
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA versatile core–shell hetero-nanostructure catalysed chemo-selective synthesis of β-enamino carbonyl compounds†
Sriparna
Dutta
ab,
Prashant
Kumar
c,
Shivani
Sharma
ad,
Sneha
Yadav
a,
Priyanka
a,
Ranjana
Dixit
ad,
Anju
Srivastava
ab and
Rakesh Kumar
Sharma
 *a
*a
aGreen Chemistry Network Centre, Department of Chemistry, University of Delhi, Delhi-110007, India. E-mail: rksharamgreenchem@hotmail.com
bDepartment of Chemistry, Hindu College, University of Delhi, Delhi-110007, India
cDepartment of Chemistry, SRM University Delhi-NCR, Sonepat, Haryana-131029, India
dDepartment of Chemistry, Ramjas College, University of Delhi, Delhi-110007, India
First published on 3rd May 2023
Abstract
A core–shell hetero-nanostructure comprising a new in situ generated naphthoquinone cobalt complex covalently immobilized on a silica coated magnetite nanosupport has been described for catalyzing the enamination of β-carbonyl compounds. The newly developed catalyst works as a nanoreactor for boosting this reaction and provides unprecedented chemoselective access to the targeted β-enaminoesters as well as β-enaminones. The synergistic covalent incorporation of an organic entity (a flexible spacer) onto an inorganic solid (surface modified support material) imparts a considerable structural rigidity to the catalyst thereby preventing leaching of the active catalytic species as well evidenced by the hot filtration experiment. Elemental survey and core level XPS spectra confirm the presence of the +II oxidation state of cobalt which is the active catalytic species. This work throws light on how catalytic activity can be tuned and “activity, selectivity and recyclability” the three desired features of a green catalyst can be accomplished on a single platform through careful manipulation of interaction between the support material and active metal complex.
Sustainability spotlightIn 2015, the entire world came to a common consensus when seventeen sustainable development goals were outlined by the various member countries of the UN that carried a vision of rendering the planet safer for all. With a very few years left to accomplish these goals, tremendous efforts will be required to transform the commitments into action. Catalysis the key principle of green chemistry provides one of the most promising and logistic routes for achieving most of the SDGs such as zero hunger, good health & well being, clean water & sanitation, affordable & clean energy, industries, innovation & infrastructure, responsible consumption & production and climate action. The present work discloses the design of a novel core–shell hetero nanostructure based catalyst comprising a new in situ generated naphthoquinone cobalt complex covalently immobilized on a silica coated magnetite nanosupport that has unveiled promising outcomes in catalyzing the enamination of β-carbonyl compounds (that synergistically integrate the nucleophilicity of the enamine and electrophilicity of the enone functionalities, thereby serving as valuable synthons for the construction of essential pharmaceutical drugs). Although literature reports document the development of several synthetic approaches involving homogeneous metal complexes that provide access to these compounds, yet appealing generality and the scope of industrial utility of these catalytic protocols are vitiated by the fact that most of them rely on homogeneous metal salts as catalysts that are not only difficult to recover from the reaction mixture but also major contributors to metal contamination. The present work is a sustainable attempt to overcome the serious limitations of previous protocols via the establishment of a catalyst that offers “activity, selectivity and recyclability” the three desired features of a green catalyst in a single platform through careful manipulation of interaction between the support material and active metal complex. |
Introduction
The drive towards cleaner and sustainable chemical manufacturing has led to a reassessment of many of the existing catalytic technologies. Today, there is a renewed focus on the development of green catalytic systems that exhibit higher activity as well as enhanced reusability as they provide a potent solution to meet the current challenges of energy and sustainability. In fact, with the booming of nanotechnology, it has been possible to design such efficient catalysts as they allow the precise engineering of finer structural details of catalysts such as size, shape and surface properties.1–5 Although efforts in this direction have enabled the successful generation of interesting nanomaterials suited for various applications, magnetically responsive nanomaterials have, in particular, shown great promise in the field of catalysis owing to their riveting physico-chemical properties.6–12 Magnetic nanoparticles work as excellent solid support materials for the design of superior and well-tuned heterogeneous nanocatalysts that exhibit high activity and selectivity similar to their homogeneous counterparts.13,14 Nanosized particles dramatically increase the net exposed surface area of the active component of the catalyst, eventually resulting in improved product yield. Besides, most importantly, these catalysts show a rapid response towards an external magnetic field that ensures their ready separation from the reaction mixture using an external magnetic device. Magnetic field assisted separation is green, economical and undoubtedly an attractive alternative to tedious, time-consuming filtration techniques. Thus, researchers have devoted an enormous amount of attention towards the synthesis of these magnetically recyclable nanocatalysts. However, an obvious deterioration in their activity as a consequence of strong agglomeration tendency of the magnetic nanoparticles has raised serious concerns.15,16 In order to address this issue, core–shell magnetic silica based nanomaterials comprising nanoparticles suitably encapsulated within a layer of silica have been developed as they uniquely combine the characteristic features of the magnetic core as well as the silica shell that screens the dipolar attraction between the individual particles, thereby preventing their aggregation.17–20 In addition, the silanaceous coat also allows free access to various chemical species from outside, rendering them suitable for catalytic applications. The efficacy of silica encapsulated magnetic nanoparticle supported catalytic systems has prompted us to come up with a new core–shell structured magnetic silica supported cobalt nanocatalyst for the synthesis of unsaturated β-carbonyl moieties such as β-enaminones and β-enaminoesters.β-Enaminones and β-enaminoesters represent well documented scaffolds that have fuelled the research interest of several organic chemists worldwide as they synergistically integrate the nucleophilicity of the enamine and electrophilicity of the enone functionalities, thereby serving as valuable synthons for the construction of essential pharmaceutical drugs.21–24 Besides, they constitute prevalent structural motifs of biologically significant natural products that are endowed with a broad spectrum of interesting pharmacological properties such as antibacterial, anticonvulsant, anti-inflammatory, antitumour etc.25–31 Thus, over the past few decades a number of captivating strategies have been reported for obtaining these heterocyclic moieties which include reactions of β-enaminoesters with organolithium reagents,32 addition of amide enolates or metallic esters to imidoyl halides,33–36 nitriles,37–39 and tosylimines40,41 and addition of enamines or ketamines to activated carboxylic acid derivatives.42–44 However, despite the existence/prevalence of a large number of intriguing protocols in the literature, the classical approach involving the direct condensation of amines with 1,3-dicarbonyls still continues to be one of the most widely pursued pathways for the synthesis of β-enamino ketones and esters because of its operational simplicity and higher atom economy.45 Consequently, a wide array of catalysts such as Sc(OTf)3,46 Zn(OAc)2·6H2O,47 ZrOCl2·8H2O,48 Ca(CF3COO)2,49 CoCl2,50 InBr3,51 I2,52 silica gel,53 CeCl3·7H2O54 and NaAuCl455 have been employed proficiently to afford this transformation. Unfortunately, the appealing generality and the scope of industrial utility of these catalytic protocols are vitiated by the fact that most of them rely on homogeneous metal salts as catalysts that are not only difficult to recover from the reaction mixture but also major contributors to metal contamination. Thus, in consideration of growing economic and industrial concerns, it is extremely imperative to develop other alternative environmentally benign, heterogeneous recyclable catalytic systems that can efficiently conquer the challenges of β-enaminone and β-enaminoester synthesis.
In a quest to design a supreme catalytic route for obtaining these industrially and biologically significant β-enamine based moieties and engaged in the development of organic–inorganic hybrid catalysts for various organic transformations, we herein report the fabrication, characterization and application of a promising core–shell magnetic silica based cobalt nanocatalyst (Co-NQ@Am-SiO2@Fe3O4). The newly developed catalytic system enables the generation of a diverse range of β-enaminones and β-enaminoesters with excellent turnover numbers and high product selectivity under neat reaction conditions at room temperature. Apart from this, the ease of catalytic retrievability via magnetic attraction and outstanding recycling efficiency (observable up to eight consecutive runs) render the present protocol a practical alternative to all previously established methodologies for the synthesis of β-enamine ketones and esters. To the best of our knowledge, this is the first report wherein a magnetically retrievable nanocatalyst has been designed and subsequently utilized for the enamination of β-ketoesters and β-ketones.
Experimental
Materials and reagents
1,2-Naphthoquinone (1,2-NQ) (97%), tetra-ethyl orthosilicate (TEOS) (99.9%) and 3-aminopropyltriethoxysilane (APTES) (98%) were purchased from Alfa Aesar, Sigma Aldrich and Fluka, respectively. Ferrous sulphate, ferric sulphate and cobalt chloride hexahydrate salts were commercially acquired from Sisco Research Laboratory (SRL). The rest of the synthetic reagents were procured from Spectrochem Pvt Ltd and were used as received without further purification.Characterization techniques
The crystallographic structure and phase of the synthesized nanomaterials were analyzed well with the help of the powder X-ray diffraction (PXRD) technique which was performed using a Bruker, D8 Advance (Karlsruhe, Bundesland, Germany) diffractometer with Cu/Kα radiation at a scanning rate of 4° min−1 in the 2θ range of 5–80° (λ = 0.15405 nm, 40 kV, 40 mA). Electron microscopic techniques such as transmission electron microscopy (TEM) and field emission scanning electron microscopy (FE-SEM) were carried out in order to determine the size, shape and purity of the nanocomposites. TEM was conducted using a JEOL 2100F, and FE-SEM images were obtained with the aid of a Tescan Mira 3 FE-SEM instrument. The energy-dispersive X-ray spectroscopic technique (equipped with the SEM instrument) was utilized for the detailed compositional analysis of the nanocomposites. Coloured elemental mapping images were also obtained using the same instrument. The XPS survey spectrum of the catalyst and the core-level spectra of Fe 2p and Co 2p were procured with the help of a K-Alpha™+ X-ray photoelectron spectrometer system. The fitting of the core level spectra was performed by adjusting the baseline relative to the signal background. The chemically distinct species were resolved using a Gaussian distribution fitting procedure with peak positions, and areas were determined. The stepwise modifications of the magnetite nanoparticles were identified by Fourier transform infrared (FT-IR) spectroscopy. The FT-IR spectra of the various stages of the nanocatalyst were acquired through a PerkinElmer Spectrum 2000 FT-IR spectrometer that utilized the KBr pellet method and was operative in the range of 4000–400 cm−1 under atmospheric conditions with a resolution of 1 cm−1. In order to extract information about the magnetic properties of the nanocomposites, a vibrating sample magnetometer (EV-9, Microsense, ADE) was used. The presence of cobalt in the final nanocatalyst was affirmed through energy dispersive X-ray fluorescence (ED-XRF) spectroscopy using a Fischerscope X-ray XAN-FAD BC. Thereafter, the amount of cobalt loaded onto surface modified magnetite nanoparticles was estimated via flame atomic absorption spectroscopy (FAAS) on a Labindia AA 7000 atomic absorption spectrometer. Prior to AAS analysis, the microwave assisted digestion of the catalyst was performed in an Anton Paar Multiwave 3000 microwave reaction system equipped with a temperature and pressure sensor. A sonicator (Tekmar Sonic Disruptor model TM300) was utilized for dispersing the nanoparticle aggregates in ethanol. The GC-MS analysis of the synthesized products was carried out on an Agilent gas chromatograph (6850 GC) with an HP-5MS 5% phenyl methyl siloxane capillary column (30.0 m × 250 μm × 0.25 μm) and a quadrupole mass filter equipped 5975 mass selective detector (MSD) using helium as carrier gas (rate 0.9 mL min−1).Catalyst preparation
 | ||
| Scheme 1 Schematic illustration of the synthesis of Co-NQ@Am-SiO2@Fe3O4 nanocatalyst. | ||
Results and discussion
Catalyst characterization
Morphological attributes of the bare and surface modified magnetite nanoparticles were investigated with the help of electron microscopic tools such as Transmission Electron Microscopy (TEM) and Scanning Electron Microscopy (SEM). The representative TEM and SEM images of the synthesized nanocomposites are depicted in Fig. 1. It is quite evident from the TEM micrograph of the core material (Fe3O4) that it consists of tiny particles of nearly spherical morphology with an average diameter lying between 8 and 12 nm. The selected area electron diffraction (SAED) pattern of Fe3O4 reveals the high crystallinity of these nanoparticles by showing the presence of a well pronounced array of diffraction rings arising from the planes of magnetite. For their detailed structural analysis, high resolution transmission electron microscopy (HR-TEM) was also conducted. The HR-TEM image of the pristine Fe3O4 nanoparticles clearly illustrates the 2D lattice fringes that correspond to the (220) plane of the inverse spinel structured magnetite with an interplanar separation value of 0.28 nm. Besides providing a significant amount of knowledge about the size, shape and crystallinity of the Fe3O4 nanoparticles, TEM and SEM also work as powerful tools for identifying the differences between the bare and modified magnetite nanoparticles. As shown in Fig. S1†, these nanoparticles acquire core–shell morphology after being suitably coated with amorphous silica. A closer examination of this image reveals that the core material is embedded deep within the silane shell, which makes it a good core–shell nanomaterial. Furthermore, the deposition of various surface modifying species on these SiO2@Fe3O4 nanoparticles is verified from the obtained TEM micrograph of the final nanocatalyst. The FE-SEM images of the various nanocomposites procured at high magnification divulge a change in the surface topography of the magnetic nanocore. On being subjected to several synthetic modifications, the smooth surface of the naked Fe3O4 nanoparticles apparently gets transformed into a rough one which may be attributed to the deposition of silica in the core–shell structured nanomaterials. However, the possibility of precipitation of primary silica nanoparticles is certainly excluded as no separate colloidal aggregates of silica can be found in the Fe-SEM images of SiO2@Fe3O4 and Co-NQ@Am-SiO2@Fe3O4. Finally, in order to elucidate that the catalyst maintains its efficiency even after being recycled for seven consecutive runs, the FE-SEM image of the recovered nanocatalyst has also been provided (Fig. S2†). | ||
| Fig. 1 (a) TEM image of nano-Fe3O4; (b) HR-TEM image of nano-Fe3O4; (c) SAED pattern of nano Fe3O4; (d) TEM image of the Co-NQ@Am-SiO2@Fe3O4 nanocatalyst; (e) FE-SEM image of Fe3O4 and (f) FE-SEM image of the Co-NQ@Am-SiO2@Fe3O4 nanocatalyst. | ||
In order to gain an insight into the crystallinity and phase composition of the synthesized nanomaterials, wide angle powder X-ray diffraction (PXRD) measurements were performed and the typical XRD profiles of Fe3O4 and SiO2@Fe3O4 are presented in Fig. 2. Interestingly, both these diffractograms display six distinguishable Bragg's peaks at 2θ = 30.1°, 35.4°, 43.1°, 53.4°, 57° and 62.6° that may be correctly assigned to the (220), (311), (400), (422), (511) and (440) crystallographic faces of the cubic inverse spinel structured magnetite. The peak positions as well as the relative intensities are in good agreement with the standard Fe3O4 XRD data as evidenced by the Joint Committee on Powder Diffraction Standards (JCPDS card: 19-629). However, the XRD pattern of silica encapsulated magnetite nanoparticles differs slightly from that of the native magnetite nanoparticles. Besides, exhibiting these well resolved diffraction peaks, the XRD spectrum of SiO2@Fe3O4 shows the appearance of a broad diffused hump centered at around 2θ = 23° which is attributed to the presence of amorphous SiO2.57 One aspect that needs to be highlighted here is that although the silica coating strategy efficiently leads to the generation of a protective layer of silica around the magnetic core, it does not affect the phase and purity of these crystalline nanoparticles, as evident from the diffractogram which clearly shows the retention of the six characteristic Bragg's peaks in this core–shelled nanomaterial. Furthermore, the mean crystallite size of the Fe3O4 nanoparticles has been evaluated using the Debye Scherrer relationship (Dhkl = kλl/β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ) where k is a constant with a typical value of 0.89 for spherical particles, D is the average diameter in A°, β is the broadening of the diffraction line measured at half of its maximum intensity in radians, λ is the wavelength of the X-rays and θ is the Bragg diffraction angle. Choosing (311) as the reference peak, this value has been estimated to be 11.9 nm which matches well with the TEM results. Furthermore, the XRD spectra of the catalyst and the recovered catalyst, exhibiting consistency with the native ferrite peaks have been provided (Fig. S3†).
cosθ) where k is a constant with a typical value of 0.89 for spherical particles, D is the average diameter in A°, β is the broadening of the diffraction line measured at half of its maximum intensity in radians, λ is the wavelength of the X-rays and θ is the Bragg diffraction angle. Choosing (311) as the reference peak, this value has been estimated to be 11.9 nm which matches well with the TEM results. Furthermore, the XRD spectra of the catalyst and the recovered catalyst, exhibiting consistency with the native ferrite peaks have been provided (Fig. S3†).
 | ||
| Fig. 2 XRD patterns of (a) Fe3O4 and (b) SiO2@Fe3O4. | ||
The elemental and compositional analyses of the bare and modified nanoparticles were performed using energy-dispersive X-ray spectroscopy (EDX), energy dispersive X-ray fluorescence spectroscopy (ED-XRF) and atomic absorption spectroscopy (AAS). Fig. S4† provides the EDX mapping images of the isolated particles of Fe3O4 and SiO2@Fe3O4. The well resolved peaks of iron (Fe) and oxygen (O) atoms in the EDX spectrum of Fe3O4 affirm the presence of an iron oxide core. Besides the elemental distribution of Fe and O, the occurrence of Si in the EDX spectrum of SiO2@Fe3O4 authenticates the successful encapsulation of the core material by silica.
Apart from this, the SEM elemental coloured mapping image of the Co-NQ@Am-SiO2@Fe3O4 nanocatalyst (Fig. 3) has been provided which shows the presence of distinct elements – “Fe, Si, O, C, N, and Co,” thus confirming the successful functionalization, ligand grafting and metallation of the silica coated magnetite nanoparticles. In addition to the elemental mapping, the ED-XRF spectrum of Co-NQ@Am-SiO2@Fe3O4 also provides a concrete proof for the existence of the metal in the catalyst by markedly showing the presence of a sharp and distinct peak of cobalt (Fig. 4). Next, the quantitative estimation of cobalt in the prepared catalyst was performed by digesting Co-NQ@Am-SiO2@Fe3O4 in a microwave oven at 400 W for 15 minutes with aqua regia and then subjecting the sample to AAS analysis. The corresponding metal content was evaluated to be 0.14 mmol g−1.
 | ||
| Fig. 3 SEM elemental mapping images of the Co-NQ@Am-SiO2@Fe3O4 nanocatalyst showing the presence of Fe, Si, O, C, N, and Co elements. | ||
 | ||
| Fig. 4 ED-XRF spectrum of the Co-NQ@Am-SiO2@Fe3O4 nanocatalyst. | ||
With the aim of clearly identifying the chemical composition and acquiring information about the oxidation state of cobalt, the surface structure of the final nanocatalyst was further characterized by X-ray photoelectron spectroscopy (XPS). Fig. 5 presents the XPS elemental survey scans and core level spectra of Fe 2p and Co 2p of the magnetic silica based organic–inorganic hybrid cobalt catalyst. The presence of elements C, O, Si, N, Fe and Co is confirmed from the survey spectrum of Co-NQ@Am-SiO2@Fe3O4. Next, the core level spectrum of Fe 2p was analyzed and it was found that two distinct bands appeared at around 710 and 725 eV along with a satellite peak at 717.8 eV (Fig. S5†). As these bands were broad, deconvolution of the same was attempted and successfully achieved. In the deconvoluted spectrum of Fe 2p, first the presence of combined peaks at binding energy values of 710.8 eV (Fe 2p3/2) and 725 eV (Fe 2p1/2) provided strong evidence of the presence of iron in the +II oxidation state in the catalyst. Furthermore, the occurrence of a satellite peak could be routinely ascribed to iron in the +II oxidation state. Apart from this, the prominent peaks appearing at binding energy values of 712 eV (Fe 2p3/2) and 725 eV (Fe 2p1/2) characterized the simultaneous existence of iron in the +III oxidation state in our sample. But since we are primarily interested in evaluating the oxidation state of cobalt which is the active catalytic species in our final catalyst, the XPS spectrum of Co 2p was plotted and investigated thoroughly. The deconvoluted spectrum revealed the existence of cobalt in the +II oxidation state. In this case, the binding energies for the two possible spin states 781.2 eV and 797.5 eV are responsible for 2p3/2 and 2p1/2 transitions, respectively and in addition, two satellite peaks were observed which primarily arose from each of the 2p transition.
 | ||
| Fig. 5 (a) Survey X-ray photoelectron spectrum (XPS) of Co-NQ@Am-SiO2@Fe3O4, (b) XPS spectra (Co 2p region) of Co in the catalyst, (c) core level XPS of Fe 2p of the sample and (d) core level XPS of Co 2p of the sample. | ||
For the structural confirmation of the various nanocomposites through functional group detection, Fourier transform infrared (FT-IR) spectroscopic analysis was carried out at room temperature. Fig. 6 depicts the FT-IR spectra of Fe3O4, SiO2@Fe3O4, Am-SiO2@Fe3O4, NQ@Am-SiO2@Fe3O4 and Co-NQ@Am-SiO2@Fe3O4.
 | ||
| Fig. 6 FTIR spectra of (a) Fe3O4; (b) SiO2@Fe3O4; (c) Am-SiO2@Fe3O4; (d) NQ@Am-SiO2@Fe3O4 and (e) Co-NQ@Am-SiO2@Fe3O4. | ||
The FT-IR spectrum of Fe3O4 exhibits a sharp and intense absorption band at 595 cm−1 indicative of typical Fe–O stretching vibrations and a relatively less intense broad band centered at 3149 cm−1 corresponding to the vibrations of OH groups present on the surface of the magnetite nanoparticles.59 These characteristic absorption bands collectively provide strong evidence for the formation of the Fe3O4 nanocore. Next, the existence of the silane shell around this core is affirmed from the spectrum of SiO2@Fe3O4 which distinctly shows the presence of Si–O–Si symmetric, Si–O–Si asymmetric and Si–O symmetric stretching modes appearing at 806, 954 and 1102 cm−1 respectively.60 Furthermore, the proof for the successful integration of the functionalizing agent “APTES” into the coated nanoparticles is provided by the appearance of two new bands at 2930 cm−1 and 1625 cm−1 that can be assigned to the CH2 and NH2 groups of the aminopropyl moiety. On moving towards NQ@Am-SiO2@Fe3O4, a distinctive band of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretch is observed at 1646 cm−1 besides the expected absorption bands which suggests that the ligand has been successfully immobilized over the surface of the amine functionalized silica coated ferrite nanoparticles. Astonishingly, as a result of complexation between the nitrogen atom and the metal, the value of the CN stretch is shifted to a lower wavenumber (1630 cm−1 from 1646 cm−1) in the case of the final nanocatalyst.
N stretch is observed at 1646 cm−1 besides the expected absorption bands which suggests that the ligand has been successfully immobilized over the surface of the amine functionalized silica coated ferrite nanoparticles. Astonishingly, as a result of complexation between the nitrogen atom and the metal, the value of the CN stretch is shifted to a lower wavenumber (1630 cm−1 from 1646 cm−1) in the case of the final nanocatalyst.
In order to examine the magnetic properties of the synthesized nanocomposites, a vibrating sample magnetometer (VSM) was employed and the resultant magnetization curves of Fe3O4, SiO2@Fe3O4 and Co-NQ@Am-SiO2@Fe3O4 recorded within the field range of −20000 to 20000 Oe at 300 K are displayed in Fig. 7. The hysteresis loops of these powdered nanomaterials reveal the absence of any remanence or co-ercivity phenomena which confirms their superparamagnetic properties at room temperature. Besides, the inset also supports this observation as the obtained plots clearly show that both the magnetization and demagnetization curves pass through the origin. Furthermore, the value of saturation magnetization (Ms) of the prepared Fe3O4 nanoparticles as estimated from the graph has been found to be 60 emu g−1 which is much lower than that of the bulk Fe3O4 nanoparticles. The significant reduction in the Ms value can be predominantly attributed to the presence of disordered spins on the surface of the nanoparticles that prevent the core spins from aligning along the field direction, thereby causing a net decrease in the saturation magnetization of the small sized nanoparticles. Additionally, the VSM plots show a gradual decrement in the Ms values on moving from the bare Fe3O4 to the surface modified Fe3O4 (i.e. Fe3O4 > SiO2@Fe3O4 > Co-NQ@Am-SiO2@Fe3O4) which may be ascribed to the existence of non-magnetic materials on the surface of the core–shelled nanoparticles.61 However, it is worth mentioning here that regardless of the lowering of the saturation magnetizations, the redispersibility and magnetic properties of the final nanocatalyst remain unaffected and the Co-NQ@Am-SiO2@Fe3O4 nanoparticles are easily retrieved from the reaction mixture with the help of a bar magnet within a very short span of time.
 | ||
| Fig. 7 Magnetization curves obtained by VSM at room temperature for (a) Fe3O4, (b) SiO2@Fe3O4, and (c) Co-NQ@Am-SiO2@Fe3O4 and (d) inset: an enlarged image near the coercive field. | ||
Investigation of catalytic activity of Co-NQ@Am-SiO2@Fe3O4 in the synthesis of β-enaminones and β-enaminoesters
The efficacy of the newly prepared magnetic silica-based cobalt nanocatalyst was evaluated in the synthesis of industrially significant β-enaminone and β-enaminoester moieties. For achieving the best catalytic results, the effects of some of the crucial thermodynamic and kinetic reaction parameters such as catalyst concentration, time, temperature, solvents and molar substrate ratio were investigated in detail. We commenced our study by choosing methyl acetoacetate and benzylamine as the model substrates.The quest for establishing optimal reaction conditions began by performing the catalytic screening test. However, at the outset, to truly assess the significance of a catalyst in the enamination reaction, a control experiment was conducted with the test substrates in the absence of a catalyst. A careful evaluation of the outcome divulged that only traces of the expected product could be detected under such experimental conditions (Table 1, entry 1). Next, a wide range of cobalt based catalysts were screened sequentially for the generation of the desired β-enaminoester scaffold with a high conversion percentage. Gratifyingly, as shown in Table 1, amongst all the tested cobalt sources, the developed Co-NQ@Am-SiO2@Fe3O4 and Co-based catalysts formed by the coordination of CoCl2 with 1,2-naphthoquinone (Co-NQ complex) prove to be the most proficient ones, exhibiting the best catalytic activity by causing a 100% conversion of the employed reactants. In comparison to the precursor CoCl2 salt utilized for the reaction which gave 95% conversion and the other salts such as CoSO4 and CoBr2 that showed very low activity (42% and 45%, respectively) the Co-NQ complex gave a higher conversion that could be ascribed to the coordinating effect of 1,2-naphthoquinone with CoCl2, leading to a robust and active complex. Furthermore, although Co-NQ and Co-NQ@Am-SiO2@Fe3O4 showed similar conversions, yet the fabricated catalyst “Co-NQ@Am-SiO2@Fe3O4” emerged as the material of choice owing to the exceptional magnetism conferred by the material which enhanced the catalytic recoverability and reusability properties. Subsequently, in order to analyse the effect of catalyst concentration, the reactions were performed by varying the amount of catalyst from 5 to 20 mg and the results have been demonstrated in Table 1. It was found that an increase in the catalyst loading caused a substantial improvement in the conversion percentage of the product from 39% to 100% which could be attributed to the simultaneous increase in the active catalytic sites. Thus, for the rest of the studies, the amount of the catalyst was fixed at 20 mg.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Yieldb (%) | Conversionc (%) | TONd | TOFe |
| a Reaction conditions: benzylamine (1 mmol), methylacetoacetate (1 mmol), catalyst (20 mg unless specified), stirring at r.t. (25 °C), no solvent. b Isolated Yield. c Conversion (%) determined by GC-MS. d TON is the number of moles of product per mole of catalyst. e TOF = TON/time (in hours). | |||||
| 1 | No catalyst | — | Traces | — | — |
| 2 | SiO2@Fe3O4 | — | Traces | — | — |
| 3 | CoSO4 | 35 | 42 | 150 | 300 |
| 4 | Co(OAc)2 | 37 | 48 | 171 | 342 |
| 5 | CoBr2 | 40 | 45 | 161 | 322 |
| 6 | CoCl2 | 82 | 95 | 339 | 678 |
| 7 | Co-NQ complex | 94 | 100 | 357 | 714 |
| 8 | Co-NQ@Am-SiO2@Fe3O4 (5 mg) | 30 | 39 | 139 | 278 |
| 9 | Co-NQ@Am-SiO2@Fe3O4 (10 mg) | 55 | 68 | 243 | 486 |
| 10 | Co-NQ@Am-SiO2@Fe3O4 (15 mg) | 88 | 95 | 339 | 678 |
| 11 | Co-NQ@Am-SiO2@Fe3O4 (20 mg) | 95 | 100 | 357 | 714 |
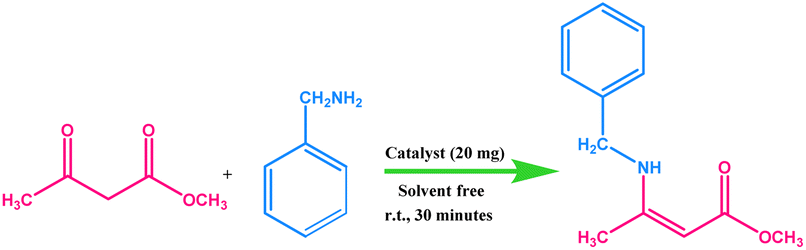
After optimization of the amount of catalyst, our next investigation was focussed on addressing the influence of the substrate molar ratio on the reaction conversions. For an accurate assessment, three different sets of reactions were performed with 1:0.5, 1:0.75 and 1:1 molar ratios of methylacetoacetate and benzylamine, respectively in the presence of the Co-NQ@Am-SiO2@Fe3O4 nanocatalyst (Fig. S6†). Apparently, the results indicated that the enamination reaction utilizing a reagent molar ratio of 1:0.5 could afford a conversion of only 70% even after a prolonged stirring (up to 6 h) period. However, with a further increase in the molar ratio of the substrates to 1:1, a complete 100% conversion of the subjected reactants could be accomplished within a shorter time (30 minutes) interval. Hence, a 1:1 substrate molar ratio was the optimum requirement for the efficient synthesis of β-enaminones and β-enaminoesters with a good conversion percentage.
The selection of an appropriate solvent is a matter of increasing importance in the field of catalytic science and research as solvents play a key role in either facilitating or hindering a reaction through their interaction with the catalyst and the solute molecules. Thus, a scrupulous examination of the solvent effect on the enamination reaction was carried out by stirring the model reactants in the presence of solvents of variable polarities. Additionally, in consideration of the current emphasis on green, waste free synthesis, solvent free conditions were also tested, while keeping the rest of the parameters fixed. Fig. S7† illustrates the influence of solvents on the conversion percentages of the reactants. In comparison to all the non-polar solvents employed, polar solvents such as ethanol, isopropanol, acetonitrile and dimethylformamide (DMF) led to a superior reaction conversion owing to the higher solubility of the substrates in them. Notably, the best results were obtained under solvent less conditions as complete (100%) conversion of the subjected reactants was achieved in this case.
Considering the significance of temperature in altering the outcome of a reaction, we decided to investigate its effect on the one pot synthesis of β-enaminones and β-enaminoesters. As a step towards accomplishing this task, at first, the enamination reaction was performed at 25 °C using 20 mg of the synthesized Co-NQ@Am-SiO2@Fe3O4 nanocatalyst. Thereafter, the temperature of the reaction was increased progressively at intervals of 10 °C and its simultaneous influence on the product conversion and selectivity was studied. The results presented in Table S1† unambiguously revealed that among all the temperatures evaluated, 25 °C was found to be the most ideal one as it afforded the desired product with 100% conversion and selectivity. However, higher reaction temperatures (35 °C, 45 °C, 55 °C, and 65 °C) showed a noticeable decrease in the product selectivity which could be attributed to the formation of the di-substituted product.
Finally, the effect of reaction time on the percentage conversion of the model reactants was monitored by varying the time interval from 5 minutes to 30 minutes at 25 °C. The experimental results are displayed in Fig. S8.†
It is well apparent from the plot that the reaction conversion increases significantly with increasing the reaction time which is due to the fact that a longer time period favours the formation of the reactive intermediate species (substrate + catalyst) that eventually facilitates the formation of the desired product. A close inspection of the results showed that a reaction time of 30 minutes was the optimal period for the complete conversion of the model substrates.
Substrate scope and versatility
Pleased by the optimization results, we set out to challenge the capabilities of the Co-NQ@Am-SiO2@Fe3O4 nanocatalyst in the synthesis of β-enaminones and β-enaminoesters (Table 2). In an endeavour towards envisaging the scope of this methodology, a wide array of β-ketoesters and β-diketones were reacted with several aromatic amines bearing electron rich as well as electron deficient substituents under the optimized reaction conditions. It was found that in general, the enamination reaction proceeded smoothly in the presence of the nanostructured cobalt catalyst via nucleophilic addition of amines to carbonyl compounds to afford the desired products with good to excellent conversion. However, the electronic factor played a key role in regulating the product conversion as evident from the results summarized in Table 2. The presence of electron withdrawing substituents such as F and Cl at the para-position of the benzene ring of the aromatic amines, in particular, resulted in a detrimental effect on the reaction conversion (Table 2, entries 6, 10, and 24). Also, in the case of aryl substituted β-ketoesters such as ethyl benzoylacetate, a moderate conversion was achieved despite continuing the reaction for a prolonged time period (Table 2, entries 13–17). Surprisingly, the rest of the β-ketoesters showed good reactivity and furnished the targeted product within a relatively shorter time. Notably, it is worth mentioning that although the conversion percentage varied drastically depending on the nature of the substrate employed, all the reactions displayed excellent chemoselectivity.|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | Conversionb (%) | Yieldc (%) | TONd | TOFe |
| a Reaction conditions: amine (1 mmol), β-ketoester (1 mmol), Co-NQ@Am-SiO2@Fe3O4 catalyst (20 mg), stirring at r.t. (25 °C). b Conversion percentages determined via GC-MS have been given in parentheses. c Isolated yield. d TON is the number of moles of the product per mole of the catalyst. e TOF = TON/time (in Hours). | |||||||
| 1 | CH3 | OCH3 | CH2Ph | 100 | 95 | 357 | 714 |
| 2 | CH3 | OCH3 | Ph | 96 | 87 | 343 | 686 |
| 3 | CH3 | OCH3 | 4-CH3Ph | 95 | 90 | 339 | 678 |
| 4 | CH3 | OCH3 | 4-OCH3Ph | 86 | 82 | 307 | 614 |
| 5 | CH3 | OCH3 | 2-OCH3Ph | 80 | 80 | 286 | 572 |
| 6 | CH3 | OCH3 | 4-FPh | 47 | 40 | 168 | 336 |
| 7 | CH3 | OCH2CH3 | CH2Ph | 98 | 91 | 350 | 700 |
| 8 | CH3 | OCH2CH3 | Ph | 98 | 93 | 350 | 700 |
| 9 | CH3 | OCH2CH3 | 4-CH3Ph | 95 | 86 | 339 | 678 |
| 10 | CH3 | OCH2CH3 | 4-ClPh | 61 | 55 | 218 | 436 |
| 11 | CH3 | OCH2CH3 | 4-OCH3Ph | 88 | 81 | 314 | 628 |
| 12 | CH3 | OCH2CH3 | 2-OCH3Ph | 67 | 59 | 239 | 478 |
| 13 | Ph | OCH2CH3 | PhCH2 | 89 | 80 | 318 | 636 |
| 14 | Ph | OCH2CH3 | Ph | 80 | 74 | 286 | 572 |
| 15 | Ph | OCH2CH3 | 4-CH3Ph | 84 | 77 | 300 | 600 |
| 16 | Ph | OCH2CH3 | 4-OCH3Ph | 78 | 69 | 279 | 558 |
| 17 | Ph | OCH2CH3 | 2-OCH3Ph | 72 | 62 | 257 | 514 |
| 18 | CH3 | OC(CH3)3 | PhCH2 | 91 | 79 | 325 | 650 |
| 19 | CH3 | OCH2Ph | PhCH2 | 65 | 59 | 232 | 464 |
| 20 | CH3 | OCH(CH3)2 | PhCH2 | 93 | 88 | 332 | 664 |
| 21 | ClCH2 | OCH2CH3 | PhCH2 | 85 | 79 | 304 | 608 |
| 22 | CF3 | OCH2CH3 | PhCH2 | 73 | 62 | 261 | 522 |
| 23 | CH3 | CH3 | PhCH2 | 100 | 95 | 357 | 714 |
| 24 | CH3 | CH3 | 4-ClPhCH2 | 72 | 72 | 257 | 514 |
| 25 | CH3 | CH3 | 4-CH3PhCH2 | 87 | 85 | 311 | 622 |

In order to investigate the chemoselectivity of the process, we carried out a simple test (Scheme 2). First, we reacted a primary amine (n-butylamine) with β-ketoester (methylacetoacetate) under the optimized reaction conditions. We obtained the desired product with 70% reaction conversion. Next, we reacted a secondary amine (morpholine) with the same substrate (i.e. methylacetoacetate) and found that no morpholine derived product was formed even after prolonged stirring of the reaction contents.
 | ||
| Scheme 2 Chemoselectivity test for the synthesis of β-enaminoesters. | ||
Hot filtration test
For organic transformations involving a supported heterogeneous catalyst, an important issue that should be taken into account is the possibility of migration of the catalytically active species from the surface of the support material into the liquid phase during the course of the reaction. In order to exclude any catalytic contribution from the leached species, a standard hot filtration experiment was performed by subjecting the model reactants to vigorous stirring at room temperature in the presence of Co-NQ@Am-SiO2@Fe3O4 (20 mg). At about half the reaction time (15 minutes), the catalyst was retrieved via magnetic decantation when the conversion percentage had reached 60% as estimated through GC-MS analysis. The resultant supernatant was allowed to react further for a prolonged time period under similar experimental conditions and the reaction progress was monitored continuously. No enhancement in the reaction conversion occurred even at extended times indicating that the active cobalt species remained tightly bound to the heterogeneous support. Additionally, atomic absorption and UV-visible spectroscopic analyses of the post reaction mixture were also carried out which showed the absence of cobalt in the supernatant as anticipated. The findings from all these studies synchronically provided concrete evidence for the heterogeneous nature of the catalyst.Recyclability test
In green chemistry approaches for catalytic transformations, recovery and reusability are the two most significant attributes of a heterogeneous catalyst that lead to its large-scale industrial applicability. To assess the long-term stability and reusability of the synthesized nanostructured catalyst, we established a set of recycling experiments for the enamination reaction which were performed sequentially under the optimized reaction conditions using the test substrates. After completion of the first cycle, the solid catalyst was separated from the reaction vessel via magnetic attraction, washed with acetone several times to remove traces of the previous reaction mixture, dried under reduced pressure and thereafter subjected to a fresh run. Strikingly, it was found that the recovered nanocatalyst did not require the addition of any external reagent for getting reactivated. The results of the recycling test as demonstrated in Fig. 8 unveiled that Co-NQ@Am-SiO2@Fe3O4 could be efficiently reused for eight consecutive trials without any substantial loss in its activity. Besides, the TEM micrograph of the recovered catalyst (obtained after the 8th run) provided key evidence for the structural durability of Co-NQ@Am-SiO2@Fe3O4. It was found that its morphological characteristics remained largely preserved during the course of the reaction, signifying that the employed reaction conditions did not have any detrimental effect on its catalytic activity. | ||
| Fig. 8 Recycling experiment for the enamination reaction [reaction conditions: benzylamine (1 mmol), methylacetoacetate (1 mmol), Co-NQ@Am-SiO2@Fe3O4 catalyst (20 mg), stirring at r.t. (25 °C)]. | ||
Comparison of the catalytic activity of Co-NQ@Am-SiO2@Fe3O4 with that of the other reported heterogeneous catalysts
A comparison of the catalytic performance of Co-NQ@Am-SiO2@Fe3O4 with the literature reported heterogeneous catalysts for the synthesis of biologically significant β-enaminones and β-enaminoesters is presented in Table 3. The results clearly demonstrate that the activity of Co-NQ@Am-SiO2@Fe3O4 is far superior to that of the different heterogeneous catalytic systems reported so far62–69 in terms of product yield, reaction conditions, durability and reusability. The reason behind the remarkable efficiency of the silica encapsulated magnetically retrievable cobalt catalyst can be attributed to the nanometer size of the support material which ensures greater binding of the substrates eventually leading to higher product yield. Furthermore, the ease of catalyst separation via simple magnetic attraction renders the protocol an attractive alternative for catalyzing the enamination of β-ketoesters and β-ketones as the magnetic field assisted separation prevents catalyst loss, reduces energy consumption and saves time in achieving catalyst recovery.| Entry | β-Ketoester/β-ketone | Amine | Catalytic system | Reaction conditions | Yield (%) | Ref. |
|---|---|---|---|---|---|---|
| 1 |
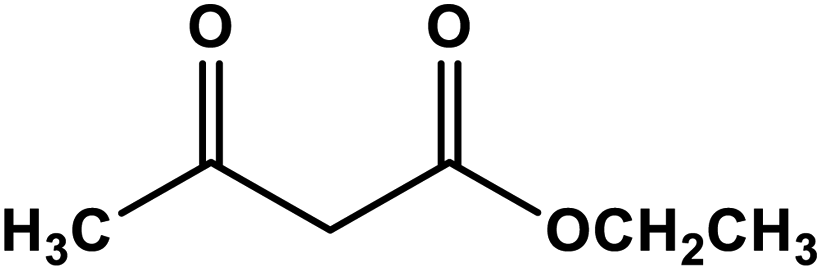
|

|
[Cu(Hnbta)(1,3-btp)]·2H2O | Solvent free r.t. | 93 | 62 |
| Metal–organic-framework | ||||||
| Recyclability: three runs | ||||||
| 2 |

|

|
SiW/c-Al2O3 | Acetonitrile r.t. | 94 | 63 |
| Supported heteropolyacid | ||||||
| Recyclability: four runs | ||||||
| 3 |

|

|
Cu nanoparticles | Methanol 45–50 °C under a N2 atmosphere | 92 | 64 |
| Recyclability: not evaluated | ||||||
| 4 |
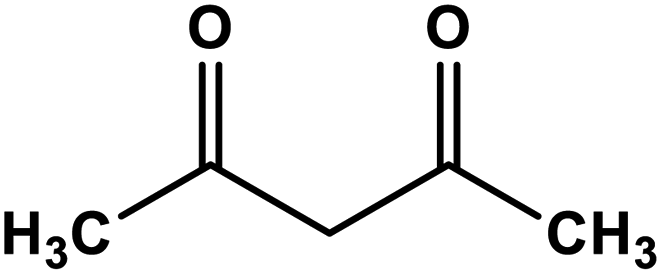
|

|
Silica sulfuric acid | Solvent-free 80 °C | 89 | 65 |
| Recyclability: three runs | ||||||
| 5 |

|

|
ZnO nanoparticles | Solvent-free 80 °C | 94 | 66 |
| Recyclability: four runs | ||||||
| 6 |

|
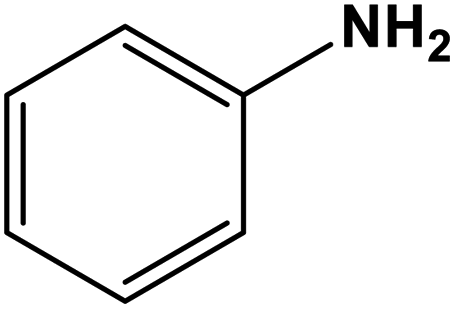
|
NiO | Ultrasound sonication 30 °C | 90 | 67 |
| Recyclability: three runs | ||||||
| 7 |
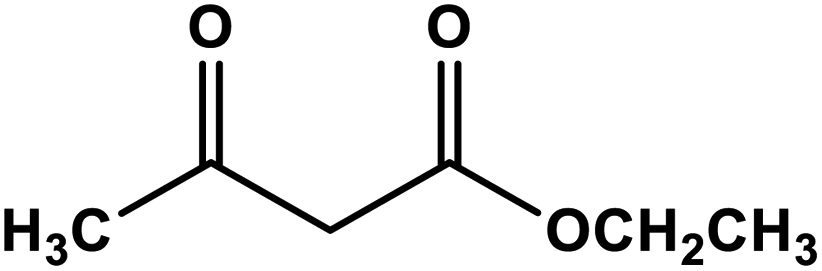
|

|
Ag nanoparticles in hollow mesoporous spheres | Methanol 60 °C | 84 | 68 |
| Recyclability: five runs | ||||||
| 8 |
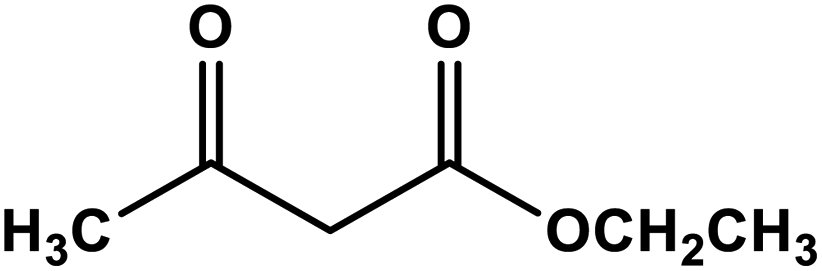
|

|
Silver nanoparticles | Methanol 60 °C | 70 | 69 |
| Recyclability: three runs | ||||||
| 9 |

|
|
Naphthoquinone cobalt complex supported on SiO2@Fe3O4 nanoparticles | Solvent free r.t. | 98 | Present work |
| Recyclability: eight runs | ||||||
| 10 |

|

|
Naphthoquinone cobalt complex supported on SiO2@Fe3O4 nanoparticles | Solvent free r.t. | 99 | Present work |
Conclusions
This work throws light on the synthesis of a new, versatile core–shell structured magnetically recoverable cobalt nanocatalyst that has been applied for the first time in the enamination of β-dicarbonyl compounds. The synthetic strategy adopted for preparing the catalyst is operationally simple and leads to the fabrication of a well configured nanostructure consisting of a naphthoquinone cobalt complex robustly anchored onto an amine functionalized silica encapsulated magnetite nanosupport. The as-synthesized nanocatalyst has been characterized well with the help of different spectroscopic and microscopic tools. XPS survey and core-level spectra of the obtained catalyst provide thorough evidence of the nature of the metal, showing that cobalt primarily existed in the +II oxidation state. This protocol impressively allows the generation of an appreciable range of the targeted molecules using the Co-NQ@Am-SiO2@Fe3O4 nanocatalyst with excellent conversion (up to 100%) and a high turnover number (up to 357) at room temperature under solvent free conditions. In contrast to literature precedents for the enamination reaction, the present methodology offers a magnetically separable nanocatalyst that can be easily recovered from the reaction mixture with the help of an external magnet and reused further for several runs without any distinct loss in its activity. Besides, some of the other factors that worked towards enhancing the economic attractiveness of the protocol were its high atom economy, use of environmentally benign and inexpensive reagents, simple work up procedure, and mild reaction conditions. We strongly believe that the design of this high performance magnetically retrievable catalytic system would pave the pathway for future research aiming at development of new organic compounds of biological and industrial significance.Author contributions
Sriparna Dutta was involved in designing, conceptualization, and experimental and writing work. Prashant Kumar was involved in writing and experimental work. Shivani Sharma and Priyanka helped in characterization and proofreading. Sneha Yadav contributed to writing and data analysis. Ranjana Dixit, Anju Srivastava, and Rakesh Kumar Sharma were involved in overall monitoring and supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
One of the authors, Sriparna Dutta, expresses her gratitude to the University Grant Commission, Delhi, India for the award of a Senior Research Fellowship. Also, the authors thank JNU for TEM analysis and USIC-CLF, DU for FTIR, XRD and VSM analyses. Also special thanks to NCL, Pune for XPS analysis. Prashant Kumar is thankful to SERB for providing funds under the scheme of Teacher Associateship for Research Excellence (TARE) (file number TAR/2021/000201).Notes and references
- S. B. Kalidindi and B. R. Jagirdar, ChemSusChem, 2012, 5, 65–75 CrossRef CAS PubMed.
- L. L. Chng, N. Erathodiyil and J. Y. Ying, Acc. Chem. Res., 2013, 46, 1825–1837 CrossRef CAS PubMed.
- V. Polshettiwar and R. S. Varma, Green Chem., 2010, 12, 743–754 RSC.
- H. Barron and A. S. Barnard, Catal. Sci. Technol., 2015, 5, 2848–2855 RSC.
- T. Mitsudome and K. Kaneda, ChemCatChem, 2013, 5, 1681–1691 CrossRef CAS.
- D. Wang and D. Astruc, Chem. Rev., 2014, 114, 6949–6985 CrossRef CAS PubMed.
- L. M. Rossi, N. J. S. Costa, F. P. Silva and R. Wojcieszak, Green Chem., 2014, 16, 2906–2933 RSC.
- V. Polshettiwar, R. Luque, A. Fihri, H. Zhu, M. Bouhrara and J.-M. Basset, Chem. Rev., 2011, 111, 3036–3075 CrossRef CAS PubMed.
- D. Zhang, C. Zhou, Z. Sun, L.-Z. Wu, C.-H. Tunga and T. Zhang, Nanoscale, 2012, 4, 6244–6255 RSC.
- R. B. N. Baig and R. S. Varma, Chem. Commun., 2013, 49, 752–770 RSC.
- R. B. N. Baig and R. S. Varma, Green Chem., 2013, 15, 398–417 RSC.
- S. Shylesh, V. Schünemann and W. R. Thiel, Angew. Chem., Int. Ed., 2010, 49, 3428–3459 CrossRef CAS PubMed.
- J. Govan and Y. K. Gunko, Nanomaterials, 2014, 4, 222–241 CrossRef PubMed.
- X. Gao, R. Liu, D. Zhang, M. Wu, T. Cheng and G. Liu, Chem. Eur. J., 2014, 20, 1515–1519 CrossRef CAS PubMed.
- S. Zheng, J. Sun, D. Song, Z. Chenab and J. Chen, Chem. Commun., 2015, 51, 11123–11125 RSC.
- Y. Zhu, L. P. Stubbs, F. Ho, R. Liu, C. P. Ship, J. A. Maguire and N. S. Hosmane, ChemCatChem, 2010, 2, 365–374 CrossRef CAS.
- Z. Wu, C. Sun, Y. Chai and M. Zhang, RSC Adv., 2011, 1, 1179–1182 RSC.
- H. Yang, G. Li and Z. Ma, J. Mater. Chem., 2012, 22, 6639–6648 RSC.
- S. Wang, Z. Zhang, B. Liu and J. Li, Catal. Sci. Technol., 2013, 3, 2104–2112 RSC.
- C. Jin, Y. Wang, H. Wei, H. Tang, X. Liu, T. Lua and J. Wang, J. Mater. Chem. A, 2014, 2, 11202–11208 RSC.
- I. O. Edafiogho, S. B. Kombian, K. V. Ananthalakshmi, N. N. Salama, N. D. Eddington, T. L. Wilson, M. S. Alexander, P. L. Jackson, C. D. Hanson and K. R. Scott, J. Pharm. Sci., 2007, 96, 2509–2531 CrossRef CAS PubMed.
- M. Abass and B. B. Mostafa, Bioorg. Med. Chem., 2005, 13, 6133–6144 CrossRef CAS PubMed.
- A. M. Ei-Shennawy, A. H. Mohamed and M. Abass, MedGenMed, 2007, 9, 15–33 Search PubMed.
- J. D. White and D. C. Ihle, Org. Lett., 2006, 8, 1081–1084 CrossRef CAS PubMed.
- N. N. Salama, K. R. Scott and N. D. Eddington, Biopharm. Drug Dispos., 2004, 25, 227–236 CrossRef CAS PubMed.
- N. D. Eddington, D. S. Cox, M. Khurana, N. N. Salama, J. P. Stables, S. J. Harrison, A. Negussie, R. S. Taylor, U. Q. Tran, J. A. Moore, J. C. Barrow and K. R. Scott, Eur. J. Med. Chem., 2003, 38, 49–64 CrossRef CAS PubMed.
- A. P. Marcus and R. Sarpong, Org. Lett., 2010, 12, 4560–4563 CrossRef CAS PubMed.
- J. P. Michael, C. B. Koning, G. D. Hosken and T. V. Stanbury, Tetrahedron, 2001, 57, 9635–9648 CrossRef CAS.
- I. O. Edafiogho, M. S. Alexander, J. A. Moore, V. A. Farrar and K. R. Scott, Curr. Med. Chem., 1994, 1, 159–175 CrossRef CAS.
- Y. F. Wang, T. Izawa, S. Kobayashi and M. Ohno, J. Am. Chem. Soc., 1982, 104, 6465–6466 CrossRef CAS.
- D. L. Boger, T. Ishizaki, J. R. Jr Wysocki, S. A. Munk, P. A. Kitos and O. Suntornwat, J. Am. Chem. Soc., 1989, 111, 6461–6463 CrossRef CAS.
- G. Cimarelli, G. Palmieri and E. Volpini, Tetrahedron Lett., 2004, 45, 6629–6631 CrossRef.
- K. Uneyama, O. Morimoto and F. Yamashita, Tetrahedron Lett., 1989, 30, 4821–4824 CrossRef CAS.
- S. Fustero, B. Pina, A. Simo and N. Fuentes, Tetrahedron Lett., 1997, 38, 6771–6774 CrossRef CAS.
- S. Fustero, B. Pina, E. Salavert, A. Navarro, C. RamTrez de Arellano and A. Simón, J. Org. Chem., 2002, 67, 4667–4679 CrossRef CAS PubMed.
- S. Fustero, B. Pina, M. GarcTa de la Torre, A. Navarro, C. Ramirez de Arellano and A. Simón, Org. Lett., 1999, 1, 977–980 CrossRef CAS.
- S. M. Hannick and Y. Kishi, J. Org. Chem., 1983, 48, 3833–3835 CrossRef CAS.
- A. S.-Y. Lee and R.-Y. Cheng, Tetrahedron Lett., 1997, 38, 443–446 CrossRef CAS.
- T. G. C. Bird and A. Olivier, Bioorg. Med. Chem. Lett., 1996, 6, 515–520 CrossRef.
- T. Fukuyama and Y. M. Yung, Tetrahedron Lett., 1981, 22, 3759–3760 CrossRef CAS.
- N. Jiang, Z. Qu and J. Wang, Org. Lett., 2001, 3, 2989–2992 CrossRef CAS PubMed.
- G. Bartoli, C. Cimarelli, R. Dalpozzo and G. Palmieri, Tetrahedron, 1995, 51, 8613–8622 CrossRef CAS.
- A. R. Katritzky, Y. Fang, A. Donkor and J. Xu, Synthesis, 2000, 2029–2032 CrossRef CAS.
- S. Fustero, M. GarcTa de la Torre, V. Jofre`, R. Pérez Carlon, A. Navarro and A. Simón Fuentes, J. Org. Chem., 1998, 63, 8825–8836 CrossRef CAS.
- D. F. Martin, G. A. Janusonis and B. B. Martin, J. Am. Chem. Soc., 1961, 83, 73–75 CrossRef CAS.
- J. S. Yadav, V. N. Kumar, R. S. Rao, A. D. Priyadarshini, P. P. Rao, B. V. S. Reddy and K. Nagaiah, J. Mol. Catal. A: Chem., 2006, 256, 234–237 CrossRef CAS.
- R. K. Vohra, J. L. Renaud and C. Bruneau, Collect. Czech. Chem. Commun., 2005, 70, 1943–1952 CrossRef CAS.
- Z. H. Zhang, T. S. Li and J. J. Li, Catal. Commun., 2007, 8, 1615–1620 CrossRef CAS.
- M. A. Harrad, R. Outtouch, M. A. Ali, L. El Firdoussi, A. Karim and A. Roucoux, Catal. Commun., 2010, 11, 442–446 CrossRef CAS.
- Z. H. Zhang and J. Y. Hu, J. Braz. Chem. Soc., 2006, 17, 1447–1451 CrossRef CAS.
- Z. H. Zhang, L. Yin and Y. M. Wang, Adv. Synth. Catal., 2006, 348, 184–190 CrossRef CAS.
- S. Gogoi, R. Bhuyan and N. C. Barua, Anal. Lett., 2005, 35, 2811–2818 CAS.
- Y. Gao, Q. Zhang and J. Xu, Synth. Commun., 2004, 34, 909–916 CrossRef CAS.
- M. M. Khodaei, A. R. Khosropour and M. Kookhazadeh, Synlett, 2004, 11, 1980–1984 CrossRef.
- A. Arcadi, G. Bianchi, S. Di Giuseppe and F. Marinelli, Green Chem., 2003, 5, 64–67 RSC.
- V. Polshettiwar, B. Baruwati and R. S. Varma, Green Chem., 2009, 11, 127–131 RSC.
- Q. Zhang, H. Su, J. Luo and Y. Wei, Green Chem., 2012, 14, 201–208 RSC.
- Z. Afrasiabi, E. Sinn, P. P. Kulkarni, V. Ambike, S. Padhye, D. Deobagakar, H. Mark, G. Chris, E. A. Christopher and A. K. Powell, Inorg. Chim. Acta, 2005, 358, 2023–2030 CrossRef CAS.
- K. Petcharoena and A. Sirivat, Mater. Sci. Eng., B, 2012, 177, 421–427 CrossRef.
- S. Wang, Z. Zhang, B. Liu and J. Li, Catal. Sci. Technol., 2013, 3, 2104–2112 RSC.
- M. Esmaeilpour, A. R. Sardarian and J. Javidi, J. Organomet. Chem., 2014, 749, 233–240 CrossRef CAS.
- Y. Zhao, D. S. Deng, L. F. Ma, B. M. Ji and L. Y. Wang, Chem. Commun., 2013, 49, 10299–10301 RSC.
- E. Rafiee, M. Joshaghani, S. Eavani and S. Rashidzadeh, Green Chem., 2008, 10, 982–989 RSC.
- M. Kidwai, S. Bhardwaj, N. K. Mishra, V. Bansal, A. Kumar and S. Mozumdar, Catal. Commun., 2009, 10, 1514–1517 CrossRef CAS.
- A. Hasaninejad, A. Zare, M. R. Mohammadizadeh, M. Shekouhy and A. R. Moosavi-Zare, J. Chem., 2010, 7, 1546–1554 CAS.
- U. U. Indulkar, S. R. Kale, M. B. Gawande and R. V. Jayaram, Tetrahedron Lett., 2012, 53, 3857–3860 CrossRef CAS.
- S. Shendage and J. Nagarkar, Curr. Chem. Lett., 2013, 2, 145–152 CrossRef CAS.
- J. Sun, Z. Dong, P. Li, F. Zhang, S. Wei, Z. Shi and R. Li, Mater. Chem. Phys., 2013, 140, 1–6 CrossRef CAS.
- K. D. Bhatte, P. J. Tambade, K. P. Dhake and B. M. Bhanage, Catal. Commun., 2010, 11, 1233–1237 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3su00073g |
| This journal is © The Royal Society of Chemistry 2023 |