
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bacterial susceptibility and resistance to modelin-5†
Sarah R.
Dennison
 *a,
Leslie HG
Morton
a,
Kamal
Badiani
b,
Frederick
Harris
a and
David A.
Phoenix
c
*a,
Leslie HG
Morton
a,
Kamal
Badiani
b,
Frederick
Harris
a and
David A.
Phoenix
c
aSchool of Pharmacy and Biomedical Sciences, University of Central Lancashire, Preston PR1 2HE, UK. E-mail: srdennison1@uclan.ac.uk
bPepceuticals Limited, 4 Feldspar Close, Warrens Park, Enderby, Leicestershire LE19 4JS, UK
cOffice of the Vice Chancellor, London South Bank University, 103 Borough Road, London SE1 0AA, UK
First published on 16th October 2023
Abstract
Modelin-5 (M5-NH2) killed Pseudomonas aeruginosa with a minimum lethal concentration (MLC) of 5.86 μM and strongly bound its cytoplasmic membrane (CM) with a Kd of 23.5 μM. The peptide adopted high levels of amphiphilic α-helical structure (75.0%) and penetrated the CM hydrophobic core (8.0 mN m−1). This insertion destabilised CM structure via increased lipid packing and decreased fluidity (ΔGmix < 0), which promoted high levels of lysis (84.1%) and P. aeruginosa cell death. M5-NH2 showed a very strong affinity (Kd = 3.5 μM) and very high levels of amphiphilic α-helical structure with cardiolipin membranes (96.0%,) which primarily drove the peptide's membranolytic action against P. aeruginosa. In contrast, M5-NH2 killed Staphylococcus aureus with an MLC of 147.6 μM and weakly bound its CM with a Kd of 117.6 μM, The peptide adopted low levels of amphiphilic α-helical structure (35.0%) and only penetrated the upper regions of the CM (3.3 mN m−1). This insertion stabilised CM structure via decreased lipid packing and increased fluidity (ΔGmix > 0) and promoted only low levels of lysis (24.3%). The insertion and lysis of the S. aureus CM by M5-NH2 showed a strong negative correlation with its lysyl phosphatidylglycerol (Lys-PG) content (R2 > 0.98). In combination, these data suggested that Lys-PG mediated mechanisms inhibited the membranolytic action of M5-NH2 against S. aureus, thereby rendering the organism resistant to the peptide. These results are discussed in relation to structure/function relationships of M5-NH2 and CM lipids that underpin bacterial susceptibility and resistance to the peptide.
Introduction
It has been predicted that infections due to pathogenic bacteria could be instrumental in up to ten million deaths a year by 2050,1,2 with the very real possibility that many infections may become untreatable.3,4 A primary focus in combatting these infections has been the ‘ESKAPE’ pathogens, which include multi-drug resistant (MDR) forms of Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species.5,6 This group of bacteria are the major cause of life-threatening, nosocomial infections in immunocompromised and critically ill patients.6,7 In response, antimicrobial peptides (AMPs) have been identified as promising pharmaceutical candidates for the prevention and treatment of infections caused by these pathogens.8,9 AMPs are naturally occurring antibiotics of the innate immune system that promote bacterial cell death through multiple mechanisms, including membrane lysis and/or attack on intracellular targets.10,11 Bacterial resistance mechanisms to the action of AMPs are known,12,13 but the absence of specific interactors or defined mechanistic pathways minimizes the likelihood of bacteria selecting for resistance to this action.14,15 Moreover, the non-specific nature of their antibacterial action allows AMPs to bypass the resistance mechanisms of ESKAPE and other MDR bacterial pathogens to conventional antibiotics, which generally result from selection driven by the single sites of action used by these drugs.8,9,16,17 This property is a major driver in the development of AMPs as alternatives or adjuvants to conventional antibiotics and as potential agents to fight the clinical challenges posed by ESKAPE pathogens.14,18 Other important properties of AMPs in this scenario are that most of these peptides act rapidly with potent, broad-spectrum antimicrobial activity but do not affect microbiota or target healthy eukaryotic cells,8–11 although many are able to kill cancer cells.10,19 Currently, a number of AMPs are either in clinical trials or therapeutic, topical use to treat a variety of infectious conditions,20,21 and progress towards their systemic application for this purpose is ongoing.22Despite the clear therapeutic potential of AMPs, a number of factors have impeded the full realisation of this potential and in response a number of strategies to improve their efficacy have been employed,23,24 including the production of synthetic, designed peptides.25 A major example of these designed AMPs is the C-terminally amidated peptide, modelin-5 (M5-NH2), which is the archetypic member of the modelin family of AMPs.26,27 Recent studies have shown that M5-NH2 is non-haemolytic28 and possesses both anticancer activity27,29,30 and the ability to kill a variety of fungi and bacteria.26,27,30–33 The present study investigates the ability of M5-NH2 to kill P. aeruginosa and S. aureus, which are ‘ESKAPE’ pathogens of critical priority on the World Health Organisation's (WHO) list of most dangerous pathogens.34,35
Experimental
Materials
M5-NH2 was supplied by Pepceuticals (Leicestershire, UK), purified by HPLC to purity greater than 99% and its sequence confirmed by MALDI mass spectrometry, as KLAKKLAKLAKLAKAL-CONH2. The phospholipids used were POPG: 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol; TOCL (1,1′,2,2′-tetraoleoyl cardiolipin): 1′,3′-bis[1,2-dioleoyl-sn-glycero-3-phospho]-glycerol; POPE: 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine; and Lys-DOPG: 1,2-dioleoyl-sn-glycero-3-[phospho-rac-(3-lysyl(1-glycerol))], all of which were purchased from Avanti Polar Lipids (Alabaster, AL). M 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) and ethylenediaminetetraacetic acid (EDTA) was purchased from Merk Sigma-Aldrich. All buffers were prepared using ultra-pure water (resistivity 18 MΩ cm). Ringer's solution, nutrient broth and nutrient agar were purchased from Thermo Fisher Scientific (Leicestershire, UK). HPLC grade solvents were obtained from VWR International Ltd (Lutterworth, UK) and all other regents were purchased from Merk Sigma-Aldrich Company Ltd (Dorset, UK).![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×g; 10 min) to form a cell pellet using a bench top centrifuge (ALC PK 120R). The resulting cell pellet was washed three times in ¼ strength Ringer's solution and then resuspended in 1 ml of a ¼ strength Ringer's solution to ensure there was starting inoculum density of circa 5.8 × 108 CFU ml−1.
000×g; 10 min) to form a cell pellet using a bench top centrifuge (ALC PK 120R). The resulting cell pellet was washed three times in ¼ strength Ringer's solution and then resuspended in 1 ml of a ¼ strength Ringer's solution to ensure there was starting inoculum density of circa 5.8 × 108 CFU ml−1.
| Bacteria | Membrane lipids | |||
|---|---|---|---|---|
| TOCL (mol%) | POPG (mol%) | POPE (mol%) | Lys-PG (mol%) | |
| P. aeruginosa | 21 | 11 | 60 | 0 |
| S. aureus | 5 | 57 | 0 | 38 |
| ΔF = (ΔFMax[A])/(Kd + [A]) | (1) |
:lipid molar ratio of 1:20. Both in the presence and absence of these SUVs, far-UV CD spectra were collected for M5-NH2, where ten scans per sample were obtained using a 10 mm path-length cell. Each scan was performed over a wavelength range of 180 nm to 260 nm at 0.5 nm intervals employing a bandwidth of 1 nm and at a speed 10 nm min−1. For all spectra obtained, the baseline acquired in the absence of peptide was subtracted and the percentage α-helical content of M5-NH2 estimated using the CDSSTR method (protein reference set 3) from the DichroWeb server.48 These experiments were repeated four times and the percentage α-helicity of M5-NH2 was averaged.
Thermodynamic analysis of these isotherms was undertaken and used to determine the Gibbs free energy of mixing (ΔGmix) of monolayers, which provides a measure of the relative stability associated with the miscibility energetics of their pure lipid components. Thermodynamically stable and thermodynamically unstable monolayers are indicated by negative and positive values of ΔGmix respectively.40,41 ΔGmix was computed according to eqn (2):
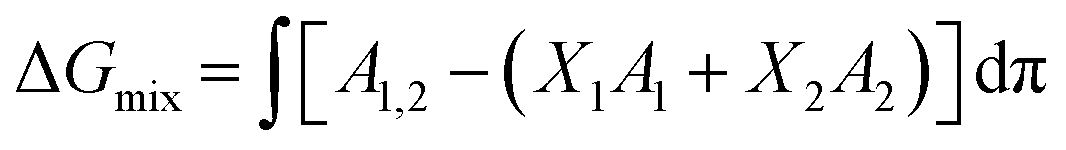 | (2) |
The calcein release assay was performed by combining 25 ml of SUVs containing entrapped calcein with 50 ml of M5-NH2 (10 mM), which was then made up to a final volume of 1 ml with HEPES (20.0 mM; pH 7.5), NaCl (150 mM) and EDTA (1.0 mM). The fluorescence intensities (FI) of calcein were monitored at 20 °C using an FP-6500 spectrofluorometer (JASCO, UK), with an excitation wavelength of 490 nm and emission wavelength of 520 nm. The fluorescence intensity induced in SUVs containing entrapped calcein by HEPES (20.0 mM; pH 7.5), NaCl (150 mM) and EDTA (1.0 mM) was taken as background leakage and that resulting from the addition of 1 μl of Triton X-100 (10%, v/v) was taken to represent 100% dye release. The percentage lysis induced by M5-NH2 was then calculated according to eqn (3):
| Lysis (%) = (([FIM5-NH2] − [FIHEPES])/([FITriton X] − [FIHEPES])) × 100 | (3) |
:57 mol%. and that of Lys-DOPG varied between 0 and 50 mol%. At each mol% of Lys-DOPG, monolayers were used to determine the membrane partitioning of M5-NH2, and SUVs used to determine the membranolytic activity of the peptide, all as described above. In both cases, these experiments were repeated four times and the average levels of membrane partitioning and lysis respectively plotted as a function of Lys-PG levels.
Results
M5-NH2 is a synthetic peptide designed to structurally mimic naturally occurring, α-helical AMPs (Fig. 1)26,27 and in the present study, this peptide is investigated for its ability to kill P. aeruginosa and S. aureus, which are on the WHO's list of most dangerous pathogens.34,35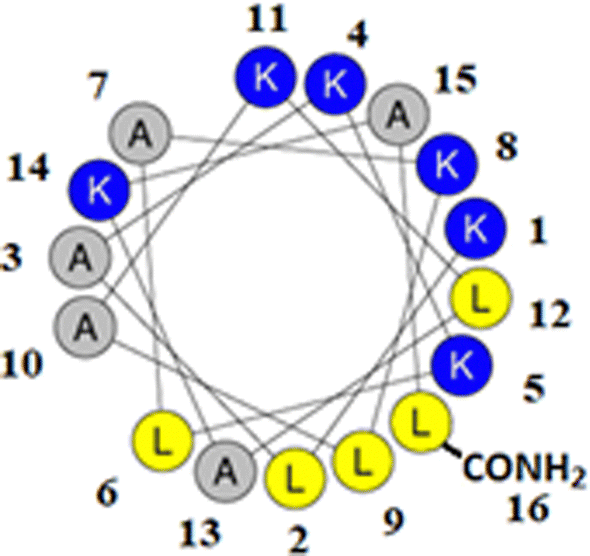 | ||
| Fig. 1 Graphical analysis of M5-NH2. Fig. 1 shows the sequence of M5-NH2 represented as a two-dimensional axial projection using software at Heliquest, where numbers associated with individual residues represent their position in this sequence (KLAKKLAKLAKLAKAL-CONH2).50 In an α-helical conformation, the peptide shows amphiphilicity with a wide, strongly cationic polar face and a narrower apolar face. The polar face of the α-helix is primarily formed from multiple lysine residues and a C-terminal, amidated leucine residue. This residue arrangement allows electrostatic/hydrophilic interactions with anionic components of the bacterial membranes studied here, including the headgroups of CL and PG. The apolar face of the α-helix comprises alanine and leucine residues, which permits hydrophobic interactions with the apolar core regions of bacterial membranes studied here, primarily formed by the acyl chains of CL, PG and PE. | ||
P. aeruginosa
M5-NH2 was found to kill P. aeruginosa with an MLC of 5.86 μM (Fig. S1; Table 2, ESI†), indicating potent activity against the organism which was comparable to that shown by the peptide against other Gram-negative bacteria.26,31 In general, the action of AMPs involves membrane interaction and a number of major steps in this process have been identified, including the early steps of membrane binding and conformational reassignment.45,51 In the first of these steps, M5-NH2 showed strong binding to SUVs mimetic of the P. aeruginosa CM (Kd = 23.5 μM; Table 2), which is consistent with the peptide's potent activity against the organism and indicative of a high affinity for these membranes (Table 2).52 To characterise this binding step, the interactions of M5-NH2 with SUVs formed from the individual lipid components of the CM of P. aeruginosa was investigated (Table 1). Anionic lipid forms around one third of the total lipid in these membranes and is predominantly formed from cardiolipin (CL) and phosphatidylglycerol (PG) (Table 1).44 M5-NH2 showed very high levels of binding to SUVs of TOCL (Kd = 3.5 μM; Table 2) and POPG (Kd = 6.7 μM; Table 2), which represented CL and PG respectively (Table 1). These data clearly suggested that the high affinity of M5-NH2 for the P. aeruginosa CM is driven by electrostatic interactions between anionic lipid in these membranes and the peptide's cationic residues. In contrast, M5-NH2 showed lower levels of binding with SUVs formed from POPE (Kd = 14.9 μM; Table 2), which represented the zwitterionic lipid phosphatidylethanolamine (PE); PE primarily forms the remaining two thirds of the total lipid in the CM of P. aeruginosa (Table 1).44 These interactions appeared to make a minor contribution to the affinity of M5-NH2 for the P. aeruginosa CM and presumably involved association of the peptide's cationic residues with phosphate groups of PE, as shown for other AMPs.53,54 Long range electrostatic attractive interactions similar to those involved in membrane binding are known to drive the initial targeting of bacterial membranes by AMPs, which is most probably the case for M5-NH2 in its action against P. aeruginosa.10,11| Membrane | K d (μM) | Lysis (%) | α-Helicity (%) | π (mN m−1) | ΔGmix |
|---|---|---|---|---|---|
| P. aeruginosa lipid mix | 23.5 | 84.1 | 75.0 | 8.0 | >0 |
| S. aureus lipid mix | 117.6 | 24.3 | 35.0 | 3.3 | <0 |
| TOCL | 3.5 | 93.1 | 96.0 | 12.3 | — |
| POPG | 6.7 | 74.5 | 86.9 | 9.6 | — |
| POPE | 14.9 | 51.0 | 53.0 | 4.9 | — |
After membrane binding, one of the most important steps in the antibacterial action of membranolytic AMPs is conformational change at the membrane interface to adopt their functional secondary structure.45 Consistent with this step, M5-NH2, which was unstructured in aqueous solution (Fig. 2), adopted high levels of α-helical structure in the presence of SUVs mimetic of the P. aeruginosa CM (α-helicity = 75.0%; Table 2 and Fig. 2). It is well established that the anisotropy of the bacterial membrane interface lowers the energy barrier for α-helix formation by AMPs45,51 and to characterise this process for M5-NH2, the role of individual P. aeruginosa CM lipids in this step was investigated (Table 1).44,55 The peptide showed very high levels of α-helical structure in the presence of SUVs formed from TOCL (α-helicity = 96.0%) and POPG (α-helicity = 86.9%), but lower α-helicity with SUVs formed from POPE (α-helicity = 53.0%) (Table 2 and Fig. 3). These data would seem to indicate that hydrophilic interactions with CL and PG make the predominant contribution to α-helix formation by M5-NH2 with the P. aeruginosa CM. It would also seem that these hydrophilic interactions are supported by a minor contribution from hydrophobic interactions between the peptide and PE in these membranes. Moreover, the involvement of both these types of interaction would appear to indicate that the α-helical structure formed by M5-NH2 at the P. aeruginosa CM interface possessed amphiphilic properties (Fig. 1). It is well established that the interplay of hydrophilic and hydrophobic interactions promotes the formation of this type of secondary structure by AMPs15,56 to facilitate their membranolytic antibacterial action.57,58
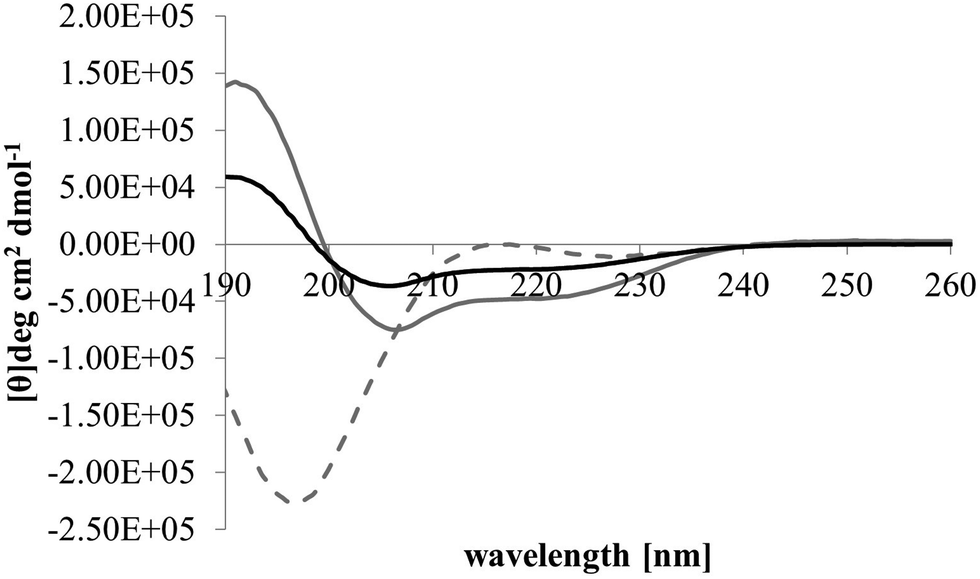 | ||
| Fig. 2 Conformational analysis of M5-NH2 in the presence of lipid mimics of the bacterial CM. Fig. 2 shows CD spectra for the conformational behaviour of M5-NH2 in solution, and in the presence of SUVs mimetic of the bacterial CM, with the lipid compositions described Table 1. In aqueous solution (dotted grey line), the peptide displayed a maximum at 215 nm and a minimum at 195 nm, indicating that the peptide was predominantly formed from random coil and β-type structures and possessed less than 10% α-helical structure. In the presence of SUVs mimetic of the S. aureus CM (black line) and the P. aeruginosa CM (dark grey line), the peptide showed minima at 208 nm and 225 nm, and a maximum at 190 nm, which is characteristic of α-helical architecture. Analysis of these spectra showed that the peptide was 75.0% α-helical in the presence of SUVs mimetic of the P. aeruginosa CM, and 35.0% α-helical in the presence of those mimetic of the S. aureus CM (Table 2). | ||
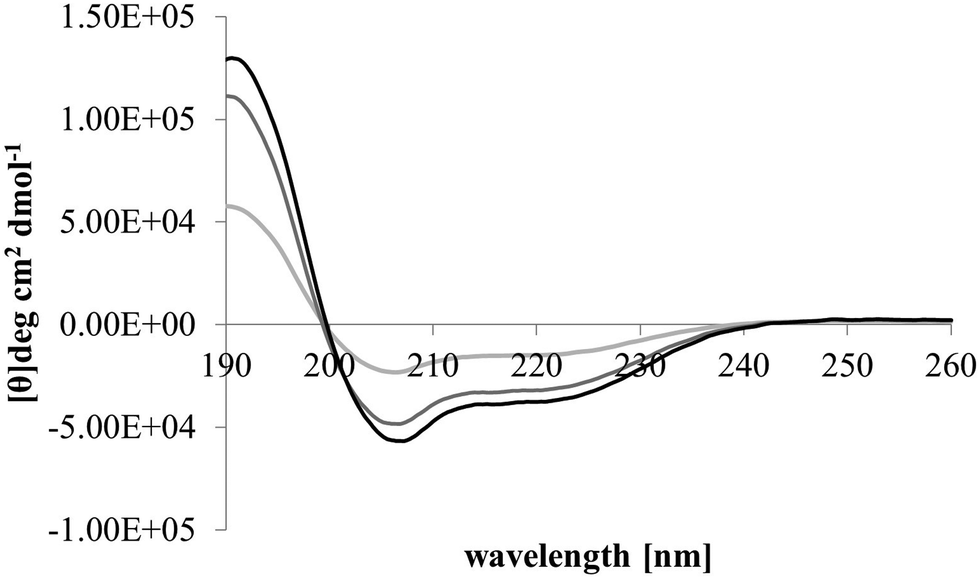 | ||
| Fig. 3 Conformational analysis of M5-NH2 in the presence of individual lipids from the bacterial CM. Fig. 3 shows CD spectra for the conformational behaviour of M5-NH2 in the presence of SUVs formed from the individual lipid components of the S. aureus and P. aeruginosa CM (Table 1). In the case of TOCL (black line), POPG (dark grey line) and POPE (light grey line) the peptide displayed minima 208 nm and 225 nm, and a maximum at 190 nm, which is characteristic of α-helical architecture. Analysis of these spectra showed that the peptide was mainly α-helical with levels of α-helicity >85.0% in the cases of TOCL and POPG, and α-helicity = 53.0% in the case of POPE (Table 2). | ||
The next major step in the antibacterial action of AMPs is membrane insertion45 and M5-NH2 partitioned into monolayers mimetic of the P. aeruginosa CM following hyperbolic kinetics (Fig. 4). In this process, the peptide showed a rapid initial rate of insertion into these monolayers over circa 100 seconds, before achieving maximal surface pressures in around 1000 seconds (Fig. 4), which is consistent with the peptide's strong affinity for these membranes (Table 2). Also consistent with this strong affinity, the high levels of these maximal surface pressure changes (π = 8.0 mN m−1) (Table 2 and Fig. 4) indicated deep levels of insertion and penetration into the monolayer hydrophobic region.40,41 Following insertion, the next major step in in the antibacterial action of AMPs is membrane lysis45 and M5-NH2 demonstrated a strong ability to compromise the integrity of SUVs mimetic of the P. aeruginosa CM (lysis = 84.1%) (Table 2). In combination, these results clearly showed that the action of the peptide against P. aeruginosa involved membranolytic action, which receives support from thermodynamic data (Fig. 5 and 6). Monolayers mimetic of the P. aeruginosa CM were thermodynamically stable with values of ΔGmix < 0; however, in the presence of the peptide, these monolayer mimics became thermodynamically unstable with values of ΔGmix > 0 (Table 2). This change in ΔGmix suggested that the insertion of M5-NH2 into these monolayers had increased their lipid packing and decreased their fluidity, which is consistent with the peptide's deep insertion into the P. aeruginosa CM40,41 (Table 2) as reported for other AMPs.59–63
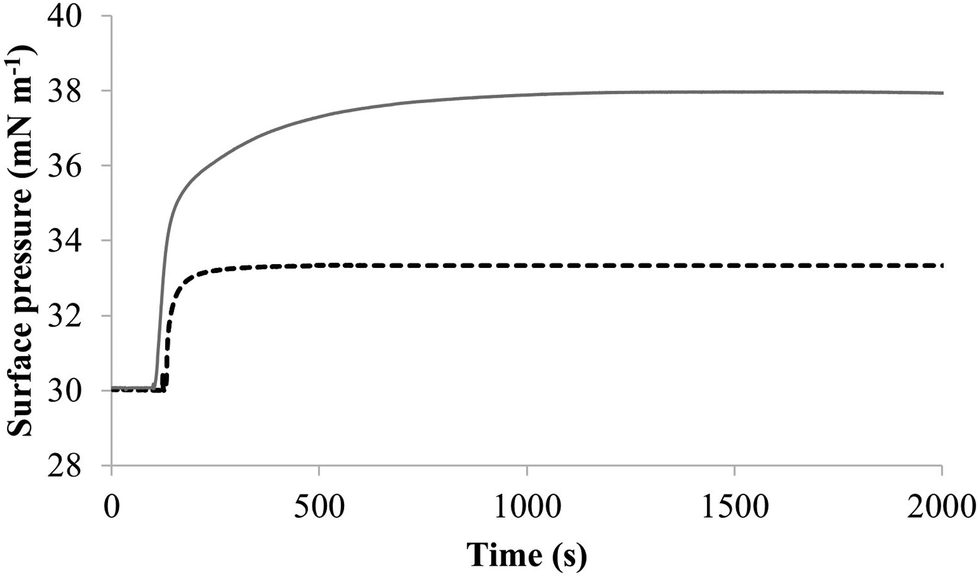 | ||
| Fig. 4 The interaction of M5-NH2 with lipid mimics of the bacterial CM. Fig. 4 shows the interaction of M5-NH2 with monolayers mimetic of the P. aeruginosa and S. aureus CM, with the lipid compositions described Table 1. The peptide inserted into these monolayers following generally similar, hyperbolic kinetics but showed widely different rates and levels of insertion. In the case of monolayer mimics of the S. aureus CM (black dotted line), the peptide took circa 2000 seconds to achieve maximal surface pressure changes of π = 3.3 mN m−1 (Table 2). However, with monolayer mimics of the P. aeruginosa CM (dark grey line), M5-NH2 showed much higher rates and levels of insertion, taking circa 1000 seconds to achieve maximal surface pressure of π = 8.0 mN m−1 (Table 2). | ||
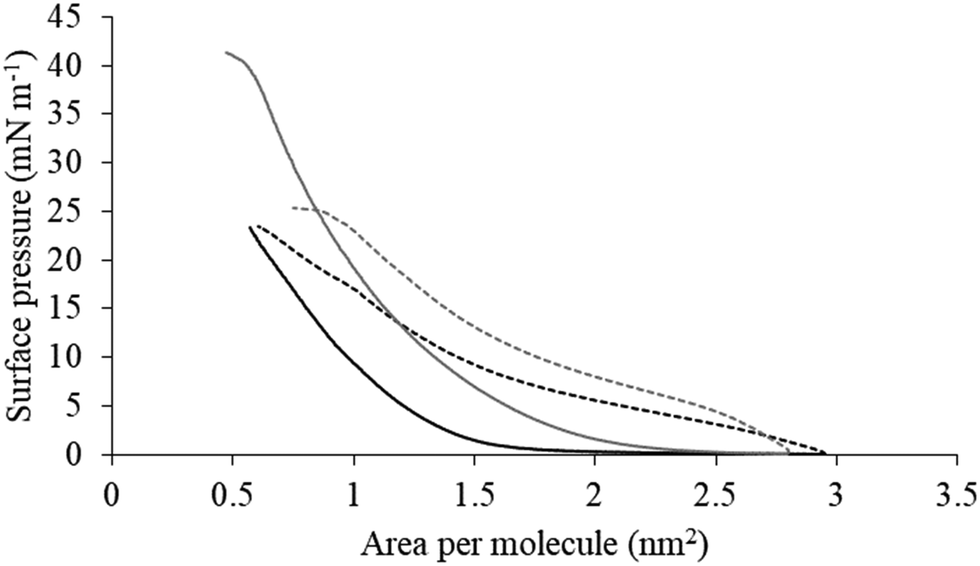 | ||
| Fig. 5 Compression isotherms for lipid mimics of the bacterial CM. Fig. 5 shows compression isotherms for monolayer mimics of the P. aeruginosa and S. aureus CM, with the lipid compositions described Table 1. Shown are compression isotherms for monolayer mimics of the S. aureus CM (dotted black line) and that of P. aeruginosa (dotted dark grey line) in the absence of M5-NH2. Also shown are these isotherms for monolayer mimics of the S. aureus CM (black line) and that of P. aeruginosa (dark grey line) in the presence of the peptide. These isotherms were analysed to determine values of ΔGmix, which are shown in Fig. 6. | ||
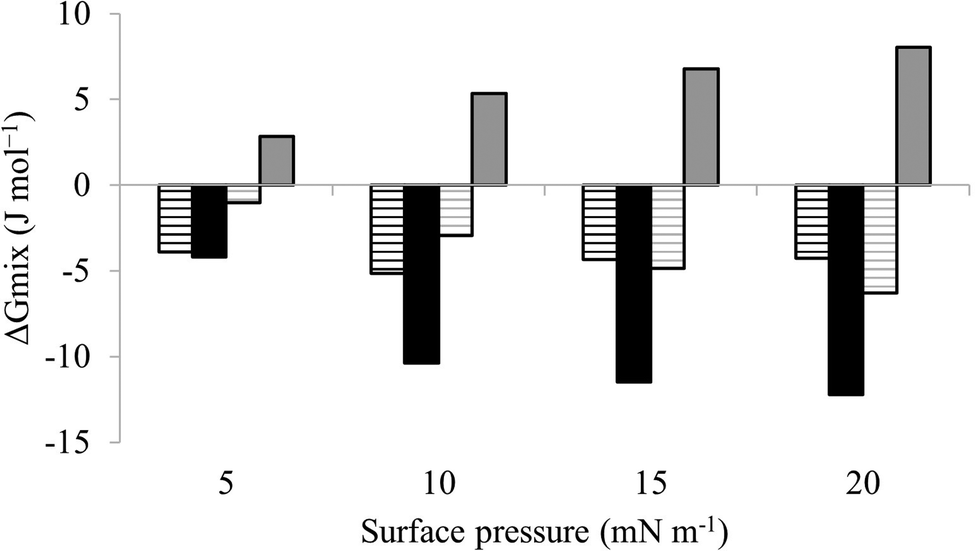 | ||
| Fig. 6 Thermodynamic analysis of compression Isotherms from lipid mimics of the bacterial CM. Fig. 6 shows values of ΔGmix derived from analysis of the compression isotherms depicted in Fig. 5. In the absence of M5-NH2, ΔGmix was < 0 for lipid monolayer mimics of the CM of both S. aureus (black striped bars) and P. aeruginosa (dark grey striped bars). In each case, ΔGmix became progressively more negative with increasing surface pressure, indicating thermodynamic stability. In the presence of M5-NH2 this trend was maintained for monolayer mimics of the S. aureus CM (black bars), except that ΔGmix was enhanced for a given surface pressure. In contrast, ΔGmix for monolayer mimics of the P. aeruginosa CM (dark grey bars) became >0 and progressively more positive with increasing surface pressure, indicating thermodynamic instability. | ||
To further investigate later steps in the membranolytic action of M5-NH2 against P. aeruginosa, its interaction with model membranes formed from individual lipid components of the organism's CM was investigated (Table 1).44,55 The peptide partitioned into monolayers formed from either TOCL or POPG following hyperbolic kinetics (Fig. 7) generally similar to those shown by M5-NH2 with monolayers mimetic of the P. aeruginosa CM (Fig. 4). M5-NH2 showed extremely rapid initial rates of insertion into these monolayers over circa 100 seconds, before achieving maximal surface pressures in around 500 seconds, (Fig. 7), which is consistent with the peptide's high affinity for these lipids (Table 2). Also consistent with this affinity, M5-NH2 induced very large maximal surface pressure changes in monolayers formed from TOCL (π = 12.3 mN m−1) and POPG (π = 9.6 mN m−1) (Table 2 and Fig. 7). The high level of these surface pressure changes reflected the involvement of strong electrostatic interactions and an ability to deeply penetrate the hydrophobic acyl chain region of these monolayers (Table 2).40,41 In contrast, although M5-NH2 partitioned into monolayers formed from POPE following hyperbolic kinetics similar to those with CL and POPG, the characteristics of this process were clearly different (Fig. 7). The peptide showed much slower rates of insertion into these monolayers, achieving maximal surface pressures in around 1500 seconds, (Fig. 7), which is consistent with its weaker affinity for these lipids (Table 2). Compared to the case of TOCL and POPG, M5-NH2 also induced lower maximal surface pressure changes in monolayers formed from POPE (π = 4.9 mN m−1), indicating a weaker but significant ability to penetrate these membranes (Table 2 and Fig. 7). The peptide also showed a strong ability to lyse SUVs formed from TOCL (lysis = 93.1%) and POPG (lysis = 74.5%) but a weaker capacity to permeabilise those formed from POPE (lysis = 51.00%) (Table 2). These data clearly showed that M5-NH2 has a strong ability penetrate and lyse model membranes formed from individual lipid components of P. aeruginosa membranes, which in both cases followed the rank order CL > POPG > POPE (Table 2). In combination, these data would seem to indicate that M5-NH2 has a strong, general preference for anionic lipid in the steps of its membranolytic action against P. aeruginosa. These data would also appear to indicate that electrostatic interactions between the peptide's cationic residues and CL and PG drive the steps of penetration and lysis in this membranolytic action. These electrostatic interactions would appear to be complemented by a minor contribution to these steps from hydrophobic interactions between M5-NH2 and PE (Fig. 1).
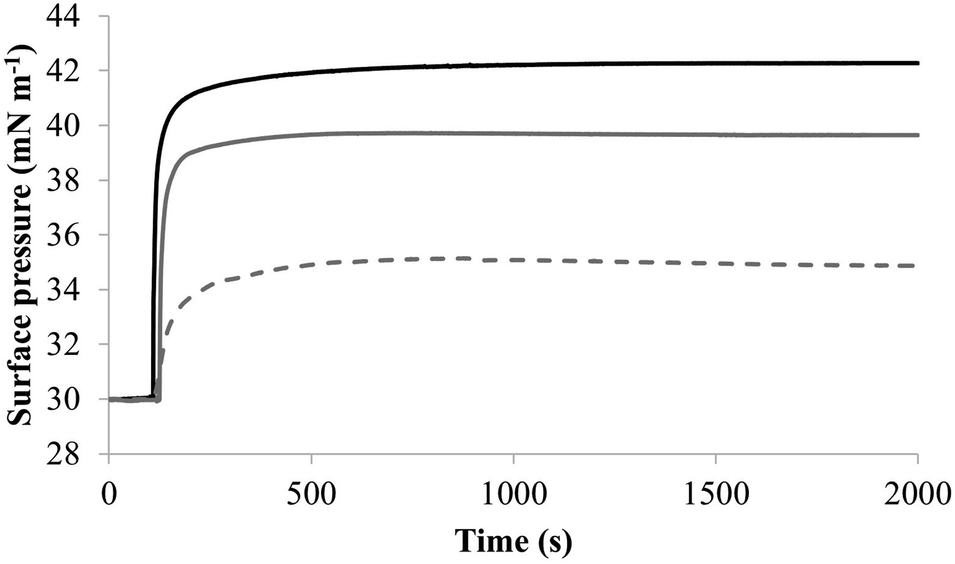 | ||
| Fig. 7 The interaction of M5-NH2 with monolayers formed from individual lipids from the bacterial CM Fig. 7 shows the interaction of M5-NH2 with monolayers formed from the individual lipid components of the S. aureus and P. aeruginosa CM (Table 1). The peptide inserted into these monolayers following hyperbolic kinetics that were similar to those with monolayers mimetic of bacterial cytoplasmic membranes, although rates of insertion were generally faster (Fig. 5). M5-NH2 peptide took <750 seconds to achieve maximal surface pressure changes with π > 9.0 mN m−1 (Table 2) for monolayers formed from TOCL (black line) and POPG (dark grey line). However, for monolayers formed from POPE (dotted light grey line), the peptide took circa 1500 seconds to achieve maximal surface pressure changes with π = 4.9 mN m−1 (Table 2). | ||
The penetration and lysis of the P. aeruginosa CM by M5-NH2 involved both electrostatic and hydrophobic interaction, which clearly reflected its use of amphiphilic structure to facilitate the later steps of its membranolytic action (Fig. 1).15 The levels of this structure formed by M5-NH2 in the presence of single lipid SUVs followed the rank order TOCL > POPG > POPE, correlating with those of its insertion and lysis with corresponding model lipid membranes (Table 2). This correlation clearly suggested that the ability of M5-NH2 to lyse and penetrate the P. aeruginosa CM was driven by the levels of its amphiphilic α-helical structure. The levels of this structure formed by the peptide with the P. aeruginosa CM accounted for three quarters of its architecture (Table 2), which equates to circa three and a half α-helical turns.11,64 The axial length of this α-helical structure is sufficient to allow depths of insertion into the P. aeruginosa CM by M5-NH2 that could penetrate the hydrophobic core of the opposing membrane leaflet.40,41,65 These levels of insertion are clearly consistent with those shown by M5-NH2 in its steps of penetration and lysis of the P. aeruginosa CM and with the associated changes to the structural properties of these membranes (Table 2). As indicated above, M5-NH2 also showed a strong preference for anionic lipid in these later steps, as well as that of adopting amphiphilic α-helical structure at the P. aeruginosa CM interface. These data clearly reflected the dominance of electrostatic interactions in these steps, which suggested that amphiphilicity is the major driver of the peptides membranolytic action against P. aeruginosa and is supported by previous work.31,33,66 Amphiphilic profiling has shown that M5-NH2 exhibits high levels of amphiphilicity along the length of its α-helical conformation33 and it has been demonstrated that this structural property drives major steps in its membranolytic action against other bacteria.31,33,66
In combination, these data would appear to indicate that in the early steps of its action against P. aeruginosa, a high affinity for anionic lipid in the CM of the organism drives binding and amphiphilic α-helix formation by M5-NH2. In the later steps of this action, the high amphiphilicity of this α-helical structure promotes deep insertion of M5-NH2 into the P. aeruginosa CM that induce high levels of lysis. These effects promote membranolytic action that leads to the death of P. aeruginosa and underpins the potent activity of M5-NH2 against the organism, which is represented schematically in Fig. 8.
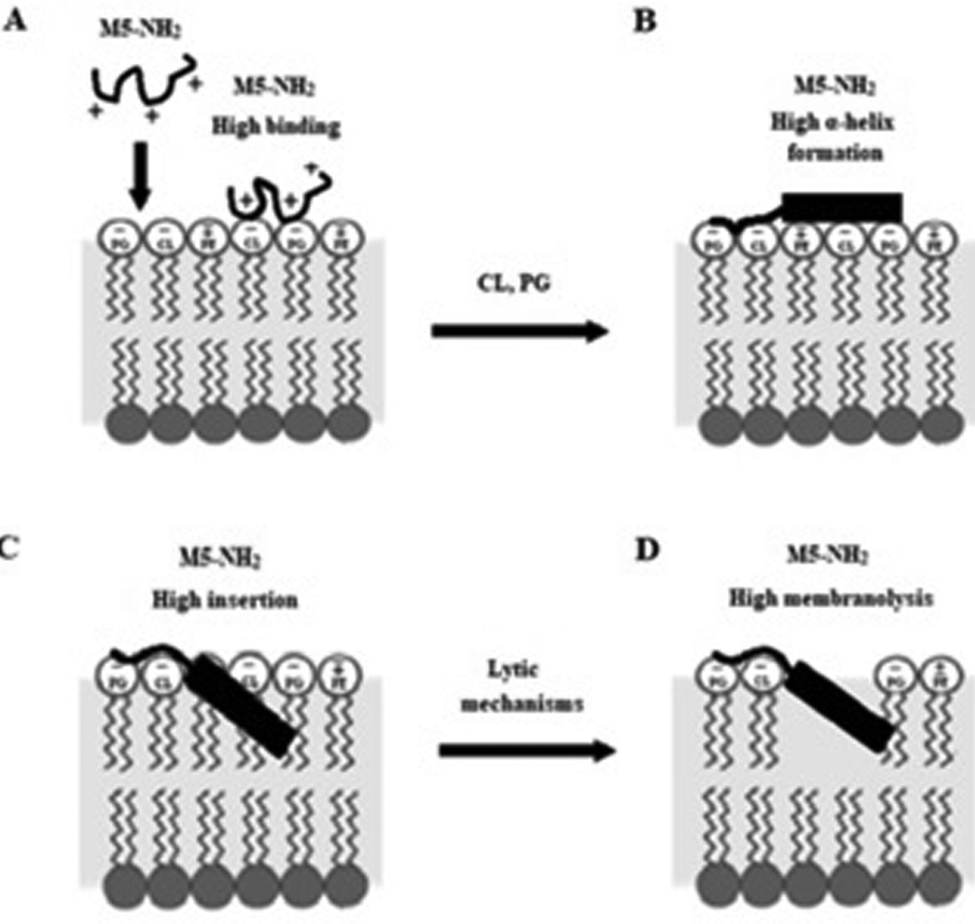 | ||
| Fig. 8 A schematic representation of the membranolytic action of M5-NH2 against P. aeruginosa. In the first major step of the action of M5-NH2 against P. aeruginosa, electrostatic attraction facilitates targeting of the CM and promotes high levels of binding to the headgroups of PG and CL by the peptide (Fig. 8A). In the next major step, the anisotropic environment of the CM interface promotes high levels of amphiphilic α-helical structure in M5-NH2, which potentially possesses a hydrophobicity gradient (Fig. 8B). The tilted characteristics and/or the amphiphilicity of this structure then drives the major step of insertion by the peptide into the P. aeruginosa CM, which leads to penetration of the hydrophobic core region of the membrane (Fig. 8C). In the final major step, the insertion of the peptide promotes a range of effects, such as increased lipid packing and decreased fluidity, that lead to destabilisation and lysis of the CM, followed by P. aeruginosa cell death (Fig. 8D). This membranolytic action is consistent with the use of pore forming mechanisms, such as toroidal pores, and potentially involves other effects, including anionic lipid clustering and tilted peptide destabilisation of the CM. | ||
S. aureus
M5-NH2 killed S. aureus with an MLC of 147.6 μM, representing activity over twenty-five times lower than that against P. aeruginosa and clearly showing S. aureus to be resistant to the peptide (Fig. S1 and Table 2, ESI†), which is consistent with previous work.26,66 The capacity for potent membranolytic activity appears to generally underpin the antibacterial activity of M5-NH2, which suggested that changes to this capacity may be involved in the resistance of S. aureus to the peptide's action.26,31,33,66 Consistent with this suggestion, M5-NH2 displayed low levels of binding to SUVs mimetic of the S. aureus CM (Kd = 117.6 μM) that were over five times lower than in the case of P. aeruginosa, reflecting the peptide's weaker activity against the former organism (Table 2). The peptide also adopted low levels of amphiphilic α-helical structure in the presence of these SUVs (Fig. 4), which accounted for around one third of its structure (35.0%) and is equivalent to circa one and a half α-helical turns (Table 2).11,64 Compared to the case of P. aeruginosa, these levels of amphiphilic α-helical structure were two thirds lower (Table 2), which would clearly be predicted to reduce the peptide's ability to penetrate and lyse the CM of S. aureus.57,58 Confirming this prediction, the insertion of M5-NH2 into monolayer mimics of these membranes induced low maximal surface pressure changes (π = 3.3 mN m−1), which were around one fifth of those with P. aeruginosa and indicated low levels of insertion into these monolayers (Table 2). Although showing similar kinetics, the peptide's insertion into monolayer mimics of the S. aureus CM was much slower than that of P. aeruginosa, taking two-fold longer to reach maximal surface pressures (Fig. 4). This slower kinetics of insertion reflected the much lower affinity of the peptide for these monolayers, as compared to the corresponding case with the P. aeruginosa CM (Table 2). Consistent with these monolayer data, M5-NH2 also displayed a weak ability to induce the lysis of SUVs mimetic of the S. aureus CM (lysis = 24.5%;), which was over two thirds lower than that in the case of P. aeruginosa (Table 2). The levels of these model membrane associations suggested that the interactions of M5-NH2 with the S. aureus CM were more associated with their head-group and upper regions than the deeper regions indicated in the case of P. aeruginosa (Table 2).40,41 This suggestion was supported by thermodynamic data (Fig. 5 and Fig. 6) showing that, in contrast to P. aeruginosa, monolayer mimics of the S. aureus CM were thermodynamically unstable with values of ΔGmix > 0 (Table 2). However, these monolayers were rendered thermodynamically stable by the presence of M5-NH2 with values of ΔGmix < 0 (Table 2) and this change in ΔGmix suggested that insertion of the peptide into monolayer mimics of the S. aureus CM had decreased their lipid packing density and increased their membrane fluidity.40,41 These changes in membrane properties are consistent with the association of the peptide with the head-group and upper regions of S. aureus membranes,40,41 and similar data have previously been reported.33,67 In combination, these data would seem to indicate that characteristics of the S. aureus CM had lowered the capacity M5-NH2 to achieve the levels of conformational change and membrane interaction associated with the major steps in its action against P. aeruginosa. The overall effect of these weaker interactions appeared to be to lower the ability M5-NH2 to engage in membranolytic activity against S. aureus, which contributed to the organism's resistance to the action of the peptide.A primary characteristic of bacterial membranes that influences the activity of AMPs is differences in the lipid composition of these membranes and a major difference between the CM of S. aureus and P. aeruginosa is the occurrence of the cationic lipid Lys-PG (Table 1).44,55 The lipid is absent from the CM of P. aeruginosa55 but accounts for around four fifths of the lipid content in the case of the S. aureus CM (Table 1)44 and is known to be an important mediator in the resistance of the organism to the action of AMPs.38,68–73 Lys-PG was represented by Lys-DOPG and the ability of the lipid to influence the interaction of M5-NH2 with the S. aureus CM was investigated by varying levels of the lipid in lipid mimics of these membranes where the CL:PG ratio (5:57) was held constant (Fig. 9 and Fig. S2, ESI†). Regression analysis of the data shown in Fig. 9 revealed a strong inverse correlation between the levels of Lys-DOPG in these S. aureus CM mimics and those of the penetration and lysis levels induced by the peptide (R2 > 0.98; Fig. 9). These results were consistent with studies on the membranolytic action of other AMPs against S. aureus and clearly indicated that Lys-DOPG was able to attenuate the interaction of M5-NH2 with these S. aureus CM mimics.38 In the absence of Lys-DOPG, M5-NH2 showed high levels of insertion (π = 10.0 mN m−1) and lysis (lysis = 76.6%) (Fig. 9) with these membrane mimics, reflecting the peptide's strong preference for anionic lipid (Table 2).26,31,33 Increasing the Lys-DOPG content of these membranes led to decreases in the levels of penetration and lysis shown by M5-NH2, although they remained high at lower levels of the lipid (Fig. 9). At 10 mol% Lys-DOPG, the peptide inserted deeply into these lipid membrane mimics (π = 7.9 mN m−1) and induced high levels of lysis (lysis = 70.5%) (Fig. 9) which is consistent with recent studies on Bacillus subtilis.33 Lys-PG forms around 10 mol% of B. subtilis membranes74,75 and M5-NH2 induced similar levels of membrane penetration and lysis in its potent membranolytic action against the organism.33 At 38 mol% POPG, which corresponds to physiological levels of Lys-PG in S. aureus membranes, the levels of insertion and lysis shown by the peptide were over three-fold lower relative to membranes lacking Lys-DOPG (Fig. 9). Increasing Lys-DOPG above physiological levels has been shown to lead to the eventual inability of AMPs to penetrate and lyse membranes of S. aureus, which presumably would be the case with M5-NH2.76
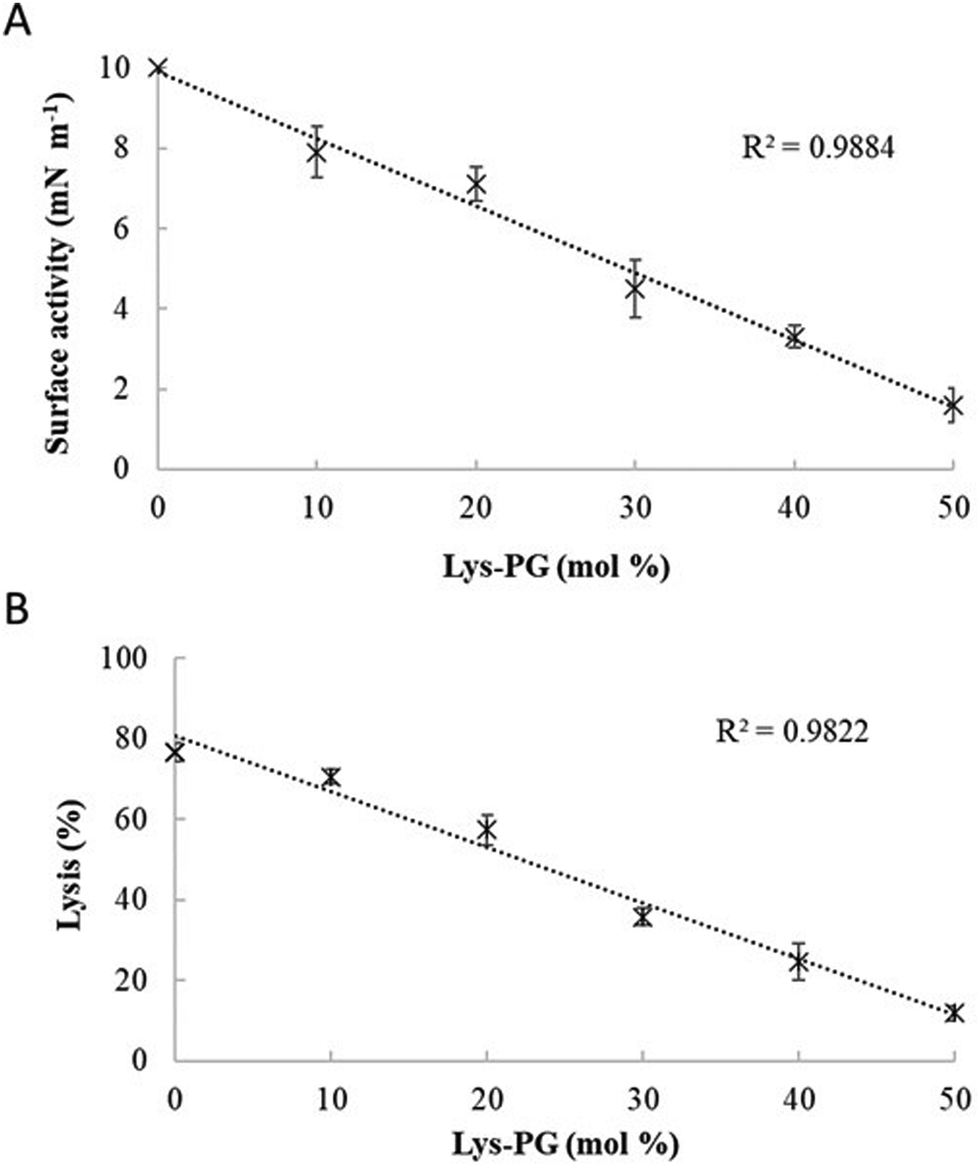 | ||
| Fig. 9 The effect of varying Lys-PG on the interaction of M5-NH2 with lipid models of S. aureus membranes. Fig. 9 shows the effect of varying Lys-PG content on the ability of M5-NH2 to penetrate lipid monolayers (Fig. 9A) and induce the lysis of SUVs (Fig. 9B) mimetic of the S. aureus CM where the TOCL:POPG content was held constant at a physiological ratio of 5:57. In both cases, regression analysis showed a strong inverse correlation between the levels of Lys-PG in these membranes and those of the penetration and lysis induced by the peptide (R2 > 0.98). As can also be seen from Fig. 9A and B, at physiological levels of the lipid (38 mol%), the levels of the membrane interactions induced by M5-NH2 (π = 3.3 mN m−1; lysis = 24.3%) were around one third of those in the case of membranes where Lys-PG was mot included (π = 10.0 mN m−1; lysis = 76.6%). Data points shown Fig. 9A and B were derived from experimental membrane insertion and lysis curves for M5-NH2 shown in Fig. S2 (ESI†). In both Fig. 9A and B, error bars are the standard deviation of the data about the mean value. | ||
In combination, these data indicate that in the early steps of its action against S. aureus, membrane binding and amphiphilic α-helix formation by M5-NH2 are attenuated by some factor in the membranes of the organism, relative to the case of P. aeruginosa. In the later steps of this action, the reduced levels and amphiphilicity of this α-helical structure promotes insertion of M5-NH2 into the upper reaches of P. aeruginosa membranes that induce low levels of lysis. These effects appear to be related to the presence of Lys-PG in the S. aureus CM and decrease the peptide's capacity for membranolytic action to the extent that they effectively promote the resistance of S. aureus to this action, which is represented schematically in Fig. 10.
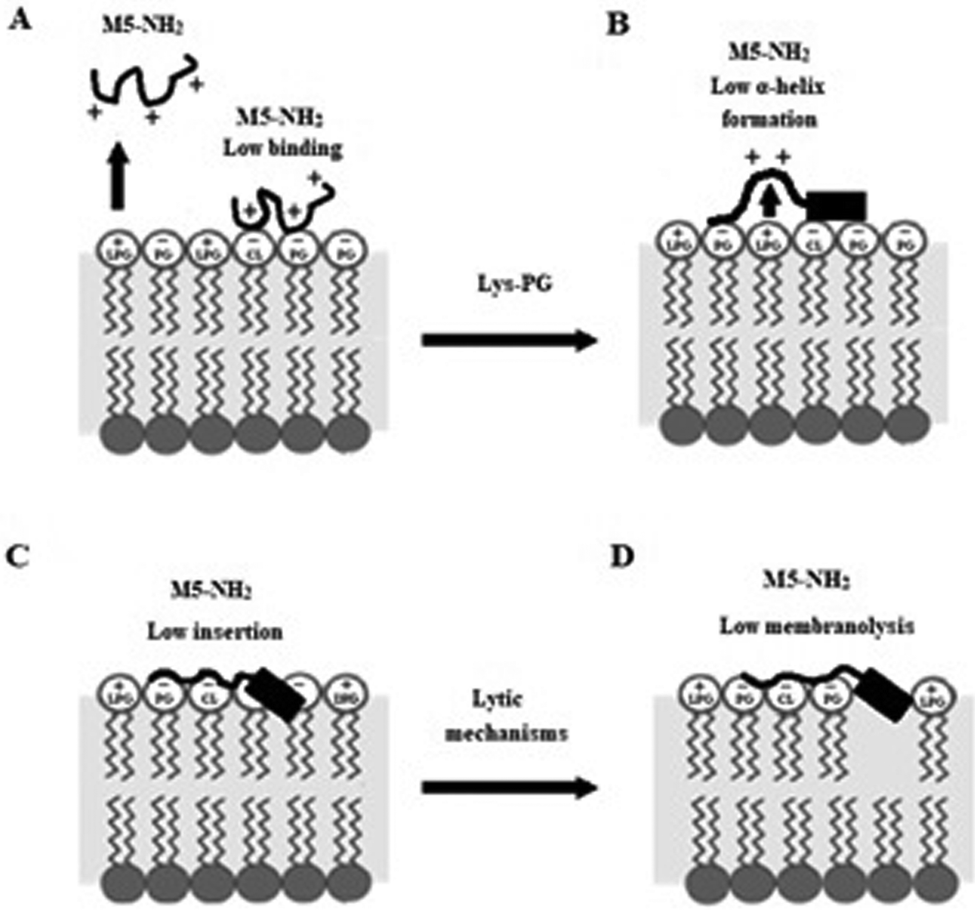 | ||
| Fig. 10 A schematic representation of S. aureus resistance of to the membranolytic action of M5-NH2 In the action of M5-NH2 against S. aureus, Lys-PG mediated effects attenuate the interactions of the peptide with the organism's CM relative to corresponding steps in the action of the peptide against P. aeruginosa (Fig. 8). In the first major step, electrostatic repulsion effects due to Lys-PG reduce the ability of the peptide to target the S. aureus CM, resulting in lower levels of binding to the headgroups of PG and CL (Fig. 10A). In the next major step, M5-NH2 adopts lower levels of α-helical structure due to Lys-PG mediated electrostatic repulsion effects that raise the energy barrier for this process (Fig. 10B). The resulting loss of tilted characteristics and/or amphiphilicity of this structure reduces insertion of the peptide into the S. aureus CM to depths that are associated with the upper regions of the membrane (Fig. 10C). In the final major step, the insertion of M5-NH2 promotes a range of effects, such as decreased lipid packing density and increased fluidity, that stabilise the CM and reduce the levels of lysis induced by the peptide, thereby rendering S. aureus resistant to M5-NH2 (Fig. 10D). The Lys-PG mediated inhibition of the peptide's membranolytic action appears to be the major mechanism used by S. aureus to resist M5-NH2, but, as described in the text, there is also the possibility that other mechanisms may make minor contributions to this resistance. | ||
Discussion
The increasing occurrence and widening antibiotic resistance of P. aeruginosa strains with MDR is critical77 and methicillin-resistant S. aureus (MRSA) is tolerant to almost all known conventional antibiotics.78 In both cases, this situation has been exacerbated by the emergence of resistance to the last-resort drugs used to eradicate infections due to these bacteria.79 AMPs show a strong potential for development as agents to treat infections due to P. aeruginosa, and S. aureus80–83 and in response, this potential was investigated for M5-NH2, which is a peptide designed to mimic naturally occurring α-helical AMPs.26,27M5-NH2 showed potent activity against P. aeruginosa that was in the very low micromolar range and appeared to involve the lysis of the organism's CM (Fig. S1 and Table 2, ESI†), clearly implying that the peptide was able to traverse the barrier posed by the outer membrane (OM) of P. aeruginosa.84,85 The OM of P. aeruginosa is essentially a bilayer formed by an external leaflet, primarily composed of anionic lipopolysaccharides (LPS), and an internal leaflet predominantly comprising phospholipids, that is spanned by specific uptake channels and non-specific porins.86 Potentially, there are three major routes by which antimicrobial compounds can traverse the OM of P. aeruginosa: diffusion directly through the OM or via porins, or translocation through electrostatic interaction with LPS and self-promoted uptake.85,87,88 Diffusion through porins is restricted to small hydrophilic molecules and, like most AMPs, the molecular weight of M5-NH2 (1.7 kDa)26 would be too high to permit use of this pathway.89 Both remaining pathways have been demonstrated for AMPs with comparable positive charge to M5-NH2, although self-promoted uptake appears to be the pathway most favoured by these strongly cationic peptides.90–93
To gain insight into the interaction of M5-NH2 with the CM of P. aeruginosa, individual, major steps in these interactions were investigated and a synthesis of these data allowed a putative scheme for the action of the peptide against P. aeruginosa to be constructed (Fig. 8). In first of these steps, M5-NH2 shows a strong affinity for the P. aeruginosa CM that is predominantly driven by high levels of binding to anionic lipids in these membranes (Table 2). This binding involves electrostatic interactions between the negative charge of phosphate moieties in CL and PG headgroups and the peptide's strong positive charge, which results from its multiple lysine residues and C-terminal amide moiety (Fig. 8A and Fig. 1).26 However, the binding of M5-NH2 to CL was the highest recorded for the peptide and was around two-fold stronger than in the case of PG, reflecting differences in their head-group charge (Table 2). CL has a charge of −2 due to its two phosphate moieties whereas that of PG has a charge of −1 due to its single phosphate group.94 Taken with the circa two-fold higher occurrence of CL over PG in the P. aeruginosa CM, this would appear to indicate that CL is the predominant mediator of M5-NH2 binding to these membranes (Table 1). The peptide showed an affinity for PE that was over four-fold lower than that for CL, which appeared to indicate that interaction with this lipid made a minor contribution to the peptide's binding of the P. aeruginosa CM (Table 2). The binding of AMPs to PE generally involves electrostatic interaction between their cationic residues and the phosphate moiety in the lipid's zwitterionic headgroup, which would appear to be the case M5-NH2.53,54 Interestingly, recent studies have suggested that the greater accessibility of phosphate moieties in PE head-groups compared to that in phosphatidylcholine (PC) head-groups may play a role in the selective binding of AMPs to bacterial membranes.53 PC is the major zwitterionic lipid in eukaryotic membranes55,95 and M5-NH2 shows negligible interaction with these membranes28,32 but strong interaction with bacterial membranes that is comparable to that found here for the P. aeruginosa CM (Table 2).31–33
The next two steps in the interaction of M5-NH2 with the P. aeruginosa CM were the closely coupled events of α-helix formation and insertion (Fig. 8B and C); the coupling of these events makes the insertion of AMPs into bacterial membranes energetically less costly.45 The process of α-helix formation by AMPs is primarily driven by the low dielectric properties and interfacial anisotropy of the bacterial CM and reduces the cost of peptide insertion through the formation of hydrogen bonds between backbone amide and carbonyl groups of these α-helices.96,97 M5-NH2 adopted high levels of α-helical structure at the interface of the P. aeruginosa CM (Table 2 and Fig. 8B) and the strong amphiphilicity of this structure (Fig. 1) appeared to be the major driver of the peptide's insertion into these membranes (Fig. 8C), which is consistent with other studies on M5-NH2.30,31,33,66
Insertion into the P. aeruginosa CM involves electrostatic interactions between the peptide's cationic polar face, comprising lysine residues and a C-terminal amide, and anionic lipid head-groups in these membranes (Fig. 1). It has previously been suggested that these positively charged residues may play a number of roles in promoting the insertion of M5-NH2 into bacterial membranes.33 These residues are evenly distributed along the α-helical long axis of the peptide (Fig. 1), which would tend to maximise their access to CL and PG headgroups in the P. aeruginosa CM (Fig. 8A). Indeed, this distribution may contribute to the very high affinity of M5-NH2 for CL (Table 2), given that the anionic moieties in the head-group of this lipid are far more accessible to AMPs than those of other membrane lipids.98
Concomitant with the electrostatic interactions of its polar face, the apolar face of M5-NH2, comprising alanine and leucine residues, engages in hydrophobic interactions with the apolar core of the P. aeruginosa CM (Fig. 1). Collectively, these interactions lead to deep penetration of this hydrophobic core region by M5-NH2, which induces increased lipid packing, decreased membrane fluidity and the destabilization of these membranes (Table 2 and Fig. 8C). Most recently, the α-helical structure formed by M5-NH2 was shown to possess an asymmetric distribution of hydrophobicity along the α-helical long axis.33 This structural feature, or hydrophobicity gradient, is characteristic of tilted AMPs and drives the deep, oblique insertion of these peptides into bacterial membranes.99 In the case of M5-NH2, the peptide's hydrophobicity gradient promoted oblique insertion into the CM of B. subtilis that led to depths of insertion and a mode of membrane destabilization that showed similarities to those found here for P. aeruginosa (Fig. 8).33 Given the high levels of α-helical structure adopted by the peptide, these observations suggest that M5-NH2 nay have the potential to promote its penetration and lysis of the P. aeruginosa CM using tilted mechanisms. Moreover, it is interesting to note that the polar face of M5-NH2 face is interrupted by hydrophobic residues, namely a leucine and two alanine residues, which is clearly a case of imperfect amphiphilicity (Fig. 1).100 This form of imperfect amphiphilicity appears able to promote high antimicrobial activity and selectivity, although the mechanisms underlying this ability are often unclear.100–103 However, in the case of M5-NH2, the leucine and alanine residues in the α-helical polar face occur at positions 7, 12 and 15 of the peptide's sequence (Fig. 1), indicating that they are distributed along the hydrophobicity gradient possessed M5-NH2.33 As shown for other residues in tilted peptides, a role of these leucine and alanine residues might be to provide the balance between amphiphilicity and hydrophobicity required to promote the oblique insertion of M5-NH2 into the CM of P. aeruginosa and other bacteria.104 It has also been suggested that the lysine residues possessed by M5-NH2 could contribute to its oblique insertion into the CM of bacteria through the snorkelling mechanism.33 Using this mechanism, the long hydrocarbon side chains of these residues would extend into the hydrophobic core region of the P. aeruginosa CM, permitting deeper levels of penetration by the peptide's tilted structure, as shown for other AMPs.66,105,106
In a final major step of the action M5-NH2 against P. aeruginosa, the insertion of the peptide into the organism's CM and the resulting destabilization leads to high levels of lysis and ultimately, cell death (Fig. 8D). It would seem that this action is unlikely to involve the formation of a membrane spanning pore by M5-NH2, such as the barrel stave model,10,11,14,15 due to the relatively short length of the peptide.33 However, studies on other bacteria, have suggested that the antibacterial action of M5-NH2 may involve mechanisms based on tilted peptide formation, the toroidal pore model and the carpet mechanism.31,33 To gain further insight into mechanisms underpinning the action of M5-NH2 against P. aeruginosa, the role of individual lipid components of the organism's membranes in the steps of α-helix formation, insertion and lysis were investigated.
M5-NH2 showed levels of α-helicity, insertion and lysis with PE membranes that were at least a third lower than those of anionic lipid, indicating a minor, but not inconsiderable, contribution to steps in the membranolytic action of M5-NH2 against P. aeruginosa (Table 2). PE is known to play a number of roles in the antibacterial action of AMPs107 and can specifically interact with α-helical AMPs to inhibit their antibacterial action.108 Some α-helical AMPs specifically target PE to promote this action;39,109 however, such specificity would seem unlikely to contribute to the action M5-NH2 against P. aeruginosa, given its strong preference for anionic lipid (Table 2). In many cases, the ability of PE to promote the antibacterial action of α-helical AMPs involves the inherent propensity of the lipid's cone shaped molecule to induce negative curvature and non-bilayer structures in membranes.110,111 This propensity has been shown to contribute to the antibacterial action of α-helical AMPs by promoting a number of membranolytic mechanisms, including the formation of toroidal-type pores and carpet-type modes of action.45,112–114 In general, each of these mechanisms would fit with the high levels of insertion and lysis found for the action of M5-NH2 against P. aeruginosa, suggesting that similar mechanisms may contribute to this action (Table 2 and Fig. 8). The ability of PE to induce non-bilayer structures also appears to facilitate bacterial membrane destabilization and lysis in response to the oblique insertion of α-helical AMPs that use tilted structure in their antibacterial action.33,39,67,115–117 This ability would appear to be consistent with the suggestion made above, which was that M5-NH2 may use tilted mechanisms to promote its membranolytic action against P. aeruginosa. Indeed, recent studies have suggested that, in these mechanisms, PE would be key to facilitating the peptide's oblique orientation and ability to perturb membrane properties such as lipid packing and membrane fluidity.33 The insertion of M5-NH2 into the CM of P. aeruginosa also appeared to lead to the perturbation of these membrane properties, thereby contributing to the peptide's membranolytic action against the organism (Table 2 and Fig. 8D).
As for binding, interactions of M5-NH2 with anionic lipid in the P. aeruginosa CM appeared to drive the remining major steps in the peptide's action against the organism with CL showing the highest levels of α-helicity, insertion and lysis recorded for the peptide (Table 2). However, whilst the levels of α-helicity adopted by the peptide with CL were only around a tenth higher than those with PG, the insertion and lysis shown by M5-NH2 with CL membranes was over a third higher than those with PG (Table 2). These observations indicated that the peptide adopted comparable levels of α-helicity with these two lipids but showed a much greater ability to penetrate and lyse CL membranes as compared to those formed by PG (Table 2). These data would seem to indicate that CL was the primary driver of α-helix formation, insertion and lysis by M5-NH2 in its action against P. aeruginosa and a number of mechanisms show the potential to support this role for the lipid.
It is well established that CL is able to influence the action of AMPs98 and in general, this ability derives from the topography of CL, which has a cone shaped molecule that promotes negative membrane curvature, along with the formation of domains and non-bilayer structures.110,111 This non-lamellar behaviour appears able to inhibit the ability of some α-helical AMPs to insert into bacterial membranes39,118–120 but in most cases, this behaviour appears to promote mechanisms of membranolytic action by these peptides.121–129 In general, these mechanisms appear to involve lipid clustering effects where CL is segregated from PE,125,130,131 which is induced by the strong positive charge of AMPs and appears to be facilitated by the formation of hydrogen bonds between the headgroups of these individual lipid types.111 In these CL mediated mechanisms, AMPs can either target naturally occurring CL domains or induce these domains in bacterial membranes, which then leads to the bacterial death through membranolytic action, although bacterial killing through non-membranolytic action has been reported.44,45,121–129,132 For example, a number of α-helical AMPs induce negatively curved CL domains in the CM of Escherichia coli,122 some of which involve clustering,126,127 to promote non-membranolytic perturbation of this membrane that leads to the lethal disruption of cellular processes.122,126,127 It is possible that such CL-mediated non-membranolytic mechanisms contribute to the action of M5-NH2 against P. aeruginosa, but, most likely, this contribution would be minor compared to membranolytic mechanisms. Potentially, the non-lamellar behaviour of CL could promote the membranolytic action of M5-NH2 against P. aeruginosa by supporting a number of established models used to describe this action for α-helical AMPs. As examples, α-helical AMPs appear to induce CL domains segregated from PE in the CM of P. aeruginosa that promote lysis via carpet-like mechanisms,128,129 and target negatively curved CL domains in the CM of E. coli to promote lysis through pore forming mechanisms.121 It is believed that lipid clustering by α-helical AMPs contributes to carpet-type and pore-forming mechanisms through the production of phase boundary defects that induce leakage in target bacterial membranes.124,125,132 Based on these observations, it would seem that lipid clustering mechanisms could contribute to the high levels of insertion and lysis found for the action of M5-NH2 against P. aeruginosa (Fig. 8C and D). Indeed, PE is strongly represented in the CM of the organism (Table 1) and M5-NH2 satisfies the two primary requirements for AMPs that induce lipid clustering effects, namely, a high positive charge and stable secondary structure (Table 2).123–125,133 Another characteristic of M5-NH2 that may show the potential to contribute to its CL-mediated membranolytic action against P. aeruginosa is its molecular shape; this property is well known to influence the interactions of AMPs with bacterial membranes.134 The molecular shape of M5-NH2 is formed by a large hydrophilic surface and a smaller hydrophobic surface, resembling an inverted cone (Fig. 1). Synthetic AMPs with a similar molecular shape to M5-NH2 appeared to kill P. aeruginosa by complementing the cone shape of CL, thereby promoting an increase in CM tension that resulted in pore formation and other effects that led to cell death.135–137 Interestingly, CL is also present in the OM of Gram-negative bacteria138 and in the case of E. coli, α-helical AMPs have been to promote lysis of this membrane via carpet-like mechanisms using CL mediated clustering effects.121 These mechanisms appeared to make only a minor contribution to the overall membranolytic action of these AMPs, but it is possible that similar mechanisms could contribute to the action of M5-NH2 against P. aeruginosa.121
M5-NH2 showed weak activity against S. aureus that was in the hundred micromolar range, which indicated resistance to the action of the peptide and was in strong contrast to its potent action against P. aeruginosa and other Gram-positive bacteria (Fig. S1; Table 2, ESI†).33 The S. aureus CM shows major differences in lipid composition to those of P. aeruginosa (Table 1) and such differences are well known to play a role in determining the specificity, susceptibility, and resistance of AMPs for bacteria.39,45,107,109 Here, these differences appeared to mediate the resistance of S. aureus to the action of M5-NH2 by decreasing the peptide's capacity for membranolytic action (Table 2). To gain insight into mechanisms underpinning this resistance, individual, major steps in the interaction of M5-NH2 with the CM of S. aureus were studied and a synthesis of these data allowed a putative scheme for the action of the peptide against the organism to be constructed (Fig. 9).
In the first major step of its action against S. aureus, M5-NH2 shows weak binding to the CM of the organism (Fig. 10A) that is orders of magnitude lower than that in the case of P. aeruginosa (Table 2). The CM of S. aureus contrasts to that of P. aeruginosa in that it possesses circa a two-fold higher content of anionic lipid which is predominantly formed from PG rather than CL (Table 1). This would seem to indicate that PG primarily drives the binding of M5-NH2 to the CM of S. aureus and that the peptide's lower affinity for this lipid relative to CL could contribute to the weaker binding of the peptide as compared to that in the case of P. aeruginosa (Table 2 and Fig. 8A). However, the major difference between the lipid compositions of the CM of S. aureus and P. aeruginosa is the presence of the positively charged lipid Lys-PG in the CM of the former organism (Table 1). It is well established that the high levels of the lipid in these membranes reduce the overall negative charge of CL and PG, which leads to the electrostatic repulsion of AMPs and effectively lowers their ability to bind these membranes.71,72,139,140 This ability is consistent with the differing levels of bacterial membrane affinity shown here by M5-NH2 and strongly suggests that similar Lys-PG-mediated electrostatic effects underpin the weak binding of the peptide to the CM of S. aureus (Table 2). Taken in combination, these data strongly suggest that the Lys-PG-mediated attenuation of membrane binding makes a major contribution to reducing the membranolytic efficacy of M5-NH2, thereby promoting the resistance of S. aureus to the action of the peptide (Fig. 10A) In support of this suggestion, Lys-PG has also been detected in the CM of other bacteria55,139–141 and appears to inhibit binding by AMPs in resistance mechanism with similarities to that mediated by the lipid in S. aureus.55,71,142–148 Interestingly, in what seems to be an evolutionary response, some creatures appear to produce anionic AMPs that bind Lys-PG to overcome S. aureus resistance mechanisms and promote their antibacterial action.149
The weak affinity of M5-NH2 for the CM of S. aureus appeared to limit rather than completely abolish the ability of the peptide to interact with these membranes. In the next two major steps of this interaction, M5-NH2 showed low levels of α-helicity and insertion with the CM of S. aureus (Fig. 9B) that were around one half of those shown by the peptide with that from P. aeruginosa (Table 2). This relative decrease in the efficacy of M5-NH2 to perform these steps appeared to be mediated by the presence of Lys-PG in the CM of S. aureus (Fig. 9B). In the case of α-helix formation by the peptide, it is known that the positive charge on Lys-PG is able to influence the energy barrier for the adoption of this structure by AMPs and this ability appears to primarily depend on the charge carried by these peptides.38,68,69 Studies on anionic α-helical AMPs with action against S. aureus, showed that Lys-PG has no effect on the levels of α-helical structure adopted by these peptides.38 In this case, it appeared that electrostatic interactions between negatively charged residues in these AMPs and the lipid neutralised its ability to influence their conformational behaviour.38 In contrast, studies on strongly cationic α-helical AMPs resisted by S. aureus reported that electrostatic repulsion between these peptides and Lys-PG in the organism's membranes inhibited the process of α-helix formation by these AMPs.68,69 These electrostatic repulsion effects appeared to raise the energy barrier for this process and counter the ability of CL and PG in the S. aureus CM to induce α-helical structure in these AMPs.68,69 Taken in combination, these observations strongly suggest that, in its action against S. aureus, Lys-PG mediated electrostatic effects inhibit the ability of M5-NH2 to adopt the higher levels of α-helicity seen with P. aeruginosa, thereby promoting the resistance of S. aureus to the peptide (Table 2 and Fig. 10B).
Lys-PG-mediated reductions in the α-helicity of M5-NH2 also appeared to promote the resistance of S. aureus to the peptide by decreasing its efficacy in the step of insertion into the organism's CM (Fig. 10C). In this step, the lower α-helicity of M5-NH2 led to levels of insertion into the S. aureus CM that were more associated with their head-group and upper regions than the deep penetration of their hydrophobic core seen with P. aeruginosa (Fig. 10C). Clearly, a major potential contributor to these decreased levels of insertion would seem to be the loss of amphiphilic structure associated with reduced α-helix formation by M5-NH2, given that, as suggested above, amphiphilicity appears to drive the peptide's membrane interactions. Another potential contributor to these decreased levels of insertion could be disruption of the peptide's hydrophobicity gradient resulting from its lower adoption of α-helical structure, as compared to the case of P. aeruginosa. Indeed, the tilted insertion of M5-NH2 into the S. aureus CM could be further compromised by their lack of PE (Table 1) and the ability of the lipid's non-lamellar behaviour to facilitate this form of bilayer penetration.33,39,67,115–117 Reduced insertion into the S. aureus CM due to Lys-PG-mediated decreased α-helicity in M5-NH2 (Table 2 and Fig. 9B) could also contribute to S. aureus resistance by inhibiting the use of novel anionic clustering mechanisms reported for the action of AMPs against Gram-positive bacteria. In these clustering mechanisms, strongly cationic AMPs promote their antibacterial action through membrane perturbation involving the induced segregation of CL from PG.123,125 However, the low levels of α-helicity shown by M5-NH2 with the S. aureus CM (Table 2 and Fig. 9B) would appear to lie outside the conformational requirements of lipid clustering mechanisms, rendering their use by the peptide in this case unlikely.123–125,133 In addition to mechanisms involving Lys-PG-mediated changes to the conformational behaviour of M5-NH2, the insertion of the peptide into the S. aureus CM could also be limited by mechanisms based on the intrinsic properties of the lipid itself. It has previously been shown that the ability of Lys-PG to reduce electrostatic repulsion between CL and PG lipid head-groups leads to decreases in the fluidity of the S. aureus CM70,150 and makes partitioning by AMPs more difficult.72,73,141,151,152
In the major steps discussed above, it was suggested that Lys-PG promoted the resistance of S. aureus to M5-NH2 by reducing the peptide's capacity for membranolytic action and that a number of mechanisms mediated by the lipid could potentially promote these effects (Fig. 10D). Strongly supporting this suggestion, in the final major step of this action, the membranolytic capacity of M5-NH2 against S. aureus was around one third of that shown against P. aeruginosa (Table 2) and this decreased membranolytic efficacy appeared to be mediated by Lys-PG (Fig. 9). Taken in combination, the Lys-PG-mediated effects found for steps in the action of M5-NH2 against S. aureus (Table 2 and Fig. 10) are consistent with those of a major intrinsic mechanism used by the organism to resist AMPs.72,151,153 This mechanism is modulated by the MprF operon and involves the catalytic lysylination of PG to produce Lys-PG, which is subsequently translocated to the outer leaflet of S. aureus membranes.140,154
Although Lys-PG-mediated reductions in membranolytic efficacy clearly appeared to be a major driver in the resistance of S. aureus to the action of M5-NH2, the peptide showed a weak ability to induce the lysis of the organism's membranes. This effect could reflect the fact that the in vivo situation of S. aureus membranes is not fully represented by the in vitro membrane systems used in the present study. One explanation for these observations is that other mechanisms play a minor role in the resistance of S. aureus to the action of M5-NH2 and it is well established that the organism uses a variety of strategies to resist AMPs.44,151,155 A frequently reported strategy would involve interactions between M5-NH2 and components of the S. aureus cell wall when the peptide was migrating to the organism's CM. The S. aureus cell wall can be considered as essentially a thick peptidoglycan (PGN) mesh that is decorated with anionic teichoic acids.156 In general, PGN appears to not significantly hinder the passage of AMPs as it is not anionic and is porous to molecules with molecular weights that would include M5-NH2 and most AMPs.44,157–159 However, the D-alanylation of teichoic acids to decrease their negative charge and promote electrostatic repulsion effects with AMPs is known to inhibit the access of these peptides to S. aureus membranes.44,158–160 Very recent studies have suggested that this access is also restricted by modifications to the polymer composition and/or synthesis of teichoic acids anchored to these membranes. These modifications appear to be modulated by MprF and function with Lys-PG electrostatic repulsion effects and teichoic acid D-alanylation to decrease the overall permeability of the S. aureus cell wall to AMPs.161
Conclusions
M5-NH2, was designed to exhibit broad antimicrobial activity26,27 and shows potent action against a spectrum of fungi and bacteria,26,27,30–33 although the mechanisms underpinning this action has been investigated in only a few cases, which has been extended in the present study.31–33 The peptide showed potent membranolytic activity against P. aeruginosa (Fig. S1 and Table 2, ESI†) and similar results have been reported for the activity of M5-NH2 against E. coli,31,32 which is increasingly regarded as an ESKAPE pathogen.35 These observations suggested that the peptide may have broad range action against Gram-negative bacteria and the potential to kill not only P. aeruginosa and E. coli but also other ESKAPE pathogens such as K. pneumoniae.162 However, although M5-NH2 showed potent membranolytic action against B subtilis,33S. aureus was resistant to the peptide (Fig. S1 and Table 2, ESI†) using mechanisms available to other Gram-positive bacteria, clearly suggesting that the peptide's activity against these bacteria may show a more limited spectrum.55,71,142–148Characterisation of the antibacterial activity of M5-NH2 showed it to possess potent CL driven membranolytic action against P. aeruginosa but was rendered ineffective against S. aureus by Lys-PG driven inhibition of this action (Table 2). These observations clearly showed the effect of differences in the lipid compositions of bacterial membranes on the activity of AMPs and reinforce the importance of taking these differences into account when designing these peptides.163 Indeed, it has been recently suggested that when designing AMPs, other often neglected factors, such as site of action pH should also be considered;164,165 changes in pH have been shown to both enhance and attenuate the response of S. aureus and P. aeruginosa to the action of AMPs.38,39,76
The Lys-PG mediated mechanisms shown here to protect S. aureus to the action of M5-NH2 most probably involve the use of an intrinsic resistance mechanism frequently reported in studies on the susceptibility of the organism to AMPs.72,151,153 The use of this intrinsic mechanism to resist M5-NH2, as opposed to those that are acquired,166 would help to explain the fact that many other strains of S. aureus show similar levels of resistance to the action of the peptide.27 Indeed, S. aureus, particularly MRSA, shows resistance to many AMPs, which has hindered progress in the development of these peptides as therapeutic agents to combat infections due to the organism.44,83,151,155 In part, this lack of progress has been due to a poor understanding of the mechanisms used by S. aureus to resist the action of AMPs and why these mechanisms work against some of these peptides and not others. It is well known that some AMPs with a similar charge to M5-NH2 are able to kill S. aureus, whilst others are resisted by the organism using Lys-PG mediated mechanisms.167 It is to be hoped that the data presented here will help to explain these differences and increase the general understanding of mechanisms underpinning the ability of S. aureus to resist AMPs.
Potentially, the CL driven membranolytic activity of M5-NH2 against P. aeruginosa involved the peptide's ability to adopt tilted α-helical structure and induce anionic lipid clustering. These mechanisms were consistent with the overall effects of the peptide's membranolytic action against P. aeruginosa and a number of models appeared able to describe this action, including toroidal-type pore formation and carpet-type mechanisms. In carpet-type mechanisms, AMPs promote their antibacterial activity using a detergent-like membranolytic action that does not generally entail the penetration of these peptide into the CM hydrophobic core region.10,11 In contrast, the use of toroidal pore formation to promote antibacterial membranolytic action by AMPs generally involves deep penetration of the bacterial CM.14,15 Given the ability of M5-NH2 to penetrate and perturb the acyl chain region of the P. aeruginosa CM, this might suggest that the peptide is more likely to use toroidal-type mechanisms than carpet-type mechanisms. Based on its action against P. aeruginosa and lack of haemolytic activity,28 M5-NH2 shows the potential for medical development in a number of areas,168,169 including biofilm inhibition170 combination therapy171 and cystic fibrosis treatment.172
Author contributions
S. R. D., F. H. and D. A. P. designed the research. K. B. synthesised the peptide. S. R. D. and L. H. G. M. performed research. S. R. D., L. H. G. M. and F. H. analysed data. S. R. D., L. H. G. M, K. B. F. H. and D. A. P. wrote the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The peptide M5-NH2 (KLAKKLAKLAKLAKALCONH2) was partially funded by the Biochemical Society Eric Reid Fund for Methodology.References
- J. O'Neill, 2016. Tackling drug-resistant infections globally: final report and recommendations. https://amr-review.org/sites/default/files/160518_Final%20paper_with%20cover.pdf.
- M. E. de Kraker, A. J. Stewardson and S. Harbarth, PLoS Med., 2016, 13, e1002184 CrossRef PubMed.
- M. Terreni, M. Taccani and M. Pregnolato, Molecules, 2021, 26 Search PubMed.
- T. M. Uddin, A. J. Chakraborty, A. Khusro, B. M. R. M. Zidan, S. Mitra, T. B. Emran, K. Dhama, M. K. H. Ripon, M. Gajdács, M. U. K. Sahibzada, M. J. Hossain and N. Koirala, J. Infection Public Health, 2021, 14, 1750–1766 CrossRef PubMed.
- D. M. P. De Oliveira, B. M. Forde, T. J. Kidd, P. N. A. Harris, M. A. Schembri, S. A. Beatson, D. L. Paterson and M. J. Walker, Clin. Microbiol. Rev., 2020, 33, e00181-19 CrossRef.
- G. Mancuso, A. Midiri, E. Gerace and C. Biondo, Pathogens, 2021, 10, 1310 CrossRef CAS PubMed.
- X. Zhen, C. S. Lundborg, X. Sun, X. Hu and H. Dong, Antimicrobial Resistance Infection Control, 2019, 8, 137 CrossRef.
- M. Rima, M. Rima, Z. Fajloun, J. M. Sabatier, B. Bechinger and T. Naas, Antibiotics, 2021, 10, 1095 CrossRef CAS PubMed.
- Y.-X. Ma, C.-Y. Wang, Y.-Y. Li, J. Li, Q.-Q. Wan, J.-H. Chen, F. R. Tay and L.-N. Niu, Adv. Sci., 2020, 7, 1901872 CrossRef CAS PubMed.
- Q.-Y. Zhang, Z.-B. Yan, Y.-M. Meng, X.-Y. Hong, G. Shao, J.-J. Ma, X.-R. Cheng, J. Liu, J. Kang and C.-Y. Fu, Military Med. Res., 2021, 8, 48 CrossRef CAS PubMed.
- S. Li, Y. Wang, Z. Xue, Y. Jia, R. Li, C. He and H. Chen, Trends Food Sci. Technol., 2021, 109, 103–115 CrossRef CAS.
- M. Abdi, S. Mirkalantari and N. Amirmozafari, J. Pept. Sci., 2019, 25, e3210 CrossRef CAS PubMed.
- R. Nuri, T. Shprung and Y. Shai, Biochim. Biophys. Acta, Biomembr., 2015, 1848, 3089–3100 CrossRef CAS.
- G. Wang, A. Verma and S. Reiling, in Antimicrobial Peptides, ed. K. Ajesh and K. Sreejith, Academic Press, 2023, pp. 237–259 DOI:10.1016/B978-0-323-85682-9.00012-X.
- T. Rončević, J. Puizina and A. Tossi, Int. J. Mol. Sci., 2019, 20, 5713 CrossRef.
- A. Pfalzgraff, K. Brandenburg and G. Weindl, Front. Pharmacol., 2018, 9, 281 CrossRef.
- M. S. Mulani, E. E. Kamble, S. N. Kumkar, M. S. Tawre and K. R. Pardesi, Front. Microbiol., 2019, 10, 539 CrossRef PubMed.
- S. Mukhopadhyay, A. S. Bharath Prasad, C. H. Mehta and U. Y. Nayak, World J. Microbiol. Biotechnol., 2020, 36, 131 CrossRef CAS PubMed.
- A. Parchebafi, F. Tamanaee, H. Ehteram, E. Ahmad, H. Nikzad and H. Haddad Kashani, Microb. Cell Fact., 2022, 21, 118 CrossRef CAS PubMed.
- G. S. Dijksteel, M. M. W. Ulrich, E. Middelkoop and B. Boekema, Front. Microbiol., 2021, 12, 616979 CrossRef PubMed.
- M. Lesiuk, M. Paduszyńska and K. E. Greber, Antibiotics, 2022, 11, 1062 CrossRef CAS PubMed.
- G. Wang and A. F. Mechesso, Admet dmpk, 2022, 10, 91–105 Search PubMed.
- B. H. Gan, J. Gaynord, S. M. Rowe, T. Deingruber and D. R. Spring, Chem. Soc. Rev., 2021, 50, 7820–7880 RSC.
- S.-J. Kang, S. H. Nam and B.-J. Lee, Antibiotics, 2022, 11, 1338 CrossRef CAS PubMed.
- P. G. Lima, J. T. A. Oliveira, J. L. Amaral, C. D. T. Freitas and P. F. N. Souza, Life Sci., 2021, 278, 119647 CrossRef CAS PubMed.
- R. Bessalle, A. Gorea, I. Shalit, J. W. Metzger, C. Dass, D. M. Desiderio and M. Fridkin, J. Med. Chem., 1993, 36, 1203–1209 CrossRef CAS PubMed.
- D. R. Owen, 2005. Short bioactive peptides. Helix Biomedix Inc: USA. The full form of this is Owen DR (2005) Short bioactive peptides in, Helix BioMedix. Inc., USA. United States Patent 6875744.https://patents.google.com/patent/US6875744B2/en.
- S. R. Dennison and D. A. Phoenix, Eur. Biophys. J., 2014, 43, 423–432 CrossRef CAS PubMed.
- S. R. Dennison, F. Harris, T. Bhatt, J. Singh and D. A. Phoenix, Mol. Cell. Biochem., 2009, 333, 129 CrossRef PubMed.
- S. R. Dennison, F. Harris, T. Bhatt, J. Singh and D. A. Phoenix, Mol. Cell. Biochem., 2009, 332, 43 CrossRef CAS PubMed.
- S. Dennison and D. Phoenix, Biochemistry, 2011, 50, 1514–1523 CrossRef CAS PubMed.
- S. R. Dennison and D. A. Phoenix, Biochemistry, 2011, 50, 10898–10909 CrossRef CAS PubMed.
- S. R. Dennison, T. Hauß, K. Badiani, F. Harris and D. A. Phoenix, Soft Matter, 2019, 15, 4215–4226 RSC.
- H. Venter, Biosci. Rep., 2019, 39, BSR20180474 CrossRef CAS PubMed.
- J. Denissen, B. Reyneke, M. Waso-Reyneke, B. Havenga, T. Barnard, S. Khan and W. Khan, Int. J. Hyg. Environ. Health, 2022, 244, 114006 CrossRef CAS PubMed.
- F. Savini, S. Bobone, D. Roversi, M. L. Mangoni and L. Stella, Pept. Sci., 2018, 110, e24041 CrossRef.
- M. Rojewska, W. Smułek, E. Kaczorek and K. Prochaska, Membranes, 2021, 11, 707 CrossRef CAS PubMed.
- S. R. Dennison, L. H. Morton, F. Harris and D. A. Phoenix, Biochemistry, 2016, 55, 3735–3751 CrossRef CAS PubMed.
- E. Malik, D. A. Phoenix, K. Badiani, T. J. Snape, F. Harris, J. Singh, L. H. G. Morton and S. R. Dennison, Biochim. Biophys. Acta, Biomembr., 2020, 1862, 183141 CrossRef CAS PubMed.
- S. R. Dennison, F. Harris and D. A. Phoenix, Protein Pept. Lett., 2010, 17, 1363–1375 CrossRef CAS PubMed.
- S. R. Dennison, F. Harris and D. A. Phoenix, in Advances in Planar Lipid Bilayers and Liposomes, ed. A. Iglič and C. V. Kulkarni, Academic Press, 2014, vol. 20, pp. 83–110 Search PubMed.
- C. Wölk, H. Youssef, T. Guttenberg, H. Marbach, G. Vizcay-Barrena, C. Shen, G. Brezesinski and R. D. Harvey, ChemPhysChem, 2020, 21, 702–706 CrossRef PubMed.
- S. Danner, G. Pabst, K. Lohner and A. Hickel, Biophys. J., 2008, 94, 2150–2159 CrossRef CAS PubMed.
- N. Malanovic and K. Lohner, Biochim. Biophys. Acta, 2016, 1858, 936–946 CrossRef CAS PubMed.
- V. Teixeira, M. J. Feio and M. Bastos, Prog. Lipid Res., 2012, 51, 149–177 CrossRef CAS PubMed.
- P. J. O'Toole, I. E. Morrison and R. J. Cherry, Biochim. Biophys. Acta, 2000, 1466, 39–46 CrossRef PubMed.
- J. Wall, C. A. Golding, M. Van Veen and P. O'Shea, Mol. Membr. Biol., 1995, 12, 183–192 CrossRef CAS PubMed.
- A. J. Miles, S. G. Ramalli and B. A. Wallace, Protein Sci., 2022, 31, 37–46 CrossRef CAS PubMed.
- J. Todd, Introduction to the Constructive Theory of Functions, Academic Press, 1963 Search PubMed.
- R. Gautier, D. Douguet, B. Antonny and G. Drin, Bioinformatics, 2008, 24, 2101–2102 CrossRef CAS PubMed.
- M. J. McKay, F. Afrose, R. E. Koeppe, 2nd and D. V. Greathouse, Biochim. Biophys. Acta, Biomembr., 2018, 1860, 2108–2117 CrossRef CAS PubMed.
- Y. He and T. Lazaridis, PLoS One, 2013, 8, e66440 CrossRef CAS PubMed.
- C. I. von Deuster and V. Knecht, Biochim. Biophys. Acta, 2012, 1818, 2192–2201 CrossRef CAS PubMed.
- A. Catte, M. R. Wilson, M. Walker and V. S. Oganesyan, Soft Matter, 2018, 14, 2796–2807 RSC.
- C. Sohlenkamp and O. Geiger, FEMS Microbiol. Rev., 2015, 40, 133–159 CrossRef PubMed.
- O. G. Travkova, H. Moehwald and G. Brezesinski, Adv. Colloid Interface Sci., 2017, 247, 521–532 CrossRef CAS PubMed.
- D. A. Phoenix and F. Harris, Mol. Membr. Biol., 2002, 19, 1–10 CrossRef CAS PubMed.
- D. A. Phoenix, F. Harris, O. A. Daman and J. Wallace, Curr. Protein Pept. Sci., 2002, 3, 201–221 CrossRef CAS PubMed.
- Q. Wang, B. Peng, M. Song, Abdullah, J. Li, J. Miao, K. Feng, F. Chen, X. Zhai and Y. Cao, Front. Nutrition, 2021, 8 DOI:10.3389/fnut.2021.768890.
- A. Morales-Martínez, B. Bertrand, J. M. Hernández-Meza, R. Garduño-Juárez, J. Silva-Sanchez and C. Munoz-Garay, Biophys. J., 2022, 121, 3034–3048 CrossRef PubMed.
- B. Zorilă, G. Necula, M. Radu and M. Bacalum, Toxins, 2020, 12, 705 CrossRef PubMed.
- S. Omardien, J. W. Drijfhout, F. M. Vaz, M. Wenzel, L. W. Hamoen, S. A. Zaat and S. Brul, Biochim. Biophys. Acta, Biomembr., 2018, 1860, 2404–2415 CrossRef CAS PubMed.
- S. Qian and P. A. Zolnierczuk, BBA Adv, 2022, 2, 100045 CrossRef CAS PubMed.
- Y. Shai, Pept. Sci., 2002, 66, 236–248 CrossRef CAS PubMed.
- W. Im and C. L. Brooks, 3rd, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 6771–6776 CrossRef CAS PubMed.
- S. R. Dennison, L. H. G. Morton and D. A. Phoenix, Biochim. Biophys. Acta, Biomembr., 2012, 1818, 2094–2102 CrossRef CAS PubMed.
- S. R. Dennison, L. H. Morton, F. Harris and D. A. Phoenix, Chem. Phys. Lipids, 2008, 151, 92–102 CrossRef CAS PubMed.
- P. W. Simcock, M. Bublitz, F. Cipcigan, M. G. Ryadnov, J. Crain, P. J. Stansfeld and M. S. P. Sansom, bioRxiv, 2020, DOI:10.1101/2020.04.24.057349.
- R. Rehal, R. D. Barker, Z. Lu, T. T. Bui, B. Demé, G. Hause, C. Wölk and R. D. Harvey, Biochim. Biophys. Acta, Biomembr., 2021, 1863, 183571 CrossRef CAS PubMed.
- E. Kilelee, A. Pokorny, M. R. Yeaman and A. S. Bayer, Antimicrob. Agents Chemother., 2010, 54, 4476–4479 CrossRef CAS PubMed.
- W. R. Miller, A. S. Bayer and C. A. Arias, Cold Spring Harbor Perspect. Med., 2016, 6, a026997 CrossRef PubMed.
- C. M. Ernst and A. Peschel, Int. J. Med. Microbiol., 2019, 309, 359–363 CrossRef CAS PubMed.
- T. Shireen, M. Singh, T. Das and K. Mukhopadhyay, Antimicrob. Agents Chemother., 2013, 57, 5134–5137 CrossRef CAS PubMed.
- J. A. den Kamp, I. Redai and L. L. van Deenen, J. Bacteriol., 1969, 99, 298–303 CrossRef CAS PubMed.
- J. R. Willdigg and J. D. Helmann, Front. Mol. Biosci., 2021, 8 DOI:10.3389/fmolb.2021.634438.
- R. Rehal, P. R. J. Gaffney, A. T. M. Hubbard, R. D. Barker and R. D. Harvey, Eur. J. Pharm. Sci., 2019, 128, 43–53 CrossRef CAS PubMed.
- A. J. Kunz Coyne, A. El Ghali, D. Holger, N. Rebold and M. J. Rybak, Infectious Diseases Therapy, 2022, 11, 661–682 CrossRef PubMed.
- M. Vestergaard, D. Frees and H. Ingmer, Microbiol. Spectrosc., 2019, 7 DOI:10.1128/microbiolspec.GPP3-0057-2018.
- E. V. K. Ledger, A. Sabnis and A. M. Edwards, Microbiology, 2022, 168 DOI:10.1099/mic.0.001136.
- S. Ruden, A. Rieder, T. Schwartz, R. Mikut and K. Hilpert, bioRxiv, 2019, DOI:10.1101/639286, 639286.
- A. J. Mulkern, L. B. Oyama, A. R. Cookson, C. J. Creevey, T. J. Wilkinson, H. Olleik, M. Maresca, G. C. da Silva, P. P. Fontes, D. M. S. Bazzolli, H. C. Mantovani, B. F. Damaris, L. A. J. Mur and S. A. Huws, npj Biofilms Microbiomes, 2022, 8, 70 CrossRef CAS PubMed.
- A. Mazumdar and V. Adam, JMCM, 2021, 4, 1–17 CrossRef.
- A. Zouhir, T. Jridi, A. Nefzi, J. Ben Hamida and K. Sebei, Pharm. Biol., 2016, 54, 3136–3150 CrossRef CAS PubMed.
- E. B. Breidenstein, C. de la Fuente-Núñez and R. E. Hancock, Trends Microbiol., 2011, 19, 419–426 CrossRef CAS PubMed.
- J. Ude, V. Tripathi, J. M. Buyck, S. Söderholm, O. Cunrath, J. Fanous, B. Claudi, A. Egli, C. Schleberger, S. Hiller and D. Bumann, Proc. Natl. Acad. Sci. U. S. A., 2021, 118 DOI:10.1073/pnas.2107644118.
- J. D. Prajapati, U. Kleinekathöfer and M. Winterhalter, Chem. Rev., 2021, 121, 5158–5192 CrossRef CAS PubMed.
- A. H. Delcour, Biochim. Biophys. Acta, 2009, 1794, 808–816 CrossRef CAS PubMed.
- P. Sharma and K. G. Ayappa, J. Membr. Biol., 2022, 255, 665–675 CrossRef CAS PubMed.
- A. Barreto-Santamaría, G. Arévalo-Pinzón, M. A. Patarroyo and M. E. Patarroyo, Antibiotics, 2021, 10, 1499 CrossRef PubMed.
- N. Klubthawee, P. Adisakwattana, W. Hanpithakpong, S. Somsri and R. Aunpad, Sci. Rep., 2020, 10, 9117 CrossRef CAS PubMed.
- M. Yasir, D. Dutta and M. D. P. Willcox, Sci. Rep., 2019, 9, 7063 CrossRef PubMed.
- H. Chai, W. E. Allen and R. P. Hicks, Int. J. Med. Chem., 2014, 2014, 809283 Search PubMed.
- L. Zhang, P. Dhillon, H. Yan, S. Farmer and R. E. Hancock, Antimicrob. Agents Chemother., 2000, 44, 3317–3321 CrossRef CAS PubMed.
- M. Sathappa and N. N. Alder, Biochim. Biophys. Acta, Biomembr., 2016, 1858, 1362–1372 CrossRef CAS PubMed.
- H. I. MacDermott-Opeskin, V. Gupta and M. L. O’Mara, Biophys. Rev., 2022, 14, 145–162 CrossRef CAS PubMed.
- P. F. Almeida, A. S. Ladokhin and S. H. White, Biochim. Biophys. Acta, Biomembr., 2012, 1818, 178–182 CrossRef CAS PubMed.
- M. E. Fealey, B. P. Binder, V. N. Uversky, A. Hinderliter and D. D. Thomas, Biophys. J., 2018, 114, 550–561 CrossRef CAS PubMed.
- R. N. Lewis and R. N. McElhaney, Biochim. Biophys. Acta, Biomembr., 2009, 1788, 2069–2079 CrossRef CAS PubMed.
- F. Harris, A. Daman, J. Wallace, S. R. Dennison and D. A. Phoenix, Curr. Protein Pept. Sci., 2006, 7, 529–537 CrossRef CAS PubMed.
- D. Juretić and J. Simunić, Expert Opin. Drug Discovery, 2019, 14, 1053–1063 CrossRef PubMed.
- X. Zhu, N. Dong, Z. Wang, Z. Ma, L. Zhang, Q. Ma and A. Shan, Acta Biomater., 2014, 10, 244–257 CrossRef CAS PubMed.
- J. Wang, S. Chou, Z. Yang, Y. Yang, Z. Wang, J. Song, X. Dou and A. Shan, J. Med. Chem., 2018, 61, 3889–3907 CrossRef CAS PubMed.
- X. Chen, S. Ji, A. Li, H. Liu and H. Fei, J. Med. Chem., 2020, 63, 1132–1141 CrossRef CAS PubMed.
- F. Harris, J. Wallace and D. A. Phoenix, Mol. Membr. Biol., 2000, 17, 201–207 CrossRef CAS PubMed.
- M. Mura, S. R. Dennison, A. V. Zvelindovsky and D. A. Phoenix, Biochim. Biophys. Acta, Biomembr., 2013, 1828, 586–594 CrossRef CAS PubMed.
- S. R. Dennison, F. Harris and D. A. Phoenix, Protein Pept. Lett., 2005, 12, 27–29 CrossRef CAS PubMed.
- D. A. Phoenix, F. Harris, M. Mura and S. R. Dennison, Prog. Lipid Res., 2015, 59, 26–37 CrossRef CAS PubMed.
- S. R. Dennison, F. Harris, M. Mura, L. H. G. Morton, A. Zvelindovsky and D. A. Phoenix, Biochemistry, 2013, 52, 6021–6029 CrossRef CAS PubMed.
- H. Schröder-Borm, R. Willumeit, K. Brandenburg and J. Andrä, Biochim. Biophys. Acta, Biomembr., 2003, 1612, 164–171 CrossRef PubMed.
- J. Jouhet, Front. Plant Sci., 2013, 4, 494 Search PubMed.
- K. Matsumoto, J. Kusaka, A. Nishibori and H. Hara, Mol. Microbiol., 2006, 61, 1110–1117 CrossRef CAS PubMed.
- R. Willumeit, M. Kumpugdee, S. S. Funari, K. Lohner, B. P. Navas, K. Brandenburg, S. Linser and J. Andrä, Biochim. Biophys. Acta, Biomembr., 2005, 1669, 125–134 CrossRef CAS PubMed.
- D. J. Paterson, M. Tassieri, J. Reboud, R. Wilson and J. M. Cooper, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E8324–E8332 CAS.
- E. F. Haney, S. Nathoo, H. J. Vogel and E. J. Prenner, Chem. Phys. Lipids, 2010, 163, 82–93 CrossRef CAS PubMed.
- L. Lins, M. Decaffmeyer, A. Thomas and R. Brasseur, Biochim. Biophys. Acta, Biomembr., 2008, 1778, 1537–1544 CrossRef CAS PubMed.
- S. R. Dennison, L. H. G. Morton, K. Brandenburg, F. Harris and D. A. Phoenix, FEBS J., 2006, 273, 3792–3803 CrossRef CAS PubMed.
- L. Lins and R. Brasseur, J. Pept. Sci., 2008, 14, 416–422 CrossRef CAS PubMed.
- D. Poger, S. Pöyry and A. E. Mark, ACS Omega, 2018, 3, 16453–16464 CrossRef CAS PubMed.
- L. Hernández-Villa, M. Manrique-Moreno, C. Leidy, M. Jemioła-Rzemińska, C. Ortíz and K. Strzałka, Biophys. Chem., 2018, 238, 8–15 CrossRef PubMed.
- N. Calderón-Rivera, J. Múnera-Jaramillo, S. Jaramillo-Berrio, E. Suesca, M. Manrique-Moreno and C. Leidy, Membranes, 2023, 13, 304 CrossRef PubMed.
- N. Rangarajan, S. Bakshi and J. C. Weisshaar, Biochemistry, 2013, 52, 6584–6594 CrossRef CAS PubMed.
- M. Makowski, M. R. Felício, I. C. M. Fensterseifer, O. L. Franco, N. C. Santos and S. Gonçalves, Int. J. Mol. Sci., 2020, 21, 9104 CrossRef CAS PubMed.
- R. F. Epand, L. Maloy, A. Ramamoorthy and R. M. Epand, Biophys. J., 2010, 98, 2564–2573 CrossRef CAS PubMed.
- R. F. Epand, W. L. Maloy, A. Ramamoorthy and R. M. Epand, Biochemistry, 2010, 49, 4076–4084 CrossRef CAS PubMed.
- R. M. Epand, in Antimicrobial Peptides: Basics for Clinical Application, ed. K. Matsuzaki, Springer Singapore, Singapore, 2019, pp. 65–71 DOI:10.1007/978-981-13-3588-4_5.
- D. Zweytick, G. Deutsch, J. Andrä, S. E. Blondelle, E. Vollmer, R. Jerala and K. Lohner, J. Biol. Chem., 2011, 286, 21266–21276 CrossRef CAS PubMed.
- D. Zweytick, B. Japelj, E. Mileykovskaya, M. Zorko, W. Dowhan, S. E. Blondelle, S. Riedl, R. Jerala and K. Lohner, PLoS One, 2014, 9, e90228 CrossRef PubMed.
- L. Lombardi, M. I. Stellato, R. Oliva, A. Falanga, M. Galdiero, L. Petraccone, G. D’Errico, A. De Santis, S. Galdiero and P. Del Vecchio, Sci. Rep., 2017, 7, 44425 CrossRef CAS PubMed.
- Y. K. Cherniavskyi, R. Oliva, M. Stellato, P. D. Vecchio, S. Galdiero, A. Falanga, S. A. Dames and D. P. Tieleman, bioRxiv, 2021, DOI:10.1101/2021.03.30.437760.
- R. M. Epand and R. F. Epand, Biochim. Biophys. Acta, 2009, 1788, 289–294 CrossRef CAS PubMed.
- R. M. Epand, S. Rotem, A. Mor, B. Berno and R. F. Epand, J. Am. Chem. Soc., 2008, 130, 14346–14352 CrossRef CAS PubMed.
- R. M. Epand and R. F. Epand, J. Pept. Sci., 2011, 17, 298–305 CrossRef CAS PubMed.
- P. Wadhwani, R. F. Epand, N. Heidenreich, J. Bürck, A. S. Ulrich and R. M. Epand, Biophys. J., 2012, 103, 265–274 CrossRef CAS PubMed.
- B. Bechinger, Curr. Opin. Colloid Interface Sci., 2009, 14, 349–355 CrossRef CAS.
- G. Sautrey, M. El Khoury, A. G. Dos Santos, L. Zimmermann, M. Deleu, L. Lins, J.-L. Décout and M.-P. Mingeot-Leclercq, J. Biol. Chem., 2016, 291, 13864–13874 CrossRef CAS PubMed.
- M. El Khoury, J. Swain, G. Sautrey, L. Zimmermann, P. Van Der Smissen, J.-L. Décout and M.-P. Mingeot-Leclercq, Sci. Rep., 2017, 7, 10697 CrossRef PubMed.
- J. Swain, M. El Khoury, J. Kempf, F. Briée, P. Van Der Smissen, J. L. Décout and M. P. Mingeot-Leclercq, PLoS One, 2018, 13, e0201752 CrossRef PubMed.
- J. E. Horne, D. J. Brockwell and S. E. Radford, J. Biol. Chem., 2020, 295, 10340–10367 CrossRef CAS.
- C. M. Ernst and A. Peschel, Mol. Microbiol., 2011, 80, 290–299 CrossRef CAS PubMed.
- G. Panda, S. Dash and S. K. Sahu, Membranes, 2022, 12 Search PubMed.
- H. Roy, IUBMB Life, 2009, 61, 940–953 CrossRef CAS PubMed.
- Y. Ramos, S. Sansone, S. M. Hwang, T. A. Sandoval, M. Zhu, G. Zhang, J. R. Cubillos-Ruiz and D. K. Morales, mBio, 2022, 13, e0229422 CrossRef PubMed.
- Y. Bao, T. Sakinc, D. Laverde, D. Wobser, A. Benachour, C. Theilacker, A. Hartke and J. Huebner, PLoS One, 2012, 7, e38458 CrossRef CAS PubMed.
- S. Samant, F. F. Hsu, A. A. Neyfakh and H. Lee, J. Bacteriol., 2009, 191, 1311–1319 CrossRef CAS PubMed.
- E. Maloney, D. Stankowska, J. Zhang, M. Fol, Q. J. Cheng, S. Lun, W. R. Bishai, M. Rajagopalan, D. Chatterjee and M. V. Madiraju, PLoS Pathog., 2009, 5, e1000534 CrossRef PubMed.
- C. Sohlenkamp, K. A. Galindo-Lagunas, Z. Guan, P. Vinuesa, S. Robinson, J. Thomas-Oates, C. R. Raetz and O. Geiger, Mol Plant Microbe Interact, 2007, 20, 1421–1430 CrossRef CAS PubMed.
- K. Thedieck, T. Hain, W. Mohamed, B. J. Tindall, M. Nimtz, T. Chakraborty, J. Wehland and L. Jänsch, Mol. Microbiol., 2006, 62, 1325–1339 CrossRef CAS PubMed.
- R. Kumariya, S. K. Sood, Y. S. Rajput, N. Saini and A. K. Garsa, Biochim. Biophys. Acta, Biomembr., 2015, 1848, 1367–1375 CrossRef CAS PubMed.
- S. R. Dennison, F. Harris, M. Mura and D. A. Phoenix, Curr. Protein Pept. Sci., 2018, 19, 823–838 CrossRef CAS PubMed.
- E. Cox, A. Michalak, S. Pagentine, P. Seaton and A. Pokorny, Biochim. Biophys. Acta, 2014, 1838, 2198–2204 CrossRef CAS PubMed.
- L. Assoni, B. Milani, M. R. Carvalho, L. N. Nepomuceno, N. T. Waz, M. E. S. Guerra, T. R. Converso and M. Darrieux, Front. Microbiol., 2020, 11, 593215 CrossRef PubMed.
- N. N. Mishra, S. J. Yang, A. Sawa, A. Rubio, C. C. Nast, M. R. Yeaman and A. S. Bayer, Antimicrob. Agents Chemother., 2009, 53, 2312–2318 CrossRef CAS PubMed.
- H. S. Joo and M. Otto, Biochim. Biophys. Acta, 2015, 1848, 3055–3061 CrossRef CAS PubMed.
- D. Song, H. Jiao and Z. Liu, Nat. Commun., 2021, 12, 2927 CrossRef CAS PubMed.
- K. L. Nawrocki, E. K. Crispell and S. M. McBride, Antibiotics, 2014, 3, 461–492 CrossRef PubMed.
- T. J. Silhavy, D. Kahne and S. Walker, Cold Spring Harbor Perspect. Biol., 2010, 2, a000414 Search PubMed.
- P. Nikolic and P. Mudgil, Microorganisms, 2023, 11, 259 CrossRef CAS PubMed.
- N. Malanovic and K. Lohner, Pharmaceuticals, 2016, 9 DOI:10.3390/ph9030059.
- N. Preußke, F. D. Sönnichsen and M. Leippe, Biosci. Rep., 2023, 43 DOI:10.1042/BSR20230474.
- R. Vaiwala, P. Sharma and K. Ganapathy Ayappa, Biointerphases, 2022, 17, 061008 CrossRef CAS PubMed.
- A. Guyet, A. Alofi and R. A. Daniel, mBio, 2023, 14, e0266722 CrossRef PubMed.
- N. Dong, X. Yang, E. W. Chan, R. Zhang and S. Chen, EBioMedicine, 2022, 79, 103998 CrossRef CAS PubMed.
- A. Hadjicharalambous, N. Bournakas, H. Newman, M. J. Skynner and P. Beswick, Antibiotics, 2022, 11, 1636 CrossRef CAS PubMed.
- D. A. Phoenix, F. Harris and S. R. Dennison, Curr. Protein Pept. Sci., 2021, 22, 775–799 CrossRef CAS PubMed.
- E. Malik, S. R. Dennison, F. Harris and D. A. Phoenix, Pharmaceuticals, 2016, 9, 31–39 CrossRef PubMed.
- D. I. Andersson, D. Hughes and J. Z. Kubicek-Sutherland, Drug Resist Updat, 2016, 26, 43–57 CrossRef CAS PubMed.
- J. Andrä, T. Goldmann, C. M. Ernst, A. Peschel and T. Gutsmann, J. Biol. Chem., 2011, 286, 18692–18700 CrossRef PubMed.
- G. S. Dijksteel, M. M. W. Ulrich, E. Middelkoop and B. K. H. L. Boekema, Front. Microbiol., 2021, 12, 37 Search PubMed.
- M. Dostert, C. R. Belanger and R. E. W. Hancock, J. Innate Immun., 2018, 11, 193–204 CrossRef PubMed.
- M. Kazemzadeh-Narbat, H. Cheng, R. Chabok, M. M. Alvarez, C. de la Fuente-Nunez, K. S. Phillips and A. Khademhosseini, Crit. Rev. Biotechnol., 2021, 41, 94–120 CrossRef CAS PubMed.
- Y. Zhu, W. Hao, X. Wang, J. Ouyang, X. Deng, H. Yu and Y. Wang, Med. Res. Rev., 2022, 42, 1377–1422 CrossRef CAS PubMed.
- F. Bugli, C. Martini, M. Di Vito, M. Cacaci, D. Catalucci, A. Gori, M. Iafisco, M. Sanguinetti and A. Vitali, Microbiol. Res., 2022, 263, 127152 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sm01007d |
| This journal is © The Royal Society of Chemistry 2023 |