
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multicompartment calcium alginate microreactors to reduce substrate inhibition in enzyme cascade reactions†
Yongkang
Xi
a,
Bradley D.
Frank
a,
Apostolos
Tatas
a,
Marko
Pavlovic
ab and
Lukas
Zeininger
 *a
*a
aDepartment of Colloid Chemistry, Max Planck Institute of Colloids and Interfaces, Am Muehlenberg 1, 14476 Potsdam, Germany. E-mail: lukas.zeininger@mpikg.mpg.de
bDepartment of Physics and John A. Paulson School of Engineering and Applied Sciences, Harvard University, Cambridge, Massachusetts 02138, USA
First published on 14th September 2023
Abstract
The formation of macromolecularly enriched condensates through associative or segregative liquid–liquid phase separation phenomena is known to play a central role in controlling various cellular functions in nature. The potential to spatially and temporally modulate multistep chemical reactions and pathways has inspired the use of phase-separated systems for the development of various synthetic colloidal micro- and nanoreactor systems. Here, we report a rational and synthetically minimal design strategy to emulate intended spatiotemporal functions in morphologically intricate and structurally defined calcium alginate hydrogel microreactors possessing multicompartmentalized internal architectures. Specifically, we implement a thermal phase separation protocol to achieve fine-control over liquid–liquid phase separation inside complex aqueous emulsion droplet templates that are loaded with hydrophilic polymer mixtures. Subsequent gelation of alginate-containing droplet templates using a novel freeze–thaw approach that can be applied to both scalable batch production or more precise microfluidic methods yields particle replicas, in which subcompartmentalized architectures can be retained. Larger active components can be enriched in the internal compartments due to their preferential solubility, and we show that selective sequestration of enzymes serves to create desired microenvironments to control and tune the reaction kinetics of a multistep enzyme cascade by reducing their mutual interference. This demonstration of mitigating substrate inhibition that is based primarily on optimizing the multicompartmentalized hydrogel particle morphology offers new opportunities for the simple and synthetically-minimal batch generation of hydrogel-based synthesis microreactors.
Introduction
Spontaneous liquid–liquid phase separation (LLPS) within crowded aqueous environments constitutes an essential capability of living cells that is fundamental to their ability to control multistep chemical transformations in time and space.1–5 Emulating the inspirational regulatory capabilities of natural systems, the construction of multi-compartment microreactors to perform multistep biochemical cascade reactions in one pot has attracted substantial attention.6–9 Spatially localizing reaction centers enables a regulation of the diffusion pathways of substrates, intermediates, and products, as well as control over constructive and destructive interference of active reaction centers, such as enzymes.10–13 In addition, higher local substrate and catalyst concentrations within the confined space of a microreactor can effectively tune reaction kinetics by overcoming unfavorable thermodynamics of the bulk environment.14–16 Consequently, having fine control over the compartmentalization of active reaction centers within multiphasic microreactors constitutes an exciting avenue to impart materials with regulatory capabilities, a concept that has been elegantly designed and investigated in a variety of colloidal structures, including liposomes,17,18 polymersomes,19 vesosomes,20,21 emulsions,22,23 and compartmentalized hydrogels.24–26The optimization of a reaction's path and performance is determined by a combination of trade-off factors, including stability of the nano- or microreactor and its encapsulation efficiency, the confinement geometry, packing, and mass transfer across phase boundaries. Whereas liposome or polymersome-based protocells can tightly confine enzymes, a mass transfer of substrates can be mitigated across the membrane boundary, which is responsible for the high stability of the system.27–29 In alternative hydrogel microreactors, the confinement efficiency correlates strongly with mesh size and crosslinking density of the hydrogel scaffold facilitating control over the mass transfer of reaction substrates, intermediates and products across phase boundaries.30,31
Cross-linked alginate-based hydrogel particles and capsules, due to their excellent biocompatibility and low cost are an attractive and widely employed matrix material for encapsulation, enrichment, and delivery of active components as well as for compartmentalization of biologicals, nutrients, or cells.32,33 Underpinned by tunable cross-linking densities that directly correlate with the mesh sizes of the hydrogel, which are similar to the extracellular matrices of tissues, a selective transport of components can be achieved.34 Consequently, alginate-based particles and capsules have been widely employed as active carriers of functional components in food, cosmetic, and medical applications, including in biomedical sequencing, drug screening assays, and controlled drug delivery.35,36 Such crosslinked alginate-based particles and core–shell structures are commonly prepared using extrusion or emulsion-templated techniques.37 Beyond uniform single phasic particles, the generation of Janus and core–shell structures has enabled a spacial localization of active catalytic components and bioactive molecules due to their phase preferential solubility.38–40 However, despite the dominant role of localized enrichment of active reaction centers for spatiotemporal control of reaction pathways with high efficiency within the non-membrane compartments in macromolecularly crowded living matter,41,42 design strategies for the targeted generation of multicompartmentalized, morphologically intricate alginate microparticles have yet been devised to exploit a local arrangement of multiple functional components to modulate chemical reaction pathways in time and space. Such investigations that provide an opportunity to exploit morphological features in order to gain spatiotemporal control over multistep cascade reaction pathways are intimately tied to the availability of accessible bulk methods for the generation of precision multicompartment hydrogel microreactors.
We report herein a thermal phase separation protocol for the continuous generation of complex calcium alginate-based enzyme-loaded biomimetic synthesis microreactors. We employ aqueous complex emulsion droplets loaded with hydrophilic polymer mixtures as colloidal tectons for the controllable generation of morphologically highly uniform complex hydrogel particles with multiple internal compartments, in which variations in temperature and time allow to achieve fine control over the state of liquid–liquid phase separation. We next introduce a freeze–thaw approach for the gelation of the complex droplet templates, where phase-selective partitioning of molecules and enzymes, such as horseradish peroxidase (HRP) and glucose oxidase (GOX), enables spatial enrichment and organization of active reaction centers within the complex hydrogel microreactors. We observe that the spatial organization significantly dictates the kinetics of a model multistep enzyme cascade reaction and can effectively suppress destructive interference of rivalling reaction centers. The resulting membraneless organelle-inspired mitigation of substrate inhibition in competitive multistep cascade reactions combined with the insights into the optimization of multicompartment morphology of artificial soft microreactors provide new insight into the facile batch generation of synthetic hydrogel cascade microreactors.
Results and discussion
Emulsion-templated freeze-melt approach for the generation of alginate hydrogel particles
For the construction of hydrogel microreactors, we opted for an emulsion-templated approach. To this end, an aqueous alginate solution (2.5 wt%) was emulsified in Span 85-containing heptane using either microfluidic or batch-scale vortex mixing at room temperature (Fig. S1a and b, ESI†). Depending on the emulsification method, the alginate-containing water-in-oil emulsions were polydisperse in size displaying an average diameter of 52.2 μm ± 55.9 μm or when generated using microfluidics were highly monodisperse with an average diameter of 161.1 μm ± 7.4 μm. The pristine droplets were then frozen at T = −20 °C, which is below the freezing temperature of the polymer-loaded aqueous droplets (−16 °C) but above the freezing point of the continuous phase oil (heptane, −91 °C). The continuous phase was removed by transferring the frozen droplets into a cooled calcium chloride solution (c(CaCl2) = 160 mM, T = −1 °C). Gradual melting of the frozen droplet precursors inside the Ca2+ solution resulted in crosslinking of the released alginate polymers yielding uniformly spherical Ca-alginate hydrogel particles.Upon gelation the size and shape of droplet templates was essentially retained (Fig. 1a and Fig. S1–S3, ESI†). Using confocal fluorescence microscopy, the internal structure of fluorescein-5-isothiocyanate (FITC)-labeled alginate hydrogel particles could be visualized (Fig. 1b). The micrographs reveal an inhomogeneous, channel-like structure in the interior of the particles, with slightly stronger fluorescence at the interface indicating a higher crosslinking density at the periphery of the particle. We attributed the skeletal structure to the crack channels formed via thermal expansion during melting, which contain similar calcium ion concentrations as the outer particle interface due to the capillary effect and thus exhibit higher crosslinking densities (Fig. S1, ESI†).
 | ||
| Fig. 1 Freeze–thaw approach for the generation of hydrogel particles from aqueous emulsion precursors. (a) Schematic for the continuous generation of Ca-alginate hydrogel particles. (b) Fluorescence micrograph of FITC-labelled Ca-alginate hydrogel particles; (c) and (d) fluorescence microscopy images of Ca-alginate hydrogel particles after exposure to a solution containing rhodamine B (c) or RITC-dextran (MW: 70 kDa) (d) and (e) fluorescence micrograph showing encapsulated FITC-labelled HRP enzymes inside a Ca-alginate hydrogel particle. | ||
To determine the molecular weight cut-off (MWCO),42 and thus the size-dependent semipermeability of the hydrogel particles, we incubated particles with FITC-labeled dextran polymers of different molecular weight and characterized the polymer size-dependent fluorescence intensity from the particles (Fig. S4, ESI†). The combined results revealed that the hydrogel particles prepared by the freeze–thaw method display good semi-permeability, that is, they retain macromolecules above the MWCO = 40 kDa while allowing small molecules to pass.
Multi-compartment hydrogels via gelation of complex aqueous emulsion droplet templates
Motivated by our findings that a combination of emulsification and phase transition is an effective strategy for large-scale generation of uniform hydrogel particles, we next employed complex droplet precursors containing aqueous mixtures of poly(ethylene glycol) (PEG) (MW: 35 kDa) and dextran (MW: 500 kDa), in addition to alginate, in order to generate multi-compartmentalized hydrogel particles (Fig. 2a). The degree of segregative phase-separation of the polymer-rich aqueous solutions can be controlled by manipulating temperature and time.43 Temperature-dependent cloud point titration of a PEG/dextran mixture in an aqueous alginate (2.5 wt%) solution revealed the temperature sensitivity of liquid–liquid phase separation (LLPS) in the aqueous dispersed phase polymer mixture (Fig. 2b). The phase diagrams showed that at low polymer concentrations such aqueous mixtures exist as single phase, while at high concentrations phase separation occurred. By altering the temperature of the system, the location at which the phase separation took place, commonly referred to as the binodal, shifted. A decrease in temperature to 4 °C promoted a slight shift of the binary towards lower polymer concentrations, indicating an exothermic phase separation process. Vice versa, increasing the temperature to 50 °C caused phase-separated aqueous polymer phases at concentrations close to the binodal at room temperature to mix and the point of phase separation shifted toward higher polymer concentrations. Using mixtures of PEG and dextran inside a sodium alginate containing aqueous solution at concentrations in between two binodals at different temperatures enabled a reversible temperature-induced LLPS of the system (Fig. 2c). | ||
| Fig. 2 Freeze–thaw approach for the generation of multi-compartment hydrogel microparticles from complex aqueous emulsion templates. (a) Schematic for the continuous generation of multi-compartment hydrogel particles. The dispersed aqueous phase of the emulsion precursors contains sodium alginate, PEG and dextran. Cooling induces phase separation to generate multi-compartment droplet templates. At different stages of the phase separation droplet molds were frozen prior to transferring them to an aqueous Ca2+-solution which induces gelation to generate the multi-compartment hydrogel particles; (b) and (c) phase diagrams of alginate (2.5 wt%) containing aqueous mixtures of PEG (35 kDa) and dextran (500 kDa) outline the temperature-dependency of the concentration at which polymer solutions de-mix to form an ATPS; (d)–(g) fluorescence and bright-field optical micrographs of complex droplet templates at different time points ((d) 20 min, (e) 40 min, (f) 70 min, (g) 180 min) after initiating phase separation; scale bar: 50 μm; (h)–(k) fluorescence micrographs of the multi-compartment hydrogel particles obtained after gelating the corresponding droplet templates. Scale bar images: top row: 300 μm. bottom row: 50 μm. | ||
To generate complex hydrogel particles, we implemented a two-step process, in which a controlled temperature-induced LLPS inside alginate, PEG and dextran containing droplets was followed by freezing and gelation of the droplet templates. Emulsification of the aqueous polymer mixture was carried out above the phase separation temperature at (T = 50 °C) to ensure a single-phase dispersed phase. The single-phase droplets were subsequently allowed to cool to room temperature (T = 20 °C), which induced slow phase separation yielding structured complex emulsion droplets. The presence of sodium alginate (2.5 wt%) within the as-formed droplets resulted in a significant increase in the time needed for phase separation to occur (Fig. S6 and Videos S1, S2, ESI†), when compared to pure PEG/dextran mixtures. While droplets comprised of pure PEG/dextran mixtures separate rapidly into two distinct aqueous phases over the course of ∼1 min, the lowered density contrast of the two aqueous phases due to the addition of sodium alginate resulted in a prolongation of this process. In this system complete phase separation toward the formation of aqueous Janus droplets was observed only after ∼180 min (Video S2, ESI†), which permitted distinct temporal control over the state of phase separation inside the droplet templates.
To visualize the internal architecture of droplets at different stages of the phase separation process, we used FITC-labelled dextran and isolated intermediate phase-separated droplet templates after 20 min, 40 min, 70 min, and 180 min. Confocal fluorescence micrographs of the droplets revealed the formation of small dextran-rich compartments inside PEG-rich droplets, which slowly merged into larger droplets that were evenly distributed throughout the droplets as phase separation continued (Fig. 2d–g). After complete phase separation, the individual compartments eventually coalesced and settled into their gravitational alignment to form a Janus droplet. The different complex droplet templates were subsequently frozen at 20 °C.
Gelation of the frozed templates inside a Ca2+-containing aqueous solution at T = −1 °C resulted in the controlled generation of structurally uniform complex hydrogel particles with a defined internal multi-compartmentalized morphology (Fig. 2a). Upon gelation, the compartmentalized internal morphology at the different stages of phase separation was retained and depending on the state of LLPS inside the droplets prior to freezing the droplet templates, hydrogel particles with controllable size and number of internal compartments were obtained (Fig. 2 and Fig. S6, S7, Videos S3–S7, ESI†). Fluorescence micrographs of the complex hydrogel particles that were synthesized employing FITC-labeled alginate indicated their internal structure. A preferred partitioning of sodium alginate within the PEG-rich phases (partitioning coefficient: Kalg = 2.15) of the droplets resulted in a higher crosslinking density of the PEG-rich hydrogel compartments after gelation as revealed through a higher FITC-labeled alginate fluorescence (Fig. S8, ESI†). The increased crosslinking density of the PEG-rich phase of the particles sufficed to block a merging of more loosely-linked dextran-rich compartments and thus provided good overall stability to the particle system, in which we observed no noticeable changes to the internal morphology over a period of several days. 3D scans of the resulting complex hydrogel particles containing FITC-labeled dextran recorded by confocal fluorescence microscopy further revealed a uniform distribution of the dextran-rich internal compartments inside the particles (Videos S6 and S7, ESI†).
Partitioning of guest molecules and enzymes
Leveraging the multi-compartmentalized nature of the hydrogel particles we next phase-selectively compartmentalized and investigated the relative partitioning of different molecules and enzymes within the individual phases (Fig. 3a). The partitioning of molecules in the PEG- and dextran-rich phases is mainly determined by electrostatic, hydrophobic, and steric interactions.44 In accordance to previous literature on the partitioning in aqueous two-phase systems comprised of PEG and dextran,45 fluorescent molecules such as FITC selectively compartmentalized into the PEG-rich phases due to their lowered hydrophilicity as revealed by the respective fluorescence micrographs of the particles (Fig. 3b). In turn, more hydrophilic Nile blue dyes partitioned into the dextran-rich phases (Fig. 3c).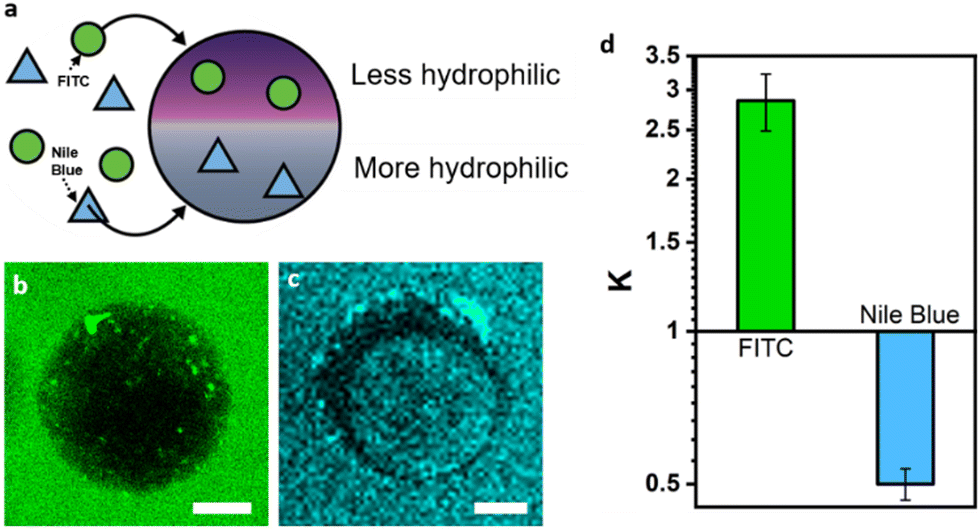 | ||
| Fig. 3 Phase preferential compartmentalization of guest molecules. (a) Scheme outlining phase-preferential partitioning of guest molecules inside Janus hydrogel particles; (b) and (c) fluorescence micrographs of hydrogel Janus particles placed inside aqueous solutions of FITC (b) and Nile blue (c), scale bar: 50 μm; (d) column graph showing the different partitioning coefficients of FITC and Nile blue in the two constituent phases of the hydrogel particles. | ||
Motivated by this finding, we investigated the self-assembly and partitioning behaviour of the enzyme catalase, an enzyme catalyzing the decomposition of hydrogen peroxide into water and oxygen. To this end, we determined the partitioning coefficient of catalase inside an alginate-containing aqueous two-phase system comprised of PEG and dextran by comparing the enzyme concentrations in the separated top and bottom phase of the ATPS using the Bradford test.46 In this system, we observed a strongly phase-selective compartmentalization inside the PEG-rich phases with a partitioning coefficient of K = 7.73 (Fig. S9, ESI†). The partitioning coefficient could be further tuned to K = 10.41 through increasing salt concentrations inside the ATPS via addition of 50 mM NaCl. Confocal fluorescence micrographs of droplet precursors containing FITC-labelled catalase, which showed a more intense fluorescence in the PEG phase corroborated these findings (Fig. 4a). Upon gelation, the resulting Janus particles exhibited FITC-catalase fluorescence almost selectively inside the PEG compartments, which we attribute to both the high partitioning coefficient of the enzymes inside droplet templates as well as potential removal of residual catalase inside the dextran-rich particle phase during washing due to the larger mesh size of the latter.
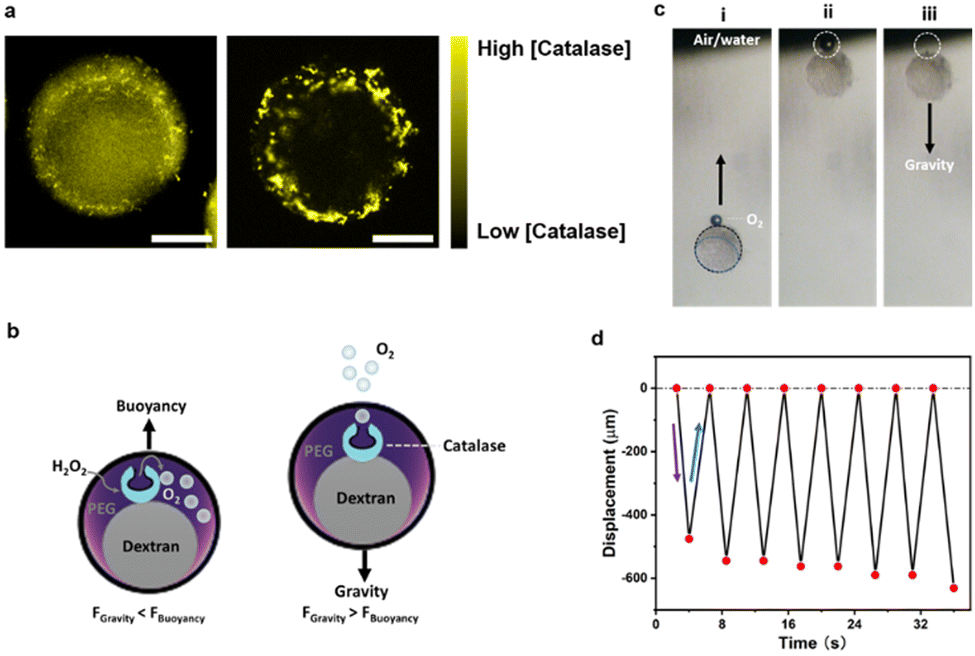 | ||
| Fig. 4 Oscillatory buoyant motion of catalase functionalized Janus hydrogel microreactors. (a) Confocal images of droplets (left) and hydrogel particles (right) with yellow fluorescence from encapsulated FITC-labeled catalase, scale bar, 50 μm. (b) Scheme illustrating oscillatory motion of catalase containing hydrogel particles upon exposure to hydrogen peroxide. (c) Time sequence optical micrographs displaying rapid changes of vertical displacement of hydrogel particles containing catalase upon producing oxygen. From left to right are the hydrogels (i) undergoing buoyant motion (ii) reaching the air–water interface of the dispersion, and (iii) sinking back into the solution due to gravity after releasing the oxygen bubble; (d) plot of the time-dependent oscillatory displacement of a hydrogel particle undergoing enzyme-mediated buoyant motion. | ||
To test the activity of the enzymes also after the gelation procedure, we added a hydrogen peroxide solution (1% v/v) to the particle dispersion. A formation of an oxygen bubbles at the upper particle interface was observed upon addition of the hydrogen peroxide (Fig. 4b) inducing a concomitant oscillatory buoyancy of these particles in the peroxide-containing aqueous dispersion (Fig. 4c and Video S8, ESI†). The generated oxygen remained at the particle interface and served to induce a buoyant motion of the particles. Upon reaching the upper air–water interface of the dispersion, the oxygen bubble was released and the particles settled with gravity until re-produced oxygen inside the particles caused a reversal of the motion direction. In the experiments, the oscillatory motion (frequency ∼0.22 s−1) continued for up to half an hour until a decreased peroxide concentration over time led to a decrease in the oxygen production rate and thus to an increase in the amplitude of the oscillatory motion (Fig. 4d and Fig. S10, Video S9, ESI†).
Controlling enzyme cascade reaction kinetics via tuning hydrogel particle morphology
Motivated by phase-selective partitioning of different molecules and enzymes into the different phases of our complex hydrogel particles, we next investigated a cross-partitioning of two different encapsulated enzymes, namely glucose oxidase (GOX) and horseradish peroxidase (HRP) (Fig. 5a). Investigation of autonomous partitioning of the two enzymes into the individual phases of the ATPS using the Bradford test revealed that GOX partitioned preferable into the dextran-rich phase (K = 0.78), while HRP was almost evenly distributed between the two phases with a slightly stronger tendency toward the PEG-rich phase (K = 1.12) (Fig. 5b). Higher cross-partitioning could be achieved via the addition of sodium chloride to the ATPS, where an addition of 50 mM resulted in K = 0.68 for GOX and K = 1.17 for HRP, respectively. However, at higher NaCl concentrations, a decrease in the K values indicated that both enzymes tended to partition in the dextran phase. This observation suggests that precise monitoring of salt concentrations was necessary to fine-tune the local distribution of enzymes (Fig. 5b). Dissolving the two enzymes labelled with different fluorescent markers (FITC-HRP and RITC-GOX) inside a bulk NaCl (50 mM)-containing ATPS showcased the phase-dominant partitioning behavior by different staining of the two phases (Fig. 5c). Similarly, in both the complex droplet templates as well as the crosslinked hydrogel particles, confocal fluorescence micrographs displayed enhanced green FITC-HRP fluorescence in the PEG-rich phase and red RITC-GOX fluorescence in the dextran-rich phase (Fig. S11, ESI†). | ||
| Fig. 5 Optimization of enzyme cascade reaction efficiency via tuning internal microreactor morphology. (a) Scheme for the cascade reaction with phase-preferentially compartmentalized enzymes, GOX and HRP; (b) partitioning coefficient (K) of HRP (yellow dots) and GOX (red squares) as a function of NaCl concentration within the dispersed phase ATPS containing sodium alginate. (c) Bulk image of the ATPS indicating phase-dominant partitioning behaviour of yellow FITC-labeled HRP and red RITC-labled GOX by different staining of the two phases; (d) time-dependent cascade reaction evolution tracked via confocal fluorescence microscopy; (e) fluorescence intensity of the cascade reaction product amplex red as a function of time for hydrogel particles with different internal morphologies; (f) reaction rates of the enzyme cascade reaction as a function of the compartment size and number inside the hydrogel microreactors. | ||
The associated understanding of enzyme cross-partitioning inside complex hydrogel particles at hand, we set out to perform a cascade reaction mediated by the parallel and independent performance of the two encapsulated enzymes GOX and HRP (Fig. 5a). In this two-step enzyme cascade reaction, glucose served as a substrate. A GOX-mediated conversion of glucose into gluconic acid generated hydrogen peroxide as a byproduct, which was then reduced under HRP catalysis to transform a second substrate, amplex red, into the release of the fluorescent marker resorufin. Using fluorescence microscopy, we tracked the time-dependent release of the reaction product after the addition of glucose. Plots of time-dependent increases in fluorescence intensity reveal large differences in the reaction rate as a function of the respective complex particle morphology.
For particles that were gelated prior to phase separation, i.e. right after droplet generation, only a very slow generation of the product was observed (green graph in Fig. 5e). In contrast to these single-phase particles with an equal distribution of the two enzymes throughout the particles, a reaction performed inside multi-compartmentalized hydrogel microreactors with many small dextran-rich domains distributed throughout the PEG-rich scaffold resulted in a twelve-fold stronger fluorescence intensity after the same reaction time (t = 2300 s), indicating a significantly pronounced conversion efficiency inside the latter. Decreasing the number of phase-separated compartments and consequently also increasing the overall size of the inner compartments, resulted in conversion rates in between those measured for highly multi-compartmentalized and homogeneously-mixed single-phase particles. Despite the greater distance between GOX and HRP and low interfacial area between the individual phases in Janus particles comprised of only two distinct compartments, the conversion efficiency was still 2.5-fold higher when compared to the single phase non-compartmentalized particle system. Thus, we concluded that particle morphology offers a significant potential to positively influence the kinetics of enzymatic cascade reactions through (i) compartmentalization and spatial separation of the enzymes and (ii) increasing the total number of internal compartments and thus the interfacial area between the compartments.
To better understand this behaviour, we prepared two different multi-compartmentalized hydrogel microreactors that displayed the same internal morphology but differed in enzyme cross-partitioning as controlled via the salt concentration in the droplet templates. As described above, hydrogel particles prepared without NaCl display enzyme partitioning into both phases, while the addition of NaCl (50 mM) prior to particle preparation significantly increases cross-separation behaviour. A substrate concentration-dependent recording of the reaction rates revealed a significantly increased Michaelis–Menten constant of Km = 0.980 mM for the cascade reaction performed inside the microreactors with higher enzyme cross-partitioning efficiency, as compared to Km = 0.517 mM for the particles with less pronounced enzyme cross-partitioning (Fig. S12, ESI†). High local concentrations of H2O2 can suppress HRP activity, an effect that has been previously studied and is known from literature.47 With only marginal differences in cross partitioning of the two substrates in the reaction, glucose and amplex red, between the two phases (Fig. S13, ESI†), we thus attributed the pronounced changes in the overall reaction rates to variations in the local concentration of the reaction intermediate hydrogen peroxide. Enzyme cross-compartmentalization effectively suppressed substrate inhibition of HRP via lowering the local hydrogen peroxide concentration, whereas, simultaneously an increased interfacial area between the two compartments led to a pronounced exchange of the reaction intermediates resulting in increased reaction rates.
Conclusions
A straightforward design strategy for the continuous generation of structurally defined complex hydrogel microreactors is demonstrated. Based on a freeze–thaw approach for the emulsion-templated gelation of alginate-containing droplet templates, which can be applied to complex aqueous two-phase system-loaded droplet templates, well-defined and uniform multi-compartment hydrogel particles could be obtained. Via controlling time and temperature to influence the liquid–liquid phase separation inside the droplet precursors, a fine control over the size and number of micro-compartments within the final particles could be achieved. Our approach enables spatially controlled encapsulation of enzymatic reaction centers, and thus the design of multi-compartmentalized hydrogel cascade microreactors. Efficient cross-partitioning of multiple enzyme reaction centers served to reduce substrate inhibition in order to tune the conversion efficiency in multistep enzyme cascade reactions. We demonstrated that the reaction rate of a model enzyme cascade reaction significantly depended on the internal particle morphology, with a multi-compartmentalized system exhibiting an up to twelve-fold increase in reaction rate. The study emphasizes the importance of multi-compartmentalized particle morphology to effectively promote the efficiency of enzymatic cascade reactions inside soft microreactors and we anticipate that the presented particle generation approach will serve as a broadly applicable tool for the development of other active soft colloidal microreactors for a variety of applications, including in biochemical process engineering, biomanufacturing, and disease treatment.Materials and methods
Materials
All chemicals were of analytical grade, and used as received without further purification. Hydrophilic polymers: Na-alginate (Sigma-Aldrich); poly(ethylene glycol) (PEG) 35k (Sigma-Aldrich), Dextran 500k (Alfa Aesar); and Span 85 (Sigma-Aldrich); enzyme: catalase, Horseradish peroxidase (HRP), glucose oxidase (GOX) (Sigma-Aldrich); dye: fluorescein isothiocyanate isomer I (FITC), Rhodamine B isothiocyanate (RITC), Rhodamine B, FITC-dextran (3k–5 kDa 10 kDa, 20 kDa and 40 kDa), RITC-dextran (70k) and reactive blue 160 are purchased from Sigma-Aldrich; continuous phase: heptane (Sigma-Aldrich). Deionized (DI) water was used in all experiments.Freeze-melt approach for preparation of alginate hydrogel particles
Alginate particles in the micrometer range were generated according to the scheme in Fig. 1a. In brief, a template W/O emulsion was made using vortex mixing or microfluidics. The emulsion was comprised of a 2.5% sodium alginate solution as the dispersed phase and 4 wt% Span85/heptane as the continuous phase. The emulsion was then passed through a freezing bath at −20 °C, resulting in a freeze-solidification of the dispersed phase. The hydrogel particles were subsequently formed by transferring the frozen droplet templates into a cooled calcium chloride solution (T = −1 °C). After centrifugation and washing with DI water, the hydrogel particles were stored in at 4 °C.FITC-labeling of polymers and enzymes: FITC-labeled dextran was prepared according to a literature procedure.45 In brief, we dissolved 1 g dextran (500k) in methyl sulfoxide (10 mL) containing a few drops of pyridine. Then 0.1 g of fluorescein isothiocyanate was added. After stirring for 5 min, dibutyltin dilaurate (20 mg) was added, and the mixture was heated at 95 °C for 2 h. After washing with ethanol to remove free dye, the FITC-dextran was filtered off and dried under vacuum at a temperature of 50 °C. The enzymes GOX and HRP were labeled with rhodamine B isothiocyanate (RITC) or fluorescein isothiocyanate (FITC) also following a literature procedure.46 In brief, 4 mg mL−1 of enzyme (GOX and HRP) solution was dissolved in a bicarbonate buffer (0.1 M, pH = 8.2) and 2.5 mg mL−1 of dye (RITC or FITC) solution were dissolved in anhydrous DMSO. Subsequently, 20 μL of the dye solution was added to 0.5 mL of the enzyme solution and mixed. The reactions were performed at room temperature and protected from light. After 60 min, the labeled enzymes were purified via dialysis for 7 days, in order to remove the free dye thoroughly. Finally, the purified enzymes labeled with dyes were stored at 4 °C until further use.
Stability of the hydrogel particles
To test the pH stability of the hydrogel particles, we exposed the pristine hydrogel particles to aqueous solutions of hydrochloric acid or sodium hydroxide with pH values of 3, 5, 6.5, 8, and 10. Micrographs of the particles recorded after 30 min allowed an indication of the stability of the particles in the pH range between 3 and 8 (Fig. S5, ESI†). Moreover, the hydrogel particles were stable up to temperatures of 90 °C. To test the resistance of the hydrogel particles to changes in salt ion concentration, we exposed freshly prepared hydrogel particles to sodium chloride solutions at concentrations of 5 mM, 20 mM and 50 mM. Here, we observed that at Na+ concentrations of 50 mM ion exchange resulted in a decrease of the crosslinking density of the hydrogel network and particles swelled or even dissolved.Freeze-melt approach for preparation of multi-compartmentalized hydrogel particles
Multicompartment particles were generated according to the scheme in Fig. 2a. The key parameters controlling the formation of hydrogel structures are temperature and time. In brief, before the emulsion is formed, the dispersed phase is heated above the phase separation temperature with a temperature-controlled microfluidic setup. (The microfluidics used to prepare the emulsion is placed in a transparent box, which contains a temperature probe, and the temperature is regulated by controlling the power of the heating blower.) The heated solution (2.5% sodium alginate, PEG 35k and dextran 500k in water and 4 wt% Span85/heptane as a continuous phase) was emulsified into the template W/O emulsion using bulk vortex mixing (speed: 250 rpm) or microfluidic methods. After emulsification, we allowed the emulsion to cool down to RT, which induced phase separation of the constituent phases (see Fig. 2C in the manuscript). After different time periods, the droplet templates were frozen in a freezing bath at −20 °C, resulting in the dispersed phase to freeze-solidify. The hydrogel particles were subsequently formed by flowing into an iced calcium chloride solution. After centrifugation and washing, the compartment particles were stored in a 4 °C refrigerator until use.Determination of partitioning coefficients
Partitioning coefficients (K) of enzymes were determined by comparing the enzyme concentration in the top and bottom phases using the Bradford test as described previously.41,42 Upon addition of Bradford's reagent to alginate containing solutions precipitation was observed. Thus, to estimate the partitioning behavior of the HRP and GOX enzymes we separately extracted the polyethylene glycol (PEG)-rich phase and the dextran-rich phase from a bulk aqueous two-phase system (ATPS) containing sodium alginate after completion of phase separation. Subsequently, each phase was diluted by a factor of 10 using deionized (DI) water before performing Bradford's assay. Partitioning coefficients of FITC and Nile blue dyes were extracted via determination of the fluorescence intensity distribution (Fiji software) in the top and bottom phases of the ATPS or hydrogel particles, respectively, according to the following formula: K = Ifluotop/Ifluobottom. For determination of the partitioning of guest molecules into pristine hydrogel particles, 5 μL of guest molecules (0.1 mg mL−1) were added to 100 μL of hydrogel particle dispersion, and the fluorescence intensity inside and outside of the hydrogel particles or compartments, respectively, was recorded. Partitioning of the substrates of the enzyme cascade reaction was determined via addition of the substrates glucose (100 mM) or amplex red (2 mM) to the bulk ATPS solution (1 mL) prior to separating the two phases. Initiation of the enzyme cascade via addition of the enzymes HRP and GOX resulted in the formation of the fluorescent marker resorufin and the fluorescence intensity of the individual phases was recorded.Cascade reaction in multi-compartmentalized hydrogel particles
GOX and HRP-containing hydrogel particles were generated according to the scheme in Fig. 2a. To this end, a template W/O emulsion was prepared at T = 40 °C inside a microfluidic channel. The emulsion was comprised of an aqueous mixture of 2.5% sodium alginate, 1.8 wt% PEG (MW: 35 kDa), 3.3 wt% dextran (MW: 500 kDa), 1 mg mL−1 GOX and 1 mg mL−1 HRP as the dispersed phase and 4 wt% Span85 in heptane as the continuous phase. After different time points of allowing phase separation to occur via placing the droplets at room temperature, the emulsion was passed through a freezing bath at −20 °C, resulting in the dispersed phase to freeze-solidify. The hydrogel particles were subsequently formed by placing the frozen droplet templates into a cooled calcium chloride solution (T = −1 °C). We added glucose (2 mM) and Amplex red (4 μM) to start the enzyme cascade reaction inside the multi-compartmentalized hydrogel particle and followed the generation of red fluorescence from the reaction product resorufin to track the progress of the reaction.Determination of the Michaelis–Menten constant (Km): the Michaelis Menten constant and the maximum reaction velocity (Vmax) were determined via plotting the evolution of the fluorescence intensity from the reaction product versus the concentration of the substrate glucose (0.002–10 mM). The initial reaction velocity was determined by recording the time required to the product intensity to reach 5% of the maximum product fluorescence intensity recorded after an infinite reaction time. A plot of the experimental data of initial reaction velocity using the Michaelis–Menten equation (using origin software) allowed to determine Km and Vmax (see Fig. S15, ESI†).
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors gratefully acknowledge funding from the Max Planck Society and through the Emmy-Noether program of the German Research Foundation (DFG; Grant No. ZE1121/3-1). Open Access funding provided by the Max Planck Society.Notes and references
- A. A. Hyman, C. A. Weber and F. Jülicher, Annu. Rev. Cell Dev. Biol., 2014, 30, 39–58 CrossRef CAS PubMed.
- Y. Shin and C. P. Brangwynne, Science, 2017, 357, eaaf4382 CrossRef PubMed.
- B. Alberts, D. Bray, K. Hopkin, A. D. Johnson, J. Lewis, M. Raff, K. Roberts and P. Walter, Essential Cell Biology, W.W. Norton & Company, New York, 2016 Search PubMed.
- S. Kumar, M. Karmacharya, I. J. Michael, Y. Choi, J. Kim and Y. K. Cho, Nat. Catal., 2021, 4, 763 CrossRef CAS.
- M. Feric, N. Vaidya, T. S. Harmon, D. M. Mitrea, L. Zhu, T. M. Richardson, R. W. Kriwacki, R. V. Pappu and C. P. Brangwynne, Cell, 2016, 165, 1686 CrossRef CAS PubMed.
- A. Bhattacharya, H. Niederholtmeyer, K. A. Podolsky, R. Bhattacharya, J. J. Song, R. J. Brea, C. H. Tsai, S. K. Sinha and N. K. Devaraj, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 18206 CrossRef CAS PubMed.
- H. Tan, S. Guo, N. D. Dinh, R. Luo, L. Jin and C. H. Chen, Nat. Commun., 2017, 8, 663 CrossRef PubMed.
- K. Zhou, K. T. Tian, C. Wang, H. Zhao, N. Gao, H. Yin, P. Wang, B. J. Ravoo and G. Li, J. Am. Chem. Soc., 2020, 142, 20605 CrossRef CAS PubMed.
- S. Jiang, L. Caire da Silva, T. Ivanov, M. Mottola and K. Landfester, Angew. Chem., Int. Ed., 2022, 61, e202113784 CrossRef CAS PubMed.
- M. Marguet, C. Bonduelle and S. Lecommandoux, Chem. Soc. Rev., 2013, 42, 512 RSC.
- J. Bauermann, S. Laha, P. M. McCall, F. Jülicher and C. A. Weber, J. Am. Chem. Soc., 2022, 144, 19294 CrossRef CAS PubMed.
- W. Mu, Z. Ji, M. Zhou, J. Wu, Y. Lin and Y. Qiao, Sci. Adv., 2021, 7, eabf9000 CrossRef CAS PubMed.
- M. Pavlovic, A. Plucinski, J. Zhang, M. Antonietti, L. Zeininger and B. V. K. J. Schmidt, Langmuir, 2020, 36, 1401–1408 CrossRef CAS PubMed.
- T. S. Cvetnić, A. Šalić, M. Benković, T. Jurina, D. Valinger, J. A. Kljusurić, B. Bruno Zelić and A. J. Tušek, Catalysts, 2023, 13, 708 CrossRef.
- T. Trantidou, M. Friddin, Y. Elani, N. J. Brooks, R. V. Law, J. M. Seddon and O. Ces, ACS Nano, 2017, 11, 6549–6565 CrossRef CAS PubMed.
- P. S. Marqués, B. D. Frank., A. Savateev and L. Zeininger, Adv. Opt. Mater., 2021, 9, 2101139 CrossRef.
- C. Guindani, L. C. da Silva, S. Cao, T. Ivanov and K. Landfester, Angew. Chem., Int. Ed., 2022, 61, e202110855 CrossRef CAS PubMed.
- N. Gao and S. Mann, Acc. Chem. Res., 2023, 56, 297–307 CrossRef CAS PubMed.
- E. Rideau, R. Dimova, P. Schwille, R. R. Wurm and K. Landfester, Chem. Soc. Rev., 2018, 47, 8572 RSC.
- L. M. Dominak and C. D. Keating, Langmuir, 2007, 23, 7148–7154 CrossRef CAS PubMed.
- N. N. Deng, M. Yelleswarapu, L. Zheng and W. T. S. J. Huck, Am. Chem. Soc., 2017, 139, 587–590 CrossRef CAS PubMed.
- M. Pavlovic, M. Antonietti and L. Zeininger, Chem. Commun., 2021, 57, 1631 RSC.
- J. Liu, L. Tian, Y. Qiao, S. Zhou, A. J. Patil, K. Wang, M. Li and S. Mann, Angew. Chem., Int. Ed., 2020, 59, 6853–6859 CrossRef CAS PubMed.
- S. Wiese, A. C. Spiess and W. Richtering, Angew. Chem., Int. Ed., 2013, 52, 567 CrossRef PubMed.
- J. G. Werner, S. Nawar, A. A. Solovev and D. A. Weitz, Macromolecules, 2018, 51, 5798 CrossRef CAS.
- M. Karg, A. Pich, T. Hellweg, T. Hoare, L. A. Lyon, J. J. Crassous, D. Suzuki, R. A. Gumerov, S. Schneider, I. I. Potemkin and W. Richtering, Langmuir, 2019, 35, 6231–6255 CrossRef CAS PubMed.
- D. Gaur, N. C. Dubey and B. P. Tripathi, Adv. Colloid Interface Sci., 2022, 299, 102566 CrossRef CAS PubMed.
- Y. Elani, R. V. Law and O. Ces, Nat. Commun., 2014, 5, 5305 CrossRef CAS PubMed.
- L. Hosta-Rigau, O. Shimoni, B. Städler and F. Caruso, Small, 2013, 9, 3573 CrossRef CAS PubMed.
- I. Maity, C. Sharma, F. Lossada and A. Walther, Angew. Chem., Int. Ed., 2021, 60, 22537 CrossRef CAS PubMed.
- M. W. Allen, J. W. Hindley, D. K. Baxani, O. Ces and Y. Elani, Nat. Rev. Chem., 2022, 6, 562 CrossRef CAS PubMed.
- S. Utech, R. Prodanovic, A. S. Mao, R. Ostafe, D. J. Mooney and D. A. Weitz, Adv. Healthcare Mater., 2015, 4, 1628 CrossRef CAS PubMed.
- K. Y. Lee and D. J. Mooney, Prog. Polym. Sci., 2012, 37, 106 CrossRef CAS PubMed.
- M. Mohajeri, M. Eskandari, Z. S. M. Ghazali and H. S. Ghazali, Biomed. Phys. Eng. Express, 2022, 8, 022001 CrossRef PubMed.
- D. M. Hariyadi and N. Islam, Adv. Pharmacol. Pharm. Sci., 2020, 2020, 8886095 CAS.
- L. Yu, C. Ni, S. M. Grist, C. Bayly and K. C. Cheung, Biomed. Microdevices, 2015, 17, 33.3 CrossRef PubMed.
- A. Letocha, M. Miastkowska and E. Sikora, Polymers, 2022, 14, 3834 CrossRef CAS PubMed.
- L. B. Zhao, L. Pan, K. Zhang, S. S. Guo, W. Liu, Y. Wang, Y. Chen, X. Z. Zhao and H. L. W. Chan, Lab Chip, 2009, 9, 2981 RSC.
- T. Kamperman, V. D. Trikalitis, M. Karperien, C. W. Visser and J. Leijten, ACS Appl. Mater. Interfaces, 2018, 10, 23433 CrossRef CAS PubMed.
- H. Sun, H. Zheng, Q. Tang, Y. Dong, F. Qu, Y. Wang, G. Yang and T. Meng, ACS Appl. Mater. Interfaces, 2019, 11, 37313 CrossRef CAS PubMed.
- L. Schoonen and J. C. M. Van Hest, Adv. Mater., 2016, 28, 1109 CrossRef CAS PubMed.
- P. Gobbo, A. J. Patil, M. Li, R. Harniman, W. H. Briscoe and S. Mann, Nat. Mater., 2018, 17, 1145 CrossRef CAS PubMed.
- M. Pavlovic, M. Antonietti, B. V. K. J. Schmidt and L. Zeininger, J. Colloid Interface Sci., 2020, 575, 88 CrossRef CAS PubMed.
- L. Shang, F. Ye, M. Li and Y. Zhao, Chem. Soc. Rev., 2022, 51, 4075 RSC.
- W. M. Aumiller, B. W. Davis, E. Hatzakis and C. D. J. Keating, J. Phys. Chem. B, 2014, 118, 10624 CrossRef CAS PubMed.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248 CrossRef CAS PubMed.
- G. R. Lopes, D. C. G. A. Pinto and A. M. S. Silva, RSC Adv., 2014, 4, 37244 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sm00816a |
| This journal is © The Royal Society of Chemistry 2023 |