
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Segmental mobility in sustainable copolymers based on poly(lactic acid) blocks built onto poly(butylene succinate) in situ
Panagiotis A.
Klonos
 *ab,
Nikolaos D.
Bikiaris
a,
Alexandra
Zamboulis
a,
Miguel Ángel
Valera
c,
Ana
Mangas
c,
Apostolos
Kyritsis
b and
Zoi
Terzopoulou
*a
*ab,
Nikolaos D.
Bikiaris
a,
Alexandra
Zamboulis
a,
Miguel Ángel
Valera
c,
Ana
Mangas
c,
Apostolos
Kyritsis
b and
Zoi
Terzopoulou
*a
aDepartment of Chemistry, Laboratory of Polymer Chemistry and Technology, Aristotle University of Thessaloniki, GR-541 24, Thessaloniki, Greece. E-mail: pklonos@central.ntua.gr; terzozoi@chem.auth.gr
bDepartment of Physics, National Technical University of Athens, Zografou Campus, 15780, Athens, Greece
cAIMPLAS, Asociación de Investigación de Materiales Plásticos Y Conexas, Carrer de Gustave Eiffel, 4, 46980 Paterna, Valencia, Spain
First published on 27th September 2023
Abstract
Two series of newly synthesized sustainable block copolymers based on poly(butylene succinate) (PBSu) and polylactide (PLA) were studied. The copolymers were synthesized by a ring-opening polymerization of PLA in the presence of two initial PBSu of low molar mass. We focused on the effects of the PBSu/PLA ratio (1/99 up to 15/85), chain length and initial PBSu length on the final thermal transitions in the copolymers with an emphasis on molecular mobility/dynamics and subsequently on crystallization. Both aspects are considered relevant to the final materials performance, as well as facilitation of polymer renewability. Calorimetry and dielectric spectroscopy were the main investigation tools. In the amorphous state (i.e., in which the direct effects of copolymer structure are assessable), the segmental mobility of neat PLA was significantly faster in the copolymers. Segmental mobility was monitored via the decrease in the calorimetric and dielectric (α relaxation) glass-transition temperatures, Tg and Tg,diel, respectively. The effect was systematic with an increase in the PBSu/PLA ratio, and was rationalized through the plasticizing role of PBSu (low-Tg component) and facilitated also by the simultaneous lowering of the chain length in the copolymers. Dielectric spectroscopy allowed evaluation of the dynamical fragility (cooperativity) of chains, which was strongly suppressed in the copolymers. This finding suggested an increase in free volume or a gradual increase of interchain distances. This phenomenon could favor the natural enzymatic degradation of the systems (compostability), which is limited in neat PLA. We recorded enhancement of nucleation and the crystalline fraction in the copolymers that was likely connected with faster chain diffusion. Further lowering of the Tg with the implementation of crystallization was noted (which seemed a controversial effect) but which indicated crystallization-induced phase separation.
1. Introduction
Polymer materials have been used extensively for almost one century, covering needs from everyday life to industry, science and academia.1,2 During recent years, environmental concerns about polymer use have arisen, including the use of fossil-based plastics. There has been growing interest in the use of non-toxic, biobased materials and polymers synthesized from renewable resources.3,4 Such materials reduce polymer accumulation in the environment,5,6 as well as the concept of a “green” economy.7,8 One of the most characteristic examples of polymers is poly(lactic acid) (PLA).9–13 PLA can be prepared from lactic acid, a renewable byproduct of fermentation of agricultural compounds via ring opening polymerization (ROP).14 Apart from its non-toxic and ecofriendly characteristics, PLA offers a combination of properties that can be exploited in numerous applications. For example, owing to a low glass transition temperature (Tg) of 50–70 °C,15–18 PLA can be processed economically. Its semicrystalline character enables tuning from a completely amorphous to highly crystalline state.19–21 Moreover, by artificially creating various semicrystalline morphologies (from dense to loose, or from large to small crystals)16,17,19,20,22–24 almost everything about its performance can be manipulated, such as mechanical strength and elasticity (e.g., three-dimensional (3D) printing, prosthetics),25–28 permeation by small molecules (e.g., oxygen, ions, water)29–31 and even thermal diffusivity.32 In general, in most polymers, crystallinity may have the most crucial role on materials performance and processing.A serious disadvantage of PLA is its weak compostability, which arises from slow physical biodegradation via enzymatic hydrolysis in the soil environment (temperature, humidity, pressure).5,21,33 This slow enzymatic hydrolysis originates, on the one hand, from the hydrophobic character of PLA and, on the other hand, from strong ester bonds in the PLA backbone. The latter subsequently precludes hydrolytic scission (physical depolymerization).21,34 This problem can be rectified via chemical routes.
Fortunately, PLA has been shown to be compatible with other polymers, such as other biobased polyesters. These include polycaplolactones (PCL)35 as well as the more recently developed polyesters based on 2,5-furan dicarboxylic acid,36 succinic acid,37 adipic acid38,39 and vanillic acid.40 In particular, polyesters prepared from succinic acid, such as poly(ethylene succinate)41 (PESu),42–44 poly(propylene succinate) (PPSu)45–47 and poly(butylene succinate) (PBSu; also called poly(tetramethylene succinate)48–53), have attracted much attention lately because they are eco-friendly materials which combine a series of attractive properties.45,46,54,55 In particular, PBSu exhibits high enzymatic degradation rates and can be manipulated readily by tuning its crystalline fraction.56
Deep understanding and mapping of molecular dynamics are extremely useful for polymer-chain topology and interchain associations, which are connected with the performance (mechanical, thermal and electrical conduction, permeability) of materials.21,35,57,58 The macroscopic performance depends on the crystallinity in a polymer. Among others, the mechanisms of crystallization (i.e., nucleation, chain entanglements, chain folding, spherulitic growth19,59) are strongly connected with the structure and mobility of the polymer chain (length, fragility, diffusion).60–65 An exceptionally useful method to study molecular mobility and dynamics in polymers is dielectric spectroscopy,66,67 which can be combined with other methods for the study of structure–property relationships.
Recently, we determined this relationship in a series of renewable copolymers based on PLA58,65,68 as well as other esters.57,69 Furthermore, we showed that, for block copolymers, the effects on crystallization related to polymer mobility could be more easily tuned but were stronger when compared with the same polymers in the form of blends.70
As noted above, PLA is a promising material for various applications in the green economy, but this polymer lacks compostability. Combining PLA with polymers with faster compostability rates has been shown to be possible.68,71,72 Comparing PLA-based systems in the form of blends or copolymers, the latter seem to be the most uniform and promising.13,70 For the materials studied in the present work, lowering of the Tg and melting point and improvement of physical enzymatic hydrolysis in soil were paramount. The humidity and enzymes present in soil can enter the copolymer and compared with neat PLA, degrade the polymer chains faster. Obviously, one would want to preserve (or even improve) some desired properties of the material (e.g., mechanical and permeation performance) for certain applications.
Motivated by the studies mentioned above, we synthesized71 two series of diblock copolymers based on PLA and PBSu. The materials were prepared by ROP of lactide in the presence (in situ) of quite short PBSu chains, a processing route that has given excellent results in the past. In this work, the emphasis was on the effects of copolymer structural chains (chain or block lengths, initial PBSu length, and PLA/PBSu ratio) on molecular mobility and, subsequently, crystallization. We employed dielectric spectroscopy and calorimetry supplemented by polarized light microscopy (PLM). This is the first report of such results for PLA/PBSu copolymers. The properties of interest were strongly connected to crystallinity, so we discuss our findings in relation to this parameter.
2. Experimental
2.1. Materials
Two series of diblock copolymers based on PBSu and PLA were studied (Table 1). The copolymers were synthesized employing known synthetic routes (as in previous works72 for similar copolyesters) and these copolymers were structurally characterized in a recent work by Terzopoulou et al.71 Briefly, samples were prepared using two forms of PBSu of relatively low intrinsic viscosity [η] (0.22 and 0.38 dL g−1) and low molar mass (Mn, 6 and 16 kg mol−1), respectively. These initial forms of PBSu were referred to as “PBSu0.22” and “PBSu0.38”, respectively. PBSu formed the first block (A) of the copolymer (Fig. 1). PLA formed the second block (B) of the copolymer. The copolymer was built via ROP of L-lactide at 180 °C (Fig. 1). The prepared PBSu was a diol (HO-PBSu-OH), so two PLA blocks were “built” at both sites of PBSu. Therefore, the block copolymers were of the BAB-type (i.e., PLA_b_PBSu_b_PLA). For simplicity, hereafter the system is referred to as “PBSu_b_PLA”. Overall, five PBSu_b_PLA copolymers were prepared for each series at a PBSu/PLA ratio of 1/99, 2.5/97.5, 5/95, 10/90 and 15/85. In the initial synthesis,71 due to the difficulty of proving the presence of only block copolymers, we could not exclude the possibility of a small amount of blend, i.e., PLA_b_PBSu_b_PLA and homo-PLA. Thus, the obtained copolymers could also be referred to as “blocky”.73 Finally, for comparison, neat PLA was synthesized by similar routes as a reference material for the copolymers.| Sample | PBSu (%) | PLA (%) | M n (g mol−1) |
|---|---|---|---|
| PBSu0.22 | 100.0 | 0 | 6k |
| PBSu0.22 (15%)_b_PLA (85%) | 15.0 | 85.0 | 32k |
| PBSu0.22 (10%)_b_PLA (90%) | 10.0 | 90.0 | 31k |
| PBSu0.22 (5%)_b_PLA (95%) | 5.0 | 95.0 | 50k |
| PBSu0.22 (2.5%)_b_PLA (97.5%) | 2.5 | 97.5 | 80k |
| PBSu0.22 (1%)_b_PLA (99%) | 1.0 | 99.0 | 76k |
| PBSu0.38 | 100.0 | 0 | 16k |
| PBSu0.38 (15%)_b_PLA (85%) | 15.0 | 85.0 | 39k |
| PBSu0.38 (10%)_b_PLA (90%) | 10.0 | 90.0 | 54k |
| PBSu0.38 (5%)_b_PLA (95%) | 5.0 | 95.0 | 80k |
| PBSu0.38 (2.5%)_b_PLA (97.5%) | 2.5 | 97.5 | 79k |
| PBSu0.38 (1%)_b_PLA (99%) | 1.0 | 99.0 | 77k |
| PLA | 0 | 100.0 | 103k |
 | ||
| Fig. 1 Chemical structures of PBSu and PLA blocks (top) and schemes of PBSu_b_PLA systems (bottom). | ||
2.2. Differential scanning calorimetry (DSC)
Calorimetric measurements were made employing a TA Q200 DSC apparatus (TA Instruments, USA). It was calibrated with sapphire for heat capacity and indium for temperature and enthalpy. Samples (∼8 mg) were placed in aluminium Tzero TA pans in a temperature range from −110 to 200 °C in a nitrogen atmosphere of high purity (99.9995%). A first scan from −110 to 195 °C was employed prior to the main DSC measurements to evaluate the degree of crystallinity of the samples as received, to erase any thermal history and to evaporate any remaining water or solvents (scan 1). At scan 2 (Fig. 2a), the melted samples were cooled at 20 K min−1 to −110 °C, and subsequently heated to 200 °C (i.e., well above the melting point) at 10 K min−1. At scan 3 (Fig. 2a), the melted samples were cooled at a fast rate to suppress crystallization and subsequently heated to 200 °C at 10 K min−1. The latter fast cooling rate was achieved by employing the “jump” command of TA software, which resulted in a non-linear cooling rate (Fig. 2b), estimated to be ∼105 K min−1 in the temperature range of the expected crystallization events. | ||
| Fig. 2 (a) Time–temperature profiles employed for DSC measurements of scans 2 and 3. (b) Estimation of the average cooling rate in the temperature range of the expected crystallization event. | ||
2.3. PLM
The distribution and morphology of formed crystals were assessed for all samples via PLM. We employed a Optiphot-1 polarizing microscope (Nikon, Japan) equipped with a THMS 600 heated stage (Linkam Scientific, UK), control unit (TP91; Linkam) and a camera (Gryphax Arktur; Jenoptic, Germany). The corresponding images were captured during hot crystallization (i.e., initially melted samples) and subsequently cooled at 20 K min−1 until the completion of crystallization.2.4. Broadband dielectric spectroscopy (BDS)
The molecular dynamics of polymers were assessed employing BDS in a nitrogen atmosphere by means of an analyzer (BDS Alpha; Novocontrol, Germany) combined with a liquid nitrogen cryosystem (temperature control better than 0.5 K; Quatro, Novocontrol) on samples of 14 mm in diameter and ∼100 μm in thickness. Samples were inserted between finely polished brass electrodes to form a “sandwich”-like capacitor, melted and quenched therein to stay amorphous. To prevent electrical contact between the metallic electrodes and to maintain the distance between electrodes, silica spacers (Novocontrol) were employed. The complex dielectric permittivity, ε* = ε′ − i·ε′′, was recorded isothermally as a function of frequency from 10−1 to 106 Hz and in a temperature range between −150 and 120 °C on heating at steps of 5 and 10 K, depending on the process followed, via WinDETA of Novocontrol.3. Results and discussion
3.1. Direct effects on glass transition and crystallization
Fig. 3 compares the DSC results for scans 2 and 3 for all samples. Beginning with the cooling at a standard rate of 20 K min−1, all samples exhibited exothermal peaks in the temperature range from ∼70 to ∼110 °C that originate from the polymer crystallization (Fig. 3a and b). Neat PBSu exhibited strong and sharp crystallization peaks at 71–72 °C. These peaks were not observed in any PBSu_b_PLA systems, which was the first indication of the sufficiently good distribution of the two polymers and, indirectly, for successful copolymer synthesis.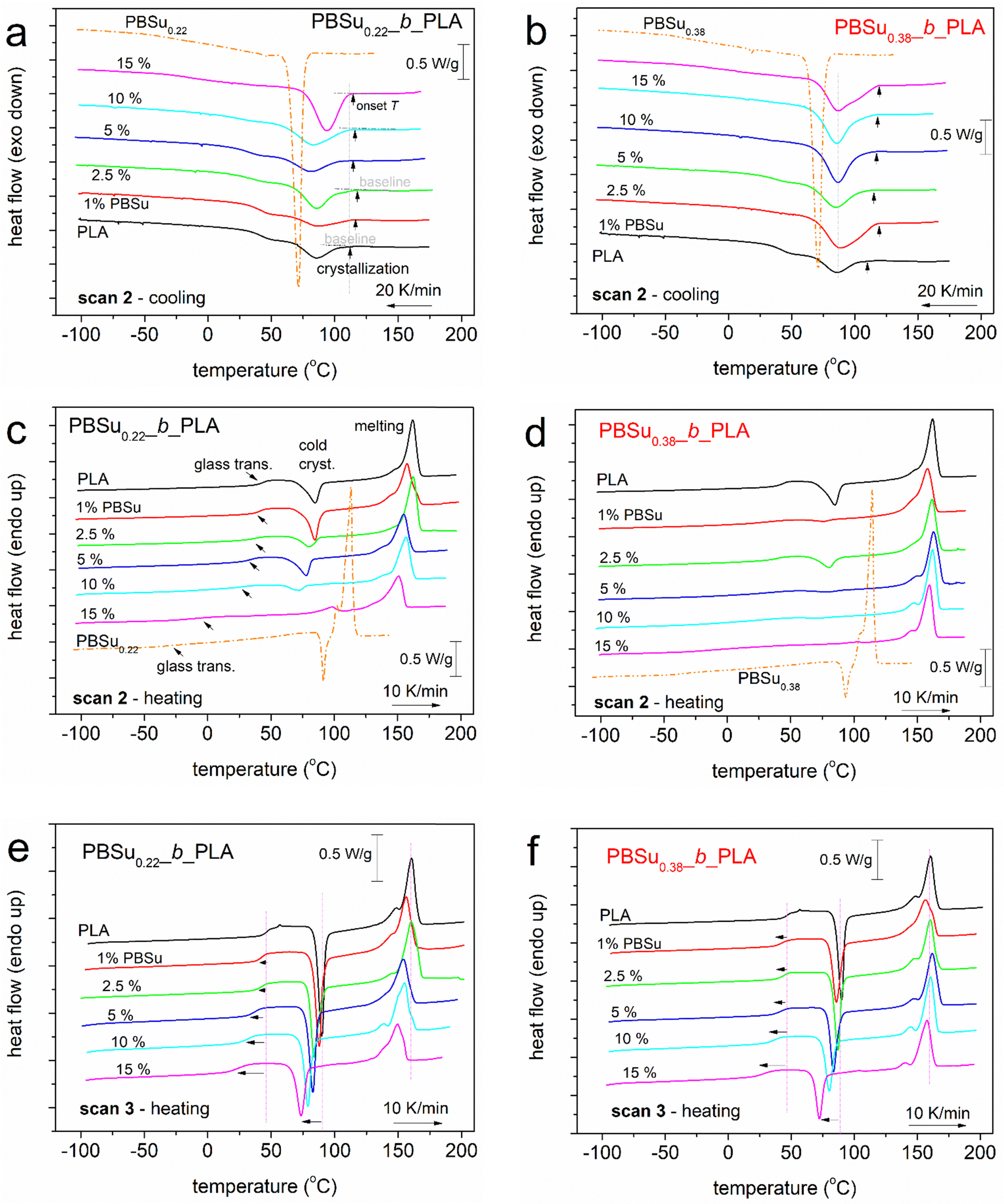 | ||
| Fig. 3 Comparative DSC traces on all samples and the thermal protocols studied. Heat flow has been normalized to the mass of each sample. Arrows mark the effects on some thermal events imposed by the copolymer composition. In particular, the black vertical arrows in a and b mark the temperature of initiation of the crystallization exotherm, Tc,onset. | ||
Upon subsequent heating, all polymers exhibited glass-transition steps (arrows in Fig. 3c). Then, cold crystallization exotherms were observed for 1–10% PBSu0.22 and 1–5% PBSu0.38. Cold crystallization is the result of incomplete crystallization during the prior cooling from hot crystallization. The samples melted between 125 and 170 °C. Neat PLA showed a single melting peak, whereas the copolymers exhibited double-structured melting peaks, which may suggest a more complex melting process and/or less homogeneous crystal structures. For copolymers with 15% PBSu, additional weak melting processes were recorded at ∼100 °C (i.e., closely below the melting point of neat PBSu). This was an indication for bulk-like crystallization of PBSu within the copolymers, and the existence of micro-metric PBSu-rich entities in the copolymers.
During fast cooling (scan 3, data not shown) from the melt state, none of the samples (including neat PLA) exhibited crystallization events. Thus, we considered these samples to be amorphous. However, by thermal treatment we expected nucleated polymers because they have been subjected to strong supercooling. During subsequent heating (Fig. 3e and f), all samples exhibited strong glass-transition steps, strong cold crystallization (as expected) and complex melting peaks.
The peaks shown in DSC traces were evaluated in terms of characteristic temperature peak temperatures and ΔH (in J g−1). Using the ΔHc of crystallization and comparing it with the theoretic values for the heat of fusion of a fully crystalline PLA, ΔH100%,PLA, taken as 93 J g−1,73 we estimated the crystalline fraction, CFc, according to eqn (1):
 | (1) |
Recently, different values for ΔH100%,PLA have been published considering the α and α′ crystal polymorphs of PLA.74,75 Finally, the characteristic temperatures for glass-transition steps, Tg, were estimated as T exhibiting a half change in heat capacity, Δcp.
Values are shown in Fig. 4 against the PBSu fraction in our systems for both series of copolymers. In the amorphous state, Tg showed a monotonic drop upon increasing of the PBSu fraction (Fig. 4a), from ∼47 °C (neat PLA) down to 23–28 °C [PBSu(15%)_b_PLA(85%]. The drop was slightly greater in the case of PBSu0.22. The Tg for PBSu is about −35 °C, which makes the Tg(PBSu) shown in Fig. 4a different due to chain–chain interactions and entanglements. Considering the copolymers as a blend of two quite miscible polymers, one with high and the other with low Tg, the results of Fig. 4a suggest that the short block (PBSu) had a plasticizing (softening) role on the large block (PLA), and this role was stronger when the PBSu block was shorter. Besides, simultaneously with an increase in the PBSu fraction, the overall length of the copolymer chain decreased. This also contributed to faster/easier chain diffusion, thereby lowering the Tg.
 | ||
| Fig. 4 PBSu dependence of characteristic values obtained by DSC from scan 3 (Tg and Tcc) and scan 2 (CF). Lines have been added as visual guides. | ||
With respect to crystallization, during scan 2 (Fig. 3a and b), the peak temperature of hot crystallization (Tc) of PLA did not change significantly in the copolymers (85–94 °C). Nevertheless, the onset T of crystallization, Tc,onset (i.e., the point at which the exothermal peak begins to form (inset vertical arrows in Fig. 3a and b)), was increased slightly in the copolymers (from 112 to 118 °C in PBSu0.22_b_PLA and from 115 to 121 °C in PBSu0.38_b_PLA). This different trend between Tc and Tc,onset was due to the change in the width of the crystallization peak. Thus, Tc,onset provided a better measure of nucleation, which seems to be facilitated in the copolymers. Also, CFc increased almost systematically from 0.16 up to 0.46 wt in in PBSu0.22_b_PLA and from 0.27 up to 0.44 wt in PBSu0.38_b_PLA (Fig. 3c). In scan 3, the peak temperature of cold crystallization, Tcc, showed a clear decrease when PBSu increased from ∼90 °C down to ∼73 °C. The increase of Tc,onset and CFc and the decrease in Tcc indicated the facilitation of crystal nucleation and the number of crystals. From the viewpoint of neat PLA (homopolymer), we would expect the opposite effect with decreasing of Mn (as what happened in the copolymers here). Therefore, the improvements in crystallization we recorded were related to the enhanced mobility and topology of chains, and alternations in free volume. These points are discussed later.
Another goal we achieved was a significant drop in the melting temperature (Fig. 3c–f), Tm, from 160–162 °C for neat PLA down to the minimum value of 149–150 °C for 15% PBSu. The result is important mainly for the processing point of view.
A final point with respect to crystallization is related to semicrystalline morphology. This was examined by the direct method of PLM employing the same thermal protocol as that in scan 2 for DSC (i.e., crystallization during cooling at 20 K min−1 beginning from the melt state). The results for all samples are shown in Fig. 5. In accordance with DSC observations, crystallization seemed to begin slightly faster (higher temperatures) in copolymers as compared with neat PLA. All samples exhibited at least two crystal populations (one with large spherulites and another with significantly smaller ones) and finally all the sample volume seemed to be filled with crystals. Thus, we could not conclude severe differences in semicrystalline morphology arising from the copolymer composition or from the length of the initial PBSu block. In recent findings using X-ray diffraction,71 samples exhibited the same number of crystallization diffraction peaks in 2θ positions, suggesting that PLA chains dominated in crystal formation. This result will be helpful with our discussion in the last section of this article.
 | ||
| Fig. 5 Representative PLM micrographs during hot crystallization for neat PLA (top) and two series of PLA_b_PBSu systems (bottom). Results are shown at initial, intermediate and final states of crystallization at the temperatures indicated on each image. Scale bar = 500 μm. | ||
3.2. Molecular mobility – segmental dynamics
Dielectric spectroscopy proffered interesting results. Raw data are shown in the form of isothermal curves of the imaginary part of dielectric permittivity (ε′′) against frequency (f) for the various temperatures studied (Fig. 6). Representative cases of neat PLA (Fig. 6a), initial PBSu0.22 (Fig. 6b) and PBSu0.22(5%)_b_PLA(95%) (Fig. 6c) are also shown. ε′′ is related to the dielectric loss,66 within which the various dipolar motions (if activated) are recorded as peaks of ε′′(f). Such peaks were followed in our samples and were usually called “dielectric relaxation processes”, or mechanisms that originated from actual molecular mobility (local and segmental).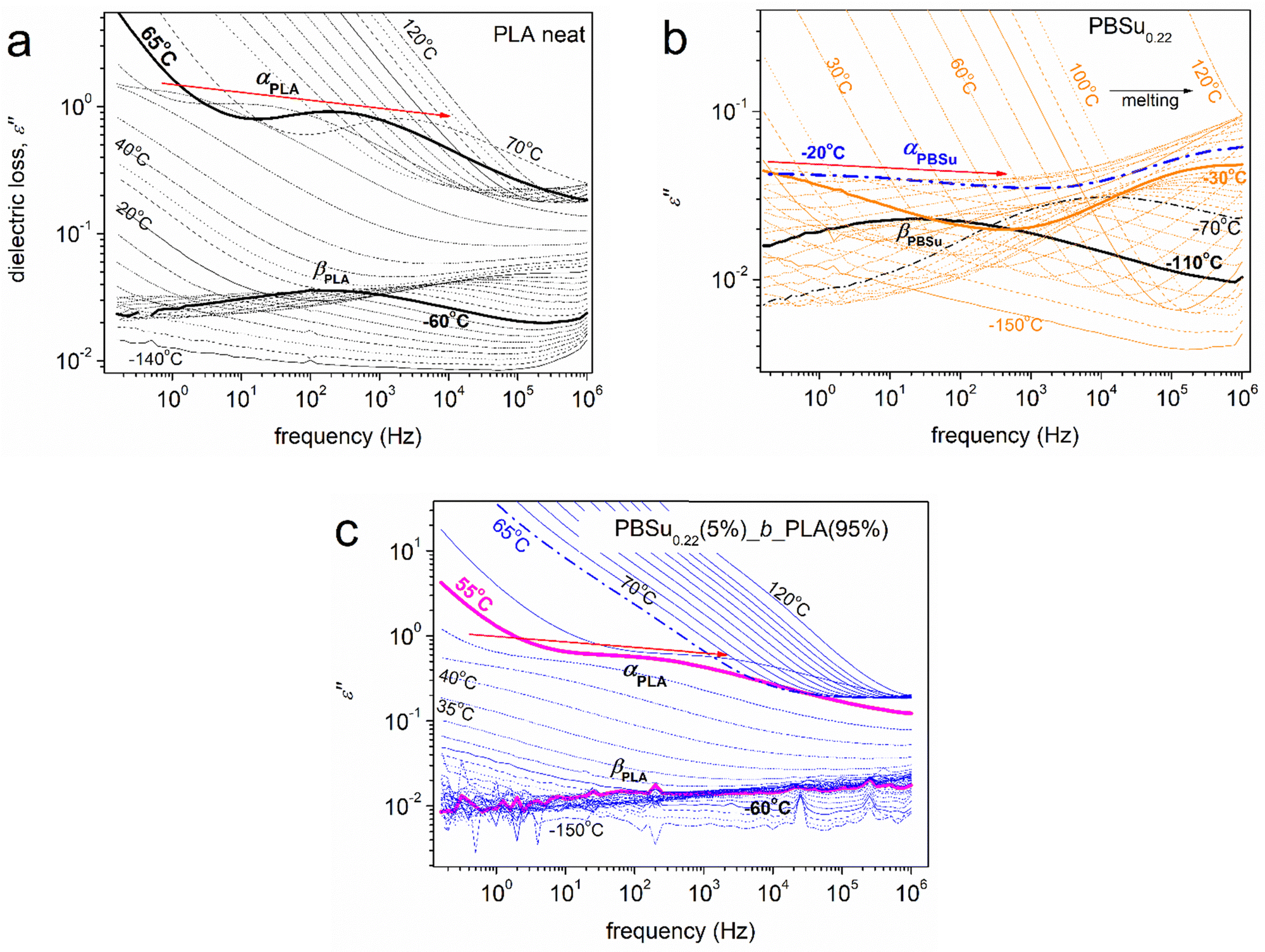 | ||
| Fig. 6 Representative BDS results (in the form of isothermal ε′′(f) curves) for selected samples described on the plots in their initially amorphous state. | ||
To facilitate a more direct comparison between dielectric results and calorimetric data, the raw ε′′(f) for various T (isothermal curves) data can be replotted as ε′′(T) for various f (isochronal curves). Such isochronal curves are presented in Fig. 7 for neat PLA and two initial PBSu, and in Fig. 8 for the two series of studied copolymers.
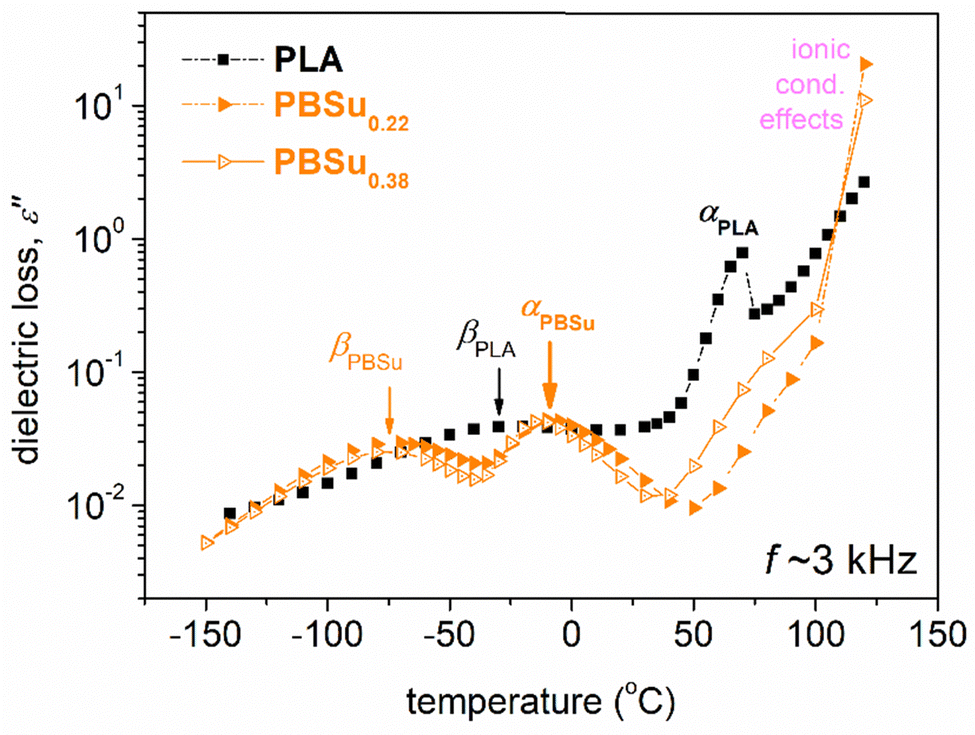 | ||
| Fig. 7 Comparative isochronal plots of ε′′ against temperature for neat PLAs and initial PBSu at a selected frequency of 3.2 kHz. The main relaxation process, recorded as peaks of ε′′(T), is indicated. | ||
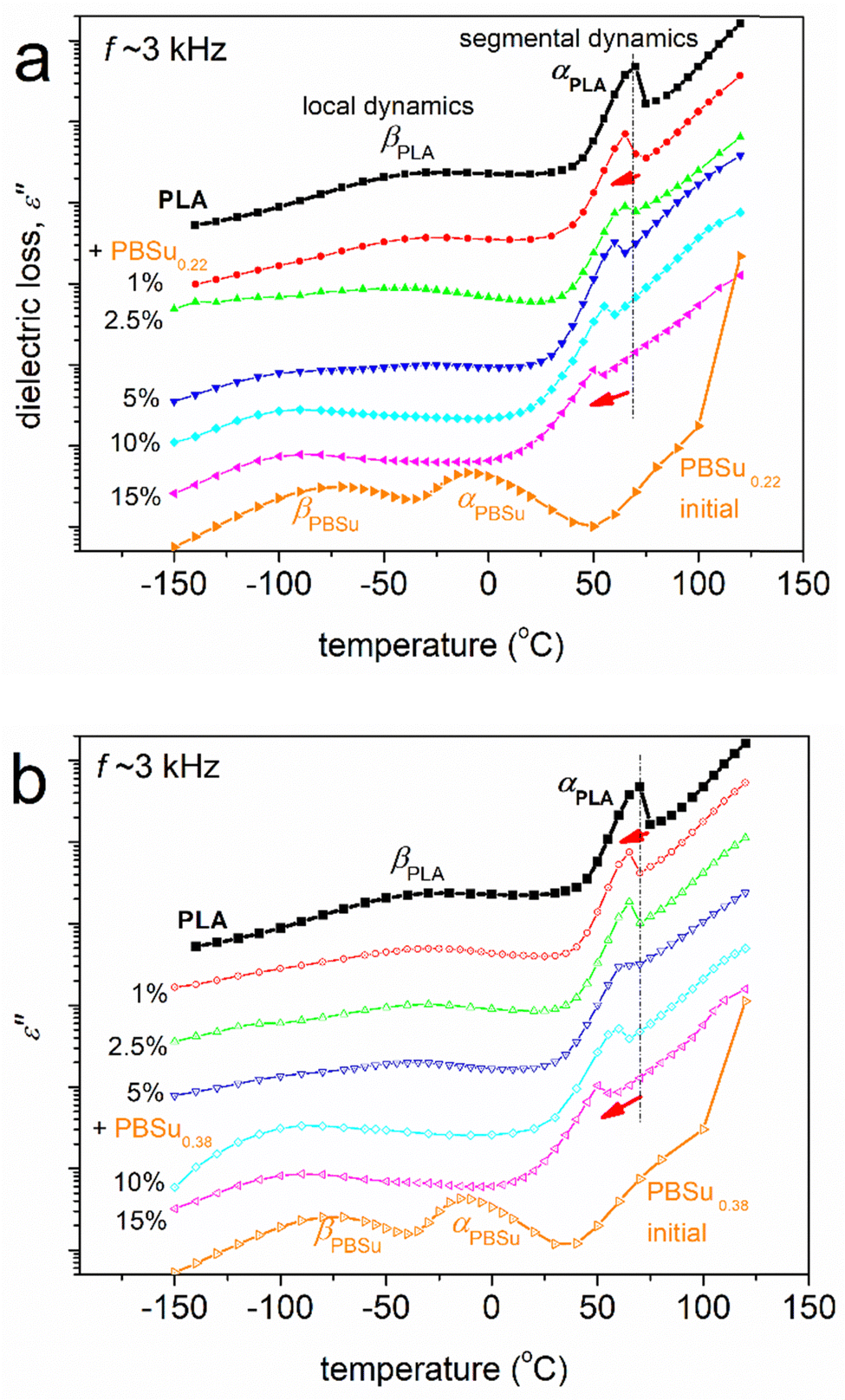 | ||
| Fig. 8 Comparative isochronal plots of ε′′ against temperature for all samples, indicated on the plots, in (a) PBSu0.22_b_PLA and (b) PBSu0.38_b_PLA. Results correspond to initially amorphous samples. The added red arrows mark the direct effect of increasing the PBSu fraction on segmental relaxation. The recorded data of ε′′ are shown upon vertical shifts. | ||
At T < Tg, the dielectric signals are usually low because the contributions to ε′′ were mainly due to local relaxations of weak localized molecular motions. Such processes are considered secondary and, thus, are usually named as β, γ, or δ relaxations.66 In our case, we recognized the local relaxations of PLA of PBSu (βPLA and βPBSu in Fig. 6a and b, respectively). The corresponding molecular origins have been described in previous works.76–81 At T ≥ Tg, the contribution to ε′′ increases strongly (overall dielectric permittivity) by more than one order of magnitude. This is due to the thermal “activation” of large-scale segmental mobility. In terms of relaxation processes, this phenomenon is recorded as the strong relaxation peak of ε′′, usually called “main segmental α relaxation”.66α relaxation is considered to be the dielectric analogue of the glass transition, occurs at more than one temperatures and is located at gradually increasing frequencies. Therefore, it is also called “glass transition dynamics”.66 The corresponding main relaxations were αPLA and αPBSu (Fig. 6). As stated above, our focus for the copolymers was on segmental α relaxations.
The raw BDS results and impact of copolymer compositions are shown in Fig. 8. With an increase in the PBSu/PLA ratio, the local dynamics of PBSu was recorded in the copolymers, whereas the αPBSu was not. Hence, segmental dynamics were dominated by αPLA relaxation. The effect was similar to the calorimetric view for Tg, and suggests zero or non-significant separation of PBSu chains. This observation provided additional evidence that the copolymer synthesis was successful. We did not believe that the segmental relaxation in the copolymers arose uniquely from the PLA block. Hence, in the following section, we refer to segmental relaxation simply as “α”.
Except for the qualitive information provided by raw dielectric data, this method enables a more in-depth evaluation. The complex dielectric spectra consist of multiple contributions: ε′′(f) peaks. Spectra were analysed by fitting of model functions, in particular, the Havriliak–Negami (HN) model66,82 (eqn (2)). In the HN function, ε∞ describes the value of the real part of dielectric permittivity: ε′. For f ≫ f0, Δε is the dielectric strength, f0 is a characteristic frequency related to the frequency of maximum dielectric loss and αHN and βHN are shape parameters, for width and symmetry, respectively. Examples of this fitting are shown in Fig. 9.
 | (2) |
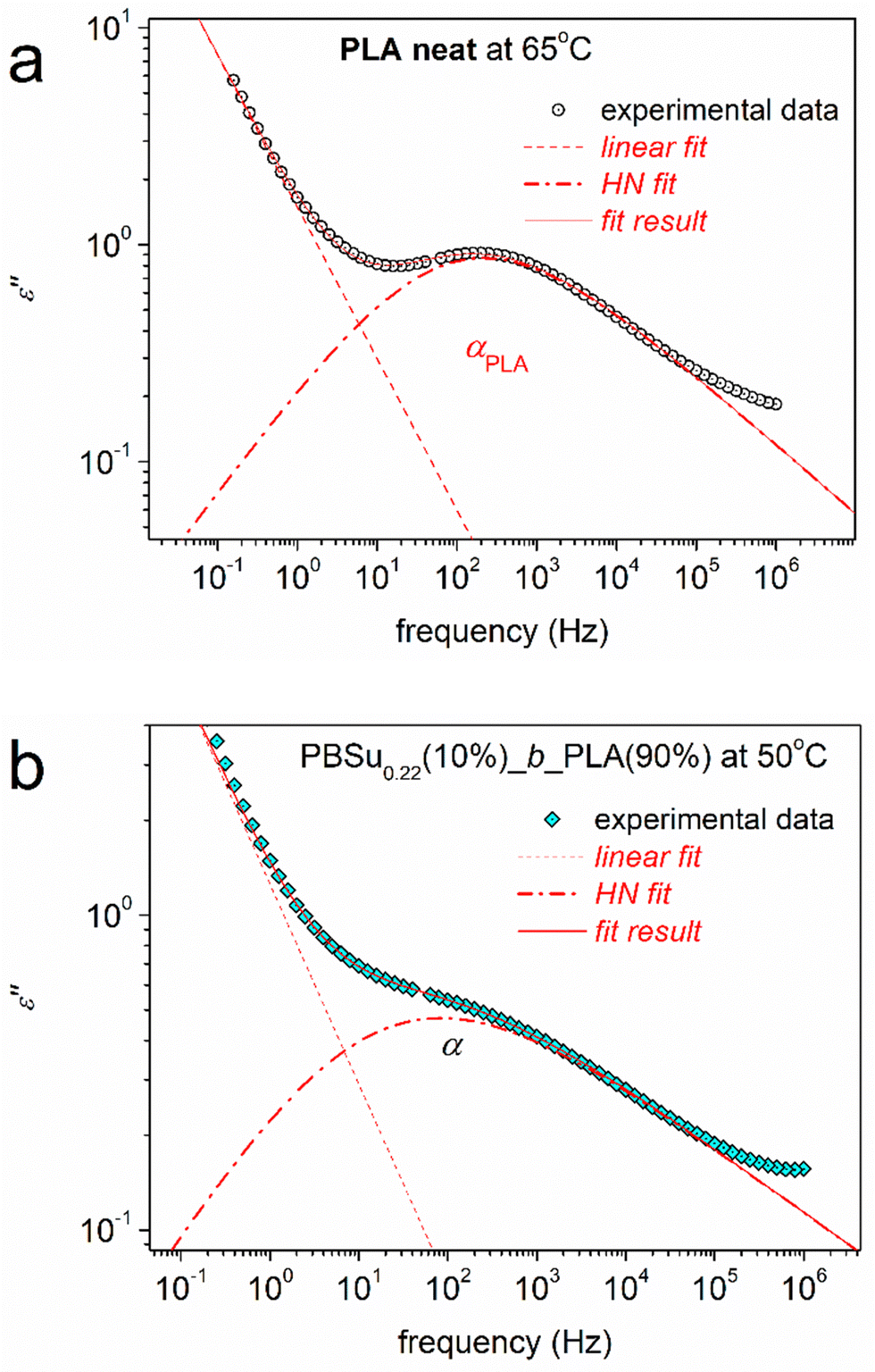 | ||
| Fig. 9 Analysis of ε′′(f) isothermal peaks (α relaxations) in terms of Havriliak–Negami (HN) terms for the samples and temperatures described on the plots. | ||
Arrhenius plots (also called “molecular dynamics maps”) for the segmental relaxations of PBSu0.22_b_PLA are shown in Fig. 10a. Fig. 10b revealed the reciprocal temperature dependence of Δε (the latter being a good measure of the population of relaxing dipoles). The results were quite similar for PBSu0.38_b_PLA (data not shown). Segmental relaxations, related to the glass transition, demonstrate a cooperative character, and are usually described by the Vogel–Fulcher–Tammann–Hesse (VFTH) expression66,83 (eqn (3)),
 | (3) |
| m = 16 + 590/D | (4) |
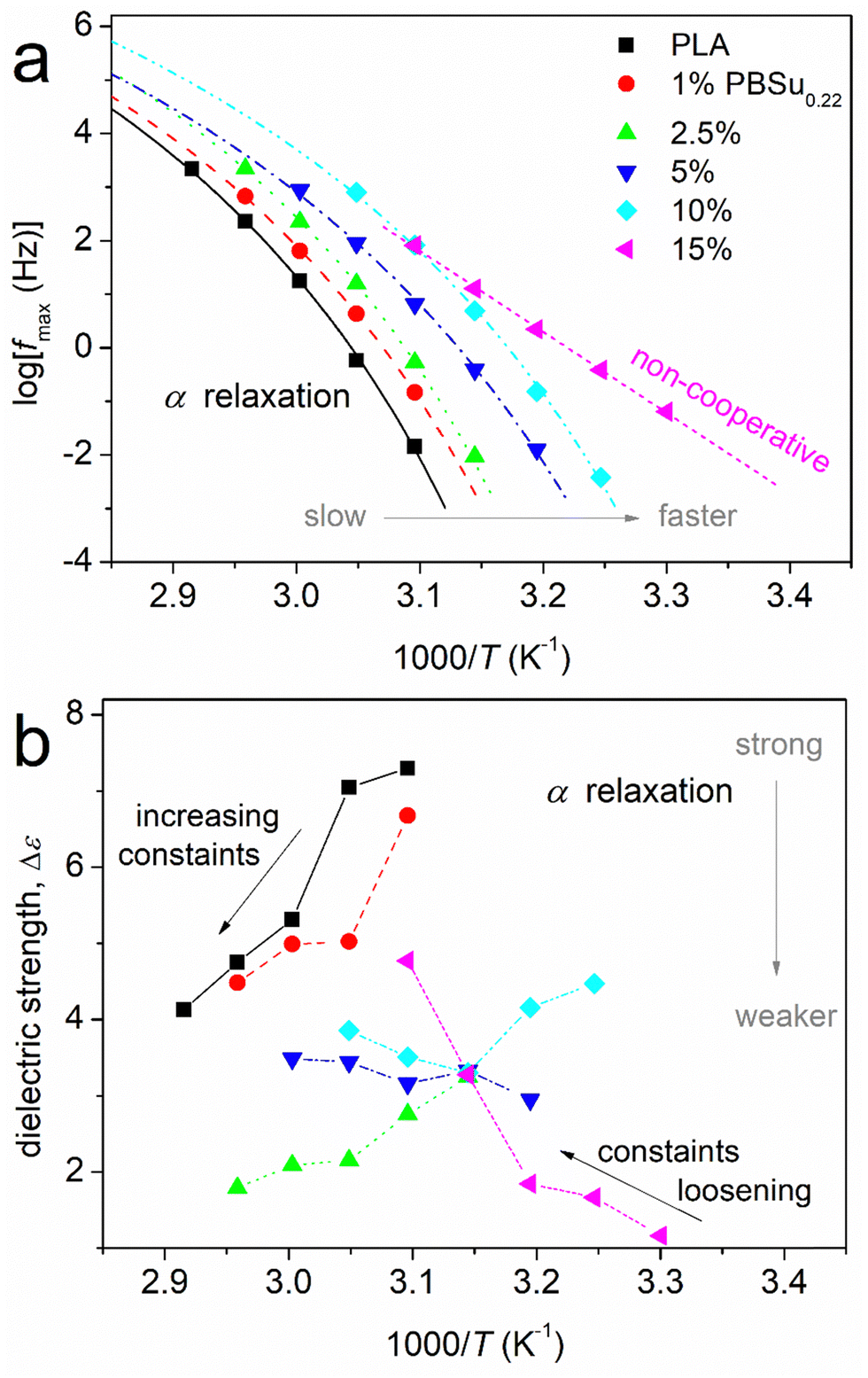 | ||
| Fig. 10 (a) Arrhenius plots and (b) dielectric strength for PBSu0.22_b_PLA. Results refer to segmental α relaxation. In (a), the curved lines connecting the data are fittings of the VFTH equation, whereas the straight line corresponds to the Arrhenius equation. | ||
Fig. 10a shows a systematic migration of the points for α relaxation toward lower temperatures/higher frequencies upon PBSu addition. These effects could be considered to be an acceleration of segmental mobility in the copolymers, and were in agreement with those from calorimetry. From the data of Fig. 10a, we could estimate the dielectric glass transition temperature (Tg,diel) as an extrapolation of the corresponding points of α relaxation to the equivalent frequency of DSC (feq ∼10−2.8 Hz, arising from the corresponding relaxation time τ ∼ 100 s).66 The estimated Tg,diel values are shown in Fig. 11a compared with those of Tg obtained by calorimetry. In general, good agreement between the two methods was recorded, but this is not always the case.65,84 For neat PLA and copolymers with 1–10 wt% PBSu, the α relaxation points shown in Fig. 10a obeyed the VFTH model function (i.e., the characteristic for cooperative dynamics). Strikingly, for 15 wt% PBSu, the fast α relaxation exhibited a linear trend (i.e., an Arrhenius-like non-cooperative behavior). This phenomenon was reflected as a gradual drop in fragility (m, eqn (3) and (4)) from ∼160 for neat PLAs down to 0 for copolymers with 15% PBSu (Fig. 11b). This drop in m suggested suppression of the cooperation capability of the polymers, most probably due to an increase in interchain distances (increasing of the cooperativity length, ξ).85,86 It is less likely that the decreased fragility arose from shortening of the chain length of the copolymers.
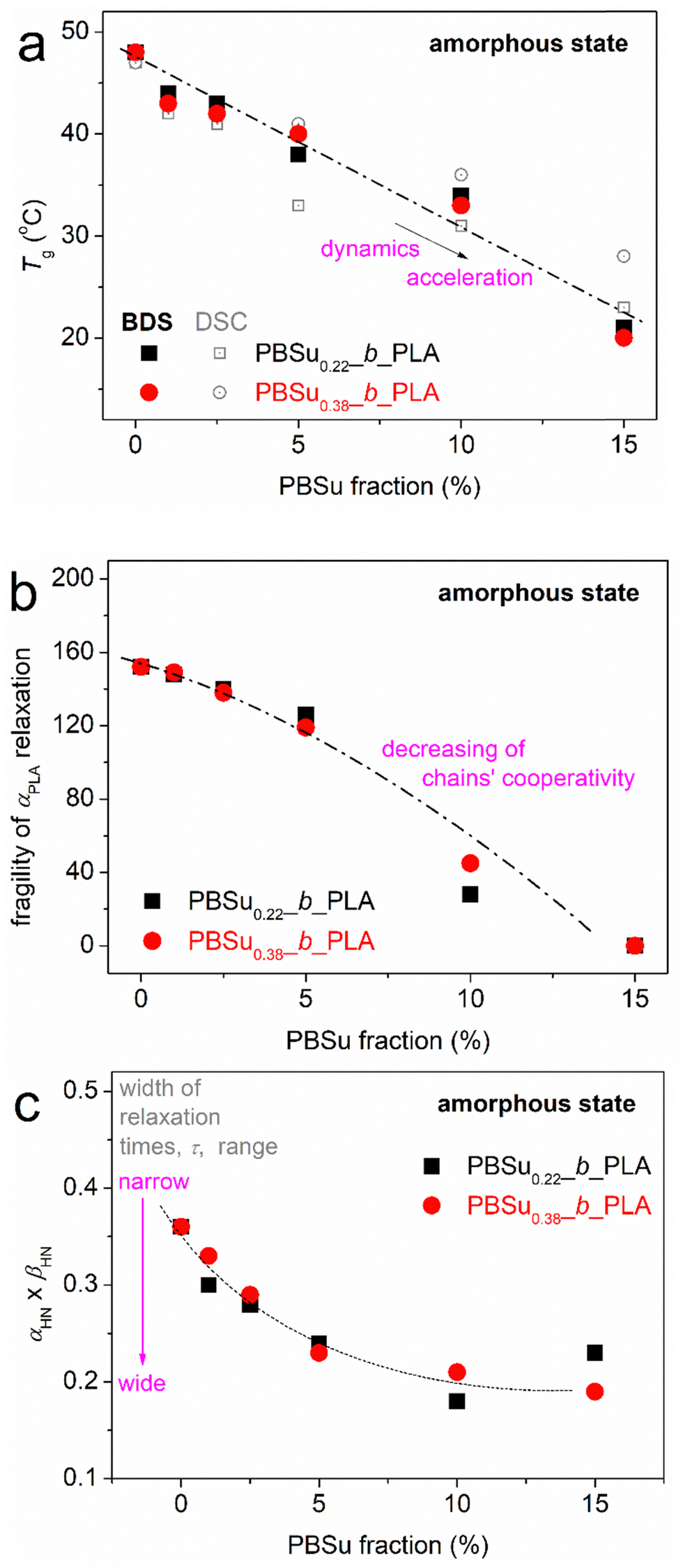 | ||
| Fig. 11 PBSu dependence of characteristic values obtained by BDS in the initially amorphous state. (a) Dielectric glass transition temperature, Tg,diel. (b) Fragility index, m, of αPLA relaxation. (c) αHN*βHN of the HN shape parameters. In (a), results on the Tg obtained by calorimetry have been added for comparison. Lines connecting the experimental points have been added as a visual guide. | ||
The effects on Tg,diel and m were compatible also with the temperature dependence of Δε for α relaxation (Fig. 10b). For neat PLA, Δε decreased with increasing temperature, which is typical for amorphous and unconstrained segmental mobility. This drop was due to a gradual increase in constraints in segmental mobility or, in topological terms, due to a decrease in cooperatively rearranging regions (CRR)66 upon an increase in T. As T increases and each individual chain becomes more mobile due to the additional thermal energy supplied, an increase in the number and complexity of chain–chain entanglements also occurs. In the copolymers, the mobility of the polymer chains is constrained ab initio due to complex structuring due to the presence of PBSu blocks or, indirectly, due to shortening of the PLA blocks. These two parameters are equivalent with each other, for example, in terms of tending to preclude the polymer chains cooperativity (recorded drop in m). Thus, the initial dielectric strength was reduced in the copolymers as compared with neat PLA. Then, the decreasing Δε(T) trend recorded for unconstrained neat PLA gradually changed in the copolymers to a milder reduction (1 and 2.5% PBSu), to almost no change (5% PBSu) and, finally, to a Δε(T) increase (10 and 15% PBSu). This effect suggested an increasing population (fraction) of copolymer chains that suffered some constraints initially which were loosened gradually when T increased.
As expected, α was, in general, asymmetric, and initially narrow. Upon critical fitting, the relaxation became wider in the copolymers. This was reflected on the shape parameters of the HN equation (Fig. 11c). The PBSu dependence of αHN*βHN dropped in the copolymers. This drop suggested that the corresponding width of the relaxation times (τ) became wider in the copolymers or, in other words, that the segmental mobility of the polymer chains (quite homogeneous in neat PLA) became more heterogeneous within the copolymers.66
3.3. Effects of crystallization on the glass transition
An interesting effect was revealed upon crystallization of samples in DSC. We compared the two series of copolymers in terms of the glass transition in the amorphous state against the semicrystalline state (Fig. 12). As expected, upon crystallization, the height of the glass-transition step decreases because fewer amorphous chains contributed to the thermal event. Unexpectedly, upon crystallization, the Tg fell for all samples (including neat PLAs). In general, for conventional homopolymers, the Tg increases in the presence of crystals because the latter act as obstacles to chains diffusion.23,87,88 Also, comparing the drop in Tg with the increase in CFc (Fig. 4c), the maximum change in the Tg coincided with the maximum increase in CFc. | ||
| Fig. 12 Glass-transition event as recorded in the amorphous (dash-dotted lines, scan 3) state against the semicrystalline (solid lines, scan 2) state. | ||
This observation is not usual, but has been noted in a recent work on PLA-based block copolymers with poly(propylene adipate), the latter being the low-Tg component65 (similar to PBSu in the present work). This effect can be interpreted via two realistic scenarios. According to the first scenario, crystallization-related spatial nano-confinement seems to be imposed on amorphous polymer chains. Such confinement should make a strong impact if the amorphous zones formed between the crystallites are nanometric and comparable with the cooperativity length, ξ.85,89 This scenario is partly compatible in the present study in the discussion on the dynamics and cooperativity for α relaxation. On the other hand, the effects recorded in Fig. 12 could be seen in another light. Upon crystal formation, the glass transition tends to approach the quite fast mobility of initial PBSu. Thus, upon crystallization, the mobility of PLA (pure αPLA) in the copolymers gradually vanishes. That is, the PLA chains mainly crystallize and, thus, become immobile, whereas the PBSu-rich segments are mobile and their corresponding weak glass transition is enhanced at the expense of αPLA.58,65,70 This second scenario implies a crystallization-driven phase separation,90,91 which is compatible with recent data using X-ray diffraction,71 denoting that the PLA chains dominate crystallization in the copolymers.
This special effect for the crystallization-induced phase can be exploited for specific applications (e.g., drug delivery). Nevertheless, more work is needed to further evaluate the origins of this effect.
4. Conclusions
We investigated the molecular mobility and crystallization in relatively new PBSu_b_PLA diblock copolymers. We studied the direct composition effects of the copolymer in the amorphous state, achieved by relatively fast cooling from the melt, because PBSu and PLA are semicrystalline. PBSu (low-Tg block) was found to have a plasticizing role in the mobility of PLA. Therefore, the overall Tg fell monotonically upon PBSu addition, suggesting easier chain diffusion in the copolymers. Segmental dynamics were followed by employing dielectric spectroscopy. With an increase in the PBSu fraction, the α relaxation of copolymers was accelerated significantly whereas, interestingly, fragility was severely suppressed. The effect denotes either an increase in the cooperativity length and/or an increase in free volume (e.g., detaching of polymer chains). These effects were compatible with the above-mentioned plasticization. These are key parameters for the facilitation of permeation of small molecules (e.g., humidity, oxygen) throughout the sample volume and, subsequently, favor compostability.The nucleation and degree of crystallinity of PLA in copolymers appeared to improve. This improvement was related with the enhanced molecular mobility of polymer chains. The difference in the length of the initial PBSu block (6 or 16 kg mol−1) did not affect the crystallization and molecular mobility of the copolymers. Finally, we exploited indications for crystallization-induced phase separation (recorded indirectly by a significant drop of Tg upon crystallization). This effect was more pronounced with an increase in the PBSu fraction. Similar results were recently reported in diblock copolymers based on PPAd_b_PLA, prepared by similar routes.65 This effect also seems promising for specific applications.
Author contributions
Panagiotis A. Klonos: conceptualization, methodology, investigation, formal analysis, and writing (original draft). Nikolaos D. Bikiaris: conceptualization, methodology, investigation, formal analysis, and writing (review and editing). Alexandra Zamboulis: validation and writing (review and editing). Miguel Ángel Valera: resources, validation, and writing (review and editing). Ana Mangas: resources and writing (review and editing). Apostolos Kyritsis: resources, validation, and writing (review and editing). Zoi Terzopoulou: investigation, data analysis, validation, and writing (review and editing).Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was funded from the European Union Horizon 2020 Research and Innovation Programme (952941, BIOMAC Project). The publication of the article in OA mode was supported financially by HEAL-Link.References
- M. Chanda and S. K. Roy, Industrial polymers, specialty polymers, and their applications, CRC Press, Boca Raton, 2008 Search PubMed.
- S. J. Rowan, ACS Macro Lett., 2021, 10, 466–468 CrossRef CAS PubMed.
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS PubMed.
- A. Gandini and T. M. Lacerda, Macromol. Eng., 2022 DOI:10.1002/9783527815562.mme0019.
- N. M. Ainali, D. Kalaronis, E. Evgenidou, G. Z. Kyzas, D. C. Bobori, M. Kaloyianni, X. Yang, D. N. Bikiaris and D. A. Lambropoulou, Sci. Total Environ., 2022, 832, 155014 CrossRef CAS PubMed.
- J. Ahmed and S. K. Varshney, Int. J. Food. Prop., 2011, 14, 37–58 CrossRef CAS.
- M. Hong and E. Y. X. Chen, Green Chem., 2017, 19, 3692–3706 RSC.
- W. Post, A. Susa, R. Blaauw, K. Molenveld and R. J. I. Knoop, Polym. Rev., 2020, 60, 359–388 CrossRef CAS.
- R. Auras, B. Harte and S. Selke, Macrom. Biosci., 2004, 4, 835–864 CrossRef CAS PubMed.
- Y. Ikada and H. Tsuji, Macromol. Rapid Commun., 2000, 21, 117–132 CrossRef CAS.
- M. Ebara, K. Uto, N. Idota, J. M. Hoffman and T. Aoyagi, Soft Matter, 2013, 9, 3074–3080 RSC.
- D. Giliopoulos, A. Zamboulis, D. Giannakoudakis, D. Bikiaris and K. Triantafyllidis, Molecules, 2020, 25, 185 CrossRef CAS PubMed.
- E. Balla, V. Daniilidis, G. Karlioti, T. Kalamas, M. Stefanidou, N. D. Bikiaris, A. Vlachopoulos, I. Koumentakou and D. N. Bikiaris, Polymers, 2021, 13, 1822 CrossRef CAS PubMed.
- O. Coulembier, J. De Winter, T. Josse, L. Mespouille, P. Gerbaux and P. Dubois, Polym. Chem., 2014, 5, 2103–2108 RSC.
- S. Saeidlou, M. A. Huneault, H. Li and C. B. Park, Prog. Polym. Sci., 2012, 37, 1657–1677 CrossRef CAS.
- R. Androsch, E. Zhuravlev and C. Schick, Polymer, 2014, 55, 4932–4941 CrossRef CAS.
- G. Z. Papageorgiou, Z. Terzopoulou, D. Bikiaris, K. S. Triantafyllidis, E. Diamanti, D. Gournis, P. Klonos, E. Giannoulidis and P. Pissis, Thermochim. Acta, 2014, 597, 48–57 CrossRef CAS.
- M. C. Righetti, M. Gazzano, N. Delpouve and A. Saiter, Polymer, 2017, 125, 241–253 CrossRef CAS.
- A. Toda, R. Androsch and C. Schick, Polymer, 2016, 91, 239–263 CrossRef CAS.
- R. Androsch, R. Zhang and C. Schick, Polymer, 2019, 176, 227–235 CrossRef CAS.
- T. Beslikas, I. Gigis, V. Goulios, J. Christoforides, G. Z. Papageorgiou and D. N. Bikiaris, Int. J. Mol. Sci., 2011, 12, 6597–6618 CrossRef CAS PubMed.
- R. Androsch, H. M. Naeem Iqbal and C. Schick, Polymer, 2015, 81, 151–158 CrossRef CAS.
- Z. Terzopoulou, P. A. Klonos, A. Kyritsis, A. Tziolas, A. Avgeropoulos, G. Z. Papageorgiou and D. N. Bikiaris, Polymer, 2019, 166, 1–12 CrossRef CAS.
- K. Pušnik Črešnar, P. A. Klonos, A. Zamboulis, Z. Terzopoulou, E. Xanthopoulou, L. Papadopoulos, A. Kyritsis, K. Kuzmič, L. Fras Zemljič and D. N. Bikiaris, Thermochim. Acta, 2021, 703, 178998 CrossRef.
- A. Constanzo, R. Spotorno, M. V. Candal, M. M. Fernández, A. J. Müller, R. S. Graham, D. Cavallo and C. McIlroy, Addit. Manuf., 2020, 36, 101415 Search PubMed.
- J. Dominguez-Robles, N. K. Martin, M. L. Fong, S. A. Stewart, N. J. Irwin, M. I. Rial-Hermida, R. F. Donnely and E. Larrenta, Pharmaceutics, 2019, 11, 165 CrossRef CAS PubMed.
- P. Georgiopoulos, E. Kontou, A. Meristoudi, S. Pispas and M. Chatzinikolaidou, J. Biomat. Appl., 2014, 29, 662–674 CrossRef CAS PubMed.
- S. Bourbigot and G. Fontaine, Polym. Chem., 2010, 1, 1413–1422 RSC.
- A. Sangroniz, A. Chaos, M. Iriarte, J. del Río, J. R. Sarasua and A. Etxeberria, Macromolecules, 2018, 51, 3923–3931 CrossRef CAS.
- V. Demchenko, Y. Mamunya, S. Kobylinskyi, S. Riabov, K. Naumenko, S. Zahorodinia, O. Povnitsa, N. Rybalchenko, M. Iurzhenko, G. Adamus and M. Kowalczuk, Molecules, 2022, 27, 3769 CrossRef CAS PubMed.
- H. Zebiri, H. Van Den Berghe, S. Sayegh, P. E. Chammas, C. Pompée, M. Chammas and X. Garric, J. Mat. Chem. B, 2021, 9, 832–945 RSC.
- P. A. Klonos, V. Peoglos, D. N. Bikiaris and A. Kyritsis, J. Phys. Chem. C, 2020, 123, 5469–5479 CrossRef.
- M. Naddeo, G. Viscusi, G. Gorrasi and D. Pappalardo, Molecules, 2021, 26, 4454 CrossRef CAS PubMed.
- Q. Zhang, M. Song, Y. Xu, W. Wang, Z. Wang and L. Zhang, Prog. Polym. Sci., 2021, 120, 101430 CrossRef CAS.
- N. Mulchandani, K. Masutani, S. Kumar, H. Yamane, S. Sakurai, Y. Kimura and V. Katiyar, Polymer Chem., 2021, 12, 3806–3824 RSC.
- G. Chen, L. Wu, H. Fan and B. Li, Ind. Eng. Chem. Res., 2018, 57(48), 16172–16181 CrossRef CAS.
- M. I. Peñas, R. A. Pérez-Camargo, R. Hernández and A. J. Müller, Polymers, 2022, 14, 1025 CrossRef PubMed.
- T. Zorba, K. Chrissafis, K. M. Paraskevopoulos and D. N. Bikiaris, Polym. Deg. Stabil., 2007, 92, 222–230 CrossRef CAS.
- T. Zhang, B. A. Howell, A. Dumitrascu, S. J. Martin and P. B. Smith, Polymer, 2014, 55, 5065–5072 CrossRef CAS.
- E. Xanthopoulou, P. A. Klonos, A. Zamboulis, Z. Terzopoulou, A. Kyritsis, P. Pissis, D. N. Bikiaris and G. Z. Papageorgiou, Polymer, 2021, 233, 124197 CrossRef CAS.
- D. N. Bikiaris and D. S. Achilias, Polymer, 2008, 49, 3677–3685 CrossRef CAS.
- Y. Ichikawa, K. Nogushi, K. Okuyama and J. Washiyama, Polymer, 2001, 42, 3703–3708 CrossRef CAS.
- R. Caminiti, A. Isopo, M. A. Orru and V. R. Albertini, Chem. Mater., 2000, 12, 369–375 CrossRef CAS.
- Z. Qiu, T. Ikehara and T. Nishi, Polymer, 2003, 44, 5429–5439 CrossRef CAS.
- E. Ranucci, Y. Liu, M. Söderqvist Lindblad and A. C. Albertson, Macromol. Rapid Commun., 2000, 21, 680–684 CrossRef CAS.
- Y. Liu, E. Ranucci, M. Soderqvist-Lindblad and A. C. Albertson, J. Polym. Sci. A Polym. Chem., 2001, 39, 2508–2519 CrossRef CAS.
- P. A. Klonos, M. Kluge, T. Robert, A. Kyritsis and D. N. Bikiaris, Polymer, 2020, 186, 122056 CrossRef CAS.
- K. J. Ihn, E. S. Yoo and S. S. Im, Macromolecules, 1995, 28, 2460–2464 CrossRef CAS.
- Z. Gan, H. Abe, H. Kurokawa and Y. Doi, Biomacromolecules, 2001, 2, 605–613 CrossRef CAS PubMed.
- Y. Ichicawa, H. Kondo, Y. Igarashi, K. Noguchi, K. Okuyama and J. Washiyama, Polymer, 2000, 41, 4719–4727 CrossRef.
- T. Miyata and T. Masuko, Polymer, 1998, 39, 1399–1404 CrossRef CAS.
- I. Arandia, A. Mugica, M. Zubitur, A. Arbe, G. Liu, D. Wang, R. Mincheva, P. Dubois and A. J. Müller, Macromolecules, 2015, 48, 43–57 CrossRef CAS.
- X. Jiang, Y. Yu, Y. Guan, T. Liu, C. Pang, J. Ma and H. Gao, ACS Sustainable Chem. Eng., 2020, 8, 3626–3636 CrossRef CAS.
- G. Z. Papageorgiou and D. N. Bikiaris, Polymer, 2005, 46, 12081–12092 CrossRef CAS.
- D. N. Bikiaris, G. Z. Papageorgiou, S. A. Papadimitriou, E. Karavas and K. Avgoustakis, AAPS PharmSciTech, 2009, 10, 138–146 CrossRef CAS PubMed.
- K. Chrissafis, K. M. Paraskevopoulos and D. N. Bikiaris, Thermochim. Acta, 2005, 435, 142–150 CrossRef CAS.
- C. Bouyahya, N. D. Bikiaris, A. Zamboulis, A. Kyritsis, M. Majdoub and P. A. Klonos, Soft Matter, 2022, 18, 9216–9230 RSC.
- P. A. Klonos, Z. Terzopoulou, A. Zamboulis, M. A. Valera, A. Mangas, A. Kyritsis, P. Pissis and D. N. Bikiaris, Soft Matter, 2022, 18, 3725–3737 RSC.
- C. Schick, Anal. Bioanal. Chem., 2009, 395, 1589–1611 CrossRef CAS PubMed.
- L. Sangroniz, B. Wang, Y. Su, G. Liu, D. Cavallo, D. Wang and A. J. Müller, Prog. Polym. Sci., 2021, 115, 101376 CrossRef CAS.
- M. Huang, X. Dong, L. Wang, L. Zheng, G. Liu, X. Gao, C. Li, A. J. Müller and D. Wang, Macromolecules, 2018, 51, 1100–1109 CrossRef CAS.
- P. Klonos, Z. Terzopoulou, S. Koutsoumpis, S. Zidropoulos, S. Kripotou, G. Z. Papageorgiou, D. Bikiaris, A. Kyritsis and P. Pissis, Eur. Polym. J., 2016, 82, 16–34 CrossRef CAS.
- P. A. Klonos, Polymer, 2018, 159, 169–180 CrossRef CAS.
- M. Safari, J. Maiz, G. Shi, D. Juanes, G. Liu, D. Wang, C. Mijangos, Á. Alegría and A. J. Müller, Langmuir, 2019, 35, 15168–15179 CrossRef CAS PubMed.
- P. A. Klonos, A. Evangelopoulou, Z. Terzopoulou, A. Zamboulis, M. A. Valera, A. Mangas, A. Kyritsis and D. N. Bikiaris, Molecules, 2022, 27, 7449 CrossRef CAS PubMed.
- F. Kremer and F. Schönhals, Broadband dielectric spectroscopy, Springer-Verlag, Berlin, 2002 Search PubMed.
- A. Schönhals and P. Szymoniak, Dynamics of composite materials, Springer, Cham, Switzerland, 2022 Search PubMed.
- V. Karava, A. Siamidi, M. Vlachou, E. Christodoulou, A. Zamboulis, D. N. Bikiaris, A. Kyritsis and P. A. Klonos, Soft Matter, 2021, 17, 2439–2453 RSC.
- E. Christodoulou, P. A. Klonos, K. Tsachouridis, A. Zamboulis, A. Kyritsis and D. N. Bikiaris, Soft Matter, 2020, 16, 8187–8201 RSC.
- P. A. Klonos, M. Lazaridou, Ch Samiotaki, A. Kyritsis and D. N. Bikiaris, Polymer, 2022, 259, 125329 CrossRef CAS.
- Z. Terzopoulou, A. Zamboulis, N. D. Bikiaris, A. Margellou, M. A. Valera, A. Mangas, S. Koltsakidis, K. Tsongas, D. Tzetzis and K. Triantafyllidis, J. Polym. Environ., 2023 DOI:10.1007/s10924-023-02981-0.
- Z. Terzopoulou, A. Zamboulis, D. N. Bikiaris, M. A. Valera and A. Mangas, Polymers, 2021, 13, 4121 CrossRef CAS PubMed.
- E. W. Fischer, H. J. Sterzel and G. Wegner, Kolloid Z. Z. Polym., 1973, 251, 980–990 CrossRef CAS.
- M. C. Righetti, M. Gazzano, M. L. Di Lorenzo and R. Androsch, Eur. Polym. J., 2015, 70, 215–220 CrossRef CAS.
- K. Jariyavidyanont, C. Schick and R. Androsch, Thermochim. Acta, 2022, 717, 179349 CrossRef CAS.
- J. Ren, O. Urakawa and K. Adachi, Polymer, 2003, 44, 847–855 CrossRef CAS.
- E. Laredo, D. Newman, R. Pezzoli, A. J. Müller and A. Bello, J. Polym. Sci. B Polym. Phys., 2016, 54, 680–691 CrossRef CAS.
- J. Leng, N. Kang, D. Y. Wang, J. Falkenhagen, A. F. Thünemann and A. Schönhals, Macrom. Chem. Phys., 2017, 218, 1700232 CrossRef.
- P. A. Klonos, L. Papadopoulos, M. Kasimatis, H. Iatrou, A. Kyritsis and D. N. Bikiaris, Macromolecules, 2021, 54, 1106–1119 CrossRef CAS.
- C. Alvarez, M. J. Capitan, N. Lotti, A. Munari and T. A. Ezquerra, Macromolecules, 2003, 36, 3245–3253 CrossRef.
- M. Soccio, A. Nogales, N. Lotti, A. Munari and T. A. Ezquerra, Polymer, 2007, 48, 4742–4750 CrossRef CAS.
- S. Havriliak and S. Negami, Polymer, 1967, 8, 161–210 CrossRef CAS.
- R. Boehmer, K. Ngai, C. A. Angell and D. J. Plazek, J. Chem. Phys., 1993, 99, 4201–4209 CrossRef CAS.
- J. Leng, P. J. Purohit, N. Kang, D. Y. Wang, J. Falkenhagen, F. Emmerling, A. F. Thüneman and A. Schönhals, Eur. Polym. J., 2015, 68, 338–354 CrossRef CAS.
- N. Delpouve, A. Saiter and E. Dargent, Eur. Polym. J., 2011, 47, 2414–2423 CrossRef CAS.
- P. A. Klonos, N. D. Bikiaris, E. Christodoulou, A. Zamboulis, G. Z. Papageorgiou and A. Kyritsis, Polymer, 2022, 242, 124603 CrossRef CAS.
- J. Dobbertin, J. Hannemann, C. Schick, M. Pötter and H. Dehne, J. Chem. Phys., 1998, 108, 9062–9068 CrossRef CAS.
- A. Wurm, M. Ismail, B. Kretzschmar, D. Pospiech and C. Schick, Macromolecules, 2010, 43, 1480 CrossRef CAS.
- G. Adam and J. H. Gibbs, J. Chem. Phys., 1965, 43, 139–146 CrossRef CAS.
- M. G. Wessels and A. Jayaraman, Soft Matter, 2019, 15, 3987–3998 RSC.
- E. Mygiakis, E. Glynos and G. Sakellariou, Eur. Polym. J., 2021, 161, 110857 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |