
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Microwave-assisted organosolv extraction for more native-like lignin and its application as a property-enhancing filler in a light processable biobased resin†
Jenevieve
Yao
 a,
Maria
Karlsson
ab,
Martin
Lawoko
ab,
Karin
Odelius
ab and
Minna
Hakkarainen
*ab
a,
Maria
Karlsson
ab,
Martin
Lawoko
ab,
Karin
Odelius
ab and
Minna
Hakkarainen
*ab
aDepartment of Fibre and Polymer Technology, KTH Royal Institute of Technology, Teknikringen 56-58, 100 44 Stockholm, Sweden. E-mail: minna@kth.se
bWallenberg Wood Science Center, KTH Royal Institute of Technology, Teknikringen 56-58, 100 44 Stockholm, Sweden
First published on 12th June 2023
Abstract
The heterogeneous nature of lignin poses significant obstacles to its practical use in material applications. Common fractionation methods employ harsh processing conditions that further exacerbate lignin's structural complexity. We propose a microwave (MW)-assisted approach for a mild organosolv extraction of structurally-intact lignin from spruce wood. The efficient energy transfer enabled by microwave irradiation facilitates the rapid extraction of lignin in 5, 10, and 20 minutes, ensuring a low level of process severity. Comparison of the 10 minutes MW-extracted lignin products with a cyclic-extracted (CE) organosolv lignin revealed that equivalent amounts of β-O-4 linkages were preserved in both processes. This is indicative of the promising potential of MW-extraction as a biomass pretreatment method for the rapid extraction of more native-like lignin. Finally, we demonstrate the utilization of both MW- and CE-extracted lignins as property-enhancing fillers in a biobased photocurable resin for digital light processing (DLP). The more native-like structures of the mildly-extracted lignins proved beneficial for functionalization with reactive methacrylate moieties, enabling the mechanical reinforcement of DLP 3D printed thermosets with improved toughness after the incorporation of only 1 wt% of the lignins. Compared to the resin without lignin, the tensile strength was improved by 15 and 41% and elongation at break by 79 and 75% in the presence of methacrylated MW- and CE-lignins, respectively. This highlights the potential of MW and CE strategies to effectively process and modify lignin, thereby enhancing its utilization in targeted material applications.
Sustainability spotlightIn spite of the wide availability and interest, commercial materials based on lignin and lignin-derived monomers are still basically non-existent. The transformation of this renewable source of aromatic compounds into value-added materials contributes to reducing fossil fuel dependency and promotes the more sustainable and efficient use of forest resources. Here we developed an efficient microwave assisted approach to rapidly extract more native-like lignin from wood and to further methacrylate this lignin and lignin derivable monomers to photocurable resins for digital light processing 3D printing, which is a low energy requiring and material saving processing method. This aligns with UN Sustainable development goals 12 (responsible consumption and production), 9 (sustainable industrialization and innovation) and 13 (climate action). |
Introduction
The first step in lignin valorization is its effective isolation from lignocellulosic biomass. To date, the most extensively developed fractionation methods are those employed in pulping industries, for which the main objectives are the delignification of fibers and the preservation of cellulose.1–3 Severe processing conditions are typically employed as the crystallinity and linearity of cellulose render it resistant to degradation.4 The lignins resulting from these processes, known as technical lignins, suffer from heavy structural modification. The structure of technical lignins is characterized by copious amounts of recalcitrant C–C bonds.5,6 This deviates from the structure of native lignin, which is rich in C–O bonds as a result of the presence of abundant β-aryl ether (i.e., β-O-4) units.7–9 This formation of C–C bonds hampers the valorization of lignin, as these bonds are more resistant to breakdown than the easily cleavable C–O bonds.10,11 Additionally, the occurrence of uncontrolled depolymerization and repolymerization also exacerbate the already complex and heterogeneous structure of lignin. Ultimately, the result is the production of lignin with significant variability in chemical and physical properties, making it difficult to predict and control its behavior in material applications.12,13In order to minimize the structural modification of lignin during its isolation, mild fractionation methods have been developed. Two such methods are flow-through (FT) organosolv extraction14–18 and cyclic extraction (CE).17–19 FT is achieved by a continuous flow through the reactor during the extraction period. In CE, the extraction is performed in cycles, with periodic complete or partial displacement of the extraction liquor. In combination with the use of a flow-through reactor in either FT or CE mode, organosolv extraction has been demonstrated to successfully isolate lignin with few structural changes.14–19 The use of a flow-through reactor, in particular, facilitates the preservation of large numbers of labile lignin interunit linkages as a result of the short solvent residence time.15,18,20 Accordingly, the extracted dissolved lignin is completely or partially removed from the system, minimizing its exposure to the reactive media. Isolation of more native-like lignin by organosolv extraction is additionally enabled by the ability of the alcohol solvent to incorporate into the β-aryl ether structure via α-alkoxylation.14,18,21 This preserves the C–O rich structure of lignin by trapping the reactive cationic intermediate that is formed under acidic conditions, thereby suppressing degradation and condensation reactions.21
Microwave irradiation enables fast, volumetric, and selective heating through the excitation of polar molecules.22,23 Specifically, as polar molecules continuously align and realign with a rapidly oscillating electric field, dipole–dipole rotation is induced. Apart from the resulting molecular collisions, frictional forces against the matrix are also generated, and both of these contribute to an almost instantaneous temperature increase of the system.24,25 The efficiency of the direct generation of heat by microwaves is in contrast to conductive heating, which is reliant on the thermal conductivity of the reaction vessel for heat transfer.26 In addition to better energy efficiency, these advantages of microwave irradiation over conventional heating have been reported to lead to higher yields and better purity under milder reaction conditions and shorter reaction times.27–30 In the context of biomass fractionation, microwave irradiation is known to induce lignocellulose-specific effects arising from the efficient breakdown of hydrogen bonds due to the influence of high heat and vibrational motion.31,32 Specifically, the hydroxy groups in biomass can absorb microwaves, causing heat and pressure build-up that give rise to the disruption of granular structures and the rearrangement of the crystallinity of cellulose.25 Microwave irradiation has thus been reported to effectively deconstruct the compact matrix of lignocellulosic biomass and facilitate the separation of lignin.31,33,34
On account of the efficient energy transfer enabled by microwave irradiation, we propose a microwave (MW)-assisted rapid extraction method to attain similar high structural quality lignin to that previously obtained by cyclic-extraction. Despite the use of a batch reactor, we hypothesized that the significantly reduced processing time in MW would facilitate the preservation of β-aryl ether units and prevent condensation reactions. Short processing times of 5, 10, and 20 minutes were implemented for the MW-extraction of lignin from spruce wood. A mildly extracted CE organosolv lignin was selected as our benchmark. From this process we also adapted the extraction conditions employed (temperature, alcohol ratio, acid catalyst concentration). The more native-like structures of the MW-and CE-lignins were exploited by utilizing their abundant hydroxy groups as chemical handles for functionalization with methacrylate moieties. These reactive methacrylate units acted as crosslinking sites that enabled the lignins to participate in the network formation during digital light processing (DLP) 3D printing of a biobased photopolymer resin. The influence of the MW- and CE-lignins as property-enhancing fillers in DLP 3D printed thermosets was then investigated.
Experimental
Materials
Wood chips from Norway spruce (Picea abies) were Wiley-milled to 40 mesh prior to removal of extractives by a 24 h acetone extraction under refluxing conditions. Vanillyl alcohol (≥98%), eugenol (99%), methacrylic anhydride (94%, inhibited with 2000 ppm topanol), 4-(dimethylamino)pyridine (DMAP; ≥99%), sulfuric acid (95.0–98.0%), sodium bicarbonate (≥99.7%), lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP, ≥95%), chromium(III) 2,4-pentanedionate (Cr(acac)3; 97%), 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (TMDP; 95%), and pyridine (anhydrous, 99.8%) were purchased from Sigma-Aldrich. Dichloromethane (≥99%), sodium hydroxide (≥99%), and magnesium sulfate (≥99%) were purchased from Fisher Scientific. Isopropanol (≥99.5%), ethanol absolute (>99.5%), and dimethylsulfoxide-d6 (DMSO-d6; 99.8%) were purchased from VWR. N-Hydroxy-5-norbornene-2,3-dicarboxylic acid imide (NHND; >99.0%) was obtained from Tokyo Chemical Industry, and chloroform-d (99.8%) from Cambridge Isotope Laboratories. All chemicals were used as received and without further purification.Microwave-assisted organosolv lignin extraction
Milled spruce wood (1 g) and the extraction solvent (50 mL, 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 liquor-to-wood ratio), which consisted of a 70:30 volume ratio of ethanol and 1.5 wt% (0.267 M) H2SO4(aq), were introduced to a 100 mL high-pressure (max. 100 bar) Teflon vessel equipped with a pressure-release valve. Extractions were carried out in a flexiWAVE MA186-001 microwave (Milestone Srl, Italy) following a programmed heating cycle, which consisted of a 5 min ramp time and isothermal heating at 160 °C for 5, 10, or 20 min. A maximum power of 1800 W for the simultaneous reaction of 8 vessels was implemented. After cooling down to room temperature, the contents of the vessels were collected and filtered to separate the cellulose-rich fibers. The filtrate was concentrated via rotary evaporation of ethanol, after which deionized water was added to precipitate the lignin product. The product was filtered, washed, and dried under vacuum at room temperature for 48 h.
:1 liquor-to-wood ratio), which consisted of a 70:30 volume ratio of ethanol and 1.5 wt% (0.267 M) H2SO4(aq), were introduced to a 100 mL high-pressure (max. 100 bar) Teflon vessel equipped with a pressure-release valve. Extractions were carried out in a flexiWAVE MA186-001 microwave (Milestone Srl, Italy) following a programmed heating cycle, which consisted of a 5 min ramp time and isothermal heating at 160 °C for 5, 10, or 20 min. A maximum power of 1800 W for the simultaneous reaction of 8 vessels was implemented. After cooling down to room temperature, the contents of the vessels were collected and filtered to separate the cellulose-rich fibers. The filtrate was concentrated via rotary evaporation of ethanol, after which deionized water was added to precipitate the lignin product. The product was filtered, washed, and dried under vacuum at room temperature for 48 h.
Cyclic organosolv lignin extraction
Cyclic organosolv extraction of lignin was performed as previously reported.18 Briefly, milled spruce wood (9.3 g) was introduced to a 66 mL extraction cell. Hot water extraction was performed for 2 h at 160 °C using a fixed volume of 70 mL and purge time of 90 s. Subsequently, organosolv extraction was performed using a solvent system composed of 70:30 v/v of ethanol to 1.5 wt% (0.267 M) H2SO4(aq) at 160 °C in 15 static cycles for 5 min each, with 100% rinse volume and purge time of 90 s. The total amount of solvent used in the organosolv extraction was 340 mL.
Microwave-assisted methacrylation of organosolv lignin
The microwave-assisted methacrylation of organosolv lignin was performed following a procedure adapted from previous work.35 MW and CE organosolv lignin (1.25 g) and methacrylic anhydride (1:8 lignin-to-anhydride mass ratio) were introduced to high-pressure Teflon vessels, and were subjected to microwave irradiation (flexiWAVE MA186-001 microwave) for isothermal heating at 110 °C for 10 min. A ramp time of 5 min and maximum power of 800 W were implemented. After the reaction, the contents of the vessels were poured into isopropanol to precipitate the methacrylated lignin product. The suspension was stirred at room temperature for 24 h prior to vacuum filtration, during which the product was washed with excess solvent to remove the unreacted anhydride and acid byproduct. The obtained methacrylated lignin was dried under vacuum at room temperature for at least 48 h.
Biobased resin preparation
The resin components eugenol and vanillyl alcohol were methacrylated based on previously reported procedures.36,37 Briefly, a reaction mixture consisting of a 1:1.1 molar ratio of eugenol (30 g) to methacrylic anhydride (30.99 g) and DMAP catalyst (2 mol% of methacrylic anhydride, 0.471 g) was stirred at 45 °C for 24 h under N2 atmosphere. A 1:2.1 molar ratio of vanillyl alcohol (30 g) to methacrylic anhydride (66 g), and the same catalytic amount of DMAP based on the amount of methacrylic anhydride (1.05 g), were reacted under the same conditions. After cooling to room temperature, the reaction mixtures were diluted with 150 mL of dichloromethane and washed sequentially with a saturated solution of NaHCO3, followed by 1 M NaOH, then 0.5 M NaOH, and lastly deionized water. Finally, the organic phase was dried over anhydrous MgSO4 prior to the removal of dichloromethane by rotary evaporation.
Resins were prepared by mixing a ratio of 80:20 (wt/wt) methacrylated eugenol (ME) and methacrylated vanillyl alcohol (MVA). Once homogeneous, 0.5 wt% (per 100 g of resin) of LAP photoinitiator was added followed by 2 h of sonication and stirring overnight. Then, 1 wt% of microwave-extracted (MW-lignin) or cyclic-extracted lignin (CE-lignin) were dispersed in the resins through overnight stirring and 2 h of sonication. All resins were stored in airtight and light-protected bottles at room temperature until use.
Digital light processing (DLP) 3D printing
Lignin-containing biobased thermosets were fabricated via digital light processing (DLP) 3D printing using an Asiga MAX X27 UV printer equipped with a 385 nm light source and pixel resolution of 27 μm. All build files (.stl) and material files (.ini) were processed on the Asiga Composer software (version 1.2.11). Material files, which contained machine- and resin-specific information, were modified with data input from experimental measurements of cure thickness (μm) as a function of exposure time (s) for each resin formulation. Suitable printing exposure times (Table 1) were then determined by the software. The prints were constructed using a layer thickness of 0.05 mm, light intensity of 28 mW cm−2, and heater temperature set to 30 °C. Uncured resin was removed from the surfaces of the prints by washing with isopropanol. Post-curing was performed in an Asiga Flash UV chamber (385 nm) for 10 min (5 min on each side in the case of the tensile bars), during which the prints were immersed in water to prevent oxygen inhibition. The 3D printed thermosets were then air-dried prior to characterization.| Resin formulation | Burn-in exposure time (s) | Exposure time (s) |
|---|---|---|
| Resin (80:20 ME:MVA) |
7 | 6 |
| Resin + 1 wt% MW-lignin | 37 | 27 |
| Resin + 1 wt% CE-lignin | 18 | 11 |
Characterization
Analysis of the lignin structures was performed via nuclear magnetic resonance (NMR) spectroscopy at 400 MHz on a Bruker Avance III HD spectrometer. All data were processed using MestReNova software (Mestrelab Research SL), through which automatic baseline and phase corrections were applied. Semi-quantitative analysis of lignin interunit linkages was carried out via heteronuclear single-quantum coherence (HSQC) NMR. Lignin (80 mg) was dissolved in 0.7 mL of DMSO-d6. Analysis was performed using pulse sequence ‘hsqcetgpsi’ with a relaxation delay of 1.5 s, and 130 scans. Spectra were referenced relative to the residual DMSO at 2.5/39.5 ppm. The regions used for the determination of aromatic units and linking motifs are presented in Table S1.† The guaiacyl C2–H cross-peak signal at (7.8–6.1)/(113.2/106.1) ppm was used as an internal standard to calculate the amount of C9 units, to which all interunit linkages were referenced.38 The determination of hydroxy group content on the extracted and methacrylated lignin samples was carried out via31P NMR spectroscopy, based on a previously reported protocol.39 Lignin (30 mg) was dissolved in 0.5 mL of solvent A, which consisted of chloroform-d and anhydrous pyridine (1:1.6 vol/vol, respectively). 0.1 mL of internal standard solution, composed of the relaxing agent Cr(acac)3 (5.0 mg mL−1) and the internal standard NHND (18.0 mg mL−1) in solvent A, was then added and stirred overnight. Prior to analysis, 0.1 mL of the phosphorylating agent TMDP was added and stirred for 30 s. The acquisition parameters included a relaxation delay of 2 s and 1024 scans. Spectra were shifted relative to the reference signal of the product of TMDP and water at 132.2 ppm. Hydroxy group content was determined by integration of peak regions assigned to aliphatic and phenolic OH groups, as shown in Table S4.† Derived integral values were then converted to mmol OH/g lignin using eqn (1), in which R is the integral ratio of the region of interest over that of the internal standard.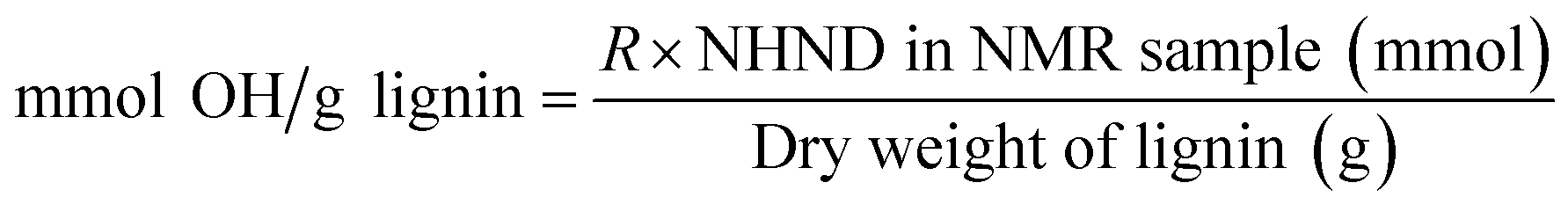 | (1) |
The degree of substitution (DS) was calculated according to eqn (2), in which C(OH)i (mmol g−1) and C(OH)f (mmol g−1) are the initial and final concentrations of OH units in lignin, respectively, and Δmmax is the weight increase (g) per g of lignin at 100% conversion.
 | (2) |
Structural elucidation of the extracted and methacrylated lignin samples was performed via Fourier transform infrared spectroscopy (FT-IR). Spectra were recorded on a PerkinElmer Spectrum 100 FT-IR spectrometer equipped with a Golden gate single reflection attenuated total reflectance (ATR) accessory system (Graseby Specac, U.K.), using 16 scans at a resolution of 4 cm−1 in the wavenumber range of 4000-600 cm−1.
Resin cure properties were determined using the Jacobs working curve model (eqn (3)), which quantifies: (1) the depth by which irradiated light can penetrate through the resin (penetration depth, Dp), and (2) the amount of light required for gel point to be reached (critical dose, Ecrit).40–42 Values for cure depth (Cd, μm) and light irradiation dosage (Emax, mJ cm−2) were measured experimentally by curing resin samples (3 mm in diameter) over the screen of the DLP printer (Asiga MAX X27 UV). The exposure time of the irradiated light (385 nm, 28 mW cm−2) was varied, and the resulting Cd were measured using a micrometer. This was performed after uncured residue had been washed off with isopropanol and post-cure (3 min, 385 nm, Asiga UV chamber). Thickness measurements were taken in triplicate. Working curves were constructed by plotting Cdvs. Emax, where Emax was calculated from the product of UV light intensity and exposure time. The cure parameters Dp and Ecrit were determined from the slope and intercept of the working curve, respectively.
 | (3) |
The thermal properties of the 3D printed thermosets were characterized via thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC). TGA was performed on a Mettler-Toledo TGA/SDTA 851e instrument with samples (5–10 mg) placed in 70 μL alumina cups. Heating was programmed to a temperature range of 30–800 °C at a rate of 10 °C min−1 in N2 atmosphere set to a flow rate of 50 mL min−1. For DSC, thermal transition temperatures were investigated using a Mettler-Toledo DSC1 STARe system with samples (5–10 mg) contained in sealed 100 μL aluminum pans. The samples were first heated from 25 to 125 °C. The temperature was held for 2 min, followed by a cooling step from 125 to −25 °C, and finally a second heating step from −25 to 125 °C. The heating–cooling cycle was performed at a rate of 10 °C min−1 under N2 atmosphere set to a flow rate of 50 mL min−1. Data analysis for both thermal characterization techniques was performed on a Mettler STARe evaluation software. Glass transition (Tg) temperature values from DSC were derived from the second heating step.
Uniaxial tensile testing of the thermosets was conducted on a single column Instron 5944 universal tensile testing machine equipped with a 500 N load cell with crosshead speed set to 5 mm min−1. Tensile bars with gauge sections of 12.5 × 8 × 1.3 mm were fabricated via DLP 3D printing. Sample conditioning (48 h) and tensile testing were carried out in a controlled environment with a temperature of 22 °C and 50% relative humidity. The reported values are averages of at least 5 measurements. Fracture surfaces were investigated by scanning electron microscopy (SEM) using a Hitachi S-4800. Samples were attached on an aluminum stub using carbon tape and sputter coated with a chromium (Cr) coater target at 2 nm thickness using a Cressington 208HR sputter coater. All SEM micrographs were taken using an acceleration voltage of 1 kV.
Results and discussion
The potential of microwave (MW)-assisted organosolv extraction as a mild fractionation method for the isolation of more native-like lignin was explored. The preservation of lignin interunit linkages, particularly β-aryl ether (i.e., β-O-4) units, the occurrence of condensation, and the extraction yield were used as criteria in the evaluation of the extraction method. A cyclic-extracted (CE) organosolv lignin with a demonstrated high degree of β-O-4 retention, minimal condensation, high purity, and high yield was used as a benchmark for comparison.18 Following extraction, the isolated lignin products from both MW and CE methods were functionalized with reactive methacrylate moieties for their utilization as value-added fillers in a biobased photocurable resin for digital light processing (DLP).Mild organosolv lignin fractionation via microwave- and cyclic-extraction methods
On account of the efficiency of cyclic-extraction in isolating lignin with few structural changes and high purity, we have selected a cyclic-extracted organosolv lignin from milled spruce wood (denoted hereafter as CE-lignin) as our benchmark. The detailed methodology for this extraction process can be found in a previously published work.18 The processing conditions implemented in the MW-extraction followed those used for the CE method (i.e., 160 °C and 70:30 vol/vol EtOH: 1.5 wt% H2SO4(aq)), with the exception of extraction time. In the CE method, 15 × 5 min static cycles were employed, whereas MW-extraction was performed for a duration of only 5 to 20 min.
Owing to the rapid and volumetric heating enabled by MW irradiation, we envisioned that a MW-assisted approach could facilitate the effective fractionation of biomass by significantly reducing processing time. As a consequence of this shortened extraction time, we anticipated the possible isolation of lignin with similarly high structural quality as that obtained from CE processes. Semi-quantitative assessment of the major interunit linkages via HSQC NMR (Fig. 1, 2a, and S1a–c†) revealed that the β-O-4 motif, the predominant β-aryl ether linkage, was preserved to a high degree through substitution of the α-OH by an ethoxy group from the alcohol solvent.21 This substitution leads to the formation of α-ethoxylated β-O-4 units, referred to as β′-O-4. This suppresses degradation and condensation reactions as reactive cationic intermediates that are formed under acidic conditions (Scheme S1a†) are trapped and replaced to produce the more acid-stable β′-O-4 (Scheme S1b†).14,21 The β′-O-4 units were found to be retained at high levels in the lignin samples isolated from the 5 and 10 min MW-extractions, whereas these units decreased in number upon prolonging the extraction time to 20 min. These results indicate that α-ethoxylated β-O-4 units (i.e., β′-O-4) remain susceptible to cleavage and this is promoted by increased process severity.20 Second, that short extraction durations of 5 and 10 min were sufficient to maximize the number of β′-O-4 units and preserve β-O-4, which for these short reaction times allow the same retention as in CE-lignin. Typically, lower degrees of β′-O-4 linkages are obtained at the beginning of organosolv extraction, as the substitution of alkoxy groups on the benzylic (α) position is insufficient at short timescales.43,44 A high degree of alcohol incorporation is a prerequisite to solubilize larger lignin fragments.14,43
 | ||
| Fig. 1 2D HSQC spectra of the native interunit linkages of the 10 minutes microwave-extracted organosolv lignin. | ||
 | ||
| Fig. 2 (a) Number of substructures semi-quantified via 2D HSQC and (b) hydroxy group content determined by 31P NMR for the microwave (MW)-extracted and cyclic-extracted (CE) lignins. | ||
Nevertheless, the prevalence of a high degree of β′-O-4 units in this work is attributed to the beneficial effects of MW irradiation. The advantages of MW irradiation in the context of biomass fractionation likely stems from its accelerated energy transfer, which has been reported to induce lignocellulose-specific effects such as the rapid depolymerization of cellulose,45,46 the enhancement of wood permeability,47 and the efficient breakdown of hydrogen bonds linking cellulose, hemicellulose, and lignin.48–51 The effectiveness of MW irradiation in lignocellulose fractionation is reflected in the high carbohydrate content, and the corresponding low lignin content, of the fiber byproducts after only 10 minutes of MW-extraction (Table S3†). Moreover, the said effects are expected to be enhanced by the presence of a strong acid (H2SO4) in the extraction solvent, as well as the use of sealed reactor vessels that could accommodate pressures up to 100 bar. First, the presence of H+ and SO42− ions is known to contribute an additional conduction mechanism to an already heat-generating dipolar system through increased kinetic energy from ionic collisions.52 Second, the application of elevated pressure, which is a consequence of the use of an extraction temperature higher than the boiling point of the solvent, has been shown to lead to more efficient defibrillation or disruption of the cell wall structure.21
Coinciding with the observed trend in β′-O-4 content, the quantification of hydroxy groups via31P NMR (Fig. 2b and S2†) revealed that the highest amount of aliphatic OH groups was obtained after 10 minutes of MW-extraction, followed by 5 minutes, and lastly by 20 minutes of extraction. Aliphatic hydroxy groups mostly originate from β-O-4 units that possess hydroxy groups in the α and γ positions.20,43,53 The slightly larger amount of aliphatic OH groups detected in lignin from the 10 minutes MW-extraction compared with the lignin from the 5 minutes extraction is likely explained by the marginally higher number of unethoxylated β-O-4 units in the former than in the latter. It is important to note that since the majority of hydroxy groups in the α-position have been ethoxylated according to the semi-quantitative results from HSQC, only the remaining non-ethoxylated α-OH groups were detected in 31P NMR.43 Although the presence of aliphatic hydroxy groups follows the prevalence of native interunit linkages, the opposite is true regarding phenolic hydroxy groups. The accumulation of phenolics occurs simultaneously with the depletion of aliphatic hydroxy groups, as the cleavage of β-O-4 linkages generates new phenolic ends.17,20 For this reason, the opposite trend of 20 min > 10 min > 5 min prevails with regard to the number of phenolic OH as a function of extraction duration. Overall, these results confirm the negative effect of prolonged extraction time on the retention of native lignin interunit linkages.16,17,54 In the case of MW-extraction, an increase in process severity by a twofold increase in extraction time from 10 to 20 min results to a consequential accelerated breakdown of β-aryl ether units.
Fragmentation reactions that result in the concurrent reduction in the level of aliphatic hydroxy groups and accumulation of phenols are considered to be the prevalent mechanism of lignin degradation in organosolv fractionation.20,55 A commonly cited product of one such fragmentation reaction is the Hibbert's ketones that can arise only from the heterolytic cleavage of β-O-4 linkages (Scheme S1d†).56 Hibbert's ketones are usually only detected in trace amounts in organosolv lignin, in which the preferred degradation route is typically the homolytic cleavage of β-O-4 linkages to produce recondensation products (e.g. 5-5 units).18,20,57 However, the formation of Hibbert's ketones has been found to be promoted under acidic conditions.17,56 Compared with CE-lignin, MW-extracted lignin exhibited higher contents of Hibbert's ketones, the amount of which increased with longer extraction time. This accumulation of Hibbert's ketones is explained by the prolonged exposure of the dissolved lignin to the acidic extraction medium, which promoted further cleavage of β-O-4 linkages as substantiated by HSQC. This was not the case with CE-lignin, however, as the constant solvent exchange in the CE process minimized the exposure of extracted lignin to the acidic environment.18 Another factor that possibly contributed to the formation of Hibbert's ketones is the high energy input by microwave irradiation that could have promoted the heterolytic pathway. Although higher levels of Hibbert's ketones were detected in the 10 minutes MW-extracted lignin compared with CE-lignin, the two lignin samples possess equivalent amounts of β-O-4 linkages, however this could be due to the higher level of β-O-4 linkages present initially in the MW-extracted lignin, that then underwent cleavage.
Structural quality, as defined by the prevalence of native linkages and the occurrence of minimal condensation, is an important consideration in the context of isolating lignin for high-value material applications. However, in addition to this criterion, the yield of extracted lignin in relation to the extraction efficiency of the fractionation method is of considerable importance. Lower yields (Fig. 3) were obtained from MW-extraction in comparison to the CE method. This can be explained by the high diffusive flux in CE, which enhances lignin extraction by preventing the saturation of the extraction solvent.15,18 However, as previously demonstrated, yield can be improved with prolonged extraction time or even by implementing higher extraction temperatures.15,17,58 As such, higher yield was obtained with increasing extraction time from 5 to 20 minutes, which is likely due to the increased fragmentation of lignin that facilitated its solubilization. Moreover, increasing the extraction temperature to 170 and 190 °C for the 10 minutes MW-extracted lignin also resulted in yields of 40 and 42%, respectively. This slightly higher yield, however, was accompanied by the undesired trade-off of aryl-ether cleavage and condensation reactions (Fig. S1d, e, S2, Tables S2 and S5†).
 | ||
| Fig. 3 Extraction yields of the microwave (MW) and cyclic-extraction (CE) methods, based on the starting lignin content of 31.4% (by mass) in spruce wood as determined from the sum of Klason and acid-soluble lignin.18 | ||
Overall, these results highlight the distinct advantages associated with MW-assisted and CE organosolv fractionation. Both methods exemplify excellent strategies for the isolation of relatively structurally-intact lignin: rapid fractionation (10 + 5 min ramp time) with acceptable yield is afforded by the MW-assisted approach, whereas greater yields and higher purity are achieved by CE extraction.
Functionalization of microwave- and cyclic-extracted organosolv lignin
The MW-extracted and CE organosolv lignins were functionalized to enable their utilization as property-enhancing fillers in additive manufacturing via digital light processing (DLP). As DLP relies on the selective curing of a photoreactive resin upon exposure to a light source, the compounds that constitute the resin must be sufficiently reactive and amenable to photo-triggered free radical polymerization. The covalent grafting of methacrylate moieties has been demonstrated to be an effective strategy to introduce reactive groups to resin components.59,60 The abundant hydroxy functionalities in the extracted organosolv lignin samples is advantageous for the purposes of this application as they can be exploited as chemical handles for functionalization with methacrylate moieties. The methacrylation of the extracted lignin serves the dual purpose of enhancing compatibility between lignin and the hydrophobic polymer matrix and, more importantly, to introduce crosslinking sites to improve the reinforcing ability of lignin.As 10 minutes of MW-extraction was found to be optimal in terms of maximizing β-O-4 preservation and preventing condensation, only the product of this extraction (hereafter referred to as MW-lignin) was utilized in succeeding investigations. Both MW-lignin and CE-lignin were methacrylated through microwave-assisted acylation reaction based on previous work by our group on acetylation of lignin.35 This approach was earlier shown to enable a solvent-free and catalyst-free acylation of hydroxy functionalities to achieve high degrees of substitution by acetyl groups using anhydride reactants.35 Through this approach, significantly shorter reaction time (10 min) and moderate reaction temperature (110 °C) were implemented, based on previous findings regarding the optimal reaction parameters to maximize the degree of substitution. Successful functionalization of both MW-lignin and CE-lignin was confirmed by FTIR (Fig. 4a) through the appearance of peaks corresponding to the presence of methacrylate groups (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch in phenolic and aliphatic esters at 1760 and 1720 cm−1, respectively, CC stretch at 1630 cm−1, and in-plane CH2 bend at 950 cm−1). The presence of an additional CO stretch at 1800 cm−1 possibly indicates the presence of residual unreacted anhydride.
O stretch in phenolic and aliphatic esters at 1760 and 1720 cm−1, respectively, CC stretch at 1630 cm−1, and in-plane CH2 bend at 950 cm−1). The presence of an additional CO stretch at 1800 cm−1 possibly indicates the presence of residual unreacted anhydride.
 | ||
| Fig. 4 (a) FTIR spectra and (b) hydroxy group content as determined by quantitative 31P NMR of methacrylated microwave (MW)- and cyclic-extracted (CE)-lignins. | ||
Degrees of substitution were quantified via31P NMR (Fig. 4b and S3†), which revealed that the hydroxy groups in MW-lignin and CE-lignin were methacrylated to 73 and 79%, respectively. The slightly higher degree of substitution by methacrylate moieties on CE-lignin signifies more crosslinking sites, potentially leading to stiffer thermosets compared to those produced from MW-lignin, as will be discussed in a later section. Notably, β-O-4 units were retained after MW-assisted methacrylation as confirmed by 2D HSQC analysis (Fig. S4†), based on the signals of β′-O-4α and β-O-4β/β′-O-4β.
Preparation of lignin-containing photocurable biobased resin
The biobased resin prepared in this work is composed of the lignin-derivable phenolic compounds eugenol and vanillyl alcohol as the building blocks. The methacrylated derivatives of the constituents were synthesized following a procedure adopted from previous work.36,37,61 As opposed to conventional esterification methods, the methacrylation route employed herein involves minimal amounts of catalyst, does not require the use of a coupling agent, and has been found to result to the full conversion of hydroxy groups in methacrylated eugenol (ME) and methacrylated vanillyl alcohol (MVA) (Fig. S5†). The use of ME as the reactive diluent eliminated the need for a solvent while maintaining low resin viscosity, which is crucial for DLP 3D printing.62 MVA, on the other hand, served as a dimethacrylated monomer. The complete resin system was thus composed of an 80:20 (wt/wt) ratio of ME to MVA, 0.5 wt% LAP photoinitiator, alone or modified with 1 wt% of methacrylated MW-lignin or CE-lignin.
The influence of 1 wt% methacrylated MW-lignin or CE-lignin on the curing properties of the resin was assessed using the Jacobs working curve model (eqn (3)), which describes: (1) the penetration depth or distance travelled by photons through a resin (Dp), and (2) the critical dose or amount of energy required to induce photopolymerization (Ecrit).40–42 Working curves (Fig. 5a), which were constructed by plotting experimentally obtained cure depths (Cd) as a function of light irradiation dosage (Emax), were used to derive the values for Dp and Ecrit for each resin formulation (Fig. 5b and c). The results reveal that the presence of lignin invariably decreased Dp and correspondingly increased Ecrit, which is consistent with the well-known ability of lignin to absorb UV light and quench free radicals.63,64 While this increase in Ecrit equates to longer print times as a result of the higher energy dosage required to polymerize a given layer, the reduction in Dp signifies improved print resolution as irradiated light is more confined to the targeted region. In essence, the lower the penetration depth, the less susceptible is the incident light to diffuse across unintended layers. This results to the overall effect of reduced tendency for overcuring, leading to enhanced print quality and improved mechanical properties, both of which will be discussed in a later section. In this regard, the presence of MW-lignin and CE-lignin in the resins does not only serve the purpose of a structural feature, but it also improves printing resolution by acting as UV absorber.
 | ||
| Fig. 5 (a) Working curves, (b) penetration depth (Dp), and (c) critical dose (Ecrit) values of the resin formulations composed of 80:20 (wt/wt) methacrylated eugenol:methacrylated vanillyl alcohol, and 1 wt% of microwave (MW)- or cyclic-extracted (CE)-lignin. | ||
A comparison of the resins containing methacrylated MW-lignin and CE-lignin shows that the latter exhibits greater photoreactivity as evidenced by the lower Ecrit. This is likely due to the larger number of reactive methacrylate moieties covalently grafted onto CE-lignin. These methacrylate groups act as crosslinking sites that enable lignin to participate in network formation. The slightly higher Dp in the presence of methacrylated CE-lignin affirms the enhanced propagation of the polymerization front in this resin formulation. These findings are corroborated by the shorter exposure time required for curing an 0.05 mm layer of the resin with methacrylated CE-lignin compared with methacrylated MW-lignin (Table 1). Aside from the increased reactivity conferred by methacrylate groups, another factor that could affect photocuring kinetics is the capacity of lignin to act as a UV absorber to convert and dissipate the absorbed light energy into the surroundings in the form of molecular thermal motion.65 Such energy dissipation has been reported to contribute to a reduction in gelation time of a resin system consisting of a UV absorber.66 However, deciphering the discrete effects of lignin's UV absorbing ability in the resin's cure behavior is complex due to the competing free radical scavenging activity of lignin. For instance, while phenolic hydroxy groups contribute to the absorption of UV light that could be converted to thermal energy, the same functional groups are also capable of quenching the free radicals that propagate photopolymerization.
3D printing of lignin-enhanced thermosets by digital light processing (DLP)
The lignin-containing biobased resins were fabricated into 3D printed thermosets via digital light processing (DLP) (Fig. 6). The resins proved to be amenable to high-resolution DLP, as manifested by the successful construction of specimens having complex and intricate features with dimensions as narrow as 250 microns. Additionally, owing to the mild fractionation methods that preserved the native lignin structure and prevented the occurrence of condensation, the resulting thermosets were light in color, which is atypical for lignin-containing materials. The MW-assisted and CE methods using the mild conditions employed herein could thereby improve the applicability of lignin in material applications, especially when the color of the end product is important. | ||
| Fig. 6 (a) Computer-aided design (CAD) model of a tree with intricate features and the resulting digital light processing (DLP) 3D printed specimens containing 1 wt% of (b) microwave (MW)-extracted lignin and (c) cyclic-extracted (CE)-lignin. | ||
Thermal characterization of the 3D printed thermosets was performed via thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC). TGA thermograms (Fig. 7a) show a two-step degradation profile. The first step (185–400 °C) is ascribed to the decomposition of non-crosslinked fragments (e.g. linear and branched chains) and uncured monomer, while the second step (400–480 °C) is attributed to the random scission of crosslinked chains and the degradation of phenolic moieties.67,68 Interestingly, the incorporation of 1 wt% of methacrylated lignin, regardless of isolation procedure, increased the thermal stability of the resin in terms of the thermal degradation temperature at 20% mass loss (Tdeg20%, Table 2). This indicates higher crosslink density in the lignin-containing resins, an evidence of the participation of covalently grafted methacrylate moieties in lignin to the network formation. This same finding is reflected by the DSC results (Fig. 7b, DTG curves in Fig. S6†), which show that both lignin-containing thermosets possess higher glass transition temperatures (Tg, Table 2) compared to the non-filled counterpart. This Tg increase signifies chain mobility restriction in the presence of methacrylated lignin, further confirming the influence of lignin and the enhanced crosslink density in the resins containing methacrylated MW-lignin and CE-lignin.
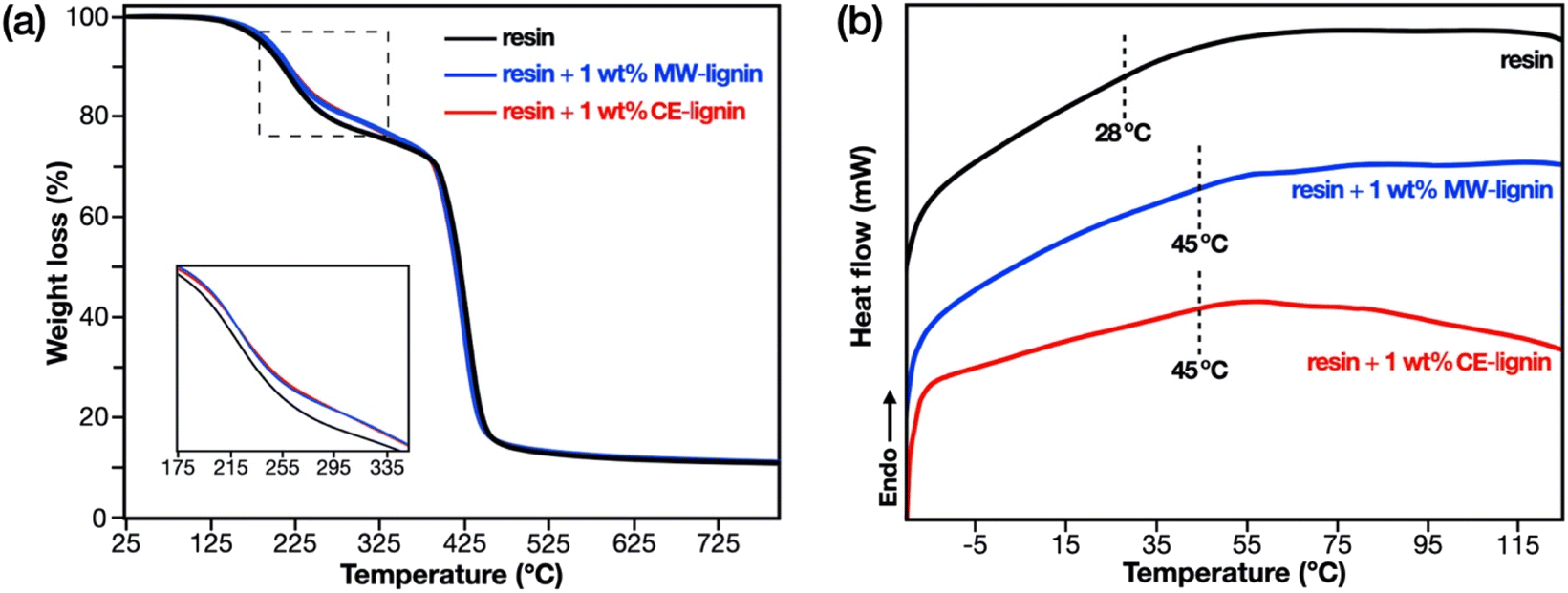 | ||
| Fig. 7 (a) TGA thermograms and (b) DSC curves of digital light processing (DLP) 3D printed thermosets composed of 80:20 (wt/wt) methacrylated eugenol:methacrylated vanillyl alcohol, and 1 wt% of microwave (MW)-extracted or cyclic-extracted (CE)-lignins. | ||
| 3D printed thermoset | T deg20% (°C) | T deg1,onset (°C) | T deg1,max (°C) | T deg2,onset (°C) | T deg2,max (°C) | T g (°C) |
|---|---|---|---|---|---|---|
| Resin (80:20 ME:MVA) |
267 | 189 | 217 | 403 | 431 | 28 |
| Resin + 1 wt% MW-lignin | 294 | 189 | 224 | 402 | 433 | 45 |
| Resin + 1 wt% CE-lignin | 295 | 187 | 227 | 404 | 431 | 45 |
The incorporation of methacrylated MW-lignin and CE-lignin to the prepared biobased resin was envisioned to enhance mechanical properties as a result of the introduction of additional crosslinking sites. Indeed, tensile testing of DLP 3D printed bars (Fig. 8) reveals that the presence of 1 wt% of methacrylated MW-lignin and CE-lignin increased tensile strength by 15 and 41%, Young's modulus by 1 and 23%, and elongation at break by 79 and 75%, respectively. The reinforcing effect of 1 wt% of methacrylated MW-lignin and CE-lignin is in contrast to the findings of a similar study, wherein the incorporation of the same amount of unmodified organosolv lignin in an acrylic resin reportedly resulted in a reduction in tensile strength, Young's modulus, and elongation at break of the printed product.69 This highlights the effectiveness of the functionalization of lignin with methacrylate moieties to enable lignin to partake in network formation. The influence of these methacrylated moieties on tensile properties is demonstrated in the higher tensile strength and Young's modulus of thermosets with CE-lignin, which is attributed to its slightly larger number of methacrylate groups. Remarkably, the enhanced tensile strength and Young's modulus, which resulted from increased crosslink density, was also complemented by a significant improvement in elongation at break. This is possibly due to the presence of abundant linkages (e.g. β-O-4) that confer flexibility in MW-lignin and CE-lignin, the effect of which is in contrast to that of recalcitrant structures with hindered free rotation.13 Hence, the incorporation of methacrylated MW-lignin and CE-lignin in the resin formulation ameliorates the previously brittle properties of the unfilled matrix towards more tough and ductile thermoset materials. Overall, the results exemplify the excellent interfacial bonding between methacrylated lignin and the matrix, which is also substantiated by the absence of a secondary phase in scanning electron micrographs of the fracture surfaces (Fig. S7†).
 | ||
| Fig. 8 (a) Tensile bars, (b) representative stress–strain curves, and the derived tensile properties – (c) ultimate tensile strength (MPa), (d) Young's modulus (MPa), and (e) elongation at break (%), of the digital light processing (DLP) 3D printed thermosets. | ||
Conclusions
The potential of microwave-assisted organosolv extraction as a mild fractionation method for the isolation of more native-like lignin was demonstrated. The efficient energy transfer enabled by microwave irradiation facilitated the deconstruction of lignocellulosic structures for the isolation of structurally intact lignin in significantly reduced processing times. As such, high degrees of native lignin interunits, particularly the β-O-4 units, were retained in the microwave-extracted lignin after 5 and 10 minutes, as determined through semi-quantitative HSQC NMR. The occurrence of minimal condensation was evidenced by the retention of large numbers of aliphatic hydroxy groups as revealed by 31P NMR. The lignin obtained from a 10 minutes microwave-extraction was therefore of similar high structural quality as our benchmark, a cyclic-extracted organosolv lignin. We further demonstrate the utilization of both MW- and CE-lignin as property-enhancing fillers in a biobased photocurable resin. Ultimately, the presence of abundant hydroxy groups in the mildly-extracted lignins proved beneficial as they acted as chemical handles for functionalization by reactive methacrylate moieties. These methacrylate moieties then enabled both MW- and CE-lignin to participate in network formation during the photopolymerization of the biobased resin, leading to the fabrication of high-resolution 3D printed thermosets with improved mechanical properties. Overall, this study highlights the value of MW-driven approaches in fine-tuning lignin characteristics to suit the requirements of diverse material applications.Author contributions
Conceptualization: J. Y., K. O., and M. H.; methodology: J. Y., M. K., M. L., K. O., and M. H.; formal analysis: J. Y., M. K., M. L., K. O., and M. H.; funding acquisition: M. H., and M. L.; supervision: M. H., K. O., and M. L.; writing-original draft preparation: J. Y.; writing-review & editing: M. H., M. K., M. L., and K. O.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Swedish Research Council, VR, (Grant No. 2018-03451) and Wallenberg Wood Science Center (WWSC) financed by the Knut and Alice Wallenberg Foundation (Grant No. KAW 2018.0452) are gratefully acknowledged for financial support. Niklas Wahlström is acknowledged for performing the carbohydrate analysis.Notes and references
- T. Renders, S. Van den Bosch, S. F. Koelewijn, W. Schutyser and B. F. Sels, Energy Environ. Sci., 2017, 10, 1551–1557 RSC.
- L. Viikari; A. Suurnäkki; S. Grönqvist; L. Raaska and A. Ragauskas, Forest Products: Biotechnology in Pulp and Paper Processing, in Encyclopedia of Microbiology, ed. Schaechter, M., Academic Press: Oxford, 3rd edn, 2009, pp. 80–94 Search PubMed.
- D. Mboowa, Biomass Convers. Biorefin., 2021 DOI:10.1007/s13399-020-01243-6.
- R. P. Wool, Lignin polymers and composites, in Bio-Based Polymers and Composites, ed. Wool, R. P. and Sun, X. S., Academic Press, Burlington, 2005, pp. 551–598 Search PubMed.
- M. V. Galkin and J. S. M. Samec, ChemSusChem, 2016, 9, 1544–1558 CrossRef CAS PubMed.
- S. Van den Bosch, T. Renders, S. Kennis, S. F. Koelewijn, G. Van den Bossche, T. Vangeel, A. Deneyer, D. Depuydt, C. M. Courtin, J. M. Thevelein, W. Schutyser and B. F. Sels, Green Chem., 2017, 19, 3313–3326 RSC.
- P. J. Deuss, M. Scott, F. Tran, N. J. Westwood, J. G. de Vries and K. Barta, J. Am. Chem., 2015, 137, 7456–7467 CrossRef CAS PubMed.
- K. Saito, Y. Makimura, H. Nishimura and T. Watanabe, ChemSusChem, 2021, 14, 2554–2563 CrossRef CAS PubMed.
- C. W. Lahive, P. C. J. Kamer, C. S. Lancefield and P. J. Deuss, ChemSusChem, 2020, 13, 4238–4265 CrossRef CAS PubMed.
- R. Parthasarathi, R. A. Romero, A. Redondo and S. Gnanakaran, J. Phys. Chem., 2011, 2, 2660–2666 CAS.
- S. Kim, S. C. Chmely, M. R. Nimos, Y. J. Bomble, T. D. Foust, R. S. Paton and G. T. Beckham, J. Phys. Chem., 2011, 2, 2846–2852 CAS.
- C. Crestini, H. Lange, M. Sette and D. S. Argyropoulos, Green Chem., 2017, 19, 4104–4121 RSC.
- C. Gioia, G. Lo Re, M. Lawoko and L. Berglund, J. Am. Chem., 2018, 140, 4054–4061 CrossRef CAS PubMed.
- D. S. Zijlstra, C. Lahive, C. A. Analbers, M. B. Figueiredo, Z. W. Wang, C. Lancefield and P. J. Deuss, ACS Sustainable Chem. Eng., 2020, 8, 5119–5131 CrossRef CAS.
- D. S. Zijlstra, C. A. Analbers, J. de Korte, E. Wilbers and P. J. Deuss, Polymers, 2019, 11, 1913 CrossRef CAS PubMed.
- D. S. Zijlstra, J. de Korte, E. P. C. de Vries, L. Hameleers, E. Wilbers, E. Jurak and P. J. Deuss, Front. Chem., 2021, 9, 655983 CrossRef CAS PubMed.
- M. Karlsson, V. L. Vegunta, R. Deshpande and M. Lawoko, Green Chem., 2022, 24, 2636–2637 RSC.
- M. Karlsson, N. Giummarella, P. A. Linden and M. Lawoko, ChemSusChem, 2020, 13, 4666–4677 CrossRef CAS PubMed.
- B. Rietzler, M. Karlsson, I. Kwan, M. Lawoko and M. Ek, Biomacromolecules, 2022, 23, 3349–3358 CrossRef CAS PubMed.
- J. R. Meyer, H. Y. Li, J. L. Zhang and M. B. Foston, ChemSusChem, 2020, 13, 4557–4566 CrossRef CAS PubMed.
- C. S. Lancefield, I. Panovic, P. J. Deuss, K. Barta and N. J. Westwood, Green Chem., 2017, 19, 202–214 RSC.
- A. M. Galan, J. Calinescu, A. Trifan, C. Winkworth-Smith, M. Calvo-Carrascal, C. Dodds and E. Binner, Chem. Eng. Process., 2017, 116, 29–39 CrossRef CAS.
- R. R. Mishra and A. K. Sharma, Composites, Part A, 2016, 81, 78–97 CrossRef CAS.
- C. O. Kappe, Chem. Soc. Rev., 2008, 37, 1127–1139 RSC.
- D. Haldar and M. K. Purkait, Chemosphere, 2021, 264, 128523 CrossRef CAS PubMed.
- E. T. Thostenson and T. W. Chou, Composites, Part A, 1999, 30, 1055–1071 CrossRef.
- A. de la Hoz, A. Diaz-Ortiz and A. Moreno, Chem. Soc. Rev., 2005, 34, 164–178 RSC.
- K. Martina, G. Cravotto and R. S. Varma, J. Org. Chem., 2021, 86, 13857–13872 CrossRef CAS PubMed.
- Á. Díaz-Ortiz; A. de la Hoz; J. R. Carrillo and M. A. Herrero, Selectivity Modifications Under Microwave Irradiation, in Microwaves in Organic Synthesis, 2012, pp. 209–244 Search PubMed.
- N. J. Hempel, M. M. Knopp, R. Berthelsen and K. Lobmann, Molecules, 2020, 25, 1068 CrossRef CAS PubMed.
- D. Mikulski, G. Klosowski, A. Menka and B. Koim-Puchowska, Bioresour. Technol., 2019, 278, 318–328 CrossRef CAS PubMed.
- Z. M. A. Bundhoo, Renewable Sustainable Energy Rev., 2018, 82, 1149–1177 CrossRef CAS.
- S. Tsubaki, K. Oono, A. Onda, K. Yanagisawa and J. Azuma, Bioresour. Technol., 2012, 123, 703–706 CrossRef CAS PubMed.
- V. L. Budarin, J. H. Clark, B. A. Lanigan, P. Shuttleworth and D. J. Macquarrie, Bioresour. Technol., 2010, 101, 3776–3779 CrossRef CAS PubMed.
- J. Yao, K. Odelius and M. Hakkarainen, ACS Appl. Polym. Mater., 2021, 3, 3538–3548 CrossRef CAS.
- Y. H. Zhang, Y. Z. Li, L. W. Wang, Z. H. Gao and M. R. Kessler, ACS Sustain. Chem. Eng., 2017, 5, 8876–8883 CrossRef CAS.
- Y. H. Zhang, V. K. Thakur, Y. Z. Li, T. F. Garrison, Z. H. Gao, J. Y. Gu and M. R. Kessler, Macromol. Mater. Eng., 2018, 303, 1700278 CrossRef.
- M. Sette, R. Wechselberger and C. Crestini, Chem. – Eur. J., 2011, 17, 9529–9535 CrossRef CAS PubMed.
- X. Z. Meng, C. Crestini, H. X. Ben, N. J. Hao, Y. Q. Pu, A. J. Ragauskas and D. S. Argyropoulos, Nat. Protoc., 2019, 14, 2627–2647 CrossRef CAS PubMed.
- P. F. Jacobs and D. T. Reid, Rapid prototyping & manufacturing : fundamentals of stereolithography, Society of Manufacturing Engineers in Cooperation with the Computer and Automated Systems Association of SME, Dearborn, MI, 1st edn, 1992, p. 434 Search PubMed.
- E. Fleck, A. Sunshine, E. DeNatale, C. Keck, A. McCann and J. Potkay, Micromachines, 2021, 12, 1266 CrossRef PubMed.
- H. Gong, M. Beauchamp, S. Perry, A. T. Woolley and G. P. Nordin, RSC Adv., 2015, 5, 106621–106632 RSC.
- M. Galkin, Curr. Opin. Green Sustainable Chem., 2021, 28, 100438 CrossRef CAS.
- A. De Santi, M. V. Galkin, C. W. Lahive, P. J. Deuss and K. Barta, ChemSusChem, 2020, 13, 4468–4477 CrossRef CAS PubMed.
- N. Sweygers, N. Alewaters, R. Dewil and L. Appels, Sci. Rep., 2018, 8, 7719 CrossRef PubMed.
- J. J. Fan, M. De Bruyn, V. L. Budarin, M. J. Gronnow, P. S. Shuttleworth, S. Breeden, D. J. Macquarrie and J. H. Clark, J. Am. Chem., 2013, 135, 11728–11731 CrossRef CAS PubMed.
- G. Torgovnikov and P. Vinden, For. Prod. J., 2010, 60, 173–182 Search PubMed.
- F. Monteil-Rivera, G. H. Huang, L. Paquet, S. Deschamps, C. Beaulieu and J. Hawari, Bioresour. Technol., 2012, 104, 775–782 CrossRef CAS PubMed.
- H. Wang, M. L. Maxim, G. Gurau and R. D. Rogers, Bioresour. Technol., 2013, 136, 739–742 CrossRef CAS PubMed.
- L. Zoia, M. Orlandi and D. S. Argyropoulos, J. Agric. Food Chem., 2008, 56, 10115–10122 CrossRef CAS PubMed.
- W. H. Chen, Y. J. Tu and H. K. Sheen, Appl. Energy, 2011, 88, 2726–2734 CrossRef CAS.
- P. Lidstrom, J. Tierney, B. Wathey and J. Westman, Tetrahedron, 2001, 57, 9225–9283 CrossRef CAS.
- B. B. Hallac, Y. Q. Pu and A. J. Ragauskas, Energy Fuels, 2010, 24, 2723–2732 CrossRef CAS.
- C. Y. Dong, X. Z. Meng, C. S. Yeung, H. Y. Tse, A. J. Ragauskas and S. Y. Leu, Green Chem., 2019, 21, 2788–2800 RSC.
- P. Sannigrahi, A. J. Ragauskas and S. J. Miller, Energy Fuels, 2010, 24, 683–689 CrossRef CAS.
- D. M. Miles-Barrett, A. R. Neal, C. Hand, J. R. D. Montgomery, I. Panovic, O. S. Ojo, C. S. Lancefield, D. B. Cordes, A. M. Z. Slawin, T. Lebl and N. J. Westwood, Org. Biomol. Chem., 2016, 14, 10023–10030 RSC.
- S. N. Obame, I. Ziegler-Devin, R. Safou-Tchima and N. Brosse, J. Agric. Food Chem., 2019, 67, 5989–5996 CrossRef CAS PubMed.
- W. J. J. Huijgen, G. Telysheva, A. Arshanitsa, R. J. A. Gosselink and P. J. de Wild, Ind. Crops Prod., 2014, 59, 85–95 CrossRef CAS.
- J. Guit, M. B. L. Tavares, J. Hul, C. N. Ye, K. Loos, J. Jager, R. Folkersma and V. S. D. Voet, ACS Appl. Polym. Mater., 2020, 2, 949–957 CrossRef CAS.
- V. S. D. Voet, T. Strating, G. H. M. Schnelting, P. Dijkstra, M. Tietema, J. Xu, A. J. J. Woortman, K. Loos, J. Jager and R. Folkersma, ACS Omega, 2018, 3, 1403–1408 CrossRef CAS PubMed.
- J. Yao and M. Hakkarainen, Compos. Commun., 2023, 38, 101506 CrossRef.
- J. W. Choi, G. J. Kim, S. Hong, J. H. An, B. J. Kim and C. W. Ha, Sci. Rep., 2022, 12, 4548 CrossRef PubMed.
- A. Garcia, M. G. Alriols, G. Spigno and J. Labidi, Biochem. Eng. J., 2012, 67, 173–185 CrossRef CAS.
- H. Sadeghifar and A. Ragauskas, Polymers, 2020, 12, 1134 CrossRef CAS PubMed.
- D. Tian, J. G. Hu, J. Bao, R. P. Chandra, J. N. Saddler and C. H. Lu, Biotechnol. Biofuels, 2017, 10, 192 CrossRef PubMed.
- Y. Li, Q. J. Mao, J. Yin, Y. F. Wang, J. Z. Fu and Y. Huang, Addit. Manuf., 2021, 37, 101716 CAS.
- J. F. Stanzione, J. M. Sadler, J. J. La Scala and R. P. Wool, ChemSusChem, 2012, 5, 1291–1297 CrossRef CAS PubMed.
- C. Zhang, S. A. Madbouly and M. R. Kessler, Macromol. Chem. Phys., 2015, 216, 1816–1822 CrossRef CAS.
- G. Arias-Ferreiro, A. Lasagabaster-Latorre, A. Ares-Pernas, P. Ligero, S. M. Garcia-Garabal, M. S. Dopico-Garcia and M. J. Abad, Polymers, 2022, 14, 2068 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3su00115f |
| This journal is © The Royal Society of Chemistry 2023 |