
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective reduction of oxalic acid to glycolic acid at low temperature in a continuous flow process†
Eric
Schuler
 a,
Lars
Grooten
a,
Paula
Oulego
b,
N. Raveendran
Shiju
a and
Gert-Jan M.
Gruter
*ac
a,
Lars
Grooten
a,
Paula
Oulego
b,
N. Raveendran
Shiju
a and
Gert-Jan M.
Gruter
*ac
aVan ‘t Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 904, 1090 GD Amsterdam, The Netherlands. E-mail: g.j.m.gruter@uva.nl
bDepartment of Chemical and Environmental Engineering, University of Oviedo, C/Julián Clavería s/n., Oviedo, E-33071, Spain
cAvantium N.V., Zekeringstraat 29, 1014 BV Amsterdam, The Netherlands
First published on 13th October 2023
Abstract
Next to biomass, CO2 is the only other carbon source to replace fossil feedstocks to produce chemicals and polymers. In a CO2 fed chemical industry, new platform chemicals will arise. Oxalic acid derived from CO2 based formate is one of them. It can be converted to a wide array of chemicals including ethylene glycol, or carboxylic acids such as glycolic acid. Glycolic acid is currently a fossil-based monomer used in the production of polyesters. Today the most common route from oxalic acid to glycolic acid proceeds via the oxalic acid di-esters as intermediates and thus requires multiple steps. Recently, we have proven that the direct reduction of oxalic acid to glycolic acid at high yields is possible. In this work, we translate this process into a stable and continuous process using industrially relevant conditions. We lowered the reaction temperature by 25 °C to 50 °C, reduced the reaction time from hours to minutes and proved the catalyst stability over 100 hours using oxalic acid derived from CO2. This research lays the foundation for an industrial continuous process for the direct reduction of oxalic acid to glycolic acid and opens an important route from CO2 to chemicals.
Sustainability spotlightIf our goal is a fossil-free society, we must also transition to a fossil-free chemical industry. We have taken a significant step towards achieving this by developing a stable and continuous process for the direct reduction of oxalic acid to glycolic acid. Glycolic acid today is fossil-based and can play a vital role in the future as a monomer or a monomer precursor for sustainable polyesters. Our innovation offers a more sustainable and efficient method of producing chemicals and polymers, which is aligned with several of the United Nations Sustainable Development Goals, including Goal 7 (Affordable and Clean Energy), Goal 9 (Industry, Innovation, and Infrastructure), Goal 11 (Sustainable Cities and Communities), Goal 12 (Responsible Consumption and Production), and Goal 13 (Climate Action). |
Introduction
Replacing fossil feedstocks to mitigate climate change is one of the pressing issues for the chemical and polymer industry.1–7 Oxalic acid fits into this scope as it can be produced sustainably from CO2 or biomass using renewable energy sources and is an interesting new platform chemical to produce mono ethylene glycol, glyoxylic acid, glyoxal, glycolaldehyde and glycolic acid.8 This work was part of the ‘OCEAN’ project which focuses on developing a process to produce sustainable polymers from CO2 as shown in Fig. 1.9 We aim to produce glycolic acid which is a promising monomer for the production of new sustainable polymers.10,11 Oxalic acid sits at the heart of this process and is obtained via the coupling of two CO2 derived formate molecules to oxalate and subsequent acidification.12–14 | ||
| Fig. 1 “OCEAN’’ process for CO2 utilization via (i) electrochemical reduction to formate, (ii) thermal formate coupling to oxalate, (iii) electrochemical oxalate acidification, (iv) catalytic reduction of oxalic acid to glycolic acid, and (v) polymer production from oxalic acid and its derivatives such as glycolic acid.12,14,15 | ||
Today, the route from oxalic to glycolic acid proceeds via esterification to a di-ester of oxalic acid and its subsequent reduction to glycolic acid.16–22 We aim to circumvent the esterification and reduce oxalic acid to glycolic acid directly to decrease the required reaction steps, complexity and cost of the overall process. Interestingly Carnahan et al. showed already in 1954 that oxalic acid could be directly reduced to ethylene glycol with ruthenium catalyst in a hydrogen atmosphere above 110–150 °C.23 In our previous work we have tested a wide array of metals known to be active in hydrogenation reactions and we tested the most active metals on various supports. Ruthenium was by far the most active and selective in the oxalic acid hydrogenation and carbon proved to be the most suitable support.15 We and others showed recently that the selective reduction to glycolic acid is also feasible with ruthenium catalysts, achieving glycolic acid yields above 90%.15,24,25 The reduction of oxalic acid requires reaction temperatures 75 °C lower than compared to the reduction of other carboxylic acids and is completed within several hours in batch reactors.31 The reaction follows a Langmuir–Hinshelwood mechanism with adsorption of oxalic acid on the active ruthenium site and hydrogen splitting on the carbon support.24 The most common side reactions, shown in Fig. 2, are the formation of acetic acid, directly formed from oxalic acid (not from glycolic acid), or the over-reduction of glycolic acid to ethylene glycol.31 At temperatures above 120 °C oxalic acid starts to decompose to volatile products, mainly CO2.
 | ||
| Fig. 2 Reaction pathways in the hydrogenation of oxalic acid. (A) Deprotonation of oxalic acid in water (pKa = 1.27). (B) Desired route to glycolic acid (17.8 kJ mol−1), (C) overreduction to ethylene glycol (16 kJ mol −1), (D) formation of acetic acid (23 kJ mol −1), (E) decomposition to volatile compounds dominant at temperatures above 120 °C.15 | ||
Catalyst stability is a concern in this reaction as the industrial use of oxalic acid as a metal-leaching agent suggests.26–28 It is desirable to perform the reaction at a low temperature to reduce the leaching of ruthenium and improve catalyst stability. Our goal is to develop a continuous process for the reduction of oxalic acid to glycolic acid which avoids the over reduction to ethylene glycol and the formation of volatiles or acetic acid. We use a trickle-bed reactor and first establish suitable conditions for the reaction using a commercially available ruthenium catalyst. One of the challenges when translating a process from batch to flow is the change of parameter settings such as reaction stoichiometry or reaction time. In a batch reactor, stoichiometry depends on the concentration of the reactants and the volumetric ratio. Whereas in a trickle bed reactor, the concentration of the reactants and the ratio of their individual residence time (determined by the flow rate) define the stoichiometry. In both batch and flow processes, the reaction time depends on how long the reactant is exposed to the catalyst at a certain temperature. In batch, this is defined by the time of the reactant in the vessel but inflow processes depend on the flow rate of the reactant through the isothermal zone of the catalyst bed. For the translation, we had to optimize four parameters: reaction temperature, hydrogen pressure and residence time which is influenced by the flow rate of the aqueous oxalic acid solution and flow of hydrogen. In a second step, we aim to test the long-term stability of the catalyst with real oxalic acid feed from the OCEAN project and investigate the structural stability of the catalyst.
Methods
We performed all fixed bed reactions at Avantium and analysed the catalysts at the University of Amsterdam and the University of Oviedo. We used a trickle-bed single flow reactor system (Fig. 3) which allows unlimited continuous operation and the automated collection of up to 8 samples per experiment without intervention. We sequentially studied the influence of reaction temperature (50–100 °C), feed-flow (0.1–0.8 ml min−1) hydrogen pressure (10–60 bar) and hydrogen flow (50–200 ml min−1). In each experiment, we duplicated the measurement for each setting to guarantee reproducibility (Fig. 3C). During the optimization, we kept the length and volume of the catalyst bed and the packing (Fig. 3B) and the reactant concentration (5 wt% oxalic acid in water) constant. For each experiment, we used a fresh catalyst and collected 8 liquid samples which we analysed on an Agilent 1260 Infinity II HPLC system for analysis of liquid samples, equipped with an autosampler, heated column compartment, diode-array detector (DAD) and refractive index detector (RID). The column was an Aminex HPX-87H (300 × 7.8 mm; dp 9 μm). 7 μL sample was injected and separated using 5 mM H2SO4 in MilliQ water (flow rate: 0.6 ml min−1, 30 °C) as the mobile phase. All conditions are listed in the table below. For the long term-stability tests, we used the most suitable reaction conditions and real feed from the upstream process in the CO2 to chemical process. All chemicals were reagent grade and obtained from Sigma Aldrich. We used 950 ± 25 mg of the same commercial catalyst 9.76 wt% Ru/C from Johnson Matthey with particle size between 105–200 μm for all experiments in this work. The catalyst was reduced ex situ at 350 °C in a hydrogen atmosphere, packed into the reactor bed and pre-reduced in situ in the flow reactor at 200 °C in a hydrogen atmosphere. To study the influence of the reaction on the catalyst we analysed the catalyst with powder X-ray diffraction (XRD), high resolution scanning tunnel electron microscopy (HRSTEM) and energy-dispersive X-ray spectroscopy (HRSTEM-EDS) before and after 100 hours exposure to the oxalic acid solution at reaction conditions. The exact conditions for each experiment can be found in the ESI.† | ||
| Fig. 3 (A) Schematic drawing of trickle-bed flow reactor system used for the development of oxalic acid to glycolic acid reduction reaction. The reactor itself is fed from an oxalic acid tank via an HPLC pump and the gas is fed individually via mass-flow controllers. After passing through the reactor, the exiting liquids and gases can either be directed to a waste container or via a selector valve to one of 8 sample vials during the experiment. (B) Packing of catalyst in the reactor. (C) Exemplary test program for reaction temperature starting with an in situ pre-reduction at 200 °C followed by two successive tests at four temperatures. | ||
Results and discussion
In our previous work,31 we established suitable reaction conditions for the reduction of oxalic acid to glycolic acid in a stirred batch reactor. Here, we aim to translate the reaction to a continuous flow process in a trickle-bed reactor (Fig. 3A, more details in ESI†). Before the optimization, we performed control experiments without catalyst and with catalyst but without hydrogen to make sure all conversions of oxalic acid are truly catalytic. We also performed an additional control experiment in which we fed glycolic acid in water to the reactor. From these pre-experiments, we can conclude that the conversion of oxalic acid requires the presence of a catalyst and pressurised hydrogen. Ethylene glycol is derived from the reduction of glycolic acid whilst acetic acid is formed from oxalic acid directly and not from glycolic acid.Fig. 4A shows that the conversion of oxalic acid increases with temperature from 50–70 °C, reaching 100% and then remains constant with further rise in temperature. At 50 °C, oxalic acid is converted to glycolic acid only (Fig. 4B). With increasing reaction temperatures, the reaction rate increases, and ethylene glycol is formed from glycolic acid and if ethylene glycol is desired as product it can be produced selectively. The formation of ethylene glycol already starts before the complete conversion of oxalic acid as the reaction at 60 °C shows. Acetic acid formation remains constant at ∼5% and is independent of temperature (at 50 and 100 °C, acetic acid was detected but was below 3%, the detection limit for quantification). No other side reactions were observed, and the carbon balance is 100% during these experiments. In comparison to the reactions performed in the stirred batch reactor we could decrease the reaction temperature and residence times required for full conversion. Improved mass transfer of hydrogen to the catalyst surface through the thinner liquid layer increases the availability of the reducing agent and allows for easier hydrogenation of oxalic acid. When we reduced the residence time by 50% (tested only in the 50–65 °C range) we observe an overall reduction in conversion (Fig. S1A†). The selectivity was not affected and the over reduction to ethylene glycol could be prevented also at higher temperatures (Fig. S1C†). In a series of feed flow experiments, we explored the influence of residence time further. We reduced the residence time from 18.2 minutes to 9, 4.5 and 2.25 minutes by increasing the feed flow (Fig. 4D–F), which reduced the overall conversion without affecting the selectivity. The acetic acid selectivity remained constant for all feed flows, in line with the observed data for the temperature experiments.
 | ||
| Fig. 4 Influence of temperature, feed flow, hydrogen pressure and hydrogen flow on the reduction of 5 wt% of oxalic acid in water in a trickle-flow reactor with 9.76 wt% Ru/C catalyst from Johnson Matthey. (A–C) Influence of temperature from 50–100 °C on (A) conversion, (B) carbon balance and (C) selectivity at a feed-flow of 0.1 ml min−1, 60 bar, 200 ml min−1. (D–F) Influence of feed-flow from 0.1–0.8 ml min−1 on (D) conversion, (E) carbon balance and (F) selectivity at 70 °C, 60 bar, and 200 ml min −1 flow of hydrogen. (G–I) Influence of hydrogen pressure from 10–25 bar on (G) conversion, (H) carbon balance and (I) selectivity at 70 °C, feed-flow of 0.1 ml min−1 and hydrogen flow of 200 ml min−1. (J–L) Influence of hydrogen flows from 50–200 ml min−1 on (J) conversion, (K) carbon balance and (L) selectivity at 80 °C, feed-flow of 0.1 ml min−1 and 60 bar. The error for all measurements was estimated to be below 5% and hence no error-bars were added. | ||
The supply of hydrogen, the co-reactant in the reaction, can be altered by changing its flow rate and pressure. At 70 °C, the conversion of oxalic acid to glycolic acid increased with increasing hydrogen pressures (Fig. 4G–I). At 80 °C the oxalic acid was already fully converted at all pressures but the over reduction to ethylene glycol increased linearly with hydrogen pressure (Fig. S2†). The production of acetic acid was not influenced by the hydrogen pressure. Interestingly, the effect of flow rate was temperature dependent. At 80 °C all oxalic acid was fully converted and the over reduction of glycolic acid to ethylene glycol reduced with increasing hydrogen flow (Fig. 4 J–L). This effect does not follow a linear trend and decreases with increasing hydrogen flow. When we lowered the temperature to 70 °C and reduced the hydrogen pressure, the overall conversion was lowered to a maximum of 85% (Fig. S3†). Interestingly we then did not observe any effect of the hydrogen flow on the conversion of oxalic acid to glycolic acid. Hence, an increasing hydrogen flow appears to reduce the residence time or reduce the adsorption of glycolic acid and therefore prevents it's over reduction to ethylene glycol. The change in gas-flow can have an influence on the turbulences in the reactor, which increase with higher gas flows. Increasing turbulence affects the mass transfer and gas–liquid flow regime. This can explain why glycolic acid, which adsorbs less to the active surface then oxalic acid, is desorbed and is not reduced to ethylene glycol at higher hydrogen flow rates. Again, the selectivity to acetic acid was constant at around 5% for all examined pressures.
Overall, longer residence time, temperature and availability of hydrogen increase the conversion of oxalic acid and glycolic acid but do not affect the selectivity. Higher residence times increase conversion and can be obtained by lowering flow rates of both oxalic acid and hydrogen. The availability of hydrogen increases with higher pressures. The formation of acetic acid was not affected by any of these parameters.
Long term stability
As this work is part of the development of an industrial process from CO2 to chemicals or polymers, we tested the reaction with the real feed coming from the electrochemical acidification reactor in which the oxalic acid is produced from potassium oxalate. The only difference to the conditions established earlier was the lower concentration of oxalic acid at 2.37 wt% in water caused by concentration limitations in the oxalate to oxalic acid acidification reactor. To prove the stability of the process, we aimed at least at a reaction time of 100 hours in a single uninterrupted reaction. Unfortunately, the conversion dropped drastically already after 16.5 hours (Fig. 5B). Earlier we had used the same catalyst for more than 100 hours without much deactivation, so we attributed the cause to the real feed and tested it for contaminants. It turned out, that not all potassium oxalate was converted to oxalic acid, leaving 9000 ppm of potassium, a known catalyst poison, in the feed.29,30 We prepared a potassium free solution with the same oxalic acid concentration for which, the conversion stabilized at 88% conversion after 56 hours and stayed constant for more than 100 hours (Fig. 5C and D). The influence of potassium ions on the reaction indicates that deactivation of the catalyst by poisoning and thus long term stability requires attention when new catalysts are developed for this reaction. The conversion could be increased by increasing the temperature, residence time or hydrogen pressure. The selectivity towards glycolic acid was 94.5% but the formation of acetic acid still reached 5.5% (4.3 min, 6.7% max). | ||
| Fig. 5 Long term testing in a flow reactor with real oxalic acid feed from the OCEAN project. (A) Selectivity and (B) carbon balance during reduction of real feed without removal of potassium at 50 °C, 60 bar, a feed-flow of 0.1 ml min−1 and hydrogen flow of 200 ml min−1. (C) Selectivity and (D) carbon balance during reduction of 2.37 wt% oxalic acid at 50 °C, 60 bar, a feed-flow of 0.1 ml min−1 and hydrogen flow of 200 ml min−1. For both reactions we used a commercial catalyst from Johnson Matthey (9.76 wt% Ru/C, 0.5 wt% moisture) as a catalyst. | ||
To gain more insight into the stability of the catalyst, we analysed it before and after reaction using XRD, HRSTEM and EDS to assess the surface structure (Fig. 6, 7 and S4†). Most of the ruthenium is present as small particles from 2–5 nm with a pseudo-spherical shape (Fig. 6). Whilst the smaller particles appear to stay in place, the bigger particles appear to agglomerate to a size of 5–15 nm during the reaction. Apart from these small well spread ruthenium particles, however, we also identified large particles bigger than 500 nm (Fig. S4†). These particles were also present after the reaction. With EDS we confirm that both small and big particles, appearing as white spots in the HRSTEM images predominantly consist of ruthenium and oxygen in a ratio of 1 to 2 which suggests the presence of RuO2. The X-ray diffraction (XRD) patterns of the fresh and used catalyst show characteristic peaks of the carbon support and RuO2 and therefore confirms the observation of ruthenium in its oxidised form (Fig. 7). Such oxidation was observed earlier for supported Ru catalysts.31 The carbon support is predominantly amorphous and stays unaltered during the reaction as confirmed by the broad peaks from 2θ 15–30° and 2θ 40–45°.32 In the EDS analysis, however, we also found significant amounts of silicon (15 wt%), iron (3 wt%) and aluminum (6 wt%) apart from RuO2 on the catalyst. We did not find any chloride or potassium on the active catalyst before and after reaction with potassium free oxalic acid as feed. With ICP-OES we analyzed the product stream and did not detect any ruthenium leached from the catalyst. Overall, we suggest that the catalyst appears to undergo some initial rearrangement which leads to a small reduction of activity which can be compensated by increasing the reaction temperature, the residence time or by increasing the catalyst amount. After this initial phase, the catalyst performs stable for more than 100 hours and the active metal species remain unaltered without any indication of leaching.
 | ||
| Fig. 6 High-resolution scanning tunnel electron microscope (HRSTEM) images and elemental mapping with energy-dispersive X-ray spectroscopy (HRSTEM-EDS) of commercially available 9.76 wt% Ru/C catalyst from Johnson Matthey before and after 100 h use in trickle-bed flow reactor for reduction of oxalic acid to glycolic acid in the absence of potassium. | ||
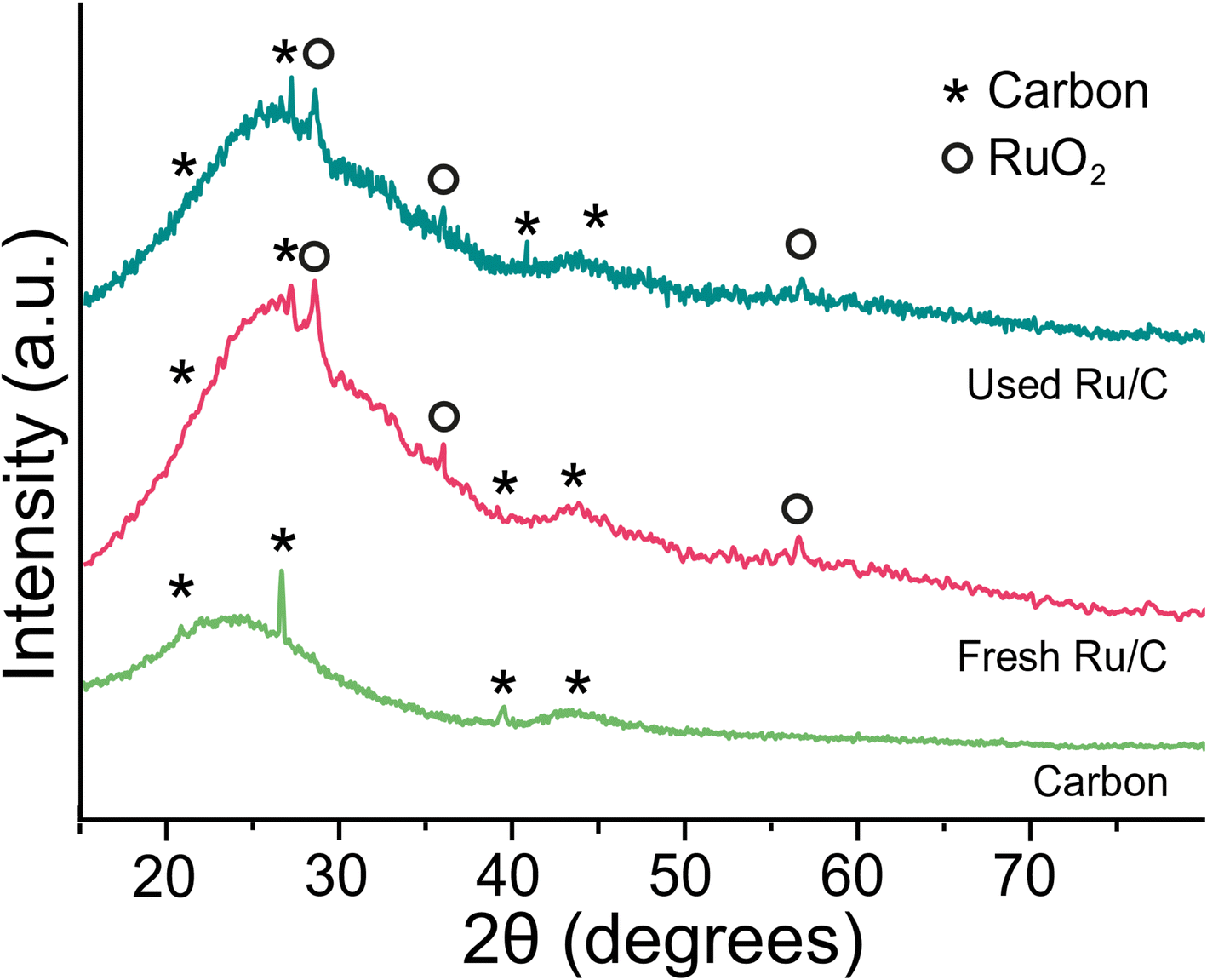 | ||
| Fig. 7 X-ray diffraction (XRD) patterns of fresh and used 9.76 wt% Ru/C catalyst from Johnson Matthey. For comparison, a diffraction pattern of carbon support is added. The diffractograms were measured with a scan rate of 1° min−1. The diffraction peaks were assigned using a spectral database within the Rigaku PDXL software. | ||
Conclusions
We conclude that oxalic acid can selectively be reduced to glycolic acid using Ru/C catalysts in a trickle-bed reactor. With this work, we extend the process to a continuous one using a commercial ruthenium catalyst from the batch process we reported earlier. We found that the increase in temperature, hydrogen pressure, and residence time increased the conversion but did not affect the acetic acid versus glycolic acid selectivity, hence also the selective production of ethylene glycol is possible. Interestingly, using the trickle-flow reactor allowed us to perform the reaction at a higher efficiency as we could reduce the reaction temperature to 50 °C and lower the reaction time significantly from hours to minutes compared to the batch reactor. Unfortunately, the production of acetic acid as a side-product could not be avoided. The structural analysis of the catalyst with ICP-MS, XRD, HRSTEM and EDS shows that the catalyst remains largely unaltered after a short induction period and no metal leaching was observed. When using real feed from the OCEAN project, where oxalic acid is produced from CO2via the acidification of potassium oxalate, we observed that the presence of potassium has a poisonous effect on the reaction. In the absence of potassium, however, our catalytic process performed stable over 100 hours with no noticeable reduction in activity or selectivity and no indication of stability problems beyond the tested period. This work is a step towards a sustainable continuous process to produce glycolic acid from oxalic acid and is crucial in the conversion of CO2 to polymers.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union’s Horizon 2020 research and innovation program under grant agreement no. 767798.Notes and references
- A. Otto, T. Grube, S. Schiebahn and D. Stolten, Closing the Loop: Captured CO2 as a Feedstock in the Chemical Industry, Energy Environ. Sci., 2015, 8(11), 3283–3297, 10.1039/c5ee02591e.
- R. Aldaco, I. Butnar, M. Margallo, J. Laso, M. Rumayor, A. Dominguez-Ramos, A. Irabien and P. E. Dodds, Bringing Value to the Chemical Industry from Capture, Storage and Use of CO2: A Dynamic LCA of Formic Acid Production, Sci. Total Environ., 2019, 663, 738–753, DOI:10.1016/j.scitotenv.2019.01.395.
- P. G. Levi and J. M. Cullen, Mapping Global Flows of Chemicals: From Fossil Fuel Feedstocks to Chemical Products, Environ. Sci. Technol., 2018, 52(4), 1725–1734, DOI:10.1021/acs.est.7b04573.
- A. Kätelhön, R. Meys, S. Deutz, S. Suh and A. Bardow, Climate Change Mitigation Potential of Carbon Capture and Utilization in the Chemical Industry, Proc. Natl. Acad. Sci. U. S. A., 2019, 116(23), 11187–11194, DOI:10.1073/pnas.1821029116.
- M. Ronda-Lloret, L. Yang, M. Hammerton, V. S. Marakatti, M. Tromp, Z. Sofer, A. Sepúlveda-Escribano, E. V. Ramos-Fernandez, J. J. Delgado, G. Rothenberg, T. Ramirez Reina and N. R. Shiju, Molybdenum Oxide Supported on Ti3AlC2 Is an Active Reverse Water-Gas Shift Catalyst, ACS Sustainable Chem. Eng., 2021, 9(14), 4957–4966, DOI:10.1021/acssuschemeng.0c07881.
- E. J. Devid, M. Ronda-Lloret, D. Zhang, E. Schuler, D. Wang, C.-H. Liang, Q. Huang, G. Rothenberg, N. R. Shiju and A. W. Kleyn, Enhancing CO2 Plasma Conversion Using Metal Grid Catalysts, J. Appl. Phys., 2021, 129(5), 053306, DOI:10.1063/5.0033212.
- M. Ronda-Lloret, Y. Wang, P. Oulego, G. Rothenberg, X. Tu and N. R. Shiju, CO2 Hydrogenation at Atmospheric Pressure and Low Temperature Using Plasma-Enhanced Catalysis over Supported Cobalt Oxide Catalysts, ACS Sustainable Chem. Eng., 2020, 8(47), 17397–17407, DOI:10.1021/acssuschemeng.0c05565.
- E. Schuler, M. Demetriou, N. R. Shiju and G.-J. M. Gruter, Towards Sustainable Oxalic Acid from CO2 and Biomass, ChemSusChem, 2021, 14(18), 3636–3664, DOI:10.1002/cssc.202101272.
- Oxalic Acid from CO2 Using Eletrochemistry at demonstratioN Scale | OCEAN Project | H2020 | CORDIS | European Commission, https://cordis.europa.eu/project/id/767798, accessed 2020-02-04 Search PubMed.
- M. A. Murcia Valderrama, R.-J. van Putten and G.-J. M. Gruter, PLGA Barrier Materials from CO2. The Influence of Lactide Co-Monomer on Glycolic Acid Polyesters, ACS Appl. Polym. Mater., 2020, 2(7), 2706–2718, DOI:10.1021/acsapm.0c00315.
- M. A. Murcia Valderrama, R.-J. van Putten and G.-J. M. Gruter, The Potential of Oxalic – and Glycolic Acid Based Polyesters (Review). Towards CO2 as a Feedstock (Carbon Capture and Utilization – CCU), Eur. Polym. J., 2019, 119, 445–468, DOI:10.1016/j.eurpolymj.2019.07.036.
- E. Schuler, P. A. Ermolich, N. R. Shiju and G. M. Gruter, Monomers from CO2 : Superbases as Catalysts for Formate-to-Oxalate Coupling, ChemSusChem, 2021, 14(6), 1517–1523, DOI:10.1002/cssc.202002725.
- E. Schuler, A. Perez de Alba Ortiz, B. Ensing, N. R. Shiju and G.-J. M. Gruter, Understanding a Key Step in CO2 to Polymers: The Role of Alkali Hydroxides in Formate Coupling Reactions, Nat. Catal., 2023 Search PubMed , submitted.
- E. Schuler, M. Stoop, N. R. Shiju and G.-J. M. Gruter, Stepping Stones in CO2 Utilization: Optimizing the Formate to Oxalate Coupling Reaction Using Response Surface Modeling, ACS Sustain. Chem. Eng., 2021, 9(44), 14777–14788, DOI:10.1021/acssuschemeng.1c04539.
- E. Schuler, L. Grooten, M. Kasireddy, S. More, N. R. Shiju, S. K. Tanielyan, R. L. Augustine and G.-J. M. Gruter, Oxalic Acid Hydrogenation to Glycolic Acid: Heterogeneous Catalysts Screening, Green Chem., 2023, 25(6), 2409–2426, 10.1039/D2GC02411J.
- J. Zhu, Y. Ye, Y. Tang, L. Chen and K. Tang, Efficient Hydrogenation of Dimethyl Oxalate to Ethylene Glycol via Nickel Stabilized Copper Catalysts, RSC Adv., 2016, 6(112), 111415–111420, 10.1039/C6RA23474G.
- H. T. Teunissen and C. J. Elsevier, Ruthenium Catalysed Hydrogenation of Dimethyl Oxalate to Ethylene Glycol, Chem. Commun., 1997, 7, 667–668, 10.1039/a700862g.
- U. Matteoli, M. Bianchi, G. Menchi, P. Frediani and F. Piacenti, Hydrogenation of Dimethyl Oxalate in the Presence of Ruthenium Carbonyl Carboxylates: Ethylene Glycol Formation, J. Mol. Catal., 1985, 29(2), 269–270, DOI:10.1016/0304-5102(85)87009-7.
- P. A. Dub and T. Ikariya, Catalytic Reductive Transformations of Carboxylic and Carbonic Acid Derivatives Using Molecular Hydrogen, ACS Catal., 2012, 2(8), 1718–1741, DOI:10.1021/cs300341g.
- U. Matteoli, M. Bianchi, G. Menchi, P. Frediani and F. Piacenti, Hydrogenation of Dimethyl Oxalate in the Presence of Ruthenium Carbonyl Carboxylates: Ethylene Glycol Formation, J. Mol. Catal., 1985, 29(2), 269–270, DOI:10.1016/0304-5102(85)87009-7.
- B.-Y. Yu and I.-L. Chien, Design and Optimization of Dimethyl Oxalate (DMO) Hydrogenation Process to Produce Ethylene Glycol (EG), Chem. Eng. Res. Des., 2017, 121, 173–190, DOI:10.1016/j.cherd.2017.03.012.
- G. Giorgianni, C. Mebrahtu, S. Perathoner, G. Centi and S. Abate, Hydrogenation of Dimethyl Oxalate to Ethylene Glycol on Cu/SiO2 Catalysts Prepared by a Deposition-Decomposition Method: Optimization of the Operating Conditions and Pre-Reduction Procedure, Catal. Today, 2021, 390–391, 343–353, DOI:10.1016/j.cattod.2021.08.032.
- J. E. Carnahan, T. A. Ford, W. F. Gresham, W. E. Grigsby, G. F. Hager, C. C. Price and D. C. Lincoln, Ruthenium-Catalyzed Hydrogenation of Acids to Alcohols, J. Am. Chem. Soc., 1955, 77(14), 3766–3768 CrossRef CAS.
- J. H. S. Santos, J. T. S. Gomes, M. Benachour, E. B. M. Medeiros, C. A. M. Abreu and N. M. Lima-Filho, Selective Hydrogenation of Oxalic Acid to Glycolic Acid and Ethylene Glycol with a Ruthenium Catalyst, React. Kinet., Mech. Catal., 2020, 131(1), 139–151, DOI:10.1007/s11144-020-01843-3.
- J. P. Lange, J. Meurs, M. Rigutto and H. Stil, A Method of Preparing Glycolic Acid, EP16154238, 2016.
- A. Verma, R. Kore, D. R. Corbin and M. B. Shiflett, Metal Recovery Using Oxalate Chemistry: A Technical Review, Ind. Eng. Chem. Res., 2019, 58(34), 15381–15393, DOI:10.1021/ACS.IECR.9B02598.
- P. Santawaja, S. Kudo, A. Mori, A. Tahara, S. Asano and J. I. Hayashi, Sustainable Iron-Making Using Oxalic Acid: The Concept, A Brief Review of Key Reactions, and An Experimental Demonstration of the Iron-Making Process, ACS Sustainable Chem. Eng., 2020, 8(35), 13292–13301, DOI:10.1021/acssuschemeng.0c03593.
- B. Pospiech and M. Warzecha, Application of Oxalic Acid as an Efficient Leaching Agent of Aluminum from Industrial Waste, Physicochem. Probl. Miner. Process., 2020, 56(2), 264–270, DOI:10.37190/PPMP19103.
- S. Cimino, G. Totarella, M. Tortorelli and L. Lisi, Combined Poisoning Effect of K+ and Its Counter-Ion (Cl− or NO3−) on MnOx/TiO2 Catalyst during the Low Temperature NH3-SCR of NO, Chem. Eng. J., 2017, 330, 92–101, DOI:10.1016/J.CEJ.2017.07.127.
- Y. Zheng, A. Degn Jensen * and J. E. Johnsson, Laboratory Investigation of Selective Catalytic Reduction Catalysts: Deactivation by Potassium Compounds and Catalyst Regeneration, Ind. Eng. Chem. Fundam., 2004, 43(4), 941–947, DOI:10.1021/IE030404A.
- T. K. Slot, P. Oulego, Z. Sofer, Y. Bai, G. Rothenberg and N. Raveendran Shiju, Ruthenium on Alkali-Exfoliated Ti3(Al0.8Sn0.2)C2 MAX Phase Catalyses Reduction of 4-Nitroaniline with Ammonia Borane, ChemCatChem, 2021, 13(15), 3470–3478, DOI:10.1002/cctc.202100158.
- H. Shang, Y. Lu, F. Zhao, C. Chao, B. Zhang and H. Zhang, Preparing High Surface Area Porous Carbon from Biomass by Carbonization in a Molten Salt Medium, RSC Adv., 2015, 5(92), 75728–75734, 10.1039/C5RA12406A.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3su00185g |
| This journal is © The Royal Society of Chemistry 2023 |