
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLocalization of Zn2+ ions affects the structural folding and mechanics of Nereis virens Nvjp-1†
Eesha
Khare‡
 ab,
Jaden
Luo‡
bcd and
Markus J.
Buehler
*b
ab,
Jaden
Luo‡
bcd and
Markus J.
Buehler
*b
aDepartment of Materials Science and Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA
bLaboratory for Atomistic and Molecular Mechanics, Massachusetts Institute of Technology, 33 Massachusetts Avenue, Cambridge, MA 02139, USA. E-mail: mbuehler@mit.edu
cDepartment of Biology, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA
dDepartment of Chemistry, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA
First published on 1st May 2023
Abstract
Several biological organisms utilize metal-coordination bonds to produce remarkable materials, such as the jaw of the marine worm Nereis virens, where metal-coordination bonds yield remarkable hardness without mineralization. Though the structure of a major component of the jaw, the Nvjp-1 protein, has recently been resolved, a detailed nanostructural understanding of the role of metal ions on the structural and mechanical properties of the protein is missing, especially with respect to the localization of metal ions. In this work, atomistic replica exchange molecular dynamics with explicit water and Zn2+ ions and steered molecular dynamics simulations were used to explore how the initial localization of the Zn2+ ions impacts the structural folding and mechanical properties of Nvjp-1. We found that the initial distribution of metal ions for Nvjp-1, and likely for other proteins with high amounts of metal-coordination, has important effects on the resulting structure, with larger metal ion quantity resulting in a more compact structure. These structural compactness trends, however, are independent from the mechanical tensile strength of the protein, which increases with greater hydrogen bond content and uniform distribution of metal ions. Our results indicate that different physical principles underlie the structure or mechanics of Nvjp-1, with broader implications in the development optimized hardened bioinspired materials and the modeling of proteins with significant metal ion content.
Introduction
Biological materials have been found to take advantage of diverse design paradigms to produce materials with high hardness and stiffness.1–3Nereis virens, a polychaete burrowing marine worm with a hard and stiff jaw used for feeding and defense in abrasive environments, is one excellent example. The hardness and stiffness of the marine worm jaw is remarkable reaching up to ∼0.8 GPa, paralleling the hardness of human cortical bone.4,5 Further, Nereis virens is able to retain its hardness even in a hydrated state.6,7 This behavior is especially intriguing, given that its jaw proteins are mostly organic compared to the mineralized calcified teeth or hard tissues in higher level organisms, which have an inorganic phase of around 75–95% by mass.4,8,9 Metal ions, and in particular Zn2+, have been found to play a role in the mechanical properties observed in these polychaete worms.4,6,8 The incorporation of these metal-coordination interactions is also unique given that these bonds have lower energies than covalent bonds, enabling them to reform after rupture and be sensitive to a number of conditions including pH or presence of other ions.10–12The isolation and structural prediction of Nvjp-1, a major component of the Nereis virens distal worm jaw extracts, has started to enable a detailed nanostructural understanding of how Zn2+ ions may contribute to the mechanical properties of the protein.13 Nvjp-1 is a histidine-rich (over 25 mol%) protein that undergoes significant hydrodynamic changes depending on pH or the presence of metal ions.13 In agreement with experiment, our group previously computationally predicted the structure of Nvjp-1 and found that the structure becomes more compact as the ratio of Zn2+ ions to protein increased.14 Through additional simulations, Bekele et al. found that pH also affects the protein structure, where metal binding happens with polar residues at low pH and is passed onto carboxylate or imidazole coordination pockets at neutral pH.15
While these research efforts have provided important insights into the role of pH and Zn2+ quantity on structural binding and mechanical properties, an understanding of how the location or distribution of Zn2+ affects these properties is missing, preventing a detailed understanding of how biological organisms use such metal ions for structural function. The larger worm jaw itself exhibits a specific metal ion gradient that directly relates with its stiffness and hardness,8,16 and a similar distribution could be expected at the nanoscale as well. Further, metal ions are known to play a role in protein folding by changing the underlying protein folding energy landscape.17,18 For example, in some biological organisms, Zn2+ has been found to induce amyloid-like conformations,19,20 including in some marine organisms.13,21 Given these observations, it is reasonable to expect the location of Zn2+ to have a strong effect on the Nvjp-1 protein structure and resulting mechanical properties. Developing such an understanding computationally would help clarify additional insights into how Zn2+ may enable the remarkable mechanical properties in Nereis virens, especially because the exact localization of Zn2+ ions in Nvjp-1 would be hard to ascertain through experiment. Given that few well-characterized protein structures with several metal-coordination bonds exist, the example with the Nvjp-1 protein provides foundational insights that likely applies to other metal-coordinated protein structures as well. This broader understanding of the location-dependent metal-ion crosslinking effect in proteins would help yield additional design principles for how sclerotized proteins could be synthetically designed to create hard structures in the way that biology does.22–24
In this work, we focus on understanding how the localization of Zn2+ impacts the structural folding and mechanical properties of Nvjp-1, with the goal of contextualizing such work for metal-coordinated proteins largely. Atomistic replica exchange molecular dynamics (REMD) simulations are performed with explicit solvent to investigate the formation of the protein structures in various coordination environments. Metal ions are initiated in different positions of the Nvjp-1 protein, previously determined by REMD in implicit solvent,14 to understand how the protein folds in the presence of metal ions. Steered molecular dynamics (SMD) experiments are conducted on the resulting protein structures to reveal how the location of metal-coordination bonds impacts mechanical tensile properties. This combination of structural prediction and mechanical properties gives information about Nvjp-1 binding with metal ions and will enable its broader use in mechanomutable engineering materials.14,22 With this information, optimized bioinspired materials can be created for a several practical applications, especially those requiring hard, sclerotized structures.
Methods
REMD is used to explore the structural effects of metal ion localization on the Nvjp-1 protein. REMD enables an efficient computational search for stable protein structures by overcoming kinetic trapping in local energy minima during protein folding.25 The starting protein structure is derived from Chou et al.,14 who used REMD to fold the primary protein sequence in implicit solvent. To simulate the system at pH 8, the protonation states of Nδ1H for histidine (His), Oδ2(−) for aspartic acid (Asp), and Oε2(−) for glutamic acid (Glu) were adjusted accordingly. Simulations were implemented with Nanoscale Molecular Dynamics (NAMD)26 and all simulations utilized the CHARMM22 force field27 with a 2 fs timestep under NVT conditions with Langevin dynamics. Zn2+ ions were added to a concentration of 8 wt% to replicate the metal ion concentration found in the tip of the Nereis virens worm jaw.8,13,28 Counterions of Cl− were added to balance the charge. A TIP3P explicit water box with a 16 Å skin was used, and periodic boundary conditions and the particle mesh Ewald full system electrostatics method were applied.After a brief energy minimization using NAMD's conjugate gradient method to avoid bad contacts and a 30 ns NPT equilibration of the water to achieve correct pressure of 1 atm, 96 replicas were used for the REMD simulation with a temperature range of 300–480 K and an exchange time of 200 fs to allow for the system relaxation under an NVT ensemble. The simulations were collectively run for ∼2 μs across all replicas. The REMD simulation in the trajectory at 300 K was analyzed using the K-means clustering algorithm in the MMTSB toolset.29 This algorithm clustered the structures based on conformational similarity within a 2 Å root-mean-square deviation (RMSD). Three representative structures within the lowest energy cluster were identified for subsequent analysis. The Visual Molecular Dynamics30 STRIDE algorithm31 was used to quantify the structural characteristics of the representative structures. The protein contact map was calculated using the Protein Contact Maps tool,32 and the metal ions were added to the contact map based on visual analysis. Chimera is used to characterize the surface properties of the protein.
Representative structures from the lowest energy clusters of the different simulation conditions were solvated in an TIP3P explicit water box extended by 60 Å for SMD simulations to account for deformation. After preliminary simulations suggested minor differences between the different pulling orientations, the terminal Cα atom on the N-terminus was fixed and the terminal Cα atom on the C-terminus was selected as the SMD atom. SMD data were collected every 10 ps at a pulling rate of 1 m s−1. Two additional pulling rates (0.2 m s−1, 20 m s−1) are also briefly explored to test the effect of strain rate on the results. The coordination bond was defined as broken at a 3 Å distance between the metal ion and coordinating polar atom.
Results and discussion
Several mechanisms have been hypothesized to promote the structural folding and formation of the worm jaw, even though the exact in vivo mechanism is unknown. The first hypothesized mechanism is a pH jump between the secreting gland cells and the extracellular matrix where the protein is formed.13 The second hypothesized mechanism is a multi-step modification process where metal ions are added to an already partially folded protein structure, which may allow both the regional control over jaw properties and the ability to modulate such properties once the jaw is already formed.33 Such multi-step phenomena have been found in other organisms with metal-ion induced sclerotized structures, such as in arthropod biting parts where a high density of channels transports metal ions to an already-mostly formed protein matrix.34 Due to computational limitations of mimicking the first mechanism, we explore the second mechanism by distributing metal ions in different positions along an already mostly-folded protein structure. Zn2+ ions are initially distributed in three orientations in the solved REMD structure from Chou et al. (Fig. 2).14 As a baseline case, named “A”, the Zn2+ ions are uniformly distributed throughout the protein, a common practice when the exact coordination geometries are unknown.14,35 In the second condition, named “B” for “beta,” Zn2+ ions are distributed around the small clusters of beta-sheets found in Nvjp-1, to determine whether this placement facilitates the formation of amyloid structures as in other proteins.13,19–21 The third condition, named “C” for compact, localizes Zn2+ ions in the compact, condensed regions of Nvjp-1. Such localization may have a reinforcing effect34,36 on the already compact regions of the protein through additional coordination bonds.The REMD simulation convergence was assessed by several criteria. As shown in Fig. 1a, the average energy of the lowest cluster energy across the simulations approached a stable plateau. The root mean square deviation of protein structure at 300 K was also analyzed for the simulations and remains relatively constant after 10 ns in Fig. 1b. Last, the coil and helix secondary structure are shown as a function of time in Fig. 1c. This analysis metric also reached a plateau around 12–13 ns per replica. While it is always possible to run the REMD simulation for longer times for more accurate structural predictions, these several criteria together indicate that the REMD simulations have reached a converged state, whereby further characterization would yield reasonable results. The initial starting point of a folded protein structure also accelerates the convergence of the REMD simulation.
 | ||
| Fig. 1 REMD simulations are converged. (a) The lowest energy cluster for simulations A, B, and C reaches a stable energy value by 20 ns per replica. (b) The root mean square deviation (RMSD) of the 300 K replica reaches a stable value. (c) The coil and helix secondary structure of the 300 K replica reaches a stable value by 20 ns per replica. | ||
To understand how metal-coordination bonds affect the protein's folding and thereafter mechanical function, three lowest energy representative structures were obtained from the REMD simulations, as shown in Fig. 2. According to our simulations results, the initial conditions of the metal ion distribution affected the resulting structure of the Nvjp-1 proteins. Though the REMD method should theoretically be independent of initial conditions, the large quantity of metal ions in specific proteins biases the exploration of different energy landscapes. As such, the practical computational resources required to overcome all the energy barriers imposed by metal ions is impractical, and indicates that simulations of metal ions in highly coordinated proteins must consider initial localization. When the metal ions were uniformly distributed throughout the protein, as is the case for simulation A, the protein structure did not deviate significantly from the initial structure. Comparing the smaller magnitude of the structural change in simulation A versus B/C suggests that the metal ions in the middle turn region of the protein contribute to locking the protein in its original structure and providing a higher energy barrier when folding. As these middle turn region metal ions are present in A, but not in B/C, less structural change in A was observed.
 | ||
| Fig. 2 Representative protein structures from REMD simulations. Protein structures are initialized with different distributions of metal ions; uniformly distributed (simulation “A”), clustered in the beta-sheet regions (simulation “B”), or clustered in the compact regions (simulation “C”). Zn2+ ions are colored in purple. Secondary structures are colored as: yellow beta-sheet, red helix, cyan coil, and iceblue turns. Structures 1–3 are three representative structures from the lowest energy cluster of each converged REMD simulation. (a) In simulation A, final protein structures do not deviate significantly from the original structure, indicating that the Zn2+ stabilizes the protein structure such that beta-sheets and helices are mostly conserved. (b and c) In simulations B and C, the proteins become more disordered. The difference in the degree of conserved structure in simulation B/C versus A suggests that the uniform distribution of Zn2+ and stabilizing effect of Zn2+ towards the middle of the protein results in additional energy barriers during protein folding. | ||
Across all three simulation conditions, the converged proteins showed a more dispersed distribution of metal ions, with fewer helixes, and the turn and coil secondary structures dominate the protein structure (Fig. 3a and Table S1, Fig. S1, ESI†). The amount of random coil increased across all simulations, where A had the most random coil content, followed by B, then C. A had more metal ions clustered towards the C-terminus region of the protein, whereas B has more metal ions clustered towards the N-terminus. The metal ions in C were more uniformly distributed throughout the protein. The proteins also all had similar radiuses of gyration within 1 Å, but A and C appeared to have a more spatially uniform distribution of amino acids, whereas B had a 10 amino acid random coil connecting two compact regions (Fig. 2).
 | ||
| Fig. 3 Characterization of representative protein structures. To quantify the structure characteristics of Fig. 2, the STRIDE secondary structure tool in VMD is used. The diagonal hash filling pattern across all sub-figures represents an increase in the % change, rather than a decrease. All % changes are evaluated from the initial structure “original” in Fig. 2 compared to the solved REMD structures. (a) All simulations have more random coil secondary structure from A > B > C, and the amount of helix structure is the same across the 3 sets. (b) Structures A, B, C have similar SASA values with an ordering of A–C > B for both the value and % increase from the original structure. (c) A reorganization of MC bonds is observed, where His and Gly lose several MC bonds, while Asp gains MC bonds. Although Gly does not coordinate with metal ions, it is demonstrated here for comparison as the protein is Gly-rich. (d) The number of hydrogen bonds and increase in hydrogen bonds in the protein changes from C > B > A. | ||
Further, there was an increase in solvent accessible surface area (SASA) across all structures (Fig. 3b and Table S2, ESI†), indicating that the proteins unfolded with the addition of the metal ions. While A and C had similar SASA values in terms of both absolute value and percent increase, B had the lowest SASA value and percent increase, indicating that it is the most compact structure.
This increase in the SASA relates directly with the trend observed for metal-coordination bonds present in the system. Fig. 3c (Table S3, ESI†) shows that the aspartate residues gained metal-coordination bonds from an increasing to decreasing order of B then A–C, and that the histidine residues lost metal-coordination bonds where A and C are similar, but B lost the fewest histidine–Zn2+ bonds. Further, the absolute number of metal-coordination bonds follows the trend where B has the most coordination bonds, followed by A, then C. This, together with the SASA indicates that more metal-coordination bonds result in a more compact structure. We expect the compactness of the structure to be directly related to hardness in compression of the protein, and further simulations of compression tests such as nanoindentation on bio–nano composites37–39 may also help reveal such connections.
In addition, the Zn2+ ions stabilize the beta-sheet clusters in B, as has been found in other proteins.13,19–21 However, surprisingly, the formation of amyloid-like structures is not observed, even though high levels of beta-sheet structure (∼30%) have been observed in the in vitro samples of recombinant Nvjp-1 by Broomell et al.13 This is likely due to both the high Zn-to-protein ratio in our work, which has been found to result in amorphous protein aggregates,13 and the lack of fiber nucleation mechanism during the folding process, such as a pH jump to induce a significant conversion of protein secondary structure.13 The increased coordination with aspartate is also in agreement with the carboxylate coordination observed in Bekele et al.15 and Schmitt et al.40 These increases in carboxylate coordination were observed in the molecular snapshots in Fig. 4b and c, and the aspartate coordination is surprising as aspartate only constitutes ∼7% of the amino acids in the protein whereas histidine constitutes ∼27%. Several carboxylate groups also coordinated Zn2+ ions on the outer surface of the protein (Fig. 4c, ∼30% of the original ions initialized in the protein). Such external coordination of Zn2+ ions (Fig. S2, ESI†) likely contributes to the intermolecular crosslinking of the Nvjp-1 proteins under higher protein concentrations representative of the worm jaw tip. While intermolecular covalent crosslinking is not explicitly possible in Nvjp-1 due to the lack of cysteine residues, Zn2+ coordination on the surface of the protein, coupled with hydrophilic and hydrophobic regions of the protein surface (Fig. S2, ESI†), could form a significant degree of noncovalent intermolecular crosslinking to enable the structural mechanics of the worm jaw.
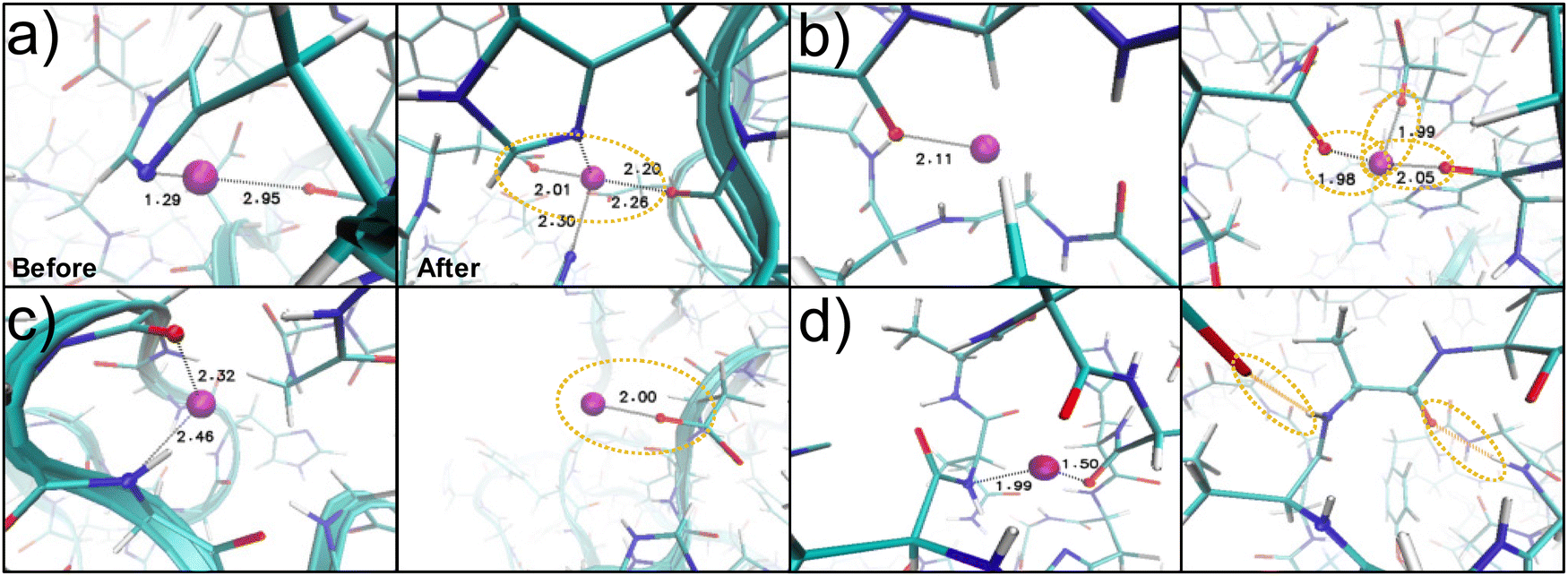 | ||
| Fig. 4 Metal binding pockets observed in REMD folding. Simulation snapshots for structure B showing the changing coordination environment before and after the REMD simulations have converged. Several changes to the coordination environment of the Zn2+ ion (purple) with the protein nitrogen groups (blue) or oxygen groups (red) are shown. The coordination site in (a) gains coordination partners to convert from a bidentate to tetradentate binding arrangement. Aspartate residues are significantly involved in coordination. (b) shows the increased coordination bonds with the carboxylate groups of the aspartate residues or (c) shows the aspartate residues keeping the Zn2+ coordinated to the outside of the protein in a monodentate binding arrangement. (d) Some coordination bonds convert to hydrogen bonds. | ||
Across all simulations, the loss of histidine–Zn2+ bonds is likely because the protein was initially oversaturated with Zn2+ as an initial concentration of 8 wt% was selected to replicate the metal ion concentration found in the tip of the Nereis virens worm jaw.8,13,28 Instead, the resulting wt% was roughly half of the initial concentration, at 4.1 ± 0.08, 4.7 ± 0.08, 4.8 ± 0.17 for A, B, and C respectively (Fig. S1, ESI†). This indicates that the additional Zn2+ ions found in the experimental measurements of the macroscopic worm jaw may be distributed in other proteins or may be located outside the protein matrices. Further simulations beyond the scope of the current study could experiment with changing the initial concentration of metal ions present, as this likely has a strong effect on the structure and mechanics of the protein.
To understand the role of hydrogen bonding in the structure, the absolute and change in the number of hydrogen bonds is plotted across the simulations (Fig. 3d and Table S4, ESI†). C had an increase in the number of hydrogen bonds and A and B had a decrease. An example of the conversion of a metal-coordination bond to a hydrogen bond in the protein structure is seen in Fig. 4d. The trend in hydrogen bonding does not correspond to the SASA or number of metal-coordination bonds in the system. This further leads us to believe that that metal-coordination bonds are more structurally important than hydrogen bonds in the protein folding and compactness of the Nereis proteins.
We find that the structural compactness of the various Nvjp-1 proteins (B > A–C) is independent from its properties under tensile SMD simulations (C > A–B), however, due to different underlying mechanistic principles. In the SMD simulations in Fig. 5a and Fig. S3a, b (ESI†), we found that C has a higher linear elastic modulus and yield strength, followed by B, then A. This trend is reproduced at high pulling speeds, but not at low pulling speeds where the coordination bonds are more likely to dissociate (Fig. S3c and d, ESI†). Further, the elastic modulus, computed up to ∼2% strain with a cross-sectional radius of ∼15 Å, is ∼1–2 GPa. Though not directly quantitatively comparable to experiment due to differences in length scale between a single protein and the entire worm jaw tip, the elastic modulus is in the same order of magnitude as the ∼10 GPa value found in experiments.4,6,8 We attribute this mechanical trend of C > A–B to the nanostructural features of the protein (location of metal-coordination bonds, hydrogen bonds, secondary structure), rather than the measure of global compactness (SASA, number of metal-coordination bonds) which impact the structural folding above. Though the exact quantitative influence of each nanostructural element cannot be clearly differentiated, three contributing mechanisms explain these mechanical differences. First, the trend of strength from C to B to A follows the trend in hydrogen bonds in Fig. 3d rather than the metal-coordination bonds. Hydrogen bonds are important for mechanical strength, and there are ∼2× the number of hydrogen bonds in the protein as there are metal-coordination bonds, resulting in a large influence of the hydrogen bonds on the mechanical properties of the protein. Second, the trend of SMD mechanical properties directly follows the trend in the amount of turn secondary structure in each of the proteins, which decreases from C to B to A, and indirectly follows the amount of coil structure, which increases from A to B to C. Thus, the random coil likely contributes minimal mechanical resistance to the pulling, whereas the turn structure stabilizes the Nvjp-1 protein against mechanical disruption. Last, and a key contribution of this work, C has an even distribution of load-bearing metal-coordination ions throughout its structure (Fig. 5b, c and Fig. S1, S3, ESI†). This uniform distribution of metal ions plays an important role in providing resistance to rupture, especially because the detailed rupture mechanisms of the proteins are heterogeneous. As further evidence to this phenomena, Fig. 5a shows that A and B have bonds breaking together towards the end, but C has a more uniform breaking of bonds through simulation time. Not only does this uniform distribution result in increased stiffness, but it also results in an increased toughness ∼25–30% in simulation C compared to simulation A or B. To further parse the exact contribution of hydrogen bond versus coordination bonds to the strength of the protein, specific protein structures beyond the scope of this work can be designed to isolate each noncovalent contribution. Nonetheless, these results together indicate the importance of hydrogen bonding, secondary structure, and the distribution of metal ions on the mechanical properties of the Nvjp-1 protein in tension.
 | ||
| Fig. 5 Steered molecular dynamics of Nvjp-1 proteins. (a) Force–displacement behavior of Nvjp-1 proteins from SMD simulations at 1 m s−1 shows differences between the various Zn2+ localization conditions. Inset is an expanded view of the force–displacement data. Circles below show when each metal ion breaks its coordination bonds to become non-loading bearing, color-coded by the initial coordinating residue types. (b) Contact map of load-bearing bonds (bi, tri, or tetradentate binding arrangement) in simulations A, B, and C shows that C has metal ions that are more uniformly distributed through the protein. (c) Screenshots of Nvjp-1 proteins show how the protein deforms for simulation C, when the N-terminus is fixed and the C-terminus is pulled. | ||
Conclusions
Our findings indicate that for proteins with high amounts of metal-coordination, such as the Nvjp-1 protein from the Nereis virens worm jaw, the initial distribution of metal ions affects the resulting folded structure, which in turn affects the mechanics of proteins by changing the secondary structures and ion distributions present in the protein. We found that while a uniform distribution of metal ions preserves much of the original structure during REMD simulations, Zn2+ ions in the beta regions help make the structure more compact by preserving or increasing the number of metal-coordination bonds present in the system. However, these structural compactness trends are independent of the mechanical trends found for these proteins due to different underlying mechanisms contributing to each phenomena. While the number of metal-coordination bonds helps determine the compactness of the protein structures, the amount of hydrogen bonding, turn secondary structure, and uniform distribution of the metal ions result in the highest elastic modulus and toughness. Understanding the difference between these underlying energy landscapes could present important insight into how metal ions can be added in different quantities or locations to tune specific folding or rupture behavior. The work presented here likely has broader implications on the role of metal-ion crosslinks on the structure and mechanics of proteins, for both biological assembly mechanisms and the design new synthetic materials.Author contributions
E. K. and M. J. B. designed the project. E. K. and J. L. conducted the experiments. All authors analyzed the results and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
E. K. would like to acknowledge the NSF Graduate Research Fellowship program and the MIT Office of Graduate Education for their financial support. The authors acknowledge support from NIH (U01EB014976, 1R01AR07779), USDA (2021-69012-35978), ARO (W911NF2220213), DOD/DOE-SERDP (WP22-3475), and ONR (N00014-19-1-2375 and N00014-20-1-2189).References
- M. A. Meyers, P.-Y. Chen, A. Y.-M. Lin and Y. Seki, Prog. Mater. Sci., 2008, 53, 1–206 CrossRef CAS.
- M. J. Buehler, MRS Bull., 2013, 38, 169–176 CrossRef CAS PubMed.
- D. J. Rubin, A. Miserez and J. H. Waite, Adv. Insect Physiol., 2010, 38, 75–130 Search PubMed.
- C. C. Broomell, M. A. Mattoni, F. W. Zok and J. H. Waite, J. Exp. Biol., 2006, 209, 3219–3225 CrossRef CAS PubMed.
- B. W. Cribb, C. L. Lin, L. Rintoul, R. Rasch, J. Hasenpusch and H. Huang, Acta Biomater., 2010, 6, 3152–3156 CrossRef CAS PubMed.
- C. C. Broomell, F. W. Zok and J. H. Waite, Acta Biomater., 2008, 4, 2045–2051 CrossRef CAS PubMed.
- H. Quan, D. Kisailus and M. A. Meyers, Nat. Rev. Mater., 2021, 6, 264–283 CrossRef CAS.
- H. C. Lichtenegger, T. Schöberl, J. T. Ruokolainen, J. O. Cross, S. M. Heald, H. Birkedal, J. H. Waite and G. D. Stucky, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 9144–9149 CrossRef CAS PubMed.
- H. C. Lichtenegger, T. Schoberl, M. H. Bartl, H. Waite and G. D. Stucky, Science, 2002, 298, 389–392 CrossRef CAS PubMed.
- E. Khare, N. Holten-Andersen and M. J. Buehler, Nat. Rev. Mater., 2021, 1–16 Search PubMed.
- S. Zechel, M. Hager, T. Priemel and M. J. Harrington, Biomimetics, 2019, 4, 20 CrossRef CAS PubMed.
- A. Srivastava, N. Holten-Andersen, G. D. Stucky and J. H. Waite, Biomacromolecules, 2008, 9, 2873–2880 CrossRef CAS PubMed.
- C. C. Broomell, S. F. Chase, T. Laue and J. H. Waite, Biomacromolecules, 2008, 9, 1669–1677 CrossRef CAS PubMed.
- C. C. Chou, F. J. Martin-Martinez, Z. Qin, P. B. Dennis, M. K. Gupta, R. R. Naik and M. J. Buehler, ACS Nano, 2017, 11, 1858–1868 CrossRef CAS PubMed.
- S. Bekele, K. Singh, E. Helton, S. Farajollahi, R. R. Naik, P. Dennis, N. Kelley-Loughnane and R. Berry, J. Phys. Chem. B, 2022, 126, 6614–6623 CrossRef CAS PubMed.
- J. H. Waite, H. C. Lichtenegger, G. D. Stucky and P. Hansma, Biochemistry, 2004, 43, 7653–7662 CrossRef CAS PubMed.
- L. Banci, I. Bertini, F. Cramaro, R. Del Conte and M. S. Viezzoli, Biochemistry, 2003, 42, 9543–9553 CrossRef CAS PubMed.
- W. Li, J. Wang, J. Zhang and W. Wang, Curr. Opin. Struct. Biol., 2015, 30, 25–31 CrossRef CAS PubMed.
- K. Pagel, T. Seri, H. von Berlepsch, J. Griebel, R. Kirmse, C. Böttcher and B. Koksch, ChemBioChem, 2008, 9, 531–536 CrossRef CAS PubMed.
- W. T. Chen, Y. H. Liao, H. M. Yu, I. H. Cheng and Y. R. Chen, J. Biol. Chem., 2011, 286, 9646–9656 CrossRef CAS PubMed.
- A. Reinecke, G. Brezesinski and M. J. Harrington, Adv. Mater. Interfaces, 2017, 4, 1600416 CrossRef.
- M. K. Gupta, K. A. Becknell, M. G. Crosby, N. M. Bedford, J. Wright, P. B. Dennis and R. R. Naik, ACS Appl. Mater. Interfaces, 2018, 10, 31928–31937 CrossRef CAS PubMed.
- S. C. Grindy and N. Holten-Andersen, Soft Matter, 2017, 13, 4057–4065 RSC.
- X. Zhang, Y. Vidavsky, S. Aharonovich, S. J. Yang, M. R. Buche, C. E. Diesendruck and M. N. Silberstein, Soft Matter, 2020, 16, 8591–8601 RSC.
- Y. Sugita and Y. Okamoto, Chem. Phys. Lett., 1999, 314, 141–151 CrossRef CAS.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kalé and K. Schulten, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS PubMed.
- A. D. MacKerell, D. Bashford, M. Bellott, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiórkiewicz-Kuczera, D. Yin and M. Karplus, J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef CAS PubMed.
- G. W. Bryan and P. E. Gibbs, J. Mar. Biol. Assoc. U. K., 1979, 59, 969–973 CrossRef CAS.
- M. Feig, J. Karanicolas and C. L. Brooks, J. Mol. Graphics Modell., 2004, 22, 377–395 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- D. Frishman and P. Argos, Proteins: Struct., Funct., Bioinf., 1995, 23, 566–579 CrossRef CAS PubMed.
- B. Rafferty, Z. C. Flohr and A. Martini, Protein Contact Maps, 2020, https://nanohub.org/resources/contactmaps.
- C. C. Broomell, R. K. Khan, D. N. Moses, A. Miserez, M. G. Pontin, G. D. Stucky, F. W. Zok and J. H. Waite, J. R. Soc., Interface, 2007, 4, 19–31 CrossRef PubMed.
- E. Degtyar, M. J. Harrington, Y. Politi and P. Fratzl, Angew. Chem., Int. Ed., 2014, 53, 12026–12044 CrossRef CAS PubMed.
- K. Mahnam, B. Saffar, M. Mobini-Dehkordi, A. Fassihi and A. Mohammadi, Res. Pharm. Sci., 2014, 9, 69–82 CAS.
- F. Jehle, E. Macías-Sánchez, P. Fratzl, L. Bertinetti and M. J. Harrington, Nat. Commun., 2020, 11, 862 CrossRef CAS PubMed.
- W. H. Roos, M. M. Gibbons, A. Arkhipov, C. Uetrecht, N. R. Watts, P. T. Wingfield, A. C. Steven, A. J. R. Heck, K. Schulten, W. S. Klug and G. J. L. Wuite, Biophys. J., 2010, 99, 1175–1181 CrossRef CAS PubMed.
- A. N. Masir, A. Darvizeh and A. Zajkani, Adv. Compos. Lett., 2019, 28, 0963693519860162 Search PubMed.
- O. Kononova, K. A. Marx and V. Barsegov, Applied Nanoindentation in Advanced Materials, John Wiley & Sons, Ltd, Chichester, UK, 2017, pp. 393–428 Search PubMed.
- C. N. Z. Schmitt, Y. Politi, A. Reinecke and M. J. Harrington, Biomacromolecules, 2015, 16, 2852–2861 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sm00360d |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2023 |