
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Revisiting organosolv strategies for sustainable extraction of valuable lignin: the CoffeeCat process†
Marie E.
Vuillemin
 a,
María Catalina
Quesada-Salas
a,
Caroline
Hadad
bd,
Jordane
Jasniewski
c,
Eric
Husson
*a and
Catherine
Sarazin
*a
a,
María Catalina
Quesada-Salas
a,
Caroline
Hadad
bd,
Jordane
Jasniewski
c,
Eric
Husson
*a and
Catherine
Sarazin
*a
aUnité de Génie Enzymatique et Cellulaire, UMR CNRS 7025, UFR des Sciences, Université de Picardie Jules Verne, 33 rue Saint Leu, 80039 Amiens Cedex 1, France. E-mail: eric.husson@u-picardie.fr; catherine.sarazin@u-picardie.fr
bLaboratoire de Glycochimie, des Antimicrobiens et des Agroressources, UMR CNRS 7378, UFR des Sciences, Université de Picardie Jules Verne, 33 rue Saint Leu, 80039 Amiens Cedex 1, France
cLaboratoire d'Ingénierie des Biomolécules (LIBio), Université de Lorraine, 2 Avenue de la Forêt de Haye, TSA40602, 54518 Vandœuvre-lès-Nancy, France
dInstitut de Chimie de Picardie FR CNRS 3085, 80039 Amiens, France
First published on 10th May 2023
Abstract
An innovative and sustainable strategy for the selective extraction of lignin from lignocellulosic biomass has been designed, namely the CoffeeCat process, in which only green solvents and reagents are required: (i) water, (ii) 2-methyltetrahydrofuran-3-one (coffee furanone) recognized as a food grade ingredient and readily biodegradable and (iii) glutamic acid. Two fractions have been isolated from Miscanthus x giganteus, the delignified fraction (DL-glu) and the enriched-lignin fraction (L-glu). Competitive extraction yields of 27% and 43% of enriched-lignin fractions were respectively obtained at 140 °C (L-glu-140) and 180 °C (L-glu-180), based on the lignin content of the original biomass. The structural properties of these lignins were characterized by spectroscopic (FTIR and NMR), microscopic (SEM) and separative (SEC-MALLS) methods. Compared to other processes described in the literature, our strategy involved the isolation of lignin fractions with high purity (up to 84%). Both fractions have been valorized: (i) the DL-glu fractions have been subjected to ionic liquid pretreatment and subsequent enzymatic hydrolysis leading to a total depolymerization of the constitutive cellulose (99%) and to an efficient conversion of hemicellulose into xylose (70%); (ii) the L-glu fractions have been used to produce lignin nanoparticles (LNPs) in a mixture of 2-methyltetrahydrofuran-3-one/water (1/110 v/v). The size distribution (272 ± 9 nm and 472 ± 6 nm), charge (−29.2 ± 0.8 mV and −20.8 ± 0.4 mV) and regular spherical shape of these LNPs have been determined using Zetasizer-DLS measurements and SEM images of L-glu-140 and L-glu-180, respectively. In addition, the possibility of easy incorporation of the L-glu fraction into polylactic acid without requiring previous lignin modification has been preliminarily explored. The CoffeeCat process was thus demonstrated as a relevant eco-solution for an integrated lignocellulosic biorefinery.
Sustainability spotlightThis work is based on the CoffeeCat process to design a sustainable strategy for a full-component refinery of lignocellulose. The strategy uses green solvents (the food approved 2-methyltetrahydrofuran-3-one and water) and reagents (glutamic acid) to selectively extract at 140 °C a nearly carbohydrate-free lignin with high β-O-4 linkage content. Synergistic ionic liquid pretreatment applied to the remaining delignified biomass led to total conversion of polysaccharides into platform sugars. The physico-chemical properties of the extracted lignin could open the way of designing biocompatible nanoparticles and facilitating the formulation of PLA–lignin biocomposites. Miscanthus is used as an example for potential biomass feedstock. This work is in line with points 12 and 13 of the UN's Sustainable Development Goals (combat climate change and ensure sustainable production patterns). |
Introduction
In the current context of environmental issues of climate change, lignocellulosic biomass (LCB), one of the most substantial renewable feedstocks, is increasingly proposed as a promising alternative1–3 to fossil resources. LCB offers potential to be converted by sustainable processing to clean energy biofuels, biomaterials and value-added chemicals,4–8 with the new value chain opening up to circular economy development.9 LCB contains mostly polymeric carbohydrates (hemicellulose, 20–30% w/w and cellulose, 30–40% w/w)10–12 and lignin, a polyphenolic polymer (15–40% w/w).11–13 The use of cellulose and hemicellulose has been intensively studied for the production of paper, sugars, bioethanol, biobutanol and other fermentation products through various biological or thermochemical pathways.4,14–22 However, lignin is frequently considered as a by-product from LCB biorefining and is still currently under-valued.4 Only less than 2% of the 1.5–1.8 billion tons of industrial lignin waste is used in the world annually.4,23,24 Most of the lignin waste is dumped or burned as low-grade fuel, which not only causes wastage of resources, but also incurs serious environmental pollution.23 Furthermore, the valorization of LCB is constrained by the highly ordered matrix of the raw material. The interactions between the LCB components are responsible for the recalcitrant properties of this biomass and considerably limit the accessibility to each compound.25–28 Different pretreatments (mechanical, chemical and biological) have been reported in the literature to fractionate the biomass, reduce its recalcitrance and access its components for further valorization.29,30 However, the implementation conditions of these strategies often generate uncontrolled depolymerization/repolymerization affecting the structural integrity of polymers, which limits the valorization potential, in particular for the lignin fraction. The complex, heterogeneous and variable structure of this is later related to the LCB source and is particularly sensitive to extraction processes (Kraft, lignosulfonate, soda, and organosolv processes).31–34 The requirement for inorganic salts, a strong acid or base, or even some organic solvents, with questionable safety to implement these extractions, is well-known to considerably affect the chemical and structural properties of lignin (sulfur content, C–C bonds, and molecular weight) and its reactivity, thus offering fewer further possibilities for chemical or enzymatic transformations.35–38 A delignification process must allow the provision of a lignin extract for high value applications in the field of food, cosmetics and medicine.39 For this kind of applications, eco-compatible conditions of implementation could be required to minimize intrinsic chemical modification and traces of incompatible solvents.Among recent literature data, the selection of a green solvent for the development of organosolv processes has emerged. For instance, the OrganoCat strategy has been reported as an economically viable approach to co-valorize the three main constitutive fractions of LCB (xylose, cellulose pulp and lignin).40–49 It consists of a biphasic system with a reactive aqueous phase supplemented in oxalic acid that selectively depolymerizes the hemicellulose and an organic phase composed of 2-methyltetrahydrofuran (MeTHF) allowing lignin solubilization.41,42,50,51 Although the OrganoCat strategy can be considered as fully bio-based,52,53 some drawbacks could be identified in terms of environmental compatibility and restriction to some field of applications. For example, high oxalic acid concentrations are required, which are not compatible with food applications.54 In addition, the recovery of highly viscous and adhesive lignin43 requires additional purification steps including petroleum-based solvents and energy consumption before valorization.43,44,55 Recently, other organosolv process studies reported the use of biobased solvents such as γ-valerolactone,56 dihydrolevoglucosenone57 or dimethyl isosorbide58 to improve the sustainability of LCB fractioning. However, the use of these solvents was often combined with sulfuric acid affecting the structure of polymers.
Therefore, in this study, we proposed to design a new organosolv process to both improve the sustainability of the extraction of an enriched-lignin fraction without residual sulfur or carbohydrate and preserve its structural integrity as much as possible. To this end, we rationally selected a food-grade and easily biodegradable solvent together with natural amino acids (i.e. proteinogenic amino acids). 2-Methyltetrahydrofuran-3-one (MeTHF-3-one) is listed as generally recognized as safe (GRAS) and readily biodegradable in agreement with the Organization for Economic Co-operation and Development (OECD) standards59 and used in food industries to mimic flavors.60 GRAS amino acids were chosen (glutamic acid and aspartic acid)61 to replace the usually used oxalic acid in the OrganoCat process, also tested for comparison. The use of these natural materials could deserve some applications requiring a “nature” label, for example, in cosmetics applications. The targeted LCB was Miscanthus x giganteus (Mxg), a dedicated crop and recognized feedstock in circular and local biorefineries.62–65 The ability of our proposed strategy to selectively extract lignin while improving the accessibility of structural polysaccharides was investigated. The generated fractions were characterized by quantitative and structural analytical techniques (FTIR, NMR, SEM, and SEC-MALLS). Possible ways of valorization for each fraction were explored: (i) the enzymatic conversion of polysaccharides into platform monomeric sugars, (ii) the conception of lignin-based nanoparticles, and (iii) the design of polylactic acid (PLA)–lignin-based composites. A flowchart of the entire process is presented in Chart 1.
 | ||
| Chart 1 Schematic of the overall CoffeeCat process. | ||
Experimental
Materials
MeTHF-3-one (>97%) was supplied by Alfa Aesar (Thermo Fisher Scientific, Heysham) and L-aspartic acid (>99%), L-glutamic acid (>99%), oxalic acid (>99%) and commercial alkali lignin (Kraft low sulfonate content, 4% sulfur, Mw ∼10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da) were purchased from Sigma-Aldrich (Germany). Miscanthus x giganteus (Mxg) was provided by INRAe (France). 1-Ethyl-3-methylimidazolium acetate ([Emim][OAc], >98%) was purchased from Solvionic S.A (Verniolle, France). Anhydrous sodium acetate (99%) was from Fluka Sigma-Aldrich (Steinheim, Germany), sodium hydroxide aqueous solution (46/48%) from Fisher Scientific (Illkirch, France), acetic acid (99%) from Carl Roth (Lauterbourg, France) and sulfuric acid 72% w/w from VWR. HPLC grade trifluoroacetic acid (99%) was purchased from Sigma-Aldrich (Germany), HPLC grade acetonitrile (>99%) was obtained from Merck (Darmstadt) and dimethyl sulfoxide D6 (99.80%) was obtained from VWR. Absolute ethanol (>99.8%) and methanol (>99.9%); standard glucose (99.5%) and xylose (99%) were purchased from Sigma-Aldrich (Steinheim, Germany). A Cellic CTec2 enzymatic cocktail including both cellulolytic and hemicellulolytic activities (specific activity ≥1000 U g−1) was supplied by Novozymes (Bagsvaerd, Denmark) and prepared with 15 FPU g−1 of lignocellulosic biomass for the production of hydrolysates rich in monomeric sugars. Glass microfibre filters (<1 μm pore size) were from Whatman, GE Healthcare Life Sciences (Boston, MA, USA). Folin & Ciocalteu's phenol reagents, anhydrous sodium carbonate and gallic acid were purchased from Sigma-Aldrich (France). 4032D PLA beads were obtained from NatureWorks PLA Ingeo (USA).
000 Da) were purchased from Sigma-Aldrich (Germany). Miscanthus x giganteus (Mxg) was provided by INRAe (France). 1-Ethyl-3-methylimidazolium acetate ([Emim][OAc], >98%) was purchased from Solvionic S.A (Verniolle, France). Anhydrous sodium acetate (99%) was from Fluka Sigma-Aldrich (Steinheim, Germany), sodium hydroxide aqueous solution (46/48%) from Fisher Scientific (Illkirch, France), acetic acid (99%) from Carl Roth (Lauterbourg, France) and sulfuric acid 72% w/w from VWR. HPLC grade trifluoroacetic acid (99%) was purchased from Sigma-Aldrich (Germany), HPLC grade acetonitrile (>99%) was obtained from Merck (Darmstadt) and dimethyl sulfoxide D6 (99.80%) was obtained from VWR. Absolute ethanol (>99.8%) and methanol (>99.9%); standard glucose (99.5%) and xylose (99%) were purchased from Sigma-Aldrich (Steinheim, Germany). A Cellic CTec2 enzymatic cocktail including both cellulolytic and hemicellulolytic activities (specific activity ≥1000 U g−1) was supplied by Novozymes (Bagsvaerd, Denmark) and prepared with 15 FPU g−1 of lignocellulosic biomass for the production of hydrolysates rich in monomeric sugars. Glass microfibre filters (<1 μm pore size) were from Whatman, GE Healthcare Life Sciences (Boston, MA, USA). Folin & Ciocalteu's phenol reagents, anhydrous sodium carbonate and gallic acid were purchased from Sigma-Aldrich (France). 4032D PLA beads were obtained from NatureWorks PLA Ingeo (USA).
Methods
| Experimental values % w/w dry weight | Reported values for Mxg % w/w dry weight | ||
|---|---|---|---|
| Water | 2.8 ± 1.0 | Not reported | |
| Ash content | 6.0 ± 0.1 | 3.1 ± 0.0 (ref. 67) | |
| Ethanol extractives | 4.4 ± 0.6 | 6.7 ± 0.2 (ref. 67) | |
| Insoluble lignin (AIL) | 24.3 ± 0.3 | 23.8 ± 2.7 (ref. 68), 24.1 (ref. 69) | |
| Soluble lignin (ASL) | 0.9 ± 0.1 | 0.9 ± 0.0 (ref. 67), 0.10 (ref. 69) | |
| Total lignin | 25.2 ± 0.2 | 24.2 (ref. 69) | |
| Structural sugars | D-Glucose | 36.7 ± 1.2 | 42.8 ± 0.2 (ref. 67) |
| D-Xylose | 18.1 ± 0.4 | 22.3 ± 1.7 (ref. 26), 23.2 ± 0.1 (ref. 67) | |
:1 was selected based on alkali lignin solubility in MeTHF-3-one/water mixtures, and the acid concentration and biomass loading were fixed at 0.1 M and 1% w/w, respectively (preliminary experiments are presented in ESI, Fig. S1 and S2†). Then, in a general procedure, freeze-dried Mxg (1% w/v) and 0.1 M acid (oxalic, aspartic or glutamic acid) were added to a 200 mL MeTHF-3-one/water mixture (volume ratio of 3:1) in a glass batch reactor equipped with a reflux condenser, then incubated under vigorous agitation in a thermostatically controlled oil bath (140 or 180 °C) for 6 h and finally cooled in an ice bath for 20 min. The pH of the suspension was controlled (2.98 ± 0.03 with oxalic acid, 5.03 ± 0.01 for aspartic acid and 5.15 ± 0.01 for glutamic acid). The liquid and solid phases were separated by vacuum filtration. The solid phase was washed with ultrapure (UP) water until a neutral pH was reached, and then freeze-dried and stored in a desiccator until further use (delignified biomass pulp namely DL-acid-temperature).
The solvent was then removed from the liquid fraction by evaporation under reduced pressure: 40 °C and 72 mbar to evaporate the water, and then the pressure was lowered to 20 mbar to evaporate the MeTHF-3-one. Structural integrity of the recovered MeTHF-3-one was verified by 1D NMR. The obtained residue was mixed with 50 mL of UP water and then centrifuged. This step was repeated at least 3 times until the water was colorless. Finally, the resulting solid was freeze-dried and stored in a desiccator until further use (enriched-lignin fraction namely L-acid-temperature). The enriched-lignin fraction yields (%) were calculated based on the weight of the latter fraction, using eqn (1).
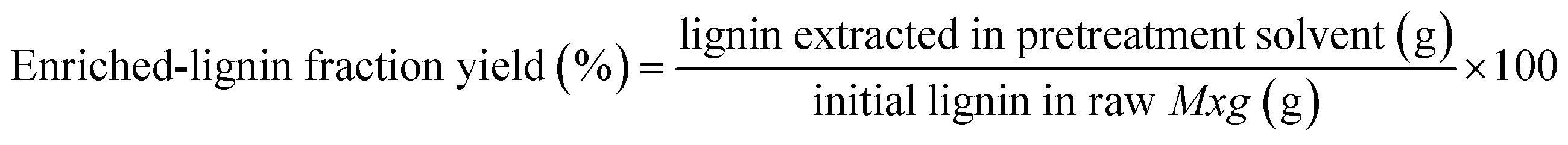 | (1) |
:1 (v/v) and stirred vigorously for 30 min at room temperature. After complete solubilization, the solution was poured into a volumetric flask and 99 mL of ultrapure water was gradually added to the lignin preparation, while stirring gently. The size, charge, and shape of the obtained nanoparticles were determined by dynamic light scattering (DLS), zeta potential analyses and scanning electron microscopy (SEM). The obtained particles were characterized for particle size and charge at 25 °C by dynamic light scattering (DLS) and zeta potential analyses using a Zetasizer Pro (Malvern, UK). Measurements were performed in triplicate, in a disposable folded capillary cell. The results were analyzed using Malvern ZS Xplorer processing software. The nanoparticles were also observed with a high-resolution environmental scanning electron microscope (ESEM, FEI Quanta 200 FEG, OR, USA). A slight drop of each sample was placed on double-sided carbon tape adhered to aluminum SEM stubs. They were dried at room temperature for 72 h, and then the secondary electron images were acquired under high-vacuum mode with an Everhart–Thornley detector (ETD) at a pressure of 0.5 Torr using a pressure limiting aperture (PLA). Dry lignin samples were also observed using the same technique, but without 72 h of drying.
:1 v/v water/IL). The mixture was vigorously mixed for 30 min, and then centrifuged at 95000 rpm for 20 min at 4 °C (Beckman coulter Allegra™ 64R Centrifuge, United States). The supernatant was removed, and the pellet was successively washed with ultrapure water (UP water) until the wash water conductivity was lower than 5 μS cm−1 (conductivity of UP water). The pellet was then freeze-dried (LABCONCO freeze dryer FreeZone 2.5, USA), and the obtained fraction was stored in a desiccator until further use.
The relative monomeric sugar yields were expressed in % according to eqn (2).
 | (2) |
Results and discussion
Extraction performances
Extraction performances of the CoffeeCat process were evaluated highlighting lignin extraction yields and are presented in Table 2, along with photographs of the recovered samples. Extraction yields were similar for all acids at 140 °C (23.8 ± 2.1%, 26.5 ± 1.9%, and 26.8 ± 3.9 for oxalic, glutamic, and aspartic acid, respectively). Pretreatment at 180 °C did not increase extraction yields for aspartic acid (26.8 ± 3.9% at 140 °C vs. 24.2 ± 3.5% at 180 °C) and for oxalic acid (23.8 ± 2.1% at 140 °C vs. 30.7 ± 3.2% at 180 °C). However, the extraction yield was nearly twice that for glutamic acid (26.5 ± 1.9% for 140 °C against 43.3 ± 2.5% at 180 °C). These first results demonstrated the possibility of substituting oxalic acid with the two amino acids proposed as alternatives. Moreover, the use of glutamic acid could improve the extraction yields by increasing the temperature. Regarding the appearance of the samples, the modification of the acid led to significant differences in the color of the lignin samples, ranging from beige-orange for oxalic acid and aspartic acid to chocolate brown for glutamic acid. In addition, increasing the temperature led to the recovery of a brighter and darker lignin. This result was in agreement with literature data, as several studies have already shown that the more severe the extraction conditions, the darker the lignins obtained.86,87 Based on these results, glutamic acid was selected for the implementation of the CoffeeCat process.| Enriched-lignin fraction yieldsa (%) | Photographs of the extracted lignin fraction | ||
|---|---|---|---|
| a Enriched-lignin fraction yields (%) calculated using eqn (1). | |||
| 140 °C | Oxalic acid | 23.8 ± 2.1 |

|
| Glutamic acid | 26.5 ± 1.9 |

|
|
| Aspartic acid | 26.8 ± 3.9 |

|
|
| 180 °C | Oxalic acid | 30.7 ± 3.2 |

|
| Glutamic acid | 43.3 ± 2.5 |

|
|
| Aspartic acid | 24.2 ± 3.5 |

|
|
Chemical compositions of isolated lignin fractions
The chemical composition of lignins obtained by the CoffeeCat process with glutamic acid depends on the process temperature. Their compositions are presented in Fig. 1. The enriched-lignin fractions are referred to as L-glu-140 and L-glu-180 for the lignins obtained at 140 °C and 180 °C, respectively. The isolated lignin fractions had very low residual sugar content (3.3 ± 1.4 and 1.4 ± 0.3 g glucose per 100 g dry matter for L-glu-140 and L-glu-180, respectively, and 1.3 ± 0.6 and 0.5 ± 0.1 g xylose per 100 g dry matter for L-glu-140 and L-glu-180, respectively). The ash content in lignins was lower than that in Mxg (Table 1) possibly indicating that silica and other inorganic materials do not solubilize during the pretreatment.88 On the other hand, L-glu-180 contained a very high proportion of acid insoluble lignin (AIL), e.g. 80 g per 100 g dry matter as well as 4 g per 100 g dry matter of acid soluble lignin (ASL). This corresponds to a total lignin content of 84% in the enriched-lignin fractions, which confirms that this process allows a lignin with a competitive purity in comparison with commercial lignins to be obtained. On the other hand, L-glu-140 contained 53 g per 100 g dry matter of AIL for 8 g per 100 g dry matter of ASL. Although the purity was lower (61%), it was interesting to note that a gentler fractioning allows the extraction of a higher amount of ASL.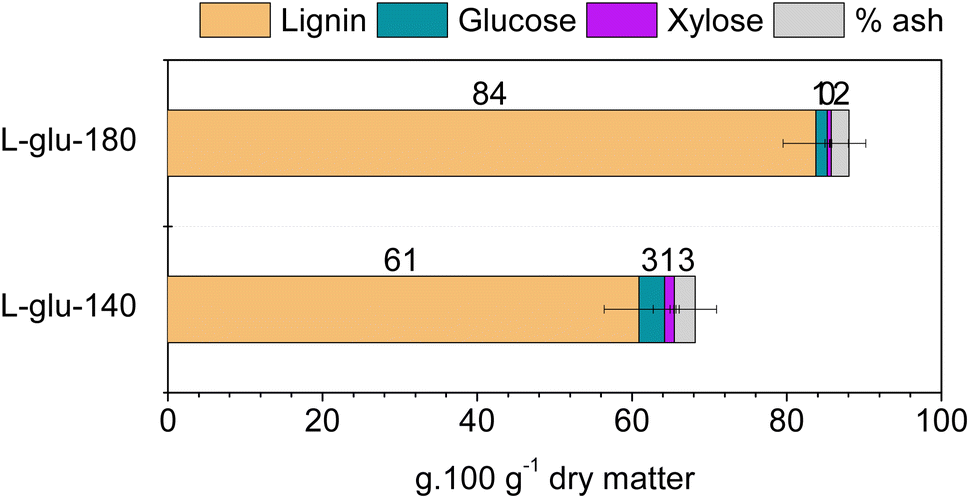 | ||
| Fig. 1 Chemical compositions of the enriched-lignin fractions obtained from the CoffeeCat process at 140 and 180 °C. | ||
A complementary analysis was performed to determine the total phenolic content of the two enriched-lignin fractions. Folin–Ciocalteu's method yielded, respectively, 14.0 ± 1.0 and 14.2 ± 0.1 mg GAE per g lignin for L-glu-140 and L-glu-180, similar to the 15.9 ± 0.2 mg of the commercially available alkali lignin.
Structural characterization of isolated lignin fractions
The SEM micrographs of the enriched-lignin fractions after freeze-drying are presented in Fig. 2. L-glu-140 and L-glu-180 presented typical lignin surface morphologies corresponding to disordered micro-structures composed of various shapes. However, L-glu-140 appeared to have a structure composed of large irregular blocks, with a rather smooth surface (Fig. 2A and C), which resembles a typical lignin residue,89–91 whereas L-glu-180 has a more brittle and compact structure (Fig. 2B), related to a “cleaner” lignin according to previous studies.88 | ||
| Fig. 2 SEM images of L-glu-140 (A), (C) and (E) and L-glu-180 (B), (D) and (F) with different magnifications. | ||
Fig. 3 shows the FTIR spectra of the enriched-lignin fractions in comparison to Mxg. Typical bands were observed between 1650 and 1730 cm−1 (Fig. 3), corresponding to the stretching of conjugated and non-conjugated carbonyls, ketones or ester groups. Bands at 1600 and 1515 cm−1 were assigned to the aromatic skeleton vibration, and bands at 1460, 1400 and 1370 cm−1 corresponded respectively to CH deformations and aromatic ring vibrations, aromatic skeleton vibration and aliphatic C–H stretching in CH3. The specific band at 1330 cm−1 is related to the aromatic ring breathing of syringyl units, a specific band rarely observed in a lignin fraction contaminated by residual carbohydrates.88 The band at 1265 cm−1 is related to aromatic ring breathing of guaiacyl units whereas the one at 1160 cm−1 corresponds to C–H stretching in a guaiacyl unit. The signal at 1130 cm−1 was assigned to aromatic in-plane bending in a syringyl unit, the signal at 1095 cm−1 to C–O deformation in aliphatic ethers, the signal at 900 cm−1 to aromatic CH out of plane deformation of a guaiacyl unit and the signal at 820 cm−1 to aromatic CH out of plane deformation in a syringyl unit. The FTIR spectra revealed that the two isolated lignins had overall the same functional groups, and that the process temperature did not seem to significantly modify lignin structures.
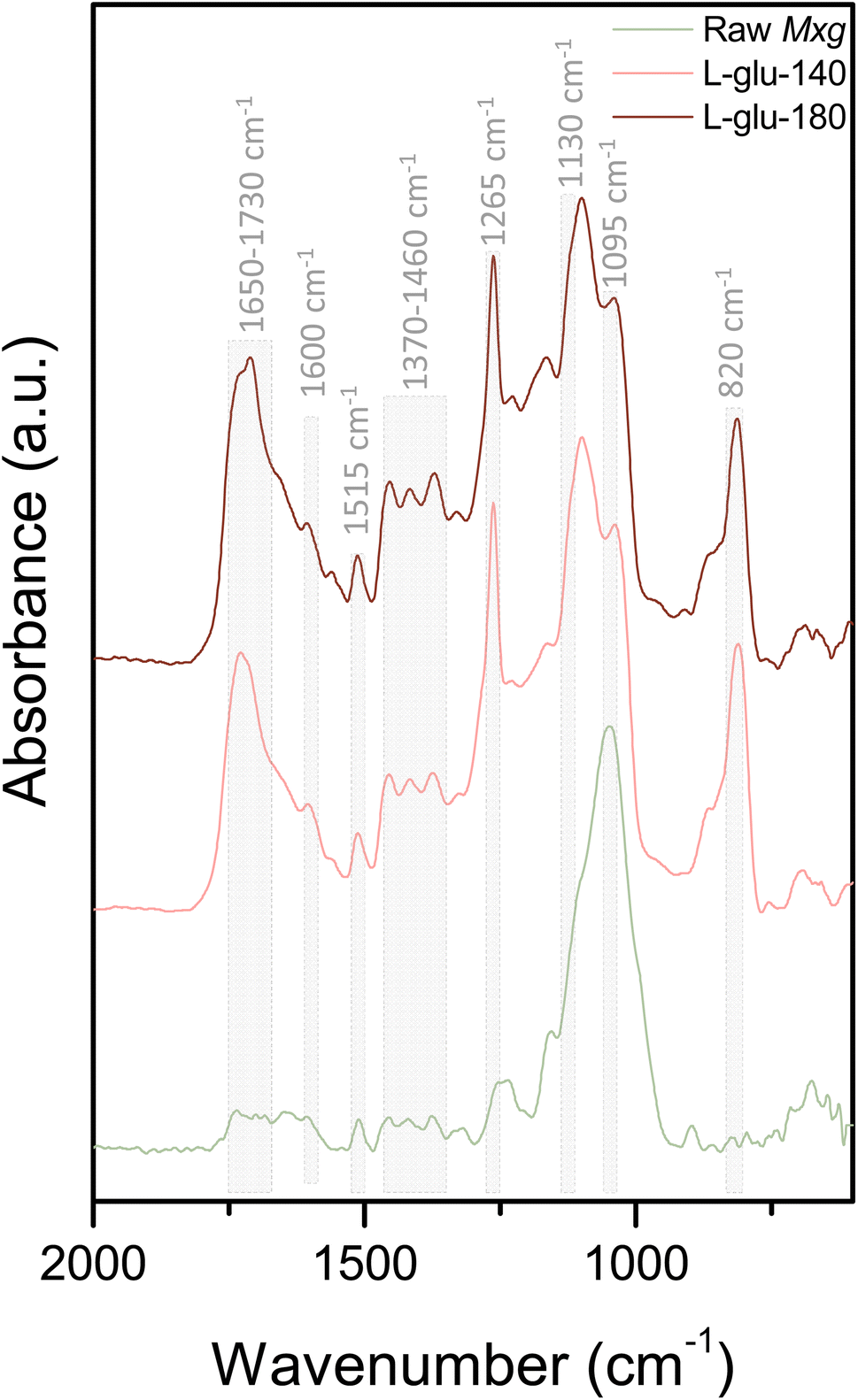 | ||
| Fig. 3 FTIR spectra of raw Mxg, L-glu-140 and L-glu-180, in the range of 600 to 2000 cm−1. The complete spectra (600 to 4000 cm−1) are presented in ESI, Fig. S3.† | ||
To get more insight into the chemical structure of the enriched-lignin fractions, 2D 1H–13C HSQC NMR analyses were performed (Fig. 4). The side chain (δC 50–90 ppm; δH 2.5–6.0 ppm) and aromatic lignin regions (δC 100–135 ppm; δH 5.0–8.5 ppm) of L-glu-140 (Fig. 4A) and L-glu-180 (Fig. 4B) were compared. The first observation is that the NMR 2D spectra confirmed the obtention of carbohydrate-depleted lignin fractions since there is no signal in the δC 70–80 ppm; δH 3–4 ppm region.92 Furthermore, the spectra of L-glu-140 show the presence of β-aryl-ether (β-O-4) linkages that are not evidenced in L-glu-180.
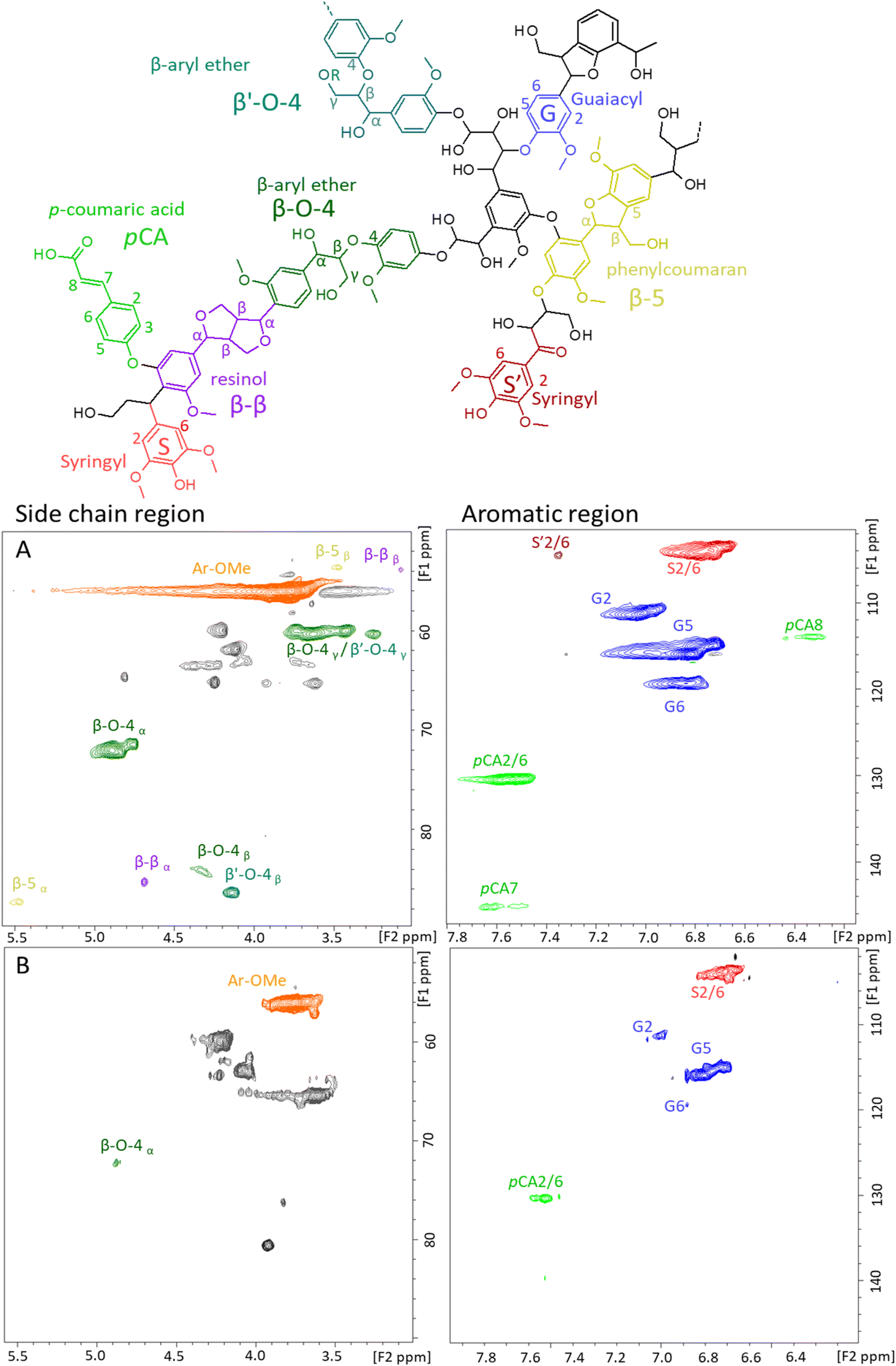 | ||
| Fig. 4 Main identified lignin substructures and 2D HSQC NMR spectra of the side chain and aromatic region of (A) L-glu-140 and (B) L-glu-180. | ||
The side chain and aromatic lignin regions of the 2D NMR spectra are of particular interest for the quantification of lignin linkages and monomer repartition as well as for the aromatic S/G/H ratio. The quantified relative amounts of the aromatic units and of the typical linkages in the lignins are listed in Table 3. Details for the calculation are provided in the ESI.†L-glu-140 and L-glu-180 had a S/G/H ratio consistent with reported values for raw Mxg (40/60/0).93–95 No significant differences were observed between L-glu-140 and L-glu-180 for the S/G/H ratio. Furthermore, the monomer repartition was not the same upon pretreatment temperature increase. Indeed, the lignin obtained at 180 °C contained less β-O-4 linkages than the one obtained at 140 °C (Table 3), and no β-5 and β-β linkages, as already reported in the literature, with a temperature increase.96
| S/G/H (%) | β-5/β-β/β-O-4 (interunit linkages/100 aromatic units) | |
|---|---|---|
| L-glu-140 | 33/67/0 | 3/2/28 |
| L-glu-180 | 42/57/0 | 0/0/9 |
Molecular weights of both L-glu-140 and L-glu-180 samples were determined using size exclusion chromatography (SEC). For each one, a single peak was detected on each detector at the same elution volume. Mw determination by light scattering gave Mw values of ∼20045 Da and ∼33179 Da for L-glu-140 and L-glu-180, respectively. These values ranged in the same order of magnitude, as illustrated by near elution volumes of 12.306 mL and 12.224 mL for L-glu-140 and L-glu-180, respectively (see ESI, Fig. S4†). Interestingly, the enriched-lignin fractions presented higher molecular weight than commercial alkali lignins, which have a Mw of ∼10000 Da,97 or organosolv lignins with Mw between 1000 and 12000 Da,58,97–99 but ranged in the Mw of “native-like-lignin” from Mxg, which has been reported to be ∼30000 Da.100 However, comparisons with literature data may be questionable as the analytical conditions and solubility of lignins may be highly variable. The Mw/Mn values were 1.148 and 1.427 for L-glu-140 and L-glu-180, respectively, suggesting a low polydispersity for these fractions. The slight difference observed between the Mw of L-glu-140 and L-glu-180 could agree with bond cleavages such as the β-O-4 linkages (Fig. 1 and Table 3), providing reactive intermediates susceptible to repolymerization with lignin. Indeed, this is phenomenon more particularly pronounced at high temperatures of extraction.101
Fraction valorization
After demonstrating the proof of concept of the CoffeeCat process, several pathways of valorization of the generated fractions were explored: (i) the enzymatic production of fermentable sugars from delignified biomass pulp; (ii) the conception of lignin-based nanoparticles and (iii) the feasibility of making polylactic acid (PLA) lignin-based composites.The enzymatic production of platform monomeric sugars was first studied. For the bioconversion of the polysaccharide fraction, one promising sustainable route is enzymatic hydrolysis.102 However, enzymatic hydrolysis of delignified biomass pulp, (DL-glu-140 and DL-glu-180) led to low yields of glucose and xylose, as was the case for raw Mxg (less than 6% xylose yield and less than 10% glucose yield, according to eqn (2)) (Fig. 5). These results suggest that the CoffeeCat process did not affect the enzymatic digestibility of the polysaccharides. They also support the current idea in the literature that enzymatic digestibility is not solely related to the lignin content, but also to the disruption of the hydrogen bond network and to some extent to the cellulose crystallinity42 or to the presence of impurities and potential inhibitors. To this end, ionic liquids can be used103–105 to overcome the persistent recalcitrance of the biomass.25–28 It was thus chosen to perform enzymatic hydrolysis after an additional mild pretreatment, the classically used 1-ethyl-3-methylimidazolium acetate ([Emim][OAc])-based pretreatment.25,26,30,68,84,85,106 Glucose and xylose yields after enzymatic hydrolysis of both raw Mxg or delignified biomass pulp before and after [Emim][OAc]-pretreatment are presented in Fig. 5.
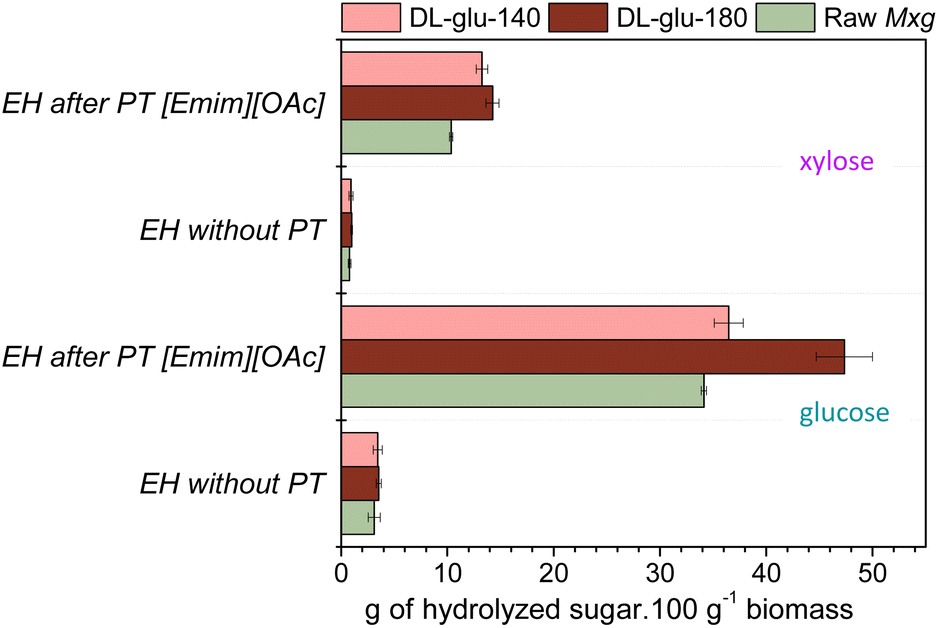 | ||
| Fig. 5 Glucose and xylose yields after enzymatic hydrolysis (EH) of raw Mxg, DL-glu-140 and DL-glu-180 with or without previous [Emim][OAc] pretreatment (PT [Emim][OAc]), in g of hydrolysed sugar per 100 g of biomass. | ||
As expected, [Emim][OAc]-pretreatment increased enzymatic digestibility of Mxg. However, an interesting result was that hydrolysis yields were significantly improved for DL-glu-140 and DL-glu-180 obtained by the CoffeeCat process. Here, a synergistic effect induced by the two subsequent pretreatments was observed, leading to hydrolysis yields higher than 80% for xylose and higher than 99% for glucose for DL-glu-140 obtained by the CoffeeCat process. This result is very promising as it supports the current trend to use combined pretreatments for biomass full valorization.107 Moreover, the FTIR spectra of the residual solid fraction evidenced the isolation of a residual left enzymatic hydrolysis lignin (see ESI, Fig. S5†). Indeed, the FTIR spectrum revealed well-resolved characteristic bands of the lignin polymer according to the FTIR spectrum of alkali lignin.
For the enriched-lignin fractions (L-glu-140 and L-glu-180) the conception of lignin-based nanoparticles (LNPs) was first proposed as a way of valorization. Indeed, a major limitation for lignin industrial processes is its insolubility in water. However, recent studies have demonstrated the possibility of preparing aqueous LNPs,108–111 that could be used for drug and gene delivery due to their biocompatibility, or for other applications such as as antibacterial and antioxidant agents, UV absorbents or hybrid nanocomposites.110,112 The synthesis of LNPs has been performed via a combination of chemical and physical methods, such as polymerization, ultrasonication, interfacial crosslinking, freeze-drying, homogenization or alkaline precipitation.112 However, many of these processes involve the use of hazardous solvents or energy-consuming techniques. The scientific community agrees on the need to create eco-friendly methods for LNP formation.110,112 One perspective currently proposed in the literature is the simple use of antisolvent precipitation, but life cycle assessment calculations have demonstrated that the thermal energy needed for the recovery of the solvents used (ethanol, acetone or THF, mainly) was a hot spot.113–115 Here, we proposed to use the antisolvent precipitation method, with a mixture of water/MeTHF-3-one. Thus, L-glu-140 and L-glu-180 were dissolved in MeTHF-3-one/water 9:1, and then diluted 100 times to reach a final concentration of 0.1 g L−1. Particle size distribution, charge and shape were determined using DLS, zeta-potential analysis and SEM. The formed nanoparticles illustrated a regular spherical shape as indicated by scanning electron microscopy (SEM) images in Fig. 6. The sizes of the LNPs observed by SEM appeared to be consistent with the size distribution obtained by DLS (Table 4). The LNPs synthetized from L-glu-140 were slightly smaller (272 ± 9 nm) than those obtained from L-glu-180 (472 ± 6 nm) but also less polydisperse (PdI < 0.2). This result is in accordance with literature data, confirming that more severe extraction conditions produce larger LNPs than milder conditions.100 The diameter of the particles is a key parameter to evaluate their properties and potential applications.116 Therefore, it is valuable to have the possibility to tailor the LNP size by controlling the temperature of the CoffeeCat process.
 | ||
| Fig. 6 SEM images of the particles synthetized from (A) and (B) L-glu-140 and (C) and (D) L-glu-180 at a concentration of 0.1 g L−1. | ||
| Z-Average (d nm) | PdI | Zeta potential (mV) | |
|---|---|---|---|
| L-glu-140 | 272 ± 9 | 0.16 ± 0.03 | −29.2 ± 0.8 |
| L-glu-180 | 472 ± 6 | 0.36 ± 0.05 | −20.8 ± 0.4 |
Zeta potential values were also measured (Table 4). The smaller particles made from L-glu-140 had a higher negative charge (−29.2 ± 0.8 mV) than the bigger particles made from L-glu-180 (−20.8 ± 0.4 mV), as reported in the literature for LNPs.100 The observed negative charge is typical of LNPs, and ensures the stability of LNPs over time.117 Preliminary experiments have shown that these particles can be stable up to 30 days (data not shown). These results suggested the possibility to produce lignin nanoparticles with tailorable sizes, depending on the CoffeeCat process temperature, which can be produced in a fully food grade mixture (MeTHF-3-one/water) if this label is required.
Another way of valorisation for lignin-enriched fractions could be their incorporation into PLA to design PLA–lignin composites. Indeed, PLA is one of the main commercially available bio-based and biodegradable polyesters used for a wide range of industrial applications, such as 3D printing applications.82,83,118 However, PLA suffers from drawbacks such as intrinsic brittleness.83,119–121 Several studies have proposed to improve its physical properties to extend its applications, especially in the field of food packaging. One strategy is to incorporate lignin to design composite polymers with improved physical and mechanical properties.82,118,119,122 However, due to the poor miscibility of commercially available lignin in a polymer matrix, the mixing of lignin with PLA often requires manual mixing and extrusion, melting, and solubilization in toxic solvents such as chloroform, or lignin chemical modification.83,119,122,123 Here we proposed for the first time to solubilise PLA in MeTHF-3-one, in which L-glu-140, the less coloured lignin-enriched fraction, is also soluble. Fig. 7A shows the complete miscibility of L-glu-140 in the PLA–MeTHF-3-one mixture. The obtained solution was treated by two different ways as described in the experimental section. Fig. 7B depicts the powder obtained after drying the solution overnight at 105 °C and grinding. The FTIR spectra of PLA, L-glu-140 and PLA-L-glu-140 powder are presented in Fig. 7D. The FTIR spectrum of PLA showed typical absorption bands at 1750 cm−1, 1180 cm−1 and 1080 cm−1 which respectively correspond to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of the carbonyl group, C–O stretching of esters and carbonyl C–O stretching.82,83 The characteristic peaks of L-glu-140 were already assigned (Fig. 3). In the PLA-L-glu-140 powder spectrum, small peaks between 1510 and 1530 cm−1 were observed, which were attributed to the CC groups of lignin aromatic rings.83,124 Furthermore, Fig. 7C shows the film obtained after drying the solution at 50 °C for 1 h. The obtained film seemed homogeneous and flexible. This suggested the feasibility of easy incorporation of lignin into PLA in MeTHF-3-one without requiring lignin modification.
O stretching of the carbonyl group, C–O stretching of esters and carbonyl C–O stretching.82,83 The characteristic peaks of L-glu-140 were already assigned (Fig. 3). In the PLA-L-glu-140 powder spectrum, small peaks between 1510 and 1530 cm−1 were observed, which were attributed to the CC groups of lignin aromatic rings.83,124 Furthermore, Fig. 7C shows the film obtained after drying the solution at 50 °C for 1 h. The obtained film seemed homogeneous and flexible. This suggested the feasibility of easy incorporation of lignin into PLA in MeTHF-3-one without requiring lignin modification.
 | ||
| Fig. 7 Photographs of (A) PLA + L-glu-140 dissolved in MeTHF-3-one (100 g L−1 PLA, 0.2 g lignin/g PLA), (B) PLA-L-glu-140 obtained after drying overnight at 105 °C and grinding. (C) a PLA-L-glu-140 flexible film obtained after heating at 50 °C for 1 h and (D) FTIR spectra of PLA, L-glu-140 and PLA-L-glu-140 powder, in the range of 600 to 2000 cm−1. The complete spectra (600 to 4000 cm−1) are presented in ESI, Fig. S6.† | ||
Conclusions
At this academic stage, the process still needs to be improved in order to be economically viable. One of the major disadvantages is certainly the use of water needed for the washing steps, although this is not exclusive to this process and commonly shared with some others such as organosolv, ionic liquid or deep eutectic pretreatments of LCB, which is additional to the cost of the solvent and the recycling of the fluids. However, the proposed strategy allowed a lignin fraction rich in β-O-4 linkages to be obtained when performed at mild temperature (140 °C) which is recovered by solvent evaporation, similar to other organosolv processes.74,98,99 This easy recovery method is clearly an advantage over the use of non-volatile ionic liquid for LCB pretreatment. Moreover, the use of a food grade solvent, already described as readily biodegradable,59 may reassure end-users and enlarge the panel of applications (e.g. cosmetics) even if traces of solvent remain after the washing step. Another point that can be highlighted and can be an advantage for subsequent transformation or direct use, is that the lignin obtained is in a powder state and not a liquor as in other processes such as IL or OrganoCat pretreatments.43,44 For example, the obtained lignin was incorporated without further modification into PLA. Finally, the lignin fraction is nearly carbohydrate-free and the validity of the process was suggested by the mass balance closure related to the three main polymers of Miscanthus (see ESI, Fig. S7†).Conflicts of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be considered a potential conflict of interest.Acknowledgements
The authors would like to thank Dr Dominique Cailleu for helping in the NMR experimentation implementation and Dr Arash Jamali for SEM observations.References
- S. Nanda, R. Azargohar, A. K. Dalai and J. A. Kozinski, Renewable Sustainable Energy Rev., 2015, 50, 925–941 CrossRef CAS.
- C. E. Wyman, Bioresour. Technol., 1994, 50, 3–15 CrossRef CAS.
- S. Fatma, A. Hameed, M. Noman, T. Ahmed, M. Shahid, M. Tariq, I. Sohail and R. Tabassum, Protein Pept. Lett., 2018, 25, 148–163 CrossRef CAS PubMed.
- L. Cao, I. K. M. Yu, Y. Liu, X. Ruan, D. C. W. Tsang, A. J. Hunt, Y. S. Ok, H. Song and S. Zhang, Bioresour. Technol., 2018, 269, 465–475 CrossRef CAS PubMed.
- P. Chen, Q. Zhang, R. Shu, Y. Xu, L. Ma and T. Wang, Bioresour. Technol., 2017, 226, 125–131 CrossRef CAS PubMed.
- S. S. Chen, T. Maneerung, D. C. W. Tsang, Y. S. Ok and C.-H. Wang, Chem. Eng. J., 2017, 328, 246–273 CrossRef CAS.
- W. J. Sagues, H. Bao, J. L. Nemenyi and Z. Tong, ACS Sustainable Chem. Eng., 2018, 6, 4958–4965 CrossRef CAS.
- I. K. M. Yu and D. C. W. Tsang, Bioresour. Technol., 2017, 238, 716–732 CrossRef CAS PubMed.
- P. Morone and G. Yilan, Acta Innovations, 2020, 36, 17–32 Search PubMed.
- A. Brandt, J. Gräsvik, J. P. Hallett and T. Welton, Green Chem., 2013, 15, 550–583 RSC.
- N. Mahmood, Z. Yuan, J. Schmidt and C. (Charles) Xu, Renewable Sustainable Energy Rev., 2016, 60, 317–329 CrossRef CAS.
- C. Sánchez, Biotechnol. Adv., 2009, 27, 185–194 CrossRef PubMed.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 1246843 CrossRef PubMed.
- J. Y. Zhu and X. J. Pan, Bioresour. Technol., 2010, 101, 4992–5002 CrossRef CAS PubMed.
- N.-Q. Ren, L. Zhao, C. Chen, W.-Q. Guo and G.-L. Cao, Bioresour. Technol., 2016, 215, 92–99 CrossRef CAS PubMed.
- Y.-S. Jang, A. Malaviya, C. Cho, J. Lee and S. Y. Lee, Bioresour. Technol., 2012, 123, 653–663 CrossRef CAS PubMed.
- C. G. Yoo, X. Meng, Y. Pu and A. J. Ragauskas, Bioresour. Technol., 2020, 301, 122784 CrossRef CAS PubMed.
- R. Younas, S. Zhang, L. Zhang, G. Luo, K. Chen, L. Cao, Y. Liu and S. Hao, Catal. Today, 2016, 274, 40–48 CrossRef CAS.
- C. A. Antonyraj and A. Haridas, Catal. Commun., 2018, 104, 101–105 CrossRef CAS.
- C. Geun Yoo, S. Zhang and X. Pan, RSC Adv., 2017, 7, 300–308 RSC.
- L. Zhang, G. Xi, J. Zhang, H. Yu and X. Wang, Bioresour. Technol., 2017, 224, 656–661 CrossRef CAS PubMed.
- S. S. Chen, L. Wang, I. K. M. Yu, D. C. W. Tsang, A. J. Hunt, F. Jérôme, S. Zhang, Y. S. Ok and C. S. Poon, Bioresour. Technol., 2018, 247, 387–394 CrossRef CAS PubMed.
- R. V. Gadhave, P. A. Mahanwar and P. T. Gadekar, Open J. Polym. Chem., 2018, 08, 1 CrossRef CAS.
- W. Yang, E. Fortunati, D. Gao, G. M. Balestra, G. Giovanale, X. He, L. Torre, J. M. Kenny and D. Puglia, ACS Sustainable Chem. Eng., 2018, 6, 3502–3514 CrossRef CAS.
- R. Alayoubi, N. Mehmood, E. Husson, A. Kouzayha, M. Tabcheh, L. Chaveriat, C. Sarazin and I. Gosselin, Renewable Energy, 2020, 145, 1808–1816 CrossRef CAS.
- T. Auxenfans, E. Husson and C. Sarazin, Biochem. Eng. J., 2017, 117, 77–86 CrossRef CAS.
- L. Hulin, E. Husson, J.-P. Bonnet, T. Stevanovic and C. Sarazin, Molecules, 2015, 20, 16334–16353 CrossRef CAS PubMed.
- E. Husson, L. Hulin, C. Hadad, C. Boughanmi, T. Stevanovic and C. Sarazin, Front. Chem., 2019, 7, 578 CrossRef CAS PubMed.
- M. Brahim, B. L. Checa Fernandez, O. Regnier, N. Boussetta, N. Grimi, C. Sarazin, E. Husson, E. Vorobiev and N. Brosse, Bioresour. Technol., 2017, 237, 11–19 CrossRef CAS PubMed.
- E. Husson, T. Auxenfans, M. Herbaut, M. Baralle, V. Lambertyn, H. Rakotoarivonina, C. Rémond and C. Sarazin, Bioresour. Technol., 2018, 251, 280–287 CrossRef CAS PubMed.
- D. S. Bajwa, G. Pourhashem, A. H. Ullah and S. G. Bajwa, Ind. Crops Prod., 2019, 139, 111526 CrossRef CAS.
- G.-H. Kim and B.-H. Um, Int. J. Biol. Macromol., 2020, 158, 443–451 CrossRef CAS PubMed.
- S. Laurichesse and L. Avérous, Prog. Polym. Sci., 2014, 39, 1266–1290 CrossRef CAS.
- M. Jiao Gan, Y. Qin Niu, X. Jing Qu and C. Hui Zhou, Green Chem., 2022, 24, 7705–7750 RSC.
- V. Patil, S. Adhikari, P. Cross and H. Jahromi, Renewable Sustainable Energy Rev., 2020, 133, 110359 CrossRef CAS.
- M. Talebi Amiri, G. R. Dick, Y. M. Questell-Santiago and J. S. Luterbacher, Nat. Protoc., 2019, 14, 921–954 CrossRef CAS PubMed.
- O. Yu and K. H. Kim, Appl. Sci., 2020, 10, 4626 CrossRef CAS.
- L. A. Zevallos Torres, A. Lorenci Woiciechowski, V. O. de Andrade Tanobe, S. G. Karp, L. C. Guimarães Lorenci, C. Faulds and C. R. Soccol, J. Cleaner Prod., 2020, 263, 121499 CrossRef CAS.
- S. Gillet, M. Aguedo, L. Petitjean, A. R. C. Morais, A. M. d. C. Lopes, R. M. Łukasik and P. T. Anastas, Green Chem., 2017, 19, 4200–4233 RSC.
- J. Viell, A. Harwardt, J. Seiler and W. Marquardt, Bioresour. Technol., 2013, 150, 89–97 CrossRef CAS PubMed.
- P. M. Grande, J. Viell, N. Theyssen, W. Marquardt, P. D. de María and W. Leitner, Green Chem., 2015, 17, 3533–3539 RSC.
- T. Damm, P. M. Grande, N. D. Jablonowski, B. Thiele, U. Disko, U. Mann, U. Schurr, W. Leitner, B. Usadel, P. Domínguez de María and H. Klose, Bioresour. Technol., 2017, 244, 889–896 CrossRef CAS PubMed.
- A. Holtz, D. Weidener, W. Leitner, H. Klose, P. M. Grande and A. Jupke, Sep. Purif. Technol., 2020, 236, 116295 CrossRef CAS.
- D. Weidener, A. Holtz, H. Klose, A. Jupke, W. Leitner and P. M. Grande, Molecules, 2020, 25(15), 3330 CrossRef CAS PubMed.
- S.-X. Li, M.-F. Li, J. Bian, S.-N. Sun, F. Peng and Z.-M. Xue, Bioresour. Technol., 2017, 243, 1105–1111 CrossRef CAS PubMed.
- E. Viola, F. Zimbardi, M. Morgana, N. Cerone, V. Valerio and A. Romanelli, Processes, 2021, 9, 2051 CrossRef CAS.
- A. Morone, R. A. Pandey and T. Chakrabarti, Ind. Crops Prod., 2017, 99, 7–18 CrossRef CAS.
- P. M. Grande, D. Weidener, S. Dietrich, M. Dama, M. Bellof, R. Maas, M. Pauly, W. Leitner, H. Klose and P. Domínguez de María, ACS Omega, 2019, 4, 14451–14457 CrossRef CAS PubMed.
- D. Weidener, M. Dama, S. K. Dietrich, B. Ohrem, M. Pauly, W. Leitner, P. Domínguez de María, P. M. Grande and H. Klose, Biotechnol. Biofuels, 2020, 13, 155 CrossRef CAS PubMed.
- T. vom Stein, P. M. Grande, H. Kayser, F. Sibilla, W. Leitner and P. D. de María, Green Chem., 2011, 13, 1772–1777 RSC.
- T. vom Stein, P. M. Grande, W. Leitner and P. Domínguez de María, ChemSusChem, 2011, 4, 1592–1594 CrossRef CAS PubMed.
- B. Cornils and P. Lappe updated by Staff, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2014, pp. 1–18 Search PubMed.
- V. Pace, P. Hoyos, L. Castoldi, P. Domínguez de María and A. R. Alcántara, ChemSusChem, 2012, 5, 1369–1379 CrossRef CAS PubMed.
- K. R. Olson and Y.-M. Hung, in Poisoning & Drug Overdose, ed. K. R. Olson, The McGraw-Hill Companies, New York, 6th edn, 2012 Search PubMed.
- F. Weinwurm, A. Drljo, W. Waldmüller, B. Fiala, J. Niedermayer and A. Friedl, J. Cleaner Prod., 2016, 136, 62–71 CrossRef CAS.
- L. Shuai, Y. M. Questell-Santiago and J. S. Luterbacher, Green Chem., 2016, 18, 937–943 RSC.
- X. Meng, Y. Pu, M. Li and A. J. Ragauskas, Green Chem., 2020, 22, 2862–2872 RSC.
- S. Yang, X. Yang, X. Meng and L. Wang, Green Chem., 2022, 24, 4082–4094 RSC.
- M. E. Vuillemin, E. Husson, S. Laclef, A. Jamali, V. Lambertyn, S. Pilard, D. Cailleu and C. Sarazin, Process Biochem., 2022, 117, 30–41 CrossRef CAS.
- D. Rowe, Chemistry and Technology of Flavours and Fragrances, John Wiley & Sons, 2009 Search PubMed.
- C. L. Burnett, B. Heldreth, W. F. Bergfeld, D. V. Belsito, R. A. Hill, C. D. Klaassen, D. C. Liebler, J. G. Marks, R. C. Shank, T. J. Slaga, P. W. Snyder and F. A. Andersen, Int. J. Toxicol., 2013, 32, 41S–64S CrossRef PubMed.
- E. Chupakhin, O. Babich, S. Sukhikh, S. Ivanova, E. Budenkova, O. Kalashnikova and O. Kriger, Energies, 2021, 14, 8368 CrossRef CAS.
- F. J. V. Gschwend, F. Malaret, S. Shinde, A. Brandt-Talbot and J. P. Hallett, Green Chem., 2018, 20, 3486–3498 RSC.
- Z. Guo, D. Li, T. You, X. Zhang, F. Xu, X. Zhang and Y. Yang, ACS Sustainable Chem. Eng., 2020, 8, 12110–12119 CrossRef CAS.
- R. Bhatia, J. B. Lad, M. Bosch, D. N. Bryant, D. Leak, J. P. Hallett, T. T. Franco and J. A. Gallagher, Bioresour. Technol., 2021, 323, 124625 CrossRef CAS PubMed.
- A. Sluiter, J. Sluiter and E. J. Wolfrum, in Catalysis for the Conversion of Biomass and Its Derivatives, Max-Planck-Gesellschaft zur Förderung der Wissenschaften, Berlin, 2013 Search PubMed.
- R. Bhatia, A. Winters, D. N. Bryant, M. Bosch, J. Clifton-Brown, D. Leak and J. Gallagher, Bioresour. Technol., 2020, 296, 122285 CrossRef CAS PubMed.
- T. Auxenfans, S. Buchoux, E. Husson and C. Sarazin, Biomass Bioenergy, 2014, 62, 82–92 CrossRef CAS.
- S. J. Kim, M. Y. Kim, S. J. Jeong, M. S. Jang and I. M. Chung, Ind. Crops Prod., 2012, 38, 46–49 CrossRef CAS.
- J. Higgins, X. Zhou, R. Liu and T. T.-S. Huang, J. Phys. Chem. A, 1997, 101, 2702–2708 CrossRef CAS.
- T.-Q. Yuan, S.-N. Sun, F. Xu and R.-C. Sun, J. Agric. Food Chem., 2011, 59, 10604–10614 CrossRef CAS PubMed.
- D. Ibarra, M. I. Chávez, J. Rencoret, J. C. Del Río, A. Gutiérrez, J. Romero, S. Camarero, M. J. Martínez, J. Jiménez-Barbero and A. T. Martínez, J. Agric. Food Chem., 2007, 55, 3477–3490 CrossRef CAS PubMed.
- J. C. del Río, J. Rencoret, G. Marques, A. Gutiérrez, D. Ibarra, J. I. Santos, J. Jiménez-Barbero, L. Zhang and Á. T. Martínez, J. Agric. Food Chem., 2008, 56, 9525–9534 CrossRef PubMed.
- D. S. Zijlstra, C. W. Lahive, C. A. Analbers, M. B. Figueirêdo, Z. Wang, C. S. Lancefield and P. J. Deuss, ACS Sustainable Chem. Eng., 2020, 8, 5119–5131 CrossRef CAS.
- S. Constant, H. L. J. Wienk, A. E. Frissen, P. de Peinder, R. Boelens, D. S. van Es, R. J. H. Grisel, B. M. Weckhuysen, W. J. J. Huijgen, R. J. A. Gosselink and P. C. A. Bruijnincx, Green Chem., 2016, 18, 2651–2665 RSC.
- C. G. Yoo, Y. Pu, M. Li and A. J. Ragauskas, ChemSusChem, 2016, 9, 1090–1095 CrossRef CAS PubMed.
- H. Wang, W. Chen, X. Zhang, Y. Wei, A. Zhang, S. Liu, X. Wang and C. Liu, Polymers, 2018, 10, 433 CrossRef PubMed.
- V. L. Singleton, R. Orthofer and R. M. Lamuela-Raventós, in Methods in Enzymology, Academic Press, 1999, vol. 299, pp. 152–178 Search PubMed.
- M. E. Vuillemin, F. Michaux, A. A. Adam, M. Linder, L. Muniglia and J. Jasniewski, Food Hydrocolloids, 2020, 107, 105919 CrossRef CAS.
- C. H. M. Camargos, R. A. P. Silva, Y. Csordas, L. L. Silva and C. A. Rezende, Ind. Crops Prod., 2019, 140, 111649 CrossRef CAS.
- A. P. Richter, B. Bharti, H. B. Armstrong, J. S. Brown, D. Plemmons, V. N. Paunov, S. D. Stoyanov and O. D. Velev, Langmuir, 2016, 32, 6468–6477 CrossRef CAS PubMed.
- C.-W. Park, W.-J. Youe, S.-J. Kim, S.-Y. Han, J.-S. Park, E.-A. Lee, G.-J. Kwon, Y.-S. Kim, N.-H. Kim and S.-H. Lee, Polymers, 2019, 11, 2089 CrossRef CAS PubMed.
- S. Wasti, E. Triggs, R. Farag, M. Auad, S. Adhikari, D. Bajwa, M. Li and A. J. Ragauskas, Composites, Part B, 2021, 205, 108483 CrossRef CAS.
- T. Auxenfans, S. Buchoux, D. Larcher, G. Husson, E. Husson and C. Sarazin, Energy Convers. Manage., 2014, 88, 1094–1103 CrossRef CAS.
- M. Araya-Farias, E. Husson, J. Saavedra-Torrico, D. Gérard, R. Roulard, I. Gosselin, H. Rakotoarivonina, V. Lambertyn, C. Rémond and C. Sarazin, Front. Chem., 2019, 7, 585 CrossRef CAS PubMed.
- O. Ajao, J. Jeaidi, M. Benali, A. M. Restrepo, N. El Mehdi and Y. Boumghar, Molecules, 2018, 23, 377 CrossRef PubMed.
- D. Piccinino, E. Capecchi, E. Tomaino, S. Gabellone, V. Gigli, D. Avitabile and R. Saladino, Antioxidants, 2021, 10, 274 CrossRef CAS PubMed.
- E. Hermiati, L. Risanto, M. A. R. Lubis, R. P. B. Laksana and A. R. Dewi, AIP Conf. Proc., 2017, 1803, 020005 CrossRef.
- M. Tayier, D. Duan, Y. Zhao, R. Ruan, Y. Wang and Y. Liu, BioResources, 2018, 13, 412–424 CAS.
- X. Zhao, Y. Zhang, M. Yang, Z. Huang, H. Hu, A. Huang and Z. Feng, Polymers, 2018, 10, 907 CrossRef PubMed.
- M. H. Nazir, M. Ayoub, I. Zahid, R. B. Shamsuddin, S. Yusup, M. Ameen, Zulqarnain and M. U. Qadeer, Biomass Bioenergy, 2021, 146, 105978 CrossRef CAS.
- J. J. Villaverde, J. Li, M. Ek, P. Ligero and A. de Vega, J. Agric. Food Chem., 2009, 57, 6262–6270 CrossRef CAS PubMed.
- I. Hita, H. J. Heeres and P. J. Deuss, Bioresour. Technol., 2018, 267, 93–101 CrossRef CAS PubMed.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- J. S. Lupoi and E. A. Smith, Appl. Spectrosc., 2012, 66, 903–910 CrossRef CAS PubMed.
- L. Shuai, M. T. Amiri, Y. M. Questell-Santiago, F. Héroguel, Y. Li, H. Kim, R. Meilan, C. Chapple, J. Ralph and J. S. Luterbacher, Science, 2016, 354, 329–333 CrossRef CAS PubMed.
- A. Ang, Z. Ashaari, E. S. Bakar and N. A. Ibrahim, BioResources, 2015, 10, 4137–4151 CAS.
- N. H. A. Latif, N. Brosse, I. Ziegler-Devin, L. Chrusiel, R. Hashim and M. H. Hussin, BioResources, 2022, 17, 469–491 CAS.
- H. Sadeghifar, T. Wells, R. K. Le, F. Sadeghifar, J. S. Yuan and A. Jonas Ragauskas, ACS Sustainable Chem. Eng., 2017, 5, 580–587 CrossRef CAS.
- L. Chen, J. Dou, Q. Ma, N. Li, R. Wu, H. Bian, D. J. Yelle, T. Vuorinen, S. Fu, X. Pan and J. J. Y. Zhu, Sci. Adv., 2017, 3, e1701735 CrossRef PubMed.
- Z. Chen, X. Bai, L. A, H. Zhang and C. Wan, ACS Sustainable Chem. Eng., 2020, 8, 9783–9793 CrossRef CAS.
- A. Orozco, M. Ahmad, D. Rooney and G. Walker, Process Saf. Environ. Prot., 2007, 85, 446–449 CrossRef CAS.
- A. M. d. C. Lopes, R. M. G. Lins, R. A. Rebelo and R. M. Łukasik, Green Chem., 2018, 20, 4043–4057 RSC.
- H. Rodríguez, Acta Innovations, 2021, 23–36 Search PubMed.
- M. C. Quesada-Salas, M. E. Vuillemin, J. Dillies, R. Dauwe, L. Firdaous, M. Bigan, V. Lambertyn, D. Cailleu, A. Jamali, R. Froidevaux, E. Husson and C. Sarazin, Ind. Crops Prod., 2023, 197, 116627 CrossRef CAS.
- T. Auxenfans, S. Buchoux, K. Djellab, C. Avondo, E. Husson and C. Sarazin, Carbohydr. Polym., 2012, 90, 805–813 CrossRef CAS PubMed.
- C. Huang, Y. Zhan, J. Wang, J. Cheng, X. Meng, L. Liang, F. Liang, Y. Deng, G. Fang and A. J. Ragauskas, Green Chem., 2022, 24, 3736–3749 RSC.
- Y. Qian, X. Zhong, Y. Li and X. Qiu, Ind. Crops Prod., 2017, 101, 54–60 CrossRef CAS.
- C. Frangville, M. Rutkevičius, A. P. Richter, O. D. Velev, S. D. Stoyanov and V. N. Paunov, ChemPhysChem, 2012, 13, 4235–4243 CrossRef CAS PubMed.
- W. Zhao, B. Simmons, S. Singh, A. Ragauskas and G. Cheng, Green Chem., 2016, 18, 5693–5700 RSC.
- M. Lievonen, J. José Valle-Delgado, M.-L. Mattinen, E.-L. Hult, K. Lintinen, M. A. Kostiainen, A. Paananen, G. R. Szilvay, H. Setälä and M. Österberg, Green Chem., 2016, 18, 1416–1422 RSC.
- P. S. Chauhan, Bioresour. Technol. Rep., 2020, 9, 100374 CrossRef.
- D. Koch, M. Paul, S. Beisl, A. Friedl and B. Mihalyi, J. Cleaner Prod., 2020, 245, 118760 CrossRef CAS.
- M. Österberg, M. H. Sipponen, B. D. Mattos and O. J. Rojas, Green Chem., 2020, 22, 2712–2733 RSC.
- R. P. B. Ashok, P. Oinas, K. Lintinen, G. Sarwar, M. A. Kostiainen and M. Österberg, Green Chem., 2018, 20, 4911–4919 RSC.
- M. Ma, L. Dai, J. Xu, Z. Liu and Y. Ni, Green Chem., 2020, 22, 2011–2017 RSC.
- Z.-H. Liu, N. Hao, S. Shinde, Y. Pu, X. Kang, A. J. Ragauskas and J. S. Yuan, Green Chem., 2019, 21, 245–260 RSC.
- Z. Xiong, X. Dai, H. Na, Z. Tang, R. Zhang and J. Zhu, J. Appl. Polym. Sci., 2015, 132(1) DOI:10.1002/app.41220.
- T. F. da Silva, F. Menezes, L. S. Montagna, A. P. Lemes and F. R. Passador, Mater. Sci. Eng., B, 2019, 251, 114441 CrossRef.
- M. Chalid, E. Yuanita and J. Pratama, Mater. Sci. Forum, 2015, 827, 326–331 Search PubMed.
- Z. Xiong, C. Li, S. Ma, J. Feng, Y. Yang, R. Zhang and J. Zhu, Carbohydr. Polym., 2013, 95, 77–84 CrossRef CAS PubMed.
- M. S. Pairon, N. A. A. Rahman, F. Ali, H. Anuar and J. Suhr, Bioresour. Technol. Rep., 2022, 20, 101244 CrossRef CAS.
- J. Guo, X. Chen, J. Wang, Y. He, H. Xie and Q. Zheng, Polymers, 2020, 12, 56 CrossRef CAS PubMed.
- M. Tanase-Opedal, E. Espinosa, A. Rodríguez and G. Chinga-Carrasco, Materials, 2019, 12, 3006 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3su00050h |
| This journal is © The Royal Society of Chemistry 2023 |