
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The effects of lignin source and extraction on the composition and properties of biorefined depolymerization products†
Natalia
Obrzut
 ,
Rob
Hickmott
,
Lily
Shure
and
Kimberly A.
Gray
*
,
Rob
Hickmott
,
Lily
Shure
and
Kimberly A.
Gray
*
Department of Civil and Environmental Engineering, Northwestern University, 2145 Sheridan Road, Evanston, Illinois 60208, USA. E-mail: k-gray@northwestern.edu
First published on 27th October 2023
Abstract
We explore the efficacy of depolymerization as a function of lignin source and extraction method under the conditions of our previously described biorefinery. Biomass source (i.e. herbaceous, softwood, and hardwood) influences the total available lignin and structural features such as the ratio of monomers (S/G) and the percentage of β-O-4 linkage. The method of extraction (i.e. Milled Wood (mild), Organosolv (medium), and Klason (harsh)) determines levels of solubilization, preservation of the intrinsic lignin structure, and distribution and properties of products. Herbaceous lignin extracted by the Organosolv process is best suited for depolymerization in our biorefinery and shows the greatest extent of solubilization (∼100% over 7 days), the highest yields of phenolics and flavonoids, and the most and smallest lignin nanoparticles. Additionally, this depolymerized product mixture has the highest antioxidant capacity. Only the harsh, Klason method successfully isolates lignin, through carbohydrate conversion, from all three sources, but the herbaceous and hardwood sources are depolymerized to a lesser extent than by the Organosolv method. With low density of labile bonds, softwood lignin is not effectively depolymerized under these conditions. Although modifying intrinsic lignin structure the least, Milled Wood lignin does not efficiently extract lignin from any of the biomass, and the extracted lignin is low in purity inhibiting further processing. These results further detail the promise of herbaceous Organosolv lignin as a renewable feedstock to high value products in a distributed-scale biorefinery.
Sustainability spotlightLignin is the only source of renewable aromatic carbon on the planet. Yet, its efficient conversion into valuable products is hindered by limited understanding of how differences in the chemical structure of native and extracted lignin affects downstream depolymerization. The aim of this work is to determine how lignin source and extraction affect the product composition and properties when depolymerized in a biorefinery utilizing the high salt and caustic effluent that is produced via a microbial electrolysis cell. Products such as phenolics, flavonoids, and nanoparticles with high antioxidant capacity have high commercial value in the pharmaceutical, nutraceutical, and cosmetic industries. Our work emphasizes the importance of the following UN sustainable development goal: Responsible Consumption and Production (SDG 12). |
Introduction
Lignin is an amorphous, aromatic, 3D biopolymer composed of three monomeric units: coniferyl alcohol (G), p-coumaryl alcohol (H), and sinapyl alcohol (S).1,2 It plays an essential structural and functional role in plants and is the largest store of renewable aromatic carbon on earth. Despite the potential chemical value of the aromatic subunits, lignocellulosic biomass is rarely processed commercially for products other than low-value combustion fuels.3 This is due, in part, to the technical challenges associated with depolymerizing this complex polymer.The ratio of monolignols varies with plant species and determines the degree of cross-linking and ease of depolymerization. Generally, hardwood lignin, such as birch and poplar, are composed of S and G monolignols, whereas softwood lignin, such as pine and spruce, are composed exclusively of G monomers. Herbaceous lignin found in switchgrass and corn stover, for example, typically contains all three monolignols, but the H content is low (below 5%).2,4 The amount of lignin varies between 10 and 35% by plant species with softwood typically containing the highest percentage of lignin and herbaceous containing the lowest percentage of lignin.1,2 The monolignols are bonded through 14 unique linkages, the most common being the β-O-4 linkage.5 The bond dissociation energy (BDE) of the β-O-4 linkage is between 221 and 295 kJ mol−1 making it easier to break compared to the 5–5 (carbon–carbon) bond with nearly double the BDE.6,7 The abundance of the β-O-4 bond, as well as the relatively low BDE, makes this bond a common target of depolymerization processes. It has been shown previously that there is a strong relationship between the number of β-O-4 linkages and the theoretical monomer yield from depolymerization.8,9
Lignin extraction or fractionation from biomass has a significant effect on its structure. Extracted lignin is often referred to as technical lignin. Some methods produce a technical lignin that conserves the natural (native) structure of lignin including milled-wood lignin (Bjorkman process), ionic liquid lignin and cellulolytic enzyme lignin.2,7 The most industrially common method of extracting lignin without severe structural modification is the Organosolv method, which is used to separate cellulose for second generation bioethanol production, producing the second largest commercially available lignin stream.10 The Kraft process, employed in paper production, is the largest commercial lignin stream comprising 85% of total technical lignin produced in the world.11,12 The Kraft process does not aim to preserve the β-O-4 linkages, resulting in lignin that varies immensely from its native structure and is highly condensed and difficult to use as the feedstock in a downstream biorefinery.9
Biorefinery research primarily focuses on “lignin first fractionation,” the goal of which is to preserve the structure of native lignin because of the difficulties working with some forms of technical lignin and the lack of β-O-4 linkages.13 Research in this area centers on: (1) analytical methods to determine and preserve the structure of native lignin,14–19 (2) genetic modification to allow easier separation and more structural preservation of lignin,20,21 (3) stabilization of the low molecular weight lignin to avoid repolymerization,22 and (4) determination of fractionation methods to better conserve the structure of native lignin.9,23–29 Aldehyde-assisted fractionation, for instance, avoids condensation through the extraction process and simultaneously functionalizes the lignin.30 Another method shows that lignin can be extracted through a flow-through solvolysis in a way that conserves aryl-ether bonds and allows lignin to be stored and processed at a later time without losing reactivity.31 A popular lignin-first method is reductive catalytic fractionation (RCF). This method cleaves ether linkages into aromatic compounds and generally avoids recondensation reactions. This method is often conducted over noble metal catalysts, non-precious metal catalysts, or novel ruthenium catalysts supported on carbon nanotubes.32
We previously demonstrated a proof-of-concept biorefinery to depolymerize lignin into high value products. In this biorefinery we coupled a microbial electrolysis cell (MEC) that oxidizes the organic content of wastewater (as modeled by acetate) at the anode reducing BOD by 60% and producing a high pH, high salt effluent at the cathode which is then used to depolymerize an extracted lignin stream.33 Under the ambient temperature and pressure conditions of our biorefinery, base catalyzed depolymerization of an Organosolv herbaceous (corn stover) lignin preserves the aromatic structure to yield discrete aromatics such as monomers (e.g., vanillin, p-coumaric acid) and flavonoids (e.g., tricin) at approximately 20% of the initial lignin mass and lignin nanoparticles (LNPs) to close mass balance. These products have wide potential commercial application in the food, pharmaceutical, nutraceutical, personal care, and agricultural industries.
Since the native structure of lignin varies as a function of its source and is further modified by the method of extraction, it is unclear how the biorefining of lignin is altered as a function of these factors. The purpose of this research, then, is to investigate how lignin source, particularly lignin content, bond type, and monolignol composition, and extraction influence our biorefinery's performance as characterized by the composition and properties of the depolymerized product mixture. These types of studies have been performed before utilizing other methods of depolymerization, such as RCF, which produces mostly monomeric compounds.32,34 Specifically, we compare three extraction methods (Milled Wood (mild), Organosolv (medium), and Klason (harsh) and study the products: phenolic content, flavonoid content, and lignin nanoparticles, and the properties of the product mixture including: shape, size, stability, polydispersity, and the antioxidant capacity.
Experimental
Materials
Sources of biomass include corn stover (herbaceous), white spruce (softwood), and bitternut hickory (hardwood) which were donated by the author's family from Cadott, Wisconsin. Archer Daniels Midland (ADM), Decatur IL kindly donated lignin extracted from corn stover via the acetosolv process (Organosolv using acetic acid). Other materials used were acetone (HPLC plus, Sigma Aldrich), sulfuric acid (certified ACS plus, Fischer), DMSO (Dimethyl sulfoxide) (certified ACS, Fischer), NMI (N-methyl Imidazole) (97%, Aldrich), sodium phosphate dibasic (Fischer), potassium phosphate monobasic (USP analytical test, JT Baker), Folin–Ciolcalteu reagent (Sigma Aldrich), sodium carbonate (ACS, VWR), gallic acid (97.5%, Sigma Aldrich), aluminum chloride hexahydrate (Alfa Aesar), sodium nitrite (98%, Alfa Aesar), sodium hydroxide (certified ACS, Fischer), phosphoric acid, rutin (>94%, TCI), Trolox (abcam), ABTS (TCI), potassium persulfate (certified ACS, Fischer), DMSO-d6 (99.9%, ACROS organics), 1,4 dioxane (>99.5%, Honeywell), Methanol (HPLC grade, Fisher), hexadecyltrimethylammonium bromide (CTAB) (>98%, Sigma Aldrich), acetic acid (ACS, EMD), potassium bromide (Spectograde, International Crystal Labs).Lignin source and extractions
Lignin was extracted from corn stover via three methods: Milled Wood (delignification), Organosolv (ADM) (delignification) and Klason (carbohydrate conversion); lignin from softwood and hardwood was isolated and quantified via the Klason method. Prior to extraction, biomass was prepared for extraction using methods: TAPPI T 257 cm-85 (2000) – “Sampling and Preparing Wood for Analysis”, and TAPPI T 264 cm-97 – “Preparation of Wood for Chemical Analysis”. The white spruce and bitternut hickory samples were acquired as 100 g blocks. They were turned into saw dust with a power saw. The saw dust was sifted through a 40-mesh screen. The corn stover was dried over night at 40 °C. It was then ground in an electric grinder and sifted through a 40-mesh screen. All samples were purified through a Soxhlet extraction. The fine material was placed in a glass extraction thimble (∼20 g), the extraction thimble was placed in position in the Soxhlet apparatus. 200 mL of solvent (95 acetone: 5 water v:v) was placed in a 250 mL extraction flask. The extraction ran for 6–8 hours with the liquid boiling briskly. After the extraction the sample was washed with boiling DI water. The sample was filtered with Whatman No. 5 paper and dried in the oven at 105 °C overnight.Lignin was extracted via the Milled Wood process according to the Bjorkman method, which involves milling biomass in a planetary ball mill and extracting lignin with an organic solvent.35 The milling was done using a SPEX 8000D Mixer/Mill equipped with a 50 mL ZrO2 grinding jar and 10 × 10 mm ball bearing set at 600 r.p.m. The milling protocol for the corn stover was 5 minute runs, 5 times, with 5 minute pauses in between for a total of 45 minutes.36 The milled corn stover was dispersed in dioxane: water (96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4, v:v) and magnetically stirred for 24 hours.37 After 24 hours the suspension was centrifuged, the supernatant was saved and the solids were resuspended in fresh dioxane:water for 24 more hours. The mixture was centrifuged and the supernatants from days 1 and 2 were combined and freeze dried (Labconco −84C 4.5L PTFE coated benchtop freeze dryer) to produce Milled Wood lignin (MWL).
:4, v:v) and magnetically stirred for 24 hours.37 After 24 hours the suspension was centrifuged, the supernatant was saved and the solids were resuspended in fresh dioxane:water for 24 more hours. The mixture was centrifuged and the supernatants from days 1 and 2 were combined and freeze dried (Labconco −84C 4.5L PTFE coated benchtop freeze dryer) to produce Milled Wood lignin (MWL).
Acid insoluble lignin was separated from carbohydrates via the Klason method for the herbaceous, hardwood, and softwood sources in accordance to TAPPI T 222 om-98 – acid insoluble lignin in wood and pulp. In a beaker, 1 g of biomass was stirred with 15 mL of 72% sulfuric acid for 2 hours. After 2 hours, the material from the beaker was transferred into a flask with 300–400 mL DI water and more water was added to a final volume of 575 mL. The solution was boiled for 4 hours, frequently adding hot water to keep the volume constant. After four hours the material was allowed to settle overnight. The supernatant was decanted off. The lignin was washed with hot water, filtered and left to dry over night at 105 °C. The total amount of acid insoluble (Klason) lignin in the biomass was calculated according to:
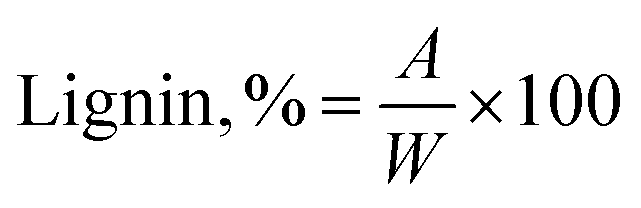
Lignin depolymerization experiments
Lignin depolymerization was carried out as described in Obrzut et al.33 Synthetic MEC catholyte was used for the experiments and prepared with a 227 mM phosphate solution (0.2 g L−1 KH2PO4·H2O and 32 g L−1 Na2HPO4) adjusted with sodium hydroxide to a final pH of 10.8. Lignin (250 mg) was combined with the phosphate solution (250 mL) in a beaker under magnetic stirring. The beakers were wrapped in aluminum foil to avoid light exposure. Two types of experiments were run; a basic set of experiments following the above protocol and a neutral set of experiments following the above protocol except the pH was adjusted to 7 with dilute phosphoric acid after 1 hour (7 mL of 10× diluted phosphoric acid). The latter experiment had previously been more successful in producing a higher yield of LNPs. Both methods had produced similar yields of phenolics and flavonoids. The experiments ran for 14 days, with 50 mL samples taken out at 60 minutes (0 days), 1 day, 4 days, 7 days, and 14 days for characterization. Each set, basic and neutral, was run in duplicate. Table 1 summarizes the source, extraction, pH, time, and name of each experiment. In bold and underlined, we show at which time each experiment reached maximum solubilization. These are the data that will be reported in Fig. 3–6. We chose maximum solubilization over the 14 day period for these values because: (1) the day of maximum solubilization corresponded with the minimum IC50 (maximum antioxidant capacity), and (2) it corresponds with a lower residual standard error than if we chose the 14 day average since both the solubilization and antioxidant capacity increase significantly over the 14 day period (solubilization under basic conditions increased 2–5 fold over 14 days, antioxidant capacity increased similarly). Tables S1 and S2† compare product and property values over a 14 days average, at maximum solubilization and maximum antioxidant capacity.| Source | Extraction | pH | Time (# days) | Experiment label |
|---|---|---|---|---|
| Herbaceous | MWL | Basic (10.8) | 0, 7 | MWL-B |
| Herbaceous | MWL | Neutral (7.0) | 0, 7 | MWL-N |
| Herbaceous | Organosolv | Basic (10.8) | 0, 1, 4, 7, 14 | HO–B |
| Herbaceous | Organosolv | Neutral (7.0) | 0, 1, 4, 7, 14 | HO–N |
| Herbaceous | Klason | Basic (10.8) | 0, 1, 4, 7, 14 | HK-B |
| Herbaceous | Klason | Neutral (7.0) | 0, 1, 4, 7, 14 | HK-N |
| Softwood | Klason | Basic (10.8) | 0, 1, 4, 7, 14 | SK-B |
| Softwood | Klason | Neutral (7.0) | 0, 1, 4, 7, 14 | SK-N |
| Hardwood | Klason | Basic (10.8) | 0, 1, 4, 7, 14 | HaK-B |
| Hardwood | Klason | Neutral (7.0) | 0, 1, 4, 7, 14 | HaK-N |
Product characterization
The initial biomass and lignin extractions were characterized by NMR and FTIR to determine the S/G ratio and β-O-4 content (NMR) and to look at changes to functional groups and aromaticity (diffuse reflectance infrared Fourier transform, DRIFT). For NMR the samples were acetylated. We followed a modified acetylation protocol by Mansfield et al.36 For both the fine, extractive free biomass, and for the extracted lignin, 100 mg of the sample were suspended in 2 mL of DMSO. An additional 1 mL of NMI was added, and the solution was stirred constantly with a magnetic stirrer for 24 hours. Then 0.5 mL of acetic anhydride was added, and the solution was stirred for another 24 hours. The solution was then transferred to 300 mL of deionized water and allowed to settle overnight. Most of the supernatant was decanted and the solution was centrifuged for 10 minutes at room temperature at 3480 rpm. The precipitate was washed with 50 mL of water, 3 times. The sample was then dried overnight at 40 °C. Once the sample was dry, 30–50 mg of acetylated biomass or lignin was dissolved in 0.5 mL of deuterated DMSO-d6 and the solution was transferred to an NMR tube (Kimble, 5 MM, 7′′, 400 MHZ). The samples were run on a Bruker Neo 600 MHz system with QCI-F cryoprobe. The software used was TopSpin 4.04. The Bruker Pulse Program for the HSQC spectra was “hsqcetgpsisp2.2”. The number of scans (NS) was set to give a total acquisition time of 3 hours for biomass and 30 minutes for lignin. DRIFT spectroscopy (Bruker Tensor 37) was run on initial biomass and lignin (diluted to 4% in KBr).The lignin depolymerization slurry samples were characterized with the following analyses: pH, total solubilization, total phenolic content, total flavonoid content, antioxidant capacity, lignin nanoparticle (LNP) concentration, LNP size, polydispersity index (PDI), and zeta potential. These characterizations were conducted on centrifuged and non-centrifuged samples. Unless indicated with a “C”, results are for the non-centrifuged samples. Centrifuging was done on 25 mL in 50 mL falcon tubes for 10 minutes at 7000 r.p.m. The pH was measured with a pH-meter (Mettler Toledo SevenCompact S210).
We determined the total solubilized lignin, total phenolic content, and total flavonoid content by measuring the UV absorbance (Eppendorf BioSpectrometer). Total solubilized lignin is defined as lignin that can be measured spectroscopically and typically includes monomer, dimer, and soluble polymers. Over time, base catalyzed depolymerization and repolymerization transforms the smaller products into flavonoids/oligomers and LNPs. Quantification was achieved through calibration curves obtained from samples of lignin dissolved in pH 13 NaOH solution (Fig. S1 in the ESI†). Peak absorbance occurred at wavelengths between 288 and 291 nm. Solubilization was measured on centrifuged samples to remove any residual solids. The total phenolic and flavonoid content was determined with the colorimetric Folin–Ciolcalteu method and the method by Zhishen et al., respectively.38,39 The former combines 100 μL of solubilized lignin with 2 mL of a 2% sodium carbonate solution. The mixture was incubated at room temperature for 5 minutes and then 100 μL of Folin–Ciolcalteu reagent was added and the mixture was incubated further for 30 minutes. The absorbance was measured at 750 nm. A calibration was performed with a solution of gallic acid (GA) (3, 4,5-trihydroxybenzoic acid) every time the experiment was performed38 (Fig. S2a in the ESI†). The results are reported as mg L−1 of GA equivalents. For the Zhishen methods, 300 μL of solubilized lignin was combined with 3.4 mL of a 30% methanol solution, 150 μL of a 0.5 mmol L−1 sodium nitrate solution and 150 μL of a 0.3 mmol L−1 aluminum chloride hexahydrate solution at room temperature. 1 mL of 1 mol L−1 sodium hydroxide was added after 5 minutes and the absorbance was immediately measured at 510 nm. The calibration curve was made using the flavonoid rutin, (3′,4′,5,7-tetrahydroxy-3-[α-L-rhamnopyranosyl-(1→6)-β-D-glucopyranosyloxy] flavone) (Fig. S2b in the ESI†). A 100 mg L−1 sample of rutin was made with each set of experiments to confirm the accuracy of the calibration curve. The results are reported as mg L−1 of rutin equivalents.
The antioxidant capacity was also determined by measuring the UV absorbance following a modified version of the ABTS method first described by Cano et al.40 The ABTS+ radical solution was prepared from 10 mL of 7 mM ABTS+ [2,2′-azino-bis(3-ethyl-benzothiazoline-6-sulfonic acid)] solution in water and 10 mL of 2 mM potassium persulfate solution in water. The mixture was incubated in the dark at room temperature overnight. The ABTS+ solution was diluted with water to an absorbance of 0.70 ± 0.1 at 734 nm. Then 1 mL of different concentrations of lignin (0, 2, 6, 10, 14, 16, 18, 20, 40, 60, 80, 100, 140, 160, 200, 300, 400, 600, 800 mg L−1) was added to 2.0 mL of ABTS+ diluted solution. The mixture was incubated in the dark for 6 minutes and were measured at 734 nm. The scavenging was calculated with the following equation:
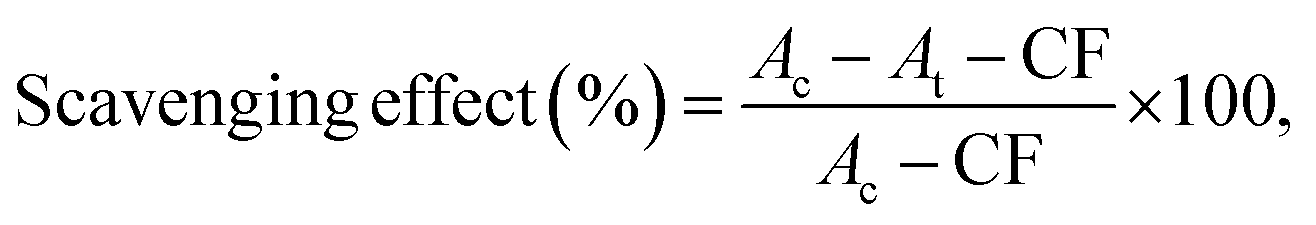
LNP concentration, size, PDI, and zeta potential were determined with Nanoparticle Tracking Analysis (NTA) and Zetasizer. The NTA (Malvern Panalytical NanoSight NS300) was used according to Obrzut et al. to estimate both concentration and size distribution.33 The limits for size distribution are between 10 and 1000 nm. The lignin solutions were diluted 50 times in water for NTA measurement. Since the NTA cannot detect LNPs larger than 1000 nm, the LNP concentration is reported for LNPs smaller than 1 micron. The concentration of LNPs larger than 1 micron is not known. The zetasizer (Malvern Panalytical Zetasizer Nano ZS) also has dynamic light scattering (DLS) capabilities to measure size and polydispersity index (PDI). DLS can measure the size up to 10.0 microns. The lignin solutions were diluted 10× for size, PDI and zeta potential measurements. Then, 1 mL of sample was placed in a DTS1070 disposable zeta potential cuvette or a plastic cuvette for DLS. Three measurements were taken for each sample with up to 100 zeta runs per sample. For these runs the refractive index of lignin was set at 1.61.42
High-performance liquid chromatography coupled to mass spectrometry was completed using previously published methods.33 Briefly, an Agilent 1260 Infinity binary LC system with an autosampler was coupled to a Bruker Amazon X ion trap with an electrospray ionization (ESI) source. The HPLC column was a Phenomenex Gemini C6-phenyl, 250 mm long, with an internal diameter of 4.6 mm and a particle size of 5 μm. The HPLC instrument was also coupled to an Agilent G4212B diode array detector (DAD). A gradient method with ultrapure water (solvent A) and HPLC-grade methanol (solvent B), both modified with 0.01% (v/v) acetic acid, at 40 °C column temperature, and a flow rate of 0.4 mL min−1 was used.
The SEM micrographs were obtained using a JEOL JSM-7900FLV microscope equipped with an Oxford Ultimax 65 energy-dispersive X-ray spectroscopy (EDS) accessory. The voltage was set to 5 kV and the current to 8 mA. The SEM was run using the lower electron detector (LED). SEM samples were prepared by diluting the suspensions 10-fold with 5% CTAB and spin-coated (Laurel Technologies) onto a silicon wafer for 1 minute with an acceleration of 1 rpm and a maximum rotation speed of 2000 rpm. For enhancing conductivity, a 10 nm AuPd layer was deposited over the sample via the Denton III Desk Sputter Coater.
Results
Structural characteristics of various lignin sources
Table 2 summarizes the structural features of interest for each lignin source: % acid insoluble lignin as determined by the Klason extraction, and the % β-O-4 linkages and S/G ratio as determined by NMR. The NMR spectra, the regions used to determine the % β-O-4 linkages and S/G ratio, as well as the calculations are detailed in the ESI (Fig S4).†36,43 The results for the biomass characterization compare well with those reported in the literature.44 Softwood biomass contains the highest lignin content, followed by hardwood, and then herbaceous, which has slightly more than half that found in softwood. In contrast, the herbaceous lignin source has the highest content of % β-O-4 linkages, followed by hardwood and then softwood, which has less than half the herbaceous proportions. S/G ratio is highest for hardwood and lowest for softwood, since softwood contains mostly the G monolignol. The herbaceous S/G ratio is around 0.5 and is the only source that contains the H monolignol. The S monolignol lacks a free ortho-position, making it unable to form 5–5 or β–β carbon–carbon bonds.45 For this reason, lignin with higher S/G ratio (hardwood) has fewer carbon–carbon bonds and has a more linear structure, whereas lignin with a low S/G ratio (softwood) has more carbon–carbon bonds and a more branched structure, making it more difficult to delignify the biomass for downstream depolymerization.46,47 The more linear structure of hardwood lignin allows more flexibility to form coiled structures, creating more density.48 The extractions (MWL, Organosolv, and Klason) do not have a major effect on the S/G ratio of herbaceous lignin, although they have a large effect on the % β-O-4 linkages. The milder extractions, Milled Wood lignin and Organosolv, remove only 19% and 32% of β-O-4 linkages, respectively. The Klason extraction was the only extraction done on all three sources because the Organosolv (acetosolv) and MWL extractions were unable to extract sufficient lignin from the samples, as previously noted in literature.49 Klason, the harshest extraction, eliminates between 80 and 90% of the β-O-4 linkages present in the initial biomass. The extractions also vary in the amount of lignin that is extracted, Klason isolates all the lignin through carbohydrate conversion and MWL extracts around 6%.2,37 We do not have data for the extraction efficacy of ADM's Organosolv, although organosolv lignin is typically recovered in a range of 20–60%, putting it between Klason and MWL.50,51| Biomass | Klason | Organosolv | Milled Wood | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Source | Latin name | Common name | % lignin (determined via Klason) | β-O-4 (%) | S/G ratio | β-O-4 (%) | S/G ratio | β-O-4 (%) | S/G ratio | β-O-4 (%) | S/G ratio |
| Hardwood | Carya cordiformis | Bitternut hickory | 23.71% | 68.67% | 1.74 | 11.90% | 1.64 | N/A | N/A | ||
| Softwood | Picea glauca | White spruce | 28.12% | 37.42% | 0.063 | 3.53% | 0.056 | N/A | N/A | ||
| Herbaceous | Corn stover | 15.60% | 83.46% | 0.55 | 14.52% | 0.52 | 56.50% | 0.55 | 67.60% | 0.50 | |
DRIFT spectra were measured to characterize structural differences among the lignin sources as a function of extraction. The DRIFT spectra between 1750–1000 cm−1 of the MWL, Organosolv, and Klason extractions of herbaceous lignin and the Klason extractions of the hardwood and softwood lignin are shown in Fig. 1. All the sources and extractions share the characteristic peaks of lignin as detailed in Table S4 in the ESI.† We point out six key peaks in Fig. 1. Peak 1 (∼1712 cm−1) and Peak 2 (∼1680 cm−1) correspond to unconjugated and conjugated carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) stretching, respectively. Peak 3 (1506 cm−1) corresponds to the aromatic skeletal vibration. Peak 4 (1220 cm−1) corresponds to phenolic O–H plus ether C–O stretching, peak 5 (1087 cm−1) corresponds to C–O deformation at Cβ and aliphatic ether and peak 6 (1033 cm−1) corresponds to C–O deformation at Cα and aliphatic ether.
O) stretching, respectively. Peak 3 (1506 cm−1) corresponds to the aromatic skeletal vibration. Peak 4 (1220 cm−1) corresponds to phenolic O–H plus ether C–O stretching, peak 5 (1087 cm−1) corresponds to C–O deformation at Cβ and aliphatic ether and peak 6 (1033 cm−1) corresponds to C–O deformation at Cα and aliphatic ether.
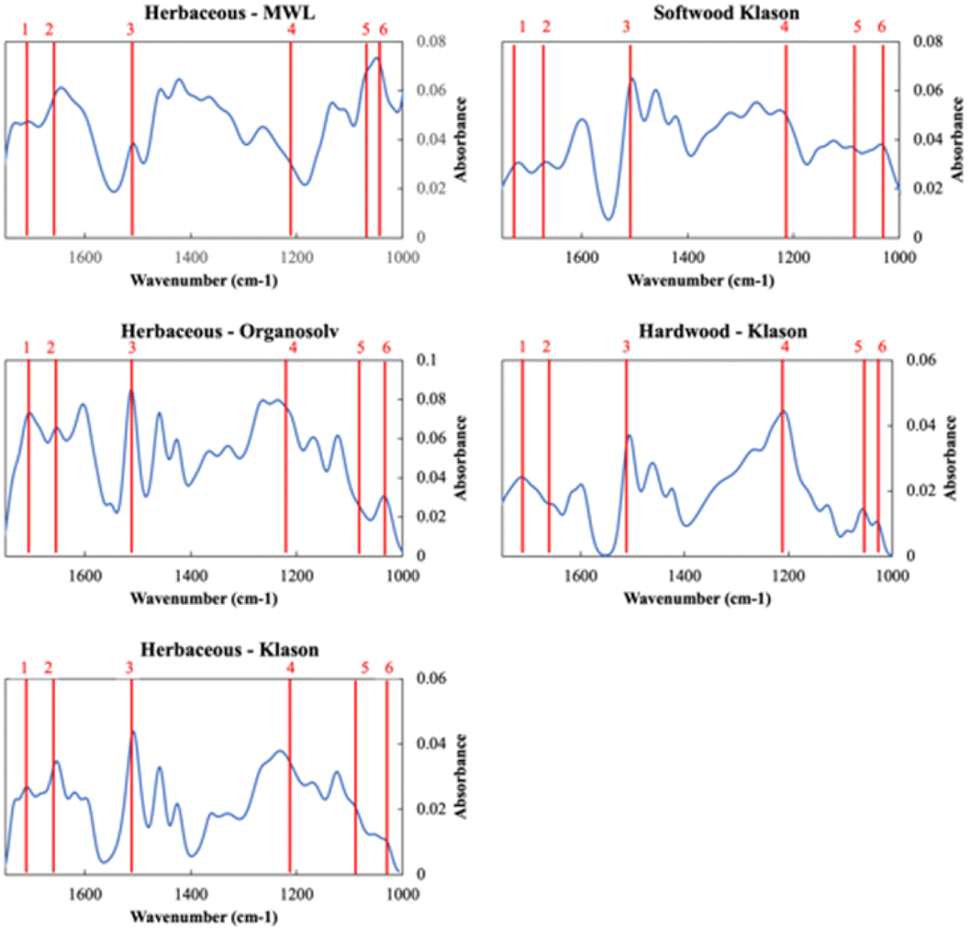 | ||
| Fig. 1 DRIFT spectra between 1750–1000 cm−1 for the five sources and extractions of lignin. 6 peaks are highlighted. Peak 1 (∼1712 cm−1): unconjugated CO stretching, Peak 2 (∼1680 cm−1): conjugated CO stretching, Peak 3 (1506 cm−1): aromatic skeletal vibration, Peak 4 (1220 cm−1): phenolic O–H plus ether C–O stretching, Peak 5 (1087 cm−1): C–O deformation at Cβ and aliphatic ether, and Peak 6 (1033 cm−1): C–O deformation at Cα and aliphatic ether. | ||
The proportions of the peaks vary between sample and extractions. In order to compare the differences between the samples we followed the analysis method of Wang et al. and Pandey which normalizes features to a common aromatic reference.52,53Fig. 2 shows the relative absorbance (Iw/Iar) of the five oxygen containing functional groups: CO conjugated, CO unconjugated, OH + C–O, C–O at Cβ and C–O at Cα. Iar corresponds to the aromatic skeletal vibration peak (Peak 3) and Iw corresponds to the intensity of a particular wavenumber. MWL (gray triangle) and Organosolv (yellow square) have the highest proportions of oxygen containing groups compared to the Klason extractions (circles). We see that MWL diverges the most, particularly with respect to conjugated CO and C–O deformations, indicating that these bonds are easily destroyed with harsher extractions. We also see that there is the least amount of deviation between extractions with respect to phenolic OH and ether C–O stretching. Generally, Klason eliminates the carbonyl structure. Of the Klason extractions, softwood (dark blue) contains the lowest proportion of carbonyl bonds. Klason produces lignin that is structurally similar regarding carbon–oxygen bonds across different sources.
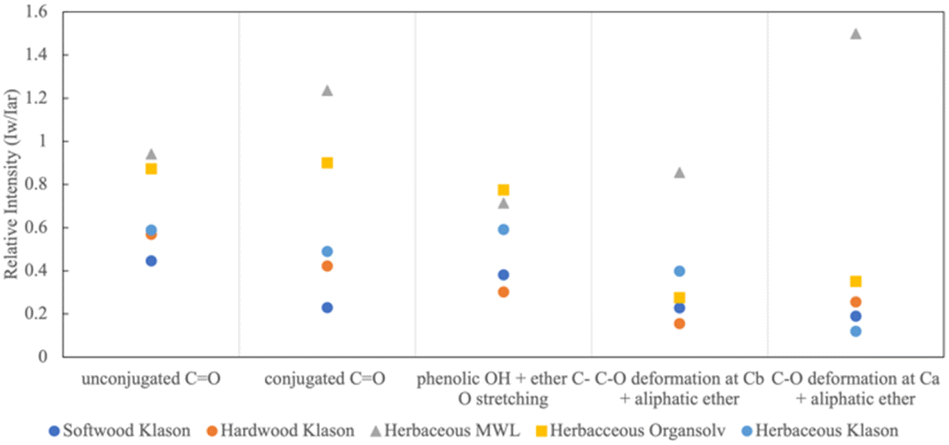 | ||
| Fig. 2 Relative intensities of typical functional groups for softwood Klason, hardwood Klason, herbaceous Klason, and herbaceous Organosolv lignins. | ||
The influence of biomass source on product characteristics of Klason lignin
To evaluate the influence of lignin source on biorefined product characteristics, we applied one extraction across all three lignin sources. The Klason extraction has a few benefits over the others: (1) the Klason extraction produces the most lignin without contamination from cellulose and hemicellulose; (2) it is the most harsh extraction, removing most of the β-O-4 linkages, which mirrors how lignin is typically extracted industrially (i.e. Kraft, soda, etc.); (3) the Klason extraction is robust and reproducible; it works on all three of the sources; and (4) there is no introduction of sulfur or other elements.In Fig. 3, we present solubilization, antioxidant capacity, phenolic content, flavonoid content, LNP concentration and LNP size for the Klason extraction of herbaceous, softwood, and hardwood lignin. Fig. 3a shows that in basic conditions, Herbaceous Klason (HK) lignin solubilizes to the greatest extent, 62% of the starting lignin concentration (1 g L−1). Hardwood Klason (HaK) reaches slightly more than half of that at 35%, and negligible amounts of softwood Klason (SK) is solubilized (around 3%) under these conditions. In neutral conditions, solubilization is much lower, 50–80% less than that in basic conditions. These data indicate that in the case of Klason extraction of lignin, neutralizing the samples at 60 minutes suppresses solubilization and subsequent product formation. Under basic conditions, HaK produces more phenolics (320 mg L−1 GA eq.) and flavonoids (230 mg L−1 rutin eq.) than either HK or SK, with HK and SK only reaching a maximum of 190 and 110 mg L−1 GA eq. phenolics and 100 and 110 mg L−1 rutin eq. flavonoids respectively (Fig. 3c and d). The flavonoid and phenolic yields of SK exceed the amount solubilized and we hypothesize that this is due to the attachment of phenolics and flavonoids to LNPs or residual solids. To confirm this, we ran the phenolic and flavonoid assays on a centrifuged sample to find that both the phenolic and flavonoid content decreased to less than 20 mg L−1 eq. Similar trends are observed with the other sources. We believe, however, that making the phenolic and flavonoid measurements without centrifugation is justified because we measure the antioxidant capacity on the entire solution and there is a relationship between the phenolics and flavonoid and antioxidant capacity, whether they are free or associated with LNPs. We measure these properties on the entire solution to avoid downstream separation and associated problems and costs.
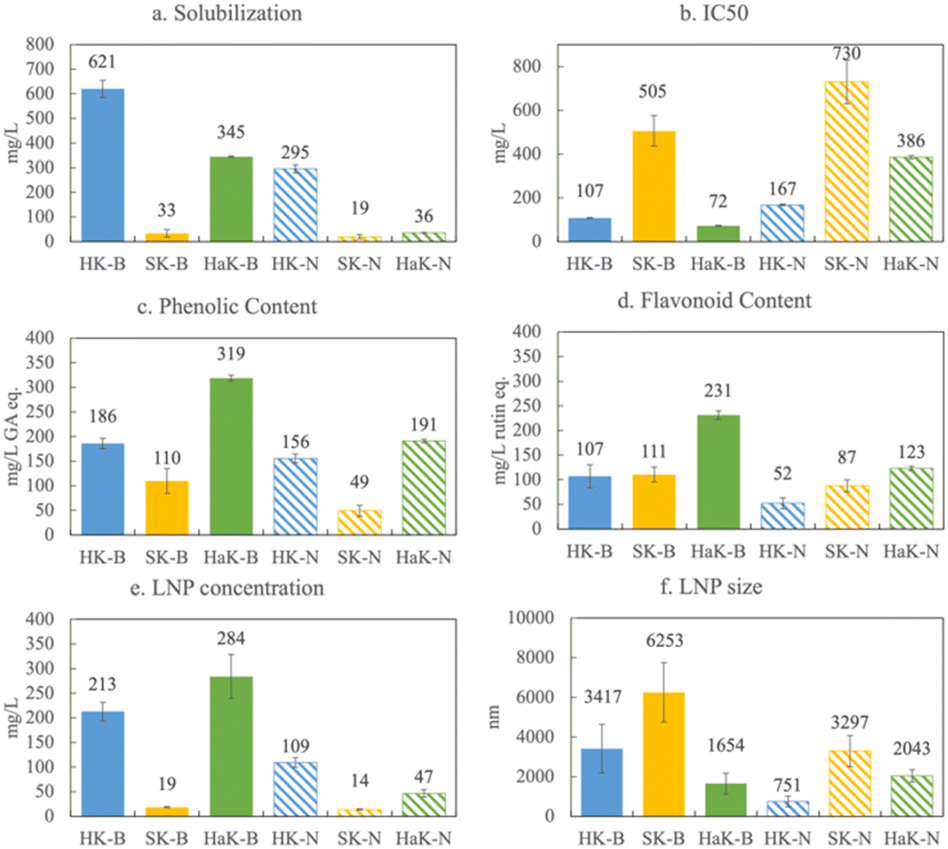 | ||
| Fig. 3 Comparison of Klason lignin extracted from herbaceous, softwood, hardwood sources: (a) solubilization (centrifuged), (b) IC50, (c) phenolic content (in gallic acid equivalents), (d) flavonoid content (in rutin equivalents), (e) LNP concentration, and (f) LNP size for the Klason extractions of herbaceous (HK), softwood (SK), and hardwood (HaK) lignin in both basic (B – solid bars) and neutral (N – hatched bars) conditions. | ||
With respect to antioxidant capacity (Fig. 3b), HaK has the lowest IC50 of 72 mg L−1, meaning that a lower concentration of HaK is required to scavenge 50% of the ABTS radical relative to the other lignin sources and thus, HaK exhibits the highest antioxidant capacity. Trolox, an analog of the industrially relevant antioxidant, vitamin E, has an antioxidant capacity of 21 mg L−1. Although HaK, shows about a third of that antioxidant capacity, there has been a push for more natural sources of antioxidants.54 The IC50 of HK is 107 mg L−1, and SK has the highest IC50 – 505 mg L−1, making it a less effective antioxidant than the other lignin types. The antioxidant capacity trend is similar to that of the phenolic content. Antioxidant capacity is thought to be influenced by many factors, including the –OH groups on phenolics and flavonoids, as well as the surface area to volume ratio of LNPs (discussed below). It is worth noting that antioxidant capacity also decreases (higher IC50) with sources under neutral pH conditions, relative to that of basic pH conditions. This is explained by the fact that less solubilized lignin yields less phenolics, flavonoids, and LNPs in neutral conditions which simply decreases the concentration of antioxidants in solution. We confirm this by conducting the antioxidant assay on the three extracted lignins in water and finding no antioxidant activity due to no solubilization (Fig. S5†). Another explanation is related to the mechanism by which solubilized lignin scavenges free radicals. Typically phenolic compounds scavenge free radicals either through a hydrogen atom transfer (HAT) reaction or a single electron transfer (SET).55 At high pH, the SET reaction is predominant, which is the specific mechanism for which the ABTS tests.40,56
The production of lignin nanoparticles follows a trend similar to that observed with other products. HaK and HK produce the most LNPs at 30% (284 mg L−1) and 20% (213 mg L−1) (Fig. 3e), respectively. The DLS data show that HaK has the smallest LNP aggregate size at around 1600 nm and HK has LNPs around 3400 nm (Fig. 3f). SK produces essentially no LNPs in the measurable size range of the NTA (up to 1000 nm) and DLS shows the size to be 6200 nm. In contrast to previous observations, neutralizing the samples fails to increase the SK LNP yield.33 The low solubilization for neutral samples, as illustrated in Fig. 3a, indicates that there is not enough lignin in solution to promote LNP formation. Fig. 4 below shows the SEM images of the LNPs. In basic conditions, HK produces small LNPs that grow to about 200 nm and at 14 days aggregate into a large, amorphous mass or the individual LNPs may be attached to a larger fragment of solid, unreacted lignin. SK produces large, disc like particles. However, it is not clear whether this is self-assembled nanoparticle or a solid lignin remnant. HaK produces spherical and non-aggregated LNPs bearing a hole (pointed out by arrows in Fig. 4c) which has been observed previously for LNPs produced from hardwood.57 The zeta potentials (Fig. S6†) for all LNPs indicate moderate stability (<−30 mV). The PDIs for all LNPs show that they are polydisperse (>0.05), although the polydispersity for SK is much greater, >0.8 (extremely high polydispersity) (Fig. S7†). Overall, despite relatively low yields of soluble lignin, HaK gives higher product yields with more favorable properties under basic biorefining conditions. In contrast, the Klason lignin extracted from softwood biomass has low product yields with unfavorable properties, indicating that this is not a suitable lignin source for these biorefining conditions.
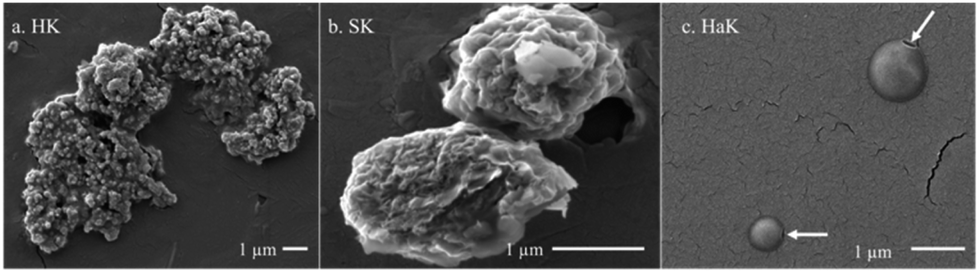 | ||
| Fig. 4 SEM images Klason extraction (basic pH) of (a) herbaceous, (b) softwood, and (c) hardwood lignin. All taken at day 14 for the basic experiments. | ||
The influence of extraction method on the product characteristics of herbaceous biomass
To evaluate the effects of extraction on product yields, Milled Wood, Organosolv, and Klason extractions of the herbaceous lignin were compared. Only the herbaceous biomass source could be extracted by the acetic acid based Organosolv and the Milled Wood methods to yield a sufficient quantity of lignin mass to be biorefined into products.Fig. 5a shows that under basic conditions Herbaceous Organosolv (HO) material solubilizes to the largest extent, up to 94% (936 mg L−1), whereas the Milled Wood Herbaceous Lignin (MWL) and HK solubilize to a lesser extent, around 65% (671 and 621 mg L−1, respectively). Solubilization decreases in all conditions at neutral pH. HO also produces the most phenolic and flavonoid equivalents, 373 mg L−1 GA eq. and 295 mg L−1 rutin eq. respectively, and these values remain constant at both basic and neutral pH. Lower bulk phenolic and flavonoid yields are observed with HK-B (186 and 107 mg L−1 eq., respectively) and they decrease in neutral conditions. Much lower product yields are observed with MWL (48 and 15 mg L−1 of phenolics and flavonoids eq., respectively) and these values do not change with pH.
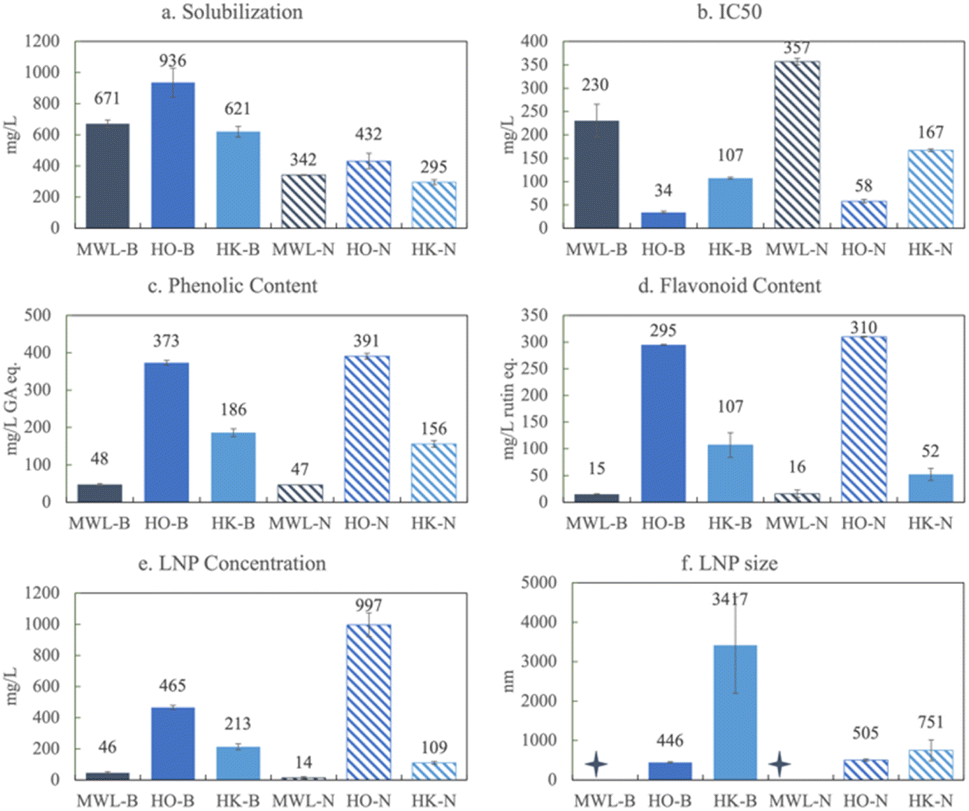 | ||
| Fig. 5 Comparison of various herbaceous lignin extractions: (a) solubilization (centrifuged), (b) IC50, (c) phenolic content (in gallic acid eq.), (d) flavonoid content (in rutin eq.), (e) LNP concentration, and (f) LNP size for the Milled Wood (MWL), Organosolv (HO), and Klason (HK) extractions of herbaceous lignin in both basic (B – solid bars) and neutral (N – hatched bars) conditions. | ||
Consistent with these trends, HO has the lowest IC50 (highest antioxidant capacity), followed by HK, and then MWL. HO and MWL are the only examples of the antioxidant capacity decreasing over time. In the case of HO, the IC50 at maximum solubilization (day 7) is 34.4 mg L−1 and at day 0 it is 29.7 mg L−1. Although the total phenolic and flavonoid content for HO increases over the 7 days (349 to 373 mg L−1 and 234 to 295 mg L−1, respectively), the centrifuged phenolics and flavonoids decrease (333 to 288 mg L−1 and 180 to 133 mg L−1). This observation indicates two things: (1) phenolics and flavonoids become incorporated into LNPs over time, and (2) phenolics and flavonoids may have a higher antioxidant capacity as free rather than attached or incorporated into LNPs. Like HaK and SK, the antioxidant capacity always decreases at neutral pH.
Fig. 5e shows that in basic conditions MWL has an LNP concentration of less than 5% (46 mg L−1) and HK has an LNP concentration of 20% (213 mg L−1), both of which decrease in neutral conditions, whereas HO has a concentration of 46% (456 mg L−1) which more than doubles (>99%) in neutral conditions, indicating that essentially all of the lignin is in the LNP form. The average size of the HO LNPs is also much smaller than HK, 446 nm in basic conditions and slightly larger at 505 nm in neutral conditions. The zeta potential of HO is slightly larger than that of HK at −39 mV, compared to −34 mV, indicating moderate stability (Fig. S8†). The PDI in basic conditions is similar between 0.47 and 0.48, indicating a polydisperse sample (Fig. S9†). In neutral conditions the PDI of HO drops to 0.38, which is in the polydispersity category but may be suitable for certain commercial applications. For instance, PDIs around 0.2–0.3 may indicate that the LNPs are applicable for drug delivery.58Fig. 6 shows SEM images of: a. MWL, b. HO, and c. HK. The SEM image for MWL fails to show the presence of LNPs, which is consistent with the DLS and zeta potential results. Given the high concentration of oxygen bonds in MWL (Fig. 2), the molecular weight of the solubilized MWL lignin may not be large enough to micellize or self-assemble into LNPs. Many of the interactions that LNP formation relies on are molecular weight dependent, such as, the hydrophobic effect, van der Waals, and chain entanglement.59 The mass spec data indicate peaks of non-aromatic compounds (Fig. S9†), and given that the MWL extraction does not exclude carbohydrates, there may be interferences that disrupt LNP formation. However, the HO LNPs are spherical and well dispersed. As previously shown (Fig. 4), the HK LNPs are highly aggregated into large clusters.
 | ||
| Fig. 6 SEM images of (a) MWL, (b) HO, and (c) HK in basic conditions. Day 7 for MWL, day 14 for HO and HK. | ||
Milled wood extraction of the herbaceous biomass produces a small lignin yield, recovering only about 4–6% of the material's lignin content. In addition, MWL has low purity as revealed by its mass spectra showing a peak at m/z of 179.055, which corresponds to glucose (Fig. S10†). Despite the recalcitrant nature of lignin as a carbon source and the antimicrobial properties of LNPs,60,61 MWL LNPs appeared to support microbial growth due to co-extracted impurities that may be as much as 15% carbohydrates.37 Although Milled Wood extraction is mild and preserves the highest amount of β-O-4 linkages, the method gives low yields of pure lignin, phenolics, flavonoids and LNPs and is not a viable route to process lignin under our biorefining conditions.
Discussion
Lignin structure is determined by its source, which is further modified by the method of extraction. Neither of these factors, however, has been rigorously interrogated relative to the type and quantity of products created by a particular depolymerization process. Our aim is to compare how, under the conditions of our biorefinery (MEC catholyte - 220 mM phosphate, pH 10.8), the product composition, yields and properties vary for representative models from each of the three types of lignin source and three examples (harsh, medium, mild) of extraction. We also probe which factors may predict product yields and properties.The three sources of lignin, softwood, hardwood, and herbaceous, differ relative to lignin content, the percentage of β-O-4 linkages, and the S/G ratio. Extraction modifies the β-O-4 content. The Milled Wood isolation process is a mild extraction technique with poor yields and low purity but it preserves most of the β-O-4 linkages. Organosolv is a medium extraction process that degrades the lignin to a limited extent, but still preserves many of the β-O-4 linkages. Organosolv is typically used in the cellulosic bioethanol industry. The Klason method is different from Milled Wood and Organosolv in that it does not isolate lignin through delignification but rather through carbohydrate conversion.2 The Klason method significantly alters the structure of lignin by removing many of the β-O-4 linkages, resulting in lignin that is structurally similar (condensed) to Kraft lignin (which is industrially produced in the paper and pulp industry alongside lignosulfonate) without introducing sulfur. Klason cleaves most of the β-O-4 linkages, but is efficient in extracting the highest quantity and purity of lignin from biomass. We found that the herbaceous biomass was the only source that could be extracted by all three methods and that the Klason extraction was the only method among those evaluated that worked well on all three sources. We then determined and compared the products and properties of the five biorefined extracted lignin samples, specifically the amount of soluble lignin, bulk phenolic/flavonoid yields, and the antioxidant capacity, as well as the concentration of LNPs and their shape, size, and polydispersity.
Table 2 presents salient characteristics of the raw biomass. The lignin content indicates the maximum amount of lignin in the biomass that can be extracted. Based on this, softwood lignin with up to 35% lignin content would seem a promising target for depolymerization. Yet, its β-O-4 bond content, which is a good indicator of the ease of depolymerization with preservation of the aromatic structure, is less than half that of the other sources, making extracted softwood lignin a poor candidate for biorefining high value aromatic products. Rather, herbaceous lignin has the greatest β-O-4 content, roughly twice that of softwood and about 20% more than that of hardwood lignin. Considering both the total lignin (8% more than herbaceous) and β-O-4 content (30% more than softwood), we might also expect hardwood lignin to be a favorable choice for depolymerization.
The ease of depolymerization and product yields, however, also depend on the monolignol composition. When we compare monolignol proportions, softwood lignin contains primarily the G monolignol, and herbaceous lignin has roughly two G monolignol for every S monolignols, with a small amount of H monolignol as well. In contrast, hardwood contains roughly two S monolignols for every G monolignol. It is significant that herbaceous lignin is the only source that contains a detectable amount of the H monolignol which is likely responsible for some of the discrete aromatic products in the depolymerized corn stover that we identified in our previous work, including the monomer p-coumaric acid (making up to 0.5% of the depolymerized lignin mixture) and the flavonoid tricin (making up to 2.3% of the depolymerized lignin mixture).33 These compounds were not detected by LCMS in depolymerized hardwood and softwood samples from our biorefinery.
The S/G ratio influences the branching and cross-linking density of the lignin structure. High methoxy content, such as that found in hardwoods with high S/G ratios, shows reduced tendency to form branched or crossed-linked structures, whereas the opposite is true of softwoods with small S/G ratios and more highly branched and cross-linked structures with fewer ether linkages (Fig. 2).48 A high S/G ratio is hypothesized to increase the β-O-4 content in lignin because the S monolignol contains a methoxy in the 5 position of the aromatic ring, which prevents the formation of 5–5 and β-5 carbon–carbon bonds45 In contrast, G units promote more carbon–carbon bonds, which are much less labile, and fewer ether linages, making softwood lignin a poor candidate for depolymerization. There is some debate, however, that the S/G is the major determinant of monomer yield in lignin depolymerization.44
The structural feature that most informs lignin solubilization and subsequent product formation under our biorefinery conditions is the β-O-4 content. In Fig. 7, a linear relationship is illustrated between the β-O-4 content and various product characteristics. A strongly positive correlation exists between increasing β-O-4 content and increasing soluble lignin, LNP concentration, phenolic content, and flavonoid content (Fig. 7a–d). An inverse relationship is observed between β-O-4 content and LNP size and polydispersity (Fig. 7e and f) both of which decrease with increasing percentage of β-O-4 linkages.
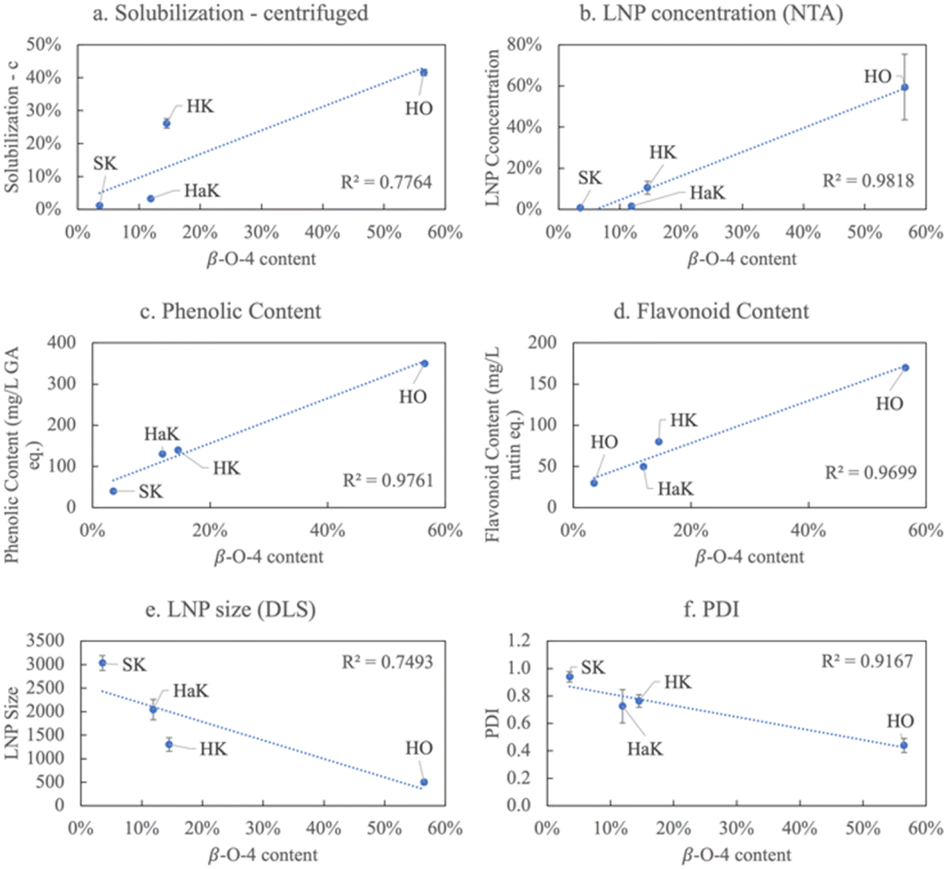 | ||
| Fig. 7 Relationships between β-O-4 content and various products in neutral conditions for each lignin source, including (a) solubilization of the centrifuged sample, (b) LNP concentration, (c) relative phenolic content, (d) relative flavoonoid content, (e) LNP size, and (f) polydispersity index. Each data point is labeled with the corresponding lignin it represents. Tables S5 and S6† show the R2 for all factors and products. | ||
Softwood, which has the highest lignin content, but the lowest β-O-4 bonding, is difficult to extract except with the harsh Klason method that further reduces the β-O-4 content to under 4% making it resistant to solubilization/depolymerization under the conditions of our biorefinery. Since the extracted softwood lignin (SK) shows limited solubilization, its product yields are also limited. NMR results confirm that the softwood lignin is composed of mostly the G monolignol linked with few β-O-4 bonds (Fig. S4†). The DRIFT data in Fig. 2 reveal that the relative intensity of oxygen containing functional groups in SK is typically less than that of the other sources extracted in the same way. These observations also explain why SK does not form nanoparticles. We have shown previously that LNPs formed in our biorefinery undergo a two-step process: (1) lignin is first depolymerized, which is catalyzed by high pH, then (2) the high salt facilitates micellization.33 There are many specific interactions responsible for the mechanisms of LNP formation including: (1) hydrophilic–lipophilic interactions; (2) π–π interactions; (3) hydrogen bonding; (4) chain entanglement; (5) van der Waals forces; and (6) hydrophobic interactions.59 The lower oxygen content in softwood hinders dissolution in a polar solvent and explains the low solubility in Fig. 3a. Low dissolution, then, inhibits LNP formation since this is the initial step of the mechanism. In addition, SK does not form LNPs due to the lack of the syringyl monolignol and relatedly, lower methoxy content. Since the non-covalent, π–π interactions between aromatic rings follow the order of syringyl (S) > guiacyl (G) > p-hydroxyphenyl (H),62 the lower methoxy content of SK diminishes π–π interactions preventing LNP formation.
As noted previously, based on the lignin content and number of β-O-4 linkages in the native biomass, hardwood appeared to be a promising lignin source for biorefining, except that it can only be extracted by the Klason (harsh) technique, which then removes most of the ether linkages. Under our biorefinery conditions, HaK lignin produces only half the soluble lignin compared to Klason extracted herbaceous lignin (HK). Yet, the soluble HaK lignin has significantly greater phenolic and flavonoid content, and hence a higher antioxidant capacity (lower IC50) than HK, suggesting that differences between the S/G ratio of hardwood and herbaceous sources may also play a role. Furthermore, we observe that the depolymerization of hardwood lignin in our biorefinery produces the most LNPs compared to the other Klason extracted material. These LNPs are spherical and dispersed but larger and fewer (failing to close mass balance) than the highly aggregated LNPs of the Klason extracted herbaceous lignin. HaK lignin can be biorefined to soluble products with antioxidant properties but in lower amounts compared with Organosolv herbaceous lignin. Thus, the biorefinery conditions do not fully solubilize HaK lignin resulting in limited product yields.
Under the conditions of our biorefinery, the Organosolv extraction of herbaceous lignin produces the maximum amount of soluble lignin (<90%) and the highest aromatic product yield (300–400 mg L−1 phenolics/flavonoids eq.) having the greatest antioxidant potential (close to the commercial standard), as well as the most LNPs accounting for all of the soluble mass. In general, the herbaceous source can be extracted by all the methods that we studied to provide higher soluble lignin than other sources. We observe the effects of extraction and processing most vividly with respect to the LNPs, which are small, spherical, and produced at the highest concentration with the Organosolv herbaceous lignin. As discussed, MWL does not form LNPs. Although the Klason extraction has a strong effect on the native structure of lignin, it preserves lignin aromaticity (see Peak 3 in Fig. 1) and gives the highest quantities of pure lignin, albeit with large losses in the β-O-4 content, which is critical to the ease of depolymerization. Furthermore, the Klason extraction of herbaceous lignin does not form small and well dispersed LNPs.
Fig. 8 illustrates the relationship between flavonoid content and IC50 (antioxidant capacity) across all sources and extractions. The phenolics, flavonoids and LNPs produced in our biorefinery have the valuable property of serving as antioxidants. Antioxidants combat oxidative stress in cells and tissues which occurs when there is an accumulation of reactive oxygen species (ROS) and can be the cause of inflammation, aging, and chronic diseases such as cancer, cardiovascular disease, neurological disease, respiratory disease, arthritis, and kidney disease.63 Antioxidants play an important role in maintaining human health. Although synthetic antioxidants have been used for decades in a variety of applications, there has been a push for more natural sources of antioxidants due to consumer demand for all natural ingredient, as well as some safety concerns about synthetic antioxidants.54 We show that although there is a relationship between flavonoids and antioxidant capacity in Fig. 8 (inset), the relationship becomes stronger when aggregated by source, indicating that the amount of bulk flavonoids produced in our depolymerization method is not the only factor responsible for antioxidant capacity. Other factors also contribute to antioxidant capacity and we observe in the Spearman correlation matrix (Fig. S11†) that soluble lignin, phenolic content, LNP concentration have a positive effect on antioxidant capacity and that LNP size and PDI have a negative effect on antioxidant capacity. These relationships can also be seen in the PCA score plot (Fig. S12 in ESI†), with solubilization, phenolic content, flavonoid content, LNP concentration, and antioxidant capacity pointing in the positive PC1 direction and LNP size and PDI pointing in the negative PC1 direction.
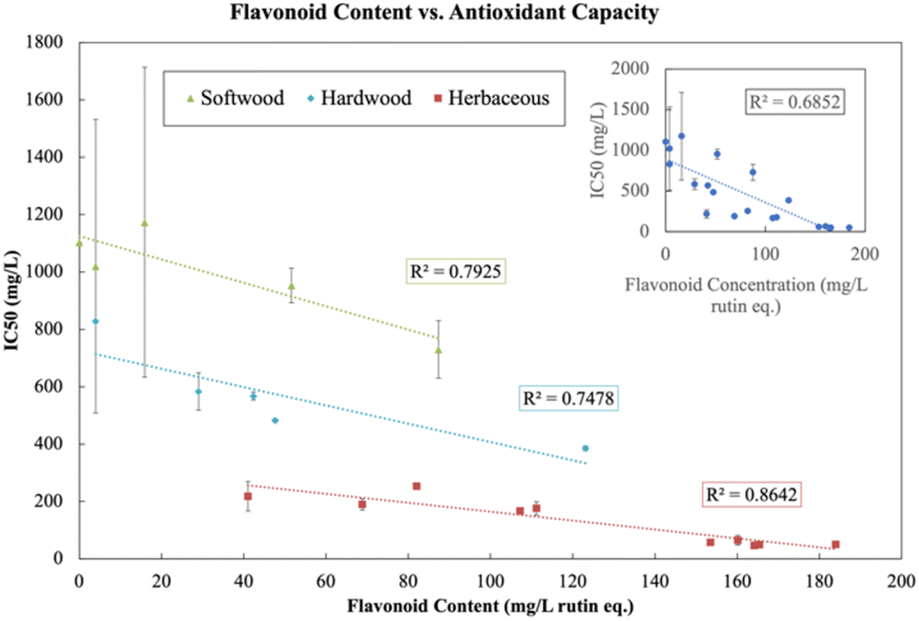 | ||
| Fig. 8 The relationship between flavonoid content and antioxidant capacity in neutral experiments. The top right inset shows the overall linear relationship. The main graph has the linear relationship segmented by the source of lignin. | ||
Conclusions
The goal of this research was to investigate if an MEC-mediated biorefinery is equally effective with different lignin streams and to determine what factors impact product yields. The biorefinery approach to depolymerizing lignin was originally employed on herbaceous Organosolv lignin, but was found to work on both different sources and extractions to varying degrees of success. Lignin source has a large effect on the efficacy of extraction methods. The Klason extraction was the only extraction to successfully extract lignin from all three sources, although it severely modifies the lignin structure by eliminating β-O-4 linkages, which ultimately diminishing lignin solubility and depolymerization.We found that under the caustic and high salt conditions of our biorefinery, herbaceous lignin extracted via the Organosolv method produces the highest amount of soluble lignin, phenolic/flavonoid content, and antioxidant capacity as well as a greater LNP concentration, with smaller and less polydispersed particles. The Organosolv extraction preserved a high amount of the β-O-4 linkages which is found to be strongly correlated with many of these products and properties.
Klason extraction of herbaceous and hardwood biomass solubilizes lignin to a lesser extent (60% and 35%, respectively) than Organosolv, but the method still produces good yields of phenolics, flavonoids, and LNPs with a moderate antioxidant capacity. Klason extracted softwood, however, is not solubilize/depolymerized in this biorefinery to produce products (phenolics, flavonoids, LNPs). This is likely due to the lack of S monolignol subunits and the paucity of β-O-4 linkages in this type of lignin. Only the herbaceous lignin could be extracted with the mild Milled Wood extraction to produce very small lignin quantities of low purity. Despite the fact that the β-O-4 linkages were highly preserved by this method, limitations in the amount of pure lignin extracted prevented the biorefining of high-value products.
In summary, a MEC-based biorefinery operating at ambient temperature and pressure is tailored to process the type of lignin recovered in the cellulosic ethanol industry from herbaceous (corn stover) biomass and extracted with the Organosolv method. At the present time, this industry produces the second largest industrial lignin stream.10 This biorefinery may also be adapted to smaller, distributed scales that target municipal herbaceous yard waste or the sustainable cultivation of such non-food crops as perennial grasses.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding for this work was provided by the Finite Earth Initiative of the McCormick School of Engineering at Northwestern University. Biological and chemical analysis was performed in the Analytical bioNanoTechnology Core Facility of the Simpson Querrey Institute at Northwestern University. The U.S. Army Research Office, the U.S. Army Medical Research and Materiel Command, and Northwestern University provided funding to develop this facility and ongoing support is being received from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-1542205). This work made use of the IMSERC MS facility at Northwestern University, which has received support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-2025633), and Northwestern University. This work also made use of the EPIC facility of Northwestern University's NUANCE Center, which has received support from the SHyNE Resource (NSF ECCS-2025633), the IIN, and Northwestern's MRSEC program (NSF DMR-1720139).Notes and references
- T. Li and S. Takkellapati, The current and emerging sources of technical lignins and their applications, Biofuels, Bioprod. Biorefin., 2018, 1–32 Search PubMed.
- W. Schutyser, et al., Chemicals from lignin: an interplay of lignocellulose fractionation, depolymerisation, and upgrading, Chem. Soc. Rev., 2018, 47(3), 852–908 RSC.
- L. Cao, et al., Lignin valorization for the production of renewable chemicals: State-of-the-art review and future prospects, Bioresour. Technol., 2018, 269, 465–475 CrossRef CAS PubMed.
- E. Feghali, et al., Convergent reductive depolymerization of wood lignin to isolated phenol derivatives by metal-free catalytic hydrosilylation, Energy Environ. Sci., 2015, 8(9), 2734–2743 RSC.
- V. M. Roberts, et al., Towards quantitative catalytic lignin depolymerization, Chemistry, 2011, 17(21), 5939–5948 CrossRef CAS PubMed.
- R. Rinaldi, et al., Paving the Way for Lignin Valorisation: Recent Advances in Bioengineering, Biorefining and Catalysis, Angew Chem. Int. Ed. Engl., 2016, 55(29), 8164–8215 CrossRef CAS PubMed.
- Z. Sun, et al., Bright Side of Lignin Depolymerization: Toward New Platform Chemicals, Chem. Rev., 2018, 118(2), 614–678 CrossRef CAS PubMed.
- T. Phongpreecha, et al., Predicting lignin depolymerization yields from quantifiable properties using fractionated biorefinery lignins, Green Chem., 2017, 19(21), 5131–5143 RSC.
- A. W. Tricker, et al., Similarities in Recalcitrant Structures of Industrial Non-Kraft and Kraft Lignin, ChemSusChem, 2020, 13(17), 4624–4632 CrossRef CAS PubMed.
- M. Taherzadeh, et al., Sustainable Resource Recovery and Zero Waste Approaches, Elsevier Science, 2019 Search PubMed.
- P. Bajpai, Chapter 12 – Pulping Fundamentals, in Biermann's Handbook of Pulp and Paper, Elsevier, 2018, pp. 295–351 Search PubMed.
- H. Chen, 3 - Lignocellulose biorefinery feedstock engineering, in Lignocellulose Biorefinery Engineering, ed. H. Chen, Woodhead Publishing, 2015, pp. 37–86 Search PubMed.
- M. Karlsson, et al., Toward a Consolidated Lignin Biorefinery: Preserving the Lignin Structure through Additive-Free Protection Strategies, ChemSusChem, 2020, 13(17), 4666–4677 CrossRef CAS PubMed.
- C. W. Lahive, et al., An Introduction to Model Compounds of Lignin Linking Motifs; Synthesis and Selection Considerations for Reactivity Studies, ChemSusChem, 2020, 13(17), 4238–4265 CrossRef CAS PubMed.
- J. R. Meyer, et al., Kinetics of Secondary Reactions Affecting the Organosolv Lignin Structure, ChemSusChem, 2020, 13(17), 4557–4566 CrossRef CAS PubMed.
- O. Musl, et al., Hydrophobic Interaction Chromatography in 2 D Liquid Chromatography Characterization of Lignosulfonates, ChemSusChem, 2020, 13(17), 4595–4604 CrossRef CAS.
- D. M. Neiva, et al., Lignin from Tree Barks: Chemical Structure and Valorization, ChemSusChem, 2020, 13(17), 4537–4547 CrossRef CAS.
- R. C. Sun, Lignin Source and Structural Characterization, ChemSusChem, 2020, 13(17), 4385–4393 CrossRef CAS PubMed.
- R. C. Sun, J. S. M. Samec and A. J. Ragauskas, Preface to Special Issue of ChemSusChem on Lignin Valorization: From Theory to Practice, ChemSusChem, 2020, 13(17), 4175–4180 CrossRef CAS PubMed.
- N. E. Thornburg, et al., Mesoscale Reaction-Diffusion Phenomena Governing Lignin-First Biomass Fractionation, ChemSusChem, 2020, 13(17), 4495–4509 CrossRef CAS PubMed.
- M. Yamamoto, et al., Importance of Lignin Coniferaldehyde Residues for Plant Properties and Sustainable Uses, ChemSusChem, 2020, 13(17), 4400–4408 CrossRef CAS PubMed.
- E. Subbotina, et al., Zeolite-Assisted Lignin-First Fractionation of Lignocellulose: Overcoming Lignin Recondensation through Shape-Selective Catalysis, ChemSusChem, 2020, 13(17), 4528–4536 CrossRef CAS PubMed.
- C. Cai, et al., Comparison of Two Acid Hydrotropes for Sustainable Fractionation of Birch Wood, ChemSusChem, 2020, 13(17), 4649–4659 CrossRef CAS PubMed.
- D. J. Cronin, et al., Deep Eutectic Solvent Extraction of High-Purity Lignin from a Corn Stover Hydrolysate, ChemSusChem, 2020, 13(17), 4678–4690 CrossRef CAS.
- A. De Santi, et al., Lignin-First Fractionation of Softwood Lignocellulose Using a Mild Dimethyl Carbonate and Ethylene Glycol Organosolv Process, ChemSusChem, 2020, 13(17), 4468–4477 CrossRef CAS PubMed.
- X. Liu, et al., Recent Advances in the Catalytic Depolymerization of Lignin towards Phenolic Chemicals: A Review, ChemSusChem, 2020, 13(17), 4296–4317 CrossRef CAS PubMed.
- R. E. Majdar, C. Crestini and H. Lange, Lignin Fractionation in Segmented Continuous Flow, ChemSusChem, 2020, 13(17), 4735–4742 CrossRef CAS PubMed.
- G. W. Tindall, et al., Fractionating and Purifying Softwood Kraft Lignin with Aqueous Renewable Solvents: Liquid-Liquid Equilibrium for the Lignin-Ethanol-Water System, ChemSusChem, 2020, 13(17), 4587–4594 CrossRef CAS PubMed.
- J. Xu, et al., Biomass Fractionation and Lignin Fractionation towards Lignin Valorization, ChemSusChem, 2020, 13(17), 4284–4295 CrossRef CAS PubMed.
- S. Bertella and J. S. Luterbacher, Simultaneous extraction and controlled chemical functionalization of hardwood lignin for improved phenolation, Green Chem., 2021, 23(9), 3459–3467 RSC.
- D. G. Brandner, et al., Flow-through solvolysis enables production of native-like lignin from biomass, Green Chem., 2021, 23(15), 5437–5441 RSC.
- S. Su, et al., Lignin-First Depolymerization of Lignocellulose into Monophenols over Carbon Nanotube-Supported Ruthenium: Impact of Lignin Sources, ChemSusChem, 2022, 15(12), e202200365 CrossRef CAS PubMed.
- N. Obrzut, et al., Valorization of Lignin under Mild Conditions: Biorefining Flavonoids and Lignin Nanoparticles, ACS Sustain. Chem. Eng., 2022, 11(2), 491–501 CrossRef.
- M. Chen, et al., Lignin Hydrogenolysis: Phenolic Monomers from Lignin and Associated Phenolates across Plant Clades, ACS Sustain. Chem. Eng., 2023, 11(27), 10001–10017 CrossRef CAS PubMed.
- A. BjÖRkman, Isolation of Lignin from Finely Divided Wood with Neutral Solvents, Nature, 1954, 174(4440), 1057–1058 CrossRef.
- S. D. Mansfield, et al., Whole plant cell wall characterization using solution-state 2D NMR, Nat. Protoc., 2012, 7(9), 1579–1589 CrossRef CAS PubMed.
- J. R. Obst and T. K. Kirk, Isolation of lignin, in Methods in Enzymology, Academic Press, 1988, pp. 3–12 Search PubMed.
- A. Khorasani Esmaeili, et al., Antioxidant Activity and Total Phenolic and Flavonoid Content of Various Solvent Extracts fromIn VivoandIn VitroGrownTrifolium pratenseL. (Red Clover), Biomed Res. Int., 2015, 2015, 1–11 CrossRef.
- J. Zhishen, T. Mengcheng and W. Jianming, The determination of flavonoid contents in mulberry and their scavenging effects on superoxide radicals, Food Chem., 1999, 64(4), 555–559 CrossRef CAS.
- A. Cano, M. Acosta and M. B. Arnao, A method to measure antioxidant activity in organic media: application to lipophilic vitamins, Redox Rep., 2000, 5(6), 365–370 CrossRef CAS PubMed.
- H. Noreen, et al., Measurement of total phenolic content and antioxidant activity of aerial parts of medicinal plant Coronopus didymus, Asian Pac. J. Trop. Med., 2017, 10(8), 792–801 CrossRef CAS PubMed.
- Y. Li, et al., Optically Transparent Wood from a Nanoporous Cellulosic Template: Combining Functional and Structural Performance, Biomacromolecules, 2016, 17(4), 1358–1364 CrossRef CAS PubMed.
- D. S. Zijlstra, et al., Mild Organosolv Lignin Extraction with Alcohols: The Importance of Benzylic Alkoxylation, ACS Sustain. Chem. Eng., 2020, 8(13), 5119–5131 CrossRef CAS.
- E. M. Anderson, et al., Differences in S/G ratio in natural poplar variants do not predict catalytic depolymerization monomer yields, Nat. Commun., 2019, 10(1), 2033 CrossRef PubMed.
- S. Van den Bosch, et al., Reductive lignocellulose fractionation into soluble lignin-derived phenolic monomers and dimers and processable carbohydrate pulps, Energy Environ. Sci., 2015, 8(6), 1748–1763 RSC.
- S. C. D. Eswaran, et al., Molecular structural dataset of lignin macromolecule elucidating experimental structural compositions, Sci. Data, 2022, 9(1), 647 CrossRef CAS PubMed.
- M. Li, Y. Pu and A. J. Ragauskas, Current Understanding of the Correlation of Lignin Structure with Biomass Recalcitrance, Front. Chem., 2016, 4, 45 Search PubMed.
- D. R. Ratnaweera, et al., The impact of lignin source on its self-assembly in solution, RSC Adv., 2015, 5(82), 67258–67266 RSC.
- A. Smit and W. Huijgen, Effective fractionation of lignocellulose in herbaceous biomass and hardwood using a mild acetone organosolv process, Green Chem., 2017, 19(22), 5505–5514 RSC.
- M. K. Gill, G. S. Kocher and A. S. Panesar, Optimization of acid-mediated delignification of corn stover, an agriculture residue carbohydrate polymer for improved ethanol production, Carbohydr. Polym. Technol. Appl., 2021, 2, 100029 CAS.
- D. S. Zijlstra, et al., Efficient Mild Organosolv Lignin Extraction in a Flow-Through Setup Yielding Lignin with High beta-O-4 Content, Polymers, 2019, 11(12), 1913 CrossRef CAS.
- K. K. Pandey, A study of chemical structure of soft and hardwood and wood polymers by FTIR spectroscopy, J. Appl. Polym. Sci., 1999, 71(12), 1969–1975 CrossRef CAS.
- S. Wang, et al., Pyrolysis behaviors of four lignin polymers isolated from the same pine wood, Bioresour. Technol., 2015, 182, 120–127 CrossRef CAS PubMed.
- F. Shahidi and Y. Zhong, Measurement of antioxidant activity, J. Funct. Foods, 2015, 18, 757–781 CrossRef CAS.
- N. F. Santos-Sanchez, et al., Antioxidant Compounds and Their Antioxdiant Mechanism, Intech Open, 2019 Search PubMed.
- A. Cano, et al., ABTS/TAC Methodology: Main Milestones and Recent Applications, Processes, 2023, 11(1), 365–370 CrossRef.
- I. V. Pylypchuk, et al., Structural and molecular-weight-dependency in the formation of lignin nanoparticles from fractionated soft- and hardwood lignins, Green Chem., 2021, 23(8), 3061–3072 RSC.
- M. Danaei, et al., Impact of Particle Size and Polydispersity Index on the Clinical Applications of Lipidic Nanocarrier Systems, Pharmaceutics, 2018, 10(2), 57 CrossRef PubMed.
- P. K. Mishra and A. Ekielski, The Self-Assembly of Lignin and Its Application in Nanoparticle Synthesis: A Short Review, Nanomaterials, 2019, 9(2), 243 CrossRef PubMed.
- J. C. Jackson, et al., Sustainable Cellulose Nanocrystals for Improved Antimicrobial Properties of Thin Film Composite Membranes, ACS Sustain. Chem. Eng., 2021, 9(19), 6534–6540 CrossRef CAS.
- V. T. Noronha, et al., Physical Membrane-Stress-Mediated Antimicrobial Properties of Cellulose Nanocrystals, ACS Sustain. Chem. Eng., 2021, 9(8), 3203–3212 CrossRef CAS.
- W. D. H. Schneider, A. J. P. Dillon and M. Camassola, Lignin nanoparticles enter the scene: A promising versatile green tool for multiple applications, Biotechnol. Adv., 2021, 47, 107685 CrossRef CAS PubMed.
- G. Pizzino, et al., Oxidative Stress: Harms and Benefits for Human Health, Oxid. Med. Cell. Longev., 2017, 8416763 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3su00262d |
| This journal is © The Royal Society of Chemistry 2023 |