
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Composition and temperature effects on the solution structure of SDS/octanol/brine by SANS, NMR and microscopy†
Liva
Donina
a,
Lionel
Porcar
b and
João T.
Cabral
 *a
*a
aDepartment of Chemical Engineering, Imperial College London, UK. E-mail: j.cabral@ic.ac.uk; Tel: +44 (0)207 594 5571
bInstitut Laue-Langevin, 71 Avenue des Martyrs, B.P. 156, F-38042 Grenoble CEDEX, France
First published on 24th October 2023
Abstract
We investigate the solution structures of model sodium dodecyl sulfate/octanol/brine ternary mixtures across the lamellar (Lα), vesicle (L4) and micellar (L1) phases employing small angle neutron scattering (SANS), optical microscopy and nuclear magnetic resonance (NMR). Specifically, we examine the effect of co-surfactant octanol (0.2–9.48 w/v%) and temperature (25–65 °C) along dilution lines at fixed octanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :SDS ratios (0.08–1.21). A transition from Lα to sponge phase (L3) above 35 °C is found along the octanol:SDS = 1.21 isopleth, with phase coexistence above ϕ ≈ 0.14 weight fraction of surfactant and co-surfactant. The lamellar bilayers swell upon dilution, with an approximately linear increase of d-spacing, accompanied by a decrease of the Caillé parameter, indicative of greater membrane rigidity. At a lower octanol:SDS ratio of 0.62, coexistence of oblate micelles and vesicles is observed with preferential formation of vesicles at low concentrations. Dilution of the L1 phase, along octanol:SDS = 0.08, results in elongated micelles, as the NaCl:SDS ratio increases, while higher temperatures favour the formation of less elongated micelles. Our results provide a detailed map of the equilibrium structures found in the Lα vicinity of this extensively investigated flow-responsive surfactant system.
:SDS ratios (0.08–1.21). A transition from Lα to sponge phase (L3) above 35 °C is found along the octanol:SDS = 1.21 isopleth, with phase coexistence above ϕ ≈ 0.14 weight fraction of surfactant and co-surfactant. The lamellar bilayers swell upon dilution, with an approximately linear increase of d-spacing, accompanied by a decrease of the Caillé parameter, indicative of greater membrane rigidity. At a lower octanol:SDS ratio of 0.62, coexistence of oblate micelles and vesicles is observed with preferential formation of vesicles at low concentrations. Dilution of the L1 phase, along octanol:SDS = 0.08, results in elongated micelles, as the NaCl:SDS ratio increases, while higher temperatures favour the formation of less elongated micelles. Our results provide a detailed map of the equilibrium structures found in the Lα vicinity of this extensively investigated flow-responsive surfactant system.
1 Introduction
Sodium dodecyl sulfate (SDS) is a common anionic surfactant, extensively employed in industry and academic research. The SDS/water phase diagram has been investigated in detail1 and, in liquid state, above the Krafft temperature (15–17 °C)2–4 and critical micelle concentration (cmc ≃6.9–8.2 mM, or 0.19–0.24 w/w%),4,5 the system yields micellar (L1), hexagonal (Hα) and lamellar phases (Lα). However, to attain ordered Hα and Lα phases, relatively high SDS concentrations of 40 and 70 w/w% and temperatures above 50 °C are required.1,6 The formation of liquid crystalline phases can be achieved at ambient temperatures and much lower surfactant concentrations (<10 w/w%) by the addition of dopants, specifically mid- to long-chain alcohols (n-butanol to decanol) and by the addition of electrolytes, such as NaCl. In general, the addition of alcohols modulates the interactions between surfactant molecules, for instance by reducing headgroup repulsions or altering packing efficiency, while salts impact electrostatic interactions between the headgroups, in turn affecting the formation of self-assembled structures.6Co-surfactants can be defined as ‘surfactants’ that, alone, are not able to form micelles, with alcohols representing an important class.7 The addition of alcohols has been well established in the phase diagram of SDS/n-alcohol/water systems, both experimentally and computationally.8 The stability of mixed micelle systems is known to be impacted by the relative matching of alkyl chain lengths.9 For example, long chain alcohols (C8 to C16) have been found to lower the critical micelle concentration (cmc) and stabilise SDS micelles, arising from the ion-dipole interactions of the SDS sulfate group and octanol –OH group. This interaction then increases packing efficiency and leads to micellar stabilisation below SDS concentrations of 150 mM.9 However, at higher SDS and alcohol concentrations, with the exception of dodecanol, micelles are destabilised due to a mismatch in the hydrocarbon chain length, where the excess length leads and thermal motion decreases packing efficiency.9,10 SANS studies on the effect of alcohols on SDS micellar phase have demonstrated that the addition of C1 to C3 alcohols lead to the formation of more spherical micelles while the addition of mid (C4 to C6) to long (C7 to C10) chain alcohols results in micelle elongation and the formation of wormlike micelles.11–13 Increasing long chain (C7 to C8) alcohol to surfactant ratio has been found to further elongate micellar structures.11
Addition of longer chain alcohols, above butanol C4, promotes the formation and stability of the lamellar phase, extending it to lower concentration ranges.7 At constant surfactant concentration, alcohol addition has been reported to lead to rich lamellar phase behaviour (Fig. 1a).14 In SDS lamellar systems, the length of the alcohol was reported to influence the bilayer thickness, with shorter chain alcohols (C5–C7) thinning the membrane. In turn this resulted in a 5-fold decrease in membrane bending rigidity kC, as compared to longer chain alcohols C8–C12.15
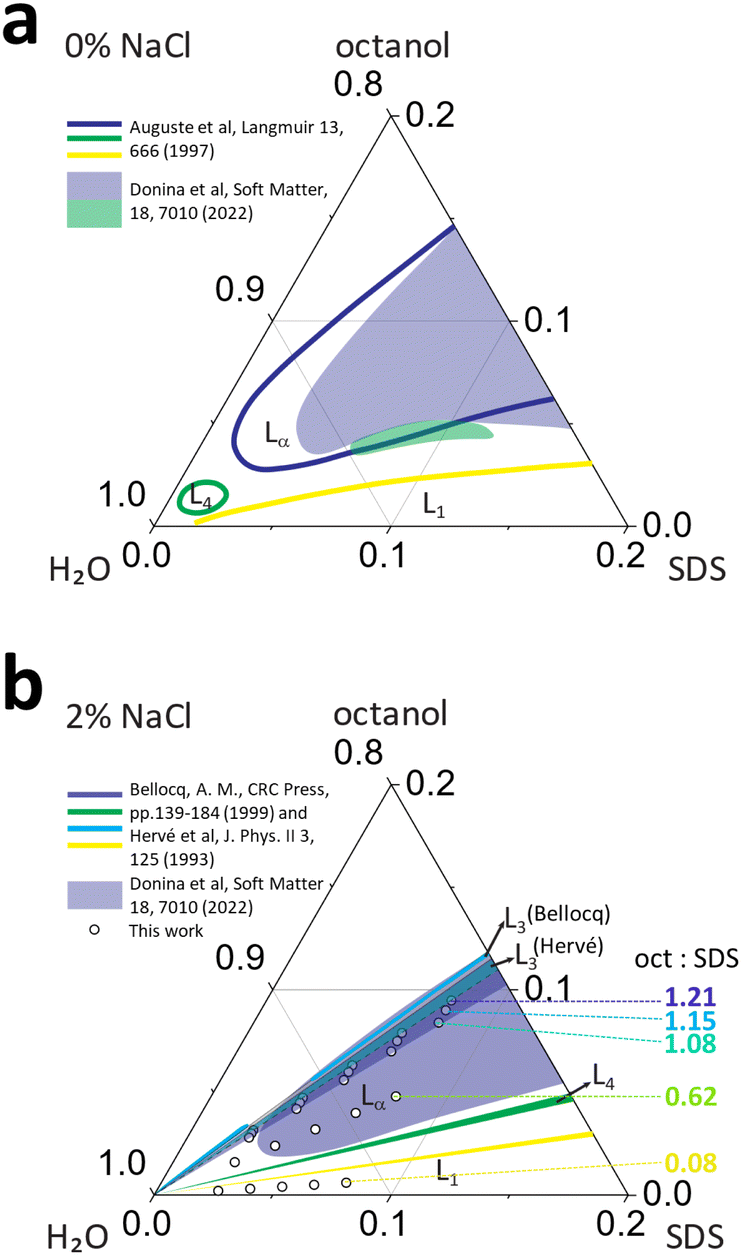 | ||
| Fig. 1 Phase maps for SDS/octanol/water with (a) 0% salt and (b) 2% NaCl (brine), compiled from literature6,16–18 shown as solid lines, and optical microscopy data from previous work18 (shaded regions), indicating micellar L1, lamellar Lα, sponge L3 and vesicle L4 phases. The data points in (b) correspond to the compositions investigated in this work, with octanol:SDS w/w ratios of 1.21, 1.15, 1.08, 0.62 and 0.08, employing SANS, microscopy and NMR, as a function of temperature (25–65 °C). | ||
The addition of electrolytes to ionic surfactants, generally increases the size of micellar aggregates due to electrostatic screening effects, by reducing headgroup repulsion, which is magnified in presence of alcohol co-surfactants.7 The addition of Na+ salts generally favours the formation of ellipsoidal SDS micelles and, above 0.4 M, the formation of wormlike micelles has been reported.13,19 Similarly, salt addition generally reduces the membrane bending rigidity in the lamellar phases of anionic surfactants, associated to charge screening of steric repulsions. It has also been demonstrated that the addition of NaCl to SDS/alcohol/water system extends the lamellar phase stability up to 95% water corner20 and promoting the formation of L4–Lα–L3 sequence of phases (Fig. 1b).10,14
In summary, the presence of mid- to long-chain (C5–C12) alcohols extends the lamellar phase of SDS aqueous solutions towards lower surfactant concentrations; the further addition of NaCl extends the narrow MLV L4 region, and opens a sponge phase L3, alongside the Lα phase, as shown in Fig. 1b. The effect of flow in this rich phase behavior has been extensively examined; specifically the lamellar-to-MLV flow-induced phase transformations in SDS/octanol/brine, generally at a concentration of 6.5% SDS/7.9% octanol/85.6% brine,17,21–28 and found to exhibit hexagonally packed vesicle formation, whose size can be modulated by temperature and shear. Rheology, microscopy and light scattering studies have enabled a detailed phase map of SDS/octanol/brine (20 g L−1 NaCl) under quiescent conditions, however detailed microscale characterisation of this system has so far not been performed.16,17,29 In this work, we employ small angle neutron scattering (SANS) to elucidate the solution structures across this rich phase diagram, investigated as a function of composition and temperature. Our data are complemented by optical microscopy and NMR to facilitate phase identification and benchmarking with previous work.
2 Methods
2.1 Sample preparation
The experimental SDS/octanol/brine concentrations series were selected based on previously reported phase maps6,16 and our own optical and NMR measurements for this system. SDS (Sigma-Aldrich BioXtra, ≥99.0%) was used within one week of opening without further purification; absence of dodecanol (a product of SDS hydrolysis) was confirmed by 1H NMR. The samples were prepared by first dissolving 7.8 g of SDS each in D2O brine (20 g L−1, Sigma-Aldrich, 99.9 atom% D) of mass 18.39, 18.50, 18.63, 19.43 and 20.37 g corresponding to the equivalent volume fraction in H2O. Substitution of H2O with D2O resulted in a small upwards shift of the phase boundaries towards octanol (well below the variation in phase boundaries reported in the literature using H2O). Dissolution was facilitated by heating the samples to 30 °C until full dissolution, and then allowed to cool to room temperature. Then, 1-octanol (Sigma-Aldrich, anhydrous, ≥99%) was added in following amounts 9.48, 9.00, 8.4, 4.8 and 0.6 g, respectively, to each of the SDS/brine solutions above, resulting in oct:SDS ratios of 1.21, 1.15, 1.08, 0.62 and 0.08 respectively, and stirred for 2 min at 1000 rpm until fully incorporated. The solutions were allowed to equilibrate for 24 h at rest, to mitigate any inadvertent shear effects during sample preparation. From these concentrations, five isopleths were prepared by dilution, mixing 5, 4, 3 and 2 mL of the stock solution with 1, 2, 3 and 4 mL of D2O brine (20 g L−1) to make up 6 mL total solution. Each sample was then stirred for 1 min at 1000 rpm until fully incorporated, and allowed to rest for 24 h.
2.2 Optical microscopy
Bright field and cross-polarised optical microscopy was carried out by placing 10 μL of sample on a cavity slide, covered by a coverslip and mounted on Linkam THMS600 stage stage at 15 °C. The sample was then heated to the desired temperature (25, 35, 45 °C) at 10 °C min−1 and allowed to equilibrate for 5 min. Images were captured using Olympus BX41 reflection microscope equipped with a Basler acA2000-165um camera controlled with PylonViewer using 10×/0.25 Ach and 50×/0.50 MPlanFLN Olympus objectives with cross-polarised filters (U-AN-2 analyser and U-PO3 polariser), and bright-field (BF) and cross-polarised data were analysed with ImageJ.2.3 1H NMR
Liquid state 1H NMR spectra were acquired for 1.21, 1.15, 1.08, 0.62 and 0.08 oct:SDS w/w ratios made with D2O (Sigma-Aldrich, 99.9 atom% D) along the respective isopleths with a Jeol 400 MHz magnetic field strength NMR spectrometer. Each spectrum was collected using a relaxation delay of 4 s and one 45° RF-pulse followed by 4.37 s acquisition time (8 scans) at 25, 35 and 45 °C temperatures leaving each sample to rest for 5 min. Pure component spectra in solution, 6.5 w/v% SDS/D2O and 7.9 w/v% octanol/CD3 Cl were collected for chemical shift identification in mixed systems (Fig. S1, ESI†). Spectra were analysed with the Mnova 14.3.0 software and calibrated using D2O residual solvent peak shift at 4.78 ppm (25 °C), 4.68 ppm (35 °C) and 4.58 ppm (45 °C).
2.4 Small-angle neutron scattering
SANS experiments were carried out at the Institut Laue Langevin, Grenoble, using the D22 spectrometer, equipped with front and rear detectors, placing the latter at two distances 17.6 m and 5.6 m to the sample, and neutron wavelength λ = 6 Å. The measured wavevector q range was 0.003 to 0.8 Å−1. The samples were loaded into ‘banjo’ Hellma cells of 1 mm path length with a needle (which induces preferred orientation in liquid crystalline phases). Sample cells were mounted on a thermostated rack and measured at 25, 35 and 45 °C. Selected samples (containing 6.5 w/v% SDS) were measured from 25–65 °C in 5 °C steps. The data were reduced and calibrated with GRASP by subtracting empty cell transmission, sample transmission, open beam transmission and D2O background scattering, and radially- or sector-averaged, as appropriate. Data fitting was carried out with SasView 5.0.5. In total, 24 samples and in excess of 100 spectra (at different temperatures) were measured and analysed.Lamellar structures Lα were fitted with the random head–tail sheet with Caillé structure factor model (SASView lamellar_HG_stack),30 parameterised by the lamellar d-spacing and thickness δ, the Caillé parameter η (characterising the membrane bending rigidity and compression modulus), and number of layers, as well as by the scattering length density profile; polydispersity pD of d-spacing was also introduced. The sponge L3 phase was fitted with a model reported by Lei et al.31,32 coded into SASView, yielding estimates of the average sponge pore size, ξ, and membrane thickness, δ, as a function of membrane volume fraction ϕv ≡ (Voctanol + VSDS)/Vtotal and temperature. A sum of flexible cylinder and lamellar models was used to describe mixed micellar-vesicle phase (L1 + L4) (in SASView, flexible_cylinder + lamellar_HG_stack for the low-q upturn), yielding micelle shape and dimensions, and characteristic low-q exponent. Finally and a core–shell cylinder model33 (SASView core_shell_cylinder) was employed to describe elongated micelles (L1). Details of scattering length density profiles (Fig. S2, ESI†), the models for analysis and interpretation (Fig. S3 and S4, ESI†) are provided in ESI.†
3 Results
3.1 Effect of octanol concentration and temperature on the lamellar Lα phase
We first consider the isopleths with oct:SDS ratio 1.21, within the Lα phase, according to the phase diagram,6,16 as shown in Fig. 2a. Composition II along the isopleth, 6.5/7.9/85.6 w/v% SDS/oct/brine corresponds to the system employed in well-known Lα-to-MLV flow induced phase transformation studies.21–27 We first look at the 1H spectra (Fig. 2a) in conjunction with microscopy images (Fig. 2b) to understand the connection between molecular and microscale behaviour.
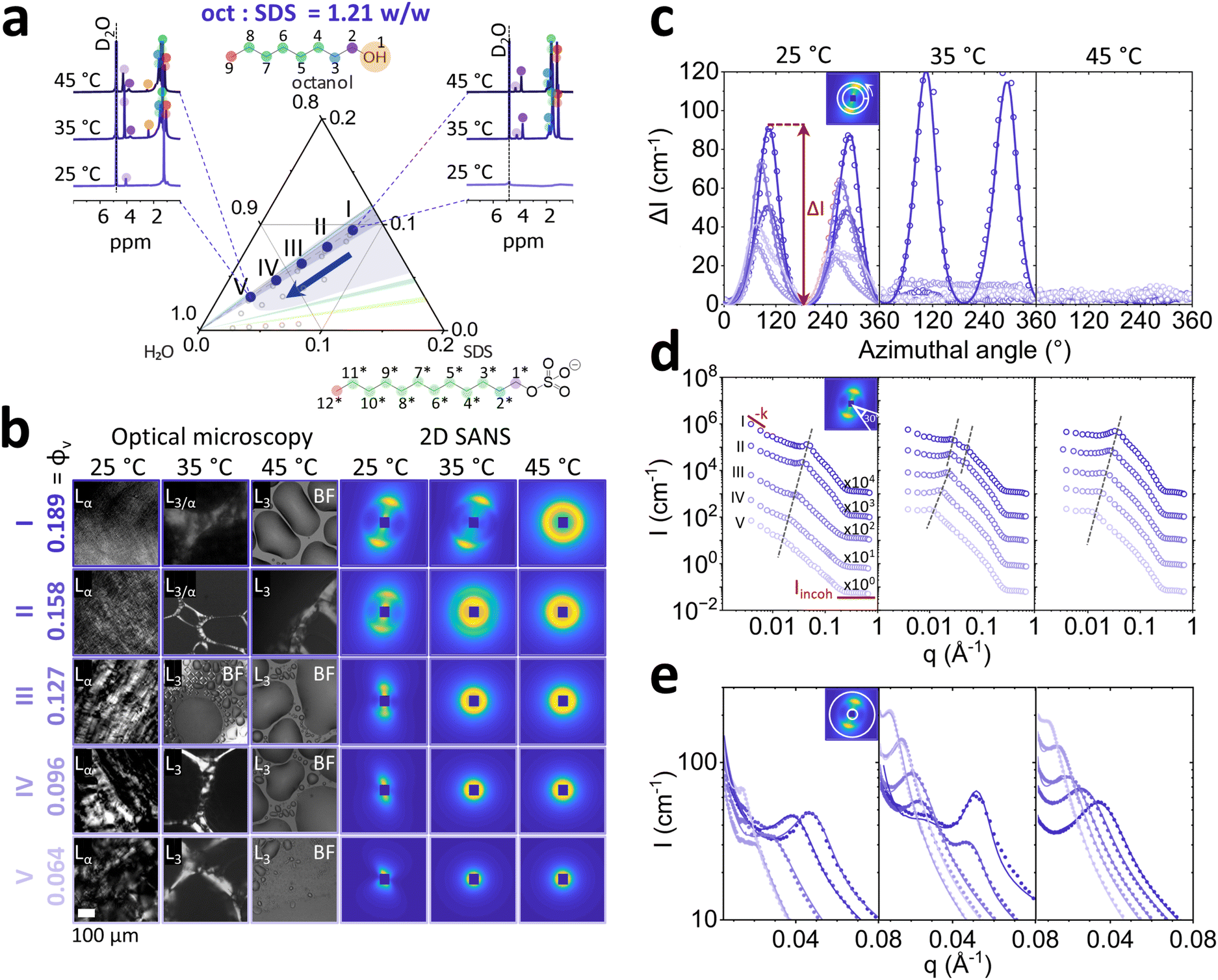 | ||
| Fig. 2 (a) Overview of the experimentally measured compositions at octanol:SDS ratio 1.21, near the Lα boundary with the L3 phase. Specifically, SDS/octanol/H2O brine: (I) 7.80/9.48/82.72, (II) 6.5/7.9/85.6, (III) 5.20/6.32/88.48, (IV) 3.90/4.74/91.36, (V) 2.60/3.16/94.24 (prepared from dilution of I); for SANS and NMR samples, D2O was used instead. Representative NMR spectra are shown for (I) and (V) as a function temperature. (b) Corresponding birefringence and brightfield (BF) optical microscopy data and SANS of samples I–V at 25, 35 and 45 °C; ϕv indicates the SDS + octanol volume fraction, i.e. total surfactant/co-surfactant content. (c) Azimuthal profile of SANS data in (b) around the respective Bragg peak for Δq = 0.014 Å1, subtracted by the respective Imin characterising scattering anisotropy. (d) Sector average I(q) with a 30° angular range, normal pattern orientation; dashed lines indicate peak position(s). Data shifted vertically (×10) for clarity. (e) Radial average of SANS data near the peak; lines are fits to the lamellar and L3 models, described in the text. | ||
1H NMR probes changes in local environment that are characteristic of molecular scale structures.34 At 25 °C, composition I (ϕv = 0.189) displays peak broadening expected in anisotropic ordered phases, such as Lα. At temperatures 35 and 45 °C, the emergence of an isotropic L3 phase is indicated by the terminal –CH3 triplet at ∼0.7–0.8 ppm (positions 9 and 12*) and hydrophobic chain –CH2 multiplet at ∼1.3 ppm (4–8 and 3*–11*) for octanol and SDS (marked ‘*’) tails. Additionally, headgroup adjacent –CH2 (2 and 1*) triplet peaks emerged further downfield at ∼3.8 ppm for octanol and ∼4 ppm for SDS following temperature increase to 45 °C and dilution. The effect of dilution along 1.21 oct:SDS isopleth to ϕv = 0.064 showed an emergence of broad singlet around 1.3 ppm belonging to –CH2 of SDS hydrocarbon tail. The absence of octanol –CH2 group near –OH chemical shift at 3.8 ppm indicates that it has been embedded at the core of the structure formed. Optical microscopy identifies a birefringent Lα phase at 25 °C, and the emergence of larger, more birefringent optical textures upon dilution (Fig. 2b). At 35 °C, the birefringence disappears as the system enters an isotropic phase, interpreted to be L3 and instead shown in brightfield microscopy, indicated as ‘BF’ in Fig. 2b, as previously reported for 6.5 SDS/7.9 octanol/86.5 H2O w/v% with Lα/L3 coexistence between 30 to 38 °C.35 For reference, microscopy images for ϕv = 0.158 (II) are shown in BF at all temperatures. At 45 °C the sample becomes fully isotropic, transitioning into the L3 phase.
SANS 2D spectra corroborate these observations, with samples at 25 °C displaying diffuse scattering closer to the beam center (Fig. 2b). Upon increasing the temperature to 35 °C, samples I and II show two scattering features, expected in phase coexistence, and further increasing temperature to 45 °C resulted in isotropic scattering, indicating the L3 phase. Analysis of normalised azimuthal intensity (ΔI) near the Bragg peak (q = 0.014 Å−1) revealed 3-fold decrease in sample anisotropy upon dilution at 25 °C (Fig. 2c). Raising the temperature to 35 °C showed an increase in anisotropy for sample I, and disappearance of anisotropy for samples II–V. At 45 °C all samples had become isotropic. Sector averaged I(q) (0.003 ≤ q ≤ 0.08 Å−1) with 30° normal to Bragg peak orientation for (Lα) at 25 °C indicated decrease of intensity and a linear shift to lower q values from of the Bragg peak upon dilution indicating increase in polydispersity and membrane swelling (Fig. 2d). At 35 °C, the coexistence of two Bragg peaks for samples I and II was quantified 2D SANS measurements: when Lα/L3 coexist, Lα Bragg peak is shifted to the higher q (0.050 to 0.055 Å−1 for sample I and 0.04 to 0.05 Å−1 for sample II) while L3 is shifted to lower q region 0.018 and 0.015 Å−1 for samples I and II respectively. At 45 °C all sample concentrations are in L3 phase and exhibit the same linear swelling upon dilution as the Lα phases. The fitted radially averaged I(q) profiles are shown in Fig. 2e. Analysis of Lα phases with oct:SDS ratios of 1.15 and 1.08 are provided in Fig. S6 and S7 (ESI†) and do not exhibit phase transformation to L3 at 35 and 45 °C. However, upon dilution to the same SDS concentrations, formation of mixed vesicle and micellar phase was observed.
Quantification of scattering anisotropy ΔI ≡ I − Imin (obtained from the azimuthal profile at the peak), exponent −k describing low q scattering via q−k, and fitted model parameters for Lα phases (d, η, pD) are shown in Fig. 3 as a function of total octanol and SDS v/v fraction ϕv, for temperatures 25, 35 and 45 °C. The ΔI profiles generally indicate an increase in Lα anisotropy with increasing ϕv, at all temperatures (Fig. 3a). After the Lα-to-L3 phase transformation, at oct:SDS = 1.21 w/w and 35–45 °C, the profile becomes isotropic. The grey shaded region corresponds to Lα–L3 phase coexistence, at ϕv = 0.189 at oct:SDS = 1.21 w/w, exhibiting highest ΔI. We observe exponents 1.5 ≤ −k ≤ 3 for Lα phases with oct:SDS = 1.08 and 1.15, at all temperatures, which generally increases upon dilution (Fig. 3b). Given the limited q-range of the powerlaw, and scatter in −k, we refrain from over-interpreting the fractal exponent, other than by qualitatively classing the Lα ordering. Upon transition to the L3 phase, the exponent −k drops to 0–0.5 (35–45 °C, beyond phase coexistence), the structure becomes largely isotropic, and prefactor A describing the contributions from in-out correlations (much larger than pore size ξ), is close to 0 (ESI,† Section 1).32 The d-spacing of the Lα phase is calculated as d = 2π/q* where q* denotes the Bragg peak position, corresponding to the repeat bilayer distance, and the average pore size in L3 phase is obtained by fitting ξ ≡ 2π/q3; for simplicity, both are denoted as d in Fig. 3c. Overall, differences within isopleths oct:SDS and various temperatures, at constant ϕv are relatively modest. Upon dilution of an Lα phase,36 the lamellar spacing is expected to follow d = Aδ/ϕv, where δ is Lα membrane thickness, A is a multiplicative prefactor, which for Lα is 1 and ϕv is the volume fraction of membrane, calculated as ϕv ≡ (Voct + VSDS)/Vtotal. Using the fitted membrane thickness δ data, we confirm that all Lα structures follow this dilution scaling. Sponge phases L3 follow the same relation, but now with A = 1.5. Our data conform to this dilution law except for the lowest concentration, at ϕv = 0.064, approaching the micellar phase. Throughout the Lα phases, we find that the Caillé parameter η ∼ 1 (the upper limit of the model), decreasing with increasing ϕv, indicating an effective stiffening of the membrane (Fig. 3d). A decrease in η also observed at the Lα–L3 coexistence, likely due to membrane compression that is reflected in a decrease in Lαd-spacing with respect to that measured at 25 °C. The polydispersity in d-spacing, pD, shown in Fig. 3e, also generally increases upon dilution.
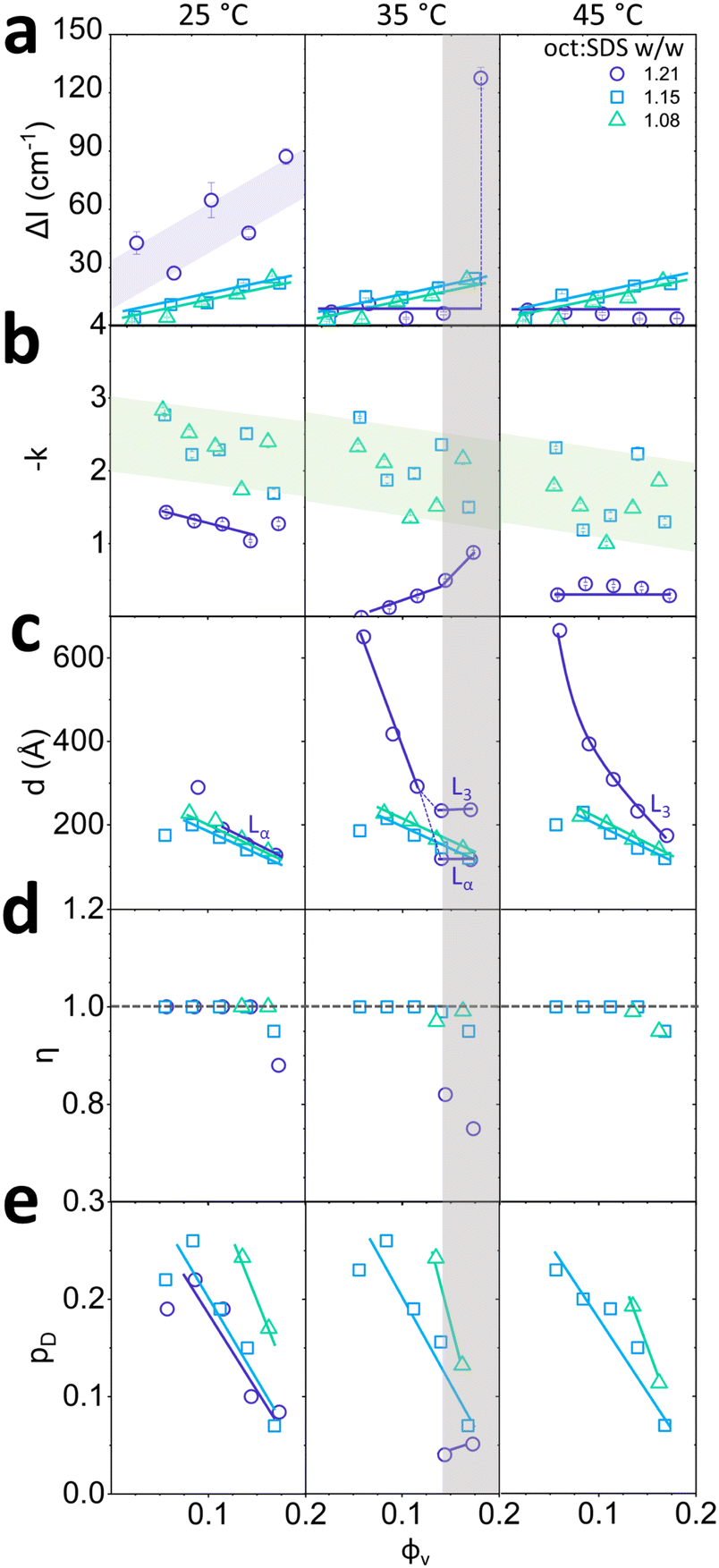 | ||
Fig. 3 Parameters of Lα phases with oct:SDS ratios of  1.21, 1.21,  1.15 and 1.15 and  1.08. (a) Normalised ΔI measured around the Bragg peak, (b) exponent fit at low-q region (0.004 < q < 0.01 Å−1), (c) d-spacing (Lα or L3 as indicated), (d) Caillé parameter η, (e) polydispersity of Lαd-spacing, as a function of ϕv for 25, 35 and 45 °C. Anisotropy to isotropy shift observed for the 1.21 oct:SDS with increasing temperature, accompanied by a transition to L3. 1.08. (a) Normalised ΔI measured around the Bragg peak, (b) exponent fit at low-q region (0.004 < q < 0.01 Å−1), (c) d-spacing (Lα or L3 as indicated), (d) Caillé parameter η, (e) polydispersity of Lαd-spacing, as a function of ϕv for 25, 35 and 45 °C. Anisotropy to isotropy shift observed for the 1.21 oct:SDS with increasing temperature, accompanied by a transition to L3. | ||
3.2 Temperature scan along representative lamellar Lα compositions
We next examine in finer detail the effect of increasing temperature for three the Lα structures, containing fixed 6.5 w/v% SDS, and varying octanol (7.9%; 7.5% and 7.0 w/v%). These correspond to 1.21, 1.15 and 1.08 oct:SDS ratios, respectively. The temperature was varied from 25 to 65 °C in 5 °C each equilibrated for 20 minutes. Thermal reversibility was tested by collecting a measurement after 20 min following a temperature quench from 65 °C to 25 °C (Fig. 4a). Red points on the temperature ramp indicate the SANS measurements.
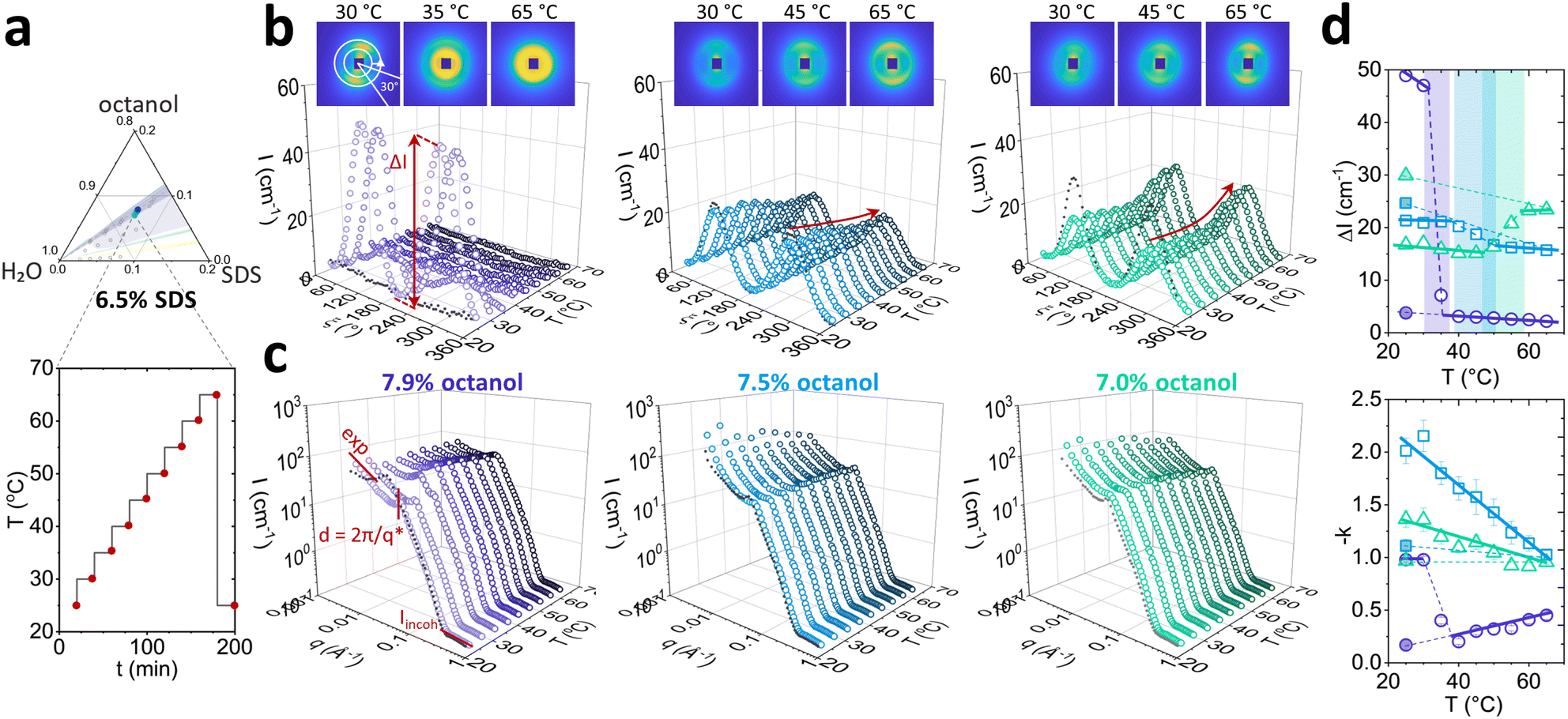 | ||
| Fig. 4 Temperature-induced changes of the 6.5 w/v% SDS samples, with 7.9%; 7.5% and 7.0 w/v% octanol, originating from the lamellar phase at room temperature. (a) Composition and temperature profile used for SANS experiments, from 25 to 65 °C, in 5 °C increments in 20 min steps; (b) azimuthal profile and (c) sector average (30° as shown) as a function of temperature. (d) Scattering anisotropy ΔI and low-q exponent as function of temperature, indicating a phase transition to L3 for 7.9 w/v% octanol sample at ≈35 °C. | ||
Azimuthal analysis of 2D SANS data reflects that the transition from Lα-to-L3 in the sample with 7.9% octanol takes place between 30 and 40 °C, in line with previous literature.35 This is indicated by a 10-fold drop (Fig. 4d top) in azimuthal intensity and the emergence of strong isotropic scattering ring in 2D with virtually no anisotropy. While the thermally-induced transition Lα to L3 occurs within minutes, the sample did not revert to Lα after 20 min following a temperature quench from 65 to 25 °C, only returning back to its original state within ∼2–7 days. This asymmetric kinetic pathway has been investigated in detail by Buchanan et al.29 Lα samples containing 7.5% and 7.0% octanol did not undergo transformation to L3 phase upon increasing temperature. However, the azimuthal signal intensity dropped for 7.5% octanol sample and increased for 7.0% octanol sample, and did not return their original state after 20 min for either samples. The anisotropy ΔI shows a nonlinear variation with temperature (Fig. 4d top): the 7.5% octanol sample shows a linear decrease of ΔI between 35 and 50 °C, and a plateau thereafter, while 7.0% octanol shows an initial decrease in ΔI from 25 to 45 °C followed by a linear increase between 45 and 55 °C.
The I(q) sector averages (30°) normal to the Bragg peak confirm that the Lα-to-L3 transformation is accompanied by an abrupt shift in peak position with temperature (7.9% octanol), contrasting with a gradual variation within the Lα (7.5% and 7.0% octanol). Changes in forward scattering were quantified by the exponent −k as a function of temperature, shown in Fig. 4d bottom; for 7.9% octanol, −k drops from 1.0 to 0.2 between 30 and 40 °C, marking the Lα-to-L3 transformation. By contrast, for 7.5% and 7.0% octanol, −k decreased linearly with temperature. Fits and parameters to the corresponding Caillé and sponge models are shown in Fig. 5. The sample containing 7.9% octanol indicated a jump in d-spacing upon transition to L3 from 150 to 225 Å between 30 and 40 °C and a subsequent decrease in L3 pore dimensions ξ from 225 to 200 Å for temperatures between 40 and 65 °C, associated with greater interfacial area accommodated at higher thermal energies. By contrast, a d-spacing increase in temperature was observed for 7.5 and 7.0 w/v% octanol samples (Fig. 5d), beyond what might be expected from variation of density alone,37,38 and suggest the presence of excess water outside the lamellar periodicity and dilution law does not apply.39 The membrane thickness δ actually decreases with temperature (Fig. 5e). The 7.9% octanol sample also shows a decrease in δ after ∼40 °C, where transition to sponge phase L3 takes place. Such decreases in δ with temperature have been previously reported (e.g., in DDABr40). A decrease in Caillé parameter η was observed for 7.5% and 7.0% octanol Lα samples at 45 and 50 °C, respectively (Fig. 5e), indicating an increase membrane bending rigidityκ and membrane compressibility modulus ![[B with combining macron]](https://www.rsc.org/images/entities/i_char_0042_0304.gif) product. While some studies have observed an opposite effect,41,42 there are many reports that corroborate our data. Freyssingeas et al.43 observed an increase in both in d and membrane bending rigidity κ in binary C12E5/water system, rationalised in terms of a decrease in polar headgroup area. While the addition of NaCl to charged surfactants has been shown to decrease the membrane bending rigidity via membrane screening effect at ambient conditions due to charge screening effect,18,44,45 the increase in temperature will facilitate the dissociation of Na+ ions from the SDS in lamellar membrane in presence of brine matrix leading to increase in headgroup repulsions and increase in membrane bending rigidity κ.46 Further, as the Bjerrum length λB increases with temperature, the spatial extent of Coulombic interactions comparatively increases, contributing to an increase in κ (see Fig. S5, ESI†),47
product. While some studies have observed an opposite effect,41,42 there are many reports that corroborate our data. Freyssingeas et al.43 observed an increase in both in d and membrane bending rigidity κ in binary C12E5/water system, rationalised in terms of a decrease in polar headgroup area. While the addition of NaCl to charged surfactants has been shown to decrease the membrane bending rigidity via membrane screening effect at ambient conditions due to charge screening effect,18,44,45 the increase in temperature will facilitate the dissociation of Na+ ions from the SDS in lamellar membrane in presence of brine matrix leading to increase in headgroup repulsions and increase in membrane bending rigidity κ.46 Further, as the Bjerrum length λB increases with temperature, the spatial extent of Coulombic interactions comparatively increases, contributing to an increase in κ (see Fig. S5, ESI†),47
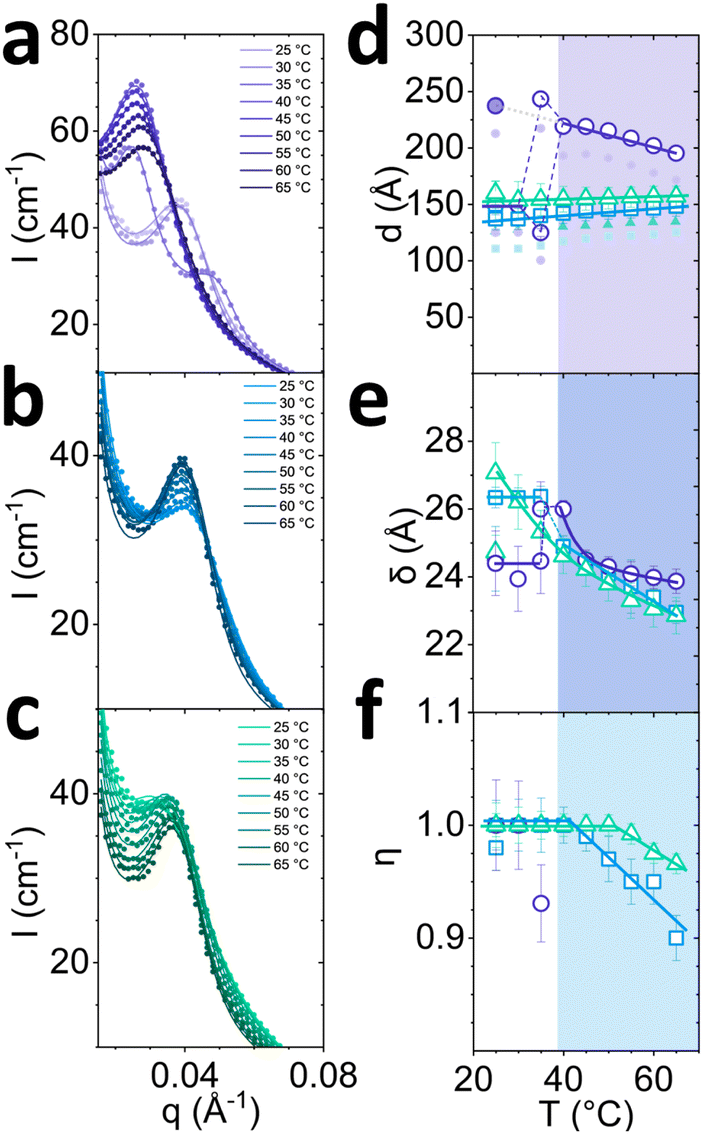 | ||
| Fig. 5 Analysis of data shown in Fig. 4 by fitting the radially-averaged SANS data near the scattering peak, as a function of temperature. Profiles for 6.5 w/v% SDS and (a) 7.9, (b) 7.5 and (c) 7.0 w/v% octanol in D2O brine. (d) Fitted Lα/L3d-spacing with Lα error bars corresponding to polydispersity, pD; filled smaller points correspond to δw thickness after membrane thickness δ has been subtracted; (e) Fitted Lα/L3 membrane thickness δ and (f) Caillé parameter η of Lα phases as a function of temperature. Coloured panels indicate onset of non-monotonic shift in parameters. | ||
3.3 Effect of temperature and octanol concentration on the mixed L4/L1 phase
We have investigated selected isopleths where the oct:SDS ratio is 0.62 w/w, corresponding to mixed vesicle/micelle L4/L1 phase (Fig. 6),6,16 and 0.08 w/w ratio corresponding to the cylindrical micellar L1 phase (Fig. 7). The mixed L4/L1 phases were analysed following the same experimental protocol as for the Lα. From 1H NMR analysis, an increase in peak resolution is observed upon dilution around ∼1 ppm corresponding to hydrocarbon tails (3–9 and 3*–12*) of octanol and SDS, indicating the formation of more spherical structures ascribed to vesicles L4 (Fig. 6a, left). The emergence of the peak at ∼4 ppm (1* Hs next to SDS SO4−) suggests that the SDS headgroups are more deshielded, compatible with an increase in surface curvature, expected for formation of vesicles. The absence of –CH2 (1) shift at 3.5 ppm implies that octanol headgroups are electrostatically and/or sterically shielded and are thus embedded within the bilayer membrane and/or micelles. Upon heating the concentrated sample containing 7.8% SDS (0.135 SDS + octanol volume fraction), a hydrocarbon peak growth was observed at ∼1 ppm suggesting a shift towards micellar structures. Crosspolarised optical microscopy (Fig. 6b) indicates the presence of vesicles as evidenced by Maltese crosses in microscopy across the isopleth. The SANS spectra are isotropic and exhibit pronounced forward scattering, along with a characteristic micellar profile. The radially averaged I(q) spectra, are shown in Fig. 6c (left). The SANS profiles were thus fitted with a sum of lamellar and flexible cylinder models (SASView lamellar_hg_stack_caille + flexible_cylinder, see individual model contributions in Fig. S8, ESI†). Data fits indicate a decrease in lamellar to micelle ratio L4/L1 at higher concentrations (Fig. 6d, left) from 4.5 to 0.5 and temperatures 45 °C and above from 2.0 to 1.0 (Fig. 6d, right). This effect with increasing temperatures has been previously reported in NaLAS/Na2SO4 system where, at similar salt concentrations (∼0.2 M), a shift towards wormlike micelles was observed at temperatures above 30 °C.48 The lamellar d-spacing d increases with dilution from 100 to 130 Å and increase with temperatures above 40 °C from 110 to 140 Å. The length of wormlike micelles decreases only slightly with dilution, from 500 to 400 Å but rapidly grows with temperature, above 45 °C, from 400 Å to 1200 Å. The data fits were compatible with fixed micellar radius R and Kuhn length b at ≃14 Å. Micellar elongation with temperature has been observed in both cationic (CTAB,49 CTAHNC,50 EHAC51) and anionic surfactant (SDS52) systems and associated with increases in headgroup area caused by counterion dissociation at temperatures ∼45 °C and above. Additionally, temperature increase facilitates octanol partitioning in water53 which leads to reduced SDS tail volume allowing for the formation of aggregates with higher curvature, including wormlike micelles.
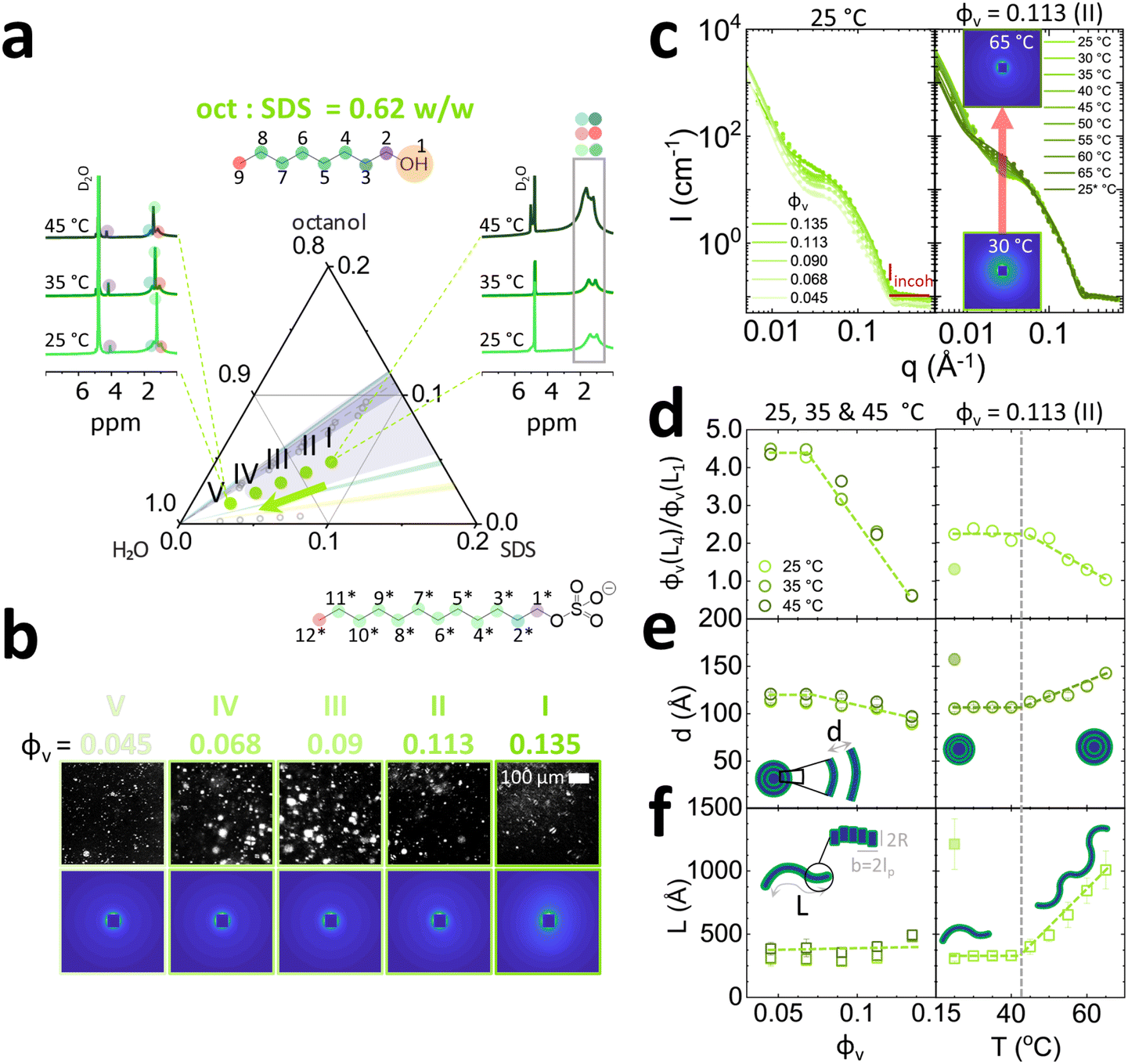 | ||
| Fig. 6 Coexistence of micellar L1 and vesicle L4 phases, along SDS (I) 7.8% SDS/4.8% octanol, (II) 6.5% SDS/4.0% octanol, (III) 5.2% SDS/3.2% octanol, (IV) 3.9% SDS/2.4% octanol, (V) 2.6% SDS/1.6% octanol, corresponding to a fixed oct:SDS = 0.62 w/w ratio. (a) Experimentally investigated compositions and illustrative NMR data at varying temperature. (b) Corresponding optical birefringence and SANS data at 25 °C. (c) Sector-averaged scattering profiles for compositions I–V at 25 °C (left) and temperature evolution of the scattering data for composition II, i.e. with ϕv (SDS + octanol) = 0.113 v/v (right) (d) fitted vesicle to micellar ratio ϕv (L4)/ϕv (L1) dependence on ϕv for 25–45 °C temperature range (left), and temperature 25–65 °C for composition II, with ϕv = 0.113. (e) Fitted dimensions of combined lamellar Caillé and flexible cylinder model, characterised by d-spacing d and (f) wormlike micelle contour length L, measured at temperatures within 25–45 °C (left); temperature dependence of lamellar and micellar dimensions for composition II (right). Kuhn length b and cylinder radius R was fixed at 14 Å. | ||
 | ||
| Fig. 7 Characterisation of the micellar phase L1, illustrated along the isopleth defined by (I) 7.8% SDS/0.6% octanol, (II) 6.5% SDS/0.5% octanol, (III) 5.2% SDS/0.4% octanol, (IV) 3.9% SDS/0.3% octanol in brine. (a) Experimentally investigated compositions and illustrative NMR data at varying temperature. (b) Corresponding SANS patterns at 25, 35, and 45 °C. (c) Radially-averaged scattering profiles, and a fine temperature scan for composition II, ϕ = 0.07. (d) Micellar dimensions obtained by fitting a core–shell cylinder model, indicated the equatorial radius Req and persistence length L, as a function of composition ϕ and temperature. The inset highlights that salt content (and salt:surfactant ratio) increases upon dilution with brine in these experiments, leading to micellar elongation. | ||
3.4 Effect of temperature and octanol concentration on the micellar L1 phase
Finally, we examine the effect of dilution and temperature in SDS/octanol/brine system following 0.08 oct:SDS dilution line, as shown in Fig. 7a. From previous literature,19,54 the addition of salt NaCl and mid-to-long chain 1-alcohol such as 1-octanol is expected to favour the formation of wormlike micelles. 1H NMR analysis revealed well resolved peaks in the hydrocarbon region between 1–2 ppm, shown in Fig. 7a. Increasing temperature results in the growth of headgroup-adjacent –CH2 (2 and 1*) peaks in 4 ppm region, implying the headgroups become more deshielded and the structures more isotropic. The 2D SANS scattering patterns are centrosymmetric as expected for L1 phases, and with greater in intensity for higher SDS/octanol concentrations (Fig. 7b). Radially averaged profiles I(q), along both composition and temperature scans, could be generally well fitted with a core shell cylinder model33 (SASView core_shell_cylinder) as shown in Fig. 7c and detailed in ESI,† yielding cylinder radii and lengths upon dilution (and accompanying increase in salt ratio) and temperature; the estimated lengths L are interpreted in terms of a persistence length. Our data show an increase in cylinder length from 50 Å to 200 Å upon dilution with brine as a consequence of increased brine:SDS ratio (Fig. 7d, left). Higher ratio of brine:SDS (Fig. 7d, left inset) leads to screening of SDS headgroup charge, promoting wormlike micelle formation. By contrast Req remains unchanged, at ∼18 Å. Increase in temperature from 25 to 65 °C leads to a linear decrease in cylinder length from 55 Å to 45 Å for the sample with ϕv =0.071 (6.5 w/v% SDS) membrane volume fraction.
4 Conclusions
In this study, we explore the effect of octanol:SDS stoichiometry, concentration and temperature on the SDS/octanol/brine (20 g L−1 NaCl) microstructures across lamellar, mixed and micellar phases, as illustrated in Fig. 8. The phase behaviour and rheology of the Lα pseudoquaternary SDS/octanol/brine system has been previously investigated, in particular due to its response to flow near the Lα–L3 phase boundary.17,18,21–28 In particular, the 6.5% SDS/7.9% octanol/85.6% brine system has been reported to undergo flow-induced phase transformations from lamellar Lα to multilamellar vesicles (MLVs), and ordered MLV arrays of prescribed dimensions at selected temperatures (near ambient) and shear rates. Generally, the addition of NaCl promotes the formation of L4–Lα–L3 sequence of phases.10 By contrast, a detailed characterisation of the solution structures formed in this region of the phase diagram, under quiescent conditions, has remained elusive, which we have sought to address.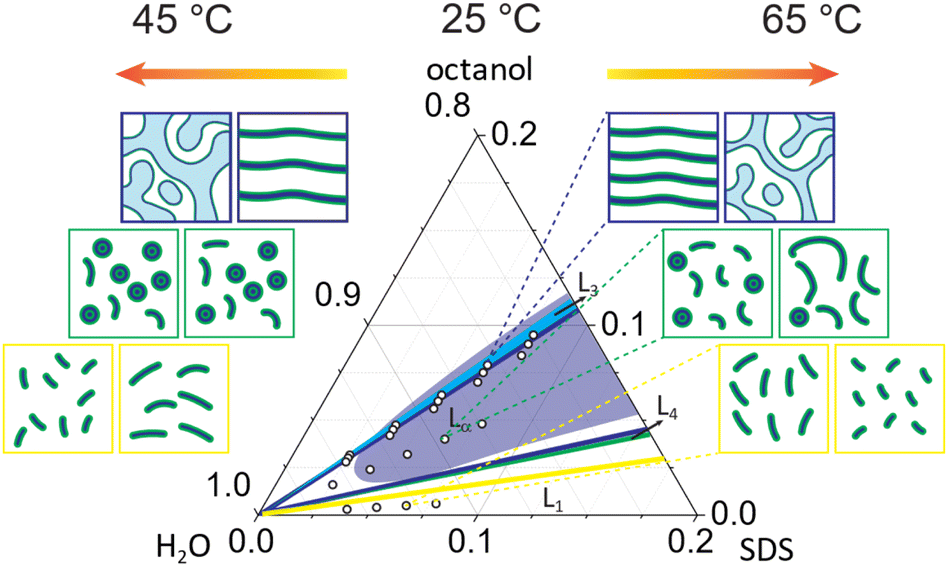 | ||
| Fig. 8 Overview of the solution structure observed in SDS/octanol/brine as a function of composition and temperature. The accompanying sketches indicate the key transformations observed: (top) membrane swelling upon dilution in all lamellar phases and Lα to L3 transformation upon increasing temperature for oct:SDS of 1.21; (middle) dilution of mixed phase promotes the formation of more isotropic phase, in this case vesicles while temperature increase results in micellar elongation; (bottom) dilution of L1 phase with brine promotes the formation of wormlike micelles while increase in temperature favours less elongated micelles. | ||
Overall, our observations confirm that the addition of co-surfactant octanol leads to the formation of more ordered phases.6,16 At the highest octanol:SDS concentration ratio studied here (1.21) at 25 °C, a lamellar phase Lα is formed which remains stable across dilution until ϕ = 0.064 with gradual increase in polydispersity pD. Upon increasing temperature above 35 °C, an (isotropic) sponge phase L3 emerges, accompanied by a loss of optical birefringence and chemical shifts corresponding to hydrocarbon chain around 1–2 ppm region in 1H NMR, isotropic SANS patterns with the Bragg peak shifted to higher d-spacing and decrease in power law exponent to −k ≤ 0.5. The sponge phase L3 remains stable across all dilution concentrations at 45 °C studied here with lamellar pore size ξ increasing at lower concentrations following the dilution law. Increasing temperature to 65 °C leads to a gradual decrease in sponge phase pore size ξ and membrane thickness δ. Lα phases with lower octanol concentrations (oct:SDS ratio 1.15 and 1.08) do not undergo transformation to L3 upon temperature increase to 35 °C. The dilution of these phases with brine leads to formation of more isotropic phases and increase in lamellar polydispersity. Increasing temperature to 65 °C increases the lamellar d-spacing, decreases membrane thickness δ, power law exponent −k and Caillé parameter η.
At lower octanol:SDS ratios (0.62), mixed micellar and vesicle phases are found at 25 °C. Here, dilution results a greater fraction of vesicles L4, while the d spacing increases slightly, and the length of wormlike micelles remains broadly unchanged. Temperature increase above 45 °C leads to a decrease in vesicle L4 fraction, accompanied by an increases in d spacing, and a significant (linear) elongation of wormlike micelles. Finally, the L1 phase (oct:SDS = 0.08) yields elongated micelles at 25 °C, which grow linearly upon dilution with brine as a consequence of decreased electrostatic repulsion between the charged SDS sulfate headgroups. In contrast, increasing temperature causes the micelles to become progressively less elongated. Overall, our results elucidate the rich solution behaviour of SDS ternary systems in presence of long chain alcohol octanol, which aids structure-formation at low surfactant content, as a function of concentration and temperature, relevant to a range of academic and industrial contexts.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Institut Laue Langevin (Grenoble) for beamtime; data available on https://doi.ill.fr/10.5291/ILL-DATA.9-10-1651 (static SANS) and https://doi.ill.fr/10.5291/ILL-DATA.9-10-1706 (flow SANS), and W. N. Sharratt for help with coding the sponge phase model. We thank EPSRC (EP/V056891/1) and Imperial College London for a PhD studentship for LD; JTC acknowledges the Royal Academy of Engineering (RAEng, UK) and Procter & Gamble for funding a Research chair.Notes and references
- P. Kekicheff, C. Grabielle-Madelmont and M. Ollivon, J. Colloid Interface Sci., 1989, 131, 112–132 CrossRef CAS.
- I. W. Hamley, Introduction to Soft Matter: Synthetic and Biological Self-Assembling Materials, 2007, pp. 161–220 Search PubMed.
- K. Holmberg, B. Jönsson, B. Kronberg and B. Lindman, Surf. Polym. Aqueous Solution, 2003, 39–66 Search PubMed.
- H. Khan, J. M. Seddon, R. V. Law, N. J. Brooks, E. Robles, J. T. Cabral and O. Ces, J. Colloid Interface Sci., 2019, 538, 75–82 CrossRef CAS PubMed.
- G. Tyagi, D. Seddon, S. Khodaparast, W. N. Sharratt, E. S. Robles and J. T. Cabral, Colloids Surf., A, 2021, 618, 126414 CrossRef CAS.
- A. Bellocq, Handbook of Microemulsion Science and Technology, CRC Press, 2018, pp. 139–184 Search PubMed.
- R. Zana, Adv. Colloid Interface Sci., 1995, 57, 1–64 CrossRef CAS.
- J. G. Méndez-Bermúdez and H. Dominguez, J. Mol. Model., 2016, 22, 1–9 CrossRef PubMed.
- A. Patist, T. Axelberd and D. O. Shah, J. Colloid Interface Sci., 1998, 208, 259–265 CrossRef CAS PubMed.
- I. Alibert, C. Coulon, A. Bellocq and T. Gulik-Krzywicki, Europhys. Lett., 1997, 39, 563 CrossRef CAS.
- E. Caponetti, D. C. Martino, M. Floriano and R. Triolo, Langmuir, 1997, 13, 3277–3283 CrossRef CAS.
- E. G. R. Putra and A. Patriati, AIP Conf. Proc., 2015, 020001 CrossRef.
- G. M. Førland, J. Samseth, M. I. Gjerde, H. Høiland, A. Ø. Jensen and K. Mortensen, J. Colloid Interface Sci., 1998, 203, 328–334 CrossRef PubMed.
- F. Auguste, A. Bellocq, D. Roux, F. Nallet and T. Gulik-Krzywicki, Trends Colloid Interface Sci. IX, 1995, 276–279 CAS.
- C. Safinya, E. Sirota, D. Roux and G. Smith, Phys. Rev. Lett., 1989, 62, 1134 CrossRef CAS PubMed.
- P. Hervé, D. Roux, A.-M. Bellocq, F. Nallet and T. Gulik-Krzywicki, J. Phys. II, 1993, 3, 1255–1270 CrossRef.
- F. Auguste, J.-P. Douliez, A.-M. Bellocq, E. J. Dufourc and T. Gulik-Krzywicki, Langmuir, 1997, 13, 666–672 CrossRef CAS.
- L. Donina, L. Porcar and J. T. Cabral, Soft Matter, 2022, 18, 7010–7019 RSC.
- L. Arleth, M. Bergström and J. S. Pedersen, Langmuir, 2002, 18, 5343–5353 CrossRef CAS.
- G. Guerin and A. Bellocq, J. Phys. Chem., 1988, 92, 2550–2557 CrossRef CAS.
- P. Sierro and D. Roux, Phys. Rev. Lett., 1997, 78, 1496 CrossRef CAS.
- O. Diat, D. Roux and F. Nallet, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1995, 51, 3296 CrossRef CAS PubMed.
- A.-S. Wunenburger, A. Colin, J. Leng, A. Arnéodo and D. Roux, Phys. Rev. Lett., 2001, 86, 1374 CrossRef CAS PubMed.
- J.-B. Salmon, A. Colin and D. Roux, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2002, 66, 031505 CrossRef PubMed.
- A. Lutti and P. Callaghan, Eur. Phys. J. E: Soft Matter Biol. Phys., 2007, 24, 129–137 CrossRef CAS PubMed.
- J. Leng, F. Nallet and D. Roux, Eur. Phys. J. E: Soft Matter Biol. Phys., 2001, 4, 77–83 CrossRef CAS.
- Z. Yatabe, R. Hidema, C. Hashimoto, R. B. Pansu and H. Ushiki, Chem. Phys. Lett., 2009, 475, 101–104 CrossRef CAS.
- L. Donina, A. Rafique, S. Khodaparast, L. Porcar and J. T. Cabral, Soft Matter, 2021, 17, 10053–10062 RSC.
- M. Buchanan, L. Starrs, S. Egelhaaf and M. Cates, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2000, 62, 6895 CrossRef CAS PubMed.
- F. Nallet, R. Laversanne and D. Roux, J. Phys. II, 1993, 3, 487–502 CrossRef CAS.
- N. Lei, C. Safinya, D. Roux and K. Liang, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1997, 56, 608 CrossRef CAS.
- L. Porcar, W. A. Hamilton, P. D. Butler and G. Warr, Langmuir, 2003, 19, 10779–10794 CrossRef CAS.
- S. R. Kline, J. Appl. Crystallogr., 2006, 39, 895–900 CrossRef CAS.
- V. Patel, D. Ray, K. Singh, L. Abezgauz, G. Marangoni, V. K. Aswal and P. Bahadur, RSC Adv., 2015, 5, 87758–87768 RSC.
- P. Panizza, L. Courbin, G. Cristobal, J. Rouch and T. Narayanan, Phys. A, 2003, 322, 38–54 CrossRef.
- G. Porte, J. Appell, P. Bassereau, J. Marignan, M. Skouri, I. Billard and M. Delsanti, Phys. A, 1991, 176, 168–186 CrossRef CAS.
- F. J. Millero, R. Dexter and E. Hoff, J. Chem. Eng. Data, 1971, 16, 85–87 CrossRef CAS.
- U. Domańska and M. Królikowska, J. Chem. Eng. Data, 2010, 55, 2994–3004 CrossRef.
- B. Demé, M. Dubois, T. Gulik-Krzywicki and T. Zemb, Langmuir, 2002, 18, 997–1004 CrossRef.
- M. Dubois, T. Zemb, N. Fuller, R. Rand and V. Parsegian, J. Chem. Phys., 1998, 108, 7855–7869 CrossRef CAS.
- J. Pan, S. Tristram-Nagle, N. Kučerka and J. F. Nagle, Biophys. J., 2008, 94, 117–124 CrossRef CAS PubMed.
- S. Purushothaman, P. Cicuta, O. Ces and N. J. Brooks, J. Phys. Chem. B, 2015, 119, 9805–9810 CrossRef CAS PubMed.
- É. Freyssingeas, F. Nallet and D. Roux, Langmuir, 1996, 12, 6028–6035 CrossRef.
- N. Delorme, J.-F. Bardeau, D. Carrière, M. Dubois, A. Gourbil, H. Mohwald, T. Zemb and A. Fery, J. Phys. Chem. B, 2007, 111, 2503–2505 CrossRef CAS PubMed.
- B.-S. Lu, S. P. Gupta, M. Belička, R. Podgornik and G. Pabst, Langmuir, 2016, 32, 13546–13555 CrossRef CAS PubMed.
- S. Mahbub, M. A. Rub, M. A. Hoque, M. A. Khan and D. Kumar, J. Phys. Org. Chem., 2019, 32, e3967 CrossRef.
- H. A. Faizi, S. L. Frey, J. Steinkühler, R. Dimova and P. M. Vlahovska, Soft Matter, 2019, 15, 6006–6013 RSC.
- A. S. Rafique, S. Khodaparast, A. S. Poulos, W. N. Sharratt, E. S. Robles and J. T. Cabral, Soft Matter, 2020, 16, 7835–7844 RSC.
- T. S. Davies, A. M. Ketner and S. R. Raghavan, J. Am. Chem. Soc., 2006, 128, 6669–6675 CrossRef CAS PubMed.
- P. Hassan, B. Valaulikar, C. Manohar, F. Kern, L. Bourdieu and S. Candau, Langmuir, 1996, 12, 4350–4357 CrossRef CAS.
- G. C. Kalur, B. D. Frounfelker, B. H. Cipriano, A. I. Norman and S. R. Raghavan, Langmuir, 2005, 21, 10998–11004 CrossRef CAS PubMed.
- S. Rajkhowa, S. Mahiuddin, J. Dey, S. Kumar, V. Aswal, R. Biswas, J. Kohlbrecher and K. Ismail, Soft Matter, 2017, 13, 3556–3567 RSC.
- B. E. Lang, J. Chem. Eng. Data, 2012, 57, 2221–2226 CrossRef CAS.
- E. Dutkiewicz and A. Jakubowska, Colloid Polym. Sci., 2002, 280, 1009–1014 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sm01098h |
| This journal is © The Royal Society of Chemistry 2023 |