DOI:
10.1039/D3SM00363A
(Paper)
Soft Matter, 2023,
19, 4686-4696
Self-assembled aggregates based on cationic amphiphilic peptides: structural insight†
Received
20th March 2023
, Accepted 22nd May 2023
First published on 23rd May 2023
Abstract
Short and ultra-short peptides have recently emerged as suitable building blocks for the fabrication of self-assembled innovative materials. Peptide aggregation is strictly related to the amino acids composing the sequence and their capability to establish intermolecular interactions. Additional structural and functional properties can also be achieved by peptide derivatization (e.g. with polymeric moieties, alkyl chains or other organic molecules). For instance, peptide amphiphiles (PAs), containing one or more alkyl tails on the backbone, have a propensity to form highly ordered nanostructures like nanotapes, twisted helices, nanotubes and cylindrical nanostructures. Further lateral interactions among peptides can also promote hydrogelation. Here we report the synthesis and the aggregation behaviour of four PAs containing cationic tetra- or hexa-peptides (C19-VAGK, C19-K1, C19-K2 and C19-K3) derivatized with a nonadecanoic alkyl chain. In their acetylated (Ac-) or fluorenylated (Fmoc-) versions, these peptides previously demonstrated the ability to form biocompatible hydrogels potentially suitable as extracellular matrices for tissue engineering or diagnostic MRI applications. In the micromolar range, PAs self-assemble in aqueous solution into nanotapes, or small clusters, resulting in high biocompatibility on HaCat cells up to 72 hours of incubation. Moreover, C19-VAGK also forms a gel at a concentration of 5 wt%.
1. Introduction
Supramolecular architectures like micelles,1–3 liposomes,4–7 nanotubes,8,9 fibers10,11 and hydrogels12–16 have been proposed for many biomedical applications including delivery of genetic materials, therapeutic and/or diagnostic agents, and as platforms for tissue engineering and regeneration. These structures are generally based on noncovalent bonds (including hydrogen bonding, metal coordination, hydrophobic, van der Waals, and electrostatic and π–π interactions) within and between molecules. Among the plethora of building blocks to generate supramolecular architectures, peptide sequences present advantages of being biocompatible, biodegradable and easy to synthesize.17–19 They usually arrange into secondary structural elements (β-sheet or α-helix), which, in certain cases, can further assemble according to a hierarchical and multiscale process. The self-assembling behavior of peptides is deeply affected by the amino acid composition and by the hydrophobic/hydrophilic balance within the sequence.20 This latter feature can be modulated by peptide functionalization with polymeric or organic moieties. For instance, derivatization of a peptide with PEG (polyethylene glycol)21,22 or with glycosyl groups23 allows increasing its solubility; in contrast, the addition of alkyl chains24,25 or aromatic moieties26–28 causes a decrease in its solubility. Based on the evidence that peptide aggregation can be opportunely modified, many bioactive peptides have been functionalized and studied. An example is the integrin peptide, RGD, which has been derivatized at the N-terminus with a fluorenyl (Fmoc-) group29,30 or with alkyl chains (C14, C16, C18)31–33 to promote its aggregation into self-supporting hydrogels. Analogously, some targeting peptides like CCK8,34 [7–14] bombesin35 and octreotide36 have been decorated with two alkyl chains with eighteen carbon atoms to generate micelles. In this context, some of us recently reported the synthesis of a small library of cationic peptides,37 which self-assemble into promising hydrogels for tissue engineering applications. In our library the original peptide sequence (Ac-K1, Ac-K2 and Ac-K3),38 containing an aliphatic region and a Lys residue within a hexapeptide sequence (ILVAGK in K1, LIVAGK in K2 and AIVAGK in K3), is kept, whereas the acetyl group at the N-terminus was replaced with a Fmoc- aromatic unit, alone or in combination with two phenylalanine residues (-FF-Fmoc). This latter motif was chosen because it represents a well-known hydrogelator. The structural characterization pointed out that only the Fmoc-derivatives are able to self-assemble in aqueous solution, albeit with a lower mechanical rigidity (557, 925 and 2526 Pa for Fmoc-K1, Fmoc-K2 and Fmoc-K3, respectively) than their acetylated precursors. Due to their soft nature, these hydrogels can be potentially used as injectable scaffolds. On the other hand, Fmoc-FF-K sequences do not gel, but are able to self-assemble into stable, injectable nanoparticles. In order to evaluate the impact of alkyl chains with eighteen carbon atoms on the aggregation behavior of K-peptides, here we synthesize their amphiphilic analogues C19-K1, C19-K2 and C19-K3 and their truncated version, C19-VAGK, never studied before (Fig. 1). The VAGK tetrapeptide, never studied before, was chosen because it represents the amino acid sequence common to K1, K2 and K3. Peptide amphiphiles (PAs) can spontaneously self-assemble in water into one-dimensional, cylindrical nanostructures with highly ordered hydrophobic and hydrophilic compartments.39 This ordered architecture is favored by the establishment of van der Waals interactions between the alkyl chains and of hydrogen bonds between the amino acid side chains.40–45 The interaction between the side chains of the peptide moiety can also favor the formation of β-sheet fibers and consequently of hydrogels that have been demonstrated to be potential biomaterial scaffolds in tissue engineering46–49 and in drug delivery applications.50–53 The propensity of the four amphiphilic peptides to self-assemble (Fig. 1) was initially evaluated using fluorescence spectroscopy. Successively, structural characterization was carried out using a set of biophysical techniques including circular dichroism (CD), Fourier transform infrared (FTIR) spectroscopy, cryogenic transmission electron microscopy (cryo-TEM), and small angle X-Ray scattering (SAXS). Their potential capability to gel was also estimated under physiological conditions. The biocompatibility of the peptide aggregates was evaluated by MTT assays using the HaCat cell line.
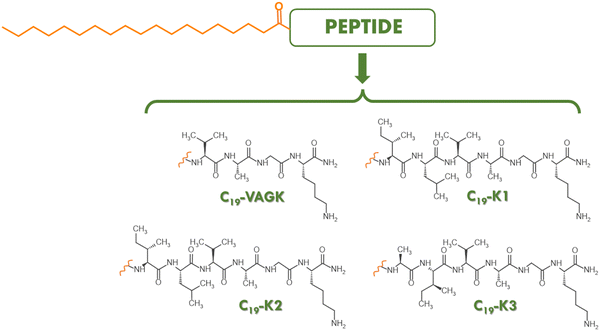 |
| | Fig. 1 Schematic representation of the four PAs: C19-VAGK, C19-K1, C19-K2 and C19-K3. | |
2. Experimental
2.1 Materials
The amphiphilic peptide sequences C19-VAGK-NH2 (C19-VAGK), C19-ILVAGK-NH2 (C19-K1), C19-LIVAGK-NH2 (C19-K2) and C19-AIVAGK-NH2 (C19-K3) were synthesized through standard SPPS (solid-phase peptide synthesis) procedures using the Fmoc/tBu strategy. To obtain amidated peptides at the C-terminus, the Rink amide MBHA resin, with a substitution rate of 0.71 mmol g−1, was used as the solid-phase support. The synthesis scale was 0.20 mmol in DMF for all the peptides. After 30 min of swelling, the Fmoc group was removed from the resin by treatment with 30% (v/v) piperidine in DMF for two cycles of 10 min each. A 2 fold molar excess of the Fmoc-protected amino acid was used for the coupling and solved in DMF together with equimolar amounts of 1-hydroxybenzotriazole (HOBt), benzotriazol-1-yl-oxytris-pyrrolidino-phosphonium (PyBOP), and a 4 fold molar excess of diisopropylethylamine (DIPEA). Each coupling was performed twice for 40 min. The coupling of nonadecanoic acid was performed by dissolving the powder in DCM in a 4 fold molar excess. This solution was then mixed with a DMF solution with 4 fold molar excess of HOBt and PyBOP and 8 fold molar excess of DIPEA. The heterogeneous phase was then left to react with the resin for 3 h. After this step, crude peptides were fully cleaved by treating the resin for 3 h at room temperature with a TFA (trifluoroacetic acid)/TIS (triisopropylsilane)/H2O (92.5/5/2.5 v/v/v) mixture. Cold ether was used to precipitate the peptides, which were freeze-dried three times.
Pure peptides were obtained from the crude ones through RP-HPLC with an LC8 Shimadzu HPLC system (Shimadzu Corporation, Kyoto, Japan) equipped with a UV lambda-Max Model 481 detector, using a Phenomenex (Torrance, CA, USA) C4 column. The flow rate was set at 20 mL min−1 and the elution solvents were H2O/0.1% TFA (A) and CH3CN/0.1% TFA (B) with (B) increasing from 30 to 80% over 15 min. The purity of the products was assessed by analytical RP-HPLC analysis using a Finnigan Surveyor MSQ single quadrupole electrospray ionization (Finnigan/Thermo Electron Corporation San Jose, CA), with a C4-phenomenex column eluting with H2O/0.1% TFA (A) and CH3CN/0.1% TFA (B) from 20 to 80% over 30 min at a flow rate of 1 mL min−1. The identity of peptides was confirmed by MS spectrometry performed by using an LTQ XL linear ion trap mass spectrometer, Sorgent ESI.
2.2 Preparation of peptide solutions and hydrogels
Peptide solutions were prepared by solving the lyophilized peptide powders in double distilled water at different concentrations. The C19-VAGK hydrogel was prepared by dissolving the peptide in 5 wt% water. The hydrogel formation was macroscopically verified by the inverted tube test.
2.3 Fluorescence spectroscopy
For the fluorescence experiments, samples were placed in a 10.0 mm × 5.00 mm quartz cell, lodged in a Varian Model Cary Eclipse spectrofluorometer. Other experimental settings were excitation and emission bandwidths of 2.5 nm and a temperature of 20 °C. The determination of the critical aggregation concentration (CAC) value for all the peptide sequences was estimated by fluorescence titration of the dye 8-anilino-1-naphthalene sulfonic acid (ANS) at a concentration of 25 μmmol L−1 in water with increasing amounts of the peptide solution.54 The measurements were recorded between 360 and 550 nm exciting the sample at 350 nm. The obtained spectra were corrected for the blank and adjusted for the dilution.
2.4 Circular dichroism (CD) spectroscopy
0.1 and 5 × 10−4 wt% water solutions of the peptides were placed in 0.1 and 10 mm quartz cells and Far-UV CD spectra were recorded using a Chirascan spectropolarimeter (Applied Photophysics, Leatherhead, UK) equipped with a thermal controller. Spectra were recorded from 280 to 180 nm. 0.5 nm step, 1 nm bandwidth, 1 s collection time per step were used as experimental conditions for the measurements. For each sample, three scans were performed, averaged, and corrected for the blank to obtain the final spectrum.
2.5 Fourier transform infrared (FTIR) spectroscopy
FTIR spectra were recorded using a Thermo-Scientific Nicolet iS5 instrument with a DTGS detector. Peptide solutions were prepared in deionized water at a concentration of 0.5 wt% and were fixed into a Specac Pearl liquid cell with CaF2 plates. Each sample was subjected to a total of 128 scans. Spectra were recorded over a range of 900–4000 cm−1.
2.6 Congo Red (CR) assay
The UV/Vis Congo Red (CR) spectroscopy assay was carried out using a freshly prepared stock solution of CR (1.75 mg mL−1) in water, filtered through 0.2 μm syringe immediately prior to use. A small aliquot (5 μL) of this solution was added to 350 μL of PAs containing solutions at a concentration of 0.1 wt%. After incubation for 15 min, the UV/Vis spectra were recorded in a 1 cm quartz cell. Background from the dye was subtracted using a Congo Red spectrum in water as a reference solution.
2.7 Thioflavin T (ThT) spectroscopic assay in the solid state
An aliquot 20 μL of solution of the C19-VAGK xerogel was air-dried and stained with 15 μL of a water solution of 50 μmol L−1 ThT. After removing excess dye using filter paper, the sample was left at room temperature overnight. Fluorescence images of the sample were recorded using a fluorescence microscope. Images were taken with a Leica DFC320 video camera (Leica, Milan, Italy) connected to a Leica DMRB microscope equipped with a 20 X objective and green fluorescent protein (GPF) filter. The software Image J (National Institutes of Health, Bethesda, MD) was used for analysis.
2.8 Cryogenic-TEM (Cryo-TEM)
A field emission cryo-electron microscope (JEOL JEM-3200FSC), operating at 200 kV, was used for imaging. Images were taken in the bright field mode using a Gatan Ultrascan 4000 CCD camera. Other experimental settings were: zero loss energy filtering (omega type) with a slit width of 20 eV and −187 °C as the specimen temperature. An automated FEI Vitrobot device and Quantifoil 3.5/1 holey carbon copper grids with a hole size of 3.5 μm were used to prepare vitrified specimens. Grids were plasma cleaned with a Gatan Solarus 9500 plasma cleaner just prior to use and then they were transferred to the environmental chamber of a FEI Vitrobot at room temperature and 100% humidity. 3 μL of sample solution was then applied on the grid and bottled twice for 5 seconds. Successively, they were vitrified in a 1/1 mixture of liquid ethane and propane at a temperature of −180 °C. Before being cryo-transferred to the microscope, the grids were maintained at liquid nitrogen temperature. The mean diameter of the thickness of nanotapes was obtained by measuring it at 5 different points.
2.9 Small angle X-ray scattering (SAXS) measurements
Synchrotron SAXS experiments on solutions were performed using a BioSAXS setup on BM29 at the ESRF (Grenoble, France).55,56 A few microlitres of samples were injected via an automated sample exchanger at a slow and very reproducible rate into a quartz capillary (1.8 nm internal diameter), in the X-ray beam. The quartz capillary was enclosed in a vacuum chamber, to avoid parasitic scattering. After the sample was injected into the capillary and reached the X-ray beam, the flow was stopped during tSAXS data acquisition. The q range was 0.005–0.48 Å−1, with λ = 1.03 Å and the images were obtained using a Pilatus Pilatus3-2M detector. Data processing (background subtraction, radial averaging) was performed using dedicated beamline software ISPYB.
2.10 Cell lines
For cell experiments with peptide solutions at concentrations below and above the CAC, aneuploid immortal keratinocyte cell line HaCat was used. Cells were obtained from the IRCCS-SDN Biobank (10.5334/ojb.26) and grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 1% GlutaMAX and 1% penicillin–streptomycin. Cells were incubated at 37 °C and 5% CO2 and seeded in 25 cm2 culture flasks.
2.11 Cytotoxicity assays
Cells were seeded in 96-well plates at a density of 0.3 × 104 cells per well. Cells were then treated with peptides dissolved in the medium at concentrations of 0.1 and 5 × 10−5 wt%. After 72 h, cell viability was assessed by using an MTT [3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide] assay. In brief, after the removal of the culture medium, MTT, dissolved in DMEM at a concentration of 0.5 mg mL−1, was added to the cells and incubated for 3 h at 37 °C. The resulting formazan crystals were dissolved by adding dimethyl sulfoxide (DMSO). Absorbance values of blue formazan were determined at 570 nm using an automatic plate reader. Cell survival was expressed as percentage of viable cells in the presence of peptides, compared to control cells grown in their absence. The assay was repeated three times and the results were averaged. Statistical significance was tested using multiple Welch's t tests. Prism 7 was used to conduct all the analyses.
2.12 Rheology
Gel rheological characterization was performed using a controlled-stress rotational rheometer (Malvern Kinexus). A 15 mm flat-plate geometry (PU20:PL61) was used for the analysis of a preformed sample (450 μL) at a concentration of 5.0 wt%. Each experiment was performed at 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C using a humidity chamber and a gap of 1 mm between the plate and geometry. Preliminary dynamic tests were carried out to identify the regime of linear viscoelasticity. The viscous elastic region was determined by oscillatory frequency (0.1–100 Hz) and strain sweep (0.01–100%) measurements. Then a time-sweep oscillatory evaluation test (using a constant 0.1% strain and 1 Hz frequency) was performed for 1000 seconds. Measurement outputs are plotted in Pascal (Pa) as shear storage or elastic modulus (G′) and the shear loss or viscous modulus (G′′).
°C using a humidity chamber and a gap of 1 mm between the plate and geometry. Preliminary dynamic tests were carried out to identify the regime of linear viscoelasticity. The viscous elastic region was determined by oscillatory frequency (0.1–100 Hz) and strain sweep (0.01–100%) measurements. Then a time-sweep oscillatory evaluation test (using a constant 0.1% strain and 1 Hz frequency) was performed for 1000 seconds. Measurement outputs are plotted in Pascal (Pa) as shear storage or elastic modulus (G′) and the shear loss or viscous modulus (G′′).
3. Results and discussion
3.1 Synthesis and aggregation behavior of PAs
The alkyl tail used in the design of PAs can deeply affect the aggregation behavior and the resulting architecture.57 Generally, the employment of long chains allows to lower the critical aggregate concentration and, consequently the in vitro and in vivo stability of the supramolecular system.31–36 Due to the commercial suitability of nonadecanoic acid, useful for the functionalization of the N-terminus of peptides, it was used to derivatize the previously reported K-peptides.37,38 Peptide amphiphiles C19-K1, C19-K2, C19-K3 and their truncated form C19-VAGK (Fig. 1), were synthesized by the solid phase peptide synthesis (SPPS) according to Fmoc/OtBu protocols. After their cleavage from the resin, the crude, lyophilized products were purified by RP-HPLC and characterized by ESI mass spectrometry (Fig. S1, ESI†).
The self-assembling capability of the four amphiphilic peptides was probed in water. The critical aggregation concentration (CAC) was calculated by titration of an ANS solution with increasing amounts of the peptide. ANS is an organic fluorescent probe widely used to study peptide and protein aggregates.54 This molecule shows, in aqueous solution, a negligible fluorescence with a maximum at 540 nm, after excitation at 350 nm. The fluorescence emission undergoes a blue shift, with a maximum at around 480 nm, and a remarkable increase in the presence of peptide aggregates. The mechanism through which this phenomenon occurs is still not fully understood. It has been suggested that this process is due to the restricted mobility and the hydrophobic environment in which ANS is located.58 Plotting the fluorescence intensity as a function of the peptide concentration (Fig. 2), the CAC, calculated at the break point, was found to be (5.90 ± 0.07) × 10−4 wt%, (1.35 ± 0.01) × 10−3 wt%, (9.83 ± 0.08) × 10−4 wt% and (2.47 ± 0.04) × 10−3 wt%, for C19-VAGK, C19-K1, C19-K2 and C19-K3, respectively (see Table 1). These values are in good agreement with the values expected for peptide amphiphiles containing an alkyl chain at nineteen carbon atoms.32 These CAC values are low enough to be compatible with their intravenous administration. From the comparison of the determined values, it can be pointed out that CACs are similar to the three hexapeptides; whereas the truncated variant (VAGK) exhibits a low CAC value, thus indicating a slightly higher tendency to self-assemble with respect to the hexapeptides.
 |
| | Fig. 2 ANS titration curves for (a) C19-VAGK, (b) C19-K1, (c) C19-K2 and (d) C19-K3. | |
Table 1 Calculated and experimentally determined molecular weights (MW) and CAC values, expressed in wt% and in mol L−1, for peptide amphiphiles. CAC values have been determined at the break point of the plots in Fig. 2
| Sample |
MWcalc. (a.m.u.) |
MWdeter. (a.m.u.) |
CAC (wt%) × 10−4 |
CAC (mol L−1) × 10−5 |
| C19-VAGK |
652.9 |
653.7 |
5.90 ± 0.07 |
0.90 ± 0.01 |
| C19-K1 |
879.3 |
879.9 |
13.5 ± 0.1 |
1.54 ± 0.01 |
| C19-K2 |
879.3 |
879.9 |
9.83 ± 0.08 |
1.12 ± 0.01 |
| C19-K3 |
837.2 |
837.9 |
24.7 ± 0.4 |
2.95 ± 0.05 |
3.2 Secondary structure characterization
CD and FTIR analyses are valuable complementary methods used to investigate the secondary structure of peptides in solutions. CD measurements were conducted at two different concentrations, one above (0.1 wt%) and one below (5 × 10−5 wt%) the CAC for all the peptide sequences (Fig. 3(a)–(d) and Fig. S2, ESI†). Due to the ionization of the amine group, the supramolecular structure generated by aggregation of lysine containing PAs can be affected by the pH value of the solution.59–61 In this context, the pH values of each peptide solution at both concentrations used for the secondary structure characterization were measured. The four peptide analogues present a similar pH value (7.0) at both concentrations. At a concentration of 5 × 10−5 wt%, the CD profile of C19-VAGK, showing positive bands with a maximum at 193 and 205 nm and a negative band with a maximum at 221 nm, indicates a mixture of α-helix like aggregates and a disordered conformation. The same mixture is also proposed for C19-K1 and C19-K3, because of the negative band centered at 205 and 203 nm, respectively. C19-K2, analyzed at the concentration below the CAC, shows a random coil organization, suggested by the presence of a positive band at around 222 nm. At a concentration of 0.1 wt%, while C19-K1 and C19-K2 show a β-sheet secondary structure, the organization seems to be a mixture between β-sheet and α-helix like that for C19-VAGK and C19-K3. These assumptions found their basis in the presence, in C19-K1 and C19-K2 CD spectra, of a positive signal centered at 203 nm and a negative one with a maximum at 220 nm. Conversely, the shape of the CD profiles belonging to C19-VAGK and C19-K3, with the positive band at around 190 nm (185 nm for C19-VAGK and 190 nm for C19-K3) and the two negative bands at around 205 and 215 nm (201 and 218 nm for C19-VAGK; 205 and 216 nm for C19-K3) suggests the α-helix like organization, with signals slightly blue-shifted compared to the classical signature of this type of arrangement. However, the two negative bands at 218 and 216 nm are characteristic of a β-sheet secondary structure. An α-helix like structure was also observed for acetylated analogues at a concentration similar to the one explored for the C19-derivatieves (0.07 wt%), while at higher concentrations (0.13 wt%) a β-turn arrangement was favored. Similarly, Fmoc- and Fmoc-FF hexapeptides were shown to have a β-sheet secondary structure at the concentration of 0.1 wt%.37 These behaviors may suggest that the peptide component partially adopts an α-helix arrangement, which is still present in part for some of the amphiphilic sequences but is mainly disfavoured by the higher concentration and by the substitution of the small acetyl group with more bulky ones like Fmoc, Fmoc-FF and nonadecanoic acid. The more evident α-helix profile observed for C19-VAGK and C19-K3 peptides may be probably attributed to the lower number of highly Cβ substituted amino acids, which notably push towards the beta sheet arrangement.62
 |
| | Fig. 3 Secondary structure characterization. Left panel: CD profiles of (a) C19-VAGK, (b) C19-K1, (c) C19-K2 and (d) C19-K3 at a concentration of 0.1 wt%. CD spectra are recorded in the range between 280 and 180 nm. Right panel: (e) IR spectra of the four peptide sequences at 0.5 wt% concentration. | |
FTIR spectra were recorded to confirm data obtained through CD measurements. FTIR spectroscopy is one of the most entrenched methods for the detection of the secondary structure of the peptide and proteins. Nine characteristic IR absorption bands (namely amide A, B, and I–VII) arise from the repeat units of the peptide skeleton. Among these, the amide I region, centered between 1700 and 1600 cm−1 and chiefly originated from the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations, is the most affected by the peptide organization. Inwardly this spectral region, each signal frequency was found to be strictly correlated to a particular element of the secondary structure. FTIR analyses were conducted on all peptides solved in deionized water at a concentration of 0.5 wt%, allowing a good signal intensity. This solution was found to be more acidic (pH = 5.7) than the previously investigated one (0.1 wt% and 5 × 10−5 wt%). The lower pH, measured for this higher peptide concentration, can be probably attributable to the higher number of residual trifluoroacetate counterions coming from the HPLC purification of the peptide.
O stretching vibrations, is the most affected by the peptide organization. Inwardly this spectral region, each signal frequency was found to be strictly correlated to a particular element of the secondary structure. FTIR analyses were conducted on all peptides solved in deionized water at a concentration of 0.5 wt%, allowing a good signal intensity. This solution was found to be more acidic (pH = 5.7) than the previously investigated one (0.1 wt% and 5 × 10−5 wt%). The lower pH, measured for this higher peptide concentration, can be probably attributable to the higher number of residual trifluoroacetate counterions coming from the HPLC purification of the peptide.
In all the spectra, reported in Fig. 3(e) as absorbance conversion, the signal at 1672 cm−1 indicates the presence of the TFA counterion.32 For C19-K1 and C19-K2, the intense signal at 1629 cm−1, together with the one at 1620 cm−1, only visible for C19-K1, suggests the preponderance of a β-sheet structure, even if minor α-helix like and random-coil components are indicated by the peaks at 1651 and 1643 cm−1, respectively. Supporting the results obtained by CD measurements, FTIR spectra seem to hint that C19-VAGK and C19-K3 sequences present an α-helix like arrangement evidenced by the peaks at 1651 and 1650 cm−1, respectively, but the main organization is still β-sheet, as proven by the more intense signals at 1632 and 1628 cm−1 for C19-VAGK, and at 1633 and 1620 cm−1 for C19-K3. Moreover, a minor random-coil component is pointed out for C19-VAGK, since a signal at 1643 cm−1 is present.
To further investigate the secondary structure of PAs in water solution we carried out the Congo Red (CR) assay, which allows to corroborate the presence of β-sheet structures in peptide solutions.63,64 This assay is generally considered positive when a colorimetric variation from light to dark red is observed for the CR solution after its incubation with peptides rich in β-sheet structures. The colorimetric change is also detectable by UV-Vis spectroscopy with a red-shift of the CR absorbance peak from 490 to 540 nm. From the inspection of Fig. 4, it can be concluded that all the PAs, except for C19-K3, are positive to the assay, thus indicating the predominance of a β-sheet conformation at a concentration of 0.1 wt%. This result agrees with the FTIR study at the same concentration. Further structural information about PA aggregates was obtained by the cryogenic transmission electron microscopy (Cryo-TEM) technique. Selected cryo-TEM images, reported in Fig. 5, show that, C19-K1, C19-K2 and C19-VAGK at a concentration of 0.1 wt% present a fibrillar network, in which the mean fiber diameter is 11.3 ± 3.4 nm, 15.4 ± 6.0 nm and 21.6 ± 6.1 nm, respectively. In contrast, C19-K3 does not form fibers, but really small clusters with a mean diameter of 3.1 ± 0.7 nm. Fiber lengths extrapolated from the cryo-TEM images seem to point out a major tendency of the VAGK peptide to laterally interact.
 |
| | Fig. 4 CR assay: (a) absorbance spectra of CR alone or co-incubated with peptide solutions. (b) Macroscopic appearance of the analysed peptide solution alone or incubated with CR. (c) UV-Vis spectra reported after the subtraction of the spectrum of the CR alone. | |
 |
| | Fig. 5 Cryo-TEM micrographs for the four peptide sequences at a concentration 0.1 wt% (scale bar: 200 nm). | |
Cryo-TEM imaging was complemented with in situ SAXS measurements for aqueous solutions, which enables the determination of the nanostructure shape and dimensions through analysis of the form factor.65 The data along with model fits to the form factor of extended nanotape fibrils with a bilayer packing of the molecules are shown in Fig. 6(a). The bilayer packing is consistent with the packing of the C19 lipid chains in the hydrophobic interior, with the peptide sequence on the exterior of the layers. The fitting parameters in the ESI,† Table S2 indicate bilayer thicknesses (t) in the range of 2.8–4.6 nm. The estimated molecular lengths are 4.1 nm for C19-K1, C19-K2 and C19-K3 and 3.4 nm for C19-VAGK. Therefore, the values of t indicate substantial interdigitation of the molecules within the bilayers in the extended β-sheet fibril structures. A schematic representation of the packing molecules in C19-VAGK nanotapes is proposed in Fig. 6(b).
 |
| | Fig. 6 (a) SAXS data – measured for 0.1 wt% samples (open symbols as indicated) along with form factor fits (solid lines) discussed in the text with fit parameters listed in the ESI,† Table S2. Some data sets are scaled by the factors indicated for ease of visualization. (b) Schematic representation of C19-VAGK nanotapes with bilayer thickness, the estimated molecular lengths and the mean fiber diameter. | |
3.3 Cell viability
The cytotoxicity of the four lipopeptides was assessed with the HaCat aneuploid immortal keratinocytes cell line. Results, in terms of cell viability (%) after 72 h, are reported in Fig. 7 and t test probability values are collected in Table S1 (ESI†). High cytocompatibility is observed when the cells are treated with peptide solutions at concentrations below the CAC (5 × 10−5 wt%). The percentage of cell viability is even slightly higher than the control (103.7 ± 5.8%, 100.4 ± 4.9%, 105.6 ± 4.8% and 103.8 ± 6.7% for C19-VAGK, C19-K1, C19-K2 and C19-K3, respectively) with no significant differences from sample to sample. For more concentrated solutions (0.1 wt%, higher than the CAC), a reduction in HaCat survival percentage is observed for all the tested lipopeptides. However, since the cell viability is around 80% for all the samples (84.8 ± 7.9%, 82.3 ± 4.5%, 78.2 ± 3.5% and 90.0 ± 5.8% for C19-VAGK, C19-K1, C19-K2 and C19-K3, respectively), it can be concluded that even the aggregates can be considered biocompatible on the tested cell line after 72 h of incubation (Fig. 7).
 |
| | Fig. 7 Cytotoxicity data from MTT assays obtained after 72 h on HaCat cells. | |
3.4 Hydrogelation test and structural and rheological characterization
Many PAs, containing a single alkyl chain, demonstrated the capability to form hydrogels under specific experimental conditions. Analogously, K1, K2 and K3 peptide sequences, in their acetylated (Ac-) and fluorenylated (Fmoc-) versions, were previously found to gel at a concentration of 2 wt% by adding few microliters PBS to the peptide water solution.37,38 The gel formation is triggered by the addition of phosphate that nullifies the repulsion among the positive charges on the lysine side chains. In this context, we investigated the potential ability of the four PAs to gel at 2 wt% upon the addition of PBS. As clearly indicated by the inverted test tube in Fig. 8(a), hydrogels are not formed. However, the formation of the self-supporting C19-VAGK hydrogel was allowed after 24 hours at room temperature for the sample in which the peptide concentration was increased up to 5 wt% (Fig. 8(b)). No syneresis phenomena were detected after a week, indicating that water is well confined in the supramolecular architecture. In contrast, no gel was observed for the longer K1, K2 and K3 variants (Fig. 8(b)) under the same conditions. As expected, at a concentration of 5 wt%, all the peptide solutions exhibited a lower pH value (∼4.0). However, due to the inability of C19-K1, C19-K2 and C19-K3 to form hydrogels, the effect of the pH on the gel formation can be excluded. On the other hand, it could be hypothesized that the increase of concentration caused an increase in the non-covalent interactions between the fibrillar structure with the consequent formation of the hydrogel. The capability of tetrapeptide PA with respect to the hexapeptide PAs can be partially justified by its slightly major tendency to self-aggregate, also testified by its lower CAC value. Fluorescence images recorded on air-dried C19-VAGK xerogel, stained with Thioflavin T (ThT), show the typical emission in the green spectral region (Fig. 8(c)), thus indicating the presence of β-sheet structures. Moreover, FTIR measurement carried out on the C19-VAGK based hydrogel confirmed the structural organization previously found for the sample in solution, thus indicating that the peptide secondary structure is retained upon the increase of the concentration from 0.5 to 5 wt% (Fig. 8(d)). To further confirm the gel state for the C19-VAGK solution, a rheological study was performed. The viscoelastic behavior was analyzed using a stress-controlled rheometer, equipped with a plate flat geometry. A time sweep measurement (1000 s, frequency ν = 1.0 Hz and 0.1% strain, Fig. 8(e)) was performed on a self-supporting sample (5.0 wt%), reporting data in terms of G′ (storage modulus) and G′′ (loss modulus). Rheological analysis was supported by preliminary tests for the identification of the measurement parameters, specifically a dynamic oscillation strain sweep (at a ν = 1.0 Hz) and dynamic frequency sweep (at 0.1% strain) (Fig. S3a and b, ESI†). A tanδ (G′/G′′) value higher than 1 indicates the formation of a hydrogel matrix. C19-VAGK gel possesses values of G′ = 104 Pa and G′′ = 21, respectively, indicating soft mechanical properties, thus comparable with other peptide-based hydrogels.
 |
| | Fig. 8 C19-VAGK gel characterization: inverted test tube at 2 wt% (a) and 5 wt% (b). (c) ThT assays: fluorescence and optical images of C19-VAGK drop-cast on a glass slide, air-dried and stained with the ThT solution. Sample is imaged in the bright field and in the spectral regions of the GFP (green fluorescent protein, λexc = 488 nm, λem = 507 nm). Scale bar = 50 μm. (d) FTIR spectrum (amide I region) of the C19-VAGK gel at 5 wt%. (e) Rheological analysis of peptide: time sweep showing the hydrogel storage modulus (G′) and loss modulus (G′′). | |
4. Conclusions
Chemical modifications on peptide building blocks can improve or decrease their capability to self-assemble. Therefore, the structural and morphological features of the aggregated nanostructures and, obviously, their functional properties change. In this perspective, de novo design of peptide derivatives can allow the development of novel biocompatible materials for a wide range of applications. For instance, peptide derivatization with alkyl chains endows them with self-assembling properties and different biological features, such as the ability to encapsulate small molecules and protect them from biodegradation, to permeate and destroy the bacterial cell membrane, and to generate hydrogels able to support cell growth. For this reason, lipopeptides have been successfully used to develop nanostructures for many applications, including drug delivery,66 antibiotic activity67 and tissue engineering.32 Starting from cationic peptides previously studied as hydrogelators, here we demonstrated that their amphiphilic derivatives, containing nonadecanoic acid as the alkyl chain in place of the acetyl group, are able to self-assemble into nanotapes, twisted ribbons or small clusters with a mean diameter of around 3 nm. The critical aggregate concentration values in the micromolar range and their very low toxicity on HaCat cells up to 72 hours of incubation suggest their potential application in biomedicine. Cryo-TEM imaging, complemented with SAXS measurement, allowed obtaining further information on the structural arrangement of the PAs into a nanostructure. In contrast to their Ac and Fmoc variants, C19-K1, C19-K2 and C19-K3 are unable to gel. On the other hand, C19-VAGK in which the peptide portion is represented by the tetrapeptide common to K1, K2 and K3 sequences, forms a hydrogel at a concentration of 5 wt%. This result is in agreement with the major tendency of VAGK to laterally interact into the fibrillar networks. By comparing these results with information obtained from the literature, different observations about the influence of both the length or the number or alkyl chains and the presence and the number of Lys or Arg as cationic residues can be done. The binding of shorter alkyl chains (C12) to peptide sequences comprising cationic residues68 has been shown to drive the structure towards an α-helix like arrangement, while the increase in the lipophilic part, which means the addiction of another C12 alkyl portion69 or the increase in the number of carbon atoms of the chain32 leads to a β-sheet structure resulting in the formation of twisted ribbons. The decrease in lipophilicity linked to the increase in the number of positively charged Lys residues in the C16-KKK lipopeptide70 was shown to disfavor the formation of fibres, by leading to the obtainment of small oligomers increasing the antimicrobial activity. Similarly, the increase in hydrophilicity obtained through the shortening of the acyl chain in arginine rich amphiphilic peptides, was shown to determine cell specificity.71 These findings point out that both the morphology of the nanostructures and the biological activity, displayed by positively charged lipopeptides, strictly depend on the hydrophilic/lipophilic balance of the monomer.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
The authors are grateful for the award of synchrotron SAXS beam time at the ESRF (proposal no. MX2422) and the support of Mark Tully during the measurements. This work was supported by EPSRC Fellowship grant EP/V053396/1 to IWH. LDM acknowledges FAPESP grant number 19/20907-7 and Fellowship 2021/10092-6, linked to FAPESP scholarship 2019/19719-1.
References
- A. M. Jhaveri and V. P. Torchilin, Front. Pharmacol., 2014, 5, 1–26 CrossRef CAS PubMed.
- M. S. Miranda, A. F. Almeida, M. E. Gomes and M. T. Rodrigues, Int. J. Mol. Sci., 2022, 23(19), 11793 CrossRef CAS PubMed.
- S. Kotta, H. M. Aldawsari, S. M. Badr-Eldin, A. B. Nair and K. Yt, Pharmaceutics, 2022, 14, 1636 CrossRef CAS PubMed.
- A. Charumathy, U. Ubaidulla, P. Sinha and G. Rathnam, Int. J. Curr. Pharm. Res., 2022, 14(3), 22–27 CrossRef CAS.
- P. Liu, G. Chen and J. Zhang, Molecules, 2022, 27(4), 1372 CrossRef CAS PubMed.
- D. Guimarães, A. Cavaco-Paulo and E. Nogueira, Int. J. Pharm., 2021, 601, 120571 CrossRef PubMed.
- M. J. W. Evers, S. I. van de Wakker, E. M. de Groot, O. J. de Jong, J. J. J. Gitz-François, C. S. Seinen, J. P. G. Sluijter, R. M. Schiffelers and P. Vader, Adv. Healthcare Mater., 2022, 11, 2101202 CrossRef CAS PubMed.
- M. Hernandez-Rivera and L. J. Wilson, Stem Cell Transl. Invest., 2016, 3, e1390 Search PubMed.
- E. Mostafavi, S. Iravani, R. S. Varma, M. Khatami and F. Rahbarizadeh, Mater. Adv., 2022, 3(12), 4765–4782 RSC.
- E. Arkan, A. H. Azandaryani, P. Moradipour and L. Behbood, Curr. Pharm. Biotechnol., 2017, 18(11), 909–924 CAS.
- C. Diaferia, E. Gianolio, T. Sibillano, F. A. Mercurio, M. Leone, C. Giannini, N. Balasco, L. Vitagliano, G. Morelli and A. Accardo, Sci. Rep., 2016, 7, 307 CrossRef PubMed.
- L. L. Lock, Y. Li, X. Mao, H. Chen, V. Staedtke, R. Bai, W. Ma, R. Lin, Y. Li, G. Liu and H. Cui, ACS Nano, 2017, 11(1), 797–805 CrossRef CAS PubMed.
- R. Binaymotlagh, L. Chronopoulou, H. F. Haghighi, I. Fratoddi and C. Palocci, Materials, 2022, 15(17), 5871 CrossRef CAS PubMed.
- M. A. Elsawy, J. K. Wychowaniec, L. A. Castillo Diaz, A. M. Smith, A. F. Miller and A. Saiani, Biomacromolecules, 2022, 23(6), 2624–2634 CrossRef CAS PubMed.
- S. Nummelin, V. Liljeström, E. Saarikoski, J. Ropponen, A. Nykänen, V. Linko, J. Seppälä, J. Hirvonen, O. Ikkala, L. M. Bimbo and M. A. Kostiainen, Chem. – Eur. J., 2015, 21, 14433–14439 CrossRef CAS PubMed.
- E. R. Draper and D. J. Adams, Chem, 2017, 3, 390–410 CAS.
- D. Tesauro, A. Accardo, C. Diaferia, V. Milano, J. Guillon, L. Ronga and F. Rossi, Molecules, 2019, 24(2), 351 CrossRef PubMed.
- R. J. Swanekamp, J. J. Welch and B. L. Nilsson, Chem. Commun., 2014, 50, 10133–10136 RSC.
- S. Fleming and R. V. Ulijn, Chem. Soc. Rev., 2014, 43(23), 8150–8177 RSC.
- S. H. Hiew, Y. Lu, H. Han, R. A. Gonçalves, S. R. Alfarano, R. Mezzenga, A. N. Parikh, Y. Mu and A. Miserez, J. Am. Chem. Soc., 2023, 145(6), 3382–3393 CrossRef CAS PubMed.
- C. Diaferia, V. Roviello, G. Morelli and A. Accardo, ChemPhysChem, 2019, 20, 2774–2782 CrossRef CAS PubMed.
- I. W. Hamley, Biomacromolecules, 2014, 15, 1543–1559 CrossRef CAS PubMed.
- R. Roytman, L. Adler-Abramovich, K. S. Ajish Kumar, T.-C. Kuan, C.-C. Lin, E. Gazit and A. Brik, Org. Biomol. Chem., 2011, 9, 5755–5761 RSC.
- L. Chen, T. O. McDonalds and D. J. Adams, RSC Adv., 2013, 3, 8714–8720 RSC.
- I. W. Hamley, Chem. Commun., 2015, 51, 8574–8583 RSC.
- K. McAulay, P. Agís Ucha, H. Wang, A. M. Fuentes-Caparrós, L. Thomson, O. Maklad, N. Khunti, N. Cowieson, M. Wallace, H. Cui, R. J. Poole, A. Seddon and D. J. Adams, Chem. Commun., 2020, 56, 4094–4097 RSC.
- Z. Yang, Y. Li, C. Shen, Y. Chen, H. Li, A. Zhou and K. Liu, Langmuir, 2021, 37, 14713–14723 CrossRef CAS PubMed.
- I. W. Hamley, ACS Appl. Bio Mater., 2023, 6, 384–409 CrossRef CAS PubMed.
- P. Chakraborty, M. Ghosh, L. Schnaider, N. Adadi, W. Ji, D. Bychenko, T. Dvir, L. Adler-Abramovich and E. Gazit, Macromol. Rapid Commun., 2019, 40, 1900175 CrossRef PubMed.
- G. Cheng, V. Castelletto, R. R. Jones, C. J. Connon and I. W. Hamley, Soft Matter, 2011, 7, 1326–1333 RSC.
- V. Castelletto, R. M. Gouveia, C. J. Connon and I. W. Hamley, Faraday Discuss., 2013, 166, 381–397 RSC.
- E. Rosa, L. de Mello, V. Castelletto, M. L. Dallas, A. Accardo, J. Seitsonen and I. W. Hamley, Biomacromolecules, 2023, 24(1), 213–224 CrossRef CAS PubMed.
- S. R. Bull, M. O. Guler, R. E. Bras, T. J. Meade and S. I. Stupp, Nano Lett., 2005, 5(1), 1–4 CrossRef CAS PubMed.
- A. Accardo, D. Tesauro, L. Aloj, L. Tarallo, C. Arra, G. Mangiapia, M. Vaccaro, C. Pedone, L. Paduano and G. Morelli, ChemMedChem, 2008, 3(4), 594–602 CrossRef CAS PubMed.
- A. Accardo, R. Mansi, G. Salzano, A. Morisco, M. Aurilio, A. Parisi, F. Maione, C. Cicala, B. Ziaco, D. Tesauro, L. Aloj, G. De Rosa and G. Morelli, J. Drug Targeting, 2013, 21(3), 240–249 CrossRef CAS PubMed.
- A. Accardo, A. Morisco, E. Gianolio, D. Tesauro, G. Mangiapia, A. Radulescu, A. Brandt and G. Morelli, J. Pept. Sci., 2011, 17(2), 154–162 CrossRef CAS PubMed.
- C. Diaferia, E. Rosa, E. Gallo, G. Smaldone, M. Stornaiuolo, G. Morelli and A. Accardo, Biomedicines, 2021, 9(6), 678 CrossRef CAS PubMed.
- Y. Loo, A. Lakshmanan, M. Ni, L. L. Toh, S. Wang and C. A. E. Hauser, Nano Lett., 2015, 15(10), 6919–6925 CrossRef CAS PubMed.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294(5547), 1684–1688 CrossRef CAS PubMed.
- D. W. P. M. Löwik and J. C. M. van Hest, Chem. Soc. Rev., 2004, 33, 234–245 RSC.
- H. G. Cui, M. J. Webber and S. I. Stupp, Biopolymers, 2010, 94, 1–18 CrossRef CAS PubMed.
- F. Versluis, H. R. Marsde and A. Kros, Chem. Soc. Rev., 2010, 39, 3434–3444 RSC.
- J. B. Matson, R. H. Zha and S. I. Stupp, Curr. Opin. Solid State Mater. Sci., 2011, 15, 225–235 CrossRef CAS PubMed.
- A. Trent, R. Marullo, B. Lin, M. Black and M. Tirrell, Soft Matter, 2011, 7, 9572–9582 RSC.
- J. B. Matson and S. I. Stupp, Chem. Commun., 2012, 48, 26–33 RSC.
- M. J. Webber and S. I. Stupp, J. Int. Med., 2010, 267(1), 71–88 CrossRef CAS PubMed.
- M. Miotto, R. M. Gouveia and C. J. Connon, J. Funct. Biomater., 2015, 6(3), 687–707 CrossRef CAS PubMed.
- F. Gelain, Z. Luo, M. Rioult and S. Zhang, npj Regener. Med., 2021, 6, 9 CrossRef CAS PubMed.
- H. Najaf, M. Jafari, G. Farahavar, S. S. Abolmaali, N. Azarpira, S. Borandeh and R. Ravanfar, Bio-Des. Manuf., 2021, 4, 735–756 CrossRef PubMed.
- D. M. Leite, E. Barbu, G. J. Pilkington and A. Lalatsa, Curr. Top. Med. Chem., 2015, 15, 2277–2289 CrossRef CAS PubMed.
- A. Altunbas and D. J. Pochan, Topics Curr. Chem., 2012, 310, 135–168 CrossRef CAS PubMed.
- C. Zhao, H. Chen, F. Wang and X. Zhang, Colloids Surf., B, 2021, 208, 112040 CrossRef CAS PubMed.
- Z. Song, X. Chen, X. You, K. Huang, A. Dhinakar, Z. Gu and J. Wu, Biomater. Sci., 2017, 5, 2369–2380 RSC.
- K. S. Birdi, H. Singh and S.-U. Dalsager, J. Phys. Chem., 1979, 83, 2733–2737 CrossRef CAS.
- P. Pernot, A. Round, R. Barrett, A. D. Antolinos, A. Gobbo, E. Gordon, J. Huet, J. Kieffer, M. Lentini, M. Mattenet, C. Morawe, C. Mueller-Dieckmann, S. Ohlsson, W. Schmid, J. Surr, P. Theveneau, L. Zerrad and S. McSweeney, J. Synchrotron Radiat., 2013, 20, 660–664 CrossRef CAS PubMed.
- M. D. Tully, J. Kieffer, M. E. Brennich, R. C. Aberdam, J. B. Florial, S. Hutin, M. Oscarsson, A. Beteva, A. Popov, D. Moussaoui, P. Theveneau, G. Papp, J. Gigmes, F. Cipriani, A. McCarthy, C. Zubieta, C. Mueller-Dieckmann, G. Leonard and P. Pernot, J. Synchrotron Radiat., 2023, 30, 258–266 CrossRef PubMed.
- C. A. E. Hauser, R. Deng, A. Mishra, Y. Loo, U. Khoe, F. Zhuang, D. W. Cheong, A. Accardo, M. B. Sullivan, C. Riekel, J. Y. Ying and U. A. Hauser, Proc. Natl. Acad. Sci. U. S. A., 2011, 108(4), 1361–1366 CrossRef CAS PubMed.
- M. Korang-Yeboah, S. Ketcham, M. Shih, A.-M. Ako-Adounvo, J. Zhang, B. M. Bandaranayake, Y. Abbey-Berko, P. Faustino and M. Ashraf, Int. J. Pharm., 2021, 604, 120677 CrossRef CAS PubMed.
- C. Gao, H. Li, Y. Li, S. Kewalramani, L. C. Palmer, V. P. Dravid, S. I. Stupp, M. Olvera de la Cruz and M. J. Bedzyk, J. Phys. Chem. B, 2017, 121(7), 1623–1628 CrossRef CAS PubMed.
- G. Zaldivar, S. Vemulapalli, V. Udumula, M. Conda-Sheridan and M. Tagliazucchi., J. Phys. Chem. C, 2019, 123(28), 17606–17615 CrossRef CAS.
- A. Dehsorkhi, V. Castelletto, I. W. Hamley, J. Adamcik and R. Mezzenga, Soft Matter, 2013, 9, 6033–6036 RSC.
- P. Koehl and M. Levitt., Proc. Natl. Acad. Sci. U. S. A., 1999, 96(22), 12524–12529 CrossRef CAS PubMed.
- A. J. Howie and D. B. Brewer, Micron, 2009, 40(3), 285–301 CrossRef CAS PubMed.
- W. E. Klunk, R. F. Jacob and R. P. Mason, Methods Enzymol., 1999, 309, 285–305 CAS.
-
I. W. Hamley, Small-Angle Scattering: Theory, Instrumentation, Data and Applications, Wiley, Chichester, 2021 Search PubMed.
- Y. Dong, K. T. Love, J. R. Dorkin, S. Sirirungruang, Y. Zhang, C. Chen, R. L. Bogorad, H. Yin, Y. Chen, A. J. Vegas, C. A. Alab, G. Sahay, K. T. Olejnik, W. Wang, A. Schroeder, A. K. Lytton-Jean, D. J. Siegwart, A. Akinc, C. Barnes, S. A. Barros, M. Carioto, K. Fitzgerald, J. Hettinger, V. Kumar, T. I. Novobrantseva, J. Qin, W. Querbes, V. Koteliansky, R. Langer and D. G. Anderson, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(11), 3955–3960 CrossRef CAS PubMed.
- O. Stachurski, D. Neubauer, I. Małuch, D. Wyrzykowski, M. Bauer, S. Bartoszewska, W. Kamysz and E. Sikorska, Bioorg. Med. Chem., 2019, 27(23), 115129 CrossRef CAS PubMed.
- A. F. Chu-Kung, K. N. Bozzelli, N. A. Lockwood, J. R. Haseman, K. H. Mayo and M. V. Tirrell, Bioconjugate Chem., 2004, 15(3), 530–535 CrossRef CAS PubMed.
- R. Qi, J. Liu, N. Zhang, X. Ji, Y. Han and Y. Wang, Langmuir, 2019, 35(18), 6154–6160 CrossRef CAS PubMed.
- A. Makovitzki, J. Baram and Y. Shai, Biochemistry, 2008, 47(40), 10630–10636 CrossRef CAS PubMed.
- E. Sikorska, O. Stachurski, D. Neubauer, I. Małuch, D. Wyrzykowski, M. Bauer, K. Brzozowski and W. Kamysz, Biochim. Biophys. Acta, Biomembr., 2018, 1860(11), 2242–2251 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence e and
Antonella
Accardo
e and
Antonella
Accardo







