
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploration of bis(pentafluorophenyl)borinic acid as an electronically saturated, bench-top stable Lewis acid catalyst†
Taylor P. L.
Cosby
 and
Christopher B.
Caputo
*
and
Christopher B.
Caputo
*
Department of Chemistry, York University, 4700 Keele St., Toronto, ON M3J 1P3, Canada. E-mail: caputo@yorku.ca
First published on 3rd August 2023
Abstract
In efforts to develop air- and moisture-stable Lewis acid “frustrated Lewis pair” catalysts, we explored the catalytic reactivity of 9-(bis(perfluorophenyl)boraneyl)-9H-carbazole (1) towards the hydrosilylation of carbonyls. The stability of 1 was followed using nuclear magnetic resonance (NMR) spectroscopy. It was found that 1 cleanly decomposed via hydrolysis to bis(pentafluorophenyl)borinic acid (2). The borinic acid 2 was shown to be stable under ambient atmospheric conditions for upwards of 3 months in the solid-state and 2 weeks dissolved in solution. The Lewis acidity of 2 was evaluated and similarly reactivity towards hydrosilylation was observed. The use of an air-stable, inexpensive, and more sustainable silane, poly(methylhydrosiloxane) (PMHS), affords the reduction of aldehydes to alcohols under atmospheric conditions using 2, emphasizing 2 as a more sustainable, bench-top stable Lewis acid catalyst.
Sustainability spotlightThe industrial mining of transition metals for use as catalysts is extremely harmful to the environment and communities involved. The abundance of precious metals is decreasing rapidly due to their demand in a variety of industries and many common metals have been assigned as ‘critical raw materials’. To combat the issues of sustainability and equity of production, researchers have explored main-group elements as more earth-abundant, cost-effective, and sustainable alternatives to the historically used metal catalysts. Many highly electrophilic boron-based catalysts have been effective in catalytic transformations; however, they are typically air- and moisture-sensitive, making large-scale implementation problematic. Within this landscape, this report focuses on air-stable Lewis acidic boranes which can be applied towards hydrosilylation of carbonyls under both aerobic and anaerobic conditions. This work aims to support industry, innovation, and infrastructure (SDG 9) and responsible consumption and production (SDG 12) through enhancing research and upgrading industrial technologies and responsible management of chemicals and waste. |
Introduction
Transition metal catalysts have historically played a major role in organic synthesis, where a myriad of cross-coupling and bond formation reactions can be catalyzed by precious metals such as platinum, palladium, rhodium, and iridium.1 Although effective, these rare metals pose environmental and equity concerns through their industrial mining and refinement.2–4 Additionally, the natural abundances of these heavier transition metals are extremely low, and several commonly used elements have been designated as critical raw materials by the European Union, emphasizing the risks associated with their supply.5 With crustal abundances of these metals ranging from 10−2 to 10−4 mg kg−1,6 there is a great deal of interest in developing more earth-abundant and sustainable alternatives to perform the same chemical transformations.3,7The use of Lewis acids to facilitate chemical transformations has been known for over a century, but has become increasingly prevalent in the last two decades.8,9 The development of frustrated Lewis pairs (FLPs) allowed for significant progress towards more sustainable metal-free catalytic pathways and has widely increased the library of accessible Lewis acids.10,11 The ubiquitous and commercially available Lewis acid tris(pentafluorophenyl)borane [B(C6F5)3], has been explored extensively as a catalyst both independently and as the partner in FLP systems, finding use in an array of organic transformations.12–15 Although many FLP and Lewis acid catalytic systems have shown comparable reactivity to their historically used transition metal counterparts, sensitivity to ambient atmosphere provides a barrier for the widespread use of highly electrophilic Lewis acid catalysts, due to an irreversible interaction with water.16 Moreover, when target substrates contain strongly Lewis basic heteroatoms, reactivity is typically impacted through formation of a classical Lewis adduct.17,18
The rigorous handling requirements for highly Lewis acidic species limits the applicability of these to be a more earth-abundant alternative to transition metal catalysts. As sustainable alternatives, the development of a robust, benchtop-stable species is key, giving equal opportunity for the use of Lewis acid catalysts by chemists without access to air- and moisture-free infrastructure. With these limitations in mind, there has been interest in designing catalysts that can maintain their Lewis acidity in the presence of water or when handled outside of inert conditions. The approaches taken to stabilize these species include steric shielding or electronic saturation of the Lewis acidic centre. However, there exists a delicate balance in the tuning of Lewis acidity; the more stable the Lewis acid, the weaker Lewis acidic character, and thus a poorer catalyst. Several Lewis acidic species have been presented that are tolerant to atmospheric conditions (Fig. 1) but their use as organic catalysts has been sparce as their synthesis is challenging, limiting their scope.19,20
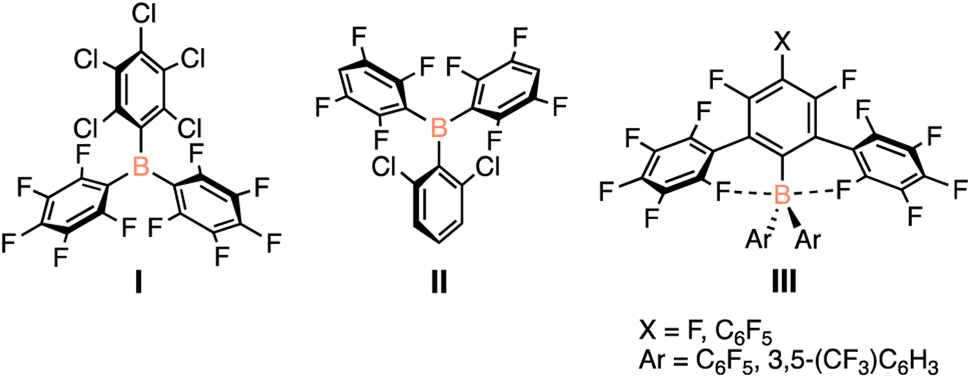 | ||
| Fig. 1 Previously reported bench-top stable electrophilic boranes. | ||
The O'Hare group reported that substituting one of the pentafluorophenyl substituents on B(C6F5)3 with a pentachlorophenyl group (Fig. 1, I) yields a bench-top stable Lewis acid, with the coordination of water shown to be reversible under reduced pressure or the addition of molecular sieves.21 In conjunction with the Lewis basic solvent tetrahydrofuran, I was shown to facilitate FLP-type hydrogenations of imines and weakly basic substrates.22 Although a glovebox-free procedure for hydrogenations was achieved, extended exposure to air still generates the water adduct, which limits catalytic efficacy.22 By decreasing the degree of halogenation of the aryl substituents around boron, the Soós group prepared a bench-top stable species that is able to facilitate catalytic hydrogenation reactions when used in tandem with a Lewis base under atmospheric conditions (Fig. 1, II).19 Finally, the Beckmann group has synthesized bulky boranes that employ a polyfluorinated terphenyl substituent as well as pentafluorophenyl rings providing significant steric shielding of the boron centre as well as strong electron withdrawing groups (Fig. 1, III).23 This species, although shown to be bench-top stable, has yet to be reported to facilitate any catalytic transformations.
An alternative method of stabilizing Lewis acidic molecules is through incorporation of heteroatoms adjacent to the electrophilic site, providing a degree of electronic saturation. The Fontaine group has elegantly shown that ammonium fluoroborates can be used as bench stable FLP pre-catalysts for heteroarene borylation.24,25 Furthermore, our group has reported a series of aminoboranes, species of the formula R2N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) BR2, that feature sterically encumbered amino groups and electron withdrawing pentafluorophenyl substituents at the boron centre.26,27 These aminoboranes have shown some stability towards atmospheric conditions and donating solvents, highlighting their potential as bench-top stable Lewis acids.27 Despite the stability derived from the nitrogen lone pair stabilizing the empty p-orbital at boron, these species are able to maintain some Lewis acidic character through employing two strongly electron withdrawing groups, as evidenced from their high fluoride ion affinities (FIA). Furthermore, the aminoboranes were found to be effective Lewis acid catalysts for stannane dehydrocoupling and a single example of hydrosilylation of acetophenone.27
BR2, that feature sterically encumbered amino groups and electron withdrawing pentafluorophenyl substituents at the boron centre.26,27 These aminoboranes have shown some stability towards atmospheric conditions and donating solvents, highlighting their potential as bench-top stable Lewis acids.27 Despite the stability derived from the nitrogen lone pair stabilizing the empty p-orbital at boron, these species are able to maintain some Lewis acidic character through employing two strongly electron withdrawing groups, as evidenced from their high fluoride ion affinities (FIA). Furthermore, the aminoboranes were found to be effective Lewis acid catalysts for stannane dehydrocoupling and a single example of hydrosilylation of acetophenone.27
Herein we expand the scope and reactivity of electronically saturated Lewis acidic boranes to borinic acids, specifically bis(pentafluorophenyl)borinic acid. This compound represents a potentially more sustainable Lewis acid catalyst, avoiding the need for stringently dry conditions. This approach not only makes Lewis acid chemistry more practical for the broader chemistry community but can reduce the energy and compressed gas consumption needed to handle species under air-free conditions. This work describes a comprehensive stability study of our electronically saturated boranes and benchmarks it to B(C6F5)3, including characterization of decomposition products, as well as a catalytic screening towards hydrosilylation of carbonyls in pursuit of a bench-top stable Lewis acid catalyst.
Results and discussion
Stability of 1 and B(C6F5)3
To begin, an investigation of the atmospheric stability of 9-(bis(perfluorophenyl)boraneyl)-9H-carbazole (1) was conducted in both solution and solid-state. To monitor the stability of 1, 1H and 19F NMR spectroscopy was employed. When dissolved in deuterated benzene (C6D6), 1 decomposed after approximately 3.5 days into a single product. The decomposition product was identified to be bis(pentafluorophenyl)borinic acid (2), which is generated through hydrolysis of 1 with water in the atmosphere (Scheme 1). The signals of the decomposition product in the 1H NMR spectrum match the signals of carbazole, with an additional resonance at 6.23 ppm (ESI Fig. S13 and S14† in C6D6) corresponding to an OH from 2. To test the stability of 1 in the solid-state, a small amount of solid was brought outside the glovebox, opened to atmosphere, and again monitored over time using NMR spectroscopy. In the solid-state, 1 was observed to be more stable than in solution, decomposing after two weeks into 2 (ESI Fig. S17 and S18†).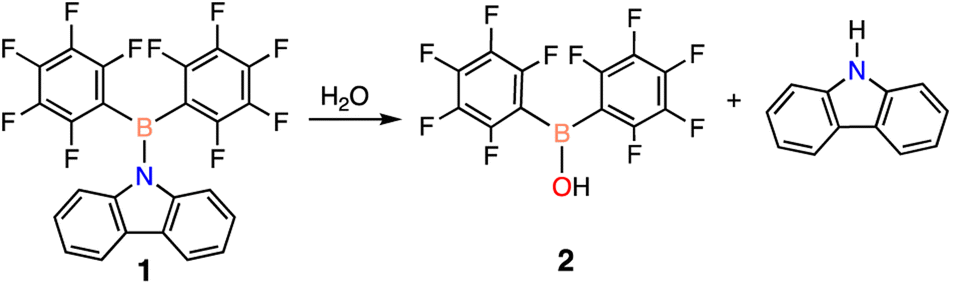 | ||
| Scheme 1 Decomposition of 1 into 2 when exposed to atmosphere. | ||
Similarly to 1, the stability of 2 was assessed in solution and in the solid-state. Again, 1H and 19F NMR spectroscopy was used to follow any potential decomposition. In solution, 2 was found to be more robust when exposed to ambient atmosphere compared to 1, forming pentafluorophenylboronic acid (3) after 14 days (Scheme 2a). However, 2 shows impressive stability compared to 1 in the solid-state with minimal decomposition under ambient conditions over 3 months (ESI Fig. S19†). It is hypothesized that the stability is due to the trimeric solid-state structure 2-t (Scheme 2b) that 2 has been previously reported to form,28 sufficiently protecting the Lewis acidic boron centre from any interactions with water.
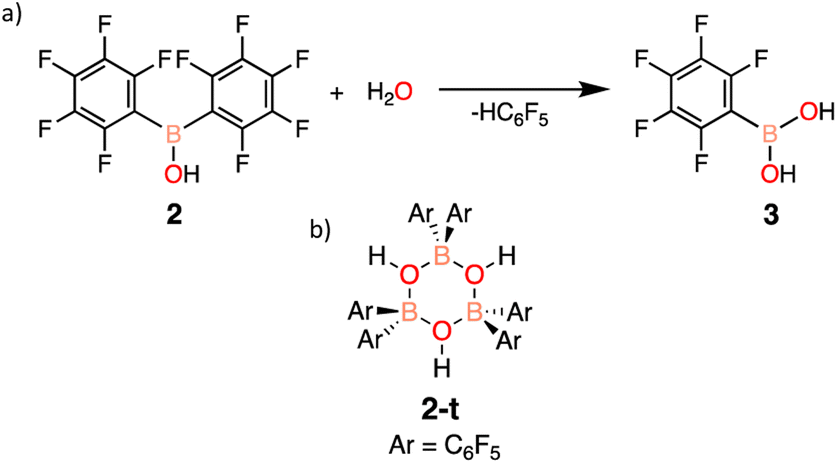 | ||
| Scheme 2 (a) Decomposition of 2 into boronic acid 3 and (b) the solid-state cyclic trimer 2-t. | ||
The stabilities of 1 and 2 were benchmarked to the ubiquitously used Lewis acid B(C6F5)3 to understand the impact of the heteroatom towards air- and moisture-stability in solution and the solid-state. Early reports using B(C6F5)3 as a catalyst suggest it has some degree of water tolerance,29,30 however, it has been shown that B(C6F5)3 can generate stable adducts with either one or two equivalents of water.31,32 The complex of one equivalent of water coordinated to B(C6F5)3, although stable and handled under atmospheric conditions, has been shown to act as a strong Brønsted acid (pKa = 8.4 in acetonitrile) rather than acting a Lewis acid.32 When heated, B(C6F5)3 is not able to be liberated, rather the borinic acid, (C6F5)2BOH, and C6F5H are observed.28 Additionally, the Ashley group has shown that the use of B(C6F5)3 dissolved in reagent grade dioxane (a non-anhydrous solvent) behaves as an FLP hydrogenation catalyst.20 The Lewis acidic reactivity is retained in this system as the coordination of water, yielding a Brønsted acidic species, is in equilibrium with the coordination of dioxane, which is the active FLP catalyst.20
A sample of B(C6F5)3 was dissolved in dry C6D6 and multiple NMR spectra were acquired after one day exposure to air. The signals in the 19F NMR spectrum show a slight upfield shift and general broadening, hypothesized to be due to a mixture of the tricoordinate parent species and tetracoordinate water adduct, [(C6F5)3B(OH2)].32 The 1H NMR spectrum shows one resonance at 4.57 ppm (free H2O = 0.4 ppm), corresponding to the bound water protons. After 4 days, the 19F NMR signals begin to sharpen and shift further upfield with concurrent reduction of the intensity of the resonance in the 1H NMR spectrum. When exposed to an ambient atmosphere for 1 week in solution, the 19F NMR chemical shifts shift further upfield (−135.0 ppm, −155.6 ppm, −163.2 ppm), matching the chemical shifts for the adduct [(C6F5)3B(OH2)]·H2O, reported by Beringhelli et al. (ESI Fig. S16†).32 There is no observable resonance in the 1H NMR spectrum as the water protons are exchanging too rapidly to be observed on the NMR timescale. Interestingly, in the solid-state, B(C6F5)3 shows a more rapid reaction with water from the atmosphere, possibly due to the immiscibility of water in C6D6. After 1 day open to atmosphere, there is evidence of [(C6F5)3B(OH2)]·H2O in the 1H and 19F NMR spectra (ESI Fig. S20†).
Lewis acidity assessment and synthesis of 2
Encouraged by the stability studies, the Lewis acidity of 2 was assessed using experimental and computational methods (Table 1). Both the Gutmann–Beckett33 and Fluorescent Lewis Adduct (FLA)34,35 methods were explored for 1 and 2, however, both species display strong fluorescence in solution and as such were not suitable candidates for the FLA method. On the other hand, the Gutmann–Beckett method was found to be compatible with 1 and 2 resulting in an acceptor number (AN) of 56 for 1 and 66 for 2. Both exhibit weaker acceptor numbers in relation to B(C6F5)3 (AN = 82).36 Computational methods were also employed to scale the Lewis acidity through the fluoride ion affinity (FIA) method using B3LYP(D3)/def2-QZVPP in both gas phase and a polarized continuum solvation model (PCM) for dichloromethane.37 Based on the FIA values, B(C6F5)3 is the strongest Lewis acid (FIA = 448 kJ mol−1) and the aminoborane 1 (413 kJ mol−1) has a higher gas phase FIA than the borinic acid 2 (344 kJ mol−1). Based on the Gutmann–Beckett Lewis acidity assessment, 2 should be more Lewis acidic than 1, however, the FIA assessment shows that 2 is less Lewis acidic.| Compound | 31P Chemical shift (ppm) {AN}a | FIAb | FIAsolvc |
|---|---|---|---|
| a NMR spectra collected in CD2Cl2, AN = 2.21(δsample − δTEPO,DCM). b B3LYP(d3)/def2-QZVPP, values given in kJ mol−1, gas phase optimizations. c B3LYP(D3)/def2-QZVPP, values given in kJ mol−1, PCM solvation model for dichloromethane. | |||
| B(C6F5)3 | 78.1 {82}36 | 448 (ref. 37) | 254 (ref. 37) |
| 1 | 75.7 {56} | 353 (ref. 27) | — |
| 2 | 80.3 {66} | 344 | 263 |
It should be noted that although a number of computational and experimental Lewis acidity assessments have been developed, correlations between such methods are not always observed. Inconsistent values can arise from steric interactions or hard–soft acid–base (HSAB) effects, however, a combination of these methods can be used qualitatively to scale and compare the strengths of Lewis acids.38
Seeing that 1 decomposes when open to ambient atmosphere to 2 and exhibits Lewis acidic character, an independent synthesis of 2 was undertaken. There are two previously reported pathways to synthesize 2, using either (C6F5)2BCl39 or (C6F5)2BPh40 and one equivalent of water. Unfortunately these routes resulted in poor and irreproducible yields of 2 (27–46%).41,42 Therefore we sought a simpler and higher yielding approach, and our revised synthesis starts with (C6F5)2BH and a sub-stoichiometric amount of water added at low temperatures to generate 2. The resulting white solid was triturated with hexanes to obtain 2 in 80% yield, with 19F NMR resonances at −132.4, −147.2, −160.5 ppm in CDCl3 (Scheme 3).
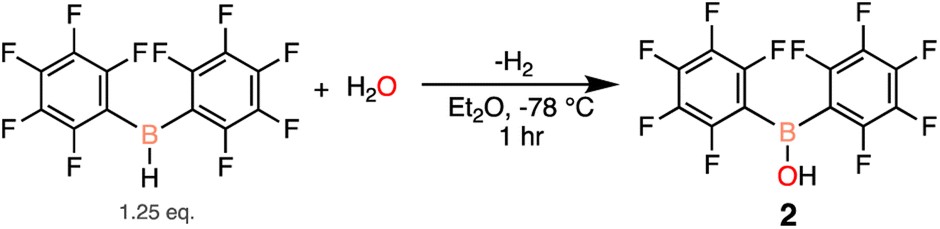 | ||
| Scheme 3 Independent synthesis of 2 starting from Piers' borane (yield = 80%). | ||
Catalytic activity of 1, 2, and B(C6F5)3
Preliminary results that we previously reported suggested that 1 was an effective catalyst for the hydrosilylation of acetophenone. Therefore, we used this as a foundation for the study of a broader scope of carbonyl containing compounds (Fig. 2). The catalytic efficacy of 2 was also explored given that it was robust towards air and moisture yet demonstrated substantial Lewis acidity through computational and experimental analysis. Finally, all the catalytic results were benchmarked to the reactivity of B(C6F5)3 to understand how 1 and 2 compare.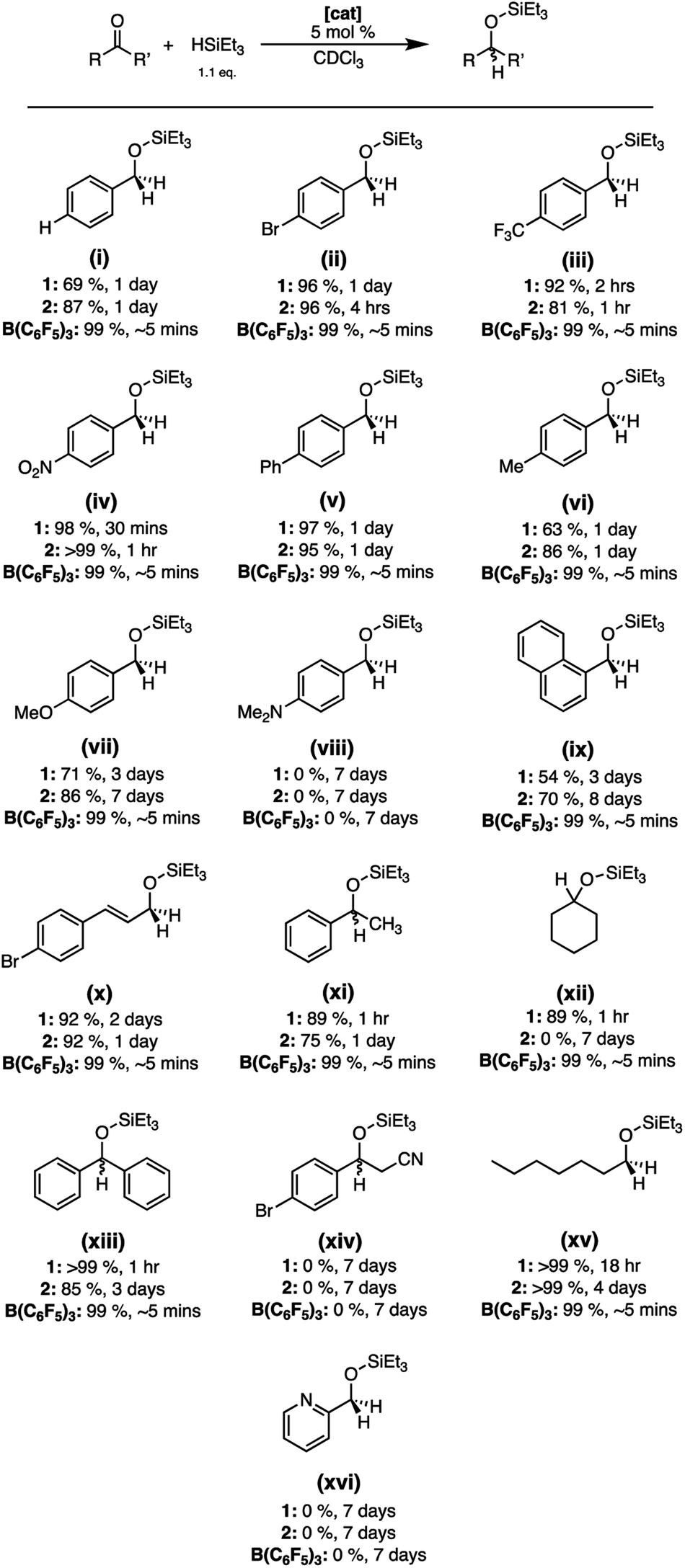 | ||
| Fig. 2 Substrate screening of hydrosilylation using 1, 2, and B(C6F5)3 under inert conditions. | ||
It was found that hydrosilylation can be achieved using 1, 2, and B(C6F5)3 under inert conditions using a 5 mol% catalyst loading in CDCl3 with most substrates, with conversions ranging from 54–99% for 1, 70–99% for 2, and 99% with B(C6F5)3 (Fig. 2). Reactions using B(C6F5)3 as the catalyst showed quantitative conversion within ∼5 minutes, whereas 1 and 2 often required longer timeframes. Benzaldehyde substrates containing electron withdrawing substituents were converted to the reduced product more quickly and had higher conversion than substrates containing electron donating groups when using 1 and 2 as catalysts. In general, 2 showed similar or higher conversions than 1 using substituted benzaldehyde substrates but needed more time to reach the end of the reaction (Fig. 2, i–xvi). Interestingly, substrates containing a Lewis basic nitrogen atom (Fig. 2, viii, xiv, xvi) did not yield the reduced product with any of the catalyst systems. In these cases, coordination of the nitrogen to the boron centre is observed, evidenced by the broadening and upfield shift of the resonances in the 1H NMR spectra. Unexpectedly, the use of 2 as a catalyst failed to show conversion when cyclohexanone was used as a substrate, yet 1 was able to reach 69% conversion after 3 days (Fig. 2, xii). Nevertheless, the catalytic data aligns well with the experimental and computational Lewis acidity assessment with 2 typically displaying higher conversions than 1, albeit less than B(C6F5)3. However, 1 tended to reach maximum conversion faster than 2, which highlights the complexity of balancing Lewis acidity with observed reactivity.21,43
After expanding the scope of substrates compatible for hydrosilylation, a solvent screening was initiated (Table 2, entries 1–3). It was found that employing solvents that are more polar, such as dichloromethane (DCM) and chloroform, the catalytic activity was increased compared to benzene. Surprisingly, using acetonitrile, a polar and Lewis basic solvent known to coordinate to Lewis acids, hydrosilylation still occurs using 1, 2, and B(C6F5)3, achieving comparable results to using DCM and chloroform. Previous literature reports state that an adduct is formed with B(C6F5)3 and acetonitrile, but this coordination is reversible which maintains some catalytic activity, albeit weaker than in non-donating solvents.44 Even more interestingly, 2 shows stronger catalytic activity using acetonitrile as a solvent than 1 and B(C6F5)3, with 99% conversion to the hydrosilylated product after 24 hours.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Condition | 1 | 2 | B(C6F5)3 | |||
| Time (h) | Yielda (%) | Time (h) | Yielda (%) | Time (h) | Yielda (%) | ||
| a All qNMR yields were calculated in reference to an internal standard, mesitylene (C9H12). | |||||||
| 1 | Benzene-d6 | 24 | >99 | — | — | ∼5 min | >99 |
| 2 | DCM-d2 | 3 | 92 | — | — | ∼5 min | >99 |
| 3 | Acetonitrile-d3 | 4 | 58 | 18 | 81 | 4 | 66 |
| 4 | Dissolved outside the glovebox | 8 | 91 | 24 | 96 | ∼5 min | >99 |
| 5 | Exposed to atmosphere for 1 week | 24 | 77 | 24 | 78 | ∼5 min | >99 |
| 6 | Exposed to atmosphere for 2 weeks | 24 | 80 | 24 | 74 | ∼5 min | 86 |
| 7 | Non-anhydrous CDCl3 | 48 | 79 | 24 | 84 | ∼5 min | >99 |
Finally, the ability of 1, 2, and B(C6F5)3 to catalyze the hydrosilylation of benzaldehyde when exposed to ambient atmosphere was explored using CDCl3 and 5 mol% catalyst (Table 2, entries 4–7). Given that no decomposition of 2 was observed in the solid-state, the catalytic activity remained consistent over time. There was no stark difference in conversion using 1 or B(C6F5)3 when dissolved under inert or atmospheric conditions. When 1 was left outside of the glovebox for 1 week and approximately 75% of the parent aminoborane remained, a decrease in efficacy of 14% was observed (Table 2, entry 5). After 2 weeks, 1 had fully decomposed into 2 and similar reactivity to the independently synthesized 2 was observed (Table 2, entry 6). Additionally, catalytic activity has persisted when 1 or 2 are dissolved in non-anhydrous solvents (Table 2, entry 7), showcasing the potential of 2 to be used catalytically and stored under atmospheric conditions. This strongly suggests that bis(pentafluorophenyl)borinic acid 2 is a capable bench-top stable catalyst for the hydrosilylation of carbonyls.
In contrast, when using B(C6F5)3 that has been left out for 1 week, there was no observable change in conversion, however, a decrease in performance by 13% was observed when B(C6F5)3 was exposed to atmosphere for 2 weeks (Table 2, entries 5 and 6). This finding provides more concrete evidence in support of the ‘water-tolerance’ that has previously been reported for B(C6F5)3; even as an electronically saturated, water-coordinated borane, B(C6F5)3 still outperforms 1 and 2 towards the hydrosilylation of benzaldehyde under these conditions. Our results indicate that B(C6F5)3 can be handled outside of inert conditions with comparable reactivity, limiting the need for strict handling and storage conditions.
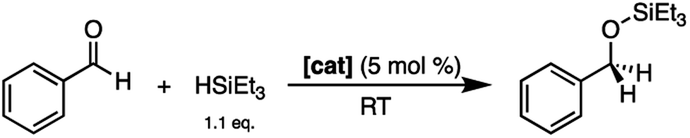
After 2 was shown to be a capable catalyst for the hydrosilylation of carbonyls using triethylsilane under ambient conditions, a cheaper and more sustainable silane source was investigated: poly(methylhydrosiloxane) (PMHS).45 PMHS is a low-cost, non-toxic reductant formed as a by-product of the Müller–Rochow process in the silicone industry, and unlike Et3SiH, is bench-top stable.46 The use of this by-product in atmospheric conditions allows for the reduction of carbonyls using a more sustainable approach than other traditional pyrophoric silanes.47 With these advantages, the reduction of 4-bromobenzaldehyde using PMHS and 2 was undertaken using wet solvents under ambient atmosphere. Upon addition of 5 mol% of 2 and four equivalents of PMHS, full conversion to the silyl ether was obtained in 24 hours. The protected carbonyl was then treated with one equivalent of methanol to yield 4-bromobenzyl alcohol in 54% isolated yield (Scheme 4). The ability of 2 to catalyze the reduction of aldehydes to alcohols using an air stable reductant further emphasizes the potential of 2 as an accessible, bench-top stable, Lewis acid catalyst.
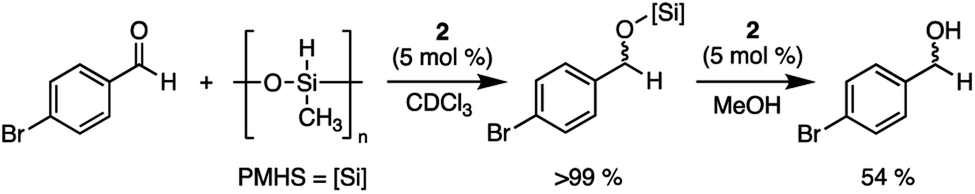 | ||
| Scheme 4 Reduction of 4-bromobenzaldehyde using PMHS and 2 to a silyl ether, followed by protonation with methanol to yield 4-bromobenzyl alcohol. | ||
There are limited reports in the literature of applications of bis(pentafluorophenyl)borinic acid in catalysis. Examples of catalytic applications of 2 have been reported for Oppenauer oxidations48 and sugar reduction chemistry.40 Although it was stated by Yamamoto and co-workers that 2 can be ‘readily handled in air’,48 these previous explorations of 2 as a catalyst apply air sensitive techniques.40,48 It has also been hypothesized recently by Chang and co-workers that 2 can act as an in situ source of Piers' borane [(C6F5)2BH] to cleave C–O bonds in sugars.40 The reactions were again performed under inert conditions and a large excess of highly reducing PhSiH3 was necessary.40
To explore whether Piers' borane was generated in our chemistry with Et3SiH, 2 was reacted with a stoichiometric amount of triethylsilane in CDCl3. Immediate effervescence of H2 was observed and the B–O–Si silyl ether was detected by 1H (0.93, 0.67 ppm), 19F (−132.6, −149.4, −161.1 ppm), and 11B (38.3 ppm) NMR spectroscopy after 24 hours (Scheme 5, 4). To further eliminate the possibility that Piers' borane was generated in situ, two equivalents of triethylsilane was added, yet there was no evidence of Piers' borane formation by 1H, 19F, and 11B NMR spectroscopy, only the formation of the B–O–Si silyl ether 4 (ESI Fig. S24–S26†).
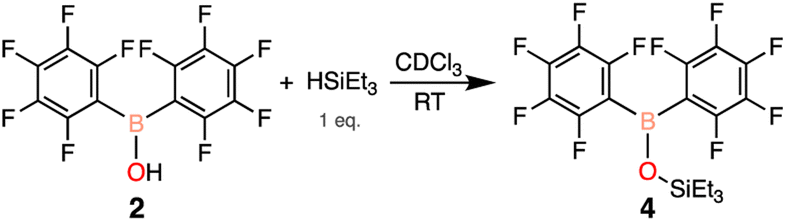 | ||
| Scheme 5 Reactivity of 2 with 1 eq. of triethylsilane after 24 hours. | ||
Finally, a kinetic investigation into the hydrosilylation of benzaldehyde using 2 was undertaken to rule out Piers' borane mediated catalysis. An induction period may be expected if the borinic acid was being converted to the hydridoborane, but the results show no induction period (ESI Fig. S23†) and thus is likely to proceed via a similar silane activated mechanism, akin to that proposed for B(C6F5)3.49
Conclusions
We found that bis(pentafluorophenyl)borinic acid is an effective and stable catalyst for the hydrosilylation of carbonyls, including under ambient conditions and using donating solvents. The use of 2 and the industrial by-product PMHS could facilitate the reduction of carbonyls into alcohols under atmospheric conditions, demonstrating the exciting potential of 2 as a bench-top stable Lewis acid catalyst. Solution and solid-state stability studies were performed on 2 showing moderate stability in solution, however, 2 is indefinitely stable towards air and moisture in the solid state. Catalytic activity towards hydrosilylation using 1, 2, and B(C6F5)3 was shown using triethylsilane and various carbonyl containing substrates. When the reactions were performed under atmospheric conditions, all three catalysts could facilitate hydrosilylation of benzaldehyde with a minimal decrease in effectiveness. Furthermore, the use of B(C6F5)3 under atmospheric conditions and in donating solvents has highlighted that it may be more water tolerant than previously suspected, showcasing the potential of B(C6F5)3 to be used outside of inert conditions.These results show exciting promise towards understanding the effects of heteroatom stabilization on the development of bench-top stable Lewis acid catalysts. Exploration of the catalytic capabilities of 2 towards a variety of organic transformations under atmospheric conditions is ongoing. Future work will continue to explore varying the electron withdrawing substituents and heteroatom groups at the boron centre to tune the Lewis acidity with a focus on developing practical, benchtop-stable, sustainable alternatives to precious metal catalysts.
Author contributions
Conceptualization, C. B. C.; methodology, T. P. L. C., and C. B. C.; formal analysis, T. P. L. C.; investigation, T. P. L. C.; writing original draft, T. P. L. C., and C. B. C.; writing – review & editing, T. P. L. C., and C. B. C.; supervision, C. B. C.; funding acquisition, C. B. C.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Natural Sciences and Engineering Research Council of Canada (NSERC), Canadian Foundation for Innovation (CFI), and Canada Research Chairs Program is thanked for their support of C. B. Caputo. Charley Garrard is thanked for her assistance with UV-Vis and fluorescence spectroscopy for the FLA method analysis. Dr Howard Hunter is thanked for his NMR assistance. T. P. L. Cosby acknowledges the support provided by the Ontario Government through an Ontario Graduate Scholarship.Notes and references
- A. E. Hughes, N. Haque, S. A. Northey and S. Giddey, Resources, 2021, 10, 93–133 CrossRef.
- Y. Seo and S. Morimoto, Resources, 2017, 6, 61–74 CrossRef.
- F. P. Carvalho, Food Energy Secur., 2017, 6, 61–77 CrossRef.
- É. Lèbre, M. Stringer, K. Svobodova, J. R. Owen, D. Kemp, C. Côte, A. Arratia-Solar and R. K. Valenta, Nat. Commun., 2020, 11, 4823–4831 CrossRef PubMed.
- E. Garbarino, F. Ardente and D. Blagoeva, Critical raw materials and the circular economy, European Commission, Joint Research Centre, Luxembourg, 2017 Search PubMed.
- W. M. Haynes, D. R. Lide and T. J. Bruno, CRC Handbook of Chemistry and Physics, CRC Press, Boca Raton, FL, 97th edn, 2016 Search PubMed.
- I. Vural Gürsel, T. Noël, Q. Wang and V. Hessel, Green Chem., 2015, 17, 2012–2026 RSC.
- V. Nori, F. Pesciaioli, A. Sinibaldi, G. Giorgianni and A. Carlone, Catalysts, 2022, 12, 5–59 CrossRef CAS.
- J. M. Bayne and D. W. Stephan, Chem. Soc. Rev., 2016, 45, 765–774 RSC.
- G. C. Welch, R. R. San Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef CAS PubMed.
- D. W. Stephan, Acc. Chem. Res., 2015, 48, 306–316 CrossRef CAS PubMed.
- D. W. Stephan, Science, 2016, 354, aaf7229 CrossRef PubMed.
- D. J. Parks and W. E. Piers, J. Am. Chem. Soc., 1996, 118, 9440–9441 CrossRef CAS.
- T. Hackel and N. A. McGrath, Molecules, 2019, 24, 432–462 CrossRef PubMed.
- A. Berkefeld, W. E. Piers and M. Parvez, J. Am. Chem. Soc., 2010, 132, 10660–10661 CrossRef CAS PubMed.
- S. Kobayashi and K. Manabe, Pure Appl. Chem., 2000, 72, 1373–1380 CrossRef CAS.
- A. Corma and H. García, Chem. Rev., 2003, 103, 4307–4366 CrossRef CAS PubMed.
- J.-M. Begouin and M. Niggemann, Chem.–Eur. J., 2013, 19, 8030–8041 CrossRef CAS PubMed.
- Á. Gyömöre, M. Bakos, T. Földes, I. Pápai, A. Domján and T. Soós, ACS Catal., 2015, 5, 5366–5372 CrossRef.
- D. J. Scott, T. R. Simmons, E. J. Lawrence, G. G. Wildgoose, M. J. Fuchter and A. E. Ashley, ACS Catal., 2015, 5, 5540–5544 CrossRef CAS PubMed.
- A. E. Ashley, T. J. Herrington, G. G. Wildgoose, H. Zaher, A. L. Thompson, N. H. Rees, T. Krämer and D. O'Hare, J. Am. Chem. Soc., 2011, 133, 14727–14740 CrossRef CAS PubMed.
- D. J. Scott, M. J. Fuchter and A. E. Ashley, Angew. Chem., Int. Ed., 2014, 53, 10218–10222 CrossRef CAS PubMed.
- C. Stoian, M. Olaru, T. A. Cucuiet, K. T. Kegyes, A. Sava, A. Y. Timoshkin, C. I. Raţ and J. Beckmann, Chem.–Eur. J., 2021, 27, 4327–4331 CrossRef CAS PubMed.
- M.-A. Légaré, É. Rochette, J. Légaré Lavergne, N. Bouchard and F.-G. Fontaine, Chem. Commun., 2016, 52, 5387–5390 RSC.
- A. Jayaraman, L. C. Misal Castro and F.-G. Fontaine, Org. Process Res. Dev., 2018, 22, 1489–1499 CrossRef CAS.
- J. N. Bentley, E. Pradhan, T. Zeng and C. B. Caputo, Dalton Trans., 2020, 49, 16054–16058 RSC.
- J. N. Bentley, S. A. Simoes, E. Pradhan, T. Zeng and C. B. Caputo, Org. Biomol. Chem., 2021, 19, 4796–4802 RSC.
- T. Beringhelli, G. D'Alfonso, D. Donghi, D. Maggioni, P. Mercandelli and A. Sironi, Organometallics, 2003, 22, 1588–1590 CrossRef CAS.
- K. Ishihara, N. Hanaki and H. Yamamoto, Synlett, 1993, 8, 577–579 CrossRef.
- K. Ishihara, N. Hanaki, M. Funahashi, M. Miyata and H. Yamamoto, Bull. Chem. Soc. Jpn., 1995, 68, 1721–1730 CrossRef CAS.
- C. Bergquist, B. M. Bridgewater, C. J. Harlan, J. R. Norton, R. A. Friesner and G. Parkin, J. Am. Chem. Soc., 2000, 122, 10581–10590 CrossRef CAS.
- T. Beringhelli, D. Maggioni and G. D'Alfonso, Organometallics, 2001, 20, 4927–4938 CrossRef CAS.
- U. Mayer, V. Gutmann and W. Gerger, Monatsh. Chem., 1975, 106, 1235–1257 CrossRef CAS.
- J. N. Bentley, S. A. Elgadi, J. R. Gaffen, P. Demay-Drouhard, T. Baumgartner and C. B. Caputo, Organometallics, 2020, 39, 3645–3655 CrossRef CAS.
- A. E. Laturski, J. R. Gaffen, P. Demay-Drouhard, C. B. Caputo and T. Baumgartner, Precis. Chem., 2023, 1, 49–56 CrossRef CAS PubMed.
- M. M. Morgan, A. J. V. Marwitz, W. E. Piers and M. Parvez, Organometallics, 2013, 32, 317–322 CrossRef CAS.
- P. Erdmann, J. Leitner, J. Schwarz and L. Greb, ChemPhysChem, 2020, 21, 987–994 CrossRef CAS PubMed.
- L. Greb, Chem.–Eur. J., 2018, 24, 17881–17896 CrossRef CAS PubMed.
- E. Le Coz, J. Hammoud, T. Roisnel, M. Cordier, V. Dorcet, S. Kahlal, J. Carpentier, J. Saillard and Y. Sarazin, Chem.–Eur. J., 2021, 27, 11966–11982 CrossRef CAS PubMed.
- J. Zhang, S. Park and S. Chang, Angew. Chem., 2017, 129, 13945–13949 CrossRef.
- A. Ueno, J. Li, C. G. Daniliuc, G. Kehr and G. Erker, Chem.–Eur. J., 2018, 24, 10044–10048 CrossRef CAS PubMed.
- P. A. Deck, C. L. Beswick and T. J. Marks, J. Am. Chem. Soc., 1998, 120, 1772–1784 CrossRef CAS.
- G. Erős, H. Mehdi, I. Pápai, T. A. Rokob, P. Király, G. Tárkányi and T. Soós, Angew. Chem., Int. Ed., 2010, 49, 6559–6563 CrossRef PubMed.
- P. Bach, A. Albright and K. K. Laali, Eur. J. Org. Chem., 2009, 2009, 1961–1966 CrossRef.
- N. J. Lawrence, M. D. Drew and S. M. Bushell, J. Chem. Soc., Perkin Trans. 1, 1999, 23, 3381–3391 RSC.
- Y. Zhang, J. Li, H. Liu, Y. Ji, Z. Zhong and F. Su, ChemCatChem, 2019, 11, 2757–2779 CrossRef CAS.
- N. M. Hein, Y. Seo, S. J. Lee and M. R. Gagné, Green Chem., 2019, 21, 2662–2669 RSC.
- K. Ishihara, H. Kurihara and H. Yamamoto, J. Org. Chem., 1997, 62, 5664–5665 CrossRef CAS.
- S. Rendler and M. Oestreich, Angew. Chem., Int. Ed., 2008, 47, 5997–6000 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3su00222e |
| This journal is © The Royal Society of Chemistry 2023 |