
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigating the complexation propensity of self-assembling dipeptides with the anticancer peptide-drug Bortezomib: a computational study†
Peter
Divanach
ab,
Eirini
Fanouraki
ab,
Anna
Mitraki
*ab,
Vagelis
Harmandaris
 cde and
Anastassia N.
Rissanou
*cef
cde and
Anastassia N.
Rissanou
*cef
aDepartment of Materials Science and Technology, University of Crete, Voutes Campus Greece, Crete, Greece. E-mail: mitraki@materials.uoc.gr
bInstitute of Electronic Structure and Laser/Foundation for Research and Technology-Hellas, (FORTH), Nikolaou Plastira 100, Vassilika Vouton, Heraklion, Crete, Greece
cInstitute of Applied and Computational Mathematics (IACM), Foundation for Research and Technology Hellas, (FORTH), IACM/FORTH, GR-71110 Heraklion, Crete, Greece. E-mail: trissanou@eie.gr
dDepartment of Mathematics and Applied Mathematics, University of Crete, GR-71409, Heraklion, Crete, Greece
eComputation-based Science and Technology Research Center, The Cyprus Institute, Nicosia 2121, Cyprus
fTheoretical & Physical Chemistry Institute, National Hellenic Research Foundation, 48 Vassileos Constantinou Avenue, 11635 Athens, Greece
First published on 20th September 2023
Abstract
The investigation of potential self-assembled peptides as carriers for the delivery of anticancer drug Bortezomib is the topic of the present study. The self-assembly of Bortezomib in water is examined using all-atom molecular dynamics simulations and corresponding experimental results from FESEM experiments. In addition, a series of dipeptides with a similar chemical formula to Bortezomib with hydrogel-forming ability are being investigated for their propensity to bind to the drug molecule. Dipeptides are divided into two classes, the protected FF (Fmoc-FF and Z-FF) and the LF-based (Cyclo-LF and LF) ones. The thermodynamic stability of the complexes formed in an aqueous environment, as well as key morphological features of the nanoassemblies are investigated at the molecular level. Binding enthalpy between Bortezomib and dipeptides follows the increasing order: LF < Cyclo-LF < Fmoc-FF < Z-FF under both van der Waals and electrostatic contributions. Protected FF dipeptides have a higher affinity for the drug molecule, which will favor its entrapment, giving them an edge over the LF based dipeptides. By evaluating the various measures, regarding both the binding between the two components and the eventual ability of controlled drug release, we conclude that the protected FF class is a more suitable candidate for drug release of Bortezomib, whereas among its two members, Fmoc-FF appears to be more promising. The selection of the optimal candidates based on the present computational study will be a stepping stone for future detailed experimental studies involving the encapsulation and controlled release of Bortezomib both in vitro and in vivo.
1. Introduction
Bortezomib (Velcade®, Millennium Pharmaceuticals, Inc. Cambridge, MA, and Johnson & Johnson Pharmaceutical Research & Development L.L.C., Raritan, NJ, abr. BTZ) is a modified, phenylalanine-boroleucine dipeptide (N-pyrazino-phe-boroleu) with a molecular mass of 384.24 Da. It was the first selective and one of the few drugs for the treatment of relapsed and refractory multiple myeloma approved by the U.S. Food and Drug Administration (FDA) in 2003. It has also shown potential in the treatment of other malignancies such as non-Hodgkin's lymphomas and solid tumors as well as prostate, breast, ovarian, colorectal, pancreatic, and non-small-cell lung cancers. Its mechanism of action lies in the inhibition of the proteasomes. Proteasomes are large multi-protease complexes, localized in the nucleus and cytosol, responsible for regulating protein expression and function through the controlled degradation of more than 80% of cellular proteins, undergoing ubiquitination.1 While once considered risky or even outright untenable therapeutic targets, proteasomes have nowadays been the focus of many inhibitors in the treatment of multiple myeloma, with sales exceeding three billion US dollars annually.2 Bortezomib is designed in order to fit into the active site of the proteasomes causing stable inhibition. The presence of an empty p-orbital on the boron atom can accommodate the lone pair of electrons from the oxygen atom of threonine, present in the chymotrypsin-like (CT-L) active site of the proteasome, forming a stable, albeit reversible, sp3 hybridized tetrahedral intermediate complex.3,4 Although with lower affinity, Bortezomib has also been reported to bind the caspase-like (C-L) and tryptic-like (T-L) subunits.4–6Despite its potency and reversibility, Bortezomib suffers from certain drawbacks, including dose-limiting toxicities7 due to its short half-life.8,9 Bortezomib has an elimination half-life of 9–15 h, before being cleared by hepatic metabolism and only lasts about 5 minutes to 2 h when administered intravenously and subcutaneously.10 This rapid elimination rate coupled with its non-specific binding to proteins in serum impairs its accumulation and penetration into solid tissues, leading to patients requiring constant intake, twice a week, over multiple cycles. Like many other cancer therapies, some patients inevitably develop resistance over time, while others end up not responding at all to Bortezomib treatment.11 As such, both combined-therapies in conjunction with Bortezomib,12,13 as well as finding the proper carriers/scaffolds towards its controlled delivery are both subjects of much attention.
Nanoparticle delivery systems (NPs) are excellent drug delivery candidates with targeted-NPs, non-targeted and triggered-NPs all being used for the treatment of multiple myeloma and various types of cancer.14 The different categories of nanoparticles, proposed in recent review articles for Bortezomib's controlled release, can mostly be divided into polymeric and lipidic drug delivery systems. Solid lipid nanoparticles (SLNs) are often used as carriers to improve the intestinal permeability of Bortezomib,15,16 alongside targeted and non-targeted liposomes,17 microemulsions18 and nanostructured lipid carriers (NLCs).19,20
Polymeric micelles are the result of self-assembly of amphiphilic block copolymers in aqueous solution. The core–shell structure they exhibit is ideal for encapsulating and improving the solubility of Bortezomib, as was demonstrated by Liu et al. through a dual pH-sensitive drug delivery system comprised of BTZ and polymer conjugates21,22 and others.23 While dendrimers24 and inorganic nanoparticles25 have also seen extensive use for Bortezomib delivery, biomimetic nanomaterials26 and nanogels27 appear especially promising. Nanogels are biodegradable, water-containing, three-dimensional, gelatinous nano-formulations with a high surface area. Amin et al.28 have developed a composite, temperature-responsive nanogel using Bortezomib-loaded dopamine nanoparticles and a photo-thermal agent, while Liu et al.29 used a dopamine-grafted hyaluronate nanogel for its encapsulation. Biomimetic nanomaterials are functional materials comprised of nanoscale components, with structural and technological similarities to those of living organisms. Min et al. have employed30 P22 viral capsids while Yu et al.31 have used a copper sulfide/carbon dot nanocomposite as a nanocarrier for Bortezomib encapsulation and delivery.
Some recurring limitations of many Bortezomib controlled-release carriers remain the possible toxicity of the side-products as well as drug-loading and target-distribution restrictions. Various computational approaches have been used in-order to identify new candidates with greater Bortezomib loading and unloading capacities as well as to compare potential candidates and understand key mechanisms involved. Junlang Chen et al. have performed molecular dynamics simulations in order to investigate the potential application of graphene as a substrate to carry and deliver Bortezomib amongst other drugs32,33 while Abdus Samad et al. have used simulations and bioinformatics approaches to identify Bortezomib as a likely inhibitor of the Minichromosome maintenance complex component 7 (MCM7) protein.
Similarly, Yadav et al. have used a combination of MD simulations and experiments to examine the binding of zoledronate with a proteasome's subunit 5 as well as assess its apoptotic and anti-proliferative activity in multiple myeloma, when combined with Bortezomib.34
In the case of peptide drugs, self-assembly can lead to gelation at high concentrations and the gels can be used for controlled release of peptide drug monomers, as long as the self-assembly process is reversible. A classic example is the octapeptide drug lanreotide, an inhibitor of growth hormone that is used for the treatment of acromegaly. The peptide self-assembles into tubes of nanometric dimensions in aqueous solutions forming a gel.35 The gel (Somatuline® autogel, https://www.ipsen.com) is used for controlled release of the drug from gel implants. Moreover, due to their ability to undergo spontaneous assembly into ordered, biocompatible nanostructures through various non-covalent interactions, self-assembling peptides constitute promising candidates for Bortezomib drug delivery. A broad range of applications has been reported for self-assembling peptides;36,37 moreover, most cyclic and protected dipeptides yield excellent hydrogels38–42 with a large surface area, high water content and adjustable pore size. The potential of self-assembled peptides as carriers for cancer drug delivery has been recently reviewed by Tamamis and Gazit,43 while Pu et al.44 have used a peptide-based supramolecular hydrogel for the delivery of Bortezomib.
In the present study, we examined the propensity of Bortezomib to self-assemble using all-atom molecular dynamics simulations and the corresponding experimental results. Furthermore, following previous simulation works related to a systematic investigation of the structural and conformational properties of biocompatible aliphatic–aromatic dipeptides,45 as well as, experimental studies on hydrogel-forming aromatic dipeptides,37,46–52 here we investigate different dipeptides as potential candidates for controlled release of Bortezomib. To our knowledge, the self-assembly of Bortezomib in water is examined computationally for the first time. Moreover, its tendency to bind to dipeptides of similar chemistry, with hydro-gelating capacities, is investigated. Cyclo-leucine-phenylalanine (Cyclo-LF) dipeptides and their linear analogs leucine-phenylalanine (LF) are juxtaposed with two types of protected diphenylalanine (FF) dipeptides, N-(carboxybenzoxyl)-L-diphenylalanine (Z-FF) and N-(fluorenylmethyloxycarbonyl)-L-diphenylalanine (Fmoc-FF), in a way that they associate with Bortezomib. The thermodynamic stability of the complexes formed in an aqueous environment, as well as key morphological features of the nanoassemblies are investigated at the molecular level. Our aim is to obtain quantitative predictions of conformational and structural properties of the Bortezomib-dipeptide complexes. Energy calculations and the kinetics of the complexation process also provide critical information. At the same time, the evaluation of the various properties will indicate the degree of efficacy for the dipeptides in order to be considered as potential candidates for Bortezomib's controlled release.
The rest of the paper is divided as follows: Section II presents the model systems and simulation details; Section III comprises the results divided into two main subsections, where reference systems of solutions including only dipeptides or Bortezomib and solutions with both the drug and dipeptides are discussed. Further division according to the properties under investigation is applied. Section IV follows with discussion and concluding remarks.
2. Systems and methods
2.1. Description of the systems and simulation details
We have examined four different dipeptides as potential complexation agents with Bortezomib (BTZ). All model dipeptides are derivatives of the phenylalanine peptide varying in the identity of the first or second amino acid, the architecture and the N-terminal modification (i.e., protection group). In more detail, we have studied Leucine-Phenylalanine (Leu-Phe), Cyclo(-Leucine-Phenylalanine-) (Cyclo(-Leu-Phe)), fluorenylmethyloxycarbonyl (Fmoc)-protected diphenylalanine (Fmoc-FF) and carboxybenzyl-protected diphenylalanine (Z-FF). Aqueous solutions of the above dipeptides with Bortezomib in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 composition ratio have been simulated in a cubic simulation box under ambient conditions (T = 300 K and P = 1 atm). The concentration (c) is 38 mg (dipeptide)/ml(solvent) for all systems whereas details for the model systems are presented in Table 1.
:1 composition ratio have been simulated in a cubic simulation box under ambient conditions (T = 300 K and P = 1 atm). The concentration (c) is 38 mg (dipeptide)/ml(solvent) for all systems whereas details for the model systems are presented in Table 1.
| System | #Molecules | # Water molec. | # atoms | LBOX(nm) | |
|---|---|---|---|---|---|
| Reference systems | |||||
| BTZ | 100 | 18530 |
59790 |
8.50 | |
| LF | 80 | 32011 |
98353 |
9.97 | |
| Cyclo-LF | 80 | 30060 |
92420 |
9.77 | |
| Z-FF | 80 | 51085 |
157415 |
11.70 | |
| FmocFF | 80 | 62375 |
192165 |
12.45 | |
| Complex systems | |||||
| BTZ – LF | 50 | 50 | 42344 |
130582 |
10.96 |
| BTZ – Cyclo-LF | 50 | 50 | 46146 |
141938 |
11.27 |
| BTZ – Z-FF | 50 | 50 | 57856 |
178318 |
12.15 |
| BTZ – FmocFF | 50 | 50 | 48379 |
148987 |
12.67 |
Fully atomistic simulations were performed using the GROMACS software.53 Solvent was described explicitly through the single point charge SPC/E water model.54 All molecules are modelled through the GROMOS54a7 force field55–57 using the Automated Topology Builder (ATB),32,57–60 which is a tool used for generating force field parameters for molecules and molecular complexes.
Non-bonded interactions were parameterized through a spherically truncated 6–12 Lennard–Jones potential and standard Lorentz–Berthelot mixing rules with a cut-off distance of 1 nm. The particle-mesh Ewald (PME) algorithm was used for the evaluation of the electrostatic interactions. The cut-off distance for PME was 1 nm, the PME-order was 4 and the Fourier spacing was 0.12. The Berendsen barostat61 and the stochastic velocity rescaling thermostat62 were used and the integration time step was 1.0 fs. After the solvation of peptide molecules in water, the starting configuration was energy minimized, while prior to the production runs all systems were equilibrated for at least 50 ns. Production runs of 100 ns followed.
2.2. Analysis methods
In order to monitor the assembly propensity of dipeptides as well as their association with Bortezomib and to characterize the formed aggregates, appropriate analysis methods were utilized. In this context, standard analysis tools of the GROMACS package53 were employed. Quantities like pair radial distribution functions (rdf), density and charge distributions as well as various structural features were investigated. Binding energies and hydrogen bonding were also calculated.For the investigation of cluster formation and the exploration of their characteristics a home-made script was developed based on the procedure described in ref. 63,64, which is outlined as follows. Initially the center of mass of each molecule (CM) is determined and considered as a potential center of a cluster (cc). The condition that was checked in order to determine whether a neighboring molecule belonged to the cluster centered at the selected (cc) was whether the CM–CM distance between the two molecules was smaller than a critical distance. The selection of this critical distance was the point where the dipeptide–dipeptide or the dipeptide–Bortezomob center of mass pair radial distribution function attained the value one (see Section 3.2.1, Fig. 6). Each time a molecule was found to belong to a cluster, it was eliminated from the pool of the system's molecules and the algorithm was repeated for the remaining particles until the pool of molecules was empty. In this way a series of non-overlapping clusters were obtained and each molecule has been encountered only in one cluster. The number of clusters and the average number of molecules per cluster were recorded. Note here that there are many algorithms for clustering and as expected a quantitative comparison in the number of clusters formed and their population will differ slightly, according to the criteria applied. However, the focus of the current analysis is mostly on the process and the result of assembly of molecules (clustering) in an aqueous environment, which becomes obvious even by optical observations.
Energetic calculations related to the estimation of the enthalpy of association were performed using the molecular mechanics/Poisson Boltzmann surface area (MM/PBSA) method, as implemented in the g_mmpbsa GROMACS utility tool,65 which uses the APBS solver66 for computing the polar part of the solvation free-energy. The entropic contributions were estimated through an in-home script, based on the procedure described by Duan et al.67
3. Results
The hydrophobic nature of parts of dipeptides, such as the phenyl rings, is expected to lead to the formation of peptide aggregates in an aqueous environment. It is therefore interesting to explore (a) the self-assembly propensity of dipeptides in water and how the presence of Bortezomib molecules affects the clustering behavior; (b) whether the characteristics of the formed complexes render the examined dipeptides as potential agents for drug delivery purposes and (c) the main driving forces for the formation of complexes. Therefore, we start with a brief investigation of the properties of Bortezomib and of the dipeptides individually in water (reference systems).3.1. Reference systems
Initially in order to examine the behavior of each dipeptide or of Bortezomib in aqueous solution, we analyze the simulations of the “reference systems” (see Table 1). We should note that LF and Cyclo-LF aqueous solutions have been studied in our previous publication45 as well; some results are used here for comparison purposes. Moreover, the Fmoc-FF and Z-FF hydrogel forming propensity has been previously established using thorough theoretical and experimental studies.68–71The chemical structures of all five molecules are presented in Fig. 1. Note that the drug substance (Bortezomib) has great similarity in terms of its chemical formula with the four peptides, which enhances the affinity between the two entities and facilitates their complexation, as we will discuss in detail in the next subsection.
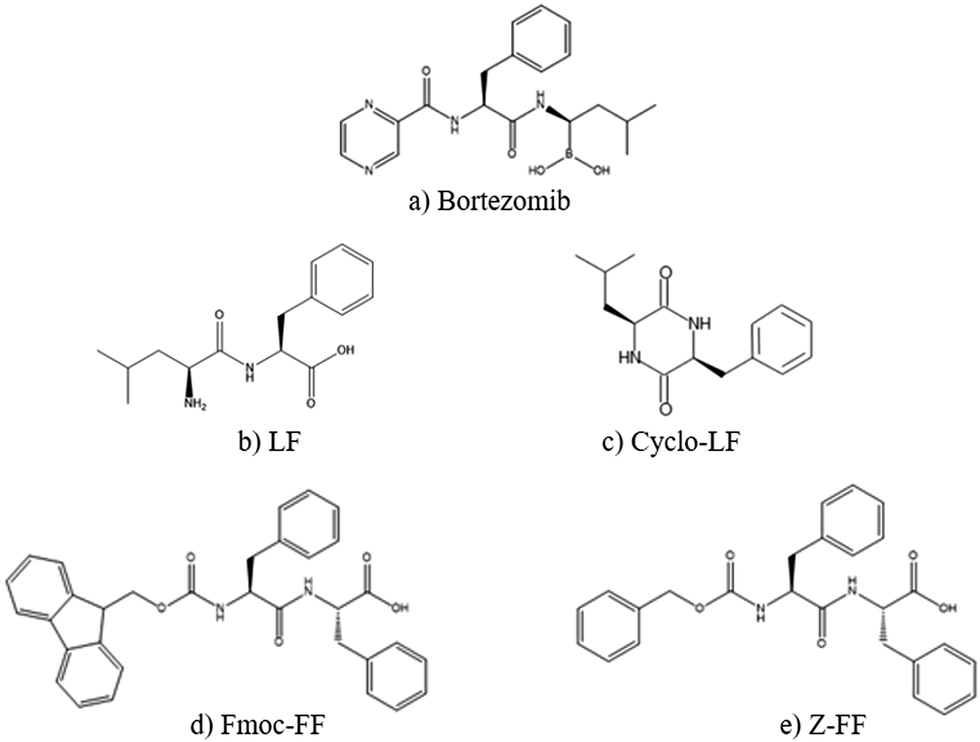 | ||
| Fig. 1 Chemical structures of all molecules examined in the present study. (a) Bortezomib; (b) LF; (c) Cyclo-LF; (d) Fmoc-FF; and (e) Z-FF. | ||
Information at the molecular level concerning the local structure of the dipeptides in water is given by the pair radial distribution function (rdf), g(r). The rdfs have been calculated between the centers-of-mass of dipeptides in all systems and the results are presented in Fig. 2. A comparison of rdf curves among the four dipeptides indicates the strongest self-assembly propensity in the Cyclo-LF as it is revealed from the highest peak. Rdfs' peaks for Fmoc-FF and Z-FF follow, whereas for LF the peak is considerably lower. The width of the peaks also varies among the dipeptides with the one of LF being the narrowest, indicating a considerably smaller size for the formed aggregates. A similar extent of the curves is observed for Fmoc-FF and Cyclo-LF, while for Z-FF it is somewhat narrower. For Bortezomib, the high peak in g(r) at short distances signifies its tendency for self-assembly in aqueous solution as well. The peak is shorter compared to the four dipeptides, but at the same time broader, indicating the formation of large aggregates.
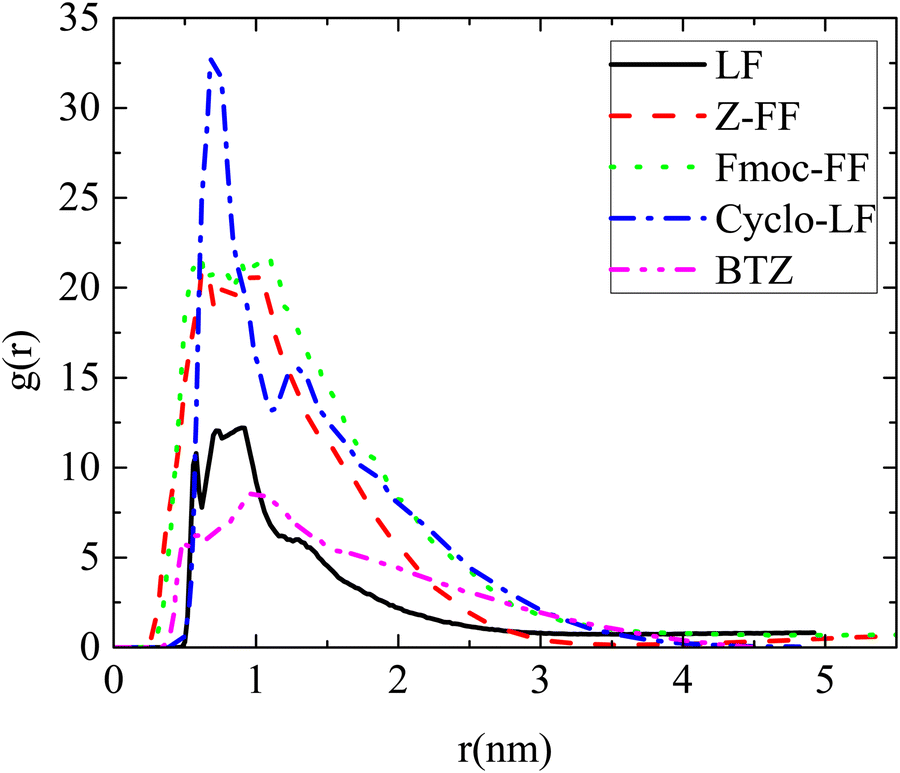 | ||
| Fig. 2 Pair radial distribution functions between the centers of mass of molecules for all reference (single component) systems. | ||
Since hydrophobicity induces self-assembly of dipeptides in water, clusters of various sizes are formed. The time evolution of this process has been recorded throughout the trajectory based on the algorithm described in Subsection 2.2 for cluster detection.
In all cases the number of the formed clusters (Ncl(t)) is a decreasing function of time, which converges to a plateau after a certain time, different for each system as presented in Fig. 3. Beyond this time, fluctuations around a fixed value are observed since a continuous restructuring of the clusters through splitting and merging occurs. Furthermore, Fig. 3 quantifies the kinetics of the self-assembly, which is quite fast for all dipeptides. For Cyclo-LF beyond ∼75 ns the number of clusters fluctuates around one, for Z-FF and Fmoc-FF fluctuations around the three and two cluster states are observed beyond ∼100 ns, whereas, in LF solution, smaller groups of dipeptides are formed and the number of clusters fluctuates around 9 beyond ∼130 ns.
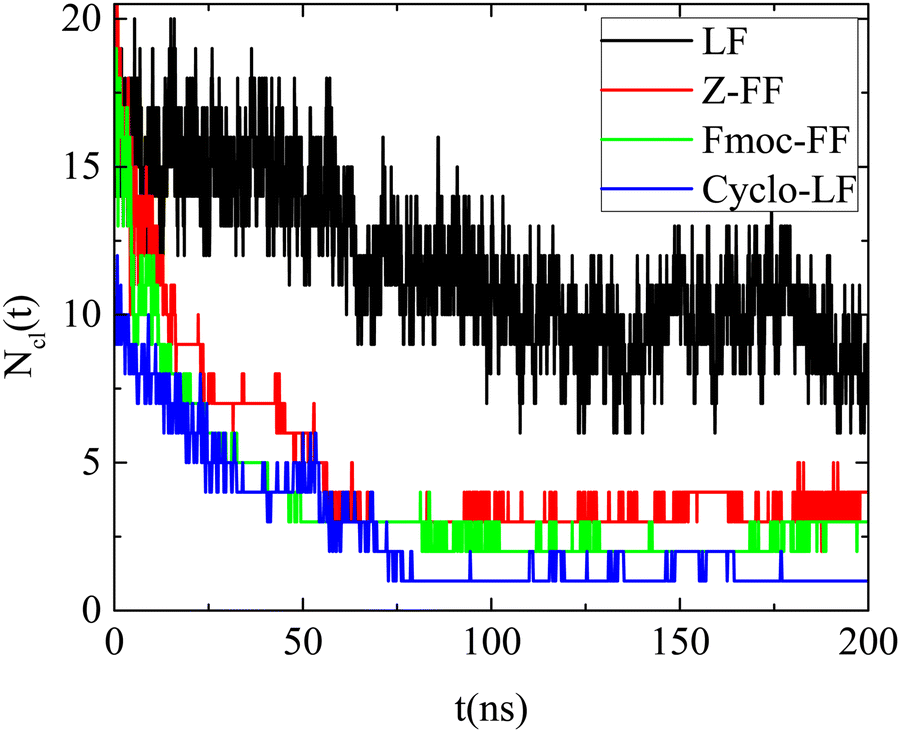 | ||
| Fig. 3 Number of clusters formed by the dipeptides as a function of time, in the examined reference (single component) systems. | ||
Histograms of cluster population are created averaging over the part of the trajectory where the clusterization process has reached to a “steady state” (i.e. beyond the time defined from Fig. 3). Fig. 4 contains the probability distributions of population within each cluster, (P〈h(t)〉), formed by dipeptides, as a function of number of molecules each one contains. Except for small groups of dipeptides (i.e., up to 10 molecules), clusters of two different populations are the most probable in Fmoc-FF, ranged in the intervals (10–28) and (50–68). For Z-FF a broad distribution in the population of clusters with occasionally stronger peaks are observed, while rarer cases of larger clusters as well as clusters consisting of few dipeptides also exist. Two well-separated populations are detected in Fig. 4 for Cyclo-LF, one in the range (1–10) indicating the case of small groups of dipeptides in aqueous solution or even individual molecules, and the other in the range (65–78) showing a significant possibility of formation of a single or a two cluster state. For LF aggregation of few peptides is the most probable case, with bigger groups of ∼15 or ∼25 peptides to be rarely formed.
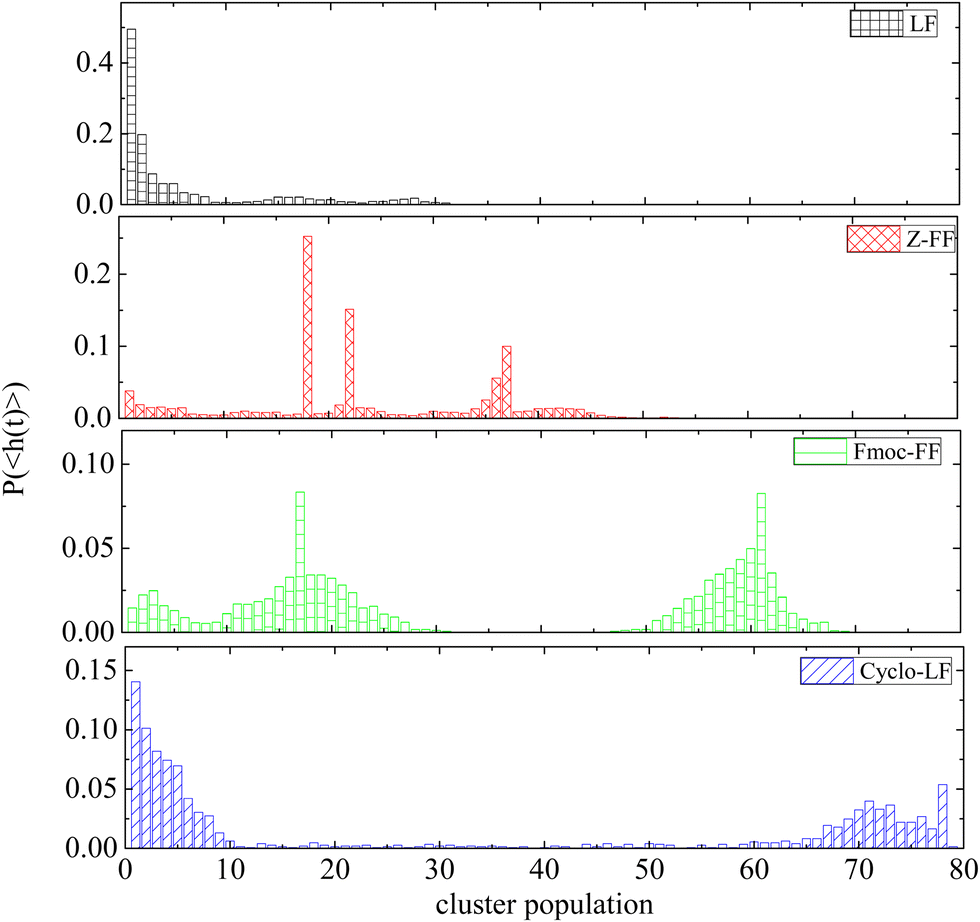 | ||
| Fig. 4 Probability distribution of population within each cluster, as a function of the number of molecules each one contains. | ||
Similarly, we explored the self-assembly propensity of Bortezomib in water, which according to Fig. 5 is faster compared to that of the four dipeptides. A rapid self-assembly process leads to the formation of a single cluster of all Bortezomib molecules. After ∼45 ns (Fig. 5a), the number of clusters varies between one and two, with the case of two being possibly one big cluster and single molecules, which are attached to and detached from the cluster spontaneously or aggregates of very few molecules according to Fig. 5b.
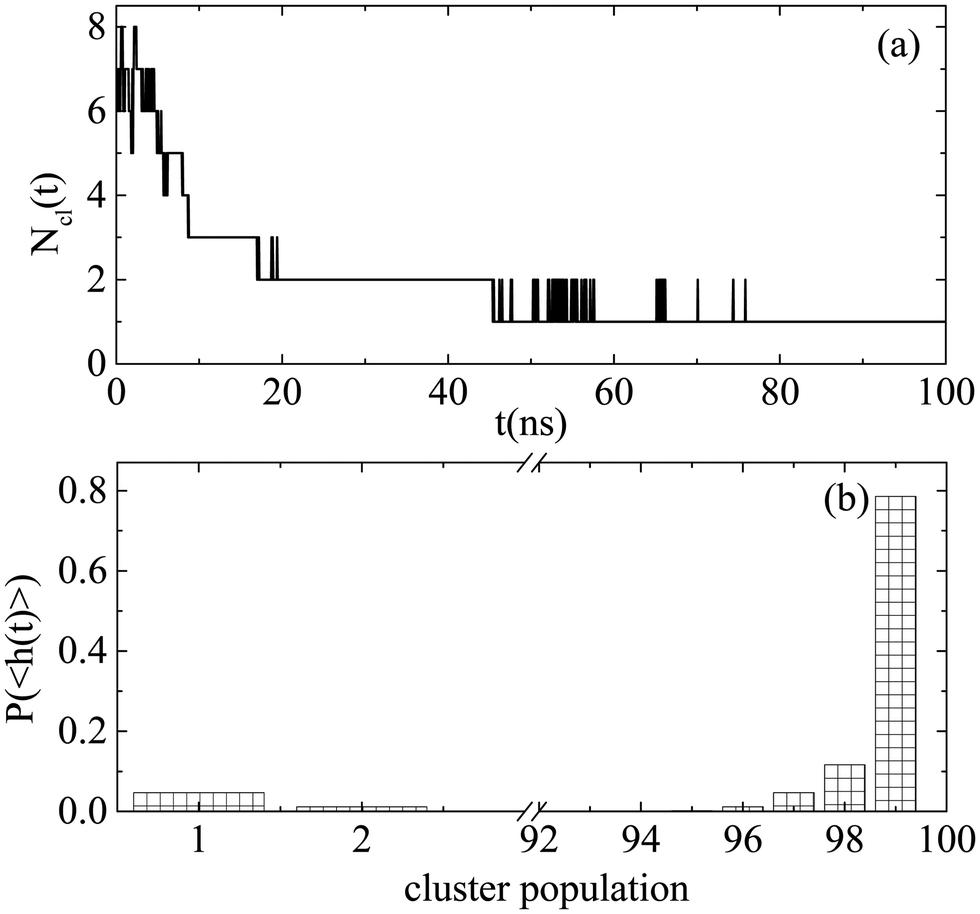 | ||
| Fig. 5 (a) Number of clusters formed by Bortezomib molecules as a function of time. (b) Probability distributions of population within each cluster as a function of number of Bortezomib molecules each one contains. | ||
Characteristic snapshots of self-assembled structures for all systems are presented in Fig. S3 (ESI†), where differences in size are also observed. Quantification of the size of the formed clusters is provided through the calculation of their radius of gyration. Based on the histograms of Fig. 4, the configurations of the most frequently encountered clusters of big size (i.e., beyond 10 molecules) are recorded. The radii of gyration of these clusters and the corresponding number of molecules they contain, expressed as a percentage of the total number of dipeptides in solution, are reported in Table 2. The radius of gyration of the individual molecules (〈Rg〉MOL) is also included. Almost all Bortezomib molecules contribute to the formation of the largest clusters. Highly populated are also the Fmoc-FF and Cyclo-LF clusters, whereas in Z-FF more clusters of less dipeptides are the most frequent. In LF the large clusters contain even fewer dipeptides and are significantly rarer. According to these recordings the size of the largest clusters in nm follows the decreasing order: BTZ > Fmoc-FF > Cyclo-LF > Z-FF > LF. Therefore, the largest clusters are between ∼(1.5–2.0) nm in all systems, however it is not only the population that determines their size, but the extent of the molecules involved in each case and the compactness of the clusters, parameters that will be further discussed in the next subsection.
| Systems | Number of molecules (∼most probable max) | 〈Rg〉 (nm) (∼most probable max) | 〈Rg〉MOL (nm) | 〈HBpep–pep〉/dipep. |
|---|---|---|---|---|
| BTZ | 99/100 | 1.9 ± 0.02 | 0.425 ± 0.001 | 0.84 ± 0.07 |
| LF | 31/80 | 1.4 ± 0.05 | 0.321 ± 0.001 | 0.52 ± 0.08 |
| Cyclo-LF | 78/80 | 1.7 ± 0.03 | 0.306 ± 0.002 | 1.11 ± 0.07 |
| Z-FF | 37/80 | 1.5 ± 0.16 | 0.472 ± 0.003 | 0.88 ± 0.08 |
| Fmoc-FF | 60/80 | 1.8 ± 0.15 | 0.513 ± 0.003 | 0.46 ± 0.07 |
Hydrogen bonds between dipeptides have also been calculated and their average number per molecule over the last part of the trajectory, where a “stabilized” state has been achieved is presented in Table 2. A big number of hydrogen bonds are formed between Cyclo-LF dipeptides, which are followed by the corresponding numbers between Z-FF and between BTZ. For LF the attenuation of hydrogen bonding is expected because of its reduced self-assembly propensity. However, it is interesting to observe the reduced number of hydrogen bonds between Fmoc-FF dipeptides, despite their strong self-assembly, which leads to cluster formation (Fig. 2–4), rendering hydrogen bonding as not the prominent driving force for clusterization.
3.2. Peptide/Bortezomib complexes
Given the above observed tendency of Bortezomib to self-assemble in aqueous solutions, we sought to experimentally examine the morphology and shape of the formed assemblies on the microscale and in the test tube. No hydrogel formation was observed in the test tube, and only spherical formation (no fibers or tubes) was observed by FESEM under all conditions tested (Fig. S1, ESI†). This precludes the possibility of gel formation by the drug itself and its use for monomer-controlled delivery, as in the case of lanreotide monomer delivery by the Somatuline® autogel. We next considered the encapsulation of the drug within the gel-forming dipeptides examined above. The experimental results are presented in the ESI† (Fig. S2); however, no conclusive results can be drawn from these as to which carrier would be optimal for Bortezomib encapsulation and subsequent release. Hence, the optimal gel-forming dipeptide carrier has to be selected from the quantitative prediction of conformational, structural, dynamic and energetic properties of the Bortezomib–dipeptide complexes. Therefore, the following analysis explores the complexation of the drug within the gel-forming dipeptides examined above, in order to assess the possibility of using them as scaffolds for delivery, based on theoretical insights.In what follows, the complexation of Bortezomib with each of the four dipeptides in aqueous solution is investigated. Various measures are calculated providing an evaluation of the aforementioned molecules as potential nanocarriers for the drug substance through an effective complexation with this. The affinity of the two components is initially expected as a result of the hydrophobicity of both moieties. Even more the chemical structure of Bortezomib is partly similar to that of the dipeptides, which is also beneficial for their assembly. However, other interactions also exist and the exploration of their combined effect will provide an important piece of information to suggest the most effective of the dipeptides.
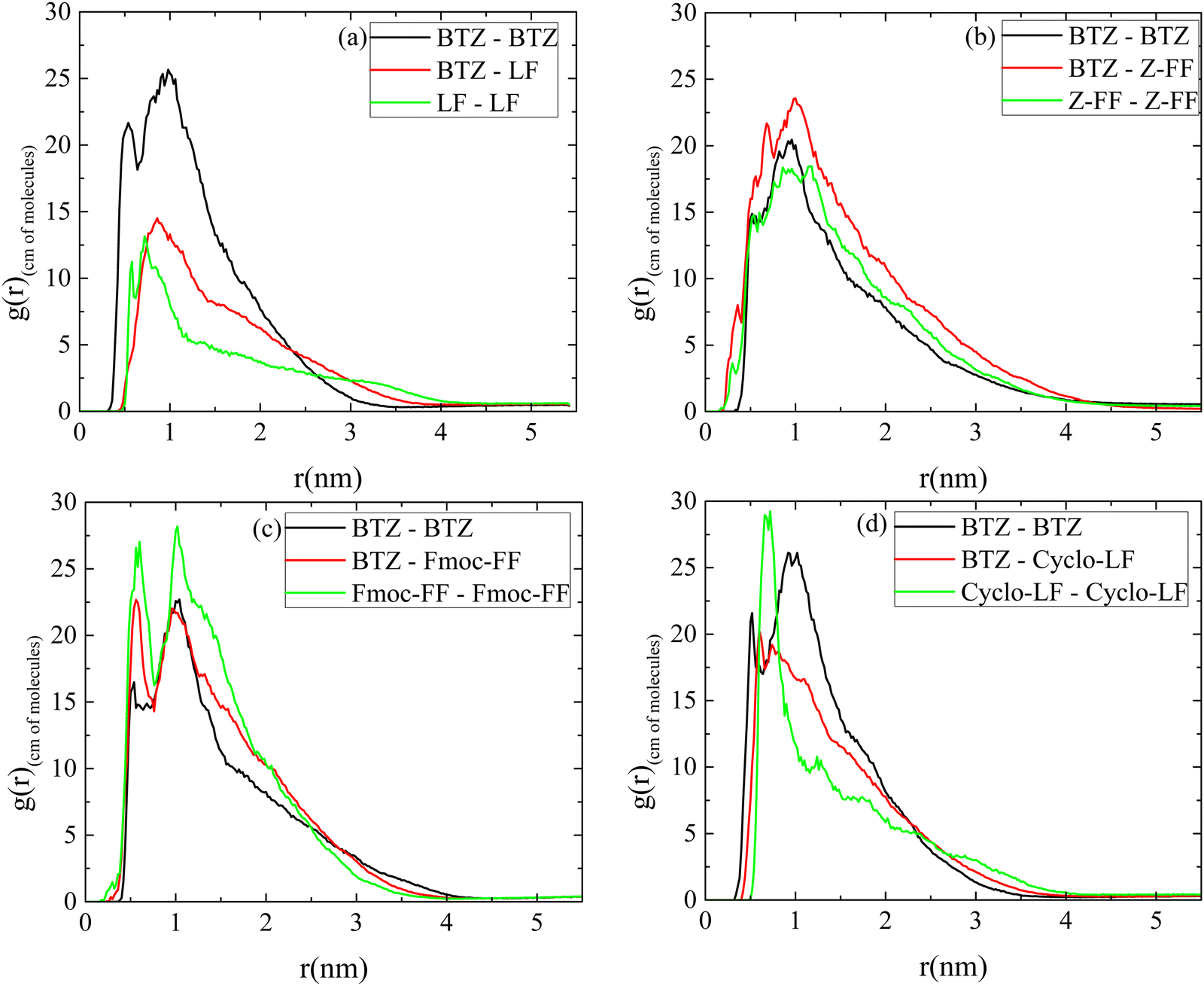 | ||
| Fig. 6 Pair radial distribution functions between the centers of mass of molecules for the pairs: Bortezomib–Bortezomib; dipeptide–dipeptide and Bortezomib–dipeptide in the aqueous solutions at T = 300 K in all systems: (a) LF system; (b) Z-FF system; (c) Fmoc-FF system; and (d) Cyclo-LF system. | ||
A comparison among BTZ–BTZ rdfs in the four systems is presented in Fig. S4a (ESI†), where in LF and Cyclo-LF solutions curves are rather similar, while a shorter peak and longer extension is observed in Z-FF and Fmoc-FF solutions. BTZ-dipeptide rdfs are compared in Fig. S4b (ESI†), where all four curves are peaked at the same distance of ∼1 nm, with that of BTZ–LF being the shorter, BTZ–Cyclo-LF follows, while BTZ–Z-FF and BTZ–Fmoc-FF rdf curves are similar, higher and broader than the other two. Overall, the rdfs indicate a tendency for all dipeptides to bind to Bortezomib, leading to clusters, which may vary in the number of dipeptides included, size and compactness.
Therefore, we proceed with a cluster analysis in order to address these features. Significant variations in cluster sizes are observed across all four systems, with broad distributions evident. Fig. 7 displays probability distributions showing how the number of molecules, encompassing both dipeptides and Bortezomib, relates to the population within each cluster. In all cases a considerable population of single or few molecule groups exist. However, crowded clusters (i.e., which include more than 10 molecules) obviously exist and big clusters of varying sizes are formed. In LF solution (Fig. 7a) the probability for big clusters is found at ~16, with a second peak at ~27. In Cyclo-LF (Fig. 7d) one high peak is detected in the range of ∼(18–28) and another one in ∼(45–60). In Z-FF solution populations are more distinct. In Fig. 7b, except for the small group (up to 10 molecules), population in the range of ∼(87–100) signifies the formation of a single big cluster of almost all molecules in the solution. For Fmoc-FF (Fig. 7c) increased population is met beyond 10, a second high peak is observed in the range of ∼(58–80), with intermediate states more sparsely populated, indicating the two-cluster state as very possible.
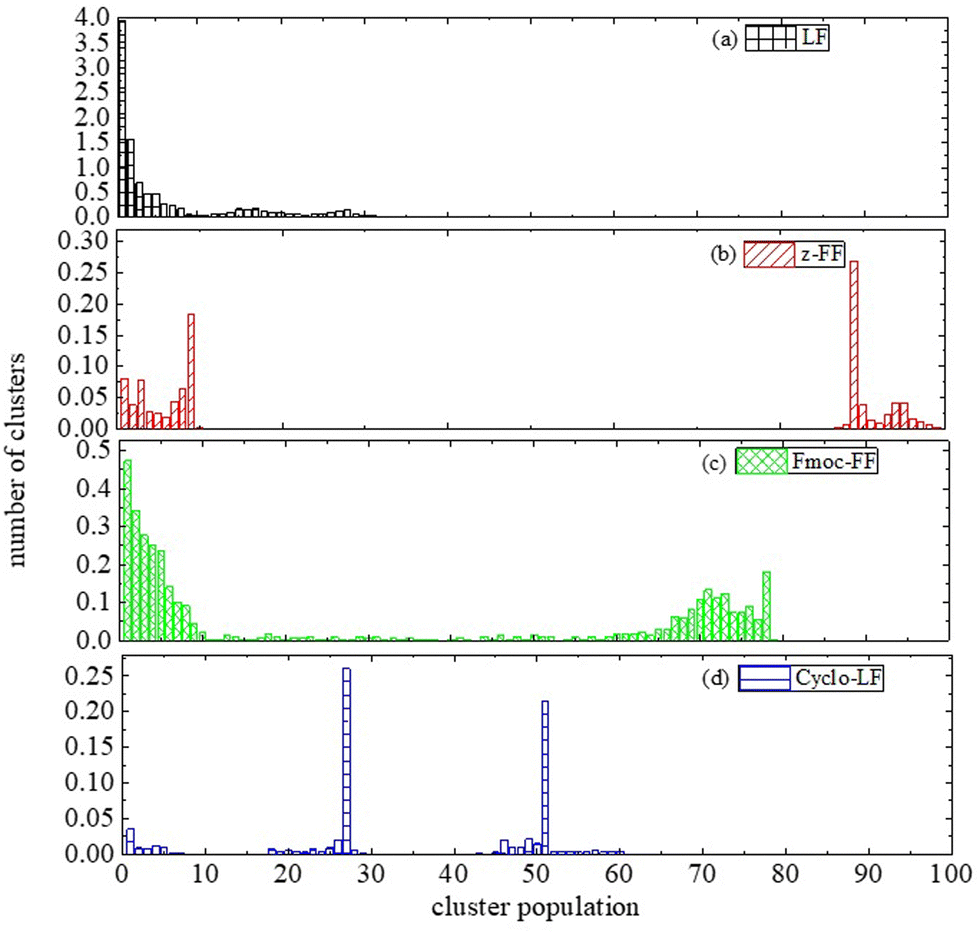 | ||
| Fig. 7 Probability distribution of population within each cluster formed by dipeptide and Bortezomib molecules, as a function of number of molecules each one contains. | ||
We have to mention here that the values given above are expected to be sensitive to the algorithm for cluster analysis (i.e., used criteria). However, the given intervals for clusters’ population are quite broad, providing a tolerance, which can cover differences among cluster definitions. Furthermore, self-assembly is a stochastic process and quantitative differences in the probability distribution of population can be observed among different pathways, yet qualitative trends toward the formation of larger or smaller clusters are revealed. Nevertheless, the main finding of this study is a comparative validation of dipeptides for their complexation with BTZ.
Information for the compactness of the formed clusters is given by the calculation of the density of atoms measured as a function of the distance from their center, as presented in Fig. 8. High density values are found in Cyclo-LF and LF, a bit higher for the former, with the peaks centered at the same position. For Z-FF the density peak is similar to that of LF but it is moved at longer distances, indicating a less compact interior of the clusters. Finally, a considerably looser structure is demonstrated from the much lower peak in the density profile of Fmoc-FF, which is centered at the same distance as that of Z-FF. Moreover, the extension of the curve at longer distances indicates a bigger average size for the formed clusters. This can be attributed to the protective side group, which in both Z-FF and Fmoc-FF imposes larger distances between atoms in their conformations. Even more pronounced is the case of Fmoc-FF where the three-ring shape of the protective group is of reduced flexibility in the way that it packs. This is an important observation because the more compact the formed structures the less water content they entrap and the less likely it is to be gel-like. This information is strongly related to the desired application of the potential drug nanocariers under investigation. Therefore, according to Fig. 7 and 8, a kind of classification of dipeptides as the protected FF and the LF-based can be assigned, rendering the former class of dipeptide-Bortezomib clusters larger in size and less compact.
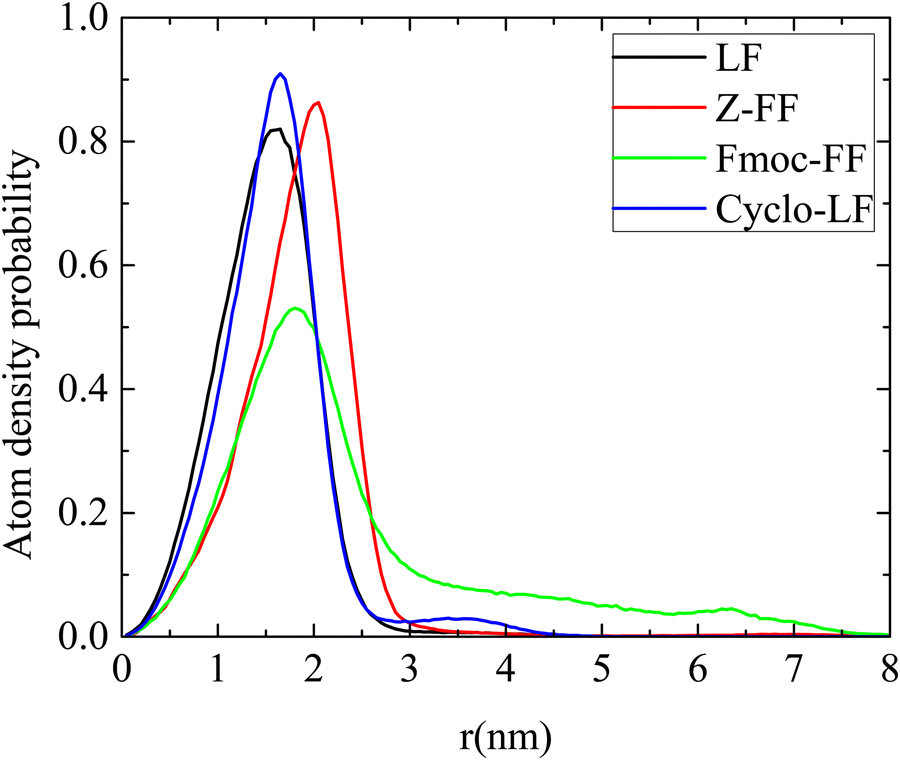 | ||
| Fig. 8 Atom density probability of clusters as a function of the distance from their center. | ||
The amount of water content inside the cluster is quantified in Fig. 9a through the calculation of the water mass density as a function of the distance from the center of the cluster. In agreement with the density profiles there is not any water content in the interior of LF and Cyclo-LF clusters up to ∼1 nm from their center. Beyond this distance a gradual increase of the water content is found reaching its bulk density at long distances. More waterproof are the Z-FF clusters which are impermeable to water up to ∼2 nm. Substantial differences are observed in the Fmoc-FF systems, where a small amount of water is entrapped in the interior of the formed clusters as a result of its looser structure. In Fmoc-FF clusters a constant amount of water is contained up to ∼1 nm, whereas beyond this distance density of water increases gradually up to 1 g cm−3.
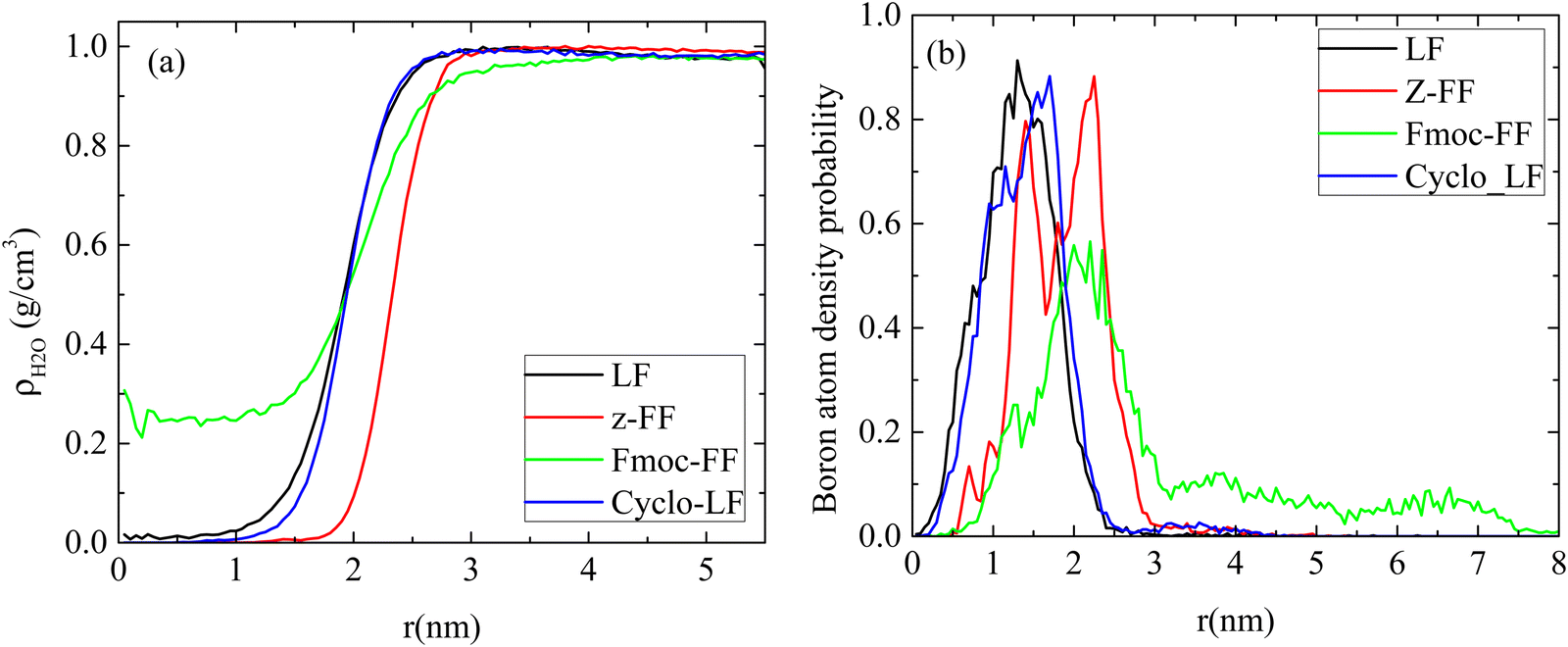 | ||
| Fig. 9 (a) Mass density of water, (b) atom density probability of boron as a function of the distance from the center of the clusters. | ||
Furthermore, boron plays a crucial role in this complexation. Boron binds to threonine and inhibits the proteasome (conjugation of electrons: empty p-orbital of boron with oxygen of threonine). In order to achieve this function boron has to be found close to the surface of the cluster. The atom density of boron as a function of the distance from the center of the cluster is presented in Fig. 9b. A small proportion of boron atoms is observed at long distances (i.e., around the surface of the cluster) whereas the majority of them are found at ∼1 nm from the center, for LF and Cyclo-LF, according to the total atom density distribution. In the Fmoc-FF system the atom density peak is at ∼2 nm whereas a considerable amount of boron atoms is extended at longer distances, while in Z-FF two peaks are detected at ∼1.5 nm and ∼2.25 nm, respectively.
A clearer picture for the arrangement of atoms within the volume of the cluster is presented in Fig. 10 for all systems. Atom density probability of the various components included in the system as a function of the distance from the center of the cluster is shown for dipeptide, Bortezomib, water and boron atoms. The concentration of Bortezomib molecules and consequently of the boron atoms on the surface of the cluster is clear only in the system with Fmoc-FF, while there is almost an equal probability of finding Bortezomib and Z-FF molecules on the surface. In both the LF and Cyclo-LF systems, the Bortezomib molecules are trapped by the corresponding dipeptide molecules in the interior of the formed clusters. Therefore, Fig. 9 and 10 point to a further classification within the protected FF dipeptides, showing Fmoc-FF as the most water permeable with more boron atoms exposed on the surface of the cluster.
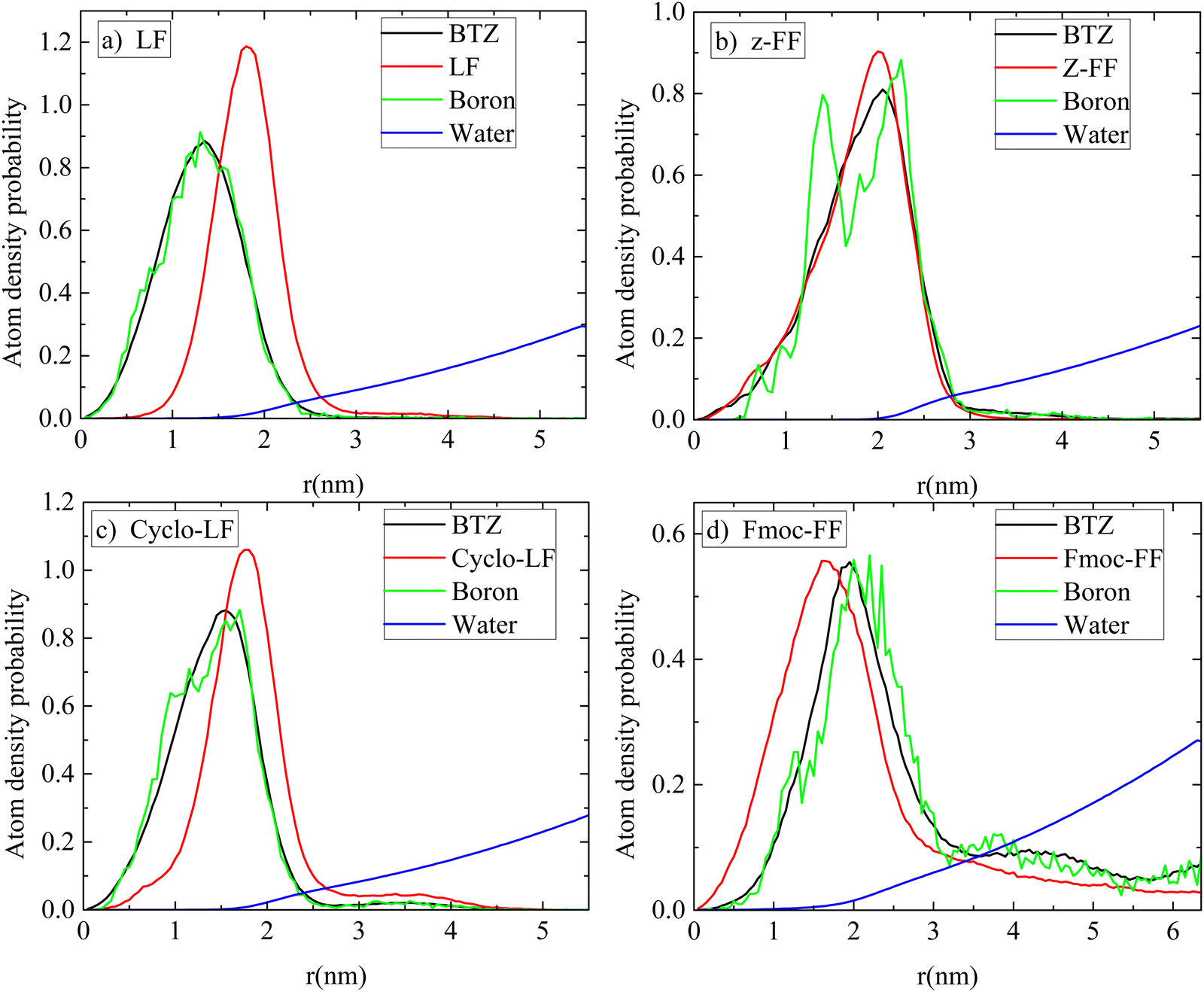 | ||
| Fig. 10 Atom density probability of peptides, BTZ, boron atoms and water as a function of the distance from the center of the cluster (a) LF; (b) Z-FF; (c) Cyclo-LF; and (d) Fmoc-FF. | ||
Characteristic snapshots of all four complexes are presented in Fig. 11, where water molecules have been omitted for clarity. Bortezomib is presented with red and dipeptide with blue. Clusters of various sizes are observed, which are classified in one or two clusters case with few individual molecules dispersed in the solution as well. Since the molecules that make up the clusters have different dimensions, the size of the complexes varies accordingly. An interesting observation concerns the LF system, where although the dipeptide presents a weaker tendency to self-assemble in water (Fig. S3, ESI†), its aqueous solution with BTZ leads to the complexation of the two components and to the formation of clusters.
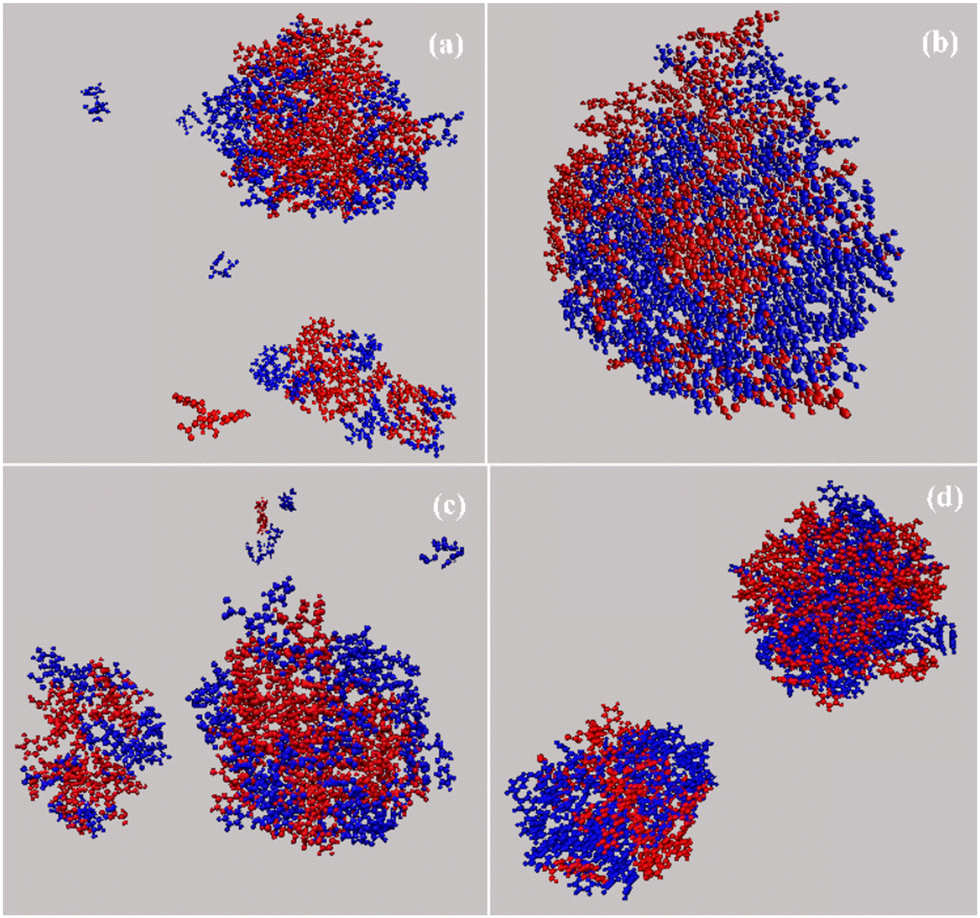 | ||
| Fig. 11 Characteristic snapshots of dipeptide–BTZ complexes (a) LF; (b) Z-FF; (c) Cyclo-LF; and (d) Fmoc-FF. Bortezomib molecules are presented in red and dipeptides in blue color. Water molecules are omitted for clarity. | ||
Binding energy. The complexation of the two molecules is determined by the energetic interactions between all the components in the aqueous solutions. The result is a combined effect of electrostatic interactions, hydrogen bonding and hydrophobicity. An important contribution to the energy quantification is provided by the calculation of the free energy of association between the dipeptide and Bortezomib. The calculation of the binding free energy can be realized through various computational approaches.65,72–77 Here we use the MM/PBSA approach75,78 in conjunction with the interaction entropy method67,79 for the calculation of the enthalpic and the entropic contributions, respectively. All the MD simulation data used for energetic calculations correspond to the last 20ns of the trajectory, within which the systems are in a “stabilized” conformational state. The variation of the enthalpic part (ΔH) of the association free energy between Bortezomib and peptide molecules per BZT molecule as a function of time is presented in Fig. 12. The lowest energy indicates the energetically most stable association. This is observed between BTZ and the two protected versions of FF (Fmoc-FF and ZF), with the attraction Z-FF even stronger. Then, the Cyclo-LF system follows, with higher enthalpy values, while for LF the attraction with Bortezomib is further attenuated.
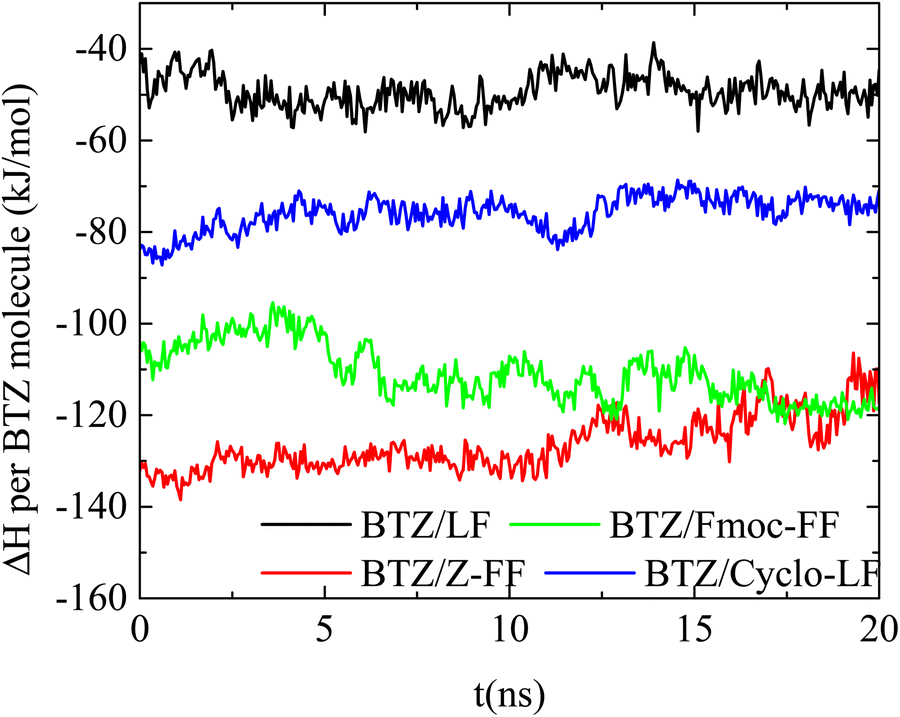 | ||
| Fig. 12 Binding enthalpy per BTZ molecule as a function of time between the dipeptides and the BTZ. | ||
The corresponding entropic contributions in the associated free energies have been calculated and are presented in Table 3, together with the different energetic components involved in the calculation of the enthalpy of association. The interaction entropy method67,79 is based on the calculation of the fluctuations of the interaction energy around its average value. This means that the more the fluctuation of the interaction energy, the greater the entropic loss in the binding free energy. For the studied systems, although highly fluctuating, the contribution of the entropy is rather small compared to the enthalpic part of the free energy.
| System | 〈EVdW〉/BTZ | 〈Eelec〉/BTZ | −TΔS/BTZ |
|---|---|---|---|
| LF | −39.15 ± 1.63 | −7.83 ± 2.16 | 6.90 ± 0.89 |
| Cyclo-LF | −55.10 ± 1.99 | −14.61 ± 2.22 | 3.90 ± 3.57 |
| Fmoc-FF | −86.86 ± 1.99 | −14.88 ± 0.97 | 6.50 ± 1.01 |
| Z-FF | −91.73 ± 2.15 | −19.93 ± 1.16 | 4.71 ± 2.99 |
Interestingly enough, the contribution of the van der Waals components is higher than that of the electrostatic components, although both act favorably towards association. Attraction between BTZ and dipeptides follows the increasing order: LF < Cyclo-LF < Fmoc-FF < Z-FF under both van der Waals and electrostatic contributions. Comparable values of the total free energy of association are derived in Fmoc-FF and Z-FF. The observed difference in Cyclo-LF arises mainly from the contribution of the van der Waals interactions, whereas in LF both electrostatic and van der Waals contributions are considerably smaller.
Hydrogen bonds. Hydrogen bonding constitutes an additional driving force of association. It is also related to the tendency of dipeptide molecules to form clusters (see Section 3.1). Hydrogen bonds are formed among all different molecules in the solution and average values over the last 20ns of the trajectory (stabilized state) are provided in Table 4. The number of hydrogen bonds between BTZ and dipeptide molecules per BTZ molecule follows an increasing order similar to that of the free energy of association: LF < Cyclo-LF < Fmoc-FF < Z-FF, providing a synergistic contribution to clustering between the two energetic components. However, differences are observed in hydrogen bonding between dipeptides and between BTZ molecules among the four systems. A big number of BTZ–BTZ hydrogen bonds are counted in the LF and Cyclo-LF aqueous solutions, while a decrease is observed in Z-FF and even more in Fmoc-FF systems. This is consistent with the entrapment of BTZ by the dipeptide molecules in the case of LF-based dipeptides, in contrast to the protected FF dipeptides where BTZ distribution is observed throughout the clusters, maintaining larger intermolecular distances. Hydrogen bonding between dipeptides is quite low for Fmoc-FF and LF, increases for Cyclo-LF and attains the highest value for Z-FF. These values reflect the arrangement of the molecules in the formed clusters by providing information whether their accessible surface is mainly exposed to themselves or to the cooperating molecule in the cluster. Compared to the corresponding reference systems, the hydrogen bonds between the dipeptide molecules are almost halved due to the intervening BZT molecules. We should note that these data are in close agreement with the tendencies observed by the analysis of the pair radial distribution functions, shown in Fig. 6.
| Systems | (BTZ–BTZ)/BTZ | (BTZ–DIPEP.)/BTZ | (DIPEP–DIPEP.)/DIPEP. |
|---|---|---|---|
| LF | 0.75 ± 0.10 | 0.16 ± 0.08 | 0.294 ± 0.08 |
| Cyclo-LF | 0.67 ± 0.10 | 0.58 ± 0.13 | 0.428 ± 0.08 |
| Fmoc-FF | 0.41 ± 0.09 | 0.69 ± 0.10 | 0.262 ± 0.07 |
| Z-FF | 0.51 ± 0.09 | 0.76 ± 0.10 | 0.524 ± 0.07 |
To further investigate binding modes during self-assembly for the different systems we performed a more detailed analysis of hydrogen bonds that is based on the separation of three active sites on BTZ as follows: (a) OH bonded to boron; (b) NH-groups; and (c) C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups. Hydrogen bonds are counted between each of these groups and the dipeptides. Results for the average number of hydrogen bonds per BTZ molecule are provided in Table S1 in the ESI,† whereas, a graphical representation is presented in Fig. 13. With the exception of LF, which is by all means the least stable case, for the other three dipeptides the NH groups participate in the majority of hydrogen bonds. The contribution of OH active sites is almost equal in all three systems, while the contribution of CO groups to hydrogen bonding is similar for Cyclo-LF and Fmoc-FF, while it is enhanced for Z-FF. Between FF-protected dipeptides the difference of the protective group seems to favor Z-FF over Fmoc-F for the hydrogen bonding between CO active sites of BTZ and the FF part of dipeptide. Overall, the larger number of hydrogen bonds of BTZ with Z-FF compared to Fmoc-FF can be attributed to the more “bulky” and less flexible form of Fmoc-protective group. The difference originates from the contribution of the backbone of the dipeptide (FF part) since no hydrogen bonds exist between the BTZ and Z-/Fmoc- groups. LF-based dipeptides have fewer hydrogen bonding active sites, resulting in fewer hydrogen bonds with BTZ.
O groups. Hydrogen bonds are counted between each of these groups and the dipeptides. Results for the average number of hydrogen bonds per BTZ molecule are provided in Table S1 in the ESI,† whereas, a graphical representation is presented in Fig. 13. With the exception of LF, which is by all means the least stable case, for the other three dipeptides the NH groups participate in the majority of hydrogen bonds. The contribution of OH active sites is almost equal in all three systems, while the contribution of CO groups to hydrogen bonding is similar for Cyclo-LF and Fmoc-FF, while it is enhanced for Z-FF. Between FF-protected dipeptides the difference of the protective group seems to favor Z-FF over Fmoc-F for the hydrogen bonding between CO active sites of BTZ and the FF part of dipeptide. Overall, the larger number of hydrogen bonds of BTZ with Z-FF compared to Fmoc-FF can be attributed to the more “bulky” and less flexible form of Fmoc-protective group. The difference originates from the contribution of the backbone of the dipeptide (FF part) since no hydrogen bonds exist between the BTZ and Z-/Fmoc- groups. LF-based dipeptides have fewer hydrogen bonding active sites, resulting in fewer hydrogen bonds with BTZ.
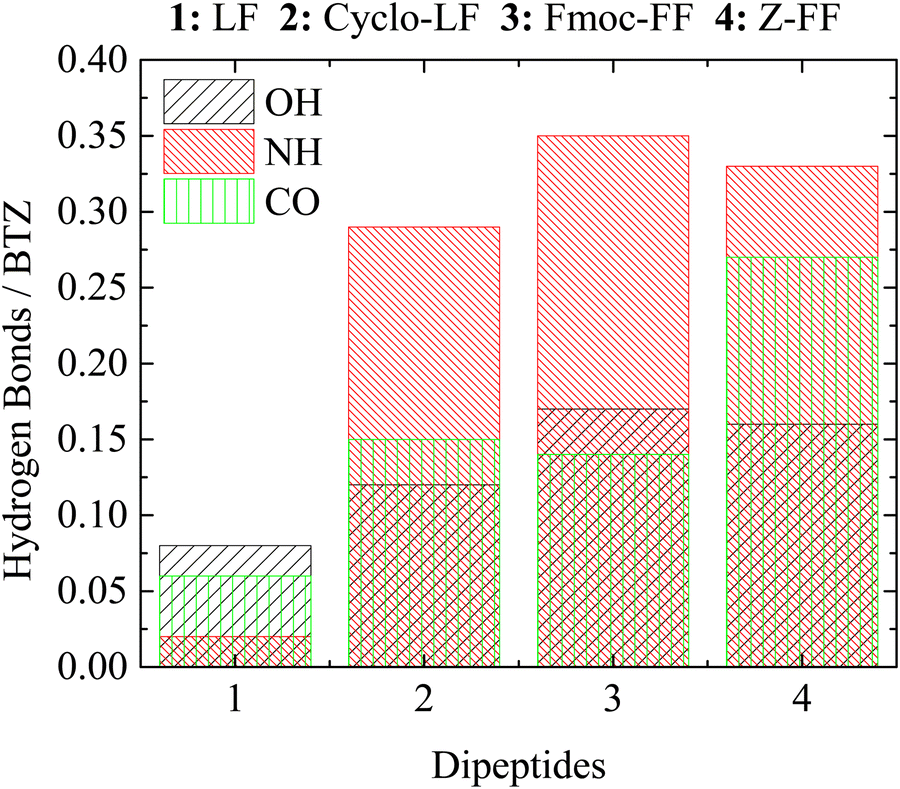 | ||
| Fig. 13 Average number of hydrogen bonds between the three groups of hydrogen bonding active sites of BTZ and dipeptide molecules. | ||
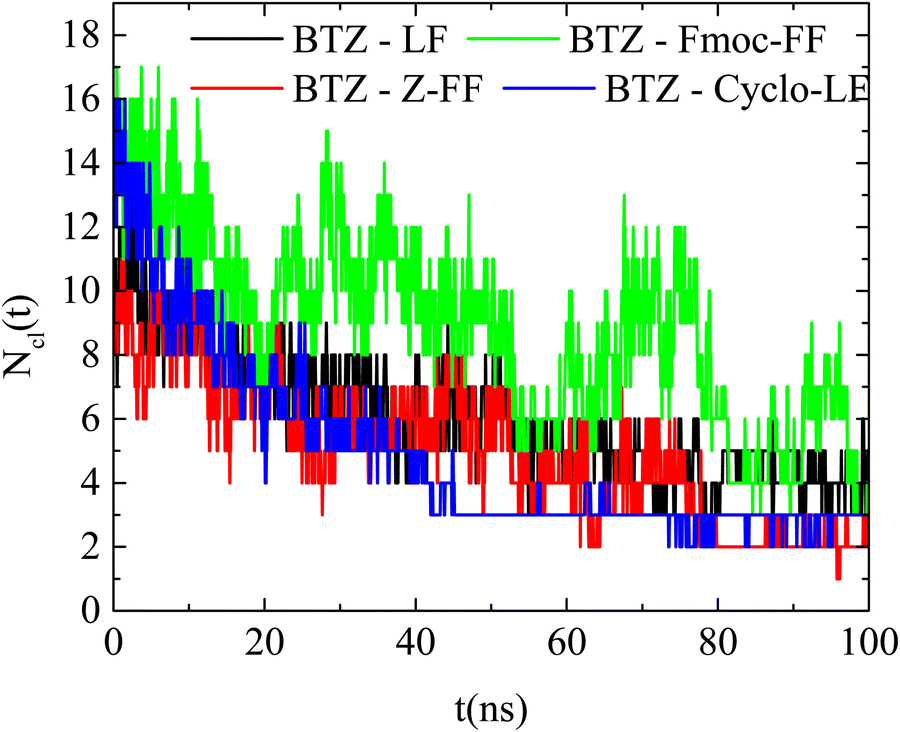 | ||
| Fig. 14 Number of clusters formed by BTZ and dipeptides as a function of time in the examined systems. | ||
Additional information can be extracted from the time dependence of the hydrogen bonding. The number of hydrogen bonds as a function of time starting from the initial configuration, where all molecules are uniformly distributed in water, is presented in Fig. S5 (ESI†). Hydrogen bonds between all pairs of components (i.e., dipeptide–dipeptide, Bortezomib–Bortezomib, and dipeptide–Bortezomib) form very quickly, as hydrophobicity forces the molecules closer to each other. In all cases the number of hydrogen bonds reaches an almost constant value beyond ∼30 ns. Moreover, no preferred sequence is detected in the approach of the molecules, so different processes in the binding of the two components towards the final clusterization cannot be reported. The large fluctuations indicate once again the continuous attachment and detachment of molecules in the formed clusters.
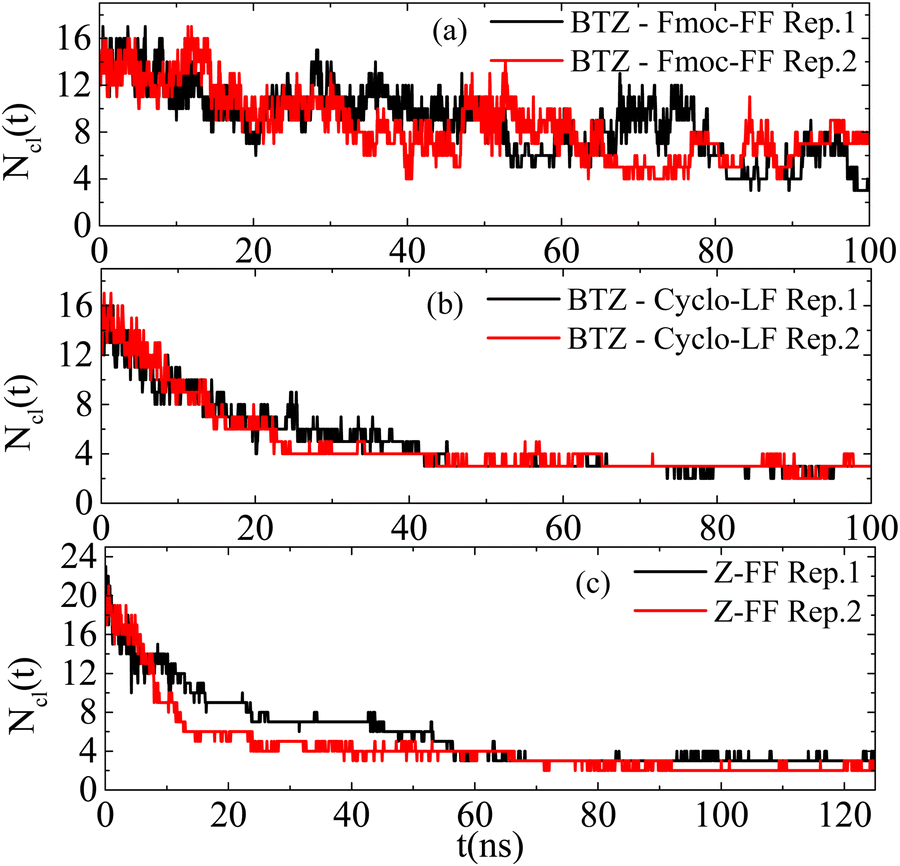 | ||
| Fig. 15 Number of clusters formed by molecules as a function of time in the (a) Fmoc-FF with BTZ; (b) Cyclo-LF with BTZ; and (c) Z-FF bulk systems. | ||
4. Conclusions
In the current work, a number of aromatic and aromatic–aliphatic dipeptides are investigated as potential candidates for the controlled release of the anticancer drug Bortezomib using all-atom molecular dynamics simulations. The self-assembly propensity of Bortezomib in water is explored initially followed by its tendency to bind to dipeptides of similar chemistry. Four dipeptides with reported hydro-gelating capacities have been examined namely Cyclo-leucine-phenylalanine (Cyclo-LF), leucine-phenylalanine (LF), N-(carboxybenzoxyl)-L-diphenylalanine (Z-FF) and N-(fluorenylmethyloxycarbonyl)-L-diphenylalanine (Fmoc-FF). Quantitative predictions of conformational, structural, dynamic and energetic properties of the Bortezomib–dipeptide complexes are revealed. At the same time, the comparison of various measures validates the degree of efficacy of the dipeptides in order to be considered as potential candidates for controlled release of Bortezomib.Starting with the neat systems of aqueous solutions of Bortezomib or dipeptides, self-assembly is observed in all cases as it is expected due to the hydrophobic parts of all molecules. Well-defined spherical clusters of BTZ are observed in water, which are formed very fast (i.e., beyond ∼45 ns, fluctuations around one or two clusters states are detected). This is also confirmed from the corresponding FESEM observations. Comparison among the aqueous solutions of the four dipeptides indicates the strongest self-assembly propensity in Cyclo-LF, Fmoc-FF and Z-FF follow, whereas for LF it is considerably weaker. The self-assembly process is accomplished quite fast for all systems (i.e., in time intervals of the order of ∼100 ns) though slower than that of BTZ. One or few cluster states are observed with the probability distributions of their average size (in terms of population) being more localized for Cyclo-LF and Fmoc-FF and broader for the two others. At the same time a considerable number of big clusters are detected for the two former dipeptides, followed by Z-FF, whereas for LF, groups of fewer dipeptides are formed. However, the order of the strength of self-assembly does not follow the number of hydrogen bonds formed between dipeptides. This suggests the synergistic effect of various driving forces resulting in the formed structures.
In aqueous solutions containing dipeptides and BTZ, pronounced self-assembly phenomena are evident, giving rise to the formation of clusters composed of both dipeptides and BTZ molecules. Through a similar systematic analysis, an initial categorization of dipeptides emerges, distinguishing between the protected dipeptides FF (Fmoc-FF and Z-FF) and the LF-based dipeptides (Cyclo-LF and LF). Notably, the former class of dipeptide–Bortezomib clusters exhibits larger dimensions in terms of cluster size. Concerning the compactness of the clusters, high density values are found in Cyclo-LF and LF, a bit higher for the former. For Z-FF the density peak is similar to that of LF but it is moved at longer distances, indicating a less compact interior of the clusters, whereas, a considerably looser structure is observed for Fmoc-FF. This is attributed to the three-ring shape of the protective group, which is of reduced flexibility in the way that it packs, imposing larger distances between atoms. As a result, an accessible area for water is found in the interior of the Fmoc-FF/BTZ cluster in contrast to the clusters of the rest of dipeptides, enhancing its hydro-gelating capacity. Moreover, Fmoc-FF/BTZ clusters are the only ones where boron atoms are located on their surface. An almost equal probability of finding Bortezomib (i.e., boron atoms) and Z-FF molecules on the surface is observed in the case of Z-FF/BTZ clusters, while in both the LF and Cyclo-LF systems, the Bortezomib molecules are trapped by the corresponding dipeptide molecules in the interior of the formed clusters. Boron acts as an inhibitor for the proteasome and its location on the surface of the clusters is critical. The above observations give an initial lead to Fmoc-FF.
In agreement with energetic measures the free energy of association between the dipeptide and Bortezomib follows an increasing order: LF < Cyclo-LF < Fmoc-FF < Z-FF (the same for van der Waals and electrostatic contributions). The enthalpic and the entropic parts of the free energy have been calculated separately although the contribution of the latter is very small. The number of hydrogen bonds between BTZ and dipeptide molecules follows a similar order to that of the free energy of association, providing their synergistic contribution to clustering. Energy-wise, the dipeptides divided into two classes show that protected FFs have higher affinity with the drug molecules, which will favor their entrapment. However, their ability to release has to be considered as well. An initial assessment is provided here; however, this will be explored extensively in a forthcoming publication.
Exploring the kinetics of the peptide/BTZ complexes, an acceleration of the process is observed compared to the reference systems of dipeptides. The number of the formed clusters approaches a steady, time-independent state after around ∼50 ns for all systems, similar to the time that Bortezomib clusters are formed in the pure BTZ aqueous solution. Comparing the time evolution of the number of clusters with the corresponding reference system, it is observed that the BTZ molecules lead to a stabilization of the clustering for the LF system in contrast to the Fmoc-FF system, where the fluctuations are enhanced, rendering the clusters less stable. This could potentially support the release of Bortezomib from Fmoc-FF clusters, since their reduced stability facilitates the dissociation of cluster segments. This process will be further explored in future studies.
Evaluating all the measures, we can claim that the protected FF category is qualified and between its two members, Fmoc-FF looks more promising. However, Z-FF also has various features that could make it a potential candidate carrier for Bortezomib and a promising candidate for future studies. The current study serves as a basis to pave the way towards optimal peptide carrier selection, for the encapsulation and controlled delivery of Bortezomib in future experimental studies.
Author contributions
Conceptualization: A. R., A. M., and V. H.; methodology: A. R.; execution of simulations and experiments: P. D., E. F., and A. R.; data analysis: A. R.; validation of results: A. R. and V. H.; writing – original draft preparation: A. R., P. D., and A. M.; and supervision, A. R. and A. M. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors would like to thank the support to this work in the form of computational time granted by Greek Research & Technology Network (GRNET) in the National HPC facility—ARIS for the Project: Peptide-Graphene nanocomposites though Molecular Simulations, BIOGRAPH. V.H. acknowledges support by the European Union's Horizon 2020 research and innovation program (Grant No. 810660). P. D's. and A. M's. research was co-financed by the European Union and Greek National Funds through the Operational Program Competitiveness, Entrepreneurship, and Innovation, under the call RESEARCH – CREATE – INNOVATE (project Acronym: EPHESIAN, project code: T1EDK-01504). MD simulations were performed at the Metropolis HPC facility of the Crete Center for Quantum Complexity and Nanotechnology. We are grateful to Ms Aleka Manousaki for assistance with FESEM experiments.References
- M. Schmidt and D. Finley, Biochim. Biophys. Acta, 2014, 1843, 13–25 CrossRef CAS PubMed.
- J. E. Park, Z. Miller, Y. Jun, W. Lee and K. B. Kim, J. Lab. Clin. Med., 2018, 198, 1–16 CAS.
- D. M. Rajagopal Appavu, Global J. Cancer Ther., 2016, 2, 005–008 CrossRef.
- F. Accardi, D. Toscani, M. Bolzoni, B. Dalla Palma, F. Aversa and N. Giuliani, BioMed Res. Int., 2015, 172458 Search PubMed.
- M. Groll, C. R. Berkers, H. L. Ploegh and H. Ovaa, Structure, 2006, 14, 451–456 CrossRef CAS PubMed.
- C. R. Berkers, M. Verdoes, E. Lichtman, E. Fiebiger, B. M. Kessler, K. C. Anderson, H. L. Ploegh, H. Ovaa and P. J. Galardy, Nat. Methods, 2005, 2, 357–362 CrossRef CAS PubMed.
- D. Chen, M. Frezza, S. Schmitt, J. Kanwar and Q. P. Dou, Curr. Cancer Drug Targets, 2011, 11, 239–253 CrossRef CAS PubMed.
- D. E. Reece, D. Sullivan, S. Lonial, A. F. Mohrbacher, G. Chatta, C. Shustik, H. Burris, 3rd, K. Venkatakrishnan, R. Neuwirth, W. J. Riordan, M. Karol, L. L. von Moltke, M. Acharya, P. Zannikos and A. Keith Stewart, Cancer Chemother. Pharmacol., 2011, 67, 57–67 CrossRef CAS PubMed.
- P. M. Voorhees, E. C. Dees, B. O'Neil and R. Z. Orlowski, Clin. Cancer Res., 2003, 9, 6316–6325 CAS.
- P. Moreau, H. Pylypenko, S. Grosicki, I. Karamanesht, X. Leleu, M. Grishunina, G. Rekhtman, Z. Masliak, T. Robak, A. Shubina, B. Arnulf, M. Kropff, J. Cavet, D. L. Esseltine, H. Feng, S. Girgis, H. van de Velde, W. Deraedt and J. L. Harousseau, Lancet Oncol., 2011, 12, 431–440 CrossRef.
- P. G. Richardson, B. Barlogie, J. Berenson, S. Singhal, S. Jagannath, D. Irwin, S. V. Rajkumar, G. Srkalovic, M. Alsina, R. Alexanian, D. Siegel, R. Z. Orlowski, D. Kuter, S. A. Limentani, S. Lee, T. Hideshima, D. L. Esseltine, M. Kauffman, J. Adams, D. P. Schenkein and K. C. Anderson, N. Engl. J. Med., 2003, 348, 2609–2617 CrossRef CAS PubMed.
- J. J. Wright, Clin. Cancer Res., 2010, 16, 4094–4104 CrossRef CAS.
- P. Kapoor, V. Ramakrishnan and S. V. Rajkumar, Semin. Hematol., 2012, 49, 228–242 CrossRef CAS PubMed.
- P. de la Puente and A. K. Azab, Eur. J. Haematol., 2017, 98, 529–541 CrossRef CAS PubMed.
- M. Mahmoudian, H. Valizadeh and P. Zakeri-Milani, Drug Dev. Ind. Pharm., 2018, 44, 1598–1605 CrossRef CAS PubMed.
- M. Mahmoudian, H. Valizadeh, R. Löbenberg and P. Zakeri-Milani, J. Microencapsulation, 2021, 38, 192–202 CrossRef CAS PubMed.
- C. Federico, K. Alhallak, J. Sun, K. Duncan, F. Azab, G. P. Sudlow, P. de la Puente, B. Muz, V. Kapoor, L. Zhang, F. Yuan, M. Markovic, J. Kotsybar, K. Wasden, N. Guenthner, S. Gurley, J. King, D. Kohnen, N. N. Salama, D. Thotala, D. E. Hallahan, R. Vij, J. F. DiPersio, S. Achilefu and A. K. Azab, Nat. Commun., 2020, 11, 6037 CrossRef CAS PubMed.
- M. Korani, S. Korani, E. Zendehdel, M. R. Jaafari, T. Sathyapalan and A. Sahebkar, Anti-Cancer Agents Med. Chem., 2020, 20, 643–650 CrossRef CAS PubMed.
- M. Rezigue, in Integrative Nanomedicine for New Therapies, ed. A. Krishnan and A. Chuturgoon, Springer International Publishing, Cham, 2020, pp. 167–230 DOI:10.1007/978-3-030-36260-7_7.
- M. Farshbaf, S. Mojarad-Jabali, S. Hemmati, A. Y. Khosroushahi, H. Motasadizadeh, A. Zarebkohan and H. Valizadeh, J. Controlled Release, 2022, 345, 371–384 CrossRef CAS PubMed.
- S. Liu, R. J. Ono, C. Yang, S. Gao, J. Y. Ming Tan, J. L. Hedrick and Y. Y. Yang, ACS Appl. Mater. Interfaces, 2018, 10, 19355–19364 CrossRef CAS PubMed.
- J. Liu, R. Zhao, X. Jiang, Z. Li and B. Zhang, Biomolecules, 2021, 12 Search PubMed.
- X. Zhang, T. Yuan, H. Dong, J. Xu, D. Wang, H. Tong, X. Ji, B. Sun, M. Zhu and X. Jiang, Colloid Polym. Sci., 2018, 296, 1827–1839 CrossRef CAS PubMed.
- A. S. Chauhan, Molecules, 2018, 23(4), 938 CrossRef PubMed.
- S. H. van Rijt, D. A. Bölükbas, C. Argyo, S. Datz, M. Lindner, O. Eickelberg, M. Königshoff, T. Bein and S. Meiners, ACS Nano, 2015, 9, 2377–2389 CrossRef CAS.
- J. H. Park, D. Dehaini, J. Zhou, M. Holay, R. H. Fang and L. Zhang, Nanoscale Horiz., 2020, 5, 25–42 RSC.
- V. A. Petrova, V. Y. Elokhovskiy, S. V. Raik, D. N. Poshina, D. P. Romanov and Y. A. Skorik, Biomolecules, 2019, 9, 291 CrossRef CAS PubMed.
- A. GhavamiNejad, M. SamariKhalaj, L. E. Aguilar, C. H. Park and C. S. Kim, Sci. Rep., 2016, 6, 33594 CrossRef CAS PubMed.
- L. Liu, S. Luan, C. Zhang, R. Wang, Y. Zhang, M. Zhang, Q. Sheng, G. Han, T. Wang and S. Song, Int. J. Biol. Macromol., 2021, 183, 369–378 CrossRef CAS PubMed.
- J. Min, H. Moon, H. J. Yang, H. H. Shin, S. Y. Hong and S. Kang, Macromol. Biosci., 2014, 14, 557–564 CrossRef CAS PubMed.
- Y. Yu, M. Song, C. Chen, Y. Du, C. Li, Y. Han, F. Yan, Z. Shi and S. Feng, ACS Nano, 2020, 14, 10688–10703 CrossRef CAS PubMed.
- S. Zeng, Y. Ji, Y. Shen, R. Zhu, X. Wang, L. Chen and J. Chen, RSC Adv., 2020, 10, 8744–8750 RSC.
- Y. Ji, R. Zhu, Y. Shen, Q. Tan and J. Chen, J. Mol. Liq., 2021, 341, 117454 CrossRef CAS.
- R. Yadav, U. K. Nath, I. Celik, S. Handu, N. Jain and P. Dhamija, Biomed. Pharmacother., 2023, 157, 113963 CrossRef CAS PubMed.
- C. Valery, M. Paternostre, B. Robert, T. Gulik-Krzywicki, T. Narayanan, J. C. Dedieu, G. Keller, M. L. Torres, R. Cherif-Cheikh, P. Calvo and F. Artzner, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10258–10262 CrossRef CAS.
- M. Reches and E. Gazit, Science, 2003, 300, 625–627 CrossRef CAS PubMed.
- L. Adler-Abramovich and E. Gazit, Chem. Soc. Rev., 2014, 43, 6881–6893 RSC.
- L. Li, L. Xie, R. Zheng and R. Sun, Front. Chem., 2021, 9 Search PubMed.
- K. Hanabusa, Y. Matsumoto, T. Miki, T. Koyama and H. Shirai, J. Chem. Soc., Chem. Commun., 1994, 1401–1402, 10.1039/C39940001401,.
- K. Hanabusa, M. Matsumoto, M. Kimura, A. Kakehi and H. Shirai, J. Colloid Interface Sci., 2000, 224, 231–244 CrossRef CAS.
- O. Bellotto, S. Kralj, R. De Zorzi, S. Geremia and S. Marchesan, Soft Matter, 2020, 16, 10151–10157 RSC.
- M. Scarel and S. Marchesan, Molecules, 2021, 26, 3376 CrossRef CAS.
- V. B. Kumar, B. Ozguney, A. Vlachou, Y. Chen, E. Gazit and P. Tamamis, J. Phys. Chem. B, 2023, 127, 1857–1871 CrossRef CAS PubMed.
- G. Pu, C. Ren, D. Li, L. Wang and J. Sun, RSC Adv., 2014, 4, 50145–50147 RSC.
- P. Divanach, E. Fanouraki, A. Mitraki, V. Harmandaris and A. N. Rissanou, J. Phys. Chem. B, 2023, 127, 4208–4219 CrossRef CAS PubMed.
- M. Reches and E. Gazit, Nano Lett., 2004, 4, 581–585 CrossRef CAS.
- A. Mahler, M. Reches, M. Rechter, S. Cohen and E. Gazit, Adv. Mater., 2006, 18, 1365–1370 CrossRef CAS.
- M. Reches and E. Gazit, Phys. Biol., 2006, 3, S10–19 CrossRef CAS PubMed.
- Z. A. Arnon, A. Vitalis, A. Levin, T. C. T. Michaels, A. Caflisch, T. P. J. Knowles, L. Adler-Abramovich and E. Gazit, Nat. Commun., 2016, 7, 13190 CrossRef CAS.
- M. Reches and E. Gazit, Science, 2003, 300, 625–627 CrossRef CAS.
- S. Gilead and E. Gazit, Supramol. Chem., 2005, 17, 87–92 CrossRef CAS.
- L. Adler-Abramovich, D. Aronov, P. Beker, M. Yevnin, S. Stempler, L. Buzhansky, G. Rosenman and E. Gazit, Nat. Nanotechnol., 2009, 4, 849–854 CrossRef CAS.
- B. Hess, C. Kutzner, D. van der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS PubMed.
- S. Chatterjee, P. G. Debenedetti, F. H. Stillinger and R. M. Lynden-Bell, J. Chem. Phys., 2008, 128, 124511 CrossRef PubMed.
- H. J. C. Berendsen, J. R. Grigera and T. P. Straatsma, J. Phys. Chem., 1987, 91, 6269–6271 CrossRef CAS.
- C. Oostenbrink, A. Villa, A. E. Mark and W. F. van Gunsteren, J. Comput. Chem., 2004, 25, 1656–1676 CrossRef CAS PubMed.
- N. Schmid, A. P. Eichenberger, A. Choutko, S. Riniker, M. Winger, A. E. Mark and W. F. van Gunsteren, Eur. Biophys. J., 2011, 40, 843–856 CrossRef CAS PubMed.
- G. Zhang, J. Zhang, Y. Wang, Y. Wu, Q. Li, Y. Liang, W. Qi, H. Rao, R. Su and Z. He, J. Colloid Interface Sci., 2020, 578, 218–228 CrossRef CAS PubMed.
- A. K. Malde, L. Zuo, M. Breeze, M. Stroet, D. Poger, P. C. Nair, C. Oostenbrink and A. E. Mark, J. Chem. Theory Comput., 2011, 7, 4026–4037 CrossRef CAS PubMed.
- C. E. Mona, É. Besserer-Offroy, J. Cabana, R. Leduc, P. Lavigne, N. Heveker, É. Marsault and E. Escher, Org. Biomol. Chem., 2016, 14, 10298–10311 RSC.
- H. J. C. Berendsen, J. P. M. Postma, W. F. V. Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- X. Daura, K. Gademann, B. Jaun, D. Seebach, W. F. van Gunsteren and A. E. Mark, Angew. Chem., Int. Ed., 1999, 38, 236–240 CrossRef CAS.
- T. F. Headen, E. S. Boek, G. Jackson, T. S. Totton and E. A. Müller, Energy Fuels, 2017, 31, 1108–1125 CrossRef CAS.
- R. Kumari, R. Kumar and A. Lynn, J. Chem. Inf. Model., 2014, 54, 1951–1962 CrossRef CAS PubMed.
- N. A. Baker, D. Sept, S. Joseph, M. J. Holst and J. A. McCammon, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10037–10041 CrossRef CAS PubMed.
- L. Duan, X. Liu and J. Z. H. Zhang, J. Am. Chem. Soc., 2016, 138, 5722–5728 CrossRef CAS PubMed.
- N. Brown, J. Lei, C. Zhan, L. J. W. Shimon, L. Adler-Abramovich, G. Wei and E. Gazit, ACS Nano, 2018, 12, 3253–3262 CrossRef CAS PubMed.
- A. M. Smith, R. J. Williams, C. Tang, P. Coppo, R. F. Collins, M. L. Turner, A. Saiani and R. V. Ulijn, Adv. Mater., 2008, 20, 37–41 CrossRef CAS.
- C. Diaferia, E. Rosa, G. Morelli and A. Accardo, Pharmaceuticals, 2022, 15, 1048 CrossRef CAS PubMed.
- C. Diaferia, E. Rosa, E. Gallo, G. Morelli and A. Accardo, Chem. - Eur. J., 2023, 29, e202300661 CrossRef CAS PubMed.
- N. Foloppe and R. Hubbard, Curr. Med. Chem., 2006, 13, 3583–3608 CrossRef CAS PubMed.
- H. Gohlke, C. Kiel and D. A. Case, J. Mol. Biol., 2003, 330, 891–913 CrossRef CAS PubMed.
- N. Homeyer and H. Gohlke, Mol. Inf., 2012, 31, 114–122 CrossRef CAS PubMed.
- P. A. Kollman, I. Massova, C. Reyes, B. Kuhn, S. Huo, L. Chong, M. Lee, T. Lee, Y. Duan, W. Wang, O. Donini, P. Cieplak, J. Srinivasan, D. A. Case and T. E. Cheatham, 3rd, Acc. Chem. Res., 2000, 33, 889–897 CrossRef CAS PubMed.
- H. Meirovitch, Curr. Opin. Struct. Biol., 2007, 17, 181–186 CrossRef CAS PubMed.
- S. P. Brown and S. W. Muchmore, J. Med. Chem., 2009, 52, 3159–3165 CrossRef CAS PubMed.
- C. Wang, D. A. Greene, L. Xiao, R. Qi and R. Luo, Front. Mol. Biosci., 2018, 4, 87 CrossRef PubMed.
- K. Huang, S. Luo, Y. Cong, S. Zhong, J. Z. H. Zhang and L. Duan, Nanoscale, 2020, 12, 10737–10750 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental data based on FESEM pictures for Bortezomib and dipeptides, snapshots from simulations, additional pair radial distribution functions and additional classification of hydrogen bonds as well as the time evolution of hydrogen bonding. See DOI: https://doi.org/10.1039/d3sm00930k |
| This journal is © The Royal Society of Chemistry 2023 |