
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA perspective on catalytic production of olefinic compounds from biomass
Yoshinao
Nakagawa
 *ab,
Mizuho
Yabushita
ab and
Keiichi
Tomishige
*abc
*ab,
Mizuho
Yabushita
ab and
Keiichi
Tomishige
*abc
aDepartment of Applied Chemistry, School of Engineering, Tohoku University, 6-6-07 Aoba, Aramaki, Aoba-ku, Sendai, 980-8579, Japan. E-mail: yoshinao@erec.che.tohoku.ac.jp; Fax: +81-22-795-7215; Tel: +81-22-795-7215
bResearch Center for Rare Metal and Green Innovation, Tohoku University, 468-1 Aoba, Aramaki, Aoba-ku, Sendai 980-0845, Japan. E-mail: tomishige@tohoku.ac.jp; Fax: +81-22-795-7214; Tel: +81-22-795-7214
cAdvanced Institute for Materials Research (WPI-AIMR), Tohoku University, 2-1-1 Katahira, Aoba-ku, Sendai, 980-8577, Japan
First published on 4th May 2023
Abstract
Chemical syntheses of industrially important olefins and unsaturated oxygenates from biomass are reviewed. Their syntheses are discussed in two aspects: common key reactions and target molecules grouped by the carbon number. Among common key reactions, dehydration of alcohols over acid–base bifunctional solid catalysts, ring-opening dehydration of tetrahydrofurans over acid catalysts, and the deoxydehydration reaction of vicinal diols are especially important. Further, catalyst development is still necessary for all these reactions. In the discussion of each target molecule, the synthesis methods are compared by considering the balance between current demand and the amount of biomass resources. Promising methods have been already developed or proposed for compounds with C4 and more and for C3 unsaturated oxygenates.
Sustainability spotlightThe chemical industry is strongly dependent on the fossil resource both as material source and energy. While there are various sources of renewable energy, biomass is the only renewable organic source. Olefins are one of the most important classes of intermediate substances in the chemical industry and the demand is very large. Here, the chemical syntheses of such important olefins from biomass are reviewed considering both feasibility of chemical conversions and potential supply of each biomass resource. Establishing the chemical synthesis routes to olefins and olefinic oxygenates in large demand from biomass aligns with the following UN sustainable development goals 7, 9, 12 and 13. |
1. Introduction
Olefins are an important class of chemicals in industry. Simple olefins such as ethylene, propylene, butadiene, and isobutene are manufactured by thermal cracking of naphtha (typical composition is shown in Table 1).1–3 The use of catalysts can change the distribution of the products of cracking. Fluidized catalytic cracking (FCC) of heavier oil such as vacuum gas oil is another main method to obtain propylene. Branched olefins as well as aromatics are co-produced in FCC. In addition to the use of the branched olefins as gasoline (FCC gasoline), the petroleum-derived simple olefins are used as-is as monomers of polyolefins or used as reactants for the synthesis of functionalized molecules via addition reactions, oxidation, hydroformylation and so on. In view of sustainability, substitution of petroleum with renewable biomass has been a hot topic, especially for the use as chemicals that cannot be replaced by other renewable energy sources.4–6 However, the oxygen content of biomass is much larger than those of petroleum and most petroleum-derived compounds. Complete removal of oxygen atoms is necessary for the production of simple olefins from biomass. Meanwhile, some products from petroleum-based olefins contain oxygenated functional groups with retention of the olefinic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond. The production of these compounds from biomass via oxygen-free olefins wastes hydrogen, and thus, the synthesis by partial deoxygenation is more atom-efficient.7,8 The construction of long-chain hydrocarbons from C1 compounds via biomass gasification such as BTL (biomass to liquid) and MTO (methanol to olefins) processes is possible; however, these processes are very energy consuming. Altogether, the synthesis of olefinic compounds from biomass requires total or partial deoxygenation reactions, which are not the main reactions in traditional industrial chemistry.
C bond. The production of these compounds from biomass via oxygen-free olefins wastes hydrogen, and thus, the synthesis by partial deoxygenation is more atom-efficient.7,8 The construction of long-chain hydrocarbons from C1 compounds via biomass gasification such as BTL (biomass to liquid) and MTO (methanol to olefins) processes is possible; however, these processes are very energy consuming. Altogether, the synthesis of olefinic compounds from biomass requires total or partial deoxygenation reactions, which are not the main reactions in traditional industrial chemistry.
| Compounds | Ref. 1 high severity cracking | Ref. 2 high severity cracking | Ref. 3 |
|---|---|---|---|
| a a–e: classified as the same group. | |||
| H2 | 3.2a | 0.94 | 1.0 |
| CH4 | 15.0 | 16.5 | 15.0 |
| C2H2 | a | 1.05 | 0.4 |
| C2H4 | 31.3 | 32.8 | 25.7 |
| C2H6 | 3.4 | 3.25 | 3.9 |
| C3H4 | a | 1.08 | 0.8 |
| C3H6 | 12.1 | 13.8 | 13.5 |
| C3H8 | a | 0.29 | 0.7 |
| C4H6 | 4.2 | 4.63 | 5.2 |
| C4H8 | 2.8b | 3.49 | 3.7 |
| C4H10 | b | 0.15 | 0.3 |
| C5 compounds | 9.0c | 2.58 | 3.5 |
| Benzene | 13.0d | 7.1 | 9.0 |
| Toluene | d | 2.35 | 4.9 |
| C8 aromatics | d | 0.73 | 2.7 |
| C6 – b.p. 473 K or 400 °F (477 K), except C6–C8 aromatics | c | 1.29 | 8.9e |
| Fuel oil | 6.0 | 4.38 | e |
Nowadays, only bio-ethylene by dehydration of bioethanol has been established and commercialized on a significant scale among biomass-derived small (≤C6) olefins.9,10 The other industrially important olefinic compounds are difficult to synthesize in low cost from biomass; however, the recent large interest in biomass conversion leads to rapid progress in this research field, especially in the development of effective catalysts. Here, we review the recent progress in the synthesis of industrially important C3–C6 olefins and unsaturated oxygenated compounds from biomass.
2. Key reactions
First, we discuss several key reactions in the conversion of highly oxygenated biomass-derived molecules to olefinic compounds, because these reactions and the catalysts for them can be commonly used for production of many compounds. The target compounds in this review are bulk chemicals, and the reactions cannot use expensive stoichiometric reagents.11,12 Discharging large amounts of wastes is also unacceptable. Therefore, some reactions commonly used in organic synthetic chemistry such as the Wittig reaction, halogenation–elimination and tosylation–elimination are inappropriate for the complete or partial removal of oxygenated functionalities.2.1. Dehydration: C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) C formation reaction
C formation reaction
Dehydration of alcohols is the simplest CC formation reaction. One OH group and hydrogen atom bonded to the neighboring carbon are removed, providing olefins (Scheme 1). Ideally, the by-product of dehydration is only water, which is acceptable in the synthesis of bulk chemicals. Dehydration of alcohols typically proceeds in the presence of strong Brønsted acid catalysts with an E2 mechanism (primary alcohols) or E1 mechanism (secondary and tertiary alcohols).13–15 Brønsted-acid-catalyzed dehydration of alcohols is a classical reaction explained in basic textbooks in general chemistry. Sulfuric acid is a typical homogeneous catalyst. Heterogeneous solid acid catalysts such as acidic resin, silica-alumina and zeolites can be also used. A fundamental issue of the dehydration with Brønsted acid catalysts is low regioselectivity. The sole dehydration product is obtained only when the substrate is ethanol, 1-propanol, 2-propanol or tert-butyl alcohol. Dehydration of other alcohols always produces multiple olefins. The distribution of the produced olefins is mostly governed by the relative thermodynamic stability and is difficult to control by the catalyst: internal olefins, especially trans-olefins, are preferably produced (cf. Saytzev rule). However, the energy difference is generally small, and a mixture of olefins is produced.14 There are additional side reactions induced by acidic catalysts. One is isomerization by the rearrangement of the carbocation intermediate and this side reaction further complicates the product distribution. Oligomerization of product olefins and C–C cracking can also occur via the carbocation intermediate at too high temperature. Highly polymerized products become coke over solid acid catalysts, which leads to severe problems as explained later.
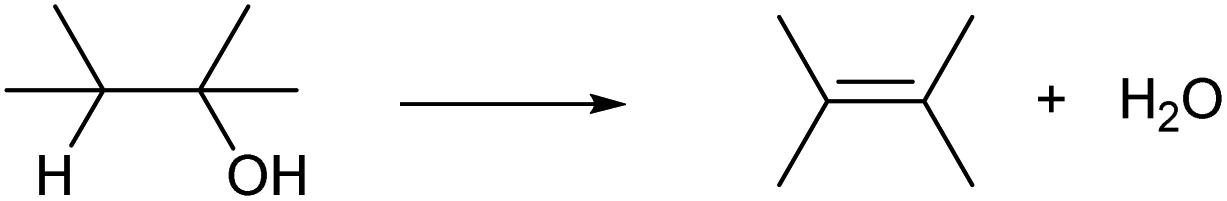 | ||
| Scheme 1 Alcohol dehydration. | ||
While the selection of catalysts is not so effective in the control of selectivity among dehydration products, the structure of the substrate much affects the selectivity patterns. The presence of additional functional groups can change the products (Scheme 2). The dehydration of vicinal diols gives carbonyl compounds via keto–enol tautomerization. Aldehydes tend to be more produced than ketones by dehydration of 1,2-diols.16,17 Aldehydes are reactive molecules and can be further converted to various products such as dioxolanes (acetal of the substrate), carboxylic acids and oligomers. While 1,3-diols do not have special reactivity, the presence of the carbonyl group at the γ-position much increases the reactivity of the OH group toward dehydration because the carbonyl group increases the acidity of the hydrogen atom at the β-position (cf. elimination reaction with the E1cB mechanism and the Michael addition reaction).18 Functional groups at the δ- or ε-position lead to the formation of cyclization products, such as 1,4-butanediol conversion to tetrahydrofuran instead of 3-buten-1-ol.19–21
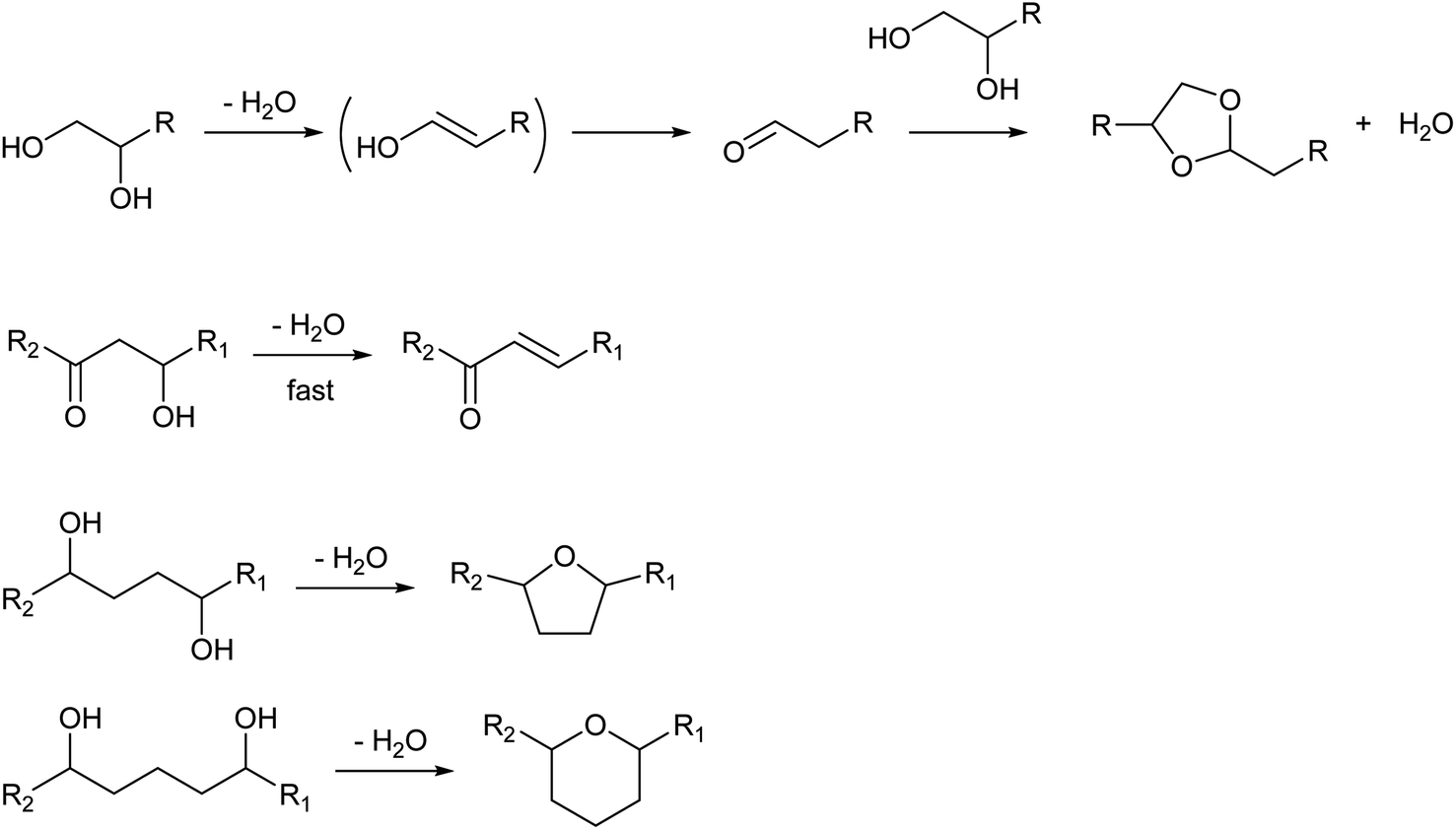 | ||
| Scheme 2 Effect of other functional groups on dehydration of alcohols. | ||
Dehydration of ethers is a variant of Brønsted-acid-catalyzed dehydration of alcohols (Scheme 3). In the presence of strong Brønsted acids, ethers are first decomposed to olefins and alcohols.13 In the cases of dialkyl ethers, the produced alcohols are then dehydrated to olefins or returned to dialkyl ethers. In the cases of cyclic ethers with a tetrahydrofuran ring, the reactivity is low and the main path is the direct formation of dienes without a release of alkenols from the catalyst surface.22,23 Side reactions such as oligomerization and C–C cracking are more likely to occur than simple dehydration of alcohols because severe reaction conditions are required.
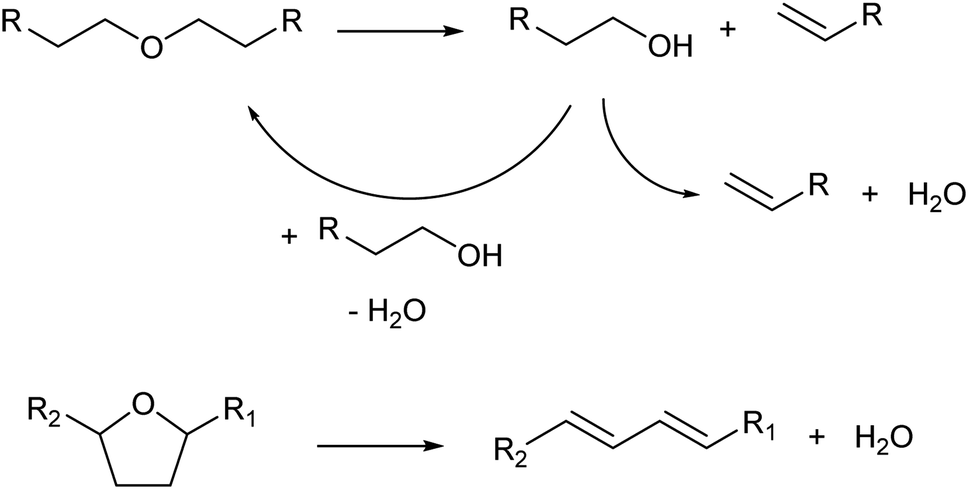 | ||
| Scheme 3 Dehydration of ethers. | ||
Apart from strong Brønsted acids, acid–base bifunctional catalysts can also promote the dehydration of alcohols. Typical catalysts are solid metal oxides such as Al2O3 (ref. 24 and 25) and rare-earth oxides.26 While Al2O3 has been widely used in mechanistic studies for a long time, especially during the 1960s and 1970s, recent reports tend to use rare-earth oxide catalysts because of their excellent performance, especially in terms of product yield. While mixing homogeneous acid and base catalysts loses these activities by neutralization, the solid oxide surface can possess acid–base bifunctionality. The proposed mechanism is shown in Scheme 4. Over such amphoteric catalysts, an alcohol substrate is adsorbed on the acid site with the OH group. The vicinal C–O and C–H bonds are dissociated simultaneously, with the acid and base sites, respectively. This is a variant of the E2 mechanism; one difference from the general E2 reaction is that the two bonds are dissociated with syn configuration,27,28 while the general E2 reaction with a strong homogeneous base is the dissociation with anti configuration. An important feature of the dehydration over bifunctional catalysts is the suppression of cyclization of diols. With strong Brønsted acid catalysts, only the C–OH group is activated, and the SN2-type cyclization easily occurs by the reaction between the activated C–OH moiety as a leaving group and another C–OH as a nucleophile. On the other hand, acid–base bifunctional catalysts activate vicinal C–O and C–H bonds simultaneously, and these sites react selectively. Side reactions over bifunctional catalysts are different from those with strong Brønsted acid catalysts. Dehydrogenation to aldehyde, reactions of the aldehydes such as condensation and decarbonylation, and the movement of CC bonds are typical side reactions. Catalyst development is necessary to suppress such side reactions.
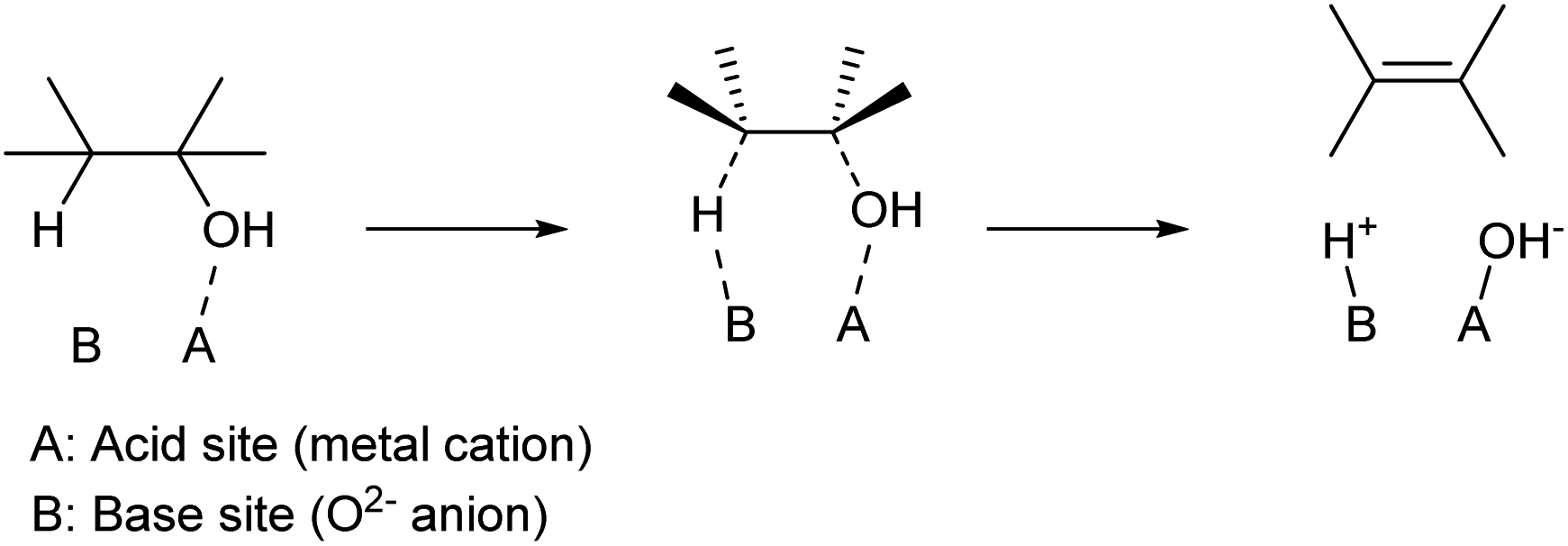 | ||
| Scheme 4 Dehydration of alcohols over acid–base bifunctional catalysts. | ||
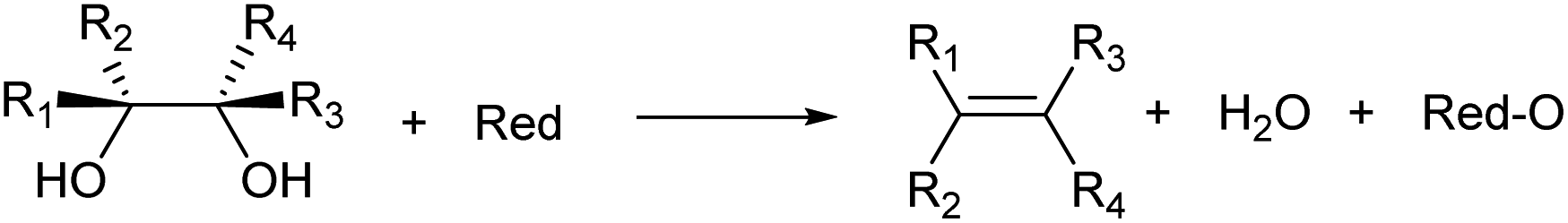
2.2. Deoxydehydration: another CC formation reaction
Deoxydehydration (commonly abbreviated as DODH) is another reaction for the formation of CC bonds. Deoxydehydration reductively converts vicinal diols to CC groups (Scheme 5).29,30 The term “deoxydehydration” only means the formal change of atoms in the substrate (−O–H2O = −2O–2H); rather, deoxydehydration can be regarded as the reverse reaction of cis-dihydroxylation of olefins (i.e. oxidation with osmium tetroxide). The merit of deoxydehydration in the production of olefinic compounds is the direct use of the polyfunctionalized substrate.6 Dehydration reaction of polyfunctionalized substrates has a large number of possible products, and it is generally difficult to obtain high selectivity. In contrast, deoxydehydration gives much simpler product distribution and is sometimes stereospecific.31 Typically, deoxydehydration is catalyzed by homogeneous Re species, and two-electron reducing agents such as PPh3 and secondary alcohols are used. The Re-catalyzed deoxydehydration proceeds with a two-electron redox cycle of high-valent monomeric Re species, composed of three steps: coordination of the vicinal diol substrate to Re as diolate species, two-electron reduction of the Re diolate species, and removal of the olefin to leave oxidized Re. The recent development of deoxydehydration systems has enabled the use of heterogeneous catalysts and H2 as the reducing agent.32,33 A variant of deoxydehydration is the stoichiometric use of formic acid.34 Formic-acid-based deoxydehydration does not use a catalyst and produces CO2 as a by-product. Although deoxydehydration has not been industrially carried out, the recent rapid development of deoxydehydration systems may lead to industrial use in the future.
 | ||
| Scheme 5 Deoxydehydration (DODH) reaction. | ||
2.3. Hydrodeoxygenation: control of the number or position of functionality
Because of the difficulty of the selectivity control in the formation of CC groups, excess functional groups in substrates should be removed before the formation of CC groups. The functional groups in biomass-derived molecules are mainly oxygen-containing ones, and their removal (deoxygenation) requires a reductive reaction. Among the reducing agents, H2 is by far the best one, in view of both atomic economy and cost. Although the use of H2 may not be convenient in laboratory experiments because of its explosive nature, this does not matter in industrial processes. The selectivity of deoxygenation with H2 as the reducing agent (hydrodeoxygenation) can be controlled by catalysts.35–37 Various catalysts with different selectivity patterns have been reported, and the development of catalysts with novel selectivity has been a hot research field in these days.
There are several types of mechanisms of hydrodeoxygenation, depending on catalysts, substrates and even reaction conditions. The mechanisms of hydrodeoxygenation include dehydration + hydrogenation, deoxydehydration + hydrogenation, and direct hydrogenolysis over a metal surface.38 The dehydration + hydrogenation mechanism over acid + metal bifunctional catalysts is the most typical route. However, it is difficult to obtain very high selectivity with dehydration + hydrogenation due to the same reason as dehydration discussed above. In addition, it is also difficult to combine “dehydration + hydrogenation” with “dehydration” to selectively obtain a single olefin because the C–O bonds that are reactive in dehydration have been removed in the former step. It is easier to obtain high olefin yield when different types of reactions are combined such as the combination of “deoxydehydration + hydrogenation” and “dehydration”.
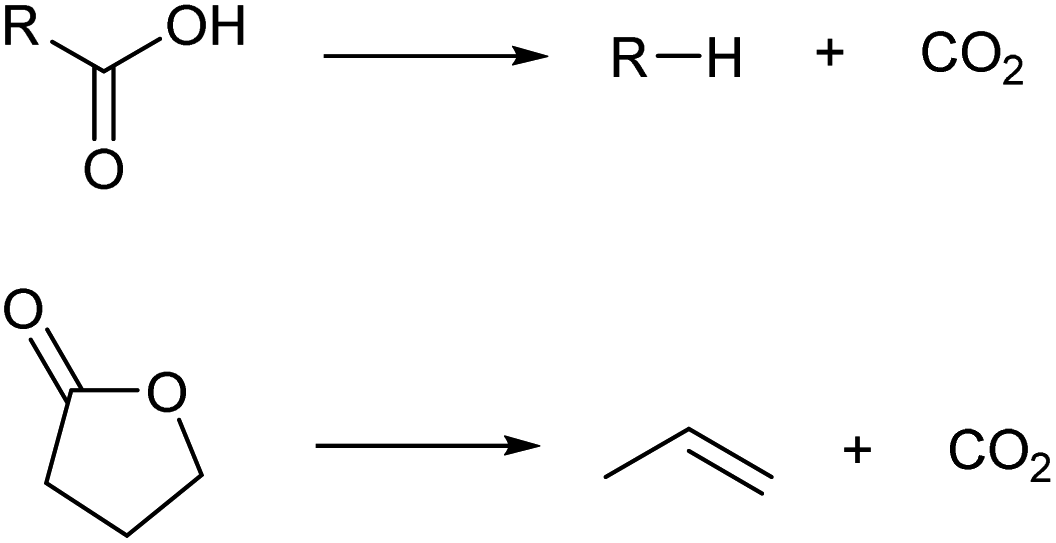
2.4. Control of the carbon chain
There are some methods to change the carbon skeletons of organic molecules. However, during the decrease of the carbon chain length, the loss of atom economy is inevitable. It is generally difficult to obtain high selectivity during the extension of the carbon chain (dimerization and oligomerization) or isomerization of the carbon chain. The low selectivity also decreases the atom economy. Considering that biomass is a precious resource in comparison with petroleum, a good atom economy is highly desirable.Among the methods to shorten a carbon chain length, decarboxylation is the most harmless one (Scheme 6).39 There is no consumption of costly reagents such as H2. The most oxidized carbon atom in the substrate is removed as carbon dioxide, and the loss of internal energy is minimal if decarboxylation proceeds ideally. The substrates of decarboxylation are carboxylic acids, esters and lactones, and there are many biomass-derived molecules in these categories.4,6 Decarboxylation of lactones is especially useful because olefins are directly produced. Various catalysts such as zero-valent metals, acids and bases are applicable to decarboxylation. The reaction temperature for decarboxylation is generally high (such as 523 K) which leads to isomerization of products.
 | ||
| Scheme 6 Decarboxylation reactions. | ||
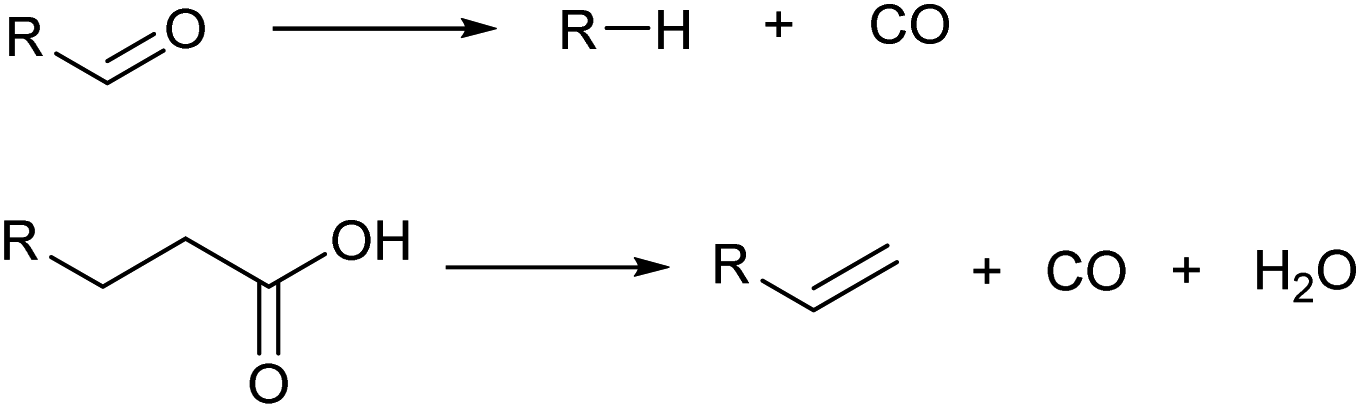
Decarbonylation also decreases the carbon number by one (Scheme 7). Although the loss of free energy is slightly larger than decarboxylation, decarbonylation is also useful in the selective synthesis of hydrocarbons from highly oxidized molecules. Decarbonylation of aldehydes is typically catalyzed by zero-valent noble metals such as Pd.40 An important variant is decarbonylative dehydration of carboxylic acids which gives olefins directly.41,42 Decarbonylative dehydration is catalyzed by noble metal complexes via pathways composed of oxidative addition, decarbonylation, β-hydride elimination and reductive elimination. Sacrificial dehydrating agents such as acetic anhydride are necessary in typical systems, and the development of systems without such requirements is desirable.
 | ||
| Scheme 7 Decarbonylation and decarbonylative dehydration. | ||
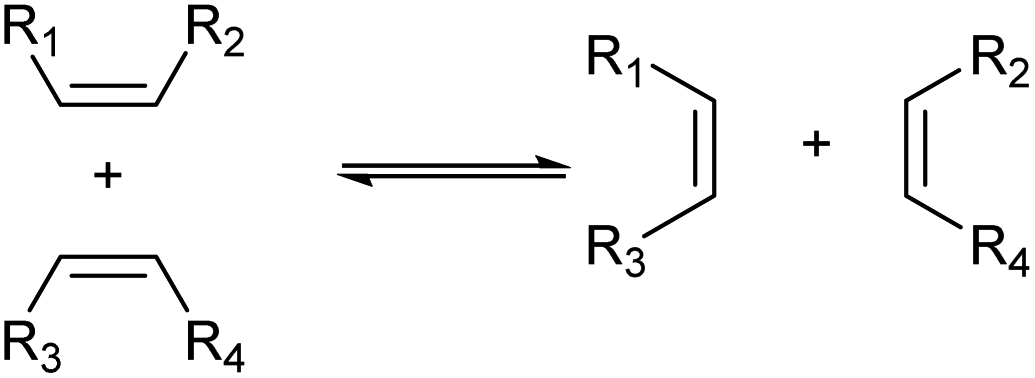
Another method worth mentioning for the control of the carbon chain is metathesis (Scheme 8).43–45 Metathesis is the mutual exchange reaction of two olefin molecules with their alkylidene units. This reaction is useful in the production of olefins with higher demand from those with lower demand and has been already industrialized for the manufacture of some olefins such as propylene from 2-butene and ethylene.44 Typical catalysts are supported high-valent transition metal oxides (MoOx, WOx and ReOx)46 and homogeneous Ru complexes.47 The selection of reactants is especially important because both products should be utilized to maximize the atom efficiency of the whole process.
 | ||
| Scheme 8 Olefin metathesis. | ||
2.5. Catalyst deactivation
Deactivation of catalysts is a frequently encountered problem in the catalytic conversions of biomass-related compounds.48,49 Coking is the most common cause of the deactivation of solid acid catalysts. Coking is the deposition of carbonaceous material (coke) on the catalyst surface, which leads to catalyst deactivation by covering the active site and/or clogging the pore. Oligomerization and dehydrogenation are the main formation routes of coke for acid catalysts, especially when highly unsaturated compounds are involved in the system. This formation mechanism is similar to the case of FCC50 where acid catalysts and unsaturated products are similarly involved. Avoiding too strong acids, applying lower reaction temperature and adding water to the feed are effective in suppression of coking; however, these methods tend to decrease the dehydration rate as well. It is difficult to completely suppress coking, and regeneration by calcination is common for solid acid catalysts. Stability during the regeneration, especially against sintering of fully oxidized phases, is also necessary. In view of stability during regeneration, too sophisticated preparation process is not advantageous. Materials obtained by calcination as the final step of preparation are usually stable during regeneration. For the supported catalysts, the selection of support is critical in the stability of the regeneration.In the liquid phase reaction, leaching of active species to the liquid phase, phase change or sintering during the reaction, and poisoning are major causes of catalyst deactivation. When deactivation is observed, it is necessary to find the cause by characterization of the deactivated catalyst. The hot-filtration test and elemental analysis of the reaction solution are effective methods to detect leaching. Countermeasures against deactivation depend on the cause for the deactivation. Selections of support, solvent and reaction conditions can affect the leaching, and the use in gas-phase reaction can be a solution. Phase change and sintering are difficult problems to solve. It is necessary to find a method to regenerate the catalysts. For poisoning, purification of the substrate, washing the used catalyst, or regeneration of used catalysts is tested depending on the type of poison.
3. Specific products
3.1. C3 compounds
While most C3 compounds in petrochemistry are manufactured from propylene, propylene cannot be obtained easily from biomass. The most accessible biomass-derived C3 compound is glycerol, which is a by-product of biodiesel manufacture from triglycerides. Glycerol is co-produced in the ratio of about 10 wt% to biodiesel. The production of waste glycerol reaches 5 million tons per year and is further increasing.51 As another biomass-based C3 compound, lactic acid has been already industrially produced by fermentation.52–54 Chemical synthesis of lactic acid from sugars is also possible.55 The current production capacity of lactic acid (<1 million tons per year as polylactic acid)56 is significantly smaller than that of the biodiesel industry. The production of acetone by ABE (acetone–butanol–ethanol) fermentation57 and that of 1,2-propanediol from sugars by hydrogenolysis58,59 are possible, but these are not regarded as main methods to obtain renewable C3 compounds at present.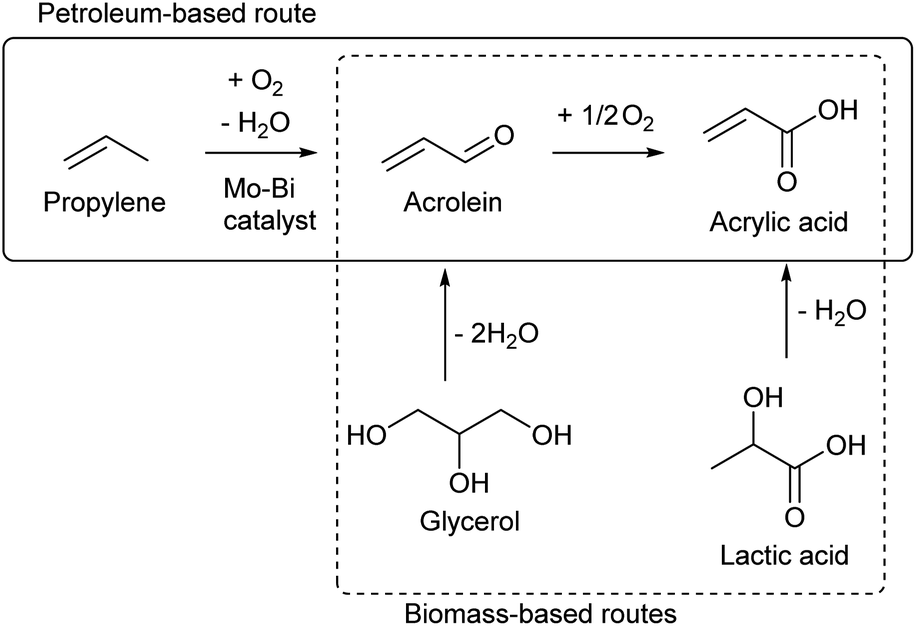 | ||
| Scheme 9 Synthesis of acrolein and acrylic acid. | ||
Acrolein can be produced relatively easily from glycerol, by acid-catalyzed double dehydration.60,62–64 Typical catalysts are W-based ones and zeolitic materials such as heteropolyacids and H-ZSM-5, respectively. The reaction temperature is around 573 K and the acrolein yield is around 80%. Water is usually added to the feed. The role of water includes the retention of Brønsted acidity of catalysts at the high reaction temperature. For zeolitic catalysts including zeolites, the control of pore size distribution is essential. Traditional zeolite crystals with only micropores tend to be deactivated rapidly by the formation of coke. Hierarchical zeolites have been reported to be more durable catalysts.65,66 Nevertheless, the long-term catalyst durability remains a challenge in the production of acrolein. Lists of reported systems can be found in recent review papers.63,64
Oxidization of acrolein produces acrylic acid, which has been an established process in petrochemistry. Therefore, glycerol-based acrylic acid can be obtained by using acrolein as an intermediate.67–69 Direct production of acrylic acid from glycerol is possible by combining oxidation and acid catalysts.70 Typical component of the former is vanadium oxide and that of the latter is tungsten oxide. The yield of acrylic acid (typically 30–60%) has been significantly lower than that of acrolein in simple dehydration systems. Although coke formation can be suppressed under oxidative conditions, stability is still a problem for the catalyst under (hydro)thermal conditions.
The demand of acrylic acid is over 5 million tons per year.67 The demand of acrylonitrile, which is also a probable derivative of biomass-based acrolein, is in a similar level to acrylic acid.71 It is unrealistic for glycerol conversions to cover all the demands of acrolein and acrylic acid, considering that there are many other valuable glycerol conversions. A report on life cycle assessment (LCA) of acrolein production from glycerol revealed that there is a very high environmental load during the agricultural stages.72 This means that an increase in vegetable oil production only to obtain acrolein is unacceptable. The production methods of acrolein or acrylic acid from sugars are also worth developing. One method is the conversion of lactic acid which is a fermentation product of sugars.68,69 Stoichiometrically, dehydration of lactic acid can give acrylic acid. This reaction is difficult among dehydration reactions, because the presence of the carbonyl group at the 1-position promotes the removal of groups at the 2-position as a cation (–H+) rather than as an anion (–OH−). High temperature is necessary for the dehydration of lactic acid, and severe conditions promote undesirable side reactions such as decarboxylation. The dehydration of lactic acid requires acid–base bifunctional catalysts such as calcium phosphate,73–75 alkali salts of zeolites76–78 and barium sulfate.79–81 The use of alkali or alkali earth metals is characteristic of the catalysts in this reaction, and these metal sites bind the carboxylic group of lactic acid. The dehydration of the bound lactic acid requires an additional pair of acid and base sites which attack the 2- and 3-positions, respectively. The reactions are typically carried out under flow conditions in the presence of water at high temperature (about 623 K). Some systems obtained about 80% yield of acrylic acid.73,75,78–81 Dehydration of alkyl lactates has been also reported.82–84 The co-fed solvent is water or the corresponding alcohol to the alkyl group. When water is used as the co-fed solvent for dehydration of alkyl lactates, the product is acrylic acid. The reactivity over hydroxyapatite (a class of calcium phosphate) was in the order of lactic acid > methyl lactate > ethyl lactate, while the selectivity to acrylic acid was in the opposite order.83 Long-term stability is a major issue in dehydration of lactic acid and alkyl lactates, like many other catalytic dehydration systems. Sels et al. recently reported that the co-feeding of methanol and water is advantageous to the inhibition of catalyst (K–ZSM-5) deactivation in methyl lactate dehydration.84 Considering that the catalyst requires three sites (alkali/alkali earth metal, acid and base) with appropriate positions, zeolite-based catalysts seem more promising because of the controllable structure.
Feringa et al. recently reported another approach to the synthesis of acrylic acid.85 This approach uses maleic anhydride as the starting compound which can be synthesized from either petroleum or biomass, as described later. Maleic anhydride is hydrolyzed to maleic acid, and then metathesis of maleic acid and ethylene gives two molecules of acrylic acid. Maleic acid was converted quantitatively to acrylic acid with an excess amount of ethylene and the commercially available Ru-based Hoveyda–Grubbs second-generation catalyst.
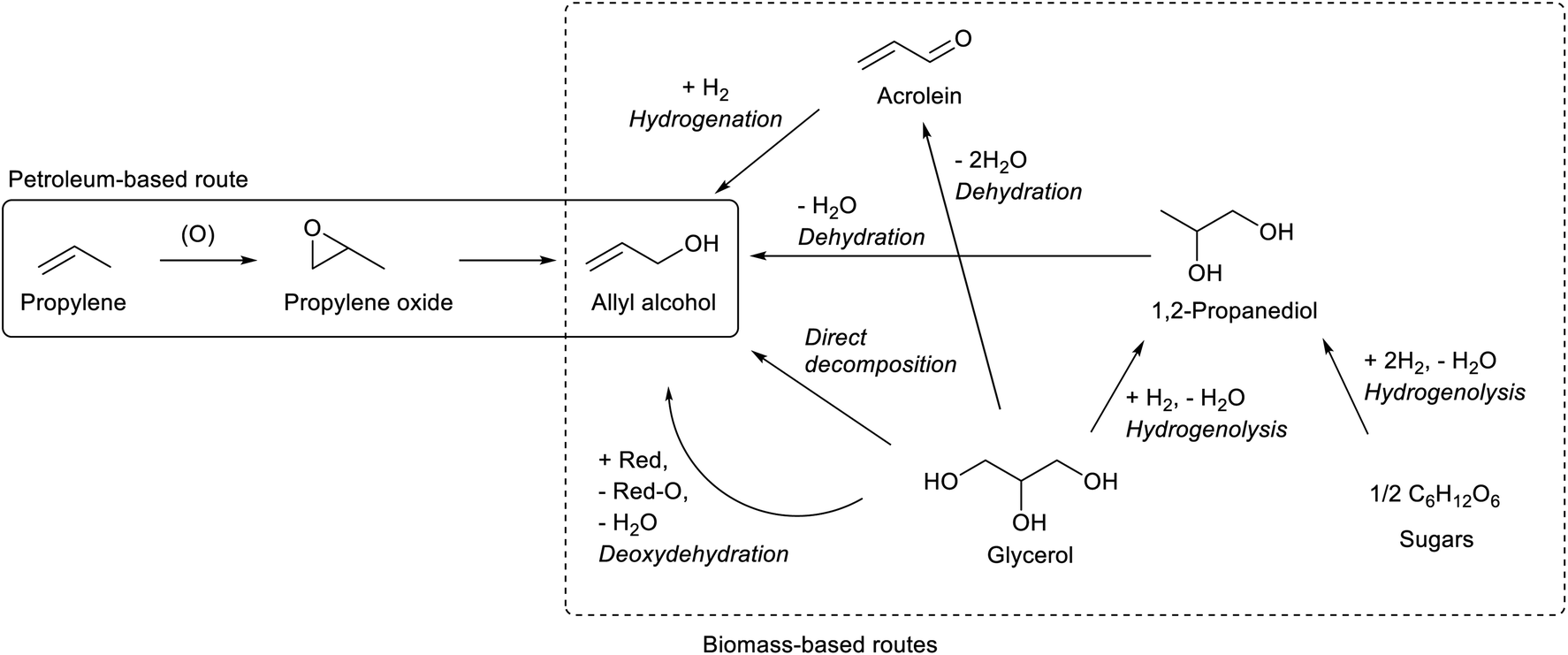
O bond out of the CC bond. Selective CO hydrogenation of conjugated α,β-unsaturated aldehydes is a hot topic and many effective systems have been reported.87 However, acrolein is the most difficult substrate to obtain a high yield of the corresponding unsaturated alcohol because the small steric hindrance around the CC bond hampers the selective recognition toward the CO bond. Among hundreds of reports for selective hydrogenation of α, β-unsaturated aldehydes,87–90 only a few papers reported over 30% selectivity to allyl alcohol, and the activity of catalysts in these reports was low.91–94 The use of non-H2 reductants (transfer hydrogenation) enables good yield of allyl alcohol more easily;95–97 but the cost of the reductant and low atom efficiency are severe problems.
 | ||
| Scheme 10 Allyl alcohol synthesis. | ||
1,2-Propanediol can be synthesized from glycerol by selective hydrogenolysis.98,99 While 1,2-propanediol has been conventionally synthesized by hydration of propylene oxide, glycerol-based 1,2-propanediol has also been already commercialized.59,98 Hydrogenolysis of sugars to 1,2-propanediol is also possible.58,59 Dehydration of 1,2-propanediol usually gives propanal; however, with an appropriate catalyst, namely K-doped ZrO2, allyl alcohol was obtained with a significant selectivity (about 50%, at 64% conversion; conditions: 673 K, gas hourly space velocity (GHSV) 9051 h−1, Ar/1,2-propanediol/H2O = 31/1/12).100 Strong basicity with the appropriate acid/base balance of catalysts is the key to selective dehydration. Severe deposition of the condensation product on the catalyst was observed. This reaction is closely related to the dehydration of 2,3-butanediol which will be discussed below. The catalysts for dehydration of 1,2-propanediol and 2,3-butanediol will be developed at the same time.
The method of thermal catalytic decomposition of glycerol is composed of glycerol dehydration to acrolein and transfer hydrogenation of acrolein with glycerol as the reducing agent. Fe-based catalysts have been mainly reported,101–104 although why Fe is effective in this reaction is not clarified yet. Other reported catalysts include vanadium- and niobium-based ones.105,106 However, the severe conditions and consumption of glycerol as the reducing agent limit the selectivity to allyl alcohol below 30% until the 2010s. In 2020, two reports showed higher selectivity by using Mo–Fe catalysts with further modification: MoFe/V-KIT-6 catalyst (31.4% selectivity to allyl alcohol at 92.1% conversion)107 and CsFeMo–ZSM-5 catalyst (52.0% selectivity at 93.8% conversion).108 In thermal decomposition, the balance of the reductant and substrate should be considered when high selectivity to allyl alcohol is obtained: the production of one allyl alcohol molecule consumes one hydrogen molecule, and hydrogenolysis such as formations of 1,2-propanediol, propanols, ethanol and acetone also consumes H2 molecules. Without co-feed of H2, the H2 molecules must be supplied by the formation of other glycerol-derived products. Except reforming reactions, it is difficult to supply more than one H2 molecule per one glycerol molecule: C3H8O3 → 3C (coke) + 3H2O + H2; C3H8O3 → 3HCHO + H2; C3H8O3 → ![[/]](https://www.rsc.org/images/entities/char_e0ee.gif) CH3COOH + H2. It is very difficult to obtain >50% selectivity without co-feed of H2, while with H2 the CC bond in allyl alcohol is easily hydrogenated over most metal catalysts. Also in 2020, Zhaoyin Hou et al. reported very promising results of high allyl alcohol yield from glycerol with H2 and the CoFe alloy catalyst.109 In this system, Fe can work as the active center for allyl alcohol formation, and Co is the hydrogenation catalyst of the CO bond in acrolein (precursor of allyl alcohol) or glyceraldehyde (formed by transfer hydrogenation). Alloying of Co with Fe further decreases the CC hydrogenation ability. Recent reports for the glycerol conversion to allyl alcohol with or without H2 are summarized in Table 2.
CH3COOH + H2. It is very difficult to obtain >50% selectivity without co-feed of H2, while with H2 the CC bond in allyl alcohol is easily hydrogenated over most metal catalysts. Also in 2020, Zhaoyin Hou et al. reported very promising results of high allyl alcohol yield from glycerol with H2 and the CoFe alloy catalyst.109 In this system, Fe can work as the active center for allyl alcohol formation, and Co is the hydrogenation catalyst of the CO bond in acrolein (precursor of allyl alcohol) or glyceraldehyde (formed by transfer hydrogenation). Alloying of Co with Fe further decreases the CC hydrogenation ability. Recent reports for the glycerol conversion to allyl alcohol with or without H2 are summarized in Table 2.
| Entry | Catalyst | Gas phase or carrier | Liquid phase or feed | Temp. [K] | Time [h] | Conv. [%] | Main products (selectivity [%]) | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Flow system. | ||||||||
| 1 | MoFe/V-KIT-6. 0.5 g | N2, 0.6 L h−1 | 35% G aq., 1 mL h−1 | 613 | —a | 92.1 | AA (31.4), acrolein (17.0), acetaldehyde (14.4) | 107 |
| 2 | CsFeMo–ZSM-5, 0.5 g | N2, 0.6 L h−1 | 10% G aq., 1 mL h−1 | 623 | —a | 93.8 | AA (52.0), acetol (18), 1,2-propanediol (7), acrolein (5) | 108 |
| 3 | CoFe(1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1)-ZIF-reduced, 0.2 g :1)-ZIF-reduced, 0.2 g |
H2, 2 MPa | 40% G, 1.2 mL h−1, H2/G = 40 | 523 | —a | 89.7 | AA (68.7), acetol (8.8), 1,2-propanediol (8.7), 1-propanol (7.5) | 109 |
| 4 | ReOx–Au/CeO2, 0.3 g | H2, 8 MPa | G 0.5 g, 1,4-dioxane 4 g | 413 | 52 | >99 | AA (91), 1-propanol (5) | 114 |
| 5 | ReOx–Ag/CeO2, 0.3 g | H2, 8 MPa | G 0.25 g, 1,4-dioxane 4 g | 413 | 12 | 93 | AA (99), 1-propanol (<1) | 115 |
Deoxydehydration of glycerol is another method of one-step production of allyl alcohol from glycerol. Glycerol to allyl alcohol is one of the most attractive applications of deoxydehydration, and there are several reports for catalyst development and the reactor setup.110–112 While earlier reports used homogeneous catalysts and secondary alcohols as reducing agents,110,113 we have recently developed heterogeneous ReOx–Au/CeO2 and ReOx–Ag/CeO2 catalysts for the synthesis of allyl alcohol from glycerol with H2 as the reducing agent.114,115 The yield of allyl alcohol reached over 90%. In these catalysts, atomically dispersed ReOx species on CeO2 works as the active site of deoxydehydration, and the added metal (Au or Ag) activates the H2 molecule to supply hydrogen species to reduce the ReOx species. The crystalline CeO2 support is essential in this catalyst, and its roles include stabilizing monomeric high-valent (≥+4) Re species and carrying activated hydrogen species as protons and electrons from added metal sites to Re species.116 For ReOx–Au/CeO2, the control of the particle size of Au is necessary: around 10 nm of Au particle size is appropriate for selective production of allyl alcohol. Too small Au particles catalyze the hydrogenation of allyl alcohol to 1-propanol in addition to H2 activation, which decreases the selectivity to allyl alcohol at a high conversion level. However, the high selectivity of ReOx–Au/CeO2 with larger Au particle size is obtained at the cost of lower activity because of the limited H2 activation ability of the Au metal surface.117 On the other hand, Ag species in ReOx–Ag/CeO2 do not have such strong structure sensitivity, and both high activity (comparable to ReOx–Au/CeO2 with small Au particles) and selectivity to allyl alcohol can be obtained without precise control of the Ag particle size.115 Considering also the lower price of Ag than that of Au, ReOx–Ag/CeO2 is a superior catalyst. Although the catalyst recyclability of ReOx–Au/CeO2 and ReOx–Ag/CeO2 has been confirmed at least three times when they are calcined after use as regeneration,114,115 long-term stability should be investigated for this type of catalyst.
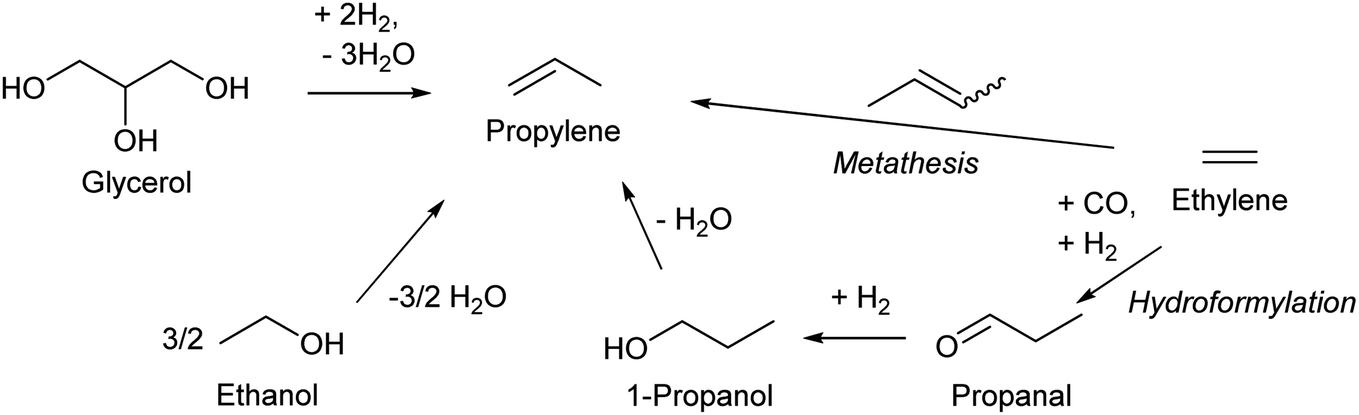 | ||
| Scheme 11 Synthesis routes of propylene. | ||
Propylene production from biomass is a sore subject in biomass conversion. Propylene is one of the most important key compounds in petrochemistry: a very large amount of propylene is manufactured by naphtha cracking or FCC of heavier oil and is utilized as an intermediate for many chemicals such as polypropylene, phenol (via cumene), acrylonitrile and acrolein. The current annual production amount is over 50 million tons for even polypropylene alone, which can be synthesized only from propylene.130 One major issue of propylene production from biomass is the inherent cost of the reductant. Production of propylene inevitably involves removal of large amounts of oxygen atoms in biomass. If C3 compounds are produced from glycerol, the price (cost) will be in the order of propanediols < propanols < propylene < propane. This order is totally opposite to that in petrochemistry. Large-scale use of glycerol as the source of propylene is thus not realistic; rather, glycerol should be converted to functionalized compounds including those currently synthesized from propylene such as acrolein, acrylic acid and acrylonitrile in view of the reductant cost. Ethanol or bio-ethylene upgrading is a more promising method for bio-propylene, and lower greenhouse gas emission by bioethanol-based propylene is suggested;131 however, the gap between the cost of bio-propylene production and the low price of petrochemical propylene is still large. The use of propylene in the chemical industry should be significantly decreased when biomass becomes the main source of organic chemicals.
3.2. C4 compounds
There are several routes for the synthesis of C4 compounds in petrochemistry. One is the direct recovery from the naphtha cracking products, and such C4 components include butenes, butadiene, and isobutene (Table 3). Yet, the main target products of naphtha cracking are ethylene and propylene, and these C4 compounds are rather minor by-products (Table 1). Therefore, the supply of C4 compounds from naphtha cracking depends on the production amount of ethylene and propylene. The use of lighter hydrocarbon resources such as associated gas than naphtha for ethylene production is a recent trend in petrochemistry, and such a process yields a lower amount of the C4 fraction.132 Therefore, the use of biomass-derived C4 compounds will be competitive even in the near future. Another synthesis route of C4 compounds in petrochemistry is the use of maleic anhydride as an intermediate. Maleic anhydride is synthesized from n-butane or benzene (the former is preferable) by selective aerobic oxidation over the vanadium phosphorous oxide catalyst.133,134 The use of biomass-derived substrates such as furfural,135 5-hydroxymethylfurfural (HMF)136 and levulinic acid137 for maleic anhydride synthesis has been confirmed to be possible (Scheme 12).138,139 Extension of the carbon chain is another approach to synthesizing C4 compounds in petrochemistry, such as acetaldehyde condensation (C2 + C2) and propylene hydroformylation (C3 + C1). The use of biomass-derived acetaldehyde is possible.| Compound | Formula | Distribution (wt%) |
|---|---|---|
| n-Butane | C4H10 | 0.4 |
| Isobutane | C4H10 | 0.75 |
| Isobutene | C4H8 | 22.83 |
| 1-Butene | C4H8 | 15.40 |
| cis-2-Butene | C4H8 | 3.41 |
| trans-2-Butene | C4H8 | 4.93 |
| 1,3-Butadiene | C4H6 | 50.54 |
| 1,2-Butadiene | C4H6 | 0.13 |
| Ethylacetylene | C4H6 | 0.18 |
| Vinylacetylene | C4H4 | 1.27 |
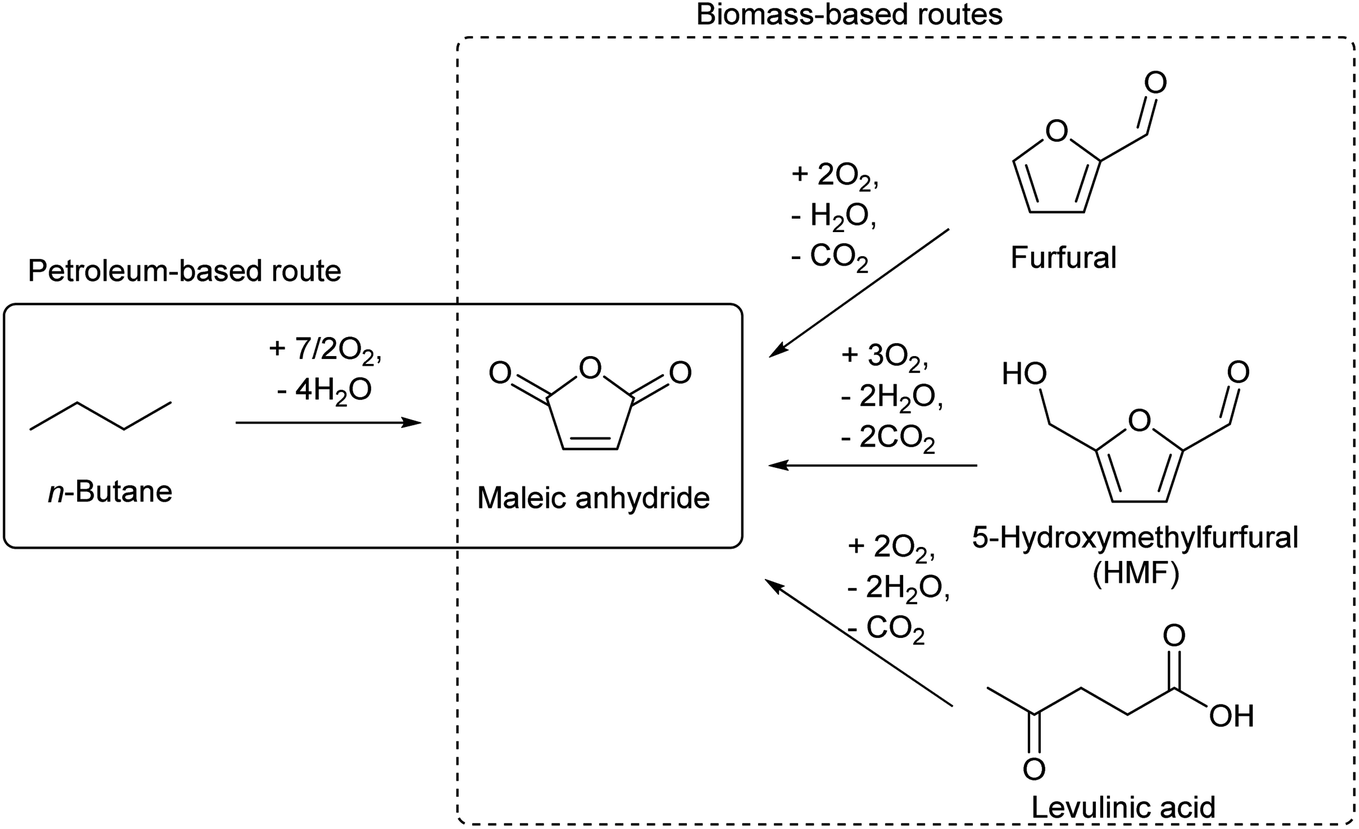 | ||
| Scheme 12 Synthesis of maleic anhydride. | ||
While chemical synthesis of C4 compounds from raw biomass is not easy, fermentation with suitable microorganisms can selectively produce C4 compounds. Such fermentation products include 1-butanol, isobutyl alcohol, butanediols, erythritol, succinic acid and fumaric acid.4–6,140,141 Fermentation uses substrates themselves as reducing agents, and therefore more oxidized products tend to be obtained in higher yields. The combination of chemical and biological conversions is an effective method to produce a less oxidized C4 compound in high yield.
Because sugar units in biomass resources have C5 and C6 carbon chains, there are several C5 oxygenates that can be relatively easily obtained from biomass. Decarboxylation of C5 compounds is also a useful reaction to produce C4 chemicals.
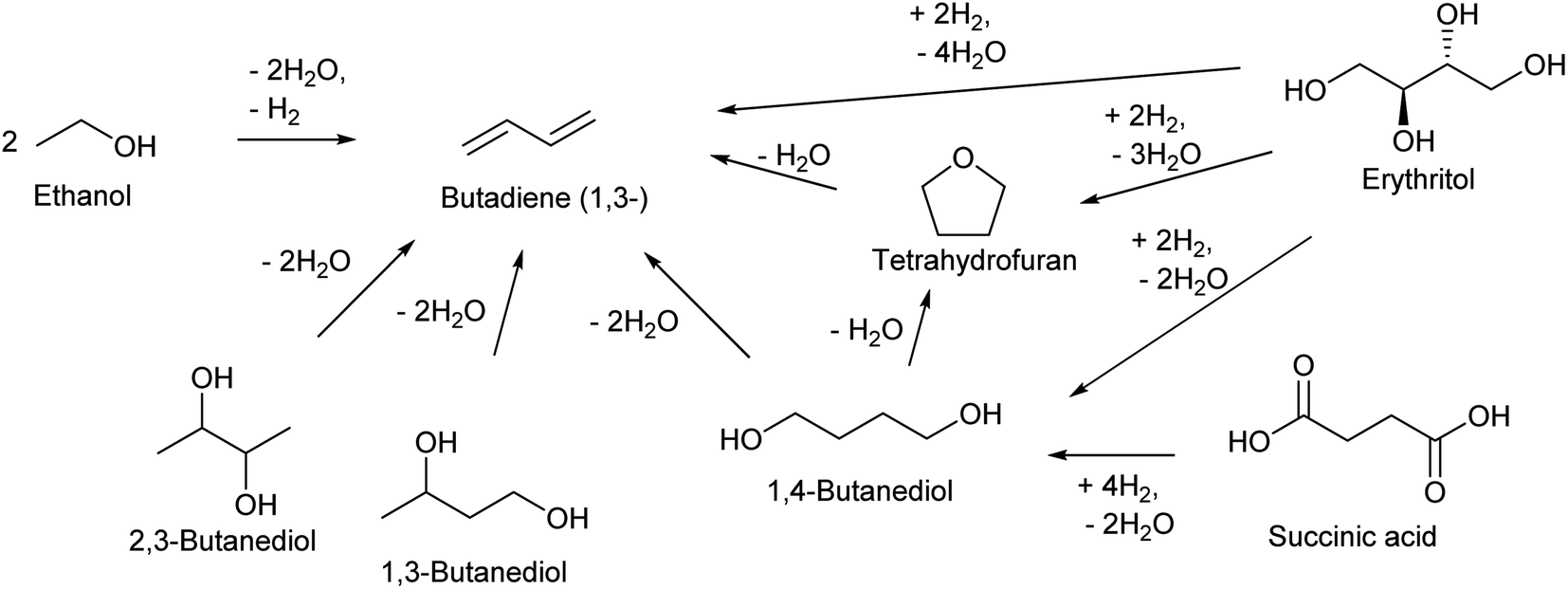 | ||
| Scheme 13 Synthesis of butadiene from biomass. The displayed compounds except butadiene and tetrahydrofuran are fermentation products. | ||
Ethanol upgrading is the most investigated route and has the longest history (over 100 years).142,144 The list of recently reported systems can be found in a review paper.142 This reaction consists of dehydrogenation, aldol condensation, hydrogenation and dehydration (Scheme 14). The hydrogenation step can be coupled with the dehydrogenation step, as transfer hydrogenation with ethanol. Basicity and acidity are necessary for the condensation step and dehydration step, respectively. Although the base sites can catalyze dehydrogenation and hydrogenation steps, metallic species such as Cu and Ag are more effective for these steps. A typical catalyst for ethanol upgrading to butadiene is the combination of MgO/SiO2 and metal.145 Supported group 4–5 (Zr, Hf and Ta) metal oxides on SiO2 with or without metals are also frequently investigated catalysts. The reaction temperature is above 523 K (typically around 623 K) because of the thermodynamic limitation of the dehydration step (crotyl alcohol to butadiene). There are many possible side reactions, such as overhydrogenation to butenes, decarbonylation to C1 + C3 products, ketone formation and polymerization. Because of the high reaction temperature and complicated reaction network, the selectivity is difficult to improve above 70%. The catalyst life is also a serious issue. Coking is a major reason of catalyst deactivation. Designing an appropriate pore structure and the co-feed of water can slow down the coking. The structural stability during hydrothermal conditions and regeneration treatment are remaining issues. Recent techno-economic and life cycle assessments demonstrated the potential of ethanol-based butadiene for a large decrease in greenhouse gas emission, and the catalyst performance in the butadiene formation step much affects the results.146
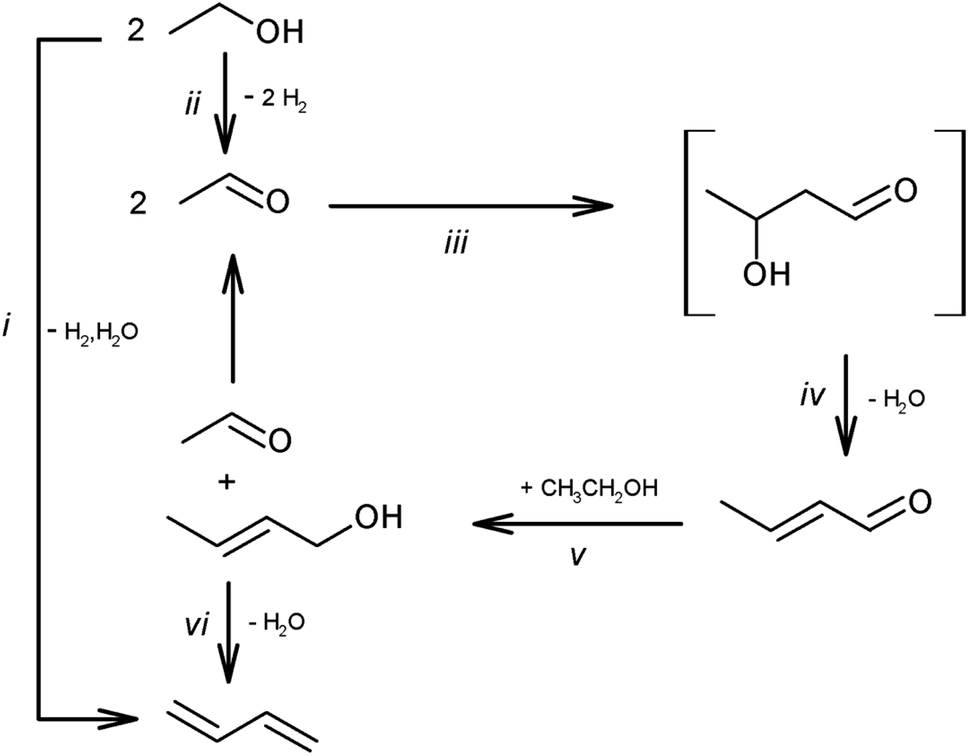 | ||
| Scheme 14 Pathways of ethanol conversion to butadiene. Reprinted from ref. 145 with permission from the Royal Society of Chemistry. | ||
Dehydration of butanediols is a simpler route to butadiene than ethanol conversion in a chemical view.147,148 The main production method of butanediols is fermentation. Each butanediol isomer (1,2-, 1,3-, 1,4- and 2,3-) can be obtained with an appropriate bacterial species. Chemical synthesis of butanediols from biomass-derived compounds is possible, such as hydrogenation of succinic acid149–152 and hydrogenolysis of erythritol6,124,153,154 and 1,4-anhydroerythritol.155–157 The chemically synthesized butanediol is usually the 1,4-isomer, although some methods produce a mixture of isomers. The difficulty and catalyst of butanediol dehydration to butadiene much depend on the position of OH groups in the substrate molecules (Scheme 15). Since dehydration of 1,2-butanediol typically produces butanal, 1,2-butanediol is not usually regarded as a source of butadiene. Most papers for butadiene synthesis by dehydration of butanediols were published within these 10 years. The recent progress in this research area is based on the effectiveness of rare-earth oxide catalysts in the dehydration of diols to unsaturated alcohols which is the first step of butadiene synthesis from butanediols. The findings date back to the report by Sato et al. in 2003,158 and his group has been leading the research field of dehydration of diols.26,147
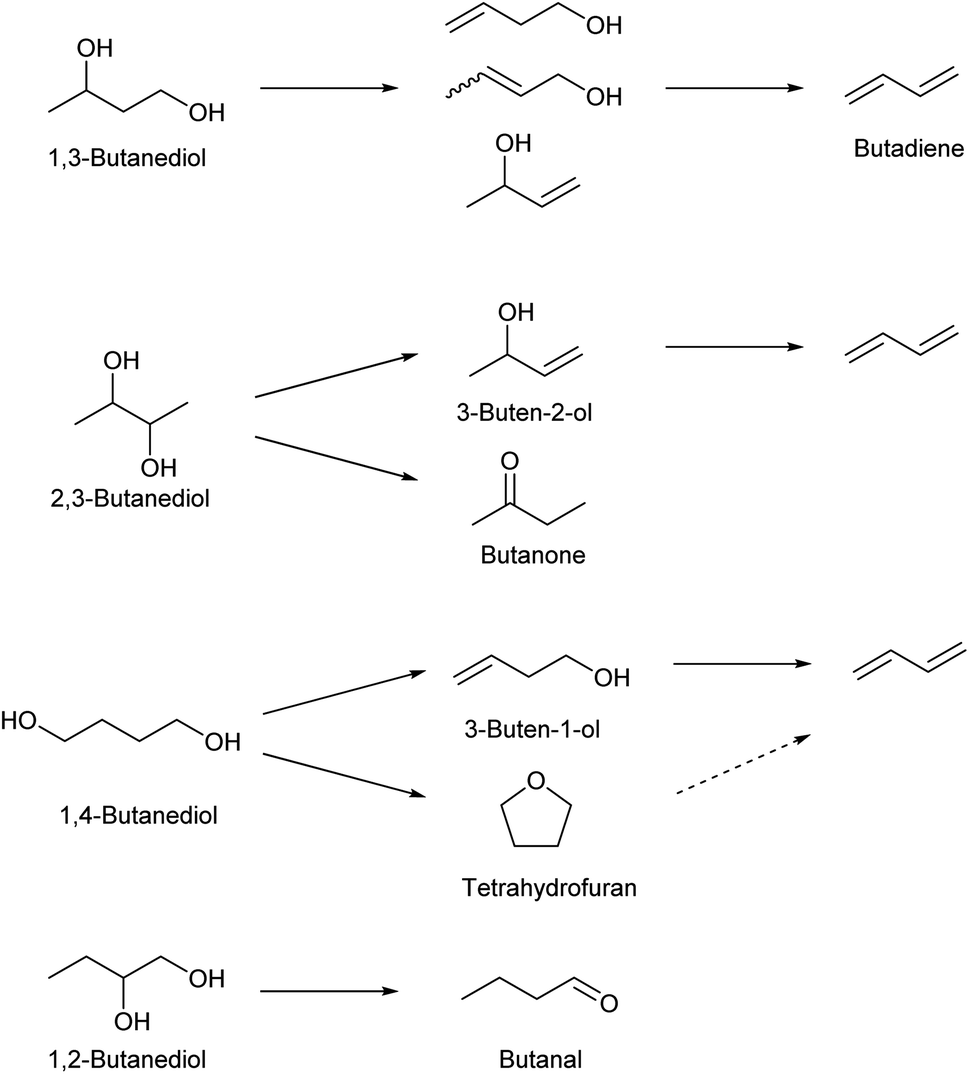 | ||
| Scheme 15 Dehydration of butanediols. Each arrow represents removal of one water molecule. | ||
1,3-Butanediol is the easiest isomer to be converted to butadiene because the dehydration itself does not generate by-products. 1,3-Butanediol dehydration proceeds over both strong acid catalysts and acid–base bifunctional catalysts. Strong acid catalysts such as zeolites tend to accompany propylene formation as a side reaction, and the selectivity to butadiene is around 60%.159,160 Bifunctional catalysts can suppress C–C dissociation reactions, and high yield (>90%; over Y2Zr2O7 catalyst) has been reported, although the reaction temperature (≥633 K) was higher than that of the systems of strong acid catalysts (around 573 K).161,162
Dehydration of 2,3-butanediol has been more intensively investigated because the fermentative production of 2,3-butanediol is relatively easy. However, simple acid-catalyzed dehydration of 2,3-butanediol produces butanone (frequently called as methyl ethyl ketone or MEK) rather than butadiene or 3-buten-2-ol intermediate.163 Dehydration to 3-buten-2-ol or butadiene requires removal of hydrogen at the terminal positions (1- and 4-). To obtain such selectivity, the reaction should be driven by a base instead of acid, and the base site is better to be sterically hindered to control the accessibility toward the terminal positions. Solid acid–base bifunctional catalysts with basic nature are effective, such as scandium oxide.163 The yield of butadiene reached about 90% with scandium oxide-based catalysts.164 Metal phosphate catalysts have been also tested. Typical phosphate catalysts are less selective than scandium oxide catalysts,165–167 while one relatively old report using CsH2PO4/SiO2 showed a very high 92% yield.168 The highest yield (94%) was obtained with a dual bed system composed of Sc2O3 and Al2O3 catalysts.169
Dehydration of 1,4-butanediol has been also investigated, since 1,4-butanediol can be produced either biologically or chemically. Both biomass and fossil resources can be the raw material for the chemical synthesis of 1,4-butanediol. However, the dehydration of 1,4-butanediol to butadiene is not easy. Brønsted acid catalysts easily cyclize 1,4-butanediol to tetrahydrofuran which is much less reactive than the substrate.170 Acid–base bifunctional catalysts can promote the CC bond formation. Because the hydrogen atoms that are removed by the base are located at inner positions, activation of more accessible hydroxy groups at the terminal positions is necessary. Therefore, the balance of acidity and basicity and/or even the positions of these sites much affect the catalytic performance.171 Rare-earth metal oxides have been reported to be effective such as CeO2 (ref. 170) and Yb2O3.161,172 Yb2O3 catalysts show excellent yields of butadiene (commercial Yb2O3: 96.6% at 633 K;161 Yb2O3 prepared through hydrothermal aging: 97.4% at 723 K (ref. 172)). The high yields were maintained at a time-on-stream of 10 h (≥95%). There was a slight increase of tetrahydrofuran selectivity at long time on stream, which is due to the accumulation of acidic carbonaceous material. The two-step method, 1,4-butanediol to 3-buten-1-ol as the first step and 3-buten-1-ol to butadiene as the second step, is an option to obtain good performance including long catalyst life.147,148 The catalyst for the first step should have weak acidity in order to suppress the cyclization to tetrahydrofuran. A high reaction temperature (typically >623 K) is necessary. In the second step, the negative effect of acidity on the selectivity is lower than that in the first step. Acidic catalysts can be used for the second step, enabling the decrease in the reaction temperature.
As described above, tetrahydrofuran can be easily obtained by acid-catalyzed dehydration of 1,4-butanediol. Other production methods of renewable tetrahydrofuran include furfural decarbonylation to furan followed by hydrogenation173 and 1,4-anhydroerythritol didehydroxylation (one-step deoxydehydration + hydrogenation).120–122 Dehydration of tetrahydrofuran to butadiene is possible with strong acids but very difficult. The ring-opening of tetrahydrofurans involves carbocation intermediates; however, from tetrahydrofuran the intermediates are unstable primary carbocation species. Tetrahydrofuran is much less reactive than its substituted forms at the 2-position such as 2-methyltetrahydrofuran and 2,5-dimethyltetrahydrofuran.22,174 The reaction temperature is high (around 623 K) among acid-catalyzed dehydration reactions because of the low reactivity of tetrahydrofuran. The high temperature and strong acidity of the catalyst promote various side reactions including coke formation, and catalyst stability is a serious issue. Recently, the phosphorous-containing siliceous self-pillared pentasil catalyst175 and ZrO2 catalyst174,176 have been reported to show high selectivity to butadiene (about 90% around 50% conversion level) at 623 K and 673 K, respectively. Both catalysts showed significant deactivation in a few hours of time on stream.
Deoxydehydration of erythritol is another production route of renewable butadiene. Erythritol is a more oxidized compound than butanediols and its fermentative production is easier. Double deoxydehydration of erythritol gives butadiene. Because of the importance of butadiene in the chemical industry, erythritol is often selected as a substrate in the reports of deoxydehydration systems including the first report of catalyzed deoxydehydration published in 1996.177 We have recently reported the deoxydehydration of erythritol with the same heterogeneous catalyst system for allyl alcohol production from glycerol and H2.115 The ReOx–Ag/CeO2 catalyst is the most effective one among investigated catalysts, and the yield reached 86% in 1,4-dioxane solvent. The reaction can be carried out even without solvent. The melt substrate (erythritol) and intermediates (butenediols) work as solvent, and the formed butadiene easily moves into the gas phase. The overhydrogenation of butadiene can be suppressed until the total conversion of butenediols. Although the reaction rate without solvent is lower than that with solvent, the selectivity can be higher without solvent, and the maximum yield of butadiene reached 90% (Fig. 1). The solventless condition gives a very good E-factor as low as 3.16, and this value is much lower than those of the other reported systems of deoxydehydration of erythritol to butadiene.
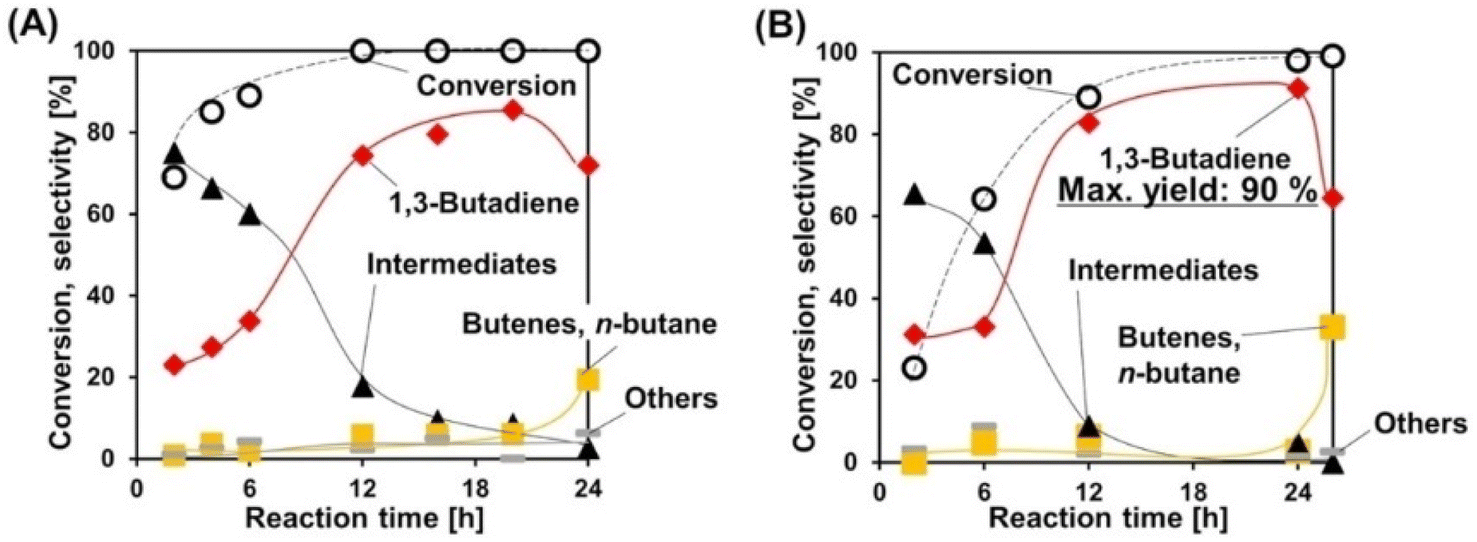 | ||
| Fig. 1 Time courses in the deoxydehydration of erythritol over ReOx–Ag/CeO2 (A) with 1,4-dioxane solvent and (B) without solvent. Reaction conditions: ReOx–Ag/CeO2 (Re: 1 wt%, Ag/Re = 0.3 [mol mol−1]) 0.3 g, erythritol 0.25 g, 1,4-dioxane 4 g (A) or 0 g (B), 413 K, H2 8 MPa. Reprinted from ref. 115 with permission from Wiley-VCH. | ||
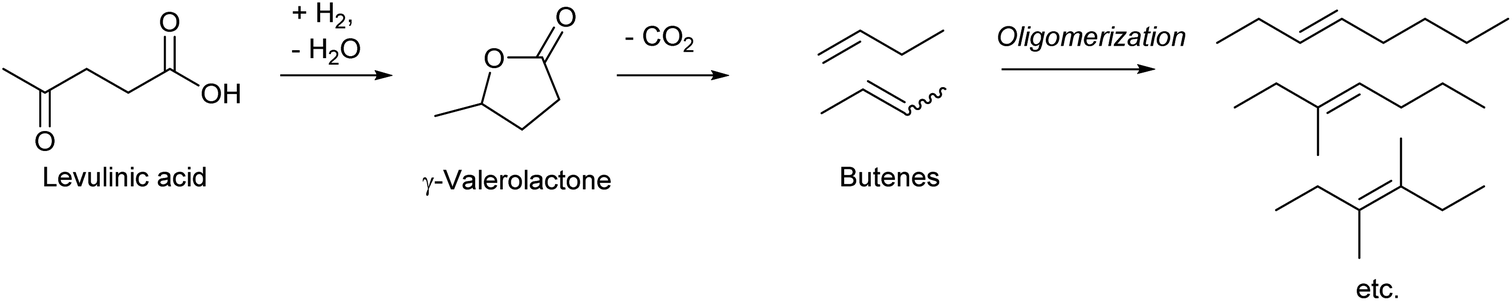
Dumesic et al. reported an epoch-making method for the production of biofuel via butenes as intermediates in 2010 (Scheme 16).195 A key step in the process is the decarboxylation of γ-valerolactone to butenes over the SiO2–Al2O3 catalyst. The yield reached 93% at 648 K, with the 1-/2-butene ratio of 33/67. Similar decarboxylation of γ-valerolactone to butenes over SiO2–Al2O3 catalysts has been reported by several research groups, and the reported yields were similarly high.196–199 The work by Mingyue Ding et al. reported more detailed distribution of produced butenes over SiO2–Al2O3:198 isobutene:1-butene:trans-2-butene:cis-2-butene = 2.5:22.7:43.6:31.2 at 648 K. Lower temperature decreased the ratio of isobutene (2.5% → 0.5% at 573 K) and increased that of trans-2-butene (43.6% → 46.6%). Recently, Sihai Yang et al. reported decarboxylation of γ-valerolactone to butenes over the Nb, Al-incorporated MFI zeolite catalyst.200 The total yield of butenes reached 99.3% (at 593 K). The isomer ratio at a 30% conversion level was isobutene:1-butene:trans-2-butene:cis-2-butene = 18:19:38:25. The ratio was more similar to the equilibrium level (45:12:27:16) than that obtained over SiO2–Al2O3, probably due to the stronger acidity of Nb, Al-incorporated zeolite. The raw sources of γ-valerolactone are sugars, cellulose and hemicellulose, and levulinic acid is a common key intermediate.201,202 The synthesis of γ-valerolactone has been intensively investigated and is becoming more and more practical.
 | ||
| Scheme 16 Biofuel synthesis via butenes. | ||
Although there are various production routes of butenes from biomass-derived platforms, all of them are energy-consuming processes because of the very low oxidation level of butenes. This contrasts with the low price of butenes in petrochemistry. Among the current utilization of butenes, the use for the synthesis of oxygenated chemicals should be replaced with direct synthesis of target oxygenated chemicals from biomass by partial deoxygenation, rather than bio-butene conversions.
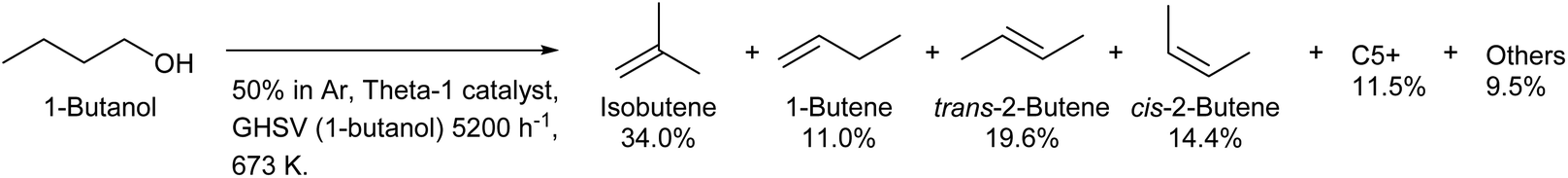
Because both lignocellulose and typical triglycerides do not have branches in the carbon chain, the production of branched compounds from biomass on a large scale is generally difficult. Only isobutyl alcohol (2-methyl-1-propanol) is produced commercially by fermentation as a branched C4 compound from biomass.208 Direct fermentative production of isobutene has been intensively investigated, and this method has the merit of easy separation.209,210 However, the efficiency of fermentation is limited because isobutyl alcohol and isobutene are less oxidized compounds. The dehydration of isobutyl alcohol to isobutene can proceed over simple acid catalysts such as H-ZSM-5 (ref. 211) and SiO2–Al2O3.212 The isomerization to linear butenes occurs to some extent, and strong acidity promotes this side reaction. Over 90% selectivity can be obtained with appropriate catalysts and conditions.212 Inversely, the production of isobutene from linear butanol by dehydration–isomerization is possible,213,214 although the isobutene formation is limited by equilibrium (theoretical limit: around 40%).200 Over 30% yield of isobutene from 1-butanol with good stability was obtained by using the Theta-1 zeolite catalyst at 673 K (Scheme 17).213,214 Theta-1 was selected by searching zeolites with 10-ring channels such as ferrierite, ZSM-23, ZSM-5 and SAPO-11; these channels are suitable for skeletal isomerization of linear alkenes with a monomolecular mechanism.
 | ||
| Scheme 17 1-Butanol conversion over the Theta-1 catalyst.214 | ||
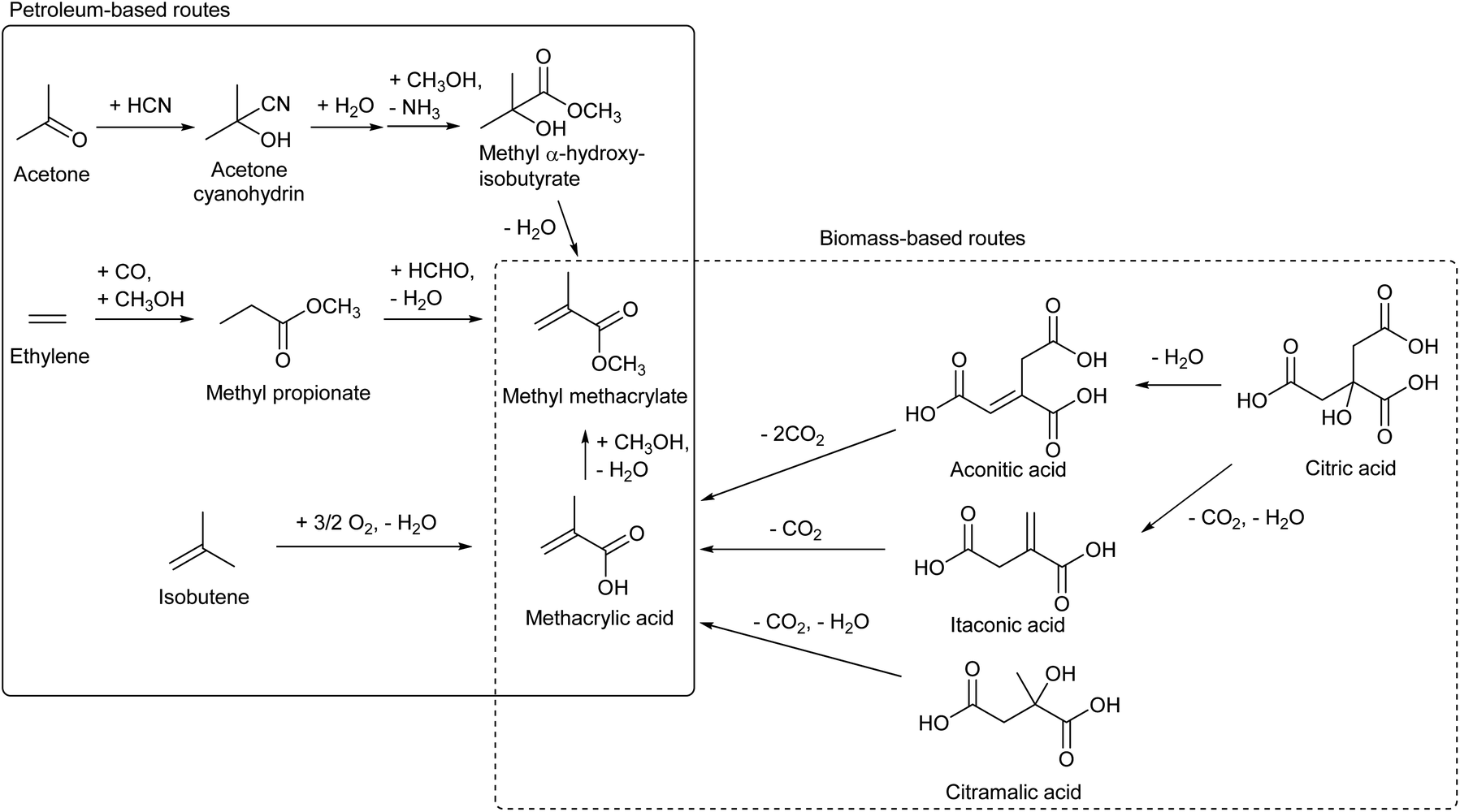
Methyl methacrylate is the monomer of PMMA, acrylic resin. The methacrylate unit is currently produced from various sources such as isobutene, ethylene (+CO, +HCHO) and acetone (+HCN) (Scheme 18).206,215 Bio-ethylene and bio-isobutene can be directly used in the current processes to replace petroleum. On the other hand, based on the principle that “from biomass, oxidized chemicals should be produced by partial deoxygenation rather than oxidation of hydrocarbons”, methacrylate synthesis from biomass without the need of oxidation is attractive. Decarboxylation reactions of itaconic acid,216–221 aconitic acid216 and citramalic acid222 have been proposed as such production methods of methacrylate. These carboxylic acids can be produced directly by fermentation or can be synthesized from citric acid which is a more common fermentation product.223 These reactions are not easy because it is necessary to suppress overdecarboxylation, and the reported yield of methacrylate was moderate (Table 4). The development of catalysts in these decarboxylation reactions is still in its infancy and better catalysts will be developed in the future.
 | ||
| Scheme 18 Syntheses of methacrylic acid and methyl methacrylate. | ||
| Entry | Substrate | Catalyst | S/C molar ratio | Solvent | Temp. [K] | Time [h] | Conv. [%] | Yield [%] | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Itaconic acid | Pt/C | 40 | Water, 1 eq. NaOH | 523 | 1 | n.r. | 68 | 216 |
| 2 | Itaconic acid | [Ru(CO)2(C2H5COO)]n + PPh3 | 1000 + 200 | Water | 498 | 1.5 | n.r. | 33.8 | 217 |
| 3 | Itaconic acid | NaOH | 5 | Water | 633 | 0.0075 | 99 | 75 | 218 |
| 4 | Itaconic acid | BaAl12O19 | 12 | Water | 523 | 3 | 100 | 50 | 219 |
| 5 | Itaconic acid | Pt/Al2O3 | 60 | Water + methanol, 1.6 eq. NaOH | 523 | 3 | 100 | 81.2 | 220 |
| 6 | Itaconic acid | Ca/Al2O3 | 20 | Water | 523 | 1.5 | 91.6 | 44.8 | 221 |
| 7 | Aconitic acid | Pt/Al2O3 | 40 | Water, 1 eq. NaOH | 523 | 1 | 100 | 47.5 | 216 |
| 8 | Citric acid | Pd/Al2O3 | 40 | Water, 1 eq. NaOH | 523 | 1 | 100 | 41 | 216 |
| 9 | Citramalic acid | Al2O3 | 0.44 (Al) | Water | 523 | 1 | 93.6 | 59.2 | 222 |
3.3. C5 and C6 compounds
Carbohydrates including sugars, hemicellulose and cellulose have C5 or C6 carbon chains. Therefore, there are many C5 and C6 biomass-derived compounds that can be used as intermediates of chemicals.4–6 Some of these compounds contain CC double bonds, such as furfural,203 HMF,224 levoglucosenone,225 angelica lactone,226 itaconic acid227,228 and muconic acid.229 The chemical conversions of these biomass-derived compounds have been well explained in recent review papers focusing on each compound. On the other hand, in the petrochemical industry, most C5 and C6 chemicals do not have olefinic (non-aromatic) CC double bonds. Unsaturated C5 and C6 chemicals, especially oxygenated ones, are mainly specialty chemicals. In this situation, we only focused on the C5 and C6 olefins without oxygen atoms in this section.
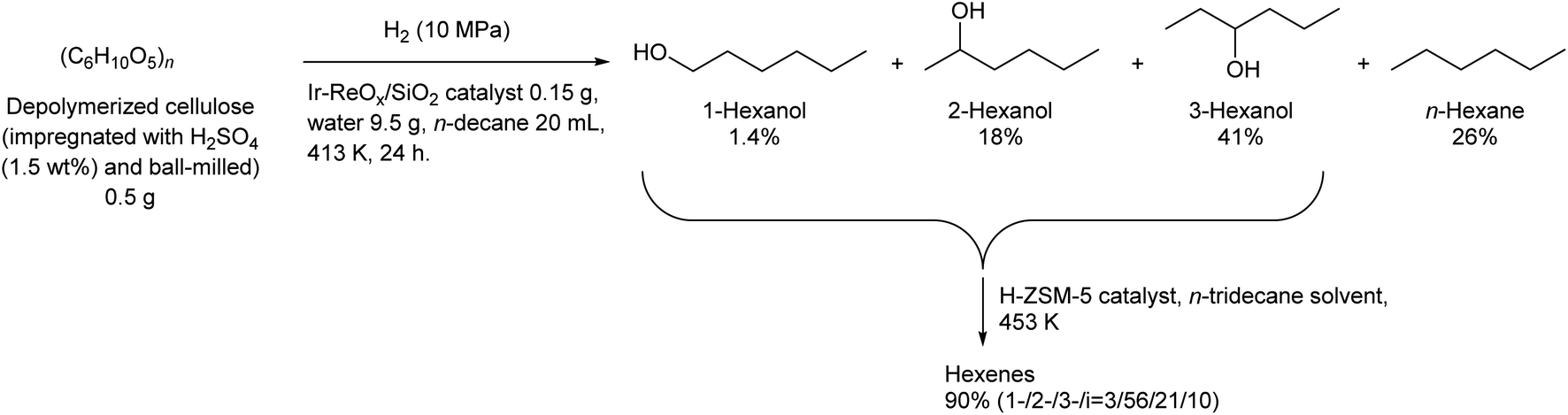
Some utilizations do not require high purity as a single isomer, such as direct use as gasoline and dimerization to jet-fuel-range hydrocarbons. Regioselectivity is not necessary in the dehydration step for this purpose, and simple Brønsted acid catalysts such as H-ZSM-5 show sufficient performance in the dehydration of secondary hexanols or pentanols.231,232 For the hydrodeoxygenation step, suppression of overhydrodeoxygenation to alkanes is a key point. Apart from the development of appropriate catalysts, the use of a biphasic solvent system is effective where the produced mono-alcohols are extracted from polar reaction solvent (typically water) to nonpolar solvent. The Ir–ReOx/SiO2 catalyst combined with H2O/alkane biphasic solvent gave 60% yield of hexanols by hydrodeoxygenation of depolymerized cellulose (Scheme 19).233 The same system was also applied to hydrodeoxygenation of xylan (hemicellulose); however, the obtained yield of pentanols was lower (≤32%).234 Deoxydehydration can be also used for the synthesis of mono-alcohols from sugar alcohols with an odd carbon number, namely xylitol. Xylitol can be converted to a mixture of 1-pentanol and 3-pentanol in 97% yield from xylitol over the ReOx–Pd/CeO2 catalyst which catalyzes both deoxydehydration and hydrogenation (Scheme 20).120 This catalyst is a variant of ReOx–Ag/CeO2 and ReOx–Au/CeO2 explained in the above sections for glycerol and erythritol conversions. The difference between ReOx–Ag/CeO2 and ReOx–Au/CeO2 is the high hydrogenation activity of Pd. This deoxydehydration + hydrogenation approach cannot be applied to sorbitol conversion to hexanols because each deoxydehydration step removes two oxygen atoms at once.
 | ||
| Scheme 19 Synthesis of hexanols and hexenes from cellulose by hydrodeoxygenation and dehydration.232,233 | ||
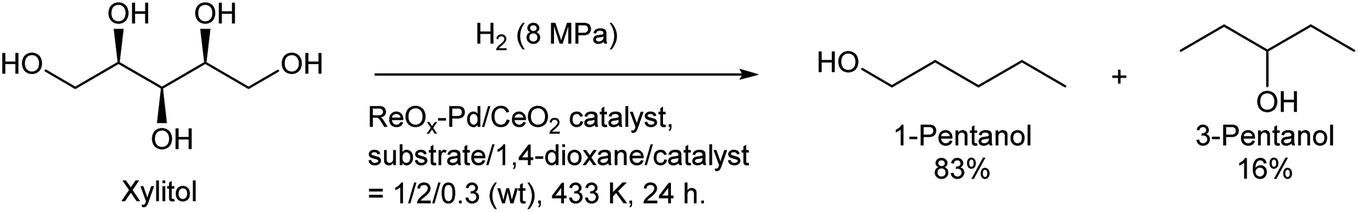 | ||
| Scheme 20 Synthesis of pentanols from xylitol by deoxydehydration + hydrogenation over the ReOx–Pd/CeO2 catalyst.120 | ||
:1.4- = 81:19, at 623 K).243 The exploration of catalysts has been much less carried out than the dehydration of butanediols. Dehydration of hexanediols to conjugated hexadiene has been rarely investigated244 because it is difficult to synthesize one specific hexanediol with appropriate positions of OH groups from biomass.
On the other hand, 2-methyltetrahydrofuran and 2,5-dimethyltetrahydrofuran can be relatively easily synthesized from biomass via hydrodeoxygenation of furfural and 5-hydroxymethylfurfural (HMF), respectively.245,246 Furthermore, dehydration of these methyl-substituted tetrahydrofurans is easier than that of simple tetrahydrofuran because the formation of the unstable primary carbocation is not necessary.22,174 Over 80% yields of 1,3-pentadiene have been reported for 2-methyltetrahydrofuran dehydration: 86% yield over the B-MWW catalyst at 658 K,247 92% yield over the ZrO2 catalyst at 723 K (ref. 248) and 91% yield over the AlNb-MCM-41 catalyst at 548 K.249 Although the reaction conditions are less severe than those for tetrahydrofuran dehydration, mild catalyst deactivation has been still observed. Dehydration of 2,5-dimethyltetrahydrofuran has been less investigated than that of 2-methyltetrahydrofuran including the distribution of product isomers; however, several comparative studies showed that the reactivity of 2,5-dimethyltetrahydrofuran is surely higher than those of tetrahydrofuran and 2-methyltetrahydrofuran.22,174 Very recently, Crossley et al. reported dehydration of 2,5-hexanediol over the H-ZSM-5 catalyst at 523 K.250 The product at short contact time was 2,5-dimethyltetrahydrofuran, and at longer contact time hexadienes were produced. Although it was not tested to fully convert 2,5-dimethyltetrahydrofuran with sufficiently long contact time, the selectivity to hexadienes ([hexadienes]/([all products] − [2,5-dimethyltetrahydrofuran])) was higher than 90%.
In comparison with the synthesis of other olefins, deoxydehydration is not a strong tool in the synthesis of pentadienes and hexadienes, because each corresponding substrate (pentanetetraol and hexanetetraol) cannot be efficiently synthesized at present.251 Production of hexatriene by deoxydehydration of sorbitol has been already reported, but not with H2 as the reducing agent.111,113
The number of studies for the synthesis of pentadienes and hexadienes has not been large. This is due to the relatively low importance of these compounds in petrochemistry, and the low importance is due to the low yield of these dienes in petrorefinery. On the other hand, the potential of these dienes in biomass conversion is not low.
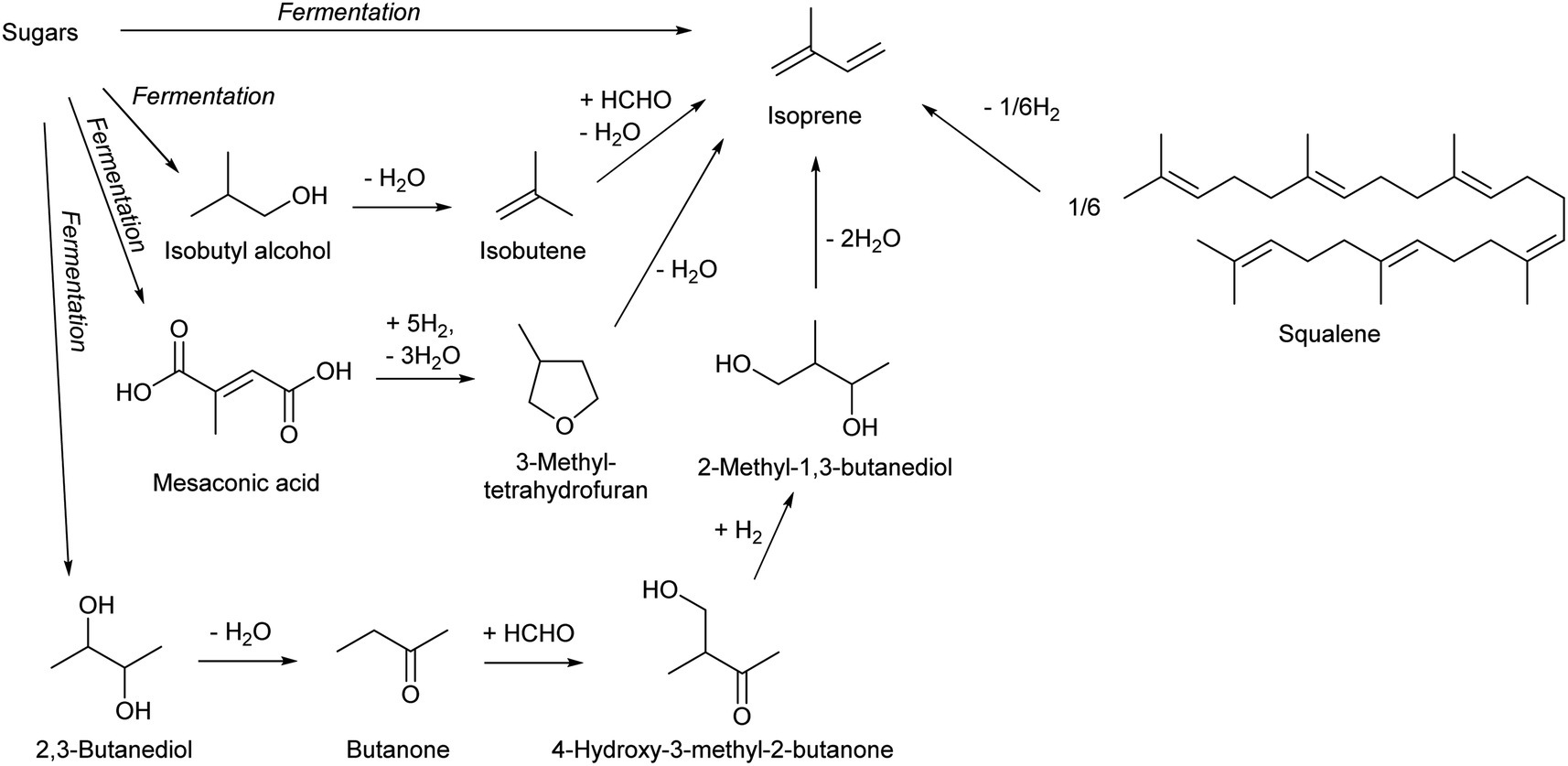 | ||
| Scheme 21 Synthesis of isoprene from biomass. | ||
It is well known that thermal decomposition of natural rubber (cis-1,4-polyisoprene) in an inert atmosphere produces isoprene.260 This reaction is not practical because natural rubber is more valuable than isoprene. Syntheses of isoprene by decomposition of other natural isoprenoids such as limonene and turpentine are also possible, which were mainly investigated in the first half of the 20th century,261 but these are also not practical because of the high price of feedstock. Much more recently, large isoprenoid hydrocarbon molecules produced by some microalgae have attracted attention.262 Typical such molecules are botryococcene (C30–37)263 and squalene (C30).264 Thermal cracking of squalene to isoprene has been reported recently, and about 50% yield (at 723 K) was obtained.265 Because the carbon chain of botryococcene does not consist of pure isoprene units, the synthesis of isoprene by decomposition of botryococcene is not realistic.
Compounds with a cyclopentane ring have been less used in industry than those with a cyclohexane ring. Nevertheless, cyclopentadiene is a relatively important chemical used as a co-monomer and a precursor of various fine chemicals.271 Cyclopentadiene has been isolated from naphtha cracking products or coal tar. The synthesis of cyclopentadiene from biomass requires the chemical formation of the cyclopentane ring because the cyclopentane ring is rare in natural compounds. A useful reaction to produce a cyclopentane ring is intramolecular aldol condensation of 1,4-diones.272 The dione structure can be formed by hydrolysis of the furan ring which is easily obtained by dehydration of sugars. Direct formation of cyclopentane ring structure from furans is possible, known as Piancatelli rearrangement.273,274 Tao Zhang et al. reported the stepwise synthesis of cyclopentadiene from biomass (Scheme 22).275 The starting compound is furfuryl alcohol which is obtained by dehydration of hemicellulose and hydrogenation and has been already synthesized on an industrial scale. Furfuryl alcohol is converted to 4-hydroxy-2-penten-1-one by aqueous phase rearrangement in the presence of a base. The hydrogenation of this unsaturated cyclic compound over the conventional Ni catalyst gives 1,3-cyclopentanediol, and its dehydration over acidic zeolite gives cyclopentadiene. The overall yield from furfuryl alcohol reached 67.5%, where the dehydration step was repeated with the recovery of the partially dehydrated product and the residual diol. As a related synthesis, that of methylcyclopentadiene has been also reported (Scheme 23).276–278 The starting compound is 2,5-hexanedione which can be obtained from hexoses such as glucose and its polymer, cellulose, via 2,5-dimethylfuran or 5-(chloromethyl)furfural. Intramolecular aldol condensation of 2,5-dimethylfuran gives 3-methyl-2-cyclopenten-1-one. Reduction and dehydration of this cyclic compound produce 1-methyl-1,3-cyclopentadiene. The yield of 1-methyl-1,3-cyclopentadiene from 2,5-dimethylfuran reached 68% with direct conversion over ZnO–MoOx catalysts.277 Zinc oxide helps the formation of oxygen vacancy in the MoOx phase, and the oxygen vacancy activates the C–O bond in the 3-methyl-2-cyclopenten-1-one intermediate. Improvement of efficiency and catalyst stability for the dehydration step is desirable for both systems.
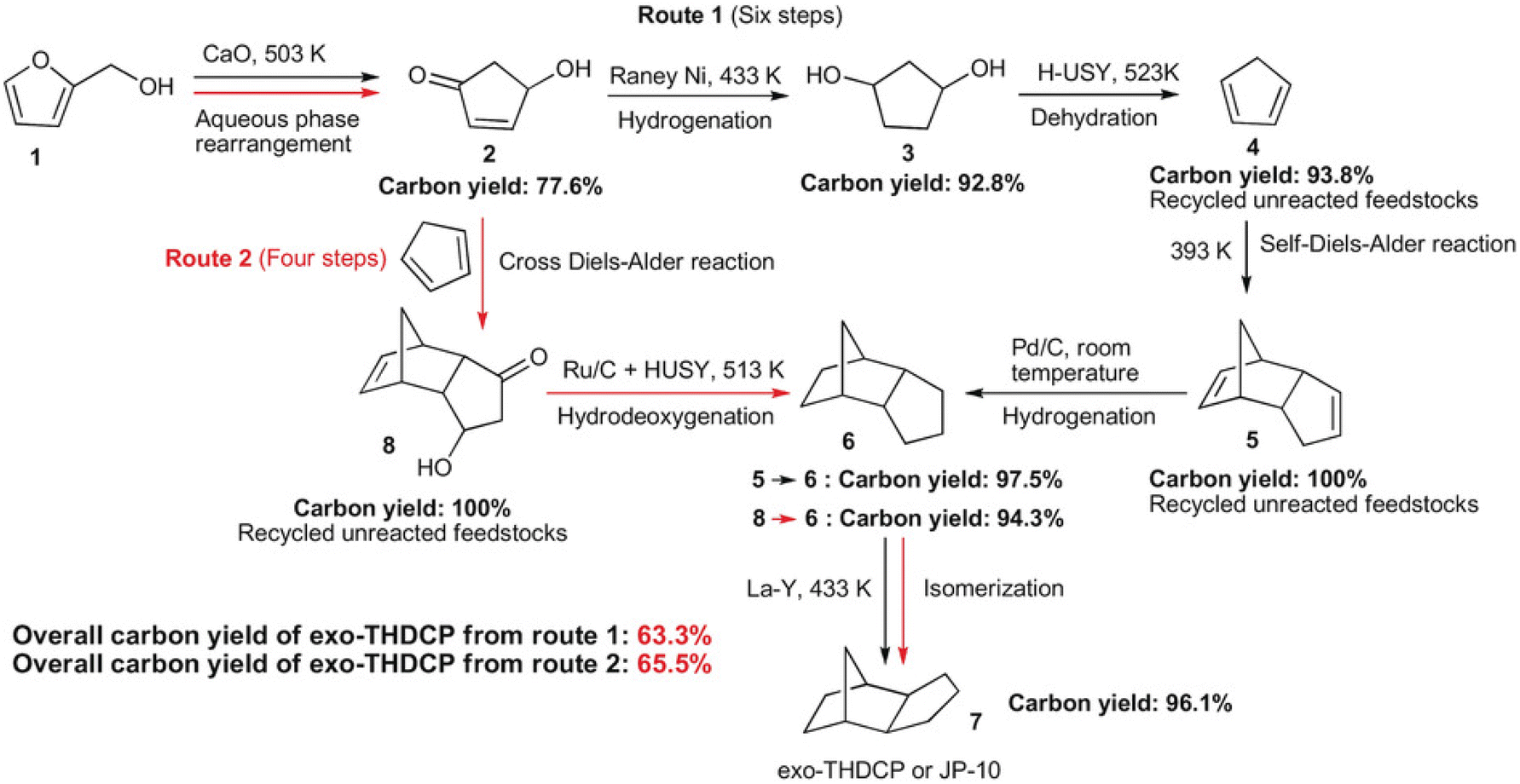 | ||
| Scheme 22 Synthesis of cyclopentadiene and the dimers from furfuryl alcohol. Reprinted from ref. 275 with permission from Wiley-VCH. | ||
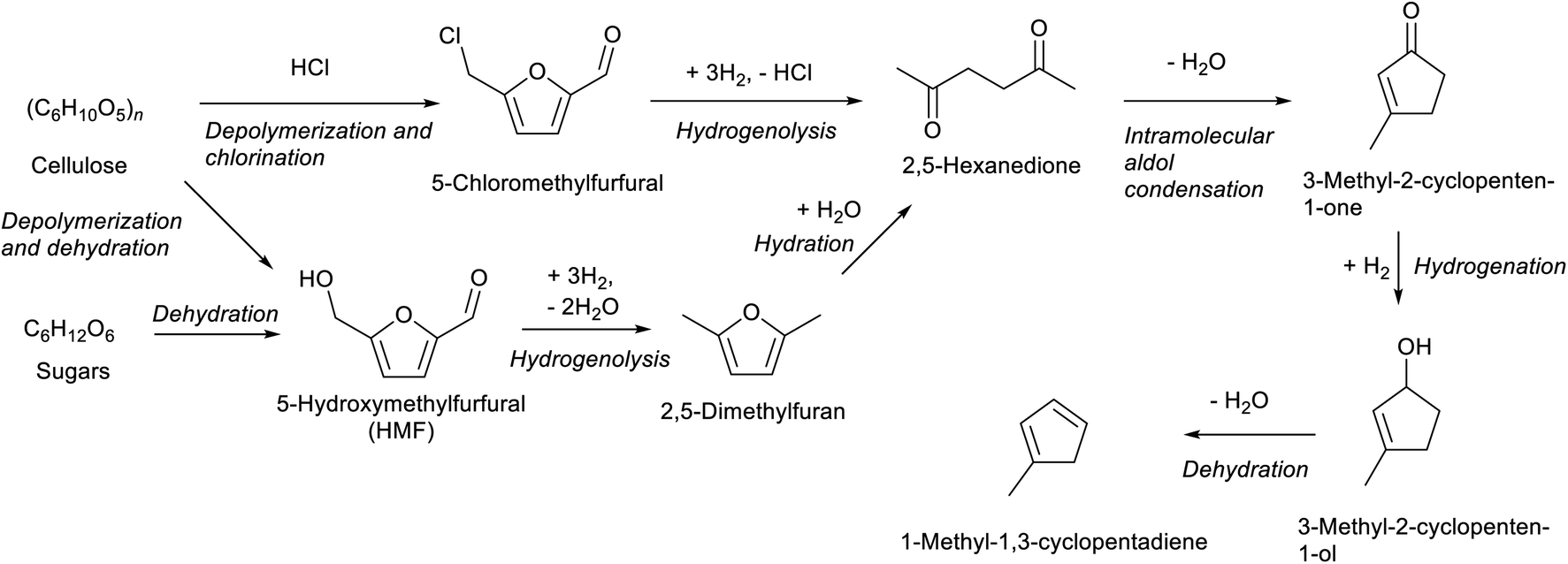 | ||
| Scheme 23 Synthesis of methylcyclopentadiene from biomass. | ||
3.4. Larger compounds
Olefinic compounds with ≥7 carbon number are specialty chemicals except alpha-olefins which can be obtained by ethylene oligomerization. As described in the section for 1-hexene, the same alpha-olefins can be synthesized by simply replacing the substrate with bio-ethylene. Another method to obtain alpha-olefins is the use of triglycerides as raw material (Scheme 24). Hydrogenation of triglycerides or free fatty acids can produce primary alcohols, and alpha-olefins can be obtained by dehydration. Catalytic hydrogenation of esters or carboxylic acids to alcohols has a long history, but there is still a large room for improvement.255–257 Decarbonylative dehydration of saturated fatty acids can also produce alpha-olefins.41,42 Hydrogenation of fatty acids or triglycerides removes the CC double bonds in the carbon chain. If one wants to utilize the CC double in the carbon chain, decarboxylation seems to be necessary. However, because of the requirement of severe conditions for the decarboxylation, it is difficult to selectively convert an unsaturated fatty acid to the corresponding olefin, such as oleic acid to cis-8-heptadecene, without isomerization, cracking or coking.279,280 Metathesis is a useful reaction for further conversion of the internal olefins which will be produced by decarboxylation of unsaturated fatty acids.281,282
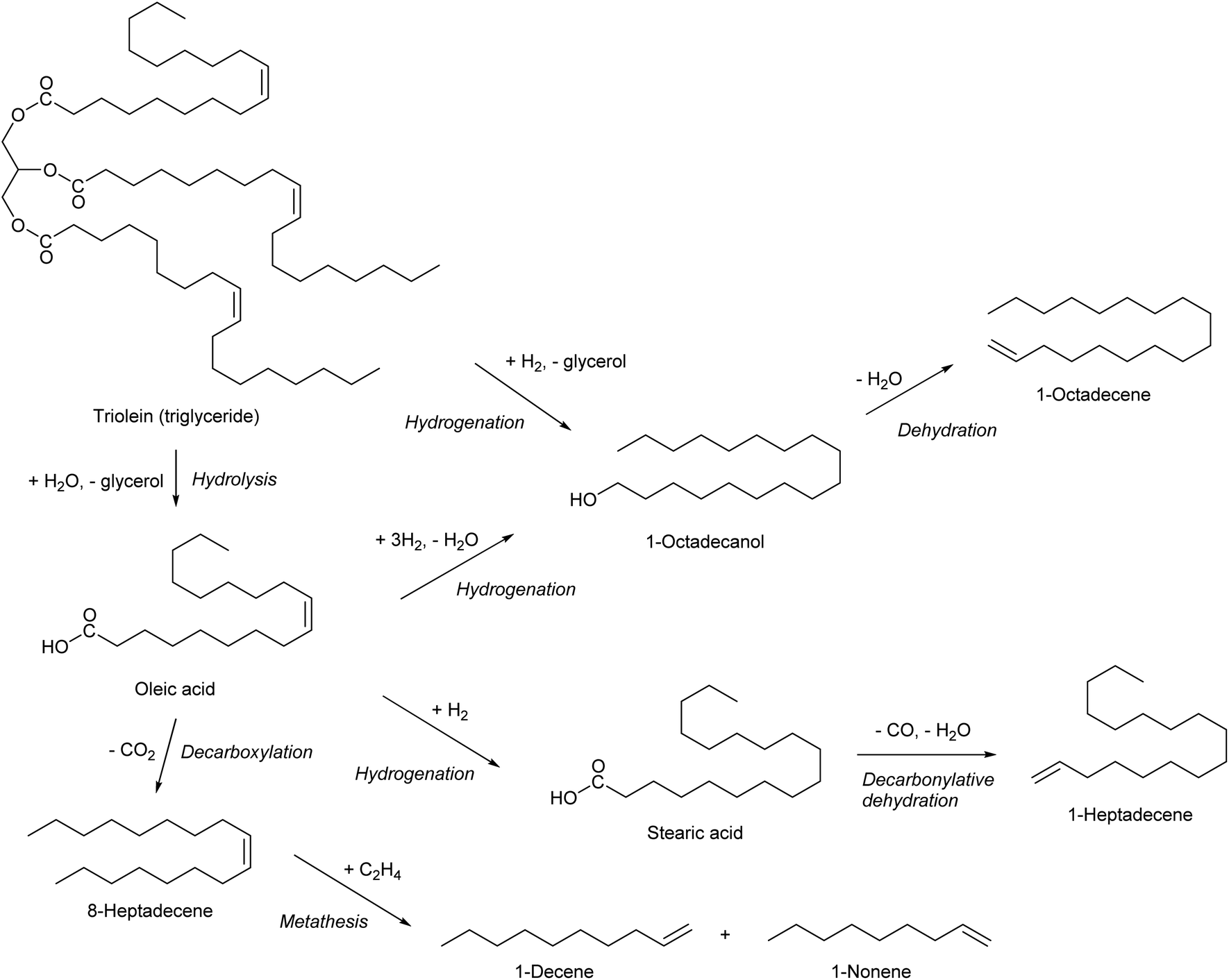 | ||
| Scheme 24 Synthesis of alpha-olefins from triolein. | ||
4. Conclusions and outlook
Most olefinic compounds used in the chemical industry can be now synthesized from biomass. Ethylene can be synthesized by dehydration of ethanol, C3 unsaturated oxygenates can be produced from glycerol, and C4 olefinic compounds can be chemically synthesized from C4 oxygenates obtained by fermentation. Cyclopentadiene production from biomass has been also reported. Catalytic dehydration is frequently used in these syntheses. Conventional Brønsted acids such as acidic zeolites can be used only in limited cases, and recently reported systems tend to use acid–base bifunctional solid catalysts such as rare-earth oxides and alkali metal salts of zeolites. The bifunctional catalysts can suppress cyclization reactions (to tetrahydrofurans and tetrahydropyrans) and rearrangement reactions of the carbon chain. Aldehyde formation and movement of the double bonds are frequently observed in dehydration over bifunctional catalysts. Tuning of catalyst properties is usually necessary for each target reaction, such as the balance of acidity and basicity as well as the pore structure. Zeolite-based catalysts including the alkali-metal salts are advantageous to precisely control the structure and properties. The control of properties of rare-earth metal oxide catalysts is more difficult; however, there is much room for improvement. In particular, mixed metal oxides containing rare-earth metals have not been explored well. Considering the large number of possible combinations, catalyst informatics may be a strong tool for the future development of rare-earth-metal-based catalysts. Another important dehydration is that of tetrahydrofurans. This reaction is catalyzed by simple acids, but the reported performance, especially for tetrahydrofuran to butadiene, is not sufficient and needs to be improved. Catalyst stability is always a major issue in dehydration over solid acid catalysts since coke formation is inevitable from olefinic compounds over solid acids. Suppressing the activity decrease during time on stream and stability during regeneration are always major topics for catalyst development. For rare-earth metal oxide catalysts, pure compounds synthesized by the calcination of precursors have been mainly used. Appropriate supports will be necessary to improve the stability during the catalytic use and regeneration.Deoxydehydration is another reaction that produces olefins from biomass-derived oxygenates. Although deoxydehydration has been less investigated than dehydration, recently developed catalysts, namely ReOx–M/CeO2 (M = Au or Ag), can very selectively produce olefins from vicinal diols. In contrast to dehydration, tuning of catalyst properties for each substrate is almost unnecessary in deoxydehydration. Although catalyst cost and long-term stability are necessary to be considered, when deoxydehydration is practically carried out, such a catalytic system has great potential to be applicable to a wide range of substrates.
Depending on the structure of target olefinic products, selective partial hydrodeoxygenation and decarboxylation reactions are key steps. These reactions are also very important in the selective synthesis of saturated compounds from biomass, and the current intensive investigation should be continued to obtain higher performance including high selectivity, long catalyst life, and low cost of catalyst.
Because of the difference in oxygen content between petroleum and biomass, simple olefins are generally easier to obtain from petroleum and difficult from biomass. This trend is most prominent in the case of propylene. Propylene is an inexpensive olefin and is utilized on a very large scale in the chemical industry. On the other hand, the production of propylene from biomass requires a large amount of reducing agents and multiple steps. The supply of the C3 biomass source is much smaller than the amount of propylene used in the chemical industry. Renewable propylene is a missing piece of the puzzle of biomass-derived chemicals. Without a drastic breakthrough such as direct high-yield synthesis of propylene by reduction of CO or CO2, the use of propylene will be much decreased when fossil resources cannot be used. Oxidized compounds are better to be synthesized by deoxygenation than oxidation of simple olefins, especially propylene, in the biomass-based chemical industry.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by JSPS KAKENHI 18H05247 and 20H02519.References
- S. B. Zdonik, E. J. Bassler and L. P. Hallee, Hydrocarbon Process., 1974, 53, 73–81 CAS.
- Shinpan Sekiyu Purosesu (Petrochemical Process, New Edition), ed. M. Matsukata, Japan Petroleum Institute, Kodansha, Tokyo, 2018, in Japanese Search PubMed.
- Z. Belohlav, P. Zamostny and T. Herink, Chem. Eng. Process., 2003, 42, 461–473 CrossRef CAS.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- A.-L. Marshall and P. J. Alaimo, Chem.–Eur. J., 2010, 16, 4970–4980 CrossRef CAS PubMed.
- K. Tomishige, M. Yabushita, J. Cao and Y. Nakagawa, Green Chem., 2022, 24, 5652–5690 RSC.
- M. Besson, P. Gallezot and C. Pinel, Chem. Rev., 2014, 114, 1827–1870 CrossRef CAS PubMed.
- K. Avasthi, A. Bohre, M. Grilc, B. Likozar and B. Saha, Catal. Sci. Technol., 2020, 10, 5411–5437 RSC.
- G. Hayes, M. Laurel, D. MacKinnon, T. Zhao, H. A. Houck and C. R. Becer, Chem. Rev., 2023, 123, 2609–2734 CrossRef CAS PubMed.
- A. Mohsenzadeh, A. Zamani and M. J. Taherzadeh, ChemBioEng Rev., 2017, 4, 75–91 CrossRef.
- J. H. Clark, Green Chem., 1999, 1, 1–8 RSC.
- H. C. Erythropel, J. B. Zimmerman, T. M. de Winter, L. Petitjean, F. Melnikov, C. H. Lam, A. W. Lounsbury, K. E. Mellor, N. Z. Janković, Q. Tu, L. N. Pincus, M. M. Falinski, W. Shi, P. Coish, D. L. Plata and P. T. Anastas, Green Chem., 2018, 20, 1929–1961 RSC.
- T. K. Phung and G. Busca, Chem. Eng. J., 2015, 272, 92–101 CrossRef CAS.
- J. B. Friesen and R. Schretzman, J. Chem. Educ., 2011, 88, 1141–1147 CrossRef CAS.
- B. J. Esselman, N. J. Hill and A. J. Ellison, J. Chem. Educ., 2021, 98, 3218–3225 CrossRef CAS.
- D. Zhang, S. A. I. Barri and D. Chadwick, Appl. Catal., A, 2011, 400, 148–155 CrossRef CAS.
- S. Ai, M. Zheng, Y. Jiang, J. Pang, C. Wang, A. Wang, X. Wang and T. Zhang, Chem. Eng. J., 2018, 335, 530–538 CrossRef CAS.
- V. Resch and U. Hanefeld, Catal. Sci. Technol., 2015, 5, 1385–1399 RSC.
- A. Yamaguchi, N. Hiyoshi, O. Sato and M. Shirai, Catal. Today, 2012, 185, 302–305 CrossRef CAS.
- V. M. Shinde, G. N. Patil, A. Katariya and Y. S. Mahajan, Chemical Engineering and Processing: Process Intensification, 2015, 95, 241–248 CrossRef CAS.
- B. Török, I. Bucsi, T. Beregszászi, I. Kapocsi and Á. Molnár, J. Mol. Catal. A: Chem., 1996, 107, 305–311 CrossRef.
- M. D. Kumbhalkar, J. S. Buchanan, G. W. Huber and J. A. Dumesic, ACS Catal., 2017, 7, 5248–5256 CrossRef CAS.
- S. Li, O. A. Abdelrahman, G. Kumar, M. Tsapatsis, D. G. Vlachos, S. Caratzoulas and P. J. Dauenhauer, ACS Catal., 2019, 9, 10279–10293 CrossRef CAS.
- H. Pines and J. Manassen, Adv. Catal., 1966, 16, 49–93 CAS.
- M. Kang and A. Bhan, Catal. Sci. Technol., 2016, 6, 6667–6678 RSC.
- S. Sato, F. Sato, H. Gotoh and Y. Yamada, ACS Catal., 2013, 3, 721–734 CrossRef CAS.
- Z. Fang, Y. Wang and D. A. Dixon, J. Phys. Chem. C, 2015, 119, 23413–23421 CrossRef CAS.
- R. Wang, Z. Zhao, P. Gao, K. Chen, Z. Gan, Q. Fu and G. Hou, J. Phys. Chem. C, 2022, 126, 10073–10080 CrossRef CAS.
- L. J. Donnelly, S. P. Thomas and J. B. Love, Chem.–Asian J., 2019, 14, 3782–3790 CrossRef CAS PubMed.
- N. N. Tshibalonza and J.-C. M. Monbaliu, Green Chem., 2020, 22, 4801–4848 RSC.
- M. Tamura, Y. Nakagawa and K. Tomishige, Catal. Sci. Technol., 2020, 10, 3805–3824 RSC.
- F. C. Jentoft, Catal. Sci. Technol., 2022, 12, 6308–6358 RSC.
- K. Tomishige, Y. Nakagawa and M. Tamura, Chin. Chem. Lett., 2020, 31, 1071–1077 CrossRef CAS.
- E. Arceo, P. Marsden, R. G. Bergman and J. A. Ellman, Chem. Commun., 2009, 3357–3359 RSC.
- A. M. Robinson, J. E. Hensley and J. W. Medlin, ACS Catal., 2016, 6, 5026–5043 CrossRef CAS.
- K. Tomishige, Y. Nakagawa and M. Tamura, Green Chem., 2017, 19, 2876–2924 RSC.
- S. Kim, E. E. Kwon, Y. T. Kim, S. Jung, H. J. Kim, G. W. Huber and J. Lee, Green Chem., 2019, 21, 3715–3743 RSC.
- Y. Nakagawa, M. Tamura and K. Tomishige, Res. Chem. Intermed., 2018, 44, 3879–3903 CrossRef CAS.
- G. J. S. Dawes, E. L. Scott, J. L. Nôtre, J. P. M. Sanders and J. H. Bitter, Green Chem., 2015, 17, 3231–3250 RSC.
- M. Mavrikakis and M. A. Barteau, J. Mol. Catal. A: Chem., 1998, 131, 135–147 CrossRef CAS.
- E. Santillan-Jimenez and M. Crocker, J. Chem. Technol. Biotechnol., 2012, 87, 1041–1050 CrossRef CAS.
- A. Chatterjee, S. H. H. Eliasson and V. R. Jensen, Catal. Sci. Technol., 2018, 8, 1487–1499 RSC.
- A. Fürstner, Angew. Chem., Int. Ed., 2000, 39, 3012–3043 CrossRef.
- J. C. Mol, J. Mol. Catal. A: Chem., 2004, 213, 39–45 CrossRef CAS.
- W. Yan, Z. You, K. Meng, F. Du, S. Zhang and X. Jin, Chin. J. Chem. Eng., 2022, 48, 44–60 CrossRef CAS.
- Q. Zhang, T. Otroshchenko and E. V. Kondratenko, Catal. Sci. Technol., 2022, 12, 5210–5216 RSC.
- P. Małecki, K. Gajda, R. Gajda, K. Woźniak, B. Trzaskowski, A. Kajetanowicz and K. Grela, ACS Catal., 2019, 9, 587–598 CrossRef.
- H. Xiong, H. N. Pham and A. K. Datye, Green Chem., 2014, 16, 4627–4643 RSC.
- I. Sádaba, M. L. Granados, A. Riisager and E. Taarning, Green Chem., 2015, 17, 4133–4145 RSC.
- H. S. Cerqueira, G. Caeiro, L. Costa and F. R. Ribeiro, J. Mol. Catal. A: Chem., 2008, 292, 1–13 CrossRef CAS.
- C. R. Chilakamarry, A. M. M. Sakinah, A. W. Zularisam, A. Pandley and D.-V. N. Vo, Environ. Technol. Innovation, 2021, 24, 101902 CrossRef CAS.
- M. Dusselier, P. V. Wouwe, A. Dewaele, E. Makshina and B. F. Sels, Energy Environ. Sci., 2013, 6, 1415–1442 RSC.
- M. A. Abdel-Rahman, Y. Tashiro and K. Sonomoto, Biotechnol. Adv., 2013, 31, 877–902 CrossRef CAS PubMed.
- R. A. de Oliveira, A. Komesu, C. E. V. Rossell and R. M. Filho, Biochem. Eng. J., 2018, 133, 219–239 CrossRef.
- S. Li, W. Deng, Y. Li, Q. Zhang and Y. Wang, J. Energy Chem., 2019, 32, 138–151 CrossRef.
- K. J. Jem and B. Tan, Adv. Ind. Eng. Polym. Res., 2020, 3, 60–70 Search PubMed.
- M. Kumar, Y. Goyal, A. Sarkar and K. Gayen, Appl. Energy, 2012, 93, 193–204 CrossRef CAS.
- M. Zheng, J. Pang, R. Sun, A. Wang and T. Zhang, ACS Catal., 2017, 7, 1939–1954 CrossRef CAS.
- A. N. Marchesan, M. P. Oncken, R. M. Filho and M. R. W. Maciel, Green Chem., 2019, 21, 5168–5194 RSC.
- L. Liu, X. P. Ye and J. J. Bozell, ChemSusChem, 2012, 5, 1162–1180 CrossRef CAS PubMed.
- J. C. Callahan, R. K. Grasselli, E. C. Milberger and H. A. Strecker, Ind. Eng. Chem. Prod. Res. Dev., 1970, 9, 134–142 CrossRef CAS.
- B. Katryniok, S. Paul, V. Bellière-Baca, P. Ray and F. Dumeignil, Green Chem., 2010, 12, 2079–2098 RSC.
- T. Ma, M. Yin, C. Su, N. Guo, X. Huang, Z. Han, Y. Wang, G. Chen and Z. Yun, J. Ind. Eng. Chem., 2023, 117, 85–102 CrossRef CAS.
- F. F. Barbosa and T. P. Braga, ChemCatChem, 2023, 15, e202200950 Search PubMed.
- H. Zhang, Z. Hu, L. Huang, H. Zhang, K. Song, L. Wang, Z. Shi, J. Ma, Y. Zhuang, W. Shen, H. Xu and Y. Tang, ACS Catal., 2015, 5, 2548–2558 CrossRef CAS.
- S. Zhao, W. D. Wang, L. Wang, W. Wang and J. Huang, J. Catal., 2020, 389, 166–175 CrossRef CAS.
- D. Sun, Y. Yamada, S. Sato and W. Ueda, Green Chem., 2017, 19, 3186–3213 RSC.
- E. V. Makshina, J. Canadell, J. van Krieken, E. Peeters, M. Dusselier and B. F. Sels, ChemCatChem, 2019, 11, 180–201 CrossRef CAS.
- X. Jin, K. Meng, G. Zhang, M. Liu, Y. Song, Z. Song and C. Yang, Green Chem., 2021, 23, 51–76 RSC.
- M. Y. Ahmad, N. I. Basir and A. Z. Abdullah, J. Ind. Eng. Chem., 2021, 93, 216–227 CrossRef CAS.
- E. M. Karp, T. R. Eaton, V. Sànchez i Nogué, V. Vorotnikov, M. J. Biddy, E. C. D. Tan, D. G. Brandner, R. M. Cywar, R. Liu, L. P. Manker, W. E. Michener, M. Gilhespy, Z. Skoufa, M. J. Watson, O. S. Fruchey, D. R. Vardon, R. T. Gill, A. D. Bratis and G. T. Beckham, Science, 2017, 358, 1307–1310 CrossRef CAS PubMed.
- D. Cespi, F. Passarini, G. Mastragostino, I. Vassura, S. Larocca, A. Iaconi, A. Chieregato, J.-L. Dubois and F. Cavani, Green Chem., 2015, 17, 343–355 RSC.
- Y. Matsuura, A. Onda, S. Ogo and K. Yanagisawa, Catal. Today, 2014, 226, 192–197 CrossRef CAS.
- B. Yan, L.-Z. Tao, Y. Liang and B.-Q. Xu, ACS Catal., 2014, 4, 1931–1943 CrossRef CAS.
- V. C. Ghantani, M. K. Dongare and S. B. Umbarkar, RSC Adv., 2014, 4, 33319–33326 RSC.
- B. Yan, L.-Z. Tao, Y. Liang and B.-Q. Xu, ChemSusChem, 2014, 7, 1568–1578 CrossRef CAS PubMed.
- G. M. Lari, B. Puértolas, M. S. Frei, C. Mondelli and J. Pérez-Ramírez, ChemCatChem, 2016, 8, 1507–1514 CrossRef CAS.
- L. Zhang, D. S. Theng, Y. Du, S. Xi, L. Huang, F. Gao, C. Wang, L. Chen and A. Borgna, Catal. Sci. Technol., 2017, 7, 6101–6111 RSC.
- J. Peng, X. Li, C. Tang and W. Bai, Green Chem., 2014, 16, 108–111 RSC.
- S. Lyu and T. Wang, RSC Adv., 2017, 7, 10278–10286 RSC.
- X. Li, Z. Chen, P. Cao, W. Pu, W. Zou, C. Tang and L. Dong, RSC Adv., 2017, 7, 54696–54705 RSC.
- B. M. Murphy, M. P. Letterio and B. Xu, J. Catal., 2016, 339, 21–30 CrossRef CAS.
- Z.-H. Liu, B. Yan, Y. Liang and B.-Q. Xu, Mol. Catal., 2020, 494, 111098 CrossRef CAS.
- E. V. Makshina, J. Canadell, J. van Krieken and B. F. Sels, ACS Sustainable Chem. Eng., 2022, 10, 6196–6204 CrossRef CAS.
- J. G. H. Hermens, A. Jensma and B. L. Feringa, Angew. Chem., Int. Ed., 2022, 61, e202112618 CrossRef CAS PubMed.
- N. Nagato, Allyl Alcohol and Monoallyl Derivatives, Kirk-Othmer Encyclopedia of Chemical Technology, 2004, DOI:10.1002/0471238961.0112122514010701.a01.pub2.
- X. Lan and T. Wang, ACS Catal., 2020, 10, 2764–2790 CrossRef CAS.
- P. Mäki-Arvela, J. Hájek, T. Salmi and D. Y. Murzin, Appl. Catal., A, 2005, 292, 1–49 CrossRef.
- M. Tamura, Y. Nakagawa and K. Tomishige, J. Jpn. Pet. Inst., 2019, 62, 106–119 CrossRef CAS.
- X. Wang, X. Liang, P. Geng and Q. Li, ACS Catal., 2020, 10, 2395–2412 CrossRef CAS.
- M. Lucas and P. Claus, Chem. Eng. Technol., 2005, 28, 867–870 CrossRef CAS.
- P. Aich, H. Wei, B. Basan, A. J. Kropf, N. M. Schweitzer, C. L. Marshall, J. T. Miller and R. Meyer, J. Phys. Chem. C, 2015, 119, 18140–18148 CrossRef CAS.
- M. Zhao, K. Yuan, Y. Wang, G. Li, J. Guo, L. Gu, W. Hu, H. Zhao and Z. Tang, Nature, 2016, 539, 76–80 CrossRef CAS PubMed.
- X. Lan, T. Wang, X. Li, N. Huang and J. Wang, Catal. Sci. Technol., 2016, 6, 7703–7707 RSC.
- R. A. Farrar-Tobar, A. Dell'Acqua, S. Tin and J. G. de Vries, Green Chem., 2020, 22, 3323–3357 RSC.
- M. Ueshima and Y. Shimasaki, Chem. Lett., 1992, 21, 1345–1348 CrossRef.
- M. Gliński and U. Ulkowska, Catal. Lett., 2011, 141, 293–299 CrossRef.
- M. R. Nanda, Z. Yuan, W. Qin and C. Xu, Catal. Rev.: Sci. Eng., 2016, 58, 309–336 CrossRef CAS.
- H. Zhao, L. Zheng, X. Li, P. Chen and Z. Hou, Catal. Today, 2020, 355, 84–95 CrossRef CAS.
- C. V. Pramod, R. Fauziah, K. Seshan and J.-P. Lange, Catal. Sci. Technol., 2018, 8, 289–296 RSC.
- Y. Liu, H. Tüysüz, C.-J. Jia, M. Schwickardi, R. Rinaldi, A.-H. Lu, W. Schmidt and F. Schüth, Chem. Commun., 2010, 46, 1238–1240 RSC.
- A. Konaka, T. Tago, T. Yoshikawa, A. Nakamura and T. Masuda, Appl. Catal., B, 2014, 146, 267–273 CrossRef CAS.
- G. Sánchez, B. Z. Dlugogorski, E. M. Kennedy and M. Stockenhuber, Appl. Catal., A, 2016, 509, 130–142 CrossRef.
- H. Lan, X. Xiao, S. Yuan, B. Zhang, G. Zhou and Y. Jiang, Catal. Lett., 2017, 147, 2187–2199 CrossRef CAS.
- H. S. Oliveira, P. P. Souza and L. C. A. Oliveira, Catal. Today, 2017, 289, 258–263 CrossRef CAS.
- R. Almeida, M. F. Ribeiro, A. Fernandes and J. P. Lourenço, Catal. Commun., 2019, 127, 20–24 CrossRef CAS.
- H. Lan, Q. Yao, Y. Zhou, B. Zhang and Y. Jiang, Mol. Catal., 2020, 498, 111279 CrossRef CAS.
- A. Kostyniuk, D. Bajec, P. Djinović and B. Likozar, Chem. Eng. J., 2020, 397, 125430 CrossRef CAS.
- H. Zhao, Y. Jiang, H. Liu, Y. Long, Z. Wang and Z. Hou, Appl. Catal., B, 2020, 277, 119187 CrossRef CAS.
- V. Canale, L. Tonucci, M. Bressan and N. d'Alessandro, Catal. Sci. Technol., 2014, 4, 3697–3704 RSC.
- J. Li, M. Lutz, M. Otte and R. J. M. K. Gebbink, ChemCatChem, 2018, 10, 4755–4760 CrossRef CAS PubMed.
- M. Lupaccini, A. Mascitti, V. Canale, L. Tonucci, E. Colacino, M. Passacantando, A. Marrone and N. d'Alessandro, Catal. Sci. Technol., 2019, 9, 3036–3046 RSC.
- M. Shiramizu and F. D. Toste, Angew. Chem., Int. Ed., 2012, 51, 8082–8086 CrossRef CAS PubMed.
- S. Tazawa, N. Ota, M. Tamura, Y. Nakagawa, K. Okumura and K. Tomishige, ACS Catal., 2016, 6, 6393–6397 CrossRef CAS.
- K. Yamaguchi, J. Cao, M. Betchaku, Y. Nakagawa, M. Tamura, A. Nakayama, M. Yabushita and K. Tomishige, ChemSusChem, 2022, 15, e202102663 CrossRef CAS PubMed.
- R. Hosaka, D. Asada, J. Cao, M. Tamura, Y. Nakagawa, K. Tomishige, J. Hasegawa and A. Nakayama, J. Phys. Chem. C, 2022, 126, 11566–11573 CrossRef CAS.
- Y. Nakagawa, S. Tazawa, T. Wang, M. Tamura, N. Hiyoshi, K. Okumura and K. Tomishige, ACS Catal., 2018, 8, 584–595 CrossRef CAS.
- X. Lin, Y. Lv, Y. Xi, Y. Qu, D. L. Phillips and C. Liu, Energy Fuels, 2014, 28, 3345–3351 CrossRef CAS.
- M. Wang, H. Yang, Y. Xie, X. Wu, C. Chen, W. Ma, Q. Dong and Z. Hou, RSC Adv., 2016, 6, 29769–29778 RSC.
- N. Ota, M. Tamura, Y. Nakagawa, K. Okumura and K. Tomishige, Angew. Chem., Int. Ed., 2015, 54, 1897–1900 CrossRef CAS PubMed.
- S. Albarracin-Suazo, L. F. L. Freitas, B. MacQueen, A. Heyden, J. A. Lauterbach, E. Nikolla and Y. J. Pagán-Torres, ACS Sustainable Chem. Eng., 2022, 10, 5719–5727 CrossRef CAS.
- K. Yamaguchi, Y. Nakagawa, C. Li, M. Yabushita and K. Tomishige, ACS Catal., 2022, 12, 12582–12595 CrossRef CAS.
- Y. Amada, S. Koso, Y. Nakagawa and K. Tomishige, ChemSusChem, 2010, 3, 728–736 CrossRef CAS PubMed.
- L. Liu, J. Cao, Y. Nakagawa, M. Betchaku, M. Tamura, M. Yabushita and K. Tomishige, Green Chem., 2021, 23, 5665–5679 RSC.
- C. T. Q. Mai, Y. Ye, G. L. Rempel and F. T. T. Ng, Catal. Today, 2023, 407, 2–10 CrossRef CAS.
- G. Ioannidou, V.-L. Yfanti and A. A. Lemonidou, Catal. Today, 2022 DOI:10.1016/j.cattod.2022.09.008.
- M. El Doukkali, F. Dumeignil and S. Paul, Appl. Catal., B, 2023, 324, 122280 CrossRef CAS.
- T. K. Phung, T. L. M. Pham, K. B. Vu and G. Busca, J. Environ. Chem. Eng., 2021, 9, 105673 CrossRef CAS.
- V. Hulea, ACS Catal., 2018, 8, 3263–3279 CrossRef CAS.
- R. Gadioli, W. R. Waldman and M. A. De Paoli, J. Appl. Polym. Sci., 2016, 133, 43566 CrossRef.
- Y. Kikuchi, Y. Oshita, K. Mayumi and M. Hirao, J. Cleaner Prod., 2017, 167, 289–305 CrossRef CAS.
- Y. Nakagawa, T. Kasumi, J. Ogihara, M. Tamura, T. Arai and K. Tomishige, ACS Omega, 2020, 5, 2520–2530 CrossRef CAS PubMed.
- T. R. Felthouse, J. C. Burnett, B. Horrell, M. J. Mummy and Y.-J. Kuo, Maleic Anhydride, Maleic Acid, and Fumaric Acid, Kirk-Othmer Encyclopedia of Chemical Technology, 2001, DOI:10.1002/0471238961.1301120506051220.a01.pub2.
- P. V. Mangili, P. G. Junqueira, L. S. Santos and D. M. Prata, Clean Technol. Environ. Policy, 2019, 21, 1073–1090 CrossRef CAS.
- X. Li, J. Ko and Y. Zhang, ChemSusChem, 2018, 11, 612–618 CrossRef CAS.
- L. Chai, X. Hou, X. Cui, H. Li, N. Zhang, H. Zhang, C. Chen, Y. Wang and T. Deng, Chem. Eng. J., 2020, 388, 124187 CrossRef CAS.
- R. Zhu, A. Chatzidimitriou, B. Liu, D. J. Kerwood and J. Q. Bond, ACS Catal., 2020, 10, 1555–1565 CrossRef CAS.
- R. Wojcieszak, F. Santarelli, S. Paul, F. Dumeignil, F. Cavani and R. V. Gonçalves, Sustainable Chem. Processes, 2015, 3, 9 CrossRef.
- Y. Wan and J.-M. Lee, ACS Catal., 2021, 11, 2524–2560 CrossRef CAS.
- Y. Su, W. Zhang, A. Zhang and W. Shao, Appl. Sci., 2020, 10, 8222 CrossRef CAS.
- J. Sebastian, K. Hegde, P. Kumar, T. Rouissi and S. K. Brar, Crit. Rev. Biotechnol., 2019, 39, 817–834 CrossRef CAS PubMed.
- G. Pomalaza, P. A. Ponton, M. Capron and F. Dumeignil, Catal. Sci. Technol., 2020, 10, 4860–4911 RSC.
- Y. Qi, Z. Liu, S. Liu, L. Cui, Q. Dai, J. He, W. Dong and C. Bai, Catalysts, 2019, 9, 97 CrossRef.
- P. I. Kyriienko, O. V. Larina, S. O. Soloviev and S. M. Orlyk, Theor. Exp. Chem., 2020, 56, 213–242 CrossRef CAS.
- E. V. Makshina, M. Dusselier, W. Janssens, J. Degrève, P. A. Jacobs and B. F. Sels, Chem. Soc. Rev., 2014, 43, 7917–7953 RSC.
- C. E. C. Camacho, B. Alonso-Fariñas, A. L. V. Perales, F. Vidal-Barrero and P. Ollero, ACS Sustainable Chem. Eng., 2020, 8, 10201–10211 CrossRef.
- H. Duan, Y. Yamada and S. Sato, Chem. Lett., 2016, 45, 1036–1047 CrossRef CAS.
- D. Sun, Y. Li, C. Yang, Y. Su, Y. Yamada and S. Sato, Fuel Process. Technol., 2020, 197, 106193 CrossRef CAS.
- C. Delhomme, D. Weuster-Botz and F. E. Kühn, Green Chem., 2009, 11, 13–26 RSC.
- Y. Takeda, M. Tamura, Y. Nakagawa, K. Okumura and K. Tomishige, Catal. Sci. Technol., 2016, 6, 5668–5683 RSC.
- D. R. Vardon, A. E. Settle, V. Vorotnikov, M. J. Menart, T. R. Eaton, K. A. Unocic, K. X. Steirer, K. N. Wood, N. S. Cleveland, K. E. Moyer, W. E. Michener and G. T. Beckham, ACS Catal., 2017, 7, 6207–6219 CrossRef CAS.
- K. Li, J. Yang, T. Song, Z. Zhu, C. Zhao, P. Wu and X. Li, Green Chem., 2023, 25, 627–638 RSC.
- A. Said, D. D. S. Perez, N. Perret, C. Pinel and M. Besson, ChemCatChem, 2017, 9, 2768–2783 CrossRef CAS.
- M. Gu, L. Liu, Y. Nakagawa, C. Li, M. Tamura, Z. Shen, X. Zhou, Y. Zhang and K. Tomishige, ChemSusChem, 2021, 14, 642–654 CrossRef CAS PubMed.
- T. Wang, M. Tamura, Y. Nakagawa and K. Tomishige, ChemSusChem, 2019, 12, 3615–3626 CrossRef CAS PubMed.
- T. Wang, Y. Nakagawa, M. Tamura, K. Okumura and K. Tomishige, React. Chem. Eng., 2020, 5, 1237–1250 RSC.
- L. Liu, T. Asano, Y. Nakagawa, M. Tamura and K. Tomishige, Green Chem., 2020, 22, 2375–2380 RSC.
- S. Sato, R. Takahashi, T. Sodesawa, N. Honda and H. Shimizu, Catal. Commun., 2003, 4, 77–81 CrossRef CAS.
- F. Jing, B. Katryniok, M. Araque, R. Wojcieszak, M. Capron, S. Paul, M. Daturi, J.-M. Clacens, F. De Campo, A. Liebens, F. Dumeignil and M. Pera-Titus, Catal. Sci. Technol., 2016, 6, 5830–5840 RSC.
- J. H. Lee and S. B. Hong, Appl. Catal., B, 2021, 280, 119446 CrossRef CAS.
- Y. Wang, D. Sun, Y. Yamada and S. Sato, Appl. Catal., A, 2018, 562, 11–18 CrossRef CAS.
- Y. Matsumura, A. Matsuda, Y. Yamada and S. Sato, Bull. Chem. Soc. Jpn., 2021, 94, 1651–1658 CrossRef CAS.
- Y. Bai, S. J. Page, J. Zhang, D. Liu and X. Zhao, Chem. Eng. J., 2020, 389, 124451 CrossRef CAS.
- K. Nakazono, R. Takahashi, Y. Yamada and S. Sato, Mol. Catal., 2021, 516, 111996 CrossRef CAS.
- W. Kim, W. Shin, K. J. Lee, H. Song, H. S. Kim, D. Seung and I. N. Filimonov, Appl. Catal., A, 2016, 511, 156–167 CrossRef CAS.
- N. T. T. Nguyen, F. Matei-Rutkovska, M. Huchede, K. Jaillardon, G. Qingyi, C. Michel and J. M. M. Millet, Catal. Today, 2019, 323, 62–68 CrossRef CAS.
- W. Kim, W. Shin, K. J. Lee, Y. Cho, H. S. Kim and I. N. Filimonov, Appl. Catal., A, 2019, 570, 148–163 CrossRef CAS.
- D. Tsukamoto, S. Sakami, M. Ito, K. Yamada and T. Yonehara, Chem. Lett., 2016, 45, 831–833 CrossRef CAS.
- H. Duan, Y. Yamada and S. Sato, Appl. Catal., A, 2015, 491, 163–169 CrossRef CAS.
- S. Sato, R. Takahashi, T. Sodesawa and N. Yamamoto, Catal. Commun., 2004, 5, 397–400 CrossRef CAS.
- A. Matsuda, Y. Matsumura, Y. Yamada and S. Sato, Mol. Catal., 2021, 514, 111853 CrossRef CAS.
- A. Matsuda, F. Sato, Y. Yamada and S. Sato, Bull. Chem. Soc. Jpn., 2022, 95, 506–512 CrossRef CAS.
- N. S. Biradar, A. A. Hengne, S. N. Birajdar, R. Swami and C. V. Rode, Org. Process Res. Dev., 2014, 18, 1434–1442 CrossRef CAS.
- Y. Ji, A. Lawal, A. Nyholm, R. J. Gorte and O. A. Abdelrahman, Catal. Sci. Technol., 2020, 10, 5903–5912 RSC.
- O. A. Abdelrahman, D. S. Park, K. P. Vinter, C. S. Spanjers, L. Ren, H. J. Cho, D. G. Vlachos, W. Fan, M. Tsapatsis and P. J. Dauenhauer, ACS Sustainable Chem. Eng., 2017, 5, 3732–3736 CrossRef CAS.
- M. Fan, Y. Ji, A. Lawal, O. A. Abdelrahman, R. J. Gorde and J. M. Vohs, Catalysts, 2022, 12, 902 CrossRef CAS.
- G. K. Cook and M. A. Andrews, J. Am. Chem. Soc., 1996, 118, 9448–9449 CrossRef CAS.
- C. P. Nicholas, Appl. Catal., A, 2017, 543, 82–97 CrossRef CAS.
- P. Berteau and B. Delmon, Catal. Today, 1989, 5, 121–137 CrossRef CAS.
- P. Berteau, B. Delmon, J.-L. Dallons and A. V. Gysel, Appl. Catal., 1991, 70, 307–323 CrossRef CAS.
- I. Veza, M. F. M. Said and Z. A. Latiff, Biomass Bioenergy, 2021, 144, 105919 CrossRef CAS.
- X. Wu, G. Fang, Y. Tong, D. Jiang, Z. Liang, W. Leng, L. Liu, P. Tu, H. Wang, J. Ni and X. Li, ChemSusChem, 2018, 11, 71–85 CrossRef CAS PubMed.
- M. Wu, M. Wang, J. Liu and H. Huo, Biotechnol. Prog., 2008, 24, 1204–1214 CrossRef CAS PubMed.
- M. Brito and F. Martins, Fuel, 2017, 208, 476–482 CrossRef CAS.
- I. Nezam, L. Peereboom and D. J. Miller, J. Cleaner Prod., 2019, 209, 1365–1375 CrossRef CAS.
- Y. Amada, H. Watanabe, Y. Hirai, Y. Kajikawa, Y. Nakagawa and K. Tomishige, ChemSusChem, 2012, 5, 1991–1999 CrossRef CAS PubMed.
- D. Penner, C. Redepenning, A. Mitsos and J. Viell, Ind. Eng. Chem. Res., 2017, 56, 3947–3957 CrossRef CAS.
- T. Arai, M. Tamura, Y. Nakagawa and K. Tomishige, ChemSusChem, 2016, 9, 1680–1688 CrossRef CAS PubMed.
- B. Liu, Y. Nakagawa, C. Li, M. Yabushita and K. Tomishige, ACS Catal., 2022, 12, 15431–15450 CrossRef CAS.
- T. Mizugaki, K. Togo, Z. Maeno, T. Mitsudome, K. Jitsukawa and K. Kaneda, Sci. Rep., 2017, 7, 14007 CrossRef PubMed.
- J. Lv, Z. Rong, L. Sun, C. Liu, A.-H. Lu, Y. Wang and J. Qu, Catal. Sci. Technol., 2018, 8, 975–979 RSC.
- L. Chen, Y. Liu, C. Gu, G. Feng, X. Zhang, J. Liu, Q. Zhang, C. Wang and L. Ma, ACS Sustainable Chem. Eng., 2021, 9, 15603–15611 CrossRef CAS.
- C. D. Baertsch, K. T. Komala, Y.-H. Chua and E. Iglesia, J. Catal., 2002, 205, 44–57 CrossRef CAS.
- J. Bedia, R. Ruiz-Rosas, J. Rodríguez-Mirasol and T. Cordero, AIChE J., 2010, 56, 1557–1568 CrossRef CAS.
- J. Q. Bond, D. M. Alonso, D. Wang, R. M. West and J. A. Dumesic, Science, 2010, 327, 1110–1114 CrossRef CAS PubMed.
- A. B. Kellicutt, R. Salary, O. A. Abdelrahman and J. Q. Bond, Catal. Sci. Technol., 2014, 4, 2267–2279 RSC.
- J. Xin, D. Yan, O. Ayodele, Z. Zhang, X. Lu and S. Zhang, Green Chem., 2015, 17, 1065–1070 RSC.
- H. Wang, Y. Wu, S. Guo, C. Dong and M. Ding, Mol. Catal., 2020, 497, 111218 CrossRef CAS.
- Y. Xiong, X. Du, P. Wang, X. Feng, J. Cui, Z. Lou, W. Shan and H. Yu, Mol. Catal., 2022, 533, 112798 CrossRef CAS.
- L. Lin, A. M. Sheveleva, I. da Silva, C. M. A. Parlett, Z. Tang, Y. Liu, M. Fan, X. Han, J. H. Carter, F. Tuna, E. J. L. McInnes, Y. Cheng, L. L. Daemen, S. Rudić, A. J. Ramirez-Cuesta, C. C. Tang and S. Yang, Nat. Mater., 2020, 19, 86–93 CrossRef CAS PubMed.
- S. Dutta, I. K. M. Yu, D. C. W. Tsang, Y. H. Ng, Y. S. Ok, J. Sherwood and J. H. Clark, Chem. Eng. J., 2019, 372, 992–1006 CrossRef CAS.
- T. Raj, K. Chandrasekhar, R. Banu, J.-J. Yoon, G. Kumar and S.-H. Kim, Fuel, 2021, 303, 121333 CrossRef CAS.
- A. Jaswal, P. P. Singh and T. Mondal, Green Chem., 2022, 24, 510–551 RSC.
- I. Agirre, I. Gandarias, M. L. Granados and P. L. Arias, Biomass Convers. Biorefin., 2020, 10, 1021–1033 CrossRef CAS.
- M. Bender, ChemBioEng Rev., 2014, 1, 136–147 CrossRef CAS.
- J. Leveau, J. P. Efromson and M. D. Lynch, Front. Bioeng. Biotechnol., 2020, 8, 207 CrossRef PubMed.
- E. S. Vasiliadou, S. Li, S. Caratzoulas and R. F. Lobo, Catal. Sci. Technol., 2018, 8, 5794–5806 RSC.
- C. Fu, Z. Li, C. Jia, W. Zhang, Y. Zhang, C. Yi and S. Xie, Energy Convers. Manage.: X, 2021, 10, 100059 CAS.
- B. N. M. van Leeuwen, A. M. van der Wulp, I. Duijnstee, A. J. A. van Maris and A. J. J. Straathof, Appl. Microbiol. Biotechnol., 2012, 93, 1377–1387 CrossRef PubMed.
- K. Fazeni-Fraisl and J. Lindorfer, Biofuels, Bioprod. Biorefin., 2023, 17, 207–225 CrossRef CAS.
- D. Gunst, K. Alexopoulos, K. V. D. Borght, M. John, V. Galvita, M.-F. Reyniers and A. Verberckmoes, Appl. Catal., A, 2017, 539, 1–12 CrossRef CAS.
- J. D. Taylor, M. M. Jenni and M. W. Peters, Top. Catal., 2010, 53, 1224–1230 CrossRef CAS.
- D. Zhang, R. Al-Hajri, S. A. I. Barri and D. Chadwick, Chem. Commun., 2010, 46, 4088–4090 RSC.
- D. Zhang, S. A. I. Barri and D. Chadwick, Appl. Catal., A, 2011, 403, 1–11 CrossRef CAS.
- M. J. D. Mahboub, J.-L. Dubois, F. Cavani, M. Rostamizadeh and G. S. Patience, Chem. Soc. Rev., 2018, 47, 7703–7738 RSC.
- J. L. Nôtre, S. C. M. W. Dijk, J. van Haveren, E. L. Scott and J. P. M. Sanders, ChemSusChem, 2014, 7, 2712–2720 CrossRef PubMed.
- J. C. Lansing, R. E. Murray and B. R. Moser, ACS Sustainable Chem. Eng., 2017, 5, 3132–3140 CrossRef CAS.
- M. Carlsson, C. Habenicht, L. C. Kam, M. J. Antal, N. Bian, R. J. Cunningham and M. Jones, Ind. Eng. Chem. Res., 1994, 33, 1989–1996 CrossRef CAS.
- A. Bohre, U. Novak, M. Grilc and B. Likozar, Mol. Catal., 2019, 476, 110520 CrossRef CAS.
- A. Bohre, K. Avasthi, U. Novak and B. Likozar, ACS Sustainable Chem. Eng., 2021, 9, 2902–2911 CrossRef CAS.
- H. Ali, S. K. Kansal and S. Saravanamurugan, ChemistrySelect, 2021, 6, 3352–3359 CrossRef CAS.
- Y. Wu, M. Shetty, K. Zhang and P. J. Dauenhauer, ACS Eng. Au, 2022, 2, 92–102 CrossRef CAS.
- Z. Li, X. Wen and H. Liu, Green Chem., 2022, 24, 1650–1658 RSC.
- Q. Hou, X. Qi, M. Zhen, H. Qian, Y. Nie, C. Bai, S. Zhang, X. Bai and M. Ju, Green Chem., 2021, 23, 119–231 RSC.
- I. Itabaiana Junior, M. A. do Nascimento, R. O. M. A. De Souza, A. Dufour and R. Wojcieszak, Green Chem., 2020, 22, 5859–5880 RSC.
- C. G. S. Lima, J. L. Monteiro, T. de Melo Lima, M. W. Paixão and A. G. Corrêa, ChemSusChem, 2018, 11, 25–47 CrossRef CAS PubMed.
- B.-E. Teleky and D. C. Vodnar, Polymer, 2019, 11, 1035 Search PubMed.
- B.-E. Teleky and D. C. Vodnar, Polymer, 2021, 13, 3574 CAS.
- I. Khalil, G. Quintens, T. Junkers and M. Dusselier, Green Chem., 2020, 22, 1517–1541 RSC.
- J. T. Dixon, M. J. Green, F. M. Hess and D. H. Morgan, J. Organomet. Chem., 2004, 689, 3641–3668 CrossRef CAS.
- K. Chen, M. Tamura, Z. Yuan, Y. Nakagawa and K. Tomishige, ChemSusChem, 2013, 6, 613–621 CrossRef CAS PubMed.
- Y. Okuyama, M. Koike, S. Sasaki, S. Liu, M. Tamura, Y. Nakagawa, A. Imai and K. Tomishige, J. Jpn. Pet. Inst., 2016, 59, 228–234 CrossRef CAS.
- S. Liu, Y. Okuyama, M. Tamura, Y. Nakagawa, A. Imai and K. Tomishige, ChemSusChem, 2015, 8, 628–635 CrossRef CAS PubMed.
- S. Liu, Y. Okuyama, M. Tamura, Y. Nakagawa, A. Imai and K. Tomishige, Green Chem., 2016, 18, 165–175 RSC.
- V. A. Rozentsvet, V. G. Kozlov, N. A. Korovina, V. P. Ivanova and S. V. Kostjuk, J. Appl. Polym. Sci., 2013, 128, 1771–1778 CAS.
- L. Porri, A. Giarrusso and G. Ricci, Prog. Polym. Sci., 1991, 16, 405–441 CrossRef CAS.
- Y. Nakagawa, M. Tamura and K. Tomishige, Catal. Surv. Asia, 2015, 19, 249–256 CrossRef CAS.
- J. Zhu and G. Yin, ACS Catal., 2021, 11, 10058–10083 CrossRef CAS.
- Z. Zhang and H. Li, Fuel, 2022, 310, 122242 CrossRef CAS.
- H. Duan, M. Unno, Y. Yamada and S. Sato, Appl. Catal., A, 2017, 546, 96–102 CrossRef CAS.
- C. Beasley, M. K. Gnanamani, M. Martinelli, K. Góra-Marek, K. Hamano, W. D. Shafer, N. Wanninayake and D. Y. Kim, ChemistrySelect, 2019, 4, 3123–3130 CrossRef CAS.
- G. Novodárszki, J. Valyon, Á. Illés, S. Dóbé and M. R. Mihályi, React. Kinet., Mech. Catal., 2017, 121, 275–292 CrossRef.
- S. Liu, Y. Qi, R. Feng, L. Cui, Q. Dai and C. Bai, ACS Sustainable Chem. Eng., 2021, 9, 8341–8346 CrossRef CAS.
- M. Stalpaert, F. G. Cirujano and D. E. De Vos, ACS Catal., 2017, 7, 5802–5809 CrossRef CAS.
- S. Chen, R. Wojcieszak, F. Dumeignil, E. Marceau and S. Royer, Chem. Rev., 2018, 118, 11023–11117 CrossRef CAS PubMed.
- S. Chen, C. Ciotonea, K. D. O. Vigier, F. Jérôme, R. Wojcieszak, F. Dumeignil, E. Marceau and S. Royer, ChemCatChem, 2020, 12, 2050–2059 CrossRef CAS.
- G. Kumar, D. Liu, D. Xu, L. Ren, M. Tsapatsis and P. J. Dauenhauer, Green Chem., 2020, 22, 4147–4160 RSC.
- Y. Ji, S. P. Batchu, A. Lawal, D. G. Vlachos, R. J. Gorte, S. Caratzoulas and O. A. Abdelrahman, J. Catal., 2022, 410, 10–21 CrossRef CAS.
- M. Fan, S. Xu, B. An, A. M. Sheveleva, A. Betts, J. Hurd, Z. Zhu, M. He, D. Iuga, L. Lin, X. Kang, C. M. A. Parlett, F. Tuna, E. J. L. McInnes, L. L. Keenan, D. Lee, M. P. Attfield and S. Yang, Angew. Chem., Int. Ed., 2022, 61, e202212164 CAS.
- H. K. Chau, Q. P. Nguyen, A. C. Jerdy, D.-P. Bui, L. L. Lobban, B. Wang and S. P. Crossley, ACS Catal., 2023, 13, 1503–1512 CrossRef CAS.
- S. H. Krishna, J. Cao, M. Tamura, Y. Nakagawa, M. De Bruyn, G. S. Jacobson, B. M. Weckhuysen, J. A. Dumesic, K. Tomishige and G. W. Huber, ACS Sustainable Chem. Eng., 2020, 8, 800–805 CrossRef CAS.
- G. M. Whited, F. J. Feher, D. A. Benko, M. A. Cervin, G. K. Chotani, J. C. McAuliffe, R. J. LaDuca, E. A. Ben-Shoshan and K. J. Sanford, Ind. Biotechnol., 2010, 6, 152–163 CrossRef CAS.
- J. Isar, D. Jain, H. Joshi, S. Dhoot and V. Rangaswamy, World J. Microbiol. Biotechnol., 2022, 38, 122 CrossRef CAS PubMed.
- D. J. Lundberg, D. J. Lundberg, K. Zhang and P. J. Dauenhauer, ACS Sustainable Chem. Eng., 2019, 7, 5576–5586 CrossRef CAS.
- M. Tamura, Y. Nakagawa and K. Tomishige, Asian J. Org. Chem., 2020, 9, 126–143 CrossRef CAS.
- L. Karam and C. N. Neumann, ChemCatChem, 2022, 14, e202200953 CrossRef CAS.
- R. Qu, K. Junge and M. Beller, Chem. Rev., 2023, 123, 1103–1165 CrossRef CAS PubMed.
- Y. Qi, S. Liu, L. Cui, Q. Dai, J. He, W. Dong and C. Bai, ACS Sustainable Chem. Eng., 2020, 8, 10323–10329 CrossRef CAS.
- R. Kobayashi, E. Kurniawan, Y. Yamada and S. Sato, Mol. Catal., 2023, 535, 112854 CrossRef CAS.
- S. Straus and S. L. Madorsky, Ind. Eng. Chem., 1956, 48, 1212–1219 CrossRef CAS.
- B. L. Davis, L. A. Goldblatt and S. Palkin, Ind. Eng. Chem., 1946, 38, 53–57 CrossRef CAS.
- D. R. Georgianna and S. P. Mayfield, Nature, 2012, 488, 329–335 CrossRef CAS PubMed.
- P. Metzger and C. Largeau, Appl. Microbiol. Biotechnol., 2005, 66, 486–496 CrossRef CAS PubMed.
- E. Naziri, F. Mantzouridou and M. Z. Tsimidou, Lipid Technol., 2011, 23, 270–273 CrossRef CAS.
- K. Kimura, K. Shiraishi, T. Kondo, J. Nakamura and T. Fujitani, Green Chem., 2020, 22, 3083–3087 RSC.
- M. Gale, C. M. Cai and K. L. Gilliard-Abdul-Aziz, ChemSusChem, 2020, 13, 1947–1966 CrossRef CAS PubMed.
- J.-M. Ha, K.-R. Hwang, Y.-M. Kim, J. Jae, K. H. Kim, H. W. Lee, J.-Y. Kim and Y.-K. Park, Renewable Sustainable Energy Rev., 2019, 111, 422–441 CrossRef CAS.
- C. Cheng, D. Shen, S. Gu and K. H. Luo, Catal. Sci. Technol., 2018, 8, 6275–6296 RSC.
- X. Wang, M. Arai, Q. Wu, C. Zhang and F. Zhao, Green Chem., 2020, 22, 8140–8168 RSC.
- S. S. Wong, R. Shu, J. Zhang, H. Liu and N. Yan, Chem. Soc. Rev., 2020, 49, 5510–5560 RSC.
- Z. Cai, B. Shen, W. Liu, Z. Xin and H. Ling, Energy Fuels, 2009, 23, 4077–4081 CrossRef CAS.
- Y. Liu, G. Li, Y. Hu, A. Wang, F. Lu, J.-J. Zou, Y. Cong, N. Li and T. Zhang, Joule, 2019, 3, 1028–1036 CrossRef CAS.
- C. Verrier, S. Moebs-Sanchez, Y. Queneau and F. Popowycz, Org. Biomol. Chem., 2018, 16, 676–687 RSC.
- S. Dutta and N. S. Bhat, ACS Omega, 2021, 6, 35145–35172 CrossRef CAS PubMed.
- G. Li, B. Hou, A. Wang, X. Xin, Y. Cong, X. Wang, N. Li and T. Zhang, Angew. Chem., Int. Ed., 2019, 58, 12154–12158 CrossRef CAS PubMed.
- Y. Liu, R. Wang, H. Qi, X. Y. Liu, G. Li, A. Wang, X. Wang, Y. Cong, T. Zhang and N. Li, Nat. Commun., 2021, 12, 46 CrossRef CAS PubMed.
- R. Wang, Y. Liu, G. Li, A. Wang, X. Wang, Y. Cong, T. Zhang and N. Li, ACS Catal., 2021, 11, 4810–4820 CrossRef CAS.
- J.-D. Woodroffe and B. G. Harvey, ChemSusChem, 2021, 14, 339–343 CrossRef CAS PubMed.
- Q. Tian, K. Qiao, F. Zhou, K. Chen, T. Wang, J. Fu, X. Lu and P. Ouyang, Energy Fuels, 2016, 30, 7291–7297 CrossRef CAS.
- K. M. Doll, G. B. Bantchev, E. L. Walter, R. E. Murray, M. Appell, J. C. Lansing and B. R. Moser, Ind. Eng. Chem. Res., 2017, 56, 864–871 CrossRef CAS.
- F. van der Klis, J. L. Nôtre, R. Blaauw, J. van Haveren and D. S. van Es, Eur. J. Lipid Sci. Technol., 2012, 114, 911–918 CrossRef CAS.
- J. Spekreijse, J. P. M. Sanders, J. H. Bitter and E. L. Scott, ChemSusChem, 2017, 10, 470–482 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |