
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanocatalytic hydrogenolysis of benzyl phenyl ether over supported nickel catalysts†
Andrew W.
Tricker
 ab,
Sean
Najmi
a,
Erin V.
Phillips
bc,
Karoline L.
Hebisch
a,
Jason X.
Kang
a and
Carsten
Sievers
*ab
ab,
Sean
Najmi
a,
Erin V.
Phillips
bc,
Karoline L.
Hebisch
a,
Jason X.
Kang
a and
Carsten
Sievers
*ab
aSchool of Chemical & Biomolecular Engineering, Georgia Institute of Technology, Atlanta, GA 30332, USA. E-mail: carsten.sievers@chbe.gatech.edu
bRenewable Bioproducts Institute, Georgia Institute of Technology, Atlanta, GA 30332, USA
cSchool of Chemistry and Biochemistry, Georgia Institute of Technology, Atlanta, GA 30332, USA
First published on 23rd January 2023
Abstract
Mechanocatalysis is a promising approach for green, solvent-free biomass deconstruction and valorization. Here, the hydrogenolysis of benzyl phenyl ether (BPE), a model lignin ether, via ball milling is demonstrated over supported nickel catalysts at nominally room temperature and atmospheric hydrogen pressure. The hydrogenolysis reaction network closely follows that of solution-based reactions, with the primary products being toluene, phenol, and cyclohexanol. The mechanical energy during milling not only drives the chemical reactions but also activates the nickel by exposing fresh metallic surfaces from passivated particles, which replaces a thermal activation step. The hydrogenolysis rate is shown to be largely insensitive to the final nickel particle size, but reactivity of the oxide support can be enhanced during milling which contributes to carbon deposition. This work demonstrates the underlying chemistry necessary for mild lignin depolymerization using reductive mechanocatalysis.
Sustainability spotlightThe production of commodity chemicals and materials from alternative feedstocks is necessary to successfully transition from a fossil-based, linear economy to a sustainable, circular economy. The deconstruction and valorization of biomass, and lignin in particular, is a promising pathway to utilize renewable carbon. However, most conventional approaches rely on the use of organic solvents, which contribute a significant portion of the process-energy and can also have adverse effects on the environment. The current research demonstrates the feasibility of the underlying chemistry necessary to depolymerize lignin under solvent-free and ambient conditions. This work aligns with US SDG #12 (responsible consumption and production) by advancing an approach to utilize renewable resources while minimizing process waste production. |
Introduction
As society shifts away from fossil-based fuels and chemicals, the necessity for complete utilization of biomass becomes ever more pressing. Lignin, a primary component of woody biomass, is an abundant renewable resource but is practically unutilized as a feedstock for chemicals and materials. Of the 60 million tonnes of lignin isolated annually, less than 2% is used for value-added products, while the rest is burned as a low value fuel.1 Lignin is a heterogeneous, amorphous polymer that is primarily comprised of three propyl-phenolic monolignols (p-coumaryl alcohol, coniferyl alcohol, and sinapyl alcohol) linked together randomly by various C–O (β-O-4, α-O-4, 4-O-5) and C–C (β–β, β–5, 5–5) bonds.2 However, the fractionation of lignin from the lignocellulosic matrix typically changes the structure of the polymer and increases the abundance of condensed (C–C) linkages.3 As the largest natural source of aromatic carbon, lignin is a promising, sustainable alternative starting point for historically benzene–toluene–xylene (BTX) derived chemicals if the polymer can be selectively depolymerized to mono-aromatics.Two of the most promising approaches to depolymerize lignin utilize hydrogen as either a cleaving or stabilizing reagent: hydroprocessing and reductive catalytic fraction (RCF).4 Hydroprocessing, generally performed with isolated lignin, combines hydrogenolysis of the linkages to produce monomers and hydrodeoxygenation (HDO) to upgrade monomers simultaneously.5 These reactions tend to produce monomer yields between 10 and 20 wt%, with harsher reaction conditions increasing monomer yields at the expense of product selectivity.4 RCF starts with complete lignocellulose and combines the isolation and depolymerization of lignin, where solvolysis formed monomers are stabilized before repolymerization can occur.6 Most reported monomer yields from RCF studies range from 10 to 30 wt% for softwood and from 30 to 60 wt% for hardwood feedstocks with very high selectivities of phenolics. Both hydroprocessing and RFC reactions employed noble metals, such as Pt, Pd, Ru, or Rh, or transition metals, primarily Ni and Cu, on either acidic or neutral supports. These reactions often require elevated temperatures (130–450 °C), elevated hydrogen pressures (10–100 bar), and large quantities of organic solvents, such as methanol, dioxane, or THF.4 The specialized equipment necessary for the combination of high hydrogen pressures and heated, flammable solvents, along with the energy demands for solvent recovery, currently stand as major barriers to scale up these processes.7
Mechanochemistry offers a novel approach for green chemical production by performing reactions under solvent-free conditions at nominally ambient temperatures and pressures.8–10 The direct input of mechanical energy, such as collisions in a ball mill, can drive reactions between solid reactants and catalysts. This energy can influence the system in many ways, such as enhancing solid–solid mixing and contact,11 creating fresh reactive surfaces,12 forming localized thermal hot spots,13 and altering reactivity of reagents14 and catalysts.15–17 The multitude of ways that mechanochemical reactions can occur has resulted in a broad range of applications, such as alloying,12 pharmaceuticals18 and absorbents19 synthesis, consumer plastics deconstruction,20,21 and waste recycling.22
Mechanical energy, without added chemicals or catalysts, has long been used as a method for pulping wood and pretreating lignocellulose for conversion to fuels and chemicals.23 In recent years, the use of mechanocatalysis for biomass valorization has seen a rise in popularity, albeit with an emphasis on cellulose or unfractionated lignocellulosic biomass.24,25 Mechanocatalytic hydrolysis of cellulose and lignocellulose impregnated with HCl, H2SO4, or solid acids, such as delaminated kaolinite, can produce high yields of water-soluble sugar oligomers within a few hours of milling.26–28 Additionally, the energy efficiency of the reaction has been shown to increase at least up to the kilogram scale while also maintaining greater than 90% yields of water-soluble oligosaccharides.29 Studies on the degradation of isolated lignin have been more limited. Early work focused on the homolytic cleavage of model compounds during ball milling.30,31 Mechanochemistry has since been utilized as a pretreatment method32 or for alkaline33,34 and oxidative35 depolymerization. However, these approaches either rely on corrosive reactants (i.e., NaOH) or use a complex, multi-component mixture of homogeneous catalysts, which may be difficult to separate and recycle.
While the mechanocatalytic hydrogenation of benzene over nickel in a vibratory ball mill was reported as far back as 1963,36 studies on reductive mechanocatalysis with gaseous hydrogen have been sparse.37 The methanation of CO2 over Ni/Fe/Ru on MgO catalysts38 and LaNi alloys39 and of CO over mixed FeCo and MgNi catalysts40 have been demonstrated. The hydrogenation of numerous alkynes, alkenes, aromatics, and various functional groups have been reported using a feed of H2 or splitting water in situ.41–43 Recently, the reduction of N2 to NH3 with H2 was reported using a batch planetary ball mill44 and in continuous-flow vibratory ball mills.16,45
Herein, the mechanocatalytic hydrogenolysis of BPE was demonstrated under solvent-free, nominally ambient conditions (room temperature, 1 bar H2) using supported nickel catalysts. Hydrogenolysis of model lignin ethers have been extensively studied under liquid phase conditions to glean insights into the underlying catalytic reactions and behavior of catalysts.46–48 Benzyl phenyl ether (BPE) is studied here as a model compound for lignin because of the similarity in C–O bond strengths between BPE and the β-O-4 linkage in lignin.49 Admittedly, BPE lacks the functional moieties of lignin (i.e., hydroxyl and methoxy groups), however the novelty and complexity of the interactions between the reagent, catalyst, and mechanical action necessitates a more simplified model system to draw meaningful insights. A process based on this approach would have a distinct advantage over traditional approaches by avoiding product-solvent separations.
Additionally, in this paper, the mechanocatalytic hydrogenolysis reaction network for BPE is elucidated. The in situ mechanical activation of the supported nickel catalysts, as well as recyclability and deactivation pathways, are investigated. Finally, the effects of the nickel support properties on the activity of the catalysts are probed. Insights gained on the catalytic system will aid in further developing processes for the solvent-free and mild depolymerization of lignin via reductive mechanocatalysis.
Experimental
Chemicals
Benzyl phenyl ether (BPE, 97%) and o-xylene (99%) were purchased from Alfa Aesar. Nickel on silica–alumina (Ni53SiAl), silicon dioxide (quartz, ∼99%), silica–alumina catalyst support (grade 135), nickel nitrate hexahydrate (Ni(NO3)2·6H2O, 99.999%), and guaiacol (>98%) were purchased from Sigma-Aldrich. Silica gel was purchased from Sorbent Technologies. Methanol (ACS grade) was purchased from VWR. Isopropyl alcohol (IPA, 99%, Lab Grade) was purchased from Ward's Science. Hydrogen (H2, UHP, Grade 5) and nitrogen (N2, UHP, Grade 5) were purchased from Airgas.Catalyst synthesis
Three 5 wt% nickel catalysts were synthesized using amorphous silica–alumina (Ni05SiAl), silica gel (Ni05Sihigh), and quartz (Ni05Silow). The catalysts were prepared by incipient wetness impregnation with an aqueous Ni(NO3)2 solution. The wetted samples were dried at 105 °C in air overnight. The dried samples were reduced in 7% H2 in Ar at 450 °C (ramp from room temperature at 10 °C min−1) for 8 h. The samples were exposed to and stored in air after returning to room temperature.Catalyst characterization
Mechanocatalytic reactions
Mechanocatalytic reactions were performed using a modified Retsch MM400 vibratory ball mill with a 25 mL stainless steel milling vessel. The milling vessel was modified to allow for continuous gas flow. The effluent line was heated to 160 °C and the effluent gas was bubbled through methanol (∼10 mL with ∼0.03 g o-xylene as an internal standard) cooled to 4 °C to trap volatile products leaving the reactor. Unless otherwise specified, reactions were conducted with 1.00 g of catalyst, 0.20 g of BPE, two 15 mm stainless steel grinding balls, and a milling frequency of 20 Hz in a continuous flow of H2. The high catalyst to BPE ratio was necessary in order to keep the substrate a powder during milling. Due to the low melting point of BPE (40 °C), lower catalyst-to-reagent ratios resulted in liquefaction of the substrate and no observed reaction. This issue would not arise when milling polymeric materials, like lignin, and lower catalyst-to-reagent ratios can be used. Additionally, this type of system could be adapted to a continuous process at larger scales, allowing for more realistic reagent-to-catalyst ratios. The vessel was left unheated but milling induced an external temperature increase to ∼30 °C within the reactor. The vessel was purged with N2 for 15 min and then with H2 for at least 15 min before milling began. After milling, the vessel was purged with N2 for 1 h to remove additional residual volatile products.Unconverted BPE and non-volatile products were measured after each reaction by removing the product-catalyst mixture from the milling vessel and sonicating in methanol (∼10 mL) for 30 min. Roughly 90% of the sample mass could be recovered from the vessel. The remaining ∼10% were left adsorbed on the vessel and balls or in the valve port of the ball mill. After sonication, o-xylene was added (∼0.03 g) as an internal standard. The yield of volatile products over the course of the reaction was measured by taking aliquots (∼0.05 mL) of the methanol trap. Product yields were determined via gas chromatography using a Varian 450-GC with a fused silica column (Supelco, SPB-1, 60 m × 0.25 mm × 1.0 μm) and a flame ionization detector (FID). The GC-FID was equipped with a PolyArc reactor (Activated Research Company), which allowed for calibration-free quantification of the products.
The long-term stability of the Ni53SiAl catalyst was studied through a series of recycle experiments. The catalyst was milled for 2 h during the initial cycle and 4 h for the second and third cycles. After being washed with methanol to recover unconverted BPE and non-volatile products, the catalyst was vacuum filtered and dried under vacuum at room temperature. Similar to the mechanocatalytic reactions with the fresh catalyst, 1.00 g of the recycled catalyst was used. The same experimental procedures were used as the reaction with fresh catalyst. After vial transfers and filtration, only ∼65% of the spent catalyst mass could be recovered (∼0.65 g). So, spent catalyst samples of several different runs were combined for each recycle stage. For example, to produce enough sample to run a third cycle, two second cycle run were performed, which required four fresh catalyst runs.
Mechanocatalytic reaction kinetics
In order to semi-quantify the activity and the deactivation of the catalysts and the rate of carbon loss, the reaction is treated as a simplified system with only two first order reactions. The first is the hydrogenolysis of BPE to toluene and the second is the coke formation from BPE. Since the toluene yield is based on products detected in the condenser, it is assumed that all the produced toluene is found in the condenser and that toluene does not participate in the coking reaction. Initially, the mechanocatalytic hydrogenolysis reactions were fit using this simplified kinetic model (eqn (1)–(4)), where XBPE is the conversion of BPE, kH is the hydrogenolysis rate constant, mNi is the mass of nickel, kC is the coking rate constant, and Ytol is the yield of toluene in the condenser. Eqn (3) and (4) are the analytical solutions to the integration of eqn (1) and (2), respectively. Since XBPE was measured for several time points for fresh Ni53SiAl, eqn (3) was fit to solve for kH·mNi + kC.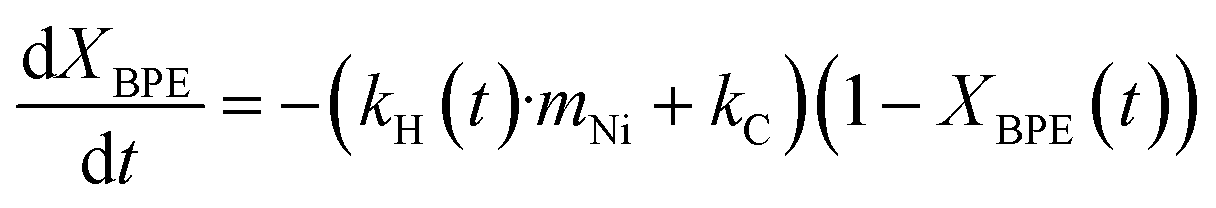 | (1) |
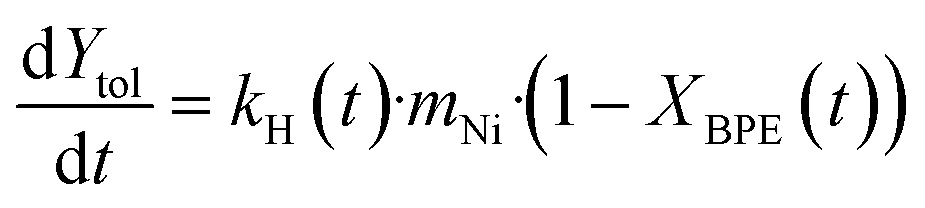 | (2) |
| XBPE(t) = 1 − exp(−(kH·mNi + kC)t) | (3) |
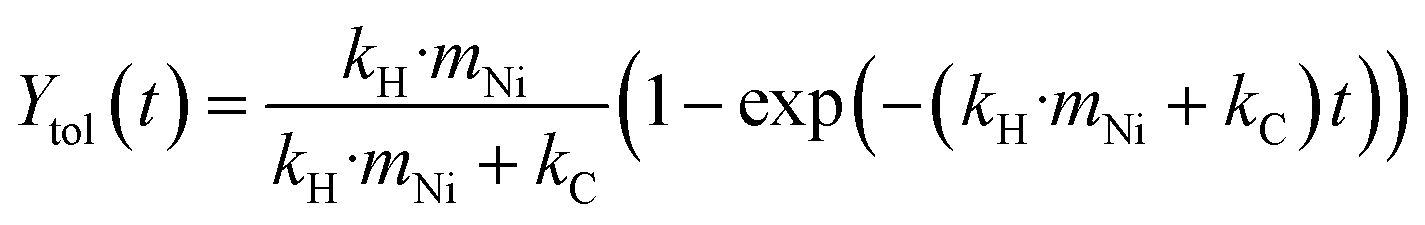 | (4) |
For experiments where the full product yields were only measured once and the conversion of BPE was incomplete, kH·mNi + kC were be determined by eqn (3) to yield eqn (5) and using the final conversion. kH was fit using Solver in Excel to minimize the error between the model and the toluene yield from the condenser. Due to the observed induction period in the condenser yield, a delay term was also fit. For Ni53SiAl-cycle 2, which was run only once and had nearly 100% conversion of BPE, calculating kC from eqn (5) would have resulted in high uncertainty and was instead approximated with the kC of Ni53SiAl-cycle 3.
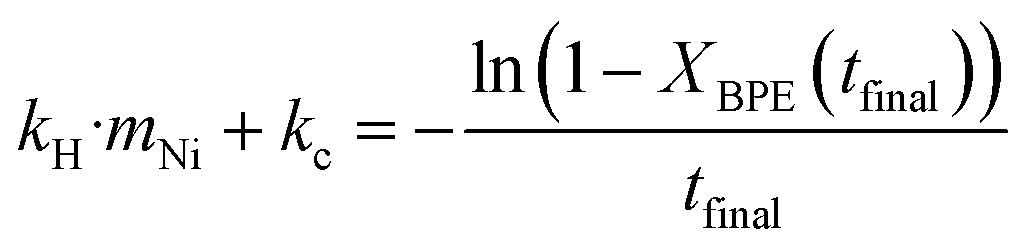 | (5) |
To assess the deactivation of Ni53SiAl, a first-order deactivation term was included in the model (eqn (6)), where kD is the deactivation rate constant. No analytical solution to eqn (S1), (S2), and (S6)† exists, so the equations were fit numerically. A Matlab script based on the built-in functions fmincon (error minimization) and ode45 (differential equation solver) was used to determine kD and the initial hydrogenolysis rate constant (kH,0). An average kC was used and assumed constant.
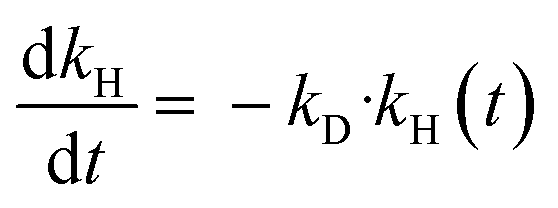 | (6) |
Results and discussion
Catalyst characterization
Inductively coupled plasma optical emission spectroscopy (ICP-OES) analysis of the nickel loadings of the catalysts showed that the commercial nickel on silica–alumina (Ni53SiAl) sample contained 53.4 wt% Ni, while the synthesized Ni catalysts with silica–alumina, high surface area silica and low surface area silica (Ni05SiAl, Ni05Sihigh, and Ni05Silow) had 4.42 wt%, 4.93 wt%, and 4.85 wt% Ni, respectively. The BET surface areas of Ni53SiAl, Ni05SiAl, Ni05Sihigh, and Ni05Silow were 85.3 m2 g−1, 499 m2 g−1, 515 m2 g−1, and 6.7 m2 g−1, respectively. Adsorption and desorption isotherms are shown in Fig. S1.† X-ray powder diffractometry (XRD) of Ni53SiAl showed the presence of both Ni and NiO, while only peaks corresponding to metallic Ni were observed in the diffractograms of Ni05SiAl, Ni05Sihigh, and Ni05Silow (Fig. S2†). Scanning transmission electron microscope (STEM) images showed that the nickel particles of Ni53SiAl were encapsulated in NiO (Fig. 1a). Similarly, STEM showed outer layers of NiO on the nickel particles for Ni05SiAl, Ni05Sihigh, and Ni05Silow (Fig. 1b–d). Energy dispersive spectrometry (EDS) mapping of Ni53SiAl showed particles uniformly coated in nickel (Fig. 1e), while the low nickel loading catalysts had discrete nickel particles (Fig. 1f–h). Ni05SiAl, Ni05Sihigh, and Ni05Silow had initial particle sizes of 18.7 nm, 15.1 nm, and 46.7 nm, respectively (Fig. S4†). The high abundance of nickel on Ni53SiAl prevented the measurement of enough particles to determine initial particle size. | ||
| Fig. 1 STEM images of individual nickel particles on (a) Ni53SiAl, (b) Ni05SiAl, (c) Ni05Sihigh, and (d) Ni05Silow. STEM images and EDS mapping of nickel for whole catalyst particles of (e) Ni53SiAl, (f) Ni05SiAl, (g) Ni05Sihigh, and (h) Ni05Silow. | ||
Hydrogenolysis reaction network
The mechanocatalytic reactions were conducted in a vibratory ball mill (Retsch, MM400) reactor, modified to allow for the continuous flow of H2 (Scheme 1). The catalyst and BPE were placed together in the milling vessel without additional pretreatment. While the vessel was not externally heated or pressurized, the milling caused the external vessel temperature to rise slightly to ∼30 °C. The reactor effluent was bubbled through methanol cooled to 4 °C to collect volatile products, and the methanol solution was sampled periodically to determine the yield of toluene during milling as a proxy for the extent of reaction. Gas chromatography was used to quantify products collected in the methanol trap and washed off the catalyst after reaction. The majority of the hydrocarbon products (toluene, benzene, cyclohexane) were recovered in the methanol trap, while oxygenated products (phenol, cyclohexanol) remained in the milling vessel and were collected from the catalyst wash.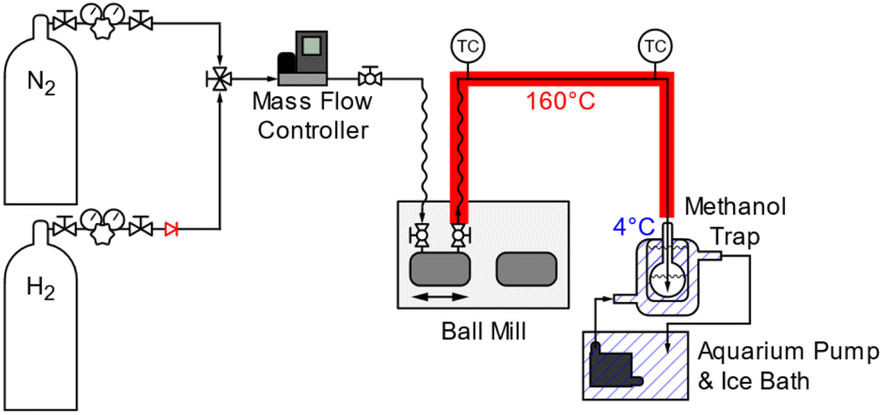 | ||
| Scheme 1 Depiction of the flow-through ball mill reactor. | ||
Analysis of reaction products formed over a commercial 53 wt% nickel on silica–alumina (Ni53SiAl) catalyst after various milling times elucidated the mechanocatalytic reaction network of benzyl phenyl ether (BPE), which closely follows reported pathways during thermally catalyzed reactions (Fig. 2).49,51–54 At early milling times, the main products were toluene (1a) and phenol (1b), which are formed by hydrogenolysis of the benzyl ether bond. Benzyl alcohol (8) was not detected, which is unsurprising since the bond dissociation energy of the benzyl ether bond (56.4 kJ mol−1)55 is much lower than that of the phenyl ether bond (95.7 kJ mol−1).56 Yields of hydrogenated ethers (4 & 5) passed through maxima (∼1%) at 60 min, suggesting that hydrogenolysis of these compounds occurred, but did not significantly contribute to the formation of the single-ring products. The formation of methyl cyclohexane (2) and cyclohexanol (3) predominantly occurred through the hydrogenation of toluene and phenol, respectively. Phenol hydrogenation to cyclohexanol was established as a reaction path based on the maximum in the phenol yield after 60 min. Low methylcyclohexane yields (<1%) can be explained by the removal of toluene vapors from the reactor prior to be being hydrogenated or a lower affinity of toluene to the active sites for hydrogenation reactions on the catalyst. Benzene (6) and cyclohexane (7) were detected after 60 min and 120 min, respectively. Their formation is attributed to hydrodeoxygenation of phenol and cyclohexanol. Since benzene and cyclohexane were only found in the condenser, benzene is expected to volatilize before hydrogenation can occur. Alkylation reactions and heavier products have been reported in thermocatalytic systems.52,57,58 The decreasing carbon balance with increasing milling time could be attributed to the formation of similar species. Over the course of the reaction, the yield of toluene consistently exceeds the combined yields of phenol and cyclohexanol, meaning the adsorption of phenolates on the catalyst surface is another source of carbon loss. Overall, the nickel catalyst behaves similarly in mechanochemical and thermochemical systems. Thus, it may be possible to extrapolate the results of other thermocatalytic reactions to predict the performance of mechanocatalytic processes for lignin conversion.
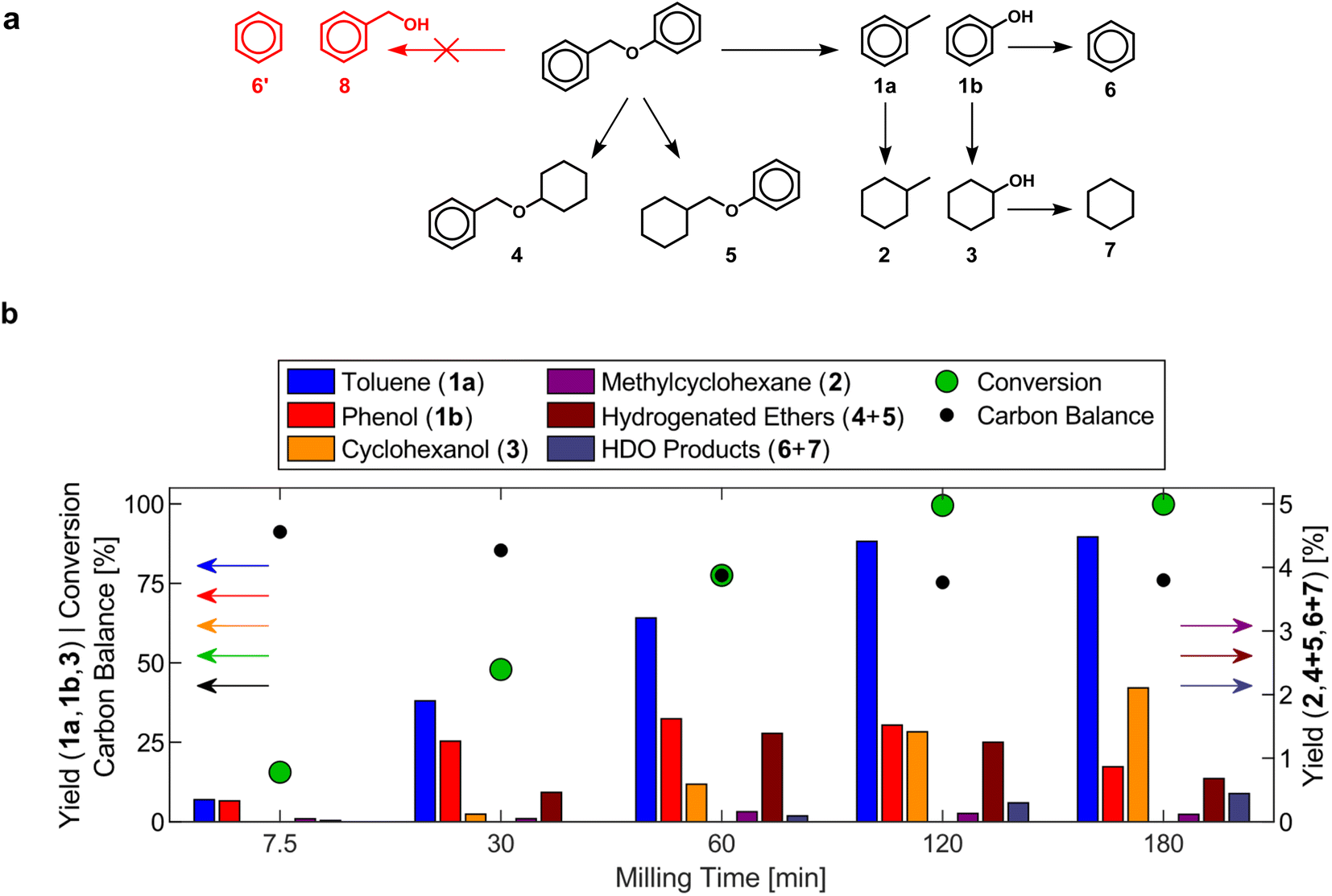 | ||
| Fig. 2 (a) Reaction network during the mechanocatalytic hydrogenolysis of BPE. (b) Product yields, conversion of BPE, and carbon balance during the mechanocatalytic hydrogenolysis of BPE with Ni53SiAl. 1.00 g Ni53SiAl, 0.20 g BPE, 25 mL S. S. vessel, 2 × 15 mm S. S. grinding balls, 20 Hz, 60 sccm H2. Numerical values are reported in Table S1.† | ||
A series of experiments was conducted to explore the catalytic transfer hydrogenolysis59,60 of BPE using isopropanol (IPA) in place of H2 (ESI Section S2.2†). IPA was added to the milling vessel (125 μL) and the reaction was conducted in a continuous flow of N2. Here, a 2.4% yield of toluene and a 1.6% yield of phenol was measured after 180 min. In principle, IPA can work as a hydrogen transfer agent for mechanocatalytic hydrogenolysis, but additional optimization and study would be necessary.
Mechanocatalyst activation
Before traditional thermocatalytic reactions, nickel must typically be reduced in situ at elevated temperatures (i.e., activated) to remove passivating oxide layers or reduce nickel oxide particles to the metal altogether. Here, thermal pretreatment of the passivated nickel catalysts was not necessary as milling in a reductive environment activated the catalyst. Initially, the nickel particles of the Ni53SiAl catalyst were encapsulated by a layer of NiO (Fig. 3a–c). However, thermally oxidized Ni53SiAl (Fig. S4†) showed significantly less activity (Fig. 3d). Attrition during milling is expected to remove the nickel oxide layer, liberating the active metallic nickel. Particularly when milling hard and brittle material, localized hot spots haven been shown to form13 which may produce temperatures high enough to directly reduce the NiO to metallic Ni in the presence of H2. The mechanochemical reduction of the NiO could explain why the rate of toluene production increased during milling with the oxidized Ni53SiAl. Importantly, the potential local thermal energy is not sufficient to break the BPE ether bond homolytically. Control experiments were conducted with BPE milled with Ni53SiAl in N2 and milled with just silica–alumina in H2, and no conversion was detected in either case. Thus, mechanical energy both drives the hydrogenolysis reaction and activates the catalyst, removing the need for high temperature pre-treatments and allowing for a fully ambient system.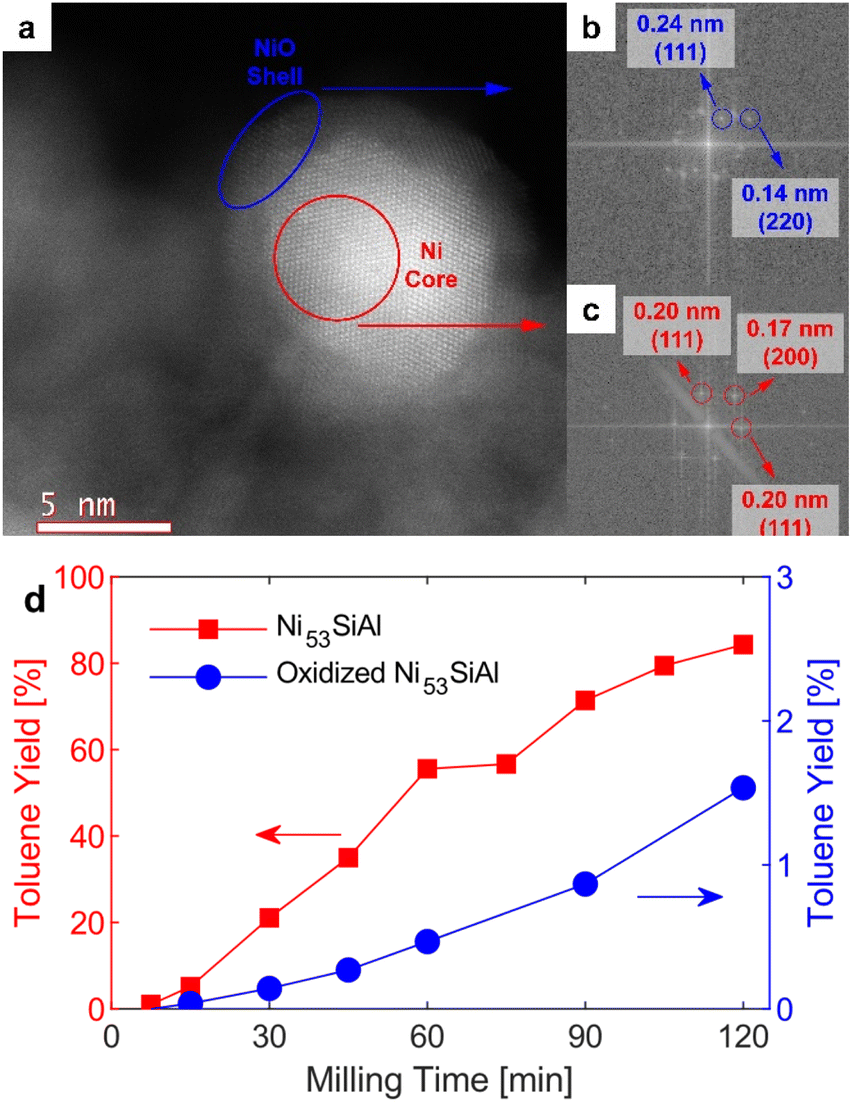 | ||
| Fig. 3 (a) STEM image of a nickel particle from Ni53SiAl showing a NiO shell around a Ni core. Fourier transformation of a region of (b) the shell (blue ellipse) and (c) the core (red circle) showing d-spacings corresponding to NiO and Ni, respectively. (d) Yields of toluene in the condenser during mechanocatalytic hydrogenolysis of BPE over Ni53SiAl (red squares) and thermally oxidized Ni53SiAl (blue circles). 1.00 g catalyst, 0.20 g BPE, 25 mL S. S. vessel, 2 × 15 mm S. S. grinding balls, 20 Hz, 60 sccm H2. | ||
Catalyst recycling and deactivation
The accumulation of toluene in the condenser allows for determining a lower limit of the hydrogenolysis rate. The toluene yield profile in the condenser during milling with fresh Ni53SiAl for 120 min (Fig. 4a – cycle 1) closely matched the progression of the toluene yield during the full product analysis (Fig. 2b). An induction period for toluene accumulation in the condenser of about 10 min was observed, which could be attributed to a catalyst activation period. However, at 7.5 min, the full toluene yield was 7% (compared to just 1% for the condenser toluene yield), suggesting the delay is an artifact of the reactor system rather than, for example, a period needed to mechanically activate the nickel.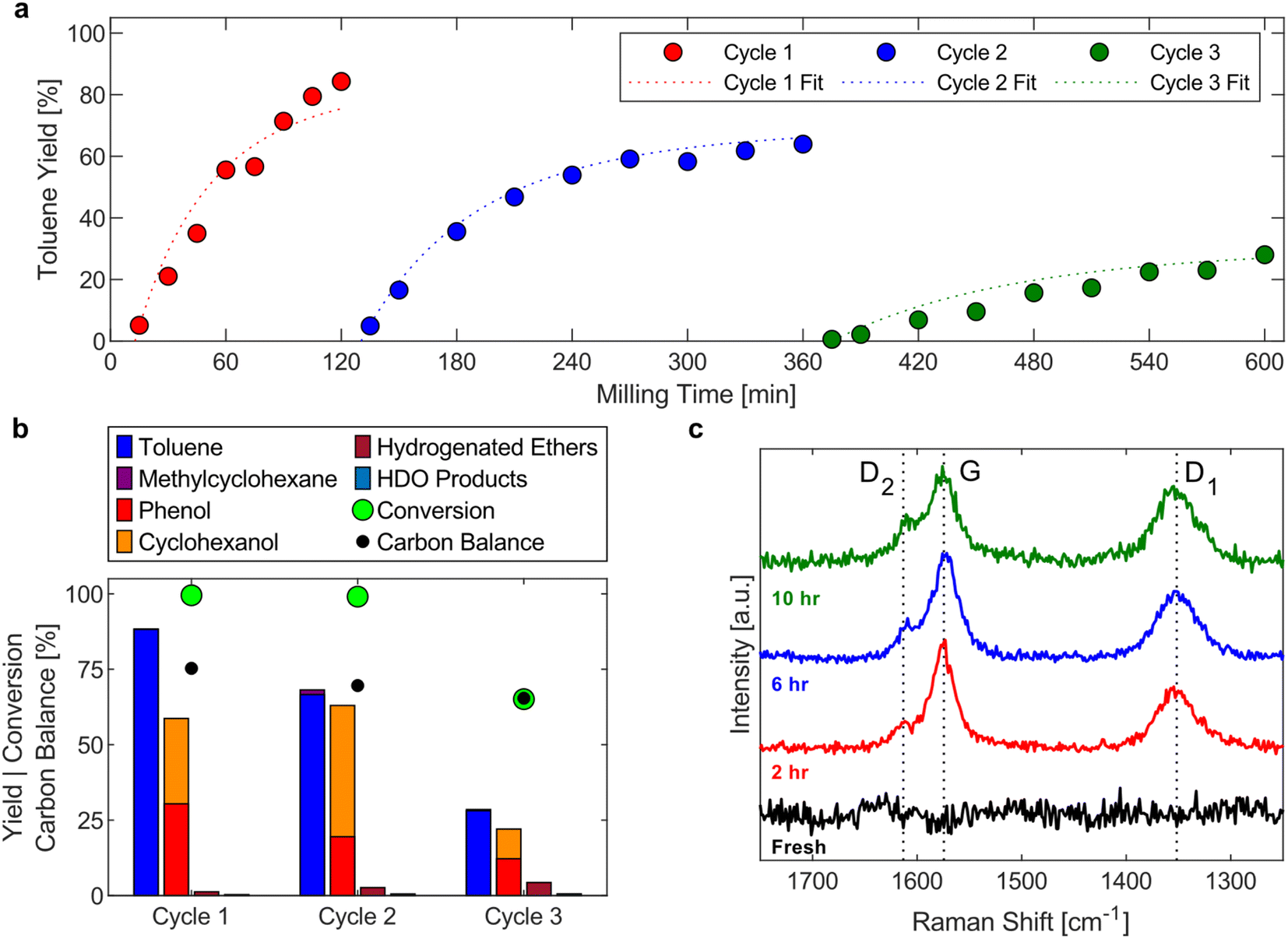 | ||
| Fig. 4 (a) Toluene yield in the condenser during the hydrogenolysis of BPE with fresh Ni53SiAl (cycle 1) and recycled Ni53SiAl (cycle 2 and cycle 3). Fits from the kinetic modelling are overlaid the toluene yields. (b) Full product yields, conversion of BPE, and carbon balance after the hydrogenolysis reaction of BPE with fresh and recycled Ni53SiAl. 1.00 g Ni53SiAl, 0.20 g BPE, 25 mL S. S. vessel, 2 × 15 mm S. S. grinding balls, 20 Hz, 60 sccm H2. (c) Raman spectra of fresh Ni53SiAl (black), Ni53SiAl after 2 h of reaction (after cycle 1) (red), Ni53SiAl after 6 h of reaction (after cycle 2) (blue), and Ni53SiAl after 10 h of reaction (after cycle 3) (green). | ||
To evaluate the longer-term stability of the catalyst under such intense conditions, the Ni53SiAl catalyst was recycled twice. After each reaction, the catalyst was washed with methanol to analyze the complete product profile, and then filtered and dried under vacuum overnight at room temperature. The toluene yields for each cycle (Fig. 4a) showed continuous deactivation of the catalyst. In the second cycle, the toluene yield flattened after three hours of milling, trending toward a maximum toluene yield of 65%. During the third cycle, the toluene yield increased over the whole 4 h experiment, but only a 30% yield was achieved. Similar to the first cycle, an induction period of about 10 min was observed in the second and third cycle. As expected from the toluene yield profiles, the full product profiles showed nearly 100% conversion of BPE by the end of cycle 2 and incomplete conversion (65%) by the end of cycle 3 (Fig. 4b). Deactivation of the nickel was also observed in the decreased selectivity of cyclohexanol to phenol between cycle 2 (69%) and cycle 3 (45%). Finally, significant carbon loss was observed for each cycle.
The carbon balance suggests carbonaceous deposits form as undesired side reactions with increasing milling time. Raman spectroscopy was performed on the spent catalyst after each cycle to qualitatively detect coke deposits (Fig. 4c). Three bands in the Raman spectra of spent catalysts were detected: the G-band (1580 cm−1; ideal graphitic lattice vibrations), the D1-band (1350 cm−1; vibrations of graphene layer edges), and the D2-band (1610 cm−1; surface graphene layers). These three bands are indicative of polyaromatic coke.61 At longer milling times, the relative intensity of the D-bands increased (compared to the G-band), which is supported by deconvolution and fitting of the bands (Table S2†). This increase of the D-bands and the decrease of the G-band suggest that the coke transformed to a more disordered and amorphous structure, which is expected with extended milling times.62 Additional changes to the catalyst between cycles were investigated as well. Agglomeration of nickel particles was not observed in STEM-EDS images of the spent catalysts (Fig. S6†) and no loss of nickel was detected between cycles (Fig. S7†). Additionally, significant oxidation of the nickel, due to handling in air, was not observed in XRD between each cycle (Fig. S8†), although this does not rule out the formation of amorphous oxide phases. These results suggest that the deactivation occurs predominantly through the formation of adsorbed phenolates, which further react to form polyaromatic coke, resulting in blockage of active sites rather than mechanical degradation of the catalyst due to milling.
Phenolates and catecholates are well known to strongly bind to catalysts,63 so the higher functionality of lignin is expected to exacerbate the formation of carbon deposits. The HDO of guaiacol, a more functionalized model compound, was briefly studied (ESI Section S4†). After 180 min, catechol, phenol, and cyclohexanol were detected at similar yields as HDO products during the hydrogenolysis of BPE (<1%), while the carbon balance was slightly lower (62% compared to 77%). Future work will focus on adapting reaction engineering strategies, such as simultaneous hydrolysis,64 to desorb phenolates or the use of redox active catalysts,65,66 to recover adsorbed products and prevent further side reactions.
Generally, the energy dose is a key descriptor for mechanochemical kinetics.21,67 However, the mechanochemical conditions were not varied in this study, so a simplified kinetic model is suitable here to provide a more quantitative description of the catalyst activity and deactivation. The system was approximated by two reactions: hydrogenolysis of BPE (kH) and deposit formation (kC), both first order with respect to BPE (eqn (1)–(5)). Initially, the conversion of BPE and yield of toluene for each cycle were fit independently to determine kH and kC (Fig. S5†). Both cycle 1 and cycle 3 had similar rates of coke formation (kC,1 = 0.17 h−1; kC,3 = 0.16 h−1), suggesting the activity of the catalyst toward coke formation is constant across all three cycles. Due to the high final conversion in cycle 2, calculating kC,2 directly would result in high uncertainty. Thus, the value for kC,3 was used during the fitting. The results of this kinetic model confirm the deactivation of the catalyst toward hydrogenolysis with a decrease in the rate constant between each cycle (kH,1 = 2.0 h−1 gNi−1; kH,2 = 0.89 h−1 gNi−1; kH,3 = 0.20 h−1 gNi−1). By the third cycle, the catalyst retained only a tenth of its initial activity. Next, all three cycles were fit simultaneously, using an average kC of 0.163 h−1, with a first order deactivation term introduced to the hydrogenolysis rate constant (eqn (6)). These fits were overlaid on the toluene yields in Fig. 5a. This fit shows an initial rate constant (kH,0) of 2.5 h−1 gNi−1 and that the catalyst deactivated at a rate of 34% per hour of milling (kD = 0.34 h−1). The good agreement between the observed activity of the catalyst and the deactivation model suggests a continuous mechanism is occurring during milling instead of discrete deactivation occurring between cycles.
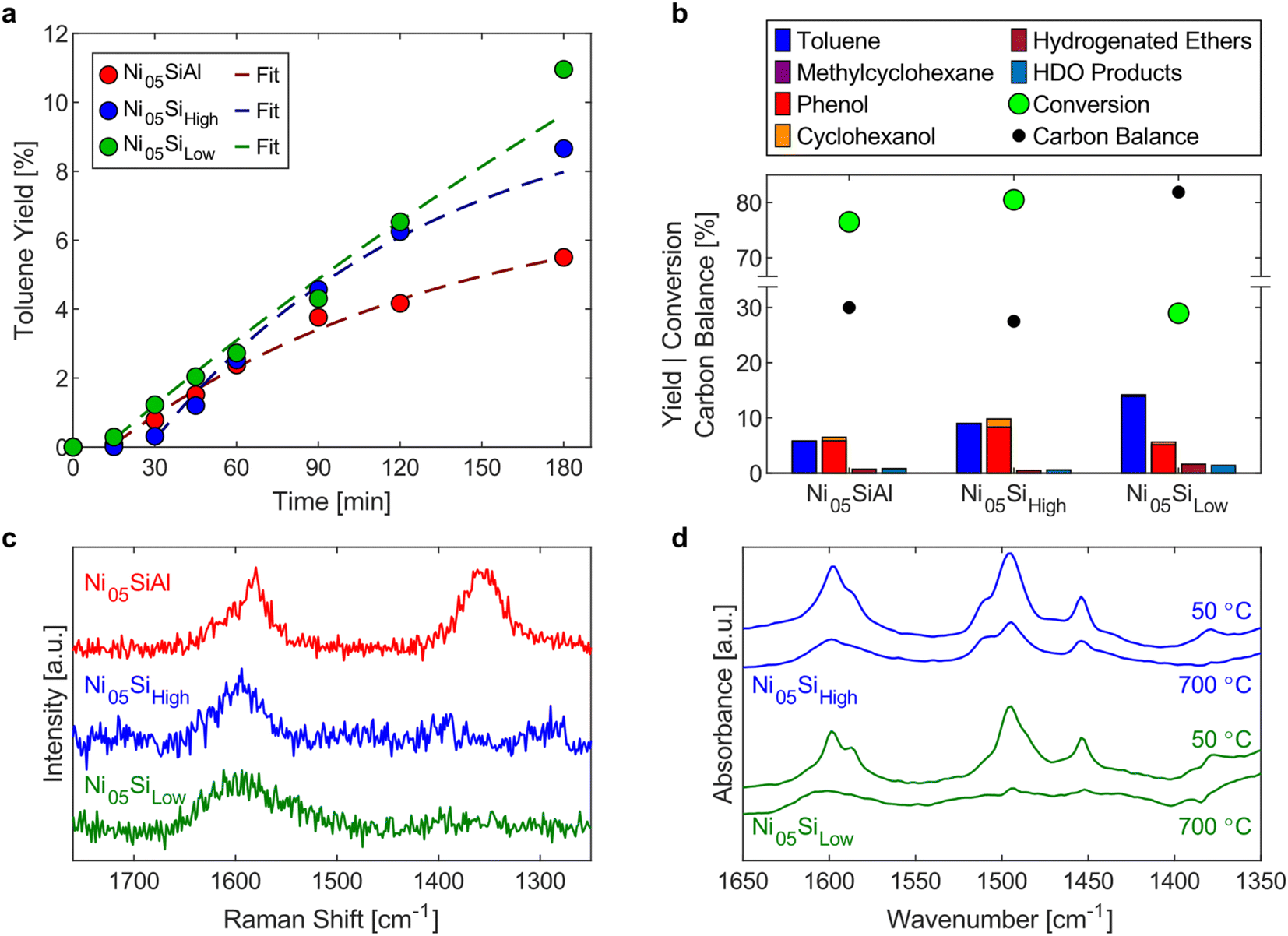 | ||
| Fig. 5 (a) Toluene yields during the hydrogenolysis of BPE with Ni05SiAl (red), Ni05Sihigh (blue), and Ni05Silow (green). Fits from the kinetic model are overlaid with the toluene yields. (b) Full product yields, conversion of BPE, and carbon balance after the hydrogenolysis reaction of BPE with Ni05SiAl, Ni05Sihigh, and Ni05Silow. 1.00 g catalyst, 0.20 g BPE, 25 mL S. S. vessel, 2 × 15 mm S. S. grinding balls, 20 Hz, 30 sccm H2. (c) Raman spectra of the spent Ni05SiAl (red), Ni05Sihigh (blue), and Ni05Silow (green). (d) DRIFTS spectra of the spent Ni05Sihigh (blue) and Ni05Silow (green) at 50 °C (top) and 700 °C (bottom). | ||
Influence of catalyst support
To explore the influence of the support properties on the reactivity, three catalysts with 5 wt% nickel loadings were synthesized using varying supports: a high surface area silica–alumina (499 m2 g−1, Ni05SiAl), high surface area silica (515 m2 g−1, Ni05Sihigh), and low surface area silica (6.7 m2 g−1, Ni05Silow). BPE hydrogenolysis was performed for 180 min with each catalyst. The yield of toluene over the course of the reaction (Fig. 5a) showed a constant increase for Ni05Silow, while the rate of toluene formation diminished with time for Ni05SiAl and Ni05Sihigh. The simplified kinetic model (eqn (4) and (5)) was applied to these reactions to determine the hydrogenolysis rate constant (fits shown in Fig. 5a). Ni05SiAl (kH = 0.81 h−1 gNi−1) and Ni05Silow (kH = 0.83 h−1 gNi−1) had similar hydrogenolysis rates, which were approximately 33% lower than the rate of Ni05Sihigh (kH = 1.21 h−1 gNi−1). The nickel particle size had a weak correlation with the activity of the catalyst. Nickel particles observed using STEM-EDS on Ni05SiAl and Ni05Silow agglomerated during milling to a size on the order of 100 nm (Fig. S9†), while the size of the nickel particles on Ni05Sihigh (15.1 nm) did not appreciably change over the course of the reaction. The reduction of the accessible nickel surface area by roughly one order of magnitude only corresponded to a 33% lower rate constant. This resistance to deactivation from agglomeration could have several explanations. The agglomeration may be indicative of more effective mechanical activation of nickel particles or larger nickel particles may more efficiently convert mechanical energy into chemical energy. The pores of the high surface area supports can also allow for nickel particles to be effectively shielded from direct mechanical forces. However, the similarity in the rate constants for the three catalysts suggests porosity does not significantly influence mechanical activation.While the hydrogenolysis rates were similar among the three catalysts, the production of undesirable side products was much higher on the high surface area catalysts. Reactions with either Ni05SiAl or Ni05Sihigh resulted in carbon balances of just 30%, while the reaction with Ni05Silow had a carbon balance of 82% (Fig. 5b). The three spent catalysts were analyzed with Raman spectroscopy to determine the nature of the carbon deposits (Fig. 5c). Spent Ni05SiAl exhibited the same characteristic bands for polyaromatic coke as the spent Ni53SiAl (Fig. 4c). The spectrum of spent Ni05Sihigh did not show polyaromatic coke, despite a similar deficit in the carbon balance. Only a single band at 1596 cm−1 was observed in the two spent silica supported catalysts, suggesting that the deposits are predominantly mono-aromatic species.68,69 The deposits on Ni05Sihigh and Ni05Silow were further analyzed using in situ DRIFTS (Fig. 5d). The region between 1400 and 1600 cm−1 corresponds to ring vibrations of aromatic species.70 After heating the samples to 700 °C, the presence of aromatics remaining on the Ni05Sihigh catalyst was much greater than the residue on the Ni05Silow or nonmilled Ni05Sihigh impregnated with BPE (Fig. S10†). This indicates chemisorption or encapsulation of the remaining aromatic species onto the Ni05Sihigh surface.
The higher surface area of the support is most likely responsible for the discrepancy in carbon balances, as opposed to catalytic activity for undesirable side reactions. As a result of milling, the Si–O bonds can break and create transient, reactive sites.71–73 Due to a larger abundance of surface Si–O bonds in the high surface area support, it is plausible that ruptures result in more reactive sites compared to the low surface area support. The higher density of active sites from rupturing can thus be responsible for more adsorbed species and a lower carbon balance on the Ni05Sihigh. Additionally, mechanochemical conditions could transform the catalyst in a way that leaves carbonaceous materials encapsulated in closed pores. A similar mechanism is expected to occur with Ni05SiAl, but with the inherent acid sites of the silica–alumina also catalyzing polyaromatic coke formation.74,75 Future work should focus on mitigating these side-reactions by optimizing milling intensity and developing supports that are less prone to rupture. Softer material, like activated carbon, should flex and bend under the mechanical impact, and harder material, like zirconium oxide, should be more resilient to rupture.
Conclusions
The mechanocatalytic hydrogenolysis of benzyl phenyl ether, a model lignin ether, was demonstrated using cost-effective, abundant catalysts under ambient H2 pressure, at room temperature, and without a solvent. The observed reaction network closely matches that for thermal hydrogenolysis of BPE, suggesting similar reactions can be expected to occur between mechanocatalytic and thermocatalytic reactions over metal catalysts. Milling not only supplied the mechanical energy to drive the reaction, but also provided metallic nickel surfaces in situ, avoiding the need for high temperature activation steps. Adsorption of phenolic species on mechanically activated domains of the catalyst support was identified as a major source for carbon loss, which provided guidance for the design of more robust mechanocatalysts. Overall, this works demonstrates the underlying chemistry necessary for developing a pathway for mild lignin depolymerization which may be able to avoid many of the safety, technical, and economic issues hindering the realization of lignin valorization at industrial scales.Author contributions
C. S. conceptualized and supervised the research. A. W. T. and E. V. P. performed mechanocatalytic reactions. K. L. H. performed catalyst characterization. S. N. and J. X. K. performed DRIFTS experiments. A. W. T. drafted the manuscript. All authors provided edits and comments for the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge The Renewable Bioproducts Institute for their support and funding of A. W. T. and E. V. P. through the Paper Science & Engineering Fellowship. S. N. was supported by the National Science Foundation Graduate Research Fellowship under Grant No. DGE-1650044. The XRD and Raman Spectroscopy work was performed in part at the Materials Characterization Facility (MCF) at Georgia Institute of Technology. The MCF is jointly supported by the GT Institute for Materials (IMat) and the Institute for Electronics and Nanotechnology (IEN), which is a member of the National Nanotechnology Coordinated Infrastructure supported by the National Science Foundation (Grant ECCS-1542174).Notes and references
- H. Luo and M. M. Abu-Omar, in Encyclopedia of Sustainable Technologies, ed. M. A. Abraham, Elsevier, Oxford, 2017, DOI:10.1016/B978-0-12-409548-9.10235-0, pp. 573–585.
- W. Boerjan, J. Ralph and M. Baucher, Annu. Rev. Plant Biol., 2003, 54, 519–546 CrossRef CAS PubMed.
- A. W. Tricker, M. J. Stellato, T. T. Kwok, N. S. Kruyer, Z. Wang, S. Nair, V. M. Thomas, M. J. Realff, A. S. Bommarius and C. Sievers, ChemSusChem, 2020, 13, 4624–4632 CrossRef CAS PubMed.
- W. Schutyser, T. Renders, S. Van den Bosch, S. F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed.
- T. Renders, G. Van den Bossche, T. Vangeel, K. Van Aelst and B. Sels, Curr. Opin. Biotechnol., 2019, 56, 193–201 CrossRef CAS PubMed.
- E. Cooreman, T. Vangeel, K. Van Aelst, J. Van Aelst, J. Lauwaert, J. W. Thybaut, S. Van den Bosch and B. F. Sels, Ind. Eng. Chem. Res., 2020, 59, 17035–17045 CrossRef CAS.
- K. J. Ardila-Fierro and J. G. Hernández, ChemSusChem, 2021, 14, 2145–2162 CrossRef CAS PubMed.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- S. Mateti, M. Mathesh, Z. Liu, T. Tao, T. Ramireddy, A. M. Glushenkov, W. Yang and Y. I. Chen, Chem. Commun., 2021, 57, 1080–1092 RSC.
- T. Friscic and W. Jones, Cryst. Growth Des., 2009, 9, 1621–1637 CrossRef CAS.
- C. Suryanarayana, Prog. Mater. Sci., 2001, 46, 1–184 CrossRef CAS.
- A. W. Tricker, G. Samaras, K. L. Hebisch, M. J. Realff and C. Sievers, Chem. Eng. J., 2020, 382, 122954 CrossRef CAS.
- S. Amirjalayer, H. Fuchs and D. Marx, Angew. Chem., Int. Ed., 2019, 58, 5232–5235 CrossRef CAS PubMed.
- S. Immohr, M. Felderhoff, C. Weidenthaler and F. Schüth, Angew. Chem., Int. Ed., 2013, 52, 12688–12691 CrossRef CAS PubMed.
- A. W. Tricker, K. L. Hebisch, M. Buchmann, Y.-H. Liu, M. Rose, E. Stavitski, A. J. Medford, M. C. Hatzell and C. Sievers, ACS Energy Lett., 2020, 5, 3362–3367 CrossRef CAS.
- Z. Liu, J. Liu, S. Mateti, C. Zhang, Y. Zhang, L. Chen, J. Wang, H. Wang, E. H. Doeven and P. S. Francis, ACS Nano, 2019, 13, 1394–1402 CAS.
- D. Tan, L. Loots and T. Friščić, Chem. Commun., 2016, 52, 7760–7781 RSC.
- B. Szczęśniak, S. Borysiuk, J. Choma and M. Jaroniec, Mater. Horiz., 2020, 7, 1457–1473 RSC.
- V. Štrukil, ChemSusChem, 2021, 14, 330–338 CrossRef PubMed.
- A. W. Tricker, A. A. Osibo, Y. Chang, J. X. Kang, A. Ganesan, E. Anglou, F. Boukouvala, S. Nair, C. W. Jones and C. Sievers, ACS Sustainable Chem. Eng., 2022, 10, 11338–11347 CrossRef CAS.
- M. Baláž, Environmental Mechanochemistry: Recycling Waste into Materials Using High-Energy Ball Milling, Springer Nature, 2021 Search PubMed.
- A. Barakat, C. Mayer-Laigle, A. Solhy, R. A. Arancon, H. De Vries and R. Luque, RSC Adv., 2014, 4, 48109–48127 RSC.
- S. Kuga and M. Wu, Cellulose, 2019, 26, 215–225 CrossRef CAS.
- Q. Zhang and F. Jerome, ChemSusChem, 2013, 6, 2042–2044 CrossRef CAS PubMed.
- F. Jerome, G. Chatel and K. D. Vigier, Green Chem., 2016, 18, 3903–3913 RSC.
- M. Kaldstrom, N. Meine, C. Fares, R. Rinaldi and F. Schuth, Green Chem., 2014, 16, 2454–2462 RSC.
- F. Schuth, R. Rinaldi, N. Meine, M. Kaldstrom, J. Hilgert and M. D. K. Rechulski, Catal. Today, 2014, 234, 24–30 CrossRef.
- M. D. K. Rechulski, M. Kaldstrom, U. Richter, F. Schuth and R. Rinaldi, Ind. Eng. Chem. Res., 2015, 54, 4581–4592 CrossRef.
- D. Y. Lee and M. Sumimoto, Holzforschung, 1990, 44, 347–350 CrossRef CAS.
- Z. H. Wu, M. Matsuoka, D. Y. Lee and M. Sumimoto, Mokuzai Gakkaishi, 1991, 37, 164–171 CAS.
- S. G. Yao, J. K. Mobley, J. Ralph, M. Crocker, S. Parkin, J. P. Selegue and M. S. Meier, ACS Sustainable Chem. Eng., 2018, 6, 5990–5998 CrossRef CAS.
- T. Kleine, J. Buendia and C. Bolm, Green Chem., 2013, 15, 160–166 RSC.
- A. D. Brittain, N. J. Chrisandina, R. E. Cooper, M. Buchanan, J. R. Cort, M. V. Olarte and C. Sievers, Catal. Today, 2018, 302, 180–189 CrossRef CAS.
- S. Dabral, H. Wotruba, J. G. Hernandez and C. Bolm, ACS Sustainable Chem. Eng., 2018, 3242–3254, DOI:10.1021/acssuschemeng.7b03418.
- G. Heinicke and I. Lischke, Z. Phys. Chem., 1963, 3, 355–356 CAS.
- C. Bolm and J. G. Hernandez, Angew. Chem., Int. Ed., 2019, 58, 3285–3299 CrossRef CAS PubMed.
- S. Mori, W. C. Xu, T. Ishidzuki, N. Ogasawara, J. Imai and K. Kobayashi, Appl. Catal., A, 1996, 137, 255–268 CrossRef CAS.
- K. Yatagai, Y. Shishido, R. Gemma, T. Boll, H.-H. Uchida and K. Oguri, Int. J. Hydrogen Energy, 2020, 45, 5264–5275 CrossRef CAS.
- G. Mulas, R. Campesi, S. Garroni, F. Delogu and C. Milanese, Appl. Surf. Sci., 2011, 257, 8165–8170 CrossRef CAS.
- Y. Sawama, T. Kawajiri, M. Niikawa, R. Goto, Y. Yabe, T. Takahashi, T. Marumoto, M. Itoh, Y. Kimura and Y. Monguchi, ChemSusChem, 2015, 8, 3773–3776 CrossRef CAS PubMed.
- Y. Sawama, N. Yasukawa, K. Ban, R. Goto, M. Niikawa, Y. Monguchi, M. Itoh and H. Sajiki, Org. Lett., 2018, 20, 2892–2896 CrossRef CAS PubMed.
- D. J. Nash, D. T. Restrepo, N. S. Parra, K. E. Giesler, R. A. Penabade, M. Aminpour, D. Le, Z. Li, O. K. Farha and J. K. Harper, ACS Omega, 2016, 1, 1343–1354 CrossRef CAS PubMed.
- G.-F. Han, F. Li, Z.-W. Chen, C. Coppex, S.-J. Kim, H.-J. Noh, Z. Fu, Y. Lu, C. V. Singh and S. Siahrostami, Nat. Nanotechnol., 2021, 16, 325–330 CrossRef CAS PubMed.
- S. Reichle, M. Felderhoff and F. Schüth, Angew. Chem., Int. Ed., 2021, 60, 26385–26389 CrossRef CAS PubMed.
- A. G. Sergeev and J. F. Hartwig, Science, 2011, 332, 439–443 CrossRef CAS PubMed.
- X. Wang and R. Rinaldi, ChemSusChem, 2012, 5, 1455–1466 CrossRef CAS PubMed.
- D. Wu, Q. Wang, O. V. Safonova, D. V. Peron, W. Zhou, Z. Yan, M. Marinova, A. Y. Khodakov and V. V. Ordomsky, Angew. Chem., Int. Ed., 2021, 60, 12513–12523 CrossRef CAS PubMed.
- L. Qi, A. Chamas, Z. R. Jones, E. D. Walter, D. W. Hoyt, N. M. Washton and S. L. Scott, J. Am. Chem. Soc., 2019, 141, 17370–17381 CrossRef CAS PubMed.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- E. M. v. Duzee and H. Adkins, J. Am. Chem. Soc., 1935, 57, 147–151 CrossRef.
- J. He, L. Lu, C. Zhao, D. Mei and J. A. Lercher, J. Catal., 2014, 311, 41–51 CrossRef CAS.
- J. He, C. Zhao and J. A. Lercher, J. Am. Chem. Soc., 2012, 134, 20768–20775 CrossRef CAS PubMed.
- L. Jiang, H. Guo, C. Li, P. Zhou and Z. Zhang, Chem. Sci., 2019, 10, 4458–4468 RSC.
- R. Parthasarathi, R. A. Romero, A. Redondo and S. Gnanakaran, J. Phys. Chem. Lett., 2011, 2, 2660–2666 CrossRef CAS.
- L. Li, H. J. Fan and H. Q. Hu, Fuel, 2015, 153, 70–77 CrossRef CAS.
- B. Güvenatam, E. H. Heeres, E. A. Pidko and E. J. Hensen, J. Mol. Catal. A: Chem., 2015, 410, 89–99 CrossRef.
- B. Guvenatam, O. Kursun, E. H. J. Heeres, E. A. Pidko and E. J. M. Hensen, Catal. Today, 2014, 233, 83–91 CrossRef.
- C. Espro, B. Gumina, T. Szumelda, E. Paone and F. Mauriello, Catalysts, 2018, 8, 313 CrossRef.
- F. Mauriello, E. Paone, R. Pietropaolo, A. M. Balu and R. Luque, ACS Sustainable Chem. Eng., 2018, 6, 9269–9276 CrossRef CAS.
- A. Sadezky, H. Muckenhuber, H. Grothe, R. Niessner and U. Pöschl, Carbon, 2005, 43, 1731–1742 CrossRef CAS.
- T. Xing, L. H. Li, L. Hou, X. Hu, S. Zhou, R. Peter, M. Petravic and Y. Chen, Carbon, 2013, 57, 515–519 CrossRef CAS.
- S. P. Pujari, L. Scheres, A. T. Marcelis and H. Zuilhof, Angew. Chem., Int. Ed., 2014, 53, 6322–6356 CrossRef CAS PubMed.
- M. J. Stellato, G. Innocenti, A. S. Bommarius and C. Sievers, Catalysts, 2021, 11, 721 CrossRef CAS.
- S. M. Schimming, O. D. LaMont, M. Konig, A. K. Rogers, A. D. D'Amico, M. M. Yung and C. Sievers, ChemSusChem, 2015, 8, 2073–2083 CrossRef CAS PubMed.
- T. Prasomsri, T. Nimmanwudipong and Y. Román-Leshkov, Energy Environ. Sci., 2013, 6, 1732–1738 RSC.
- M. Kessler and R. Rinaldi, Front. Chem., 2022, 1099, DOI:10.3389/fchem.2021.816553.
- U. P. Agarwal, J. D. McSweeny and S. A. Ralph, J. Wood Chem. Technol., 2011, 31, 324–344 CrossRef CAS.
- F. Madzharova, Z. Heiner and J. Kneipp, J. Phys. Chem. C, 2020, 124, 6233–6241 CrossRef CAS PubMed.
- G. S. Foo, A. K. Rogers, M. M. Yung and C. Sievers, ACS Catal., 2016, 6, 1292–1307 CrossRef CAS.
- F. Delogu, J. Phys. Chem. C, 2011, 115, 21230–21235 CrossRef CAS.
- F. Delogu, Int. J. Hydrogen Energy, 2011, 36, 15145–15152 CrossRef CAS.
- G. Innocenti, D. J. Benkeser, J. E. Dase, X. Wirth, C. Sievers and K. E. Kurtis, Fuel, 2021, 299, 120892 CrossRef CAS.
- D. M. Bibby, R. F. Howe and G. D. McLellan, Appl. Catal., A, 1992, 93, 1–34 CrossRef CAS.
- M. Guisnet and P. Magnoux, Appl. Catal., A, 2001, 212, 83–96 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2su00089j |
| This journal is © The Royal Society of Chemistry 2023 |