
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-supported MOF/cellulose-nanocrystals materials designed from ultrafiltration†
Lorenzo
Metilli
 *a,
Héloïse
Ugo
b,
William
Chèvremont
c,
Cyril
Picard
b and
Frédéric
Pignon
a
*a,
Héloïse
Ugo
b,
William
Chèvremont
c,
Cyril
Picard
b and
Frédéric
Pignon
a
aUniversity Grenoble Alpes, CNRS, Grenoble INP (Institute of Engineering Univ. Grenoble Alpes), LRP, Grenoble F-38000, France. E-mail: lorenzo.metilli@univ-grenoble-alpes.fr
bUniv. Grenoble Alpes, CNRS, LIPhy, 38000 Grenoble, France
cESRF, The European Synchrotron, CS 40220, Grenoble F-38043, France
First published on 16th October 2023
Abstract
Metal–organic-frameworks (MOFs) are promising materials for addressing critical issues such as petrochemical separation, water purification, energy storage and drug delivery. Their large-scale deployment, however, is hampered by a limited processability due to their powdery nature. Recently, the hybridization of MOFs with biopolymers has emerged as a greener, biocompatible strategy to shape MOFs composites into more processable membranes, films, and porous materials. In this work, cellulose nanocrystals (CNCs) were used in combination with ZIF-8 (a widely used synthetic zeolite) to produce hybrid composites through ultrafiltration. Results showed that small quantities of CNCs (1 to 20 CNC:ZIF-8 volume ratio) were sufficient to form a self-supported, dense deposit with high ZIF-8 loadings. Compared to classical MOF in situ growth strategies, this approach allowed the tuning of the composition of the final nanocomposite by controlling the nature and quantities of particles in the suspension. The fabrication of the deposit was strongly dependent on the physiochemical properties of the suspension, which were fully characterized with a set of complementary techniques, including in situ SAXS. This technique was employed to investigate the filtration process, which exhibited a homogeneous deposition of ZIF-8 particles mediated by CNC self-assembly. Finally, the available pore volume and integrity of the internal porosity of ZIF-8 were characterized by water porosimetry, demonstrating that the presence of CNCs did not alter the properties of the supported ZIF-8.
1 Introduction
Metal–organic-frameworks (MOFs) are a class of “coordination network compounds with inorganic ligands, containing potential voids” as defined by IUPAC,1 which has received enormous interest over the last 20 years, due to their appealing potential for several industrial applications, ranging from petrochemical separation, gas storage, catalysis, sensing, and drug delivery. The attractiveness of these materials lies in the precise architecture of nano-sized pores and channels found in MOFs, which endow them with variable levels of porosity, surface area, tuneable reactivity and stability.2,3 The immense combinatorial library of metal nodes and organic ligands allowed the synthesis of almost 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 MOFs, as reported in the Cambridge Structural Database (CSD).4
000 MOFs, as reported in the Cambridge Structural Database (CSD).4
Despite the tremendous research efforts on MOFs at the lab scale, only a few dozen compounds are currently mass-produced and employed in real-world applications.2 Such significant gap is due to both economic and technological bottlenecks, as the majority of MOFs feature high production costs, exhibit low yield and/or purity, and require synthesis conditions which do not meet current sustainability standards. Moreover, processability of MOFs constitutes an often overlooked issue for their application:2,5,6 in fact, MOFs are typically produced as (sub)micron powders, which suffer from low packing density and dispersibility, hence their deployment in industrial applications is challenging.7 Shaping of MOFs as pellets or monoliths requires energy-intensive processes (such as direct coating8 or extrusion9), and it is usually accompanied by loss in surface area and selectivity.10 The processability of MOFs is ameliorated through the blending with polymeric materials, which enables the production of mixed matrix membranes (MMMs), hydrogel and aerogels.11,12 Nevertheless, the use of synthetic polymers derived from hydrocarbons raises sustainability concerns, as well as biocompatibility issues.
An alternative strategy, reported first almost a decade ago,13 involves the replacement of synthetic polymers with biopolymers, and their hybridization with MOF particles. Due to their hydrophilicity and variety of polar groups, biopolymers provide physiochemical interactions with MOF particles and allow their shaping into films, gels and foams.14 Amongst many polysaccharides, cellulose nanomaterials such as nanofibers (CNF) and nanocrystals (CNC) are a popular choice for bio-MOF composites.15,16 Cellulose is the most abundant biopolymer on Earth, and its derived anisotropic particles possess high mechanical resistance, self-assembling abilities, and diverse surface chemistry functionalization. These properties endow CNFs and CNCs with high compatibility with inorganic nanoparticles, and subsequently the formation of novel hybrid materials with outstanding stability, optical and barrier properties.17 The preparation of MOF-cellulose structures proceeds either by in situ synthesis of the MOF particles on the surface of the cellulose nanomaterials, or by the direct mixing of pre-formed MOFs and cellulose. The former enables high control over the size of the nanoparticles and a homogeneous distribution of the two components, and the possibility of creating layer-by-layer materials. The latter, however, guarantees high MOF loadings, at the expenses of cellulose-MOF structural homogeneity.16
Zeolitic Imidazolate Framework 8 (ZIF-8) is a synthetic zeolite containing zinc centres connected by 2-methylimizatolate units. Its mild synthesis conditions, large pore volume (0.663 cm3 g−1) and surface area (as large as 1947 m2 g−1), and small pore gate size (3.4 Å) grant ZIF-8 excellent molecular sieving properties, as well as catalytic, sensing and energy storage abilities.18–21 Several publications detail the in situ synthesis of ZIF-8 particles on cellulose nanomaterials to overcome the drawbacks of its manufactured pristine form. Recent examples include composite membranes and aerogels for gas separation,22–25 sequestration of pollutants from water,26–32 as well as nanocomposites for catalysis33 and electrically conductive materials34 or drug delivery systems.35 While these composite structures have made their mark in terms of applicability, their preparation falls back on the limitation imposed by laboratory-scale in situ synthesis of MOFs. Direct mixing of cellulose and pre-prepared MOFs enables simpler fabrication procedure with controlled, high loadings of nanoporous particles into the cellulose matrix, and above all, the usage of mass-produced MOFs. Zhu et al. (2022) and Qiu et al. (2022) developed composite membranes for water purification of CNFs and ZIF-8, by mixing and vacuum filtering the two components.36,37 Another example was reported by Zhu et al. (2016), where CNCs and ZIF-8 where mixed and then freeze-dried to yield an aerogel for heavy metal sequestration from water.38 The authors were able to incorporate as much as 50% by weight of MOFs into the structure, which was strengthened by cross-linking of chemically modified CNCs.
In the above examples, the cellulose/ZIF-8 final material was produced by evaporation or freeze-drying, without a tight control over the structure and orientation of the cellulose and ZIF-8 components. Several properties, including mechanical resistance, mass transport and therefore performance were not optimized. Ultrafiltration of anisotropic colloidal suspension represents an efficient method to fabricate concentrated assemblies of nanoparticles with defined orientation.39–41 For instance, in the work of Semeraro et al. (2020), an aqueous suspension of CNCs (0.04% volume fraction) was filtered to yield a concentrated deposit (25% volume fraction) with an oriented, chiral nematic organization of the CNCs.41 The study of this filtration process with in situ Synchrotron Radiation Small Angle X-Ray Scattering (SAXS) provided valuable information on the time and spatial evolution of the concentration and particle orientation at the nanoscale, enabling the fine-tuning of the filtration parameters in order to control the nanocomposite structure. Up to now, ultrafiltration of mixed cellulose-MOF suspension has not yet been reported as a processing method to fabricate hybrid nanocomposites.
In this work, the interaction between CNCs and ZIF-8 was exploited, together with ultrafiltration, to prepare a self-supported nanostructured material with densely packed ZIF-8 particles. The dispersant ability of CNCs towards ZIF-8 particles was first demonstrated, by thorough characterization of the physiochemical properties of the mixed suspension. Afterwards, the filtration process was characterized using in situ SAXS, with a particular focus on the formation of the deposit. Finally, the nanostructure of the resulting composite material was observed ex situ using electron microscopy, while the available pore volume of ZIF-8 was measured using a bespoke water porosimeter.42,43
2 Materials and methods
2.1 Materials
Cellulose nanocrystals (CNCs) were purchased from CelluForce (CelluForce, Canada) as a spray-dried powder. Suspensions were prepared by dispersing a specific weight of CNCs in ultrapure water, to which NaCl (Sigma-Aldrich, France) was added to reach a controlled ionic strength (1 mM). The sample was mixed with a magnetic stirrer for 2 hours, and then sonicated with a Branson Digital Sonifier (Emerson Electric Company, USA) for 15 minutes at 30% duty cycle. ZIF-8 (Basolite Z1200) was purchased from Sigma-Aldrich (Sigma-Aldrich, France). Mixtures of ZIF-8 and CNCs were prepared by adding ZIF-8 to a suspension of sonicated CNCs and sonicated further for 15 minutes at 30% duty cycle. To simplify the nomenclature of samples, mixtures of CNCs and ZIF-8 were labelled according to the volume ratio between CNCs and ZIF-8, expressed as the parameter α, and the total mass fraction of particles in the suspension, expressed as w. For example, a suspension containing CNCs and ZIF-8 in a 1:10 volume ratio, and a total mass fraction of 3%, was termed α10w3.
From this nomenclature, the mass and volume fraction of the individual components can be calculated with eqn (1) and (2):
 | (1) |
 | (2) |
2.2 Particle size measurements
The particle size distribution of the suspensions was determined by combining static light scattering (SLS) and dynamic light scattering (DLS), due to the high polydispersity found in the samples. In particular, SLS was used to study the size of the particles aggregates, in the range between 1 and 1000 μm, whereas DLS was most efficient in measuring individual particles size, in the range within 1 nm to 10 μm. SLS was carried out using a Malvern Mastersizer 2000 (Malvern, UK) equipped with an aqueous dispersion cell. For each sample, the obscuration level was set to 20%, and the measurement was carried out in triplicates. DLS was performed using a Vasco Particle Size Analyzer (Cordouan technologies, France). The samples were diluted with ultrapure water to 0.1% by weight before the measurements, and measured in triplicates at 20 °C.2.3 Surface charge
The surface charge of suspended particles was measured with a ZEN6200 Zetasizer (Malvern, UK). The concentration of the suspensions was set to 0.1 wt%, and the measurements were conducted in five repeats at 20 °C.2.4 Microscopy
Optical microscopy images of the suspensions were obtained using a Nikon Eclipse Ti2 (Nikon, Japan) using a ×20 magnification objective. Higher magnification images were obtained with scanning and transmission electron microscopes (SEM and TEM). For SEM, the filtration deposits were fractured, and placed on top of a metallic stub covered with carbon tape. The surface of the sample was coated with Au/Pd in a Baltec MED 020 sputter coater (Baltec, Switzerland), and observed using a FEI Quanta-FEG 250 microscope (FEI, USA) operating at an acceleration voltage of 2.5 kV. An estimate of the crystal size distribution was obtained by manually measuring the particles in 10 images using ImageJ 1.53t (National Institute for Health, USA). For TEM, a droplet of diluted suspension was placed on a glow-discharged carbon-coated copper grid and allowed to dry. The samples were then imaged using a JEOL JEM-2100-Plus microscope (JEOL, Japan) operating at an accelerating voltage of 120 kV. Images were acquired with a Gatan Rio 16 camera (Ametek, USA).2.5 Rheological measurements
The rheology of the samples was investigated using a stress-controlled MCR301 rheometer (Anton Paar, Austria). For oscillatory rheology, a cone-and-plate geometry of 50 mm in diameter (truncation height 149 μm, angle 0.991°) was used. Amplitude sweeps were carried out at a fixed 1 Hz oscillation frequency, between 1% and 1000% deformation. Viscosity curves were instead measured with a 75 mm cone-and-plate system (truncation height 149 μm, angle 0.991°). The samples were sheared from 1000 s−1 to 0.01 s−1, and the resulting data was fitted to either a Herschel–Bulkley model44 (eqn (3)):σ = σy + K![[small gamma, Greek, dot above]](https://www.rsc.org/images/entities/i_char_e0a2.gif) n n | (3) |
the shear rate (s−1), and n the dimensionless flow index. If the sample does not exhibit shear stress, i.e. σy = 0, this model reduces to a power law (eqn (4):| σ = Kn | (4) |
In both setups, measurements were carried out at 20 °C in triplicates. For the rheo-optical measurements, the geometry was changed to a quartz parallel-plate, 25 mm in diameter, and a gap of 300 μm. The samples were sheared at 1000 s−1 for 15 s, followed by a shear rate of 1 s−1 for 5 minutes. The appearance of the sample under shear was then recorded using a Basler Ace acA2500 digital camera (Basler, Germany).
2.6 Suspension stability
The stability of the suspension against phase separation was determined using a LuMiFuge Stability Analyzer (LUM, Germany). The instrument measured the extinction of light transmission in sample tubes during centrifugation, accelerating the kinetics of separation. The suspensions (2 mL) were placed in plastic tubes and spun at 1000 rpm for 15 minutes. The cumulative light transmission was measured for each sample, and its evolution over time was linked to the samples' stability.2.7 Small angle X-ray scattering (SAXS)
SAXS measurements were performed at beamline TRUSAXS of the European Synchrotron Radiation Facility (ESRF, Grenoble, France). The samples were analysed using a wavelength λ = 0.095 nm, and a sample-detector-distance (SDD) of 10 and 1.5 m, corresponding to a q (scattering vector in nm−1) range between 0.007 and 6 nm−1. Aqueous suspensions of CNCs and ZIF-8 were analysed both in static conditions and under frontal filtration (in situ). For all samples, background and solvent were subtracted from the signal prior to analysis, thus obtaining the absolute scattering intensity I(q) (mm−1). Regarding the static measurements, a set of CNC suspensions were measured to obtain the position of the interparticle distance peak as a function of concentration. To do so, the structure factor S(q) was calculated by dividing the azimuthally-averaged scattering intensities by the form factor F(q) of CNCs. This was calculated by fitting the scattering intensity of a diluted CNC suspension (0.14 wt%) with a parallelepiped fit using the software Sasview (https://www.sasview.org). The position of the peak in q space was converted to real space with the relationship d = 2π/q, plotted against the sample concentration, and fitted using an exponential decay (Fig. S1 of the ESI†).45 The equation was later used to calculate the concentration of CNCs during filtration from the position of the S(q) peak.The filtration was characterized in situ using a custom-made cell in polycarbonate similar to the one described in Semeraro et al. (2020).41 Briefly, a channel of 100 × 4 × 8 mm was separated by a metal support and a PVDE membrane with 0.2 μm pore size (Orelis Environment, France) (see Fig. 1). The suspension was filtered by applying a transmembrane pressure of 1.2 bar for 30 minutes; during this time, 2D X-ray scattering patterns were acquired through the cell window at different heights positions in steps of 25 μm continuously. The starting point (Δz = 0) corresponded to the closest position to the membrane where the signal was not affected by the presence of the PVDE. The 2D scattering patterns were integrated azimuthally over the whole q range to yield 1D scattering plots using SAXSUtilities software.46 The corresponding structure factor profiles S(q) were obtained by dividing them by the CNCs form factor F(q). The interparticle distance peak in S(q) was then used to probe the evolution of the concentration of CNCs during filtration. For samples containing anisotropic particles, the degree of anisotropy was calculated with the software SASET,47 using a model-free principal component analysis (PCA) method; the value of anisotropy is comprised between fully isotropic (0) and fully oriented particles (1). The permeate was collected in a recipient and its weight measured using a Precisa 400 M balance over time (Precisa, France).
 | ||
| Fig. 1 Schematic depiction (a) and photograph (b) of the in situ SAXS filtration setup at beamline TRUSAXS, ESRF (Grenoble). The suspension enters the upper chamber following the applied transmembrane pressure ΔP, and it is forced through the PVDF membrane, which is supported by a metallic mesh. The X-Ray beam passes through the polycarbonate windows, analysing the filtration deposit along the z direction. The permeate is then collected from the outlet in a container, and its mass measured during the filtration process. | ||
2.8 Fabrication of CNC/ZIF-8 deposits
Larger filtration deposits of CNCs and ZIF-8 were prepared through frontal filtration under the same conditions, using however a circular cell, 25 mm in diameter. The filtration was applied for 48 hours, and the permeate mass was measured over time with a Precisa 400 M balance over time (Precisa, France). At the end of the filtration, the deposit was recovered and sealed in a plastic container for further analysis. The specific deposit resistance, which provides information on permeability of the deposit, was calculated by measuring the slope of the t/V vs. V plot, where t is the time and V is the volume of permeate.482.9 Thermogravimetric analysis (TGA)
A small quantity of the filtration deposit (ca. 30 mg) was placed into the TGA furnace and was heated from 30 °C to 900 °C at 5 °C min−1 in oxygen atmosphere. The measurement was repeated in triplicates, and the ratio between CNC and ZIF-8 in the deposit was estimated from the area under the mass loss curve of the two components.2.10 Water porosimetry
The water intrusion–extrusion properties of ZIF-8 and CNCs deposits were assessed by using a custom-made liquid porosimeter.42,49 Owing to the unique property of water to access the pores of ZIF-8 above a specific pressure, this techniques has been widely reported to study the accessibility and integrity of ZIF-8 particles, under many experimental condition.50,51 Briefly, the sample was placed into a dedicated deformable high-pressure cell, immersed in distilled water as a pressure-transmitting fluid. The pressure was monitored using a dedicated sensor placed inside the sample chamber. The experiments were conducted at room temperature and fixed compression-dilatation speed, by means of a piston moving at constant velocity of 1 mm s−1. The piston was activated by an electro-mechanical press (MTS C45, USA). Upon displacement of the piston, the variation of the sample chamber was calculated, and the pressure measured accordingly. Several sensors were used to record the pressure (T.E.I. PHP167, France), the axial displacement of the piston (MicroEpsilon LD1630-10, Germany) and the temperature (T.C. PT100, France) in the chamber versus time. After subtraction of the elastic contribution due to liquid compressibility and system finite rigidity, the intruded and extruded volume was normalized by multiplying the theoretical ZIF-8 available pore volume (350 mm3 g−1) by the estimated ZIF-8 mass in the samples. Finally, the data was plotted as a function of normalized volume and pressure for different samples.3 Results and discussion
3.1 Physiochemical properties of CNCs/ZIF-8 aqueous suspensions
A first experiment based on the frontal filtration of a ZIF-8 suspension without and with a small amount of CNCs is show in Fig. 2. The comparison of these two situations is particularly instructive. | ||
| Fig. 2 Photographs of the frontal filtration of two different suspensions: (a) pure ZIF-8 with mass fraction of 1.4%, showing a water depletion layer between the membrane and the floating ZIF-8, and (b) a mixture of ZIF-8 and CNCs with the same total mass fraction of 1.4%, and a volume fraction of CNCs of 1/20 relative to ZIF-8 (α20w1.4), exhibiting no phase separation, and the formation of a filtration deposit. | ||
In the case of ZIF-8 aqueous suspension, upon application of the transmembrane pressure, the ZIF-8 particles started aggregating in macroscopic, floating flocs (see Movie M1 in the ESI†) which could not be deposited at the membrane surface during filtration (Fig. 2a). The addition of a small quantity of CNC particles (1:20 volume ratio) significantly affected the hydrodynamic behaviour of the system, enabling the formation of a compact deposit (Fig. 2b). The suspension above the membrane appeared homogeneous through the filtration process (see Movie M2 in the ESI†). In fact, ZIF-8 is known for its high hydrophobicity and relatively low density (0.9 g mL−1), making its dispersion in water challenging.52–54 Consequently, aqueous suspensions of ZIF-8 are unstable and tend to phase-separate quickly under gravity (see Fig. S2 of the ESI†). Fig. 3a and b shows the appearance of ZIF-8 in its powder and suspension state, respectively.
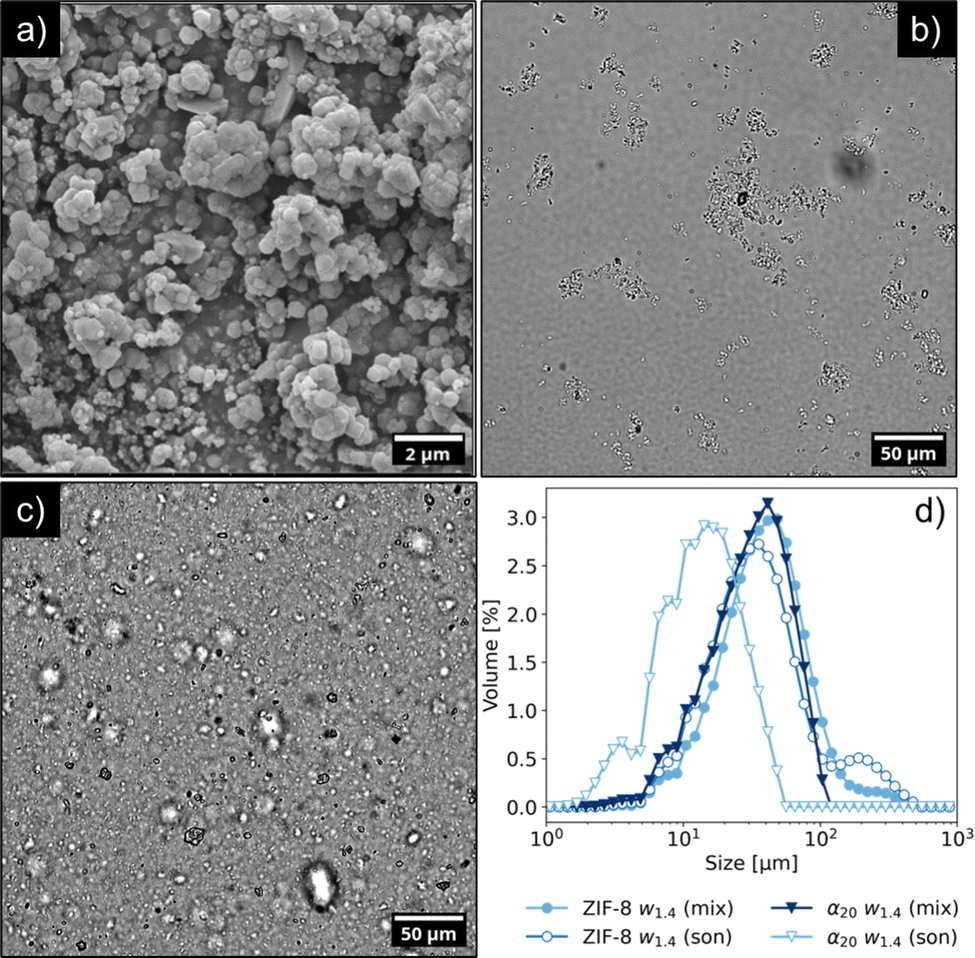 | ||
| Fig. 3 (a) SEM image of Basolite Z1200 powder, illustrating the polydisperse nature of ZIF-8 crystals, (b) optical microscopy image of an aqueous suspension of ZIF-8, 1.4 wt%, showing extended aggregation between particles. (c) Aqueous suspension of CNCs and ZIF-8 in 1:20 volume ratio (α20w1.4) after sonication, demonstrating the ability of small quantities of CNCs to reduce the size of the ZIF-8 aggregates, and (d) static light scattering (SLS) measurements of ZIF-8 suspension stirred for 1 hour (filled circles) and after sonication (empty circles), compared with the size distribution of the α20w1.4 sample stirred for 1 hour (filled downward triangles) and after sonication (empty downward triangles). | ||
SEM analysis showed that ZIF-8 crystals displayed a polydisperse size distribution, ranging between 100 nm and 1 μm, and appeared to be aggregated. The average diameter was estimated to be 311 ± 159 nm. While some crystals exhibited a defined, rhombic dodecahedron morphology, other particles featured smoother edges, and for smaller crystals the shape could not be determined unambiguously. The presence of such smaller crystals on the surface of the larger ones suggests the occurrence of secondary nucleation. The XRD pattern of the powder agreed with the data reported in literature for Basolite Z1200®9 (Fig. S3 of the ESI†). When mixed with water (Fig. 3b), the ZIF-8 particles retained their aggregated state, owing to strong van der Waals forces.52 Furthermore, the lack of colloidal stability was also highlighted by the low zeta potential in water (+22 mV). Attempts to separate the ZIF-8 particles through sonication were not effective, resulting only in a slight variation of the size distribution (see Fig. S4 of the ESI†). However, as shown in Fig. 3c, the impact of sonication was completely different in presence of CNCs. In particular, Fig. 3d details the impact on the size distribution of ZIF-8 in absence or presence of CNCs.
Microscopy images revealed that the branched, ZIF-8 aggregates were replaced with either individual ZIF-8 particles or smaller, rounder aggregates with average size of 10 μm. CNCs acted as a dispersant for ZIF-8, reducing its aggregation (although not completely) and by stabilizing the resulting suspension for at least 24 h (see Fig. S2 in the ESI†). In the case of a α1w1.4 sample, the surface charge of the sample was −49 mV, a value close to the one of CNC aqueous suspension. This value suggests that the ZIF-8 particles were covered with a layer of CNCs, owing to the electrostatic interactions between opposite charges.15,16
The morphology of the particles is shown at higher magnification in Fig. 4. From the TEM images, it could be noted that excess amounts of CNCs were visible in the background rather than surrounding the ZIF-8 particles, suggesting that ZIF-8 suspensions might be stabilized by fewer CNCs than a 1:1 CNC:ZIF-8 volume ratio. Secondly, the CNCs formed a dense layer around the ZIF-8 particles, as well as an inter-connecting network between aggregates. Further insight about the size of the particles could be inferred from DLS measurements, which were complementary to SLS (Fig. 5). The size distribution of CNCs displayed an average value of 231 nm, in good agreement with the TEM images (see Fig. S5 in the ESI†). The size distribution of ZIF-8 ranged from 1 to 10 μm, approaching however the upper limit of detection of the DLS instrument. It is worth noticing that this increase in intensity matched the distribution curve observed in Fig. 3 for ZIF-8 by static light scattering. The size distribution of sample α1w0.1 was between 0.2 and 2 μm, which corresponded to smaller aggregates or individual ZIF-8 crystals, possibly surrounded by a layer of CNCs. This size range was in agreement with the TEM images of the ZIF-8 clusters shown in Fig. 4a and b.
 | ||
| Fig. 4 Transmission electron microscopy (TEM) images of a dried α1w1.4 sonicated sample at low magnification (a) and high magnification (b). The darker objects are the ZIF-8 clusters, whereas the CNCs display lower contrast and are visible in the background. The ZIF-8 clusters could be found within an extended network of CNCs, which bind together the ZIF-8 crystals, and form tethers between clusters of ZIF-8 crystals as well. The scale bars are 10 μm (a) and 300 nm (b), respectively. | ||
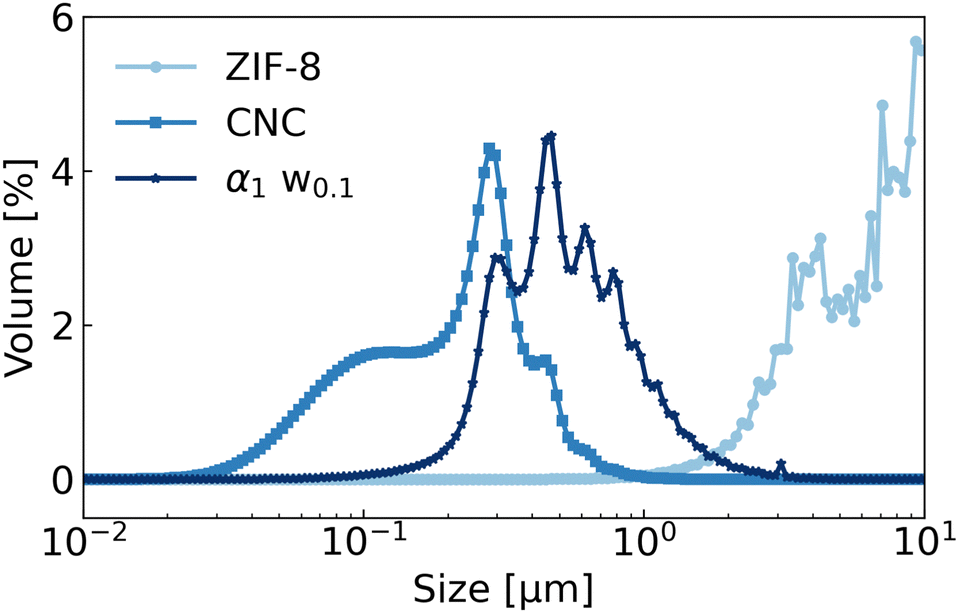 | ||
| Fig. 5 Dynamic Light Scattering (DLS) measurements of ZIF-8, CNCs and a sonicated α1w0.1 sample. The measured volume distribution for ZIF-8 suspensions approached the upper limit of detection for the DLS instrument, at 10 μm. | ||
3.2 Rheology of CNC/ZIF-8 suspensions
The flow behaviour of the suspensions was also investigated. The results of oscillatory rheometry are presented in Fig. 6.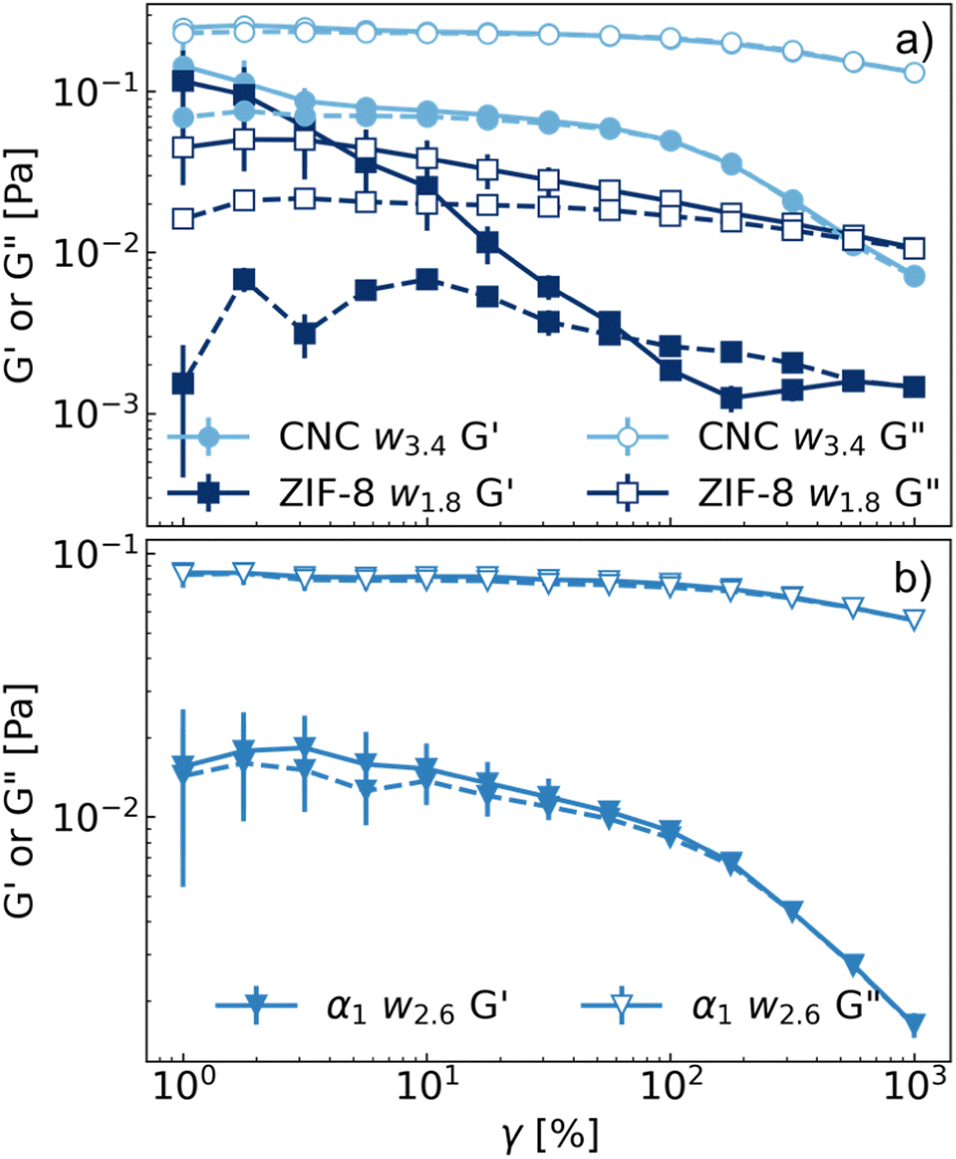 | ||
| Fig. 6 (a) Oscillatory rheometry experiments of CNCs (circles), ZIF-8 (squares) and (b) α1w2.6 (downward triangles) suspensions with corresponding volume fraction of ϕ = 0.02. Solid lines represent oscillations starting from from low (1%) to high deformations (1000%), whereas dashed lines from high to low deformations. Values of G′ are represented by filled symbols, values of G′′ in empty symbols. | ||
CNC suspensions were liquid-like (G′′ > G′) in the explored range of deformation; when shearing from low to high deformation, a small decrease in G′ could be observed, suggesting the occurrence of a weak yield stress behaviour, related to the presence of liquid crystalline phases of CNCs.45,55–57 ZIF-8 suspensions exhibited a crossing point between G′ and G′′ at low strain rates, which could account for the disruption of ZIF-8 aggregates during shear. In fact, both G′ and G′′ showed a lower value during the oscillations from high to low deformation, with G′ < G′′. Concerning the CNC/ZIF-8 mixture, it could be appreciated that the G′ and G′′ were intermediate with respect to the pure CNC and ZIF-8 dispersions (Fig. 6b). Most importantly, no hysteresis effect was observed during the oscillation for G′ or G′′, suggesting that the network of ZIF-8 aggregates was not present anymore. This hypothesis will be discussed in further detail in the following paragraphs.
To investigate the effect of composition on the physiochemical properties of the suspensions, samples with different α ratios were prepared (see Table 1). Stability analysis by centrifugation revealed that the minimum ratio of CNC to ZIF-8 required to prevent phase-separation was 1 to 10 (Fig. S6a of the ESI†). Sample α20w3 displayed some degree of phase separation through partial creaming of the suspension, while for sample α30w3 the amount of CNCs was clearly insufficient to stabilize the mixture, displaying a similar profile to the pure ZIF-8 water suspension. Concerning the zeta potential (Fig. S6b of the ESI†), it remained negative and close to the value of CNCs-only suspensions (between −45 and −50 mV) up to sample α5w3. At higher ratios, it decreased in absolute value (−27 mV for α20w3) albeit remaining negative, suggesting that the surface of ZIF-8 aggregates were still covered in CNCs. The stability and negative charge persisting with small amounts of CNCs could be explained by the particularly high aspect ratio of CelluForce nanocrystals,58 which enabled a sufficient surface coverage of ZIF-8 aggregates. In line with this observation, lesser amounts of CNCs (i.e., larger α values) were equally effective in reducing the size of ZIF-8 clusters during sonication, converging towards an average size of 20 μm (Fig. S7b of the ESI†). Therefore, the suspensions contained aggregates of particles with comparable size distribution, varying only in the relative amounts of the two components. The viscosity profiles of these samples were then analysed. The results are presented in Fig. 7.
n) or Power Law (Kn), shown in Fig. S8 of the ESI
| Sample | v c [%] | v c [%] | σ y [Pa] | K [Pa sn] | n |
|---|---|---|---|---|---|
| CNC w5.1 | 3 | 0 | — | 1.38 | 0.42 |
| α 0.1 w 4.9 | 2.74 | 0.27 | — | 0.73 | 0.49 |
| α 0.2 w 4.7 | 2.50 | 0.50 | — | 0.21 | 0.60 |
| α 1 w 3.9 | 1.50 | 1.50 | — | 0.046 | 0.70 |
| α 5 w 3.1 | 0.50 | 2.50 | — | 0.0029 | 0.94 |
| α 10 w 2.9 | 0.27 | 2.74 | — | 0.0019 | 0.98 |
| ZIF-8 w2.7 | 0 | 3 | 0.021 | 0.0028 | 0.91 |
 | ||
| Fig. 7 Viscosity profiles of CNC, ZIF-8, and mixed suspensions with different α values, at a fixed volume fraction of ϕ = 0.03: CNC w5.1 (filled circles), α0.1w4.9 (filled upwards triangles), α0.2w4.7 (empty upwards triangles), α1w3.9 (filled squares), α5w3.1 (filled downwards triangles), α10w2.9 (empty downwards triangles) and ZIF-8 w2.7 (filled stars). The individual volume fractions, vc and vz, are listed in Table 1. On the sides, schematic depiction of the proposed suspension microstructure under shear, showing the effect of the presence of CNCs on the clusters of ZIF-8 crystals. | ||
The flow curve of the CNC suspension was similar to the reported data in the literature,45,55–57,59,60 showing the characteristic shear-thinning profile. The sample containing ZIF-8 also exhibited a shear-thinning nature, although the decrease in viscosity was significantly steeper. The replacement of CNCs particles with ZIF-8 caused a significant decrease in the viscosity of the sample, owing to two simultaneous contributions: a decrease in the volume fraction of CNCs and therefore in their mutual organisation, as well as a decrease in the CNC–CNC interactions by the presence of ZIF-8 clusters. Nevertheless, the shear-thinning behaviour was still visible until sample α1. Suspensions containing larger amounts of ZIF-8 (α5 and α10), instead, exhibited a Newtonian viscous behaviour close to water (1 mPa s), except for a small increase in viscosity for sample α5 at low shear rates. When compared with the profile of ZIF-8 aqueous suspension, it is evident that the presence of lesser amounts of CNCs drastically affected the rheology of the sample. Further analysis of the stress vs. shear rate plot (Fig. S8 in the ESI†) showed that the presence of CNCs removed the yield stress exhibited by ZIF-8 aqueous suspension, at least in the current experimental conditions explored in this work. In summary, CNCs stabilized the ZIF-8 aggregates, preventing their phase separation and “fluidifying” the suspension. This hypothesis was confirmed by rheo-optical measurements (see videos M3, M4 and Fig. S9 in the ESI†). Upon decreasing the shear rate from 1000 s−1 to 1 s−1, the ZIF-8 particles formed a percolated, extended aggregate network within 30 seconds. This phenomenon was also reported for other dispersion under shear, such as carbon black gels.61 Sample α10, on the other hand, remained homogeneous. Such change in the rheology affected the hydrodynamic behaviour of CNC/ZIF-8 suspensions, enabling their filtration as firstly presented in Fig. 2.
3.3 SAXS analysis of CNC/ZIF-8 suspensions
Further analysis of the CNC/ZIF-8 suspensions was conducted with Small Angle X-Ray scattering (SAXS) (Fig. 8). Table 2 details the amounts of particles found in each sample. | ||
| Fig. 8 Azimuthally averaged scattering intensities of aqueous suspensions in static conditions of ZIF-8 (-·-), CNC (–) and CNC/ZIF-8 samples (continuous lines) at different α values, with a fixed total mass fraction of 6%. The individual mass fraction values of CNC and ZIF-8 are presented in Table 2. Results from a Beaucage fit (red line) of the pure ZIF-8 suspension returned a slope of −3.2, and a radius of gyration Rg = 143 nm. | ||
| Sample | w c [%] | w z [%] |
|---|---|---|
| CNC w4 | 4.00 | 0 |
| α 1 w 6 | 3.92 | 2.08 |
| α 5 w 6 | 1.65 | 4.35 |
| α 10 w 6 | 0.94 | 5.05 |
| α 20 w 6 | 0.52 | 5.48 |
| ZIF-8 w3 | 0 | 3 |
The aqueous suspension of ZIF-8 showed an increase in the scattering intensity in the Porod region (0.1 < q < 1 nm−1) with a slope of −3.2, which is close to the data reported in literature for a similar zeolite system;62 this value in particular describes the scattering of particles with rough surfaces. Upon fitting the scattering intensity with a Beaucage model,63 a radius of gyration of 143 nm was found, which accounted likely for the isolated ZIF-8 particles in the suspension. This value agreed with the average size measured from SEM images. The increasing scattering intensity at low q values, however, implies that larger clusters were also present in the sample. Concerning the CNC suspension, the scattering intensity featured a broad shoulder around 0.2 nm−1, which represents the inter-particle distance between individual CNC rods.64,65
For this particular sample (CNC w4), such distance corresponded to d = 31 nm. The scattering from the sonicated mixture of CNC and ZIF-8 (α1) contained features from the two components, i.e., the intensity with slope −3.2 from the ZIF-8 and the interparticle distance of the CNCs. Samples containing larger amounts of ZIF-8 (from α5w6 to α20w6), however, no longer displayed the CNCs structure factor, but retained the form factor between 0.1 and 1 nm−1. Considering the amounts of CNCs present in the suspensions (Table 2), the absence of the structure factor peak in the mixtures could be explained by the following. The position of the structure factor peak was similar between sample α1w6 and CNC w4, which indicated that the corresponding concentration of free, interacting CNCs did not differ significantly in presence of ZIF-8. This agrees with the hypothesis that only low amounts of CNC were necessary to stabilize the ZIF-8 particles by electrostatic interaction. The CNCs in α5w6, instead, were either partially or completely adsorbed to the surface of ZIF-8 particles, reducing the amount of free CNC available in the suspension, and therefore not displaying any structure factor peak. This could be however also due to larger contribution from the quantity of ZIF-8 to the X-Ray scattering intensity. Samples α10w6 and α20w6 showed a progressive decrease in the CNC F(q) relative intensity, due to the decreasing number of cellulose nanocrystals in the suspensions.
3.4 In situ SAXS analysis of CNC/ZIF-8 suspension filtration
Fig. 9 shows the scattering profiles of a α1w3 suspension measured during frontal filtration. | ||
| Fig. 9 (a) Azimuthally averaged scattering intensities along the z-axis of a α1w3 suspension, obtained from in situ SAXS during frontal filtration, at rest (ΔP = 0), and (b) the respective calculated S(q) (b). The different plots represent the scattering intensity at different z positions, each one separated by 25 μm, along the height of the filtration cell window, as shown in Fig. 1. (c) Azimuthally averaged scattering intensities at the same z positions and (d) the corresponding S(q) after 9 minutes of filtration (ΔP = 1.2 bar). | ||
At rest, the suspension showed a slight gradient in intensity and position of the S(q) peaks, in proximity with the membrane (Δz = 0) (Fig. 9a and b). This could be ascribed to the shear induced by loading the sample into the filtration cell. After applying the transmembrane pressure, the scattering intensities measured close to the membrane displayed both an increase in intensity and a shift of the CNC S(q) peak towards higher q values, following an increase in concentration of CNCs and ZIF-8 particles (Fig. 9c and d). The concentration of CNCs, calculated from the position of the S(q) peak, and the anisotropy of the 2D scattering patterns, as a function of time and distance from the membrane, are shown in Fig. 10.
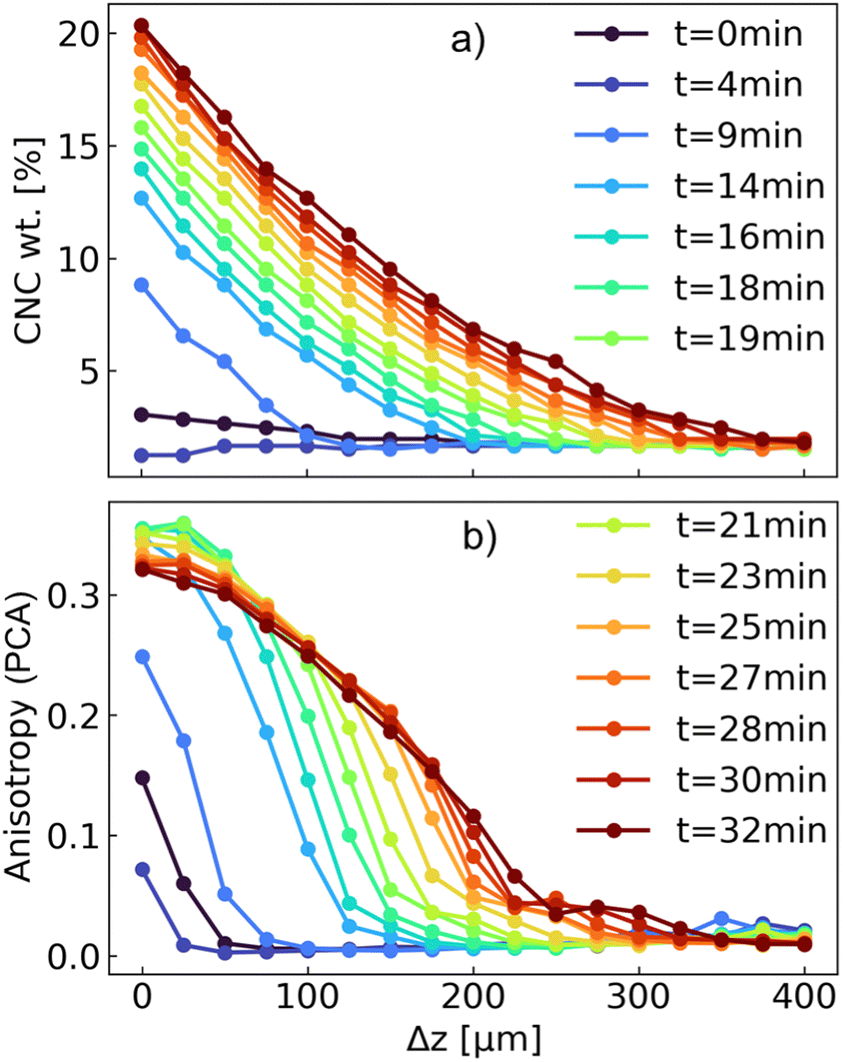 | ||
| Fig. 10 (a) Evolution of the weight concentration of CNC and (b) anisotropy, calculated from PCA, as a function of filtration time and distance from the membrane in the region of interest (up to 400 μm from the surface of the membrane) for the α1w3 suspension. The weight concentration of CNC was calculated from the S(q) peak, as demonstrated in Fig. S1 of the ESI,† whereas the anisotropy was computed from the 2D scattering intensities, using the SASET software. | ||
After 9 minutes of frontal filtration, a significant increase in the CNC concentration was observed: the nominal CNC concentration in the starting suspension was 1.96 wt% and increased almost by an order of magnitude for the closest layers to the PVDF membrane. After 30 minutes, the concentration at the interface reached ca. 20 wt%, with the formation of a deposit of ca. 350 μm in thickness. A similar trend was observed for the azimuthally-averaged scattering intensity at q = 1 nm−1, to which both CNC and ZIF-8 contributed (Fig. S10 of the ESI†). At the same time, the anisotropy increased rapidly from 0 (totally isotropic) to 0.25 within 9 minutes. Interestingly, the anisotropy did not increase significantly further with the CNC concentration, which could be explained by several factors. Considering the presence of the ZIF-8 particles and their concomitant deposition during filtration, the overall anisotropy of free CNCs, which were oriented perpendicular to the filtration direction was counteracted by CNCs oriented parallel to the surface of ZIF-8 crystals, as well as the isotropic scattering of ZIF-8 itself. In fact, it could be seen that, beyond 19 minutes of filtration, the anisotropy at the surface of the membrane started decreasing slightly. This was visible also in the 2D scattering pattern, which displayed only a moderate, vertical orientation during the filtration process (Fig. S11 of the ESI†).
While the in situ filtration was monitored for a relatively short amount of time, similar results were obtained by probing the nanostructure of a deposit filtered for 72 h, starting from a α1w1.4 suspension (Fig. S12 of the ESI†). The initial suspension contained 0.92 wt% of CNCs and 0.48 wt% of ZIF-8. From the S(q) plot, it could be seen that the azimuthally-averaged scattering intensities displayed the same profile as Fig. 9d, with a slight shift in the S(q) of CNCs along the height of the deposit. In this case, the gradient of CNCs concentration ranged from ca. 9 wt%, close to the membrane, to 6 wt% along the rest of the deposit. The anisotropy decreased accordingly with the concentration of CNCs and ranged between 0.2 and 0.05. Within the assumption that the ratio between CNCs and ZIF-8 was maintained during the formation of the deposit, the concentration of CNCs increased from 0.92 wt% to an average 6 wt%, and the ZIF-8 from 0.48 wt% to an estimated 3.13 wt%. Therefore, a six-fold increase in concentration was possible for CNC/ZIF-8 suspensions under frontal filtration.
To obtain sufficiently large filtration deposits, additional samples were prepared using the circular filtration cell. The filtration profiles of the samples with different α values are presented in Fig. 11.
 | ||
| Fig. 11 Ultrafiltration processes, carried out in the circular cell, of CNC/ZIF-8 suspensions with different α values, and a fixed mass fraction of 3%. Evolution of the permeate volume over time (a) and inverse flow rate plot (b). The slopes of t/V vs. V for the samples correspond to the deposit resistance, which is reported in Table 3, together with the mass fractions of CNCs and ZIF-8 for each sample. | ||
For all samples, the permeate mass (volume) of the suspensions exhibited a constant decline in flux over the filtration time. In general, in the first phase of filtration, a deposit is formed rapidly on the surface of the membrane, which causes then a decline in flux.66 It can be noticed that pure CNC, α0.1w3 and α0.2w3 displayed a similar profile, with the presence of small quantities of ZIF-8 not affecting the hydrodynamic properties of the suspension. Instead, an increasing permeate mass was recorded with larger quantities of ZIF-8 in the mixture. As the resulting filtration deposit contained more ZIF-8 particles in the structure, the layered CNC deposit became more permeable to water. Such trend was reported also in other mixed membrane materials (MMMs).67 This was reflected also in the inverse flow rate plot, which showed how the resistance decreased with increasing quantities of ZIF-8 in the suspension (see Table 3). Furthermore, the slope of the curves, which represents the specific deposit resistance, was constant for each samples, suggesting that the filtration proceeded by accumulating particles with a homogeneous composition, and the same resistance to flow.48 In fact, TGA measurements showed that the filtration deposits contained ZIF-8 and CNCs in a close ratio to the original feed suspension (Fig. S13 of the ESI†). Here, a first decrease in mass at ca. 300 °C corresponded to the degradation of CNCs,58 whereas the ZIF-8 started decomposing just above 600 °C.21
| Sample | w c [%] | w z [%] | Deposit Resistance (s mL−2) |
|---|---|---|---|
| CNC w3 | 3.00 | 0.00 | 1301.65 |
| α 0.1 w 3 | 1.96 | 1.04 | 1729.57 |
| α 0.2 w 3 | 2.85 | 0.15 | 1664.43 |
| α 1 w 3 | 2.71 | 0.29 | 1064.81 |
| α 5 w 3 | 0.82 | 2.18 | 526.32 |
| α 10 w 3 | 0.48 | 2.52 | 221.23 |
| α 20 w 3 | 0.26 | 2.74 | 99.94 |
3.5 Ex situ analysis of CNC/ZIF-8 deposits
The filtrated deposits were therefore dried (Fig. 12) and analysed with scanning electron microscopy (SEM) (Fig. 13). | ||
| Fig. 12 Photographs of a dried, self-supported α10w3 deposit obtained by ultrafiltration process after 48 h, using the circular filtration cell. | ||
 | ||
| Fig. 13 SEM images, at low and high magnification, of CNC/ZIF-8 filtration deposits, obtained from the filtration of the suspensions presented in Fig. 11, at increasing α values: (a) and (b) α1, (c) and (d) α5, and (e) and (f) α10, (g) and (h) α20. The position of the membrane is shown in the images at lower magnification (a), (c), (e) and (g). | ||
On a larger length scale, sample α1 displayed a layered structure, perpendicular to the direction of filtration (Fig. 13a). Such feature was induced by the self-assembly of CNCs during frontal filtration, as already reported in literature.41 At higher magnification, it could be seen that the ZIF-8 particles and aggregates were embedded within the layer of CNCs, which were distributed also around the ZIF-8 particle surface (Fig. 13b). No segregation of the two components was observed, confirming that the filtration deposit was homogeneous. Samples α5, α10 and α20, on the other hand, exhibited a progressive disappearance of the CNC layers, as CNCs in the feed suspensions were replaced by ZIF-8. The filtration deposit appeared as a sponge-like structure with microscopic voids between ZIF-8 particles, which were covered in thin layers of CNCs. These images demonstrate the high affinity between the two particles, and the ability of CNC to stabilize ZIF-8 particles with as low as 1:20 volume ratio.
Finally, the pore volume of ZIF-8 in the deposits, which is directly linked to the functionality of ZIF-8, and many other nanoporous materials, was measured using the custom-made porosimeter described in the Materials and Methods. The results are shown in Fig. 14.
 | ||
| Fig. 14 Water intrusion (filled symbols) and extrusion cycles (empty symbols) in pure ZIF-8 and CNC/ZIF-8 filtration deposits, used to measure the available pore volume and intrusion/extrusion pressures in ZIF-8 particles. The horizontal dashed line marks the intrusion pressure at 250 bar, while the two vertical lines mark the theoretical pore volume available in ZIF-8 crystals, with a maximum at 350 mm3 g−1. The photograph shows the measurement cell, containing a α1 filtration deposit. Samples are labelled in the following way: pure ZIF-8 (circles), α1 (upward triangles), α10 (squares) and α20 (downward triangles). | ||
The filling of and emptying of the pores is represented by the two plateaus, characterized by an average intrusion pressure of Pint = 250 bar, and an average extrusion pressure Pext = 200 bar. Both values were consistent with the data obtained from a sample made of ZIF-8 and pure water only, meaning that CNCs did not resist significantly to either the intrusion or extrusion process. The normalized intrusion volume, which compares samples with different ZIF-8 loadings, showed that the presence of CNCs did not block the access to the internal pores of ZIF-8. In fact, it can be seen from Fig. 14 that all the curves were aligned at a specific volume of the order of 350 mm3 g−1, in agreement with the porosity measured with water and pure ZIF-8.21 These results implied also that the electrostatically bound layer of CNCs on the surface of ZIF-8 did not alter the intrusion/extrusion pressures. Furthermore, as ZIF-8 was found in the form of aggregates, whether just mixed with water or in presence of CNCs, the same intrusion pressure was able to overcome the van der Waals forces that caused the aggregation and enable the entrance of water molecules in the pores of ZIF-8.
4 Conclusions
In this work, the use of cellulose nanocrystals (CNCs) to improve the processing of a commercial ZIF-8 with ultrafiltration was demonstrated for the first time. Small quantities of CNCs (as low as 1:20 CNC:ZIF-8 volume ratio) were sufficient to reduce the ZIF-8 clusters size (from 40 to 20 μm), and to provide remarkable stability against phase separation to the suspensions, thus enabling the formation of a compact deposit via frontal filtration. The origin of the stabilization was found in the opposite electrostatic charge between ZIF-8 and CNCs, with the high aspect ratio of CNCs particles improving their interaction with the ZIF-8 surfaces. From the rheological point of view, the presence of CNCs prevented aggregation of the ZIF-8 particles under shear, removing the yield stress observed for ZIF-8 aqueous suspensions, making the mixture Newtonian with a viscosity close to the one of water.
The filtration, monitored in situ with synchrotron radiation SAXS, showed that the deposition of CNCs was accompanied by a modest increase in anisotropy, owing to the concurrent presence of isotropic, ZIF-8 particles. Within the investigated filtration conditions, the concentration in the deposit increased six-fold compared to the feed suspension within 72 hours. Electron microscopy images revealed that the ZIF-8 particles were covered in a thin layer of CNCs, even at high α values, which functioned as a natural binder. Finally, both available ZIF-8 porosity and the required water intrusion pressure were not affected by the presence of the cellulose nanocrystals, thus retaining its efficiency as a mesoporous material.
While the present work focused on ZIF-8, the findings are applicable to the stabilization and processing of other MOF or zeolite materials which suffer from poor processability. Fine-tuning of the suspension properties and filtration conditions are pivotal in the production of hybrid biopolymer-inorganic nanocomposites, enhancing their deployment in scalable, industrial applications.
Author contributions
Lorenzo Metilli: conceptualization, data curation, formal analysis, investigation, writing – original draft. Héloïse Ugo: data curation, formal analysis, investigation, methodology, validation. WIlliam Chèvremont: investigation, methodology. Cyril Picard: conceptualization, funding acquisition, project administration, supervision, writing – review and editing. Frédéric Pignon: conceptualization, funding acquisition, project administration, supervision, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Didier Blésès, Frédéric Hugenell and François Caton (Laboratoire Rhéologie et Procédés) for their technical assistance. Christine Lancelot-Pin and Jean-Luc Puteaux (Cermav) are also acknowledged for the electron microscopy images and scientific discussion. ESRF is acknowledged for provision of synchrotron beamtime (proposal SC-5319). The author(s) acknowledge(s) the support of the French Agence Nationale de la Recherche (ANR), under grant ANR-20-CE43-0015 (ANISOFILM). LabEx Tec21 (Investissements dAvenir – grant agreement ANR-11-LABX-0030) is acknowledged for providing the funding for the project. LRP and CERMAV are part of Institut Carnot PolyNat (Investissements dAvenir – grant agreement ANR-16-CARN-0025-01), and the Glyco@Alps programme (Investissements dAvenir – grant agreement ANR-15-IDEX-02).Notes and references
- S. R. Batten, N. R. Champness, X.-M. Chen, J. Garcia-Martinez, S. Kitagawa, L. Öhrström, M. O'Keeffe, M. Paik Suh and J. Reedijk, Pure Appl. Chem., 2013, 85, 1715–1724 CrossRef CAS.
- R. Freund, O. Zaremba, G. Arnauts, R. Ameloot, G. Skorupskii, M. Dinca, A. Bavykina, J. Gascon, A. Ejsmont, J. Goscianska, M. Kalmutzki, U. Lächelt, E. Ploetz, C. S. Diercks and S. Wuttke, Angew. Chem., Int. Ed., 2021, 60, 23975–24001 CrossRef CAS PubMed.
- T. Rasheed, K. Rizwan, M. Bilal and H. M. N. Iqbal, Molecules, 2020, 25, 1598 CrossRef CAS PubMed.
- D. Ongari, L. Talirz and B. Smit, ACS Cent. Sci., 2020, 6, 1890–1900 CrossRef CAS PubMed.
- Y. Belmabkhout and K. Cordova, Reticular Chemistry and Applications: Metal-Organic Frameworks, De Gruyter, 2023 Search PubMed.
- M. Rubio-Martinez, C. Avci-Camur, A. W. Thornton, I. Imaz, D. Maspoch and M. R. Hill, Chem. Soc. Rev., 2017, 46, 3453–3480 RSC.
- P. Cheng, C. Wang, Y. V. Kaneti, M. Eguchi, J. Lin, Y. Yamauchi and J. Na, Langmuir, 2020, 36, 4231–4249 CrossRef CAS PubMed.
- N. A. H. M. Nordin, A. F. Ismail, A. Mustafa, R. S. Murali and T. Matsuura, RSC Adv., 2014, 4, 52530–52541 RSC.
- A. Almasoudi and R. Mokaya, J. Mater. Chem., 2012, 22, 146–152 RSC.
- Z. Wang, L. Liu, Z. Li, N. Goyal, T. Du, J. He and G. K. Li, Energy Fuels, 2022, 36, 2927–2944 CrossRef CAS.
- Y. Cheng, S. J. Datta, S. Zhou, J. Jia, O. Shekhah and M. Eddaoudi, Chem. Soc. Rev., 2022, 51, 8300–8350 RSC.
- A. Knebel, A. Bavykina, S. J. Datta, L. Sundermann, L. Garzon-Tovar, Y. Lebedev, S. Durini, R. Ahmad, S. M. Kozlov, G. Shterk, M. Karunakaran, I. D. Carja, D. Simic, I. Weilert, M. Klüppel, U. Giese, L. Cavallo, M. Rueping, M. Eddaoudi, J. Caro and J. Gascon, Nat. Mater., 2020, 19, 1346–1353 CrossRef CAS PubMed.
- E. Laurila, J. Thunberg, S. P. Argent, N. R. Champness, S. Zacharias, G. Westman and L. Öhrström, Adv. Eng. Mater., 2015, 17, 1282–1286 CrossRef CAS.
- S. El Hankari, M. Bousmina and A. El Kadib, Prog. Mater. Sci., 2019, 106, 100579 CrossRef CAS.
- Y. Lu, C. Liu, C. Mei, J. Sun, J. Lee, Q. Wu, M. A. Hubbe and M.-C. Li, Coord. Chem. Rev., 2022, 461, 214496 CrossRef CAS.
- K. Tu, Y. Ding and T. Keplinger, Carbohydr. Polym., 2022, 291, 119539 CrossRef CAS PubMed.
- K. De France, Z. Zeng, T. Wu and G. Nyström, Adv. Mater., 2021, 33, 2000657 CrossRef CAS PubMed.
- G. Confalonieri, T. J. Daou, H. Nouali, R. Arletti and A. Ryzhikov, Molecules, 2020, 25, 2145 CrossRef CAS PubMed.
- V. Hoseinpour and Z. Shariatinia, Tissue Cell, 2021, 72, 101588 CrossRef CAS PubMed.
- S. Kouser, A. Hezam, M. J. N. Khadri and S. A. Khanum, J. Porous Mater., 2022, 29, 663–681 CrossRef CAS.
- K. S. Park, Z. Ni, A. P. Côté, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186–10191 CrossRef CAS PubMed.
- M. Jia, X.-F. Zhang, Y. Feng, Y. Zhou and J. Yao, J. Membr. Sci., 2020, 595, 117579 CrossRef CAS.
- H. Ma, Z. Wang, X.-F. Zhang, M. Ding and J. Yao, Carbohydr. Polym., 2021, 270, 118376 CrossRef CAS PubMed.
- Z. Su, M. Zhang, Z. Lu, S. Song, Y. Zhao and Y. Hao, Cellulose, 2018, 25, 1997–2008 CrossRef CAS.
- J. Thunberg, S. C. Zacharias, M. Hasani, O. A. Oyetunji, F. M. A. Noa, G. Westman and L. Öhrström, Inorganics, 2021, 9, 84 CrossRef CAS.
- L. Chen, F. Wang, W. Yang, D. Gu, M. Li and Q. Pan, ChemistrySelect, 2020, 5, 4078–4084 CrossRef CAS.
- Y. Song, J. Y. Seo, H. Kim and K.-Y. Beak, Carbohydr. Polym., 2019, 222, 115018 CrossRef CAS PubMed.
- S. Ma, M. Zhang, J. Nie, J. Tan, S. Song and Y. Luo, Carbohydr. Polym., 2019, 208, 328–335 CrossRef CAS PubMed.
- W. Meng, S. Wang, H. Lv, Z. Wang, X. Han, Z. Zhou and J. Pu, BioResources, 2022, 17, 2615–2631 CAS.
- H. Nasser Abdelhamid and A. P. Mathew, Chem. Eng. J., 2021, 426, 131733 CrossRef CAS.
- L. Zhu, L. Zong, X. Wu, M. Li, H. Wang, J. You and C. Li, ACS Nano, 2018, 12, 4462–4468 CrossRef CAS PubMed.
- Q. Zhou, J. Chen, B. Jin, S. Chu and R. Peng, Cellulose, 2021, 28, 5241–5256 CrossRef CAS.
- S. Zhang, Y. Xu, D. Zhao, W. Chen, H. Li and C. Hou, Molecules, 2019, 25, 124 CrossRef PubMed.
- Q. Li, X. An and X. Qian, Polymers, 2022, 14, 332 CrossRef CAS PubMed.
- Y. Liu, Y. Huo, M. Li, C. Qin and H. Liu, Cellulose, 2022, 29, 379–393 CrossRef.
- W. Zhu, M. Han, D. Kim, Y. Zhang, G. Kwon, J. You, C. Jia and J. Kim, Environ. Res., 2022, 205, 112417 CrossRef CAS PubMed.
- Z. Qiu, X. Shao, Y. Chen, J. Pan, F. Qiu and T. Zhang, J. Membr. Sci., 2022, 646, 120255 CrossRef CAS.
- H. Zhu, X. Yang, E. D. Cranston and S. Zhu, Adv. Mater., 2016, 28, 7652–7657 CrossRef CAS PubMed.
- Y. Jin, N. Hengl, S. Baup, F. Pignon, N. Gondrexon, M. Sztucki, A. Romdhane, A. Guillet and M. Aurousseau, Carbohydr. Polym., 2015, 124, 66–76 CrossRef CAS PubMed.
- F. Pignon, M. Abyan, C. David, A. Magnin and M. Sztucki, Langmuir, 2012, 28, 1083–1094 CrossRef CAS PubMed.
- E. F. Semeraro, N. Hengl, M. Karrouch, L. J. Michot, E. Paineau, B. Jean, J.-L. Putaux, C. Lancelon-Pin, L. Sharpnack and F. Pignon, Colloids Surf., A, 2020, 584, 124030 CrossRef CAS.
- L. Guillemot, A. Galarneau, G. Vigier, T. Abensur and É. Charlaix, Rev. Sci. Instrum., 2012, 83, 105105 CrossRef PubMed.
- M. Michelin-Jamois, C. Picard, G. Vigier and E. Charlaix, Phys. Rev. Lett., 2015, 115, 036101 CrossRef PubMed.
- V. W. H. Herschel and R. Bulkley, Kolloid-Z., 1926, 39, 291–300 CrossRef.
- E. Gicquel, J. Bras, C. Rey, J.-L. Putaux, F. Pignon, B. Jean and C. Martin, Cellulose, 2019, 26, 7619–7634 CrossRef CAS.
- M. Sztucki, SAXSutilities2: A Graphical User Interface for Processing and Analysis of Small-Angle X-ray Scattering Data, 2021 Search PubMed.
- M. Muthig, S. Prévost, R. Orglmeister and M. Gradzielski, J. Appl. Crystallogr., 2013, 46, 1187–1195 CrossRef CAS.
- P. Bacchin, P. Aimar and R. Field, J. Membr. Sci., 2006, 281, 42–69 CrossRef CAS.
- C. Picard, V. Gérard, L. Michel, X. Cattoën and E. Charlaix, J. Chem. Phys., 2021, 154, 164710 CrossRef CAS PubMed.
- I. Khay, G. Chaplais, H. Nouali, C. Marichal and J. Patarin, RSC Adv., 2015, 5, 31514–31518 RSC.
- Y. Grosu, S. Gomes, G. Renaudin, J.-P. E. Grolier, V. Eroshenko and J.-M. Nedelec, RSC Adv., 2015, 5, 89498–89502 RSC.
- C. Avci, I. Imaz, A. Carné-Sánchez, J. A. Pariente, N. Tasios, J. Pérez-Carvajal, M. I. Alonso, A. Blanco, M. Dijkstra, C. López and D. Maspoch, Nat. Chem., 2018, 10, 78–84 CrossRef CAS PubMed.
- Z. Jia, G. Wu, D. Wu, Z. Tong and W. S. Winston Ho, J. Porous Mater., 2017, 24, 1655–1660 CrossRef CAS.
- P. Song, X. Mao, Y. Ren, H. Zeng and Q. Lu, Langmuir, 2020, 36, 2322–2329 CrossRef CAS PubMed.
- S. Shafiei-Sabet, W. Y. Hamad and S. G. Hatzikiriakos, Langmuir, 2012, 28, 17124–17133 CrossRef CAS PubMed.
- M. Fazilati, S. Ingelsten, S. Wojno, T. Nypelö and R. Kádár, J. Rheol., 2021, 65, 1035–1052 CrossRef CAS.
- F. Pignon, M. Challamel, A. De Geyer, M. Elchamaa, E. F. Semeraro, N. Hengl, B. Jean, J.-L. Putaux, E. Gicquel, J. Bras, S. Prevost, M. Sztucki, T. Narayanan and H. Djeridi, Carbohydr. Polym., 2021, 260, 117751 CrossRef CAS PubMed.
- M. S. Reid, M. Villalobos and E. D. Cranston, Langmuir, 2017, 33, 1583–1598 CrossRef CAS PubMed.
- C. Schütz, M. Agthe, A. B. Fall, K. Gordeyeva, V. Guccini, M. Salajková, T. S. Plivelic, J. P. F. Lagerwall, G. Salazar-Alvarez and L. Bergström, Langmuir, 2015, 31, 6507–6513 CrossRef PubMed.
- K. J. De France, K. G. Yager, T. Hoare and E. D. Cranston, Langmuir, 2016, 32, 7564–7571 CrossRef CAS PubMed.
- V. Grenard, N. Taberlet and S. Manneville, Soft Matter, 2011, 7, 3920 RSC.
- S. Saha, S. Springer, M. E. Schweinefuß, D. Pontoni, M. Wiebcke and K. Huber, Cryst. Growth Des., 2016, 16, 2002–2010 CrossRef CAS.
- G. Beaucage, H. K. Kammler and S. E. Pratsinis, J. Appl. Crystallogr., 2004, 37, 523–535 CrossRef CAS.
- W. J. Orts, L. Godbout, R. H. Marchessault and J.-F. Revol, Macromolecules, 1998, 31, 5717–5725 CrossRef CAS.
- F. Cherhal, F. Cousin and I. Capron, Langmuir, 2015, 31, 5596–5602 CrossRef CAS PubMed.
- D. Antelmi, B. Cabane, M. Meireles and P. Aimar, Langmuir, 2001, 17, 7137–7144 CrossRef CAS.
- J. Dai, S. Li, J. Liu, J. He, J. Li, L. Wang and J. Lei, J. Membr. Sci., 2019, 589, 117261 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sm00798g |
| This journal is © The Royal Society of Chemistry 2023 |