
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
3D printable, thermo-responsive, self-healing, graphene oxide containing self-assembled hydrogels formed from block copolymer wormlike micelles†
Qi
Yue
 ab,
Zhidong
Luo
ab,
Xueyuan
Li
ab and
Lee A.
Fielding
*ab
ab,
Zhidong
Luo
ab,
Xueyuan
Li
ab and
Lee A.
Fielding
*ab
aDepartment of Materials, School of Natural Sciences, University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: lee.fielding@manchester.ac.uk
bHenry Royce Institute, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK
First published on 1st August 2023
Abstract
Graphene oxide (GO) containing block copolymer nanocomposite hydrogels formed from poly(glycerol monomethacrylate-block-hydroxypropyl methacrylate) (PGMA-PHPMA) wormlike micelles were prepared by either mixing GO and copolymer at low temperature or via in situ reversible addition-fragmentation chain-transfer (RAFT) polymerisation-induced self-assembly (PISA) of HPMA in the presence of a PGMA macromolecular chain-transfer agent and GO flakes. Hydrogels containing 15–25% w/w copolymer and 0 and 8% w/w GO, based on copolymer, were investigated and the maximum gel strength measured was ∼33 kPa for a 25% w/w copolymer gel prepared by in situ polymerisation and containing 2% w/w GO based on copolymer. This gel strength represents a fifteen-fold increase over the same copolymer gel without the addition of GO. The nanocomposite gels were found to recover efficiently after the application of high shear, with up to 98% healing efficiency within seconds. These gels are also 3D printable, self-healing, adhesive and temperature responsive on cooling and re-heating. The observed properties were both GO and copolymer concentration dependent, and tensile testing demonstrated that the nanocomposite gels had higher moduli, elongation at break and toughness than gels prepared without GO.
Introduction
Graphene oxide (GO) composites are widely studied and often have improved properties over their non-GO containing equivalents.1,2 For example, GO can simply be mixed into polymers to form re-enforced nanocomposites or with gel forming polymers to form hydrogels with improved stiffness and other mechanical properties.3,4 This facile preparation method has advantages such as simplicity, mild conditions and fast, reversible gelation transitions.5–7 In addition, composite hydrogels prepared by mixing a pre-formed material and GO have been demonstrated to be responsive to near-infrared light,8 biocompatible,9–11 and pH responsive.12 GO-containing hydrogels can also be prepared whereby the hydrogel matrix chemically reacts with the GO through the oxygen-containing functional groups on its surface.13–16 In such cases improved properties are often observed. In situ polymerisation describes a method of preparing nanocomposites where a polymerisation reaction occurs in the presence of e.g., nanoparticles.17–20 Typically, the nanoparticles are uniformly dispersed in a reaction mixture which is subsequently polymerised to obtain a nanocomposite material. During this process, polymer chains can be physically adsorbed or chemically grafted to the nanoparticles and composite materials with good particle dispersion and improved properties such as strength, toughness and self-healing ability have been reported.21–26In previous work, our group investigated the preparation of GO-containing block copolymer worm-like micelle (worm) gels prepared by polymerisation-induced self-assembly (PISA) by exploiting thermal worm-to-sphere morphological transitions.27 GO was mixed with pre-formed co-polymer and the gel was allowed to reform on heating or cooling, depending on the formulation. Specifically, the diblock copolymer worm hydrogel composition which allowed nanocomposite preparation at low temperature was based on poly(glycerol monomethacrylate)-b-poly(2-hydroxypropyl methacrylate) (PGMA-PHPMA) prepared via RAFT aqueous dispersion polymerisation.28–32 These PGMA-PHPMA gels are well known to undergo a reversible de-gelation transition when the temperature is reduced from room temperature to ∼4 °C due to a ‘worm-to-sphere’ morphological transition causing the loss of worm-like micelle entanglement.28,33 Furthermore, it was demonstrated that GO could readily be dispersed in the reformed worm gels and provided moderate mechanical reinforcement within specific concentration ranges (15% w/w copolymer concentration and below 1.5% w/w GO concentration).27
PISA-based nanocomposites have also been reported whereby the polymerisation is conducted in the presence of an inorganic component.34–36 For instance, silica nanoparticles have been encapsuled by copolymer vesicles.37,38 This alternative route can reduce the number of synthetic steps, improve the properties of the composites formed and can enable the production of nanocomposite materials that are not attainable through other methodologies.
In the current work, the preparation of GO-containing block copolymer worm gels via in situ polymerization (as opposed to mixing GO with a pre-formed co-polymer) is reported for the first time, whereby the diblock copolymer is synthesised directly in the presence of the GO nano-filler. Specifically, the preparation of GO-containing nanocomposite worm gels via in situ RAFT dispersion polymerisation of HPMA in the presence of GO was investigated (Fig. 1). The effect of GO sheet size and gel preparation method (mixing of copolymer with GO at low temperature versus in situ polymerisation) was examined. Furthermore, the mechanical and rheological properties were investigated for a series of nanocomposite gels prepared by in situ polymerisation using a range GO and copolymer concentrations. For optimised GO loadings the 3D printability, temperature-responsive, and self-healing behaviour of these gels was demonstrated.
 | ||
| Fig. 1 Preparation of graphene oxide (GO) containing block copolymer worm gels via RAFT in-situ polymerization. These nanocomposite gels can be 3D printed, are self-healing at room temperature and are thermo-responsive on cooling to 2 °C. | ||
Experimental
Materials
All reagents were purchased from Sigma-Aldrich (UK) and used as received, unless otherwise noted. Poly(glycerol monomethacrylate) with a mean degree of polymerisation of 58 (PGMA58) was prepared in-house using previously published methods and is described in the Supporting Information.28,29 2-hydroxypropyl methacrylate (HPMA, 97%), was purchased from Alfa Aesar (UK). Graphene oxide aqueous dispersion was purchased from Graphena (Spain) and purified before use. Deionised water was used in all experiments.GO dispersion treatment
GO dispersions (4 mg mL−1) were purified by dialysis against water for 5 days and centrifuged to remove excess water. The collected GO dispersions (∼15 mg mL−1) were sonicated twice using a probe sonicator (Fisher Scientific, maximum output power: 500 W, USA) for 15 min each time before use (80% amplitude and 2 s on/off cycles). The mean intensity-average particle diameter for the GO dispersion after probe sonication was ∼227 nm, as determined by dynamic light scattering (DLS). After 1 week, the intensity-average particle diameter remained below 250 nm (Fig. S5, ESI†). AFM images confirmed the smaller size of the GO sheets after sonication, with GO sheet size distribution being relatively narrow compared with GO dispersions prepared using bath sonication (Fig. S4, ESI†).Preparation of PGMA-PHPMA-x% GO nanocomposite worm gels by in situ polymerisation
PGMA58-PHPMA170-x% GO nanocomposite worm gels were prepared by in situ RAFT aqueous dispersion polymerisation of HPMA in the presence of a PGMA58 macromolecular chain-transfer agent and GO. Specifically, 15, 20 and 25% w/w PGMA58-PHPMA170-x% GO (G58-H170-x% GO, x = 2–8) composite worm gels were prepared. An example protocol for the synthesis of a 20% PGMA58-PHPMA170-4% GO gel is as follows. PGMA58 macro-CTA (2.0 g, 0.208 mmol) and HPMA monomer (5.09 g, 35.3 mmol; target DP = 170) were weighed into a 100 mL round bottomed flask and purged with N2 for 20 min. 4,4-Azobis(4-cyanovaleric acid) (ACVA) was added (19.4 mg, 0.07 mmol, CTA/ACVA molar ratio = 3.0) and purged with N2 for a further 5 min. GO dispersion 18.6 mL (15 mg mL−1) and deionised water (10 mL, producing a 20.0% w/w dispersion in total), which had been previously purged with N2 for 30 min, was then added and the dispersion was purged for a further 5 min prior to immersion in an oil bath set at 70 °C. The reaction was stirred for 3 h before the polymerisation was quenched by exposure to air. The product was a soft free-standing gel (Fig. S7, ESI†). Subsequent composite gels with various compositions were prepared by varying the amount of HPMA, GO dispersion and water to the reaction mixture. 1H NMR analysis was conducted using MeOD after removal of GO via centrifugation (13k rpm, 20 min) and filtration through a 0.2 μm syringe filter. A representative PGMA58-PHPMA170 copolymer worm gel prepared in the absence of GO had an Mn of 45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 and an Mw/Mn of 1.11, as determined by GPC analysis in DMF eluent (Fig. S3, ESI†).
000 g mol−1 and an Mw/Mn of 1.11, as determined by GPC analysis in DMF eluent (Fig. S3, ESI†).
Characterisation
Results and discussion
PGMA-PHPMA-x% GO nanocomposite worm gels prepared via physical mixing copolymer at low temperature in the presence of different sized GO flakes
In our previous work,27 GMA58-PHPMA170-x% GO (G58-H170-x% GO) nanocomposite gels were prepared by initially synthesising a PGMA-PHPMA (G-H) block copolymer worm gel and subsequently mixing an aqueous dispersion of GO with the free-flowing copolymer at low temperature. On re-heating to room temperature, nanocomposite worm gels with improved mechanical properties were obtained. It was hypothesised that the size of the GO flakes would have a direct impact on the observed gel properties. For example, Bai et al.,12,40 reported that GO-polymer network formation required the use of relatively large GO flakes. Hence, GO dispersions were subjected to either bath or probe sonication and the resulting flakes were analysed by AFM (Fig. S4, ESI†). The latter technique clearly results in smaller GO flakes (approximately 200 nm) with a narrower flake size distribution. In both cases the thickness of the measured GO flakes was approximately 1 nm, indicating sonication only affected the flake size and not thickness. DLS measurements taken over a period of 8 days (Fig. S5, ESI†) indicated that the GO dispersions remained colloidally stable with a Z-average diameter of approximately 230 nm. Hence, various G58-H170-x% GO composite gels were synthesised using each GO flake size by physically mixing them with 15% w/w G58-H170 at low temperature and subsequent re-heating. For gels prepared with 0.7 to 2.5% w/w GO, based on copolymer, free-standing nanocomposite gels were obtained (Fig. S6, ESI†). These gels were analysed using oscillatory rheology to determine their gel strengths.Generally, the measured gel strength for the gels prepared with smaller flakes was higher than for those prepared with larger flakes, as demonstrated by Fig. 2. Furthermore, the use of smaller flakes allows the formation of gels over a much wider GO concentration range. For example, at 2.0% w/w GO, the gel strength was ∼5.0 kPa for the nanocomposite gel prepared with small GO flakes whereas with the large flakes a free-flowing liquid was obtained at the same GO concentration. Thus, it seems reasonable to conclude that the presence of relatively large GO flakes has a disrupting effect on worm-gel reformation whereas a relatively uniform population of ∼200 nm flakes facilitates gel-formation and provides a more consistent re-enforcing effect. However, this physical mixing strategy has some important limitations for the preparation of worm-gels with higher GO and G-H concentrations. This is because certain concentrations of worm-gels lose their gel-liquid response at high concentrations and the addition of GO suspension to a pre-formed copolymer necessarily dilutes the overall dispersion.
 | ||
| Fig. 2 Strain-sweep oscillatory rheology data for nanocomposite worm gels prepared by mixing 15% w/w PGMA-PHPMA copolymer at low temperature with various concentrations of GO (based on copolymer) processed using probe (blue data points) and bath (red data points) sonication. The average storage modulus was measured in the linear viscoelastic reign and all measurements were conducted at 10 rad s−1 at room temperature. | ||
G58-H170-x% GO composite gels prepared via in situ polymerisation
The preparation of G58-H170-x% GO composite gels by in situ polymerisation was investigated by chain extending a PGMA58 macromolecular chain-transfer agent (see Fig. S1–S3, ESI†) with HPMA via RAFT aqueous dispersion polymerisation at 15, 20 and 25% w/w copolymer concentration in the presence of GO (0–8.0% w/w GO based on copolymer). The target degree of polymerisation (DP) of the core-forming PHPMA block was fixed at 170 to obtain free-standing worm-gels in the absence of GO (see Fig. S7a and c, ESI†).28 Free-standing gels were obtained in the presence of GO up to 6.0% w/w. At concentrations exceeding 6.0% w/w GO, free-flowing dispersions were obtained. This behaviour is similar to G-H-x% GO nanocomposite gels prepared by physical mixing (Fig. 2)27 but the GO concentration range for obtaining free-standing gels prepared by in situ polymerisation is higher. In addition, these n% G58-H170-x% GO (x ≤ 6) nanocomposite gels exhibit long shelf-life and can remain as free-standing homogeneous gels in their storage vials for at least 6 months (Fig. S7b and d, ESI†).The properties of the nanocomposite gels prepared by in situ polymerisation were subsequently investigated using oscillatory rheology (summarised in Fig. 3d). The measured gel strengths (G′) varied as the GO loading was increased from 0 to 6.0% w/w. For instance, for 15% G58-H170-x% GO the measured gel strength increased from 1.1 kPa to 6.1 kPa as the GO concentration was increased from 0 to 4.0% w/w, based on copolymer (Fig. 3a). On increasing the GO content from 4.0 to 6.0% w/w, the measured gel strength decreased to 3.5 kPa. A similar trend was observed for the 20% G58-H170-x% GO series (Fig. 3b) whereby the gel strength significantly increased from 1.5 kPa to 20.5 kPa when the GO concentration was increased from 0 to 4.0% w/w and then decreased to 3.7 kPa for 6.0% w/w GO. When increasing to a copolymer concentration of 25% w/w (Fig. 3c), the maximum gel strength achieved was with 2.0% GO and resulted in a nanocomposite gel with G′ approximately 33 kPa.
 | ||
| Fig. 3 (a)–(c) Storage modulus (G′) versus % strain for graphene oxide containing PGMA58-PHPMA170 block copolymer dispersions with graphene oxide concentrations between 0 and 6.0% w/w: (a) 15% w/w G58-H170; (b) 20% w/w G58-H170; (c) 25% w/w G58-H170. (d) Average storage moduli measured in the linear viscoelastic region for the range of GO-containing G58-H170 copolymer dispersions studied. All measurements were conducted at 10 rad s−1 at a controlled temperature of 25 °C. | ||
The observed decrease in gel strength above a certain GO concentration occurs for gels prepared by physical mixing and for gels prepared by in situ polymerisation. This was previously hypothesised to be due to higher concentrations of GO flakes affecting copolymer self-assembly into worms through absorption of copolymer chains and/or hindering sphere–sphere fusion.27 The mechanism of this process is not yet fully understood but it is clear that there is an optimal GO concentration for forming composite worm-gels for a given copolymer concentration and gel preparation route. Furthermore, the maximum G′ of a G-H-x% GO gel prepared by physical mixing to-date is ∼5.0 kPa (Fig. 2). Angular frequency rheology measurements (Fig. S8, ESI†) demonstrated that these G58-H170-x% GO composites retain their gel-like behaviour with increasing shear rate, with G′ remaining greater than G′′ up to at least 10 Hz. Thus, in situ polymerisation yields nanocomposite gels with increased strengths by allowing higher copolymer concentrations to be used and for free-standing gels to form at higher GO loadings.
The differences observed between gels prepared by physical mixing and in situ polymerisation are likely due to a combination of improved mixing efficiency and higher copolymer concentrations being used. However, it is noteworthy that Voylov et al.41 utilised GO as a radical initiator and demonstrated that surface-initiated polymerisation can occur from GO. Thus, the improved properties of the gels prepared by in situ polymerisation of HPMA in the presence of GO are potentially due to chain grafting of polymer to GO occurring during radical polymerisation resulting in covalent bonding between the two materials.42 To investigate whether chain grafting occurred during the in situ polymerisations reported herein, Raman spectroscopy was conducted. Fig. S9 (ESI†) shows an increase in the ratio between the D and G band for gels prepared by in situ polymerisation compared to pristine GO and gels prepared by mixing copolymer with GO at low temperature. Additionally, the rate of HPMA polymerisation in the presence of GO was reduced in comparison to chain extension of PGMA in the absence of GO (Fig. S10, ESI†). Thus, these Raman and 1H NMR spectroscopy studies may imply that transformation from sp2 C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C to sp3 C–C occurs on the GO sheets during in situ polymerisation through chain grafting during polymerisation.43 Unfortunately, additional data to determine whether chain grafting during polymerisation occurred could not be obtained and as such this hypothesis remains the subject of further investigations. Nevertheless, the enhanced rheological properties afforded by conducting GO composite gel preparation via in situ polymerisation results in them being interesting materials to study further in terms of their mechanical and responsive properties.
C to sp3 C–C occurs on the GO sheets during in situ polymerisation through chain grafting during polymerisation.43 Unfortunately, additional data to determine whether chain grafting during polymerisation occurred could not be obtained and as such this hypothesis remains the subject of further investigations. Nevertheless, the enhanced rheological properties afforded by conducting GO composite gel preparation via in situ polymerisation results in them being interesting materials to study further in terms of their mechanical and responsive properties.
3D printing and self-healing of G58-H170-x% GO composite gels prepared by in situ polymerisation
The shear-thinning and recovery behaviour of the nanocomposite gels were investigated by oscillatory rheology experiments whereby the shear strain was varied between 0.2% and 100% in 5 minute intervals. This data is summarised in Table S1 (ESI†) and the data for the nanocomposites with the highest gel strength at each copolymer concentration is shown in Fig. 4, middle row. In all cases, when the initial shear strain was 0.2%, the nanocomposite gels were within the linear viscoelastic region and showed solid-like behaviour (G′ > G′′). When the shear strain increased to 100%, the composite gels shear thinned and had liquid-like behaviour (G′′ > G′). After removal of the high shear strain, the samples recovered almost immediately to a solid-like state. | ||
| Fig. 4 (a)–(c) Photographs of GO nanocomposite worm gels after 3D printing. Upper images show samples immediately after 3D printing and being cut and the lower images are after self-healing for 6 h at room temperature. (d)–(f) Oscillatory rheology recovery experiments for GO nanocomposite worm gels measured using a continuous strain sweep with alternating strain (γ = 0.2% and 100%) at 25 °C with an angular frequency of 10 rad s−1. (g)–(i) Healing efficiency determined from oscillatory rheology recovery experiments. | ||
The gel strength after 5 minutes of low shear recovered to between 80 and 98% of the original gel strength, with higher gel strength samples recovering slightly slower than the low gel-strength samples. However, after the initial cycle the recovery remained consistent and after 7 cycles the recovery efficiency did not decrease further for all samples. Interestingly, nanocomposite gels prepared with higher copolymer concentrations had significantly improved healing efficiencies (Fig. 4, bottom row, and Table S1, ESI†). The range of gel strengths achieved and the shear thinning properties of these nanocomposite gels makes them ideal materials for 3D gel printing. Thus, the 15% G58-H170-4% GO, 20% G58-H170-4% GO and 25% G58-H170-2% GO composite gels (the maximum G′ achieved for each copolymer concentration) were printed layer-by-layer using an extrusion-based 3D printer at room temperature. Initially, 3-layer rectangular slabs were printed. Immediately after printing these slabs, they were cut into two pieces and separated by approximately 2 mm (Fig. 4a–c, top images). For the 20 and 25% w/w nanocomposite gels the individual lines from the printing process were still observable whereas for the 15% gel they were not. This correlates with the shear-thinning recovery behaviour described above. Importantly, the individual layers for all samples were already fused together before cutting the slab and no significant extrusion deformation was observed. After 5 h at room temperature (lower images in Fig. 4a–c) the two pieces of composite gel were completely fused together in all cases, and no observable layers or printing artefacts could be seen suggesting excellent self-healing behaviour. Furthermore, the overall shape and size of the printed slab remained relatively constant over this period, with the 15% sample spreading out to a limited extent. These re-healed gels were able to easily adhere-to and suspend samples of wood, plastic, metal, rubber and glass without deformation (Fig. S11, ESI†).
Mechanical properties of G58-H170-x% GO composite gels prepared by in situ polymerisation
Tensile testing data for the G58-H170-x% GO composite gels prepared by in situ polymerisation before and after healing are shown in Fig. 5 and summarised in Table S2 (ESI†). Most of the stress–strain curves had an initial linear gradient up to ∼0.5% strain before yielding. The gels then underwent strain hardening as they were extended until fracture. For example, the 15% w/w copolymer gel with the best performance contained 4% w/w GO based on copolymer. This sample had a Young's modulus of 95 kPa, a fracture strain of 3.5% and a toughness of 486 kJ mm−3 (Fig. 5a). On increasing the copolymer concentration to 20% and 25% w/w the observed properties improved (Fig. 5b and c), with the 25% G58-H170-2% GO having the best overall mechanical properties (modulus = 447 kPa, fracture strain = 13%, toughness = 42000 kJ mm−3) from the gels tested. These observations followed the same trends observed by oscillatory rheology studies (Fig. 3d). In all cases the inclusion of GO resulted in improved properties in comparison to copolymer gels without GO (Fig. S12, ESI†). The data for the best performing gel at each copolymer concentration is summarised in Fig. 5d and e. As expected, increasing the copolymer concentration results in improved properties, but the optimal GO concentration is not entirely consistent between copolymer concentrations.
 | ||
| Fig. 5 (a)–(c) Tensile testing data for 15%, 20% and 25% w/w G58-H170-x% GO composite worm gels. Solid lines show data for initial samples and dashed lines after self-healing at room temperature for 6 h. (d)–(f) Summary of fracture strain, Young's modulus, and toughness of the GO nanocomposite worm gels. | ||
After fracturing the samples, they were re-cast in moulds and allowed to heal at room temperature for several hours before re-testing (dotted lines and orange bars in Fig. 5 for composite gels and Fig. S12 for copolymer gels, ESI†). For the 15 and 20% w/w copolymer gels, the original stress–strain curves appear very similar to the curves obtained after self-healing. This is consistent with the rheology data shown in Fig. 4. It is worth noting that the curves of the healed 20% G58-H170-x% GO gels overlapped almost completely with the original gels suggesting that this is the optimal copolymer composition for rapid self-healing. However, the 25% G58-H170-x% GO samples had distinctly different stress–strain behaviour after healing, showing more plastic deformation when tested, indicating less efficient healing of these samples after undergoing failure under tensile strain. Nevertheless, the re-healed samples still demonstrated better overall tensile testing performance than the lower copolymer concentrations.
Influence of graphene oxide concentration on nanocomposite gel temperature response
It is well-known that G-H copolymer gels undergo a reversible (de)gelation transition at ca. 5–10 °C due to a worm-to-sphere morphological transition.44,45 Thus the thermo-responsive properties of the G-H-x% GO nanocomposite gels with the best mechanical properties were investigated. TEM images of the composite gels diluted at room temperature (Fig. 6a–c) demonstrates that copolymer worms were present and associated with GO flakes. Digital photographs of 3D printed letters also demonstrate that the gels retain their shape and height at room temperature. Upon cooling to 2 °C, the 15% G58-H170-4% GO and 20% G58-H170-4% GO samples underwent the expected phase transition, with only spheres observable in the TEM image of the 15% G58-H170-4% GO sample (diluted at 2 °C) and the 3D printed letters losing their form as the gels transition to a liquid state (Fig. 6d and e).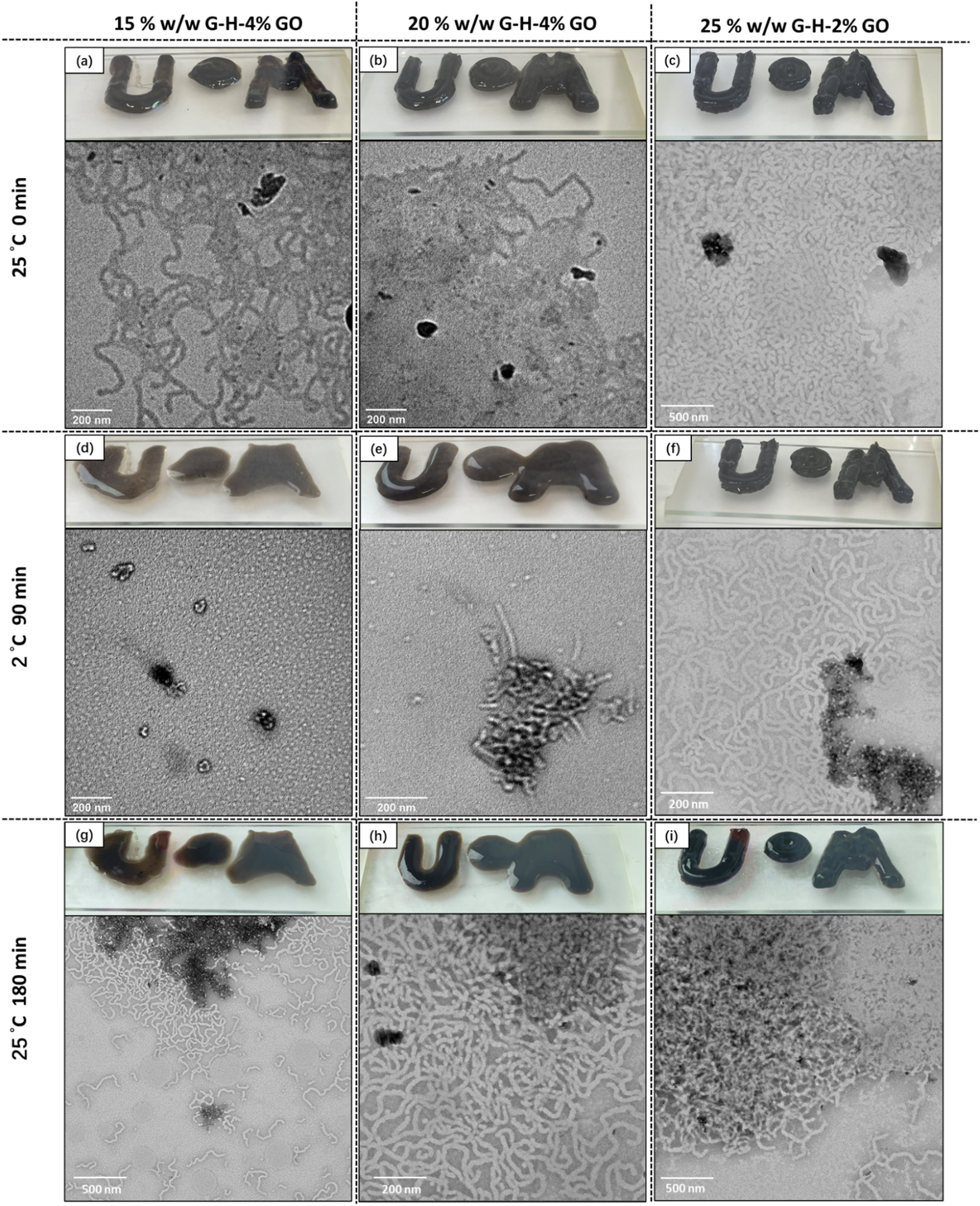 | ||
| Fig. 6 TEM images and 3D printed letters of GO nanocomposite worm gels (a)–(c) at 25 °C; (d)–(f) after being cooled to 2 °C for 90 min and (g)–(i) after returning to 25 °C for 90 min. All TEM samples were diluted to 0.01% w/w before being deposited on to carbon-coated TEM grids at room temperature, expect samples (d)–(f), which were prepared at 2 °C. | ||
On returning to room temperature for 90 min, the 15% G58-H170-4% GO and 20% G58-H170-4% GO samples returned to being a gel and TEM images (Fig. 6g and h) confirmed that the worm-like micelles had re-assembled. Interestingly, the 25% G58-H170-2% GO gel (the sample with the highest gel strength) did not show signs of de-gelation on cooling as the printed letters retained their shape and relatively straight edges (Fig. 6f). Furthermore, TEM investigations of the sample confirmed the presence of worm-like-micelles even after cooling. On reheating and standing for 90 min, worms can clearly be observed by TEM and, whilst the printed letters are now more curved, the layers are still observable after 90 min (Fig. 6i).
Variable temperature rheology studies (Fig. 7 and Table S1, ESI†) supported these observations, with the 15% and 20% w/w copolymer nanocomposite gels having a measurable critical gelation temperature (CGT) on cooling between 2 °C and 6 °C. As expected, these samples showed an increase in G′ and G′′ on heating, with CGT values measured between 2 and 9 °C. Furthermore, on cooling the 25% G58-H170-x% GO gels (Table S1 and Fig. 7c, ESI†) significant decreases in G′ and G′′ and a CGT were not observed. The reversible de-gelation behaviour over several heating–cooling cycles (2 h for each step) was investigated (Fig. 7, bottom row). Similar to the shear-thinning recovery tests in Fig. 4, the 15% G58-H170-4% GO and 20% G58-H170-4% GO gels showed highly reversible re/de-gelation behaviour. The G′ of 15% G58-H170-4% GO gradually reduced from 6 kPa to 4.3 kPa over 8 h of alternating temperature cycles. After being returned to room temperature for 12 h, the value of G′ recovered somewhat but was still lower than the original measured value (Fig. 7c). In comparison, the best performing composite gel prepared by physical mixing to-date (15% G58-H170-2.0% GO) had similar behaviour to the 15% w/w G58-H170-4% gel prepared by in situ polymerisation but had a lower gel strength and recovery efficiency (Fig. S13, ESI†). On increasing the copolymer concentration to 20%, the 20% G58-H170-4% GO gel prepared by in situ polymerisation had improved recovery behaviour after repeated cooling cycles and after 8 h of temperature cycling G′ was only slightly reduced from 20.3 kPa to 17.7 kPa. After 12 h standing at room temperature the original gel strength (20.6 kPa) was recovered. Although, de-gelation was not observed for the 25% G58-H170-2% GO gel, the measured value of G′ did begin to decrease over the course of repeated cooling–heating cycles, from 33 kPa to 28 kPa after 8 h. Nevertheless, after 12 h at room temperature G′ increased back to a slightly higher value than originally measured (35 kPa). This is probably due to a small amount of water evaporation over the course of the measurement. The unique combination of smart properties afforded by these gels allows them to be potentially used in various applications such as responsive information protection. For example, utilising their 3D printability and inherently black coloration, a series of relatively simple morse code (Fig. S14, ESI†) and complex QR codes were 3D printed using 15% w/w G58-H170-4% GO; 20% w/w G58-H170-4% GO and 25% G58-H170-2% GO (Fig. 8, top row). All three samples were suitable for producing these codes and a video of the QR code printing process at 6× speed is available as supporting information (Video S1, ESI†). Furthermore, the response of these QR codes to low temperature varies based on the composition of the gel used to prepare them. After cooling to 2 °C for 90 minutes, the 25% G58-H170-2% GO sample was still readable (Fig. 8, bottom row) However, after being cooled to 2 °C the 15% w/w G58-H170-4% GO and 20% w/w G58-H170-4% GO gels underwent de-gelation. Thus, the originally discrete regions of the QR codes began to lose their shape and coalesce so that they could not be scanned (Fig. 8). This occurred more rapidly for the 15% gel (unreadable after 30 minutes, Fig. S15, ESI†) than the 20% gel (unreadable after 60 minutes, Fig. S15, ESI†). This simple demonstration of thermo-response and 3D printability shows that these nanocomposite gels could potentially be utilised for triggered, or environment-dependent information protection.
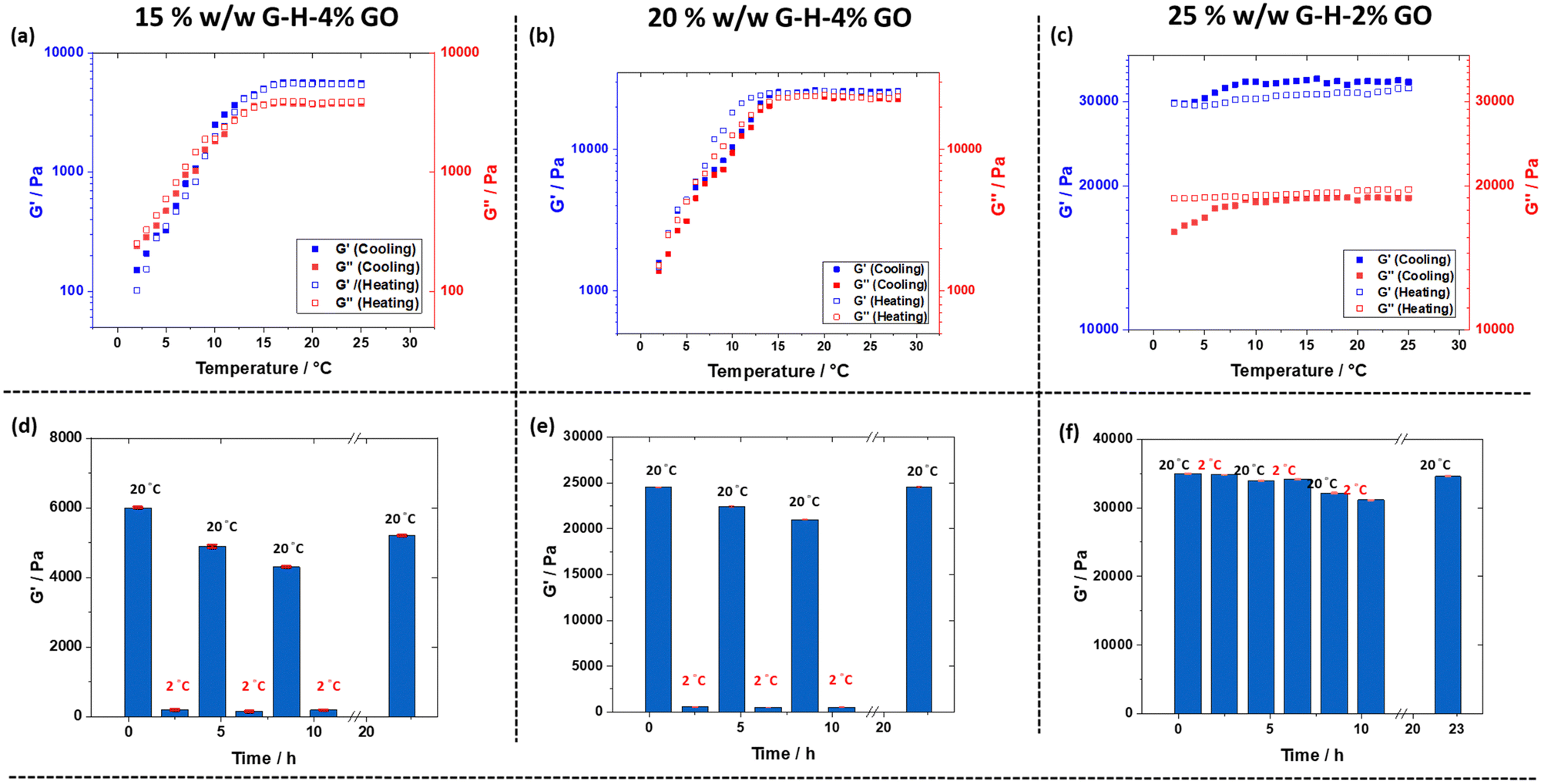 | ||
| Fig. 7 Temperature-dependent oscillatory rheology studies obtained for aqueous dispersions of: (a) 15% G58-H170-4% GO; (b) 20% G58-H170-4% GO; (c) 25% G58-H170-2% GO. The temperature was varied from 25 °C to 2 °C to 25 °C in 1 °C steps with 3 minutes equilibration at each step. (d–f) Storage modulus (G′) measured during temperature-dependent oscillatory rheology studies: (d) 15% G58-H170-4% GO; (e) 20% G58-H170-4% GO and (f) 25% G58-H170-2% GO. The temperature was varied from 20 °C to 2 °C to 20 °C s with 2 h equilibration at each step. The final step was a 12 h equilibration at 20 °C. All measurements were conducted at an angular frequency of 10 rad s−1 and applied strain amplitude of 1.0. | ||
 | ||
| Fig. 8 3D printed QR codes (each 12.5 cm2) using 15% G58-H170-4% GO; 20% G58-H170-4% GO and 25% G58-H170-2% GO nanocomposite worm gels. Upper row: images of QR codes after printing at room temperature. Bottom row: images of QR codes after being held at 2 °C for 90 min. | ||
Conclusions
GO-containing worm gels can be prepared by mixing aqueous dispersions of GO with pre-synthesised PGMA-PHPMA copolymer at low temperature, followed by re-heating the mixture to room temperature. These nanocomposite gels have improved gel strengths in comparison to equivalent PGMA-PHPMA worm-gels prepared without any additive. The gel strength of these gels can be optimised by using relatively small GO flakes in addition to having an appropriate copolymer and GO concentration. The properties of these nanocomposite gels can be further improved by conducting in situ RAFT dispersion polymerisation of HPMA in the presence of GO at relatively high copolymer concentrations. Maximum gel strengths were observed for GO concentrations between 2 and 4% w/w, based on copolymer. Increasing the copolymer concentration from 15 to 25% w/w further increased gel properties such as Young's modulus and toughness, with a maximum recorded gel strength of ∼33 kPa for a 25% G58-H170-2% GO composite worm gel.At copolymer concentrations of 15 and 20% w/w the nanocomposite gels retain their thermo-responsive behaviour which is imparted by the PHPMA core-forming block of the self-assembled worms undergoing a worm-to-sphere morphological transition on cooling. However, upon increasing the copolymer concentration to 25% w/w de-gelation is not observed for these composite gels. The G58-H170-x% GO composite gels prepared by in situ polymerisation were 3D printable due to their shear-thinning behaviour and rapid gel recovery rate after the removal of shear. Due to the reinforcing effect of the added GO, the optimised composite gels retained their 3D printed shape over relatively long periods of time at room temperature without deformation. For the 15 and 20% w/w copolymer gels, printed shapes can lose their form on cooling whereas the 25% w/w copolymer gel retains its shape. Furthermore, these nanocomposite gels have self-healing properties at room temperature, as demonstrated by their recovery behaviour after tensile fracture and from cutting 3D printed gel samples.
Due the multi-responsive nature, tunability, and 3D printability of these nanocomposite gels, they potentially have materials science applications in areas ranging including soft-robotics,46 information printing, encryption and protection,47 and biomaterials.48–52 Furthermore, this approach towards the preparation of nanomaterial containing block copolymer gels is likely not limited to GO as a reinforcing additive and the inclusion of additional additives to provide additional functionality is currently under active investigation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The University of Manchester Electron Microscopy Centre is acknowledged for access to electron microscopy facilities. This work was supported by the Henry Royce Institute for Advanced Materials, funded through EPSRC grants EP/R00661X/1, EP/S019367/1, EP/P025021/1, and EP/P025498/1 and the Sustainable Materials Innovation Hub, funded through the European Regional Development Fund OC15R19P.Notes and references
- S. Park and R. S. Ruoff, Chemical methods for the production of graphenes, Nat. Nanotechnol., 2009, 4(4), 217–224 CrossRef CAS PubMed.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Graphene and graphene oxide: synthesis, properties, and applications, Adv. Mater., 2010, 22(35), 3906–3924 CrossRef CAS PubMed.
- B. N. Tran, S. C. Thickett, V. Agarwal and P. B. Zetterlund, Influence of Polymer Matrix on Polymer/Graphene Oxide Nanocomposite Intrinsic Properties, ACS Appl. Polym. Mater., 2021, 3(10), 5145–5154 CrossRef CAS.
- M. M. Gudarzi and F. Sharif, Self assembly of graphene oxide at the liquid–liquid interface: A new route to the fabrication of graphene based composites, Soft Matter, 2011, 7(7), 3432–3440 RSC.
- H. Guo, T. Jiao, Q. Zhang, W. Guo, Q. Peng and X. Yan, Preparation of graphene oxide-based hydrogels as efficient dye adsorbents for wastewater treatment, Nanoscale Res. Lett., 2015, 10(1), 1–10 CrossRef CAS PubMed.
- R. Bissessur, P. K. Liu, W. White and S. F. Scully, Encapsulation of polyanilines into graphite oxide, Langmuir, 2006, 22(4), 1729–1734 CrossRef CAS PubMed.
- R. Abdollahi, M. T. Taghizadeh and S. Savani, Thermal and mechanical properties of graphene oxide nanocomposite hydrogel based on poly (acrylic acid) grafted onto amylose, Polym. Degrad. Stab., 2018, 147, 151–158 CrossRef CAS.
- J. Duan, F. Liu, Y. Kong, M. Hao, J. He, J. Wang, S. Wang, H. Liu and Y. Sang, Homogeneous chitosan/graphene oxide nanocomposite hydrogel-based actuator driven by efficient photothermally induced water gradients, ACS Appl. Nano Mater., 2020, 3(2), 1002–1009 CrossRef CAS.
- C. Huang, H. Bai, C. Li and G. Shi, A graphene oxide/hemoglobin composite hydrogel for enzymatic catalysis in organic solvents, Chem. Commun., 2011, 47(17), 4962–4964 RSC.
- A. F. de Faria, D. S. T. Martinez, S. M. M. Meira, A. C. M. de Moraes, A. Brandelli, A. G. Souza Filho and O. L. Alves, Anti-adhesion and antibacterial activity of silver nanoparticles supported on graphene oxide sheets, Colloids Surf., B, 2014, 113, 115–124 CrossRef PubMed.
- X.-F. Sun, J. Qin, P.-F. Xia, B.-B. Guo, C.-M. Yang, C. Song and S.-G. Wang, Graphene oxide–silver nanoparticle membrane for biofouling control and water purification, Chem. Eng. J., 2015, 281, 53–59 CrossRef CAS.
- H. Bai, C. Li, X. Wang and G. Shi, A pH-sensitive graphene oxide composite hydrogel, Chem. Commun., 2010, 46(14), 2376–2378 RSC.
- J. T. Paci, T. Belytschko and G. C. Schatz, Computational studies of the structure, behavior upon heating, and mechanical properties of graphite oxide, J. Phys. Chem. C, 2007, 111(49), 18099–18111 CrossRef CAS.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, The chemistry of graphene oxide, Chem. Soc. Rev., 2010, 39(1), 228–240 RSC.
- K. P. Loh, Q. Bao, G. Eda and M. Chhowalla, Graphene oxide as a chemically tunable platform for optical applications, Nat. Chem., 2010, 2(12), 1015–1024 CrossRef CAS PubMed.
- X. Huang, X. Qi, F. Boey and H. Zhang, Graphene-based composites, Chem. Soc. Rev., 2012, 41(2), 666–686 RSC.
- Q. He, S. Wu, Z. Yin and H. Zhang, Graphene-based electronic sensors, Chem. Sci., 2012, 3(6), 1764–1772 RSC.
- C. Zhao, Q. Wang, H. Zhang, S. Passerini and X. Qian, Two-dimensional titanium carbide/RGO composite for high-performance supercapacitors, ACS Appl. Mater. Interfaces, 2016, 8(24), 15661–15667 CrossRef CAS PubMed.
- A. Roy and S. Patra, Development of An All Vanadium Redox Flow Battery For Efficient Utilization of Renewable Energy, IIEST, Shibpur, 2018.
- B. K. Kim, Y. J. Shin, S. M. Cho and H. M. Jeong, Shape-memory behavior of segmented polyurethanes with an amorphous reversible phase: The effect of block length and content, J. Polym. Sci., Part B: Polym. Phys., 2000, 38(20), 2652–2657 CrossRef CAS.
- M. S. Rama and S. Swaminathan, Influence of structure of organic modifiers and polyurethane on the clay dispersion in nanocomposites via in situ polymerization, J. Appl. Polym. Sci., 2010, 118(3), 1774–1786 CAS.
- S. Stankovich, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Synthesis and exfoliation of isocyanate-treated graphene oxide nanoplatelets, Carbon, 2006, 44(15), 3342–3347 CrossRef CAS.
- Y.-Y. Zheng, Preparation and characterization of functionalized graphene oxide nanoribbons/EVA composite films, J. Mater. Eng., 2015, 43(2), 96–102 CAS.
- W.-B. Yang, L. Zhang, J.-W. Liu, H.-R. Liu and B.-H. Tang, Progress in research on preparation and application of graphene composites, J. Mater. Eng., 2015, 43(3), 91–97 CAS.
- J. Shen, B. Yan, T. Li, Y. Long, N. Li and M. Ye, Mechanical, thermal and swelling properties of poly (acrylic acid)–graphene oxide composite hydrogels, Soft Matter, 2012, 8(6), 1831–1836 RSC.
- M. Zhong, Y.-T. Liu and X.-M. Xie, Self-healable, super tough graphene oxide–poly (acrylic acid) nanocomposite hydrogels facilitated by dual cross-linking effects through dynamic ionic interactions, J. Mater. Chem. B, 2015, 3(19), 4001–4008 RSC.
- Q. Yue, S.-P. Wen and L. A. Fielding, Preparation and characterisation of graphene oxide containing block copolymer worm gels, Soft Matter, 2022, 18(12), 2422–2433 RSC.
- N. J. Warren, M. J. Derry, O. O. Mykhaylyk, J. R. Lovett, L. P. Ratcliffe, V. Ladmiral, A. Blanazs, L. A. Fielding and S. P. Armes, Critical dependence of molecular weight on thermoresponsive behavior of diblock copolymer worm gels in aqueous solution, Macromolecules, 2018, 51(21), 8357–8371 CrossRef CAS PubMed.
- M. Williams, N. Penfold, J. Lovett, N. Warren, C. Douglas, N. Doroshenko, P. Verstraete, J. Smets and S. Armes, Bespoke cationic nano-objects via RAFT aqueous dispersion polymerisation, Polym. Chem., 2016, 7(23), 3864–3873 RSC.
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, Mechanistic insights for block copolymer morphologies: how do worms form vesicles?, J. Am. Chem. Soc., 2011, 133(41), 16581–16587 CrossRef CAS PubMed.
- W. Cai, W. Wan, C. Hong, C. Huang and C. Pan, Morphology transitions in RAFT polymerization, Soft Matter, 2010, 6(21), 5554–5561 RSC.
- M. Semsarilar, E. R. Jones, A. Blanazs and S. P. Armes, Efficient Synthesis of Sterically-Stabilized Nano-Objects via RAFT Dispersion Polymerization of Benzyl Methacrylate in Alcoholic Media, Adv. Mater., 2012, 24(25), 3378–3382 CrossRef CAS PubMed.
- J. Lovett, L. Ratcliffe, N. Warren, S. Armes, M. Smallridge, R. Cracknell and B. Saunders, A robust cross-linking strategy for block copolymer worms prepared via polymerization-induced self-assembly, Macromolecules, 2016, 49(8), 2928–2941 CrossRef CAS PubMed.
- X. Qiao, P.-Y. Dugas, B. Charleux, M. Lansalot and E. Bourgeat-Lami, Synthesis of multipod-like silica/polymer latex particles via nitroxide-mediated polymerization-induced self-assembly of amphiphilic block copolymers, Macromolecules, 2015, 48(3), 545–556 CrossRef CAS.
- J. Tan, D. Liu, X. Zhang, C. Huang, J. He, Q. Xu, X. Li and L. Zhang, Facile preparation of hybrid vesicles loaded with silica nanoparticles via aqueous photoinitiated polymerization-induced self-assembly, RSC Adv., 2017, 7(37), 23114–23121 RSC.
- C. J. Mable, R. R. Gibson, S. Prevost, B. E. McKenzie, O. O. Mykhaylyk and S. P. Armes, Loading of silica nanoparticles in block copolymer vesicles during polymerization-induced self-assembly: Encapsulation efficiency and thermally triggered release, J. Am. Chem. Soc., 2015, 137(51), 16098–16108 CrossRef CAS PubMed.
- C. J. Mable, M. J. Derry, K. L. Thompson, L. A. Fielding, O. O. Mykhaylyk and S. P. Armes, Time-resolved saxs studies of the kinetics of thermally triggered release of encapsulated silica nanoparticles from block copolymer vesicles, Macromolecules, 2017, 50(11), 4465–4473 CrossRef CAS PubMed.
- A. Czajka, S. J. Byard and S. P. Armes, Silica nanoparticle-loaded thermoresponsive block copolymer vesicles: a new post-polymerization encapsulation strategy and thermally triggered release, Chem. Sci., 2022, 13(33), 9569–9579 RSC.
- L. A. Fielding, J. A. Lane, M. J. Derry, O. O. Mykhaylyk and S. P. Armes, Thermo-responsive diblock copolymer worm gels in non-polar solvents, J. Am. Chem. Soc., 2014, 136(15), 5790–5798 CrossRef CAS PubMed.
- H. Bai, C. Li, X. Wang and G. Shi, On the gelation of graphene oxide, J. Phys. Chem. C, 2011, 115(13), 5545–5551 CrossRef CAS.
- D. Voylov, T. Saito, B. Lokitz, D. Uhrig, Y. Wang, A. Agapov, A. Holt, V. Bocharova, A. Kisliuk and A. P. Sokolov, Graphene oxide as a radical initiator: free radical and controlled radical polymerization of sodium 4-vinylbenzenesulfonate with graphene oxide, ACS Macro Lett., 2016, 5(2), 199–202 CrossRef CAS PubMed.
- Y. Cai, Y. Fadil, F. Jasinski, S. C. Thickett, V. Agarwal and P. B. Zetterlund, Miniemulsion polymerization using graphene oxide as surfactant: In situ grafting of polymers, Carbon, 2019, 149, 445–451 CrossRef CAS.
- M. Shi, T. Lin, Y. Hu, J. Peng, J. Li and M. Zhai, Functionalization of graphene oxide by radiation grafting polyhedral oligomeric silsesquioxane with improved thermal stability and hydrophilicity, J. Mater. Sci., 2020, 55(4), 1489–1498 CrossRef CAS.
- R. Verber, A. Blanazs and S. Armes, Rheological studies of thermo-responsive diblock copolymer worm gels, Soft Matter, 2012, 8(38), 9915–9922 RSC.
- M. Kocik, O. Mykhaylyk and S. Armes, Aqueous worm gels can be reconstituted from freeze-dried diblock copolymer powder, Soft Matter, 2014, 10(22), 3984–3992 RSC.
- Z. Wang, H. Cui, M. Liu, S. L. Grage, M. Hoffmann, E. Sedghamiz, W. Wenzel and P. A. Levkin, Tough, Transparent, 3D-Printable, and Self-Healing Poly (ethylene glycol)-Gel (PEGgel), Adv. Mater., 2022, 34(11), 2107791 CrossRef CAS PubMed.
- W. Huang, C. Qi and Y. Gao, Injectable self-healable nanocomposite hydrogels with mussel-inspired adhesive properties for 3D printing ink, ACS Appl. Nano Mater., 2019, 2(8), 5000–5008 CrossRef CAS.
- T. Chen, X. Qiao, P. Wei, G. Chen, I. T. Mugaanire, K. Hou and M. Zhu, Tough Gel-Fibers as Strain Sensors Based on Strain–Optics Conversion Induced by Anisotropic Structural Evolution, Chem. Mater., 2020, 32(22), 9675–9687 CrossRef CAS.
- B. Wang, J. R. Moon, S. Ryu, K. D. Park and J. H. Kim, Antibacterial 3D graphene composite gel with polyaspartamide and tannic acid containing in situ generated Ag nanoparticle, Polym. Compos., 2020, 41(7), 2578–2587 CrossRef CAS.
- R. Cao, M. Qin, C. Liu, S. Li, P. Guo, G. Han, X. Hu, W. Feng and L. Chen, Photo-and Thermosensitive Polymer Membrane with a Tunable Microstructure Doped with Graphene Oxide Nanosheets and Poly (N-isopropylacrylamide) for the Application of Light-Cleaning, ACS Appl. Mater. Interfaces, 2020, 12(12), 14352–14364 CrossRef CAS PubMed.
- C. Ligorio, M. Zhou, J. K. Wychowaniec, X. Zhu, C. Bartlam, A. F. Miller, A. Vijayaraghavan, J. A. Hoyland and A. Saiani, Graphene oxide containing self-assembling peptide hybrid hydrogels as a potential 3D injectable cell delivery platform for intervertebral disc repair applications, Acta Biomater., 2019, 92, 92–103 CrossRef CAS PubMed.
- C. Ligorio, A. Vijayaraghavan, J. A. Hoyland and A. Saiani, Acidic and basic self-assembling peptide and peptide-graphene oxide hydrogels: Characterisation and effect on encapsulated nucleus pulposus cells, Acta Biomater., 2022, 143, 145–158 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic route and schemes of PGMA macro-CTA. 1H NMR spectra and GPC chromatograms of PGMA macro-CTA and PGMA-PHPMA copolymer. Preparation route of PGMA-PHPMA-x% GO by physical mixing. AFM and DLS of the GO average flake size. Tables of Oscillatory rheology information of the GO containing composite worm gel and tensile test information of the GO containing composite worm gel. Digital photographs of (co)polymers and self-healing tensile test progress; Angular frequency of GO containing copolymer gels via in situ polymerisation. Variable temperature rheology studies on copolymers; Tensile test for different copolymer concentration of PGMA-PHPMA-0% GO composite worm gels. Recovery, various temperature rheology and G′ recorded over several heating–cooling cycles experiments by physical mixing. Critical gelation temperature recorded over several heating–cooling cycles for 15% G58-H170-2% GO. Raman spectra of GO and GO-PGMA-PHMA (physical mixing and in situ). Digital photographs of GO containing copolymer via in situ polymerisation after self-healing adheres to various materials. Digital photographs of 3D printed international morse code and QR codes. Movie of 3D printing QR code. See DOI: https://doi.org/10.1039/d3sm00737e |
| This journal is © The Royal Society of Chemistry 2023 |