
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tailoring lixiviant properties to optimise selectivity in E-waste recycling†
Jennifer M.
Hartley
 *a,
Sean
Scott
a,
Rodolfo
Marin Rivera
a,
Phil
Hunt
a,
Anthony J.
Lucio
a,
Philip
Bird
b,
Robert
Harris
a,
Gawen R. T.
Jenkin
b and
Andrew P.
Abbott
a
*a,
Sean
Scott
a,
Rodolfo
Marin Rivera
a,
Phil
Hunt
a,
Anthony J.
Lucio
a,
Philip
Bird
b,
Robert
Harris
a,
Gawen R. T.
Jenkin
b and
Andrew P.
Abbott
a
aSchool of Chemistry, University of Leicester, Leicester, LE1 7RH, UK. E-mail: jmh84@le.ac.uk
bSchool of Geography, Geology and the Environment, University of Leicester, Leicester, LE1 7RH, UK
First published on 4th November 2022
Abstract
The scale of E-waste production makes the selective recovery of technology metals an important research topic. It has previously been shown that deep eutectic solvents, DESs, can be used to rapidly digest gold and in the current study the effect of varying water and ethylene glycol content on the ability to selectively recover metals in DESs was investigated. It was found that increased water content resulted in an increase in metal etching rates for copper due to the decreasing viscosity of the solution, but etching rates of nickel, silver and gold were decreased due to the competition between chloride and oxide/hydroxide chemistry causing passivating films to form. Iodine was used as a catalyst and speciation was investigated in different solvent compositions. The ratio of the two trihalide species was mostly unaffected by solvent composition, but the presence of molecular iodine was detected with 20 wt% water due to decreased iodine solubility resulting from lower chloride concentration. It was determined that solvent physical properties were most important for metal etching rates at low water content, but at high water content the effect of oxide/hydroxide chemistry was more significant, resulting in the possibility of selective metal etching of copper from PCBs.
Sustainability spotlightDue to the ever-increasing demand for electronic goods, the requirement for raw materials also increases, alongside the generation of large amounts of E-waste from discarded electronic goods. In order to satisfy this demand without opening new mines, the E-waste must be recycled rather than being sent to landfill. The sustainable advancement of the present work is that it provides a method of selectively etching gold from printed circuit boards through simple addition of water to a deep eutectic solvent. The present work aligns with Goal 12 “Responsible Consumption and Production” because it is helping to form a circular economy, by providing a selective recovery route for technology-critical metals. |
1 Introduction
Waste electrical and electronic equipment (E-waste) was estimated to be approximately 53.6 Mt in 2019 and potentially growing to 74.7 Mt by 2030.1 It is of concern for several reasons, including hazardous materials which can leach from landfill,2 the production of toxins resulting from its poorly controlled processing, and the large use of critical metals which are disparately distributed into complex architectures. Only about 17% of E-waste is collected and processed by a variety of controlled physical, hydrometallurgical and pyrometallurgical methods. The remainder is unofficially processed, usually in Asian and African countries,3 resulting in localised pollution of heavy metals, halogenated organic compounds and acidic lixiviants.Consequently, there has been a great interest in developing more energy-efficient and environmentally-compatible processes. One of these methods is via the electrochemical oxidation of metals using a redox catalyst that can be regenerated in situ, such as Cl−/Cl2, O2/H2O2, Cu2+, Fe3+, or iodine complexes.4–6 Iodine is a powerful, yet moderately benign, oxidising agent that has been used for metal and mineral oxidation, in both aqueous7–9 and IL/DES environments,10–13 often with a focus on the precious metal (high value) components of the system. A key property of ILs and DESs is that oxide/hydroxide chemistry can be avoided, as the anionic components of the solvent will dominate the chemistry.14 This results in metal speciation being unaffected by small amounts of water.15 However, these non-aqueous systems are often characterised by very high viscosity of the solvent. For example, choline chloride-based DESs commonly have viscosities greater than 30 mPa s at room temperature.16,17 For the phosphonium liquids, the values are 8167 > X > 303 mPa s.18 This is obviously problematic for anything where mass transport or conductivity can be a limiting factor.
In materials processing, a lower viscosity is a desirable property for increased mass transport properties and higher conductivity values. It is possible to decrease the viscosity of DESs significantly by either modifying the HBD![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ChCl ratios, or by adding small amounts of water. For example, the addition of only ca. 6 wt% water to a DES formed from choline chloride and ethylene glycol decreases the solution viscosity at 20 °C from 52.1 mPa s to 21.0 mPa s, i.e. by almost half.19 This decrease in viscosity improves the quality of metal plating without altering the copper species in solution.20,21 However, above 40 wt% water, the solvent matrix breaks down to form an aqueous solution of DES components.22 One of the main benefits of using an IL or DES is the minimisation or lack of oxide/hydroxide chemistry, allowing the accessibility of metals and their alloys that generally could not be achieved in aqueous systems, including aluminium, magnesium, and germanium.14,23–25 By adding water to decrease the viscosity, there is a chance that passivating oxide or chloride layers may form on the more reactive metals during processing, and hinder dissolution processes, which may make the process unusable, or might instead impart some selectivity of dissolution. The most commonly used version of the DES formed from ethylene glycol and choline chloride has a 2:1 molar ratio of EG:ChCl. Systems formed with a higher ethylene glycol content display lower viscosity, and lower tendency for the choline chloride to crystallise out at low ambient temperatures.26
:ChCl ratios, or by adding small amounts of water. For example, the addition of only ca. 6 wt% water to a DES formed from choline chloride and ethylene glycol decreases the solution viscosity at 20 °C from 52.1 mPa s to 21.0 mPa s, i.e. by almost half.19 This decrease in viscosity improves the quality of metal plating without altering the copper species in solution.20,21 However, above 40 wt% water, the solvent matrix breaks down to form an aqueous solution of DES components.22 One of the main benefits of using an IL or DES is the minimisation or lack of oxide/hydroxide chemistry, allowing the accessibility of metals and their alloys that generally could not be achieved in aqueous systems, including aluminium, magnesium, and germanium.14,23–25 By adding water to decrease the viscosity, there is a chance that passivating oxide or chloride layers may form on the more reactive metals during processing, and hinder dissolution processes, which may make the process unusable, or might instead impart some selectivity of dissolution. The most commonly used version of the DES formed from ethylene glycol and choline chloride has a 2:1 molar ratio of EG:ChCl. Systems formed with a higher ethylene glycol content display lower viscosity, and lower tendency for the choline chloride to crystallise out at low ambient temperatures.26
It has recently been determined that the HBD plays a critical role for the species of iodine that are formed in DES media, with three general speciation types observed: mixed I2Cl− and I3−, mixed I2 and I3−, and I3− alone.27 The amount of I2Cl− present was linked to the activity of the chloride anion, with high chloride activity resulting in higher amounts of I2Cl−. The chloride activity itself is related to the strength of the interactions between chloride and the HBD used. However, the alteration of solvent composition via addition of water or using a different EG:ChCl ratio may result in different speciation (and hence reactivity) of iodine, and also for dissolved metal species. It is therefore critical to understand any iodine speciation changes and whether or not these will affect the oxidising power of the system. In this work, we present how the water and ethylene glycol content of a DES formed from EG:ChCl affects the oxidation of E-waste metals when iodine is used as an oxidising agent. It was found that differences in oxidation behaviour were not related to a change in iodine species, instead being due to an increase in oxide chemistry resulting in selective passivation of the metal substrates. The metals investigated within the present work were selected due to their common presence in printed circuit boards. Copper is the major conducting component, and nickel is used as a barrier layer between the copper and silver/gold layers to prevent the diffusion of metals. Silver and gold are used to protect the copper from oxidation and also to provide contact points and edge connectors.
2 Experimental
2.1 Solvent preparation
The DES was made from 1 molar equivalent of choline chloride (ChCl) (Scientific Laboratory Supplies, 99%) and 2 mol eq. of ethylene glycol (EG) (98%, Sigma Aldrich) by stirring the two components at 80 °C in a sealed vessel until a homogenous liquid had formed, hereafter referred to as EG:ChCl. Due to the hygroscopic nature of the components, the as-prepared DES will contain ca. 1–2 wt% water.28 This was then mixed with deionised water to form solutions containing 5, 10, 15, and 20 wt% water, before sufficient iodine (Fisher Scientific, ≥95%) was added to obtain a nominal 0.1 mol kg−1 concentration solution. It must be noted that due to the increasing water content disrupting the chloride activity of the DESs, not all the iodine dissolved. For the systems made from EG:ChCl with different EG contents (ratios of 2:1, 3:1, 4:1, and 10:1), the same preparation procedure was followed.
2.2 Metal digestion as a function of water content experiments
Copper, silver, gold, and nickel wires with diameters of 1.25, 1.0, 0.25, and 1.0 mm, respectively, were mounted for the etching experiments by embedding into epoxy resin (block with a cylindrical shape, Fig. S1†) and polished with SiC abrasive paper down to 1200 grit size, followed by polishing with diamond paste (6, 3, and 1 μm). Etching was carried out by submerging the surface of the block in a well-stirred solution of the DES at 50 °C with a nominal iodine concentration of 0.1 mol kg−1. The constant agitation was applied to maintain a homogeneous and continuous flow of oxidising species across the sample surface. The block was held stable with a clamp so that the stirrer bar did not contact the etching surface. The temperature was controlled using a hotplate stirrer with a glass-coated thermocouple placed directly in the etching solution.The sample was etched step-wise in 50 mL of solvent. Etching was performed for an effective period of 1 hour at 50 °C. After each step, the sample was removed from the solvent, rinsed with acetone to remove any residual iodine, followed by deionised water, and dried in air. After each step, 2D and 3D optical images of the sample were captured by using a Zeta Instruments Zeta 2000 optical profiler using the inbuilt Zeta 3D software version 1.8.5. Images were constructed by determining the features of an image that are in focus at different heights. These were then analysed to produce a reconstructed 3D topography of the surface of each one of the metal wires. Line profiles were measured across the surface, where the dissolution rate was measured relative to a flat surface of an insoluble phase, i.e. the resin holding the metallic wires. After the corresponding images were obtained, the sample was returned to the solvent. The experiments were carried out in triplicate to ensure the reproducibility of the results. The errors were determined as the standard deviations of the etch depths.
Printed circuit board samples were leached in EG:ChCl with no added water and EG:ChCl with 40 wt% water at 50 °C in 0.1 mol kg−1 iodine in 80 mL of EG:ChCl, with gentle stirring over the course of 5 hours. After each hour, the samples were removed from the etching solution, rinsed with acetone and deionised water to remove the solvent and iodine, and imaged to qualitatively identify how long it took to completely remove all metals from the surface. Each individual gold rectangle is 2.5 × 1 mm, and is composed of 47 μm copper, 7 μm nickel, and 0.75 μm gold.4
2.3 Speciation
The speciation of iodine in EG:ChCl as a function of water and ethylene glycol content was determined via extended X-ray absorption fine structure (EXAFS). Spectra were measured at B18 of the Diamond Light Source synchrotron, with measurements recorded at the iodine K-edge, nominally 33169 eV. Spectra were recorded in transmission mode, using ionisation chambers, with the monochromator being a Si(311) double crystal. Reference spectra for amplitude calibration were made using a reference sample made from potassium iodide ground into a pellet with cellulose. The samples were contained in plastic cuvettes with a path length of 1 cm, which were mounted in the beam, and aligned at approximately 55° with respect to the X-ray beam. Nine spectra were recorded for each sample in quick EXAFS mode. These were then averaged, calibrated and background subtracted with the program Athena.29 The EXAFS spectra were fitted using the program Artemis to determine type and number of coordinating atoms, and calculate the interatomic distances and their root-mean-square variations (σ2). Quoted uncertainties on fitted parameters are equal to two standard deviations. The fitting range was 3.9 to 12.0 Å−1, and series fits were made using all water contents or all EG:ChCl ratios. Coordination number, reference energy (E0) and interatomic distances were always allowed to vary. Initial fits were made with σ2 for each element remaining the same, but allowed to vary during secondary fits to test if the additional parameters significantly improved the fit index.
The UV-vis spectra were recorded with a UV5Bio (Mettler Toledo) UV-vis spectrometer, between wavelengths of 190 and 1100 nm. These spectra were obtained using a 0.1 mm glass slide cuvette and 0.0025 mol kg−1 iodine for optimised peak resolution. Data presented in this study was normalised to the 260 nm peak to aid in determining species ratios.
3 Results and discussion
3.1 Kinetics of metal oxidation – addition of water
It was previously shown that Ni and Au could be recovered from printed circuit boards using CuII and FeIII as selective etchants for copper. Etch rates of up to 27 μm h−1 and 73 μm h−1 were measured for copper etching at 25 and 50 °C respectively using 1 mol kg−1 CuII in a calcium chloride ethylene glycol eutectic.4 Similar etch rates were also determined for gold using iodine in the EG:ChCl system.10 In the present study the effect of water in the EG:ChCl system was studied and found to have a significant impact on the etching rates of copper, nickel, silver and gold. Fig. 1a shows the effect of water content on the etching rates after 1 hour, while the full kinetic experiments can be found in Fig. S2.† It can be seen that increased water content results in the etching rates increasing for copper and gold, but decreasing for nickel and silver. The fastest etching was observed during the first 10 minutes, indicating that an equilibrium is developing between the diffusion of ions into and out of the double layer. This is also the timeframe within which any passivating layers are most likely to develop. In EG:ChCl, these passivating layers are likely to be chloride or glycolate complexes, as was seen after the anodic dissolution of copper.30 Additionally, nickel ions are known to be sensitive to the type of HBD present in solution, including water. For example, nickel ions in EG:ChCl coordinate with EG to form [Ni(EG)3]2+, whereas in a urea: choline chloride system a [Ni(urea)6]2+ species is formed.15 This sensitivity to the HBD continues in other choline chloride-based DESs.31
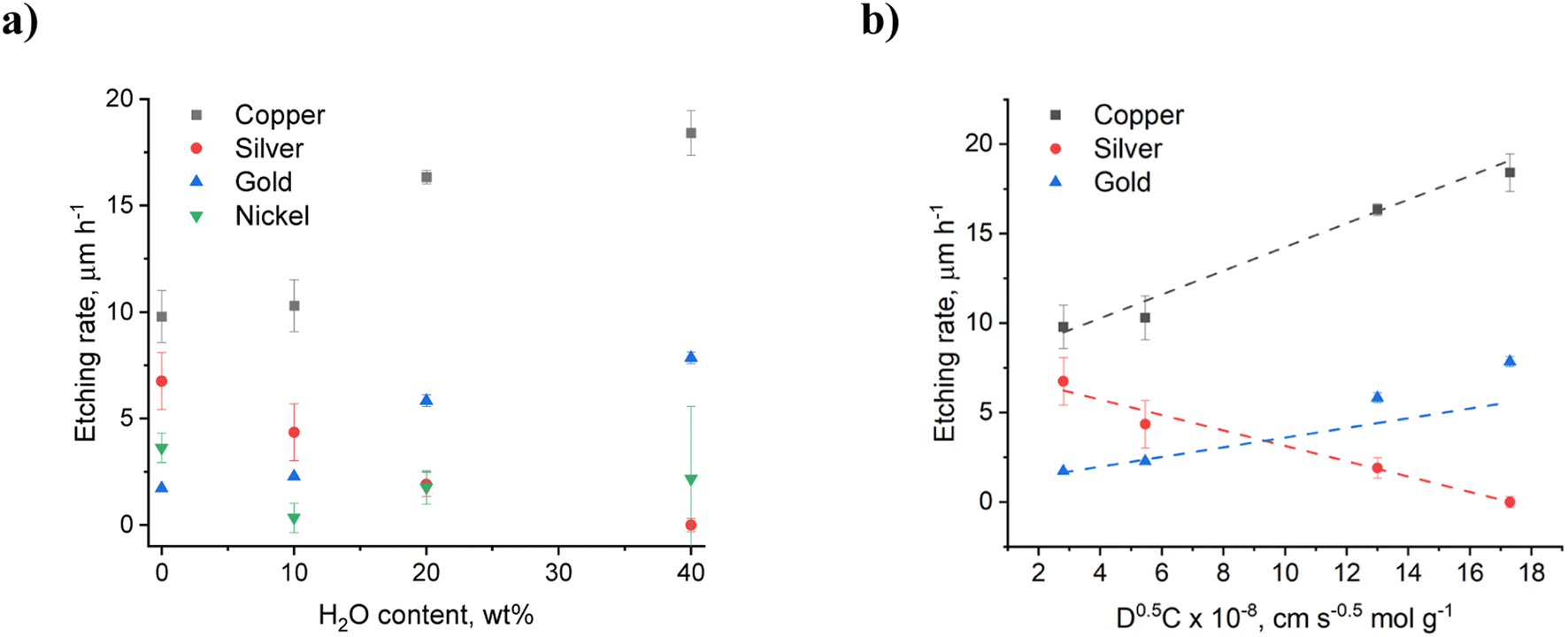 | ||
| Fig. 1 (a) Etching rates for copper, silver, gold, and nickel wires after 1 hour, (a) as a function of water content in EG:ChCl containing a nominal concentration of 0.1 mol kg−1 iodine at 50 °C, and (b) as a function of diffusion coefficient and iodine concentration (Ni excluded due to complex behaviour). | ||
Assuming that the etching rate is at steady state, then the current for dissolution, i, should be given by:
| i(t) = nFAC*(D/πt)0.5 | (1) |
The etching rate of copper in EG:ChCl with 0.1 mol kg−1 iodine does not vary significantly between 0 wt% and 10 wt% water content, remaining at around 10 μm h−1. However, the etching rate is almost doubled from a water content of 20 wt% and higher. 3DM images of the copper wire after 1 hour show that etching in dry EG:ChCl results in the enhancement of grain boundaries, whereas etching in the system containing 40 wt% water results in a smooth polished surface (Fig. 2). 3DM images of the unetched wires are available in Fig. S3.† The oxidation of gold was predicted to be slow in EG:ChCl due to the unfavourable thermodynamics of the reaction,33 and was the metal that etched the slowest. Etching takes place in the dry system at a rate of approximately 2 μm h−1, and followed a similar trend to that observed during copper etching, i.e. the addition of 10 wt% water has little impact on etching rate, but the etching rate increases significantly when the water content is 20 wt% or greater. As observed for copper, etching in the 0 wt% added water system took place at the grain boundaries, however the presence of 40 wt% water did not result in a polished gold surface.
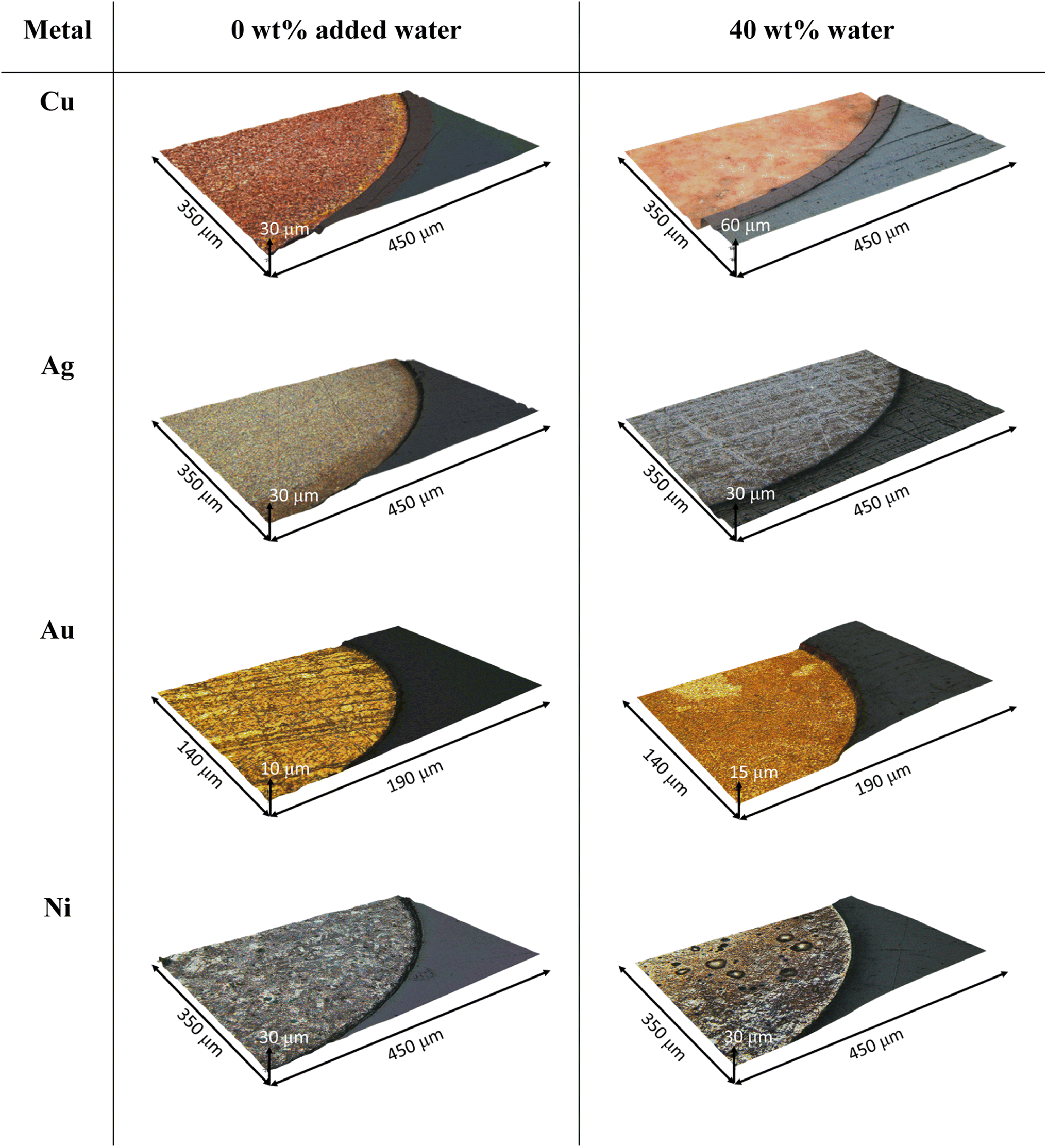 | ||
| Fig. 2 3DM optical microscope images (Cu, Ag, Ni = 20× magnification, Au = 50× magnification) of the four metals after 1 hour of etching in EG:ChCl containing a nominal 0.1 mol kg−1 iodine concentration, with different water content at 50 °C. Left: 0 wt% added water, right: 40 wt% water. | ||
It is expected that CuI and AuI are generated by the chemical oxidation of metallic copper and gold with iodine, based on previously determined electrochemistry and relative redox potentials. Cyclic voltammetry shows that both the CuI and CuII oxidation states are stable in EG:ChCl with no added water, with formal electrode potentials for the CuI/0 and CuII/I couples being −0.338 V and 0.349 V vs. [Fe(CN)6]3−/4−, respectively.33 In EG:ChCl containing iodine, two redox couples are observed which can be assigned to the [X3]−/3X− and I2/[X3]− couples, where X is iodine or chlorine, with formal electrode potentials of 0.361 V and 0.5 V vs. [Fe(CN)6]3−/4−, respectively. The trihalide species is stable in solution and hence, based on these electrode potentials, the most dominant copper species produced via oxidation will be CuI, as oxidation of CuI to CuII with iodine is only slightly thermodynamically favourable. Voltammetry shows the AuI/0 redox couple, with an electrode potential similar to iodine.12 While the Cu+ and Au+ ions generated by chemical oxidation of metallic copper and gold are stable in a dry DES due to the formation of [MCl2]− species,15 they are less stable in aqueous environment due to the formation of poorly soluble CuCl (Ksp = 1.2 × 10−6) or AuCl (Ksp = 2.0 × 10−13).34 However, if only small amounts of M+ are generated within the timeframe of the reaction (i.e. the etching rate is slower than [MCl2]− diffusion into the bulk solution), then the surface is less likely to passivate. Solid CuCl or AuCl were not observed at the wire surface. Critical differences in etching rate as a function of water content are seen for silver, where the increased water content resulted in a decrease in etching rate, and nickel, where the etching rates do not follow an identifiable trend. In dry EG:ChCl, silver etched at a rate of ca. 7 μm h−1, i.e. the second fastest etching rate of the metals investigated here. However, increased water content resulted in a decrease in etching rate to almost zero when 40 wt% water was present. A greyish-white film was observed on the surface of the wire after etching in the systems containing additional water content (Fig. 2), which was most likely composed of a silver halide salt, as solubility of silver ions decreases with increased water content when in the presence of chloride ions. For example, even in the presence of 5 mol dm−3 NaCl, silver ions are only soluble in aqueous systems up to 6.05 × 10−3 mol dm−3 at 25 °C.35 This is in comparison to at least 0.2 mol dm−3 solubility in dry EG:ChCl, which has a chloride content of ca. 4.25 mol dm−3 (ca. 3.8 mol kg−1).36 These observations regarding the relative etching rate of the group 11 elements in the 0 wt% water EG:ChCl system match trends seen in other halide-containing ionic liquids, such as [C6mim][Br2I].37
The slowest etching rate was of nickel, which is consistently low for all water contents investigated here, with a wide range of etch depths recorded. This variation in etch depth profile could be related to either the presence of an unevenly thick passivation layer, or from anisotropy in the etching process itself through pitting or selective etching at grain boundaries. In the 0 wt% water system, 3DM images (Fig. 2) show a patchy appearance to the nickel surface, which looks like the individual crystallites are being defined. In the 40 wt% water system, distinct pits in the nickel wire are present, of approximately 50 μm diameter. During etching, these pits may be filled with supersaturated nickel chloride solution,38,39 or nickel hydroxide/glycolate salts, formed from reaction of dissolved nickel ions with the DES components.40 The following reactions have been proposed, with the electrons being generated from the oxidation of nickel to form Ni2+ ions:41
| C2H6O2 + 2e− → [C2H4O2]2− + H2 | (2) |
| 2H2O + 2e− → 2OH− + H2 | (3) |
In summary, selectivity towards the oxidation of different metals can be introduced via tailoring the water content of EG:ChCl to induce surface passivation. A process could be envisaged where printed circuit boards could be treated in an aqueous DES to etch out the less valuable copper circuits, and effectively delaminate the nickel–gold protective layers that could then be recovered in the metallic form. Mass transport is clearly important to the etching process because oxidation rates for the non-passivating metals increase as solvent viscosity decreases. However, the effect of changing water content on iodine speciation must also be considered, especially as significantly decreased iodine solubility was observed in the systems containing the greatest water content.
3.2 Kinetics of metal oxidation – addition of ethylene glycol
While the presence of water in EG:ChCl may be beneficial for decreasing solution viscosity and imparting a measure of selectivity into the etching of different metals, there is however the issue of decreasing iodine solubility or of adversely affecting the solution species generated during oxidation. An alternative method would be to vary the ethylene glycol: choline chloride ratios, which will not compromise the main benefit of using a DES, i.e. avoiding water chemistry. Fig. 3 shows the effect of EG content on metal etching. Unexpectedly, there were no clear trends relating to solvent viscosity. For example, the systems containing 20 wt% water or EG:ChCl in a 4:1 molar ratio both have viscosity values of ca. 20 mPa s. There was a decrease in etching rate for copper and gold in the EG:ChCl 4:1 system compared to 20 wt% water, whereas there was a slight increase in the etching rates of silver and nickel. Therefore, other factors must be considered, including relative chloride content, and iodine speciation.
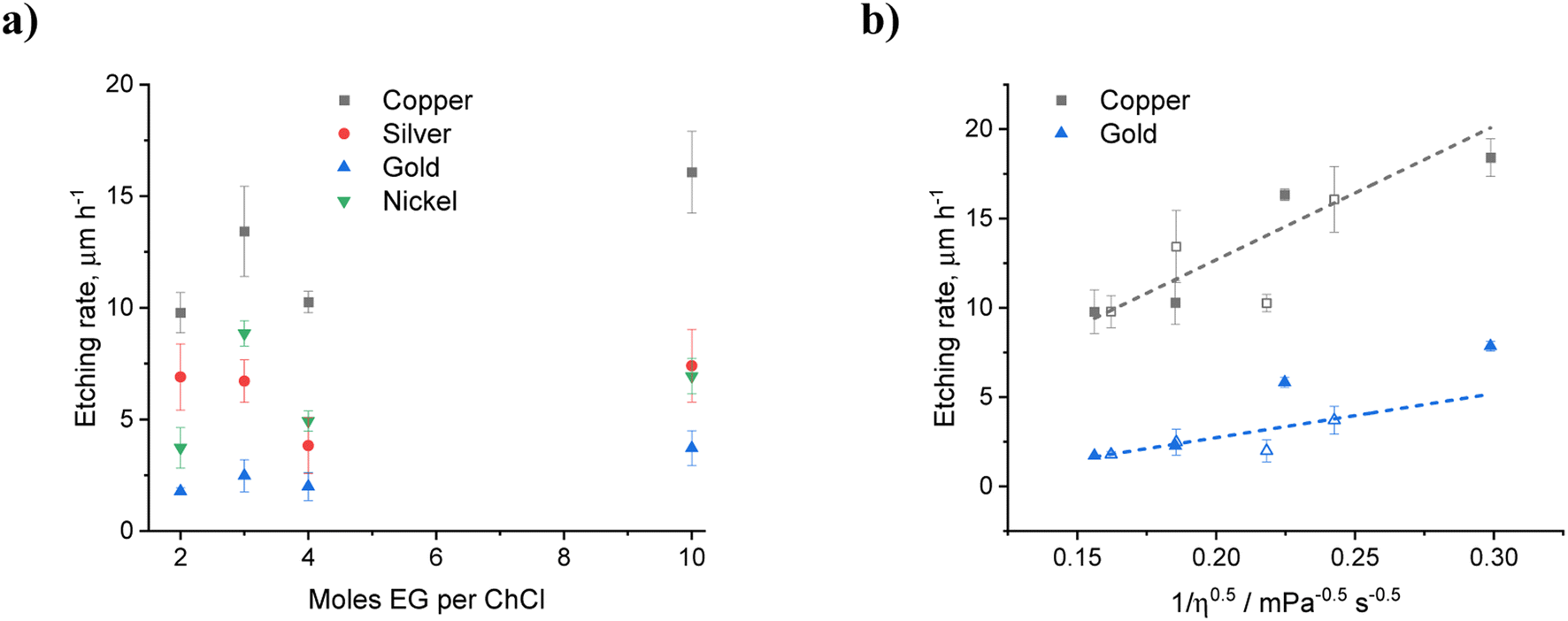 | ||
| Fig. 3 (a) Etching rates for copper, silver, gold, and nickel wires after 1 hour, as a function of EG content in EG:ChCl containing a nominal concentration of 0.1 mol kg−1 iodine. (b) Etch rates for copper and gold with water (filled symbols) and ethylene glycol (open symbols) as additives and as a function of liquid viscosity. Viscosity values taken from Al-Murshedi et al.20 and Harris42 (see Table S4†). | ||
As the amount of HBD relative to ChCl is increased, the chloride content will inevitably decrease. This can result in a decrease in the stability of the dissolved species, for both the metal complexes and the oxidising agent. While EG and water will both dilute the chloride content, the differing sizes of the molecules makes comparison on a mass and molar perspective difficult. The most likely effect of adding a diluent is that it decreases the viscosity and therefore increases the diffusion of oxidising agent to the metal surface. Fig. 3b shows the etch rate for copper and gold with iodine as the oxidising agent in EG:ChCl with different amounts of diluent. It can be seen that there is a roughly linear increase in etch rate with (viscosity)−1/2 showing that dissolution is diffusion controlled. This highlights the effect that water can have in either increasing dissolution rate through increased mass transport, or decreasing it through passivation of metal surfaces via the formation of insoluble chloride or oxide species.
3.3 Iodine speciation in EG![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :ChCl as a function of water or EG content
:ChCl as a function of water or EG content
Previous work has shown that iodine forms I2Cl− and I3− complexes in EG:ChCl with no additional water.27 The presence of water or different ethylene glycol content may affect the formation of these species, as the solubility of iodine in ethylene glycol is 0.28 mol dm−3, whereas the solubility in water is only 1.32 × 10−3 mol dm−3 when no additional complexing agents are present.43 Therefore, extended X-ray absorption fine structure (EXAFS) spectroscopy was carried at the iodine K-edge out on samples containing nominally 0.1 mol kg−1 iodine, with the DES containing a range of water contents up to 20 wt%, and also for four different molar ratios of EG:ChCl.
Data fits of iodine in the DES systems containing different amounts of water shows that a higher water content results in the average I*–I coordination number decreasing from ca. 1.4 to 1.1 atoms, along with a decrease in mean square relative disorder (Fig. S4a, b and Table S2†). The corresponding average I*–I scattering path lengths also decrease from ca. 2.80 to 2.75 Å. These observations indicate a decrease in the relative amount of triiodide in comparison to either I2Cl− or I2. As the average number of I*–Cl scattering paths decreased with increasing water content, with no change to scattering path length or mean square disorder, it could be inferred that, at these concentrations of iodine, a mixture of I2Cl− and I2 is present in the most aqueous (20 wt%) solutions in a roughly 1:1 ratio. This easily-released molecular iodine is likely to be the source of the brown staining of the storage container lids, and will be a potential route to the loss of oxidising agent if not kept within a sealed vessel during oxidative leaching. Data fits of iodine in EG:ChCl with different EG:ChCl molar ratios showed that the average I*–I coordination remained the same throughout the series, indicating the continual presence of I3− regardless of EG content (Fig. S4c, d and Table S3†). A decrease in average I*–Cl coordination can be inferred at EG:ChCl ratios of greater than 4:1, indicating that the relative proportion of [I2Cl]− to the other iodine species has decreased. A decrease in the I*–I scattering path lengths decrease from 2.791 to 2.761 Å could imply the presence of I2 molecules in the higher EG-content systems.
UV-vis spectroscopy was used to analyse dilute systems containing only 2.5 mmol kg−1 iodine to support the observation that the I2Cl−:I3− ratios remain more stable with changing EG:ChCl ratios in comparison to when additional water content is present (Fig. 4). The use of these dilute systems avoids the formation of molecular iodine due to decreased solubility. It can be seen that the presence of water content has only a slight effect on the spectra, indicating a small increase in I2Cl− content relative to I3−, as shown by the decrease in the relative absorbance values at the 290 nm and 360 nm maxima. The absorbance maxima are shown in Table S4† with minimal solvatochromic shift. In contrast, the presence of extra EG has minimal effect on the type of iodine species present and their relative ratios, except at extremely high EG content and with higher iodine concentrations. It appears that the main effect of changing solvent composition is the solubility of iodine and not the type of dissolved species present.
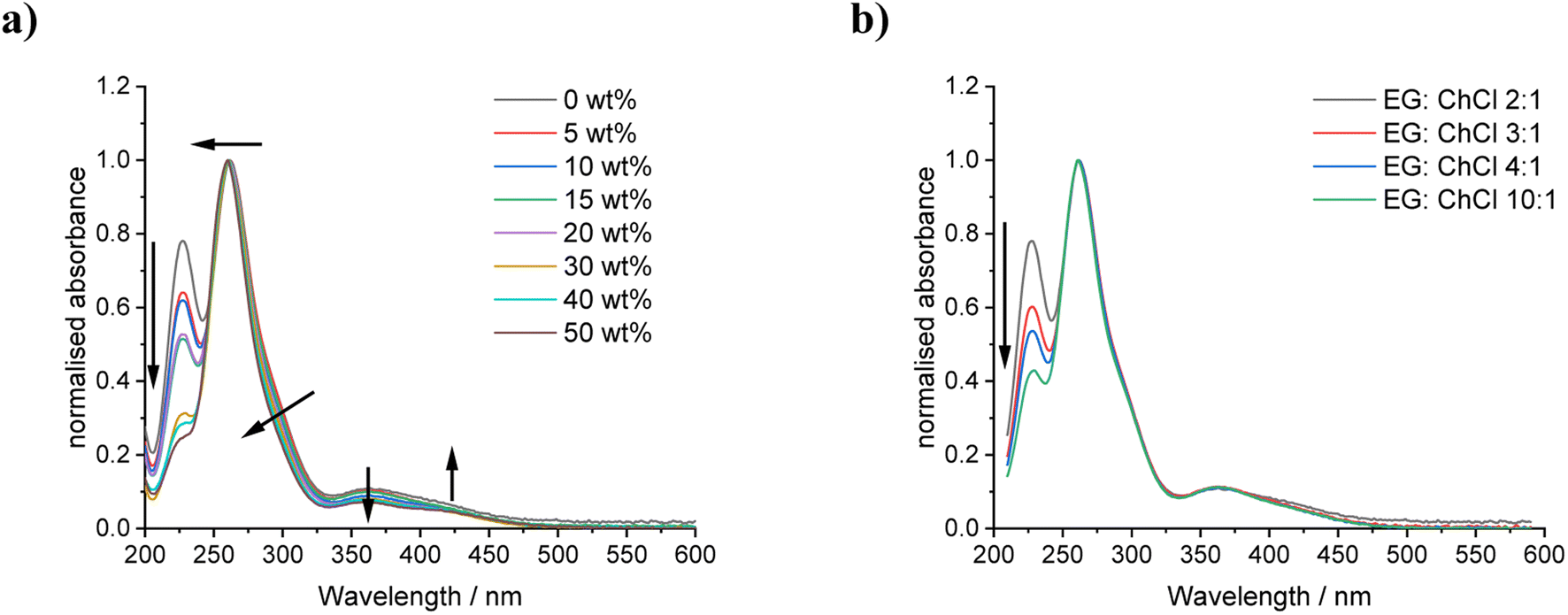 | ||
| Fig. 4 UV-vis spectra of iodine in EG:ChCl with (a) varying water content, and (b) EG:ChCl ratio. Absorbances normalised to the 260 nm peak. Arrows indicate the effect of increasing water or EG content on the spectra. | ||
Therefore, if a low viscosity system of EG:ChCl is desired, without altering iodine speciation significantly and while maintaining a high solubility of iodine, it would be preferable to modify the solvent physical properties with additional EG content rather than the addition of water. This would also minimise the possibility of oxide/hydroxide chemistry adversely influencing the dissolution mechanism of metals (e.g. via the formation of passivating layers) or resulting in the formation of solution species with different reactivity or solubility. If, however, the selective formation of passivation layers is beneficial, water is preferable.
3.4 Printed circuit board etching
As different etching behaviours for copper, silver, gold, and nickel in EG:ChCl containing a nominal concentration of 0.1 mol kg−1 iodine were observed as a function of water content, an experiment was envisaged where one could test this selective etching using printed circuit boards. The printed circuit boards used within the present work contained layers of copper, nickel, and gold on a polymer substrate. While all the board fragments were slightly different in size as they were taken from a larger board, their overall layer composition and thicknesses could be assumed to be constant.
The two systems tested contained either 0 wt% or 40 wt% additional water content, with experiments carried out at 50 °C under gentle stirring conditions. Based on the relative etching rates of the pure metals, it was hypothesised that the significantly faster etching of copper in the 40 wt% water system compared to the 0 wt% water system would result in the delamination of metallic gold and nickel from the substrate. These would then be easily recoverable via gravity filtration, rather than requiring additional extraction steps for the individual elements to be recovered from a mixed metal solution. However, it was observed that the addition of water actually hindered the copper etching/delamination process. After 1 hour of etching in the 40 wt% water system, all the gold had been dissolved, leaving behind a grey nickel-rich layer at the PCB surface (Fig. 5). This nickel layer passivates, hindering dissolution of the copper. After 5 hours, the majority of the nickel and copper was removed from the polymer substrate. In comparison, only slight dissolution of the gold layer after 1 hour was observed in the 0 wt% water system, but once the gold layer had been removed, dissolution of nickel and copper was rapid because there was no passivation of the nickel.
 | ||
| Fig. 5 Etched printed circuit board samples before and after leaching at 50 °C in 0.1 mol kg−1 iodine in EG:ChCl, with “gentle” stirring. Note that the stirrer bar was located at the bottom right corner of the sample. Each individual gold rectangle is 2.5 × 1 mm. | ||
Therefore overall, the use of dry EG:ChCl is the more effective strategy for the complete extraction of metals from this PCB, while the use of a 40 wt% water system permits selective leaching of the gold because the nickel acts as a temporary barrier to further dissolution. A two-step process could be developed in which the gold is selectively removed in one bath, followed by copper in a second. Alternatively it may be possible to undercut and lift off the outer two layers and vastly increase recycling turnover. This could be enhanced via use of ultrasonic stimulation to aid in crack propagation, as seen for the processing of photovoltaic cells.5
4 Conclusions
From these experiments, it has been determined that the addition of water to a solvent formed from ethylene glycol and choline chloride has a multifaceted effect on metal oxidation with iodine. The addition of water improves mass transport by decreasing the solvent viscosity, as was best observed for copper oxidation, where the etching rate consistently increases. However, the presence of water also increases the chance for oxide/hydroxide chemistry to occur, which results in the formation of passivating species on nickel and silver. Depending on the metals to be oxidised, this effect may be either beneficial as metal selectivity could be imparted into a process, or detrimental if all the target metals have passivated. Additionally, the higher the water content, the poorer the iodine solubility. The effect of different molar ratios of EG:ChCl on the etch rate of metals was complex and could not be related directly to viscosity, or chloride content.
This poorer iodine solubility is highlighted by the EXAFS data fits of the nominally 0.1 mol kg−1 iodine solutions, with increased water content affecting the average iodine–iodine coordination to be more like molecular iodine. In dilute systems of 2.5 mmol kg−1 iodine, where the decreased solubility of iodine is no longer significant, UV-vis spectroscopy shows an increase in I2Cl− content relative to I3−. The ethylene glycol: choline chloride ratio did not have a significant effect on iodine speciation.
These findings were applied to the etching of metals from printed circuit boards, and it was found that in systems with no added water, all of the metals tested could be etches, whereas with 40 wt% water added, gold was selectively etched within 1 hour, leaving a passivated nickel layer that protected the underlying copper. Eventually, this layer dissolved, but at a slower rate than with no water added. Critically, these results show that the changing metal etching rates are strongly dependent on the physical and chemical properties of the solvent and not on the iodine speciation as was previously suspected.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would like to thank the Faraday Institution (Faraday Institution grant codes FIRG027, project website https://relib.org.uk), the UKRI Interdisciplinary Circular Economy Centre for Technology Metals, Met4Tech project (EP/V011855/1) for funding this work, and the European Union's Horizon 2020 research and innovation program under the Marie Sklodowska-Curie grant agreement number 101026159. This work was carried out with the support of Diamond Light Source, instrument B18 (proposal SP26622). The authors would also like to thank Nitya Ramanan for assistance at the beamline during remote working conditions.References
- R. Ahirwar and A. K. Tripathi, Environ. Nanotechnol., Monit. Manage., 2021, 15, 100409 CAS.
- K. Grant, F. C. Goldizen, P. D. Sly, M.-N. Brune, M. Neira, M. van den Berg and R. E. Norman, Lancet Global Health, 2013, 1, e350–e361 CrossRef PubMed.
- V. Forti, C. P. Balde, R. Kuehr and G. Bel, The Global E-Waste Monitor 2020: Quantities, Flows and the Circular Economy Potential, United Nations University/United Nations Institute for Training and Research, International Telecommunication Union, and International Solid Waste Association, Bonn, Geneva and Rotterdam, 2020 Search PubMed.
- R. Marin Rivera, G. Zante, J. M. Hartley, K. S. Ryder and A. P. Abbott, Green Chem., 2022, 24, 3023–3034 RSC.
- G. Zante, R. Marin Rivera, J. M. Hartley and A. P. Abbott, J. Cleaner Prod., 2022, 370, 133552 CrossRef CAS.
- Y. Xue and Y. Wang, Green Chem., 2020, 22, 6288–6309 RSC.
- A. Serpe, A. Rigoldi, C. Marras, F. Artizzu, M. Laura Mercuri and P. Deplano, Green Chem., 2015, 17, 2208–2216 RSC.
- M. Sahin, A. Akcil, C. Erust, S. Altynbek, C. S. Gahan and A. Tuncuk, Sep. Sci. Technol., 2015, 50, 2587–2595 CAS.
- B. Altansukh, K. Haga, N. Ariunbolor, S. Kawamura and A. Shibayama, Eng. J., 2016, 20, 29–40 CrossRef CAS.
- G. R. T. Jenkin, A. Z. M. Al-Bassam, R. C. Harris, A. P. Abbott, D. J. Smith, D. A. Holwell, R. J. Chapman and C. J. Stanley, Miner. Eng., 2016, 87, 18–24 CrossRef CAS.
- A. Van den Bossche, N. Rodriguez Rodriguez, S. Riaño, W. Dehaen and K. Binnemans, RSC Adv., 2021, 11, 10110–10120 RSC.
- A. P. Abbott, R. C. Harris, F. Holyoak, G. Frisch, J. Hartley and G. R. T. Jenkin, Green Chem., 2015, 17, 2172–2179 RSC.
- D. Jones, J. Hartley, G. Frisch, M. Purnell and L. Darras, Palaeontol. Electron., 2012, 15, 1–7 Search PubMed.
- A. P. Abbott, G. Frisch, J. Hartley and K. S. Ryder, Green Chem., 2011, 13, 471–481 RSC.
- J. M. Hartley, C. M. Ip, G. C. Forrest, K. Singh, S. J. Gurman, K. S. Ryder, A. P. Abbott and G. Frisch, Inorg. Chem., 2014, 53, 6280–6288 CrossRef CAS PubMed.
- C. D'Agostino, R. C. Harris, A. P. Abbott, L. F. Gladden and M. D. Mantle, Phys. Chem. Chem. Phys., 2011, 13, 21383–21391 RSC.
- L. Bahadori, M. H. Chakrabarti, F. S. Mjalli, I. M. AlNashef, N. S. A. Manan and M. A. Hashim, Electrochim. Acta, 2013, 113, 205–211 CrossRef CAS.
- A. Van den Bossche, E. De Witte, W. Dehaen and K. Binnemans, Green Chem., 2018, 20, 3327–3338 RSC.
- F. S. Mjalli and H. Mousa, Chin. J. Chem. Eng., 2017, 25, 1877–1883 CrossRef.
- A. Y. M. Al-Murshedi, J. M. Hartley, A. P. Abbott and K. S. Ryder, Trans. IMF, 2019, 97, 321–329 CrossRef CAS.
- P. E. Valverde, T. A. Green and S. Roy, J. Appl. Electrochem., 2020, 50, 699–712 CrossRef CAS.
- O. S. Hammond, D. T. Bowron and K. J. Edler, Angew. Chem., Int. Ed. Engl., 2017, 56, 9782–9785 CrossRef CAS PubMed.
- F. Liu, Y. Deng, X. Han, W. Hu and C. Zhong, J. Alloys Compd., 2016, 654, 163–170 CrossRef CAS.
- T. Tsuda, Y. Ikeda, A. Imanishi, S. Kusumoto, S. Kuwabata, G. R. Stafford and C. L. Hussey, J. Electrochem. Soc., 2015, 162, D405–D411 CrossRef CAS.
- K. K. Maniam and S. Paul, Coatings, 2021, 11, 80 CrossRef CAS.
- C. Lei, H. F. Alesary, F. Khan, A. P. Abbott and K. S. Ryder, Surf. Coat. Technol., 2020, 403, 126434 CrossRef CAS.
- J. M. Hartley, S. Scott, Z. Dilruba, A. J. Lucio, P. J. Bird, R. C. Harris, G. R. T. Jenkin and A. P. Abbott, Phys. Chem. Chem. Phys., 2022, 24, 24105–24115 RSC.
- N. Frenzel, J. Hartley and G. Frisch, Phys. Chem. Chem. Phys., 2017, 19, 28841–28852 RSC.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- A. P. Abbott, G. Frisch, J. Hartley, W. O. Karim and K. S. Ryder, Prog. Nat. Sci., 2015, 25, 595–602 CrossRef CAS.
- J. T. M. Amphlett, M. D. Ogden, W. Yang and S. Choi, J. Mol. Liq., 2020, 318, 114217 CrossRef CAS.
- A. Y. M. Al-Murshedi, Deep Eutectic Solvent-Water Mixtures, University of Leicester, 2018 Search PubMed.
- A. P. Abbott, G. Frisch, S. J. Gurman, A. R. Hillman, J. Hartley, F. Holyoak and K. S. Ryder, Chem. Commun., 2011, 47, 10031–10033 RSC.
- J. Bjerrum, G. Schwarzenbach and L. G. Sillen, Stability Constants of Metal-Ion Complexes, with Solubility Products of Inorganic Substances/Pt.II, Inorganic Ligands, Chemical Society, London, 1958 Search PubMed.
- J. J. Fritz, J. Solut. Chem., 1985, 14, 865–879 CrossRef CAS.
- A. P. Abbott, G. Frisch, H. Garrett and J. Hartley, Chem. Commun., 2011, 47, 11876–11878 RSC.
- B. May, M. Lexow, N. Taccardi, H. P. Steinruck and F. Maier, ChemistryOpen, 2019, 8, 15–22 CrossRef CAS PubMed.
- J. A. Hammons, A. J. Davenport, S. M. Ghahari, M. Monir, J. P. Tinnes, M. Amri, N. Terrill, F. Marone, R. Mokso, M. Stampanoni and T. Rayment, J. Phys. Chem. B, 2013, 117, 6724–6732 CrossRef CAS PubMed.
- T. Rayment, A. J. Davenport, A. J. Dent, J.-P. Tinnes, R. J. K. Wiltshire, C. Martin, G. Clark, P. Quinn and J. F. W. Mosselmans, Electrochem. Commun., 2008, 10, 855–858 CrossRef CAS.
- S. Spathariotis, Recovery of Metals Using Deep Eutectic Solvents, University of Leicester, 2019 Search PubMed.
- K. Haerens, E. Matthijs, K. Binnemans and B. Van der Bruggen, Green Chem., 2009, 11, 1357–1365 RSC.
- R. C. Harris, Physical Properties of Alcohol Based Deep Eutectic Solvents, University of Leicester, 2009 Search PubMed.
- A. Osol and C. C. Pines, J. Am. Pharm. Assoc., 1952, 41, 634–637 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2su00038e |
| This journal is © The Royal Society of Chemistry 2023 |