, 2nd edn, 2001. ch. 13.7 Search PubMed.
- N. J. J. de Klerk and M. Wagemaker, ACS Appl. Energy Mater., 2018, 1, 5609–5618 Search PubMed.
- T. Fuchs, B. Mogwitz, S. Otto, S. Passerini, F. H. Richter and J. Janek, Batteries Supercaps, 2021, 4, 1145–1155 CrossRef.
- M. Watanabe, M. L. Thomas, S. Zhang, K. Ueno, T. Yasuda and K. Dokko, Chem. Rev., 2017, 117, 7190–7239 CrossRef PubMed.
- E. J. Cheng, M. Shoji, T. Abe and K. Kanamura, iScience, 2022, 25, 103896 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Tables with the values used for the plots used to generate Fig. 3 and 4, plot of the ratio of ionic conductivities as a function of the relative polarity of the solvent, equations for the estimation of the energy density and round-trip efficiency loss calculations. See DOI: https://doi.org/10.1039/d2ta02179j |
| This journal is © The Royal Society of Chemistry 2023 |