
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Growth kinetics of the adsorbed layer of poly(2-vinylpyridine) – an indirect observation of desorption of polymers from substrates†
Marcel
Gawek
,
Hassan
Omar
 ,
Paulina
Szymoniak
and
Andreas
Schönhals
*
,
Paulina
Szymoniak
and
Andreas
Schönhals
*
Bundesanstalt für Materialforschung und-prüfung (BAM), Unter den Eichen 87, 12205 Berlin, Germany. E-mail: Andreas.Schoenhals@bam.de; Fax: +49 30/8104-73384; Tel: +49 30/8104-3384
First published on 1st May 2023
Abstract
The growth kinetics of the adsorbed layer of poly(2-vinylpiridine) on silicon oxide is studied using a leaching technique which is based on the Guiselin brushes approach. The adsorbed layer is grown from a 200 nm thick P2VP film for several annealing time periods at different annealing temperatures. Then the film is solvent-leached, and the height of the remaining adsorbed layer is measured by atomic force microscopy. At the lowest annealing temperature only a linear growth regime is observed, followed by a plateau. Here, the molecular mobility of segments is too low to allow for a logarithmic growth. At higher annealing temperatures, both linear and logarithmic growth regimes are observed, followed by a plateau. At even higher annealing temperatures, the growth kinetics of the adsorbed layer changes. A linear growth followed by logarithmic growth kinetics is observed for short annealing time periods. For longer annealing time periods, an upturn of the growth kinetics is observed. At the highest annealing temperature, only a logarithmic growth regime is found. The change in the growth kinetics is discussed by an alteration in the structure of the adsorbed layer. Moreover, the interaction between the polymer segments and the substrate becomes weaker due to both enthalpic and entropic effects. Therefore, at high annealing temperatures the polymer segments might more easily desorb from the substrate.
Introduction
The performance of many innovative multiphase materials involving polymers like polymer-based nanocomposites and hybrid nanomaterials including also thin films with thicknesses of several nanometres is determined by the interaction of the macromolecules with inorganic substrates.1–5 Especially, polymer films with thicknesses below 100 nm are important in materials protection, as functional layers in sensors, and in novel (opto-)electronic devices (see for instance ref. 6). For non-repulsive interactions of the polymer with the substrate, Granick et al. showed that an irreversible adsorbed layer with a nanometric thickness is expected to develop which is stabilized by the connectivity of the polymer chain.1,7–11 The adsorbed polymer segments on the substrate surface can have a reduced molecular mobility or in some cases can even be immobilized.12 Temperature modulated differential scanning calorimetry (TMDSC) experiments employing a high molecular weight poly(vinyl acetate) (PVAc) sample on a silica substrate demonstrated that the adsorbed polymer (which can be even grafted) show a thermal activity with three different regions corresponding to interfacial layers:13,14 (I) a “Tightly bound” region, corresponding to segments directly located on the surface of the substrate. These segments undergo a glass transition located at higher temperatures (characterized by the glass transition temperature Tg) compared to bulk. (II) A “loosely bound” region which is related to polymer segments spatially arranged away from the substrate surface. (III) A “mobile region”, where segments are located at the polymer/air interface, showing a decreased Tg compared to bulk. This decrease of Tg is due to missing segment/segment interactions.13,14 Recently this effect was confirmed for thin films by dielectric measurements and molecular dynamic simulations.15 Similar results were found for instance for poly(methyl methacrylate) (PMMA) on silica substrates. PMMA has a stronger interaction with silicon oxide, compared to PVAc, which is due to hydrogen bonding.13Housmans et al.11 unveiled the growth kinetics of a spatially heterogeneous structure within the adsorbed layer for high Mw atactic polystyrene (PS) employing a leaching approach. The growth kinetics of the adsorbed layer has a two-step adsorption regime. At short time periods, the time dependence of the thickness of the adsorbed layer is linear. Here the polymer chains pin as many segments as possible to the substrate, at an energy cost of kBT (kB is Boltzmann's constant). This leads to a “strongly bound” layer,16,17 with a higher density10,18,19 and reduced molecular mobility. Form the theoretical point of view, this could be understood by a first-order adsorption mechanism. In this approach, chains in contact with the surface pin onto the substrate via kinetics limited by the characteristic time for the attachment of one segment.20,36 The number of adsorbed segments, and therefore the thickness of the adsorbed layer hads, increases linearly with time. Considering that the statistics of the chains obey a reflected random walk close to non-repulsive walls, at a given time, it holds hads ∼ N1/2 ∼ radius of gyration (Rg). The structure of this part of the adsorbed layer consists mainly of trains.10 However, at a certain time, a crowding of segments at the substrate prevents the first-order adsorption mechanism. Housmans et al. proposed a description of the irreversible adsorption mechanism including a crossover between linear and logarithmic growth.11 For a more detailed discussion see ref. 36. In the logarithmic regime, the thickness of the adsorbed layer increases further by diffusion of segments and/or changing its conformation, through the already existing layer. This process will take place at the expense of their entropy. This yields a “loosely bound layer” with a different segment mobility, compared to that of the strongly bound one. At this stage of the growth of the adsorbed layer, loops and tails are formed. In addition, the growth kinetics of this layer also depends on other parameters, for instance, annealing time, molecular weight, packing frustration, and processing conditions.21 For a detailed review of these parameters, the reader is referred to ref. 22. Earlier, it was shown that polymers can be adsorbed on metal oxide, glass or carbon surfaces (for instance see ref. 11, 23–35). Reviews about adsorbed layers in general are given in ref. 36 and 37 which also report the adsorption kinetics of the adsorbed layer for different polymers. Recently, in addition to the leaching approach, chip calorimetry has been employed to investigate the growth kinetics of the adsorbed layer.38
This work attempts to address the growth kinetics of the adsorbed layer of poly(2-vinylpiridine) (P2VP) on a flat silicon oxide substrate. Some results of the growth kinetics of P2VP have been published by Koga et al. considering only one temperature.10 Here a series of different temperatures are employed here. To the best of our knowledge, no temperature-dependent data for the growth kinetics of the adsorbed layer have been reported. These temperature-dependent data lead to the conclusion of a possible desorption of P2VP segments from the substrate.
Materials and methods
P2VP was purchased from PSS Polymer Standards Service GmbH (Mw = 1020 kg mol−1, Mn = 766 kg mol−1, PDI = 1.33). The glass transition temperature was estimated to be 371 K (98 °C) by differential scanning calorimetry (heating rate: 10 K min−1, second heating run).A wafer with a size of 10 × 10 mm2 was used as the substrate. The thickness of the native silicon oxide layer was about 1.7 nm as estimated by ellipsometry. The substrate was cleaned with acetone to remove the protecting photoresist layer. A CO2 snow jet gun was employed to further clean the surface of the substrate down to the microscale. Then it was placed in a plasma oven with an air atmosphere (60 watt) for 600 s to further clean the substrate and to activate the natural silica. A film with a thickness of ca. 200 nm was spin coated from a diluted solution of P2VP in chloroform (a rotational speed of 3000 rpm for 60 s). Then the polymer film was annealed at several temperatures (383 K, 403 K, 413 K and 433 K) for different time periods. These annealing temperatures correspond to Tg + 12 K, Tg + 32 K, Tg + 42 K and Tg + 62 K. The degradation temperature of P2VP was reported to be ca. 660 K.39 The considered annealing temperatures are much lower. Therefore, degradation of P2VP during annealing is unlikely.
It is noteworthy that the question of how the thickness of the initial film influences adsorption was investigated in ref. 33 for poly(vinyl methyl ether) by some of the authors. It was found that the thickness of the resulting adsorbed layer is independent of the initial film down to ca. 120 nm. Then with decreasing initial film thickness the thickness of the adsorbed layer decreases. At a critical thickness of the initial film of ca. 30 nm no stable adsorbed layer is formed anymore.
After the annealing procedure of the 200 nm thick films, the solvent-leaching experiments (also called Guiselin brushes) were employed. An irreversibly adsorbed layer was obtained with this procedure. For this purpose, the samples were soaked in chloroform for 30 min. Since chloroform is a good solvent for P2VP it can be expected that all polymer chains which are not adsorbed to the substrate will be washed away. In that sense the leaching process is dissolution of the polymer which is not adsorbed. After the leaching process, the films were rinsed using chloroform and fast dried under a dry nitrogen stream. Finally, the samples were annealed for further 20 min at the chosen annealing temperature. For a more detailed discussion of the process, the reader is referred to ref. 11. In addition to a leaching time of 30 min, few experiments were carried out where the leaching time was varied to investigate its influence on the thickness of the adsorbed layer.
A Cypher atomic force microscope (AFM) (Asylum Research, Santa Barbara, CA, USA) equipped with a silicon cantilever with a reflective aluminum coating (AC160TS, Oxford Instruments) was employed to measure the thicknesses of the adsorbed layers. For this purpose, the resulting adsorbed layer was scratched using a sharp knife and the step height of the scratch was measured by the AFM. Three scratches were measured per sample and arithmetically averaged. The error of the obtained values was found to be around 1 nm. In addition, AFM measurements were employed to control film topography and to confirm that no dewetting occurred.
Results
Fig. 1 depicts an AFM image of the adsorbed layer leached from a 200 nm thick film annealed at 413 K (140 °C, Tg + 42 K) for 336 h as an example of how the thickness of the irreversible adsorbed layer was estimated. The figure shows that the adsorbed layer has a relatively smooth surface and that no dewetting takes place even at relatively high temperatures. The phase/friction contrast image of the AFM image given in Fig. 1 is given in Fig. S1 in the ESI.† The figure evidences that really the scratch goes through the whole film thickness. Besides Fig. 1 the result shows that no dewetting was observed for all topographic AFM images in the considered temperature and annealing time range although dewetting is a common feature of thin polymer films (see for instance ref. 40–42). Moreover, the thickness of the adsorbed layer could be estimated accurately.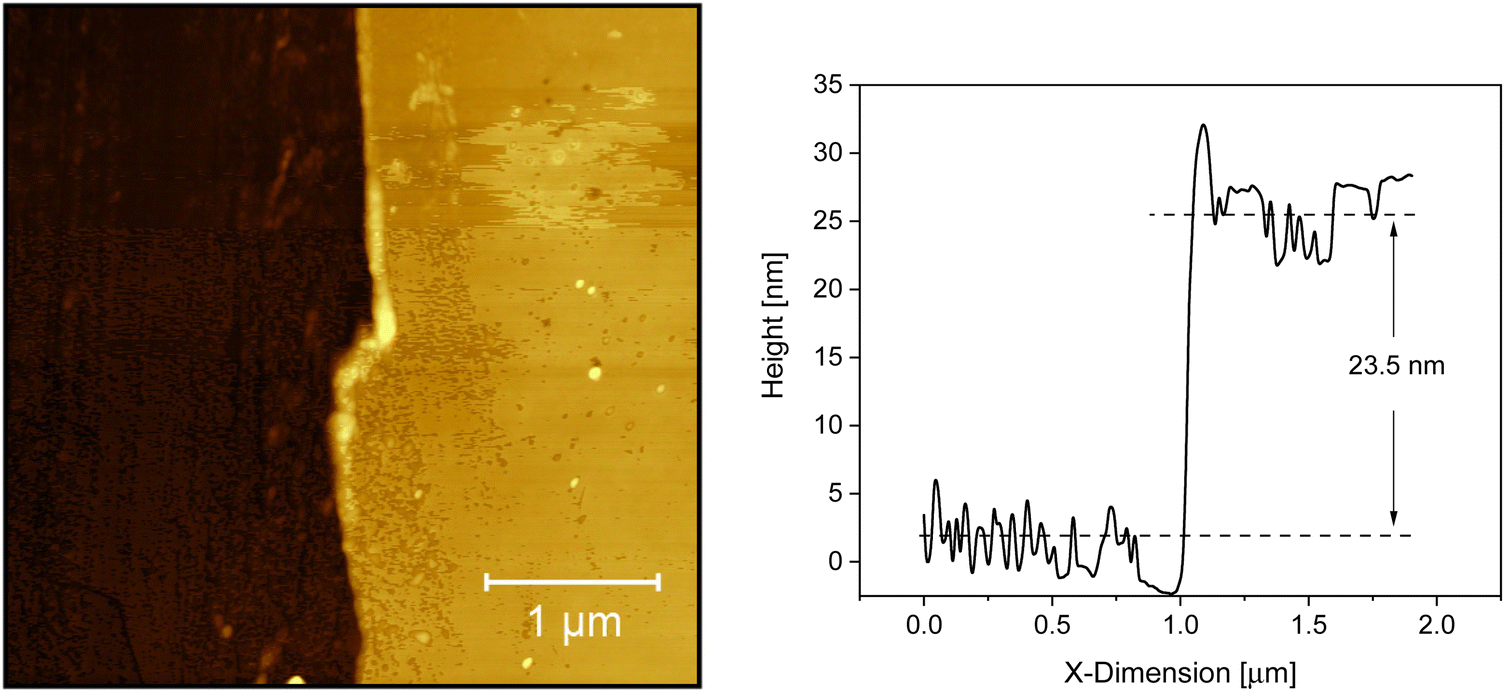 | ||
| Fig. 1 AFM image for the adsorbed layer of P2VP solvent leached from a ca. 200 nm thick film which was annealed at 413 K (140 °C, Tg + 42 K) for 336 h. The right side of the figure shows the thickness profile of a scratch through the adsorbed layer. | ||
Fig. 2 shows the time dependencies of the thickness of the irreversibly adsorbed layer hads leached from a 200 nm thick P2VP film, which were annealed at temperatures of 383, 403, 413 and 433 K (Tg + 12 K, Tg + 32 K, Tg + 42 K and Tg + 62 K). First, the thickness of the adsorbed layer is larger than previously reported.10 This might be due to the different treatment of the substrate which leads to a different interaction of the polymer with the solid surface. Moreover, the molecular weight of P2VP employed here is much higher (1020 kg mol−1) than that considered in ref. 10 (200 kg mol−1). It is known that the thickness of the adsorbed layer depends also on the molecular weight of the studied polymer.11 Second, the raw data in the figure reveal the differences in the growth kinetics of the adsorbed layer for the different annealing temperatures without employing a theoretical model. In ref. 10 the initial growth kinetics of the adsorbed layer of P2VP measured at one temperature was analyzed using a power law 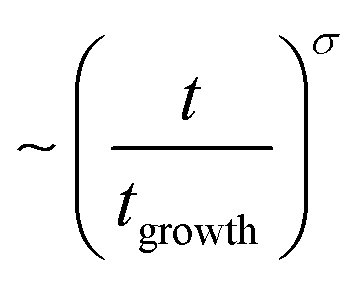 , where tgrowth is a time constant which characterizes the growth kinetics and σ is an exponent. In a first attempt, this approach is employed here also to analyze the growth kinetics of P2VP in the initial stages at different temperatures, although no theoretical basis is discussed for that approach in ref. 10. Fig. S2 (ESI†) depicts that the data at the annealing temperature of 383 K (Tg + 12 K) can be described using a power law with an exponent of one. This corresponds to a linear growth regime discussed in the introduction. The errors of the fit parameters are given in the caption of Fig. S2 (ESI†). At first glance at an annealing temperature of 403 K (Tg + 32 K) the growth kinetics can be approximated using a power law where the exponent is decreased to 0.49 (see Fig. S3a, ESI†). Fig. S3b (ESI†) shows an enlarged image of the growth kinetics at the initial time range. This figure shows that the fit with a power law has large systematical deviations from the data. These deviations are further proved by the residuals of the fit depicted in Fig. S3c (ESI†). The residual displays rather a structured pattern than a statistical distribution of the data points around the zero line. This pattern indicates that the power law is not a perfect description of the data. Fig. S3b (ESI†) shows that a linear fit provides a better description of the initial stages of the growth kinetics. Fig. S4a (ESI†) depicts that the time dependence of growth kinetics at an annealing temperature of 433 (Tg + 42 K) might also follow a power law with a further decrease in exponent to 0.32. Fig. S4b (ESI†) provides the residual plot of the power law fit to the growth kinetic data obtained at an annealing temperature of 413 K. Like for 403 K, this plot shows that a power law fit probably does not provide the best analysis of the data. The growth kinetics at an annealing temperature of 413 K (Tg + 62 K) cannot be described using a power law. Fig. S5 (ESI†) shows that the exponent of the power law decreases approximately linearly with the annealing temperature. As currently the power law approach to the growth kinetics has no theoretical basis the temperature dependence of the exponent cannot be discussed further. Besides the exponent σ, the time constant tgrowth can be extracted from the fits and is plotted versus inverse temperature as shown in Fig. 3. The data can be approximated using the Arrhenius equation which reads
, where tgrowth is a time constant which characterizes the growth kinetics and σ is an exponent. In a first attempt, this approach is employed here also to analyze the growth kinetics of P2VP in the initial stages at different temperatures, although no theoretical basis is discussed for that approach in ref. 10. Fig. S2 (ESI†) depicts that the data at the annealing temperature of 383 K (Tg + 12 K) can be described using a power law with an exponent of one. This corresponds to a linear growth regime discussed in the introduction. The errors of the fit parameters are given in the caption of Fig. S2 (ESI†). At first glance at an annealing temperature of 403 K (Tg + 32 K) the growth kinetics can be approximated using a power law where the exponent is decreased to 0.49 (see Fig. S3a, ESI†). Fig. S3b (ESI†) shows an enlarged image of the growth kinetics at the initial time range. This figure shows that the fit with a power law has large systematical deviations from the data. These deviations are further proved by the residuals of the fit depicted in Fig. S3c (ESI†). The residual displays rather a structured pattern than a statistical distribution of the data points around the zero line. This pattern indicates that the power law is not a perfect description of the data. Fig. S3b (ESI†) shows that a linear fit provides a better description of the initial stages of the growth kinetics. Fig. S4a (ESI†) depicts that the time dependence of growth kinetics at an annealing temperature of 433 (Tg + 42 K) might also follow a power law with a further decrease in exponent to 0.32. Fig. S4b (ESI†) provides the residual plot of the power law fit to the growth kinetic data obtained at an annealing temperature of 413 K. Like for 403 K, this plot shows that a power law fit probably does not provide the best analysis of the data. The growth kinetics at an annealing temperature of 413 K (Tg + 62 K) cannot be described using a power law. Fig. S5 (ESI†) shows that the exponent of the power law decreases approximately linearly with the annealing temperature. As currently the power law approach to the growth kinetics has no theoretical basis the temperature dependence of the exponent cannot be discussed further. Besides the exponent σ, the time constant tgrowth can be extracted from the fits and is plotted versus inverse temperature as shown in Fig. 3. The data can be approximated using the Arrhenius equation which reads
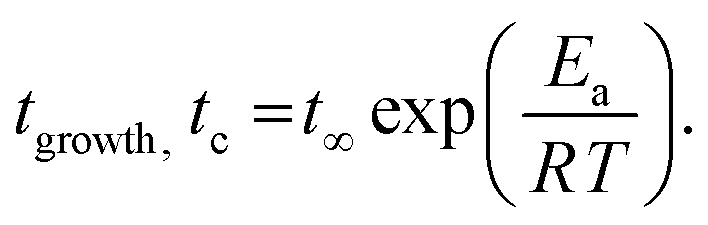 | (1) |
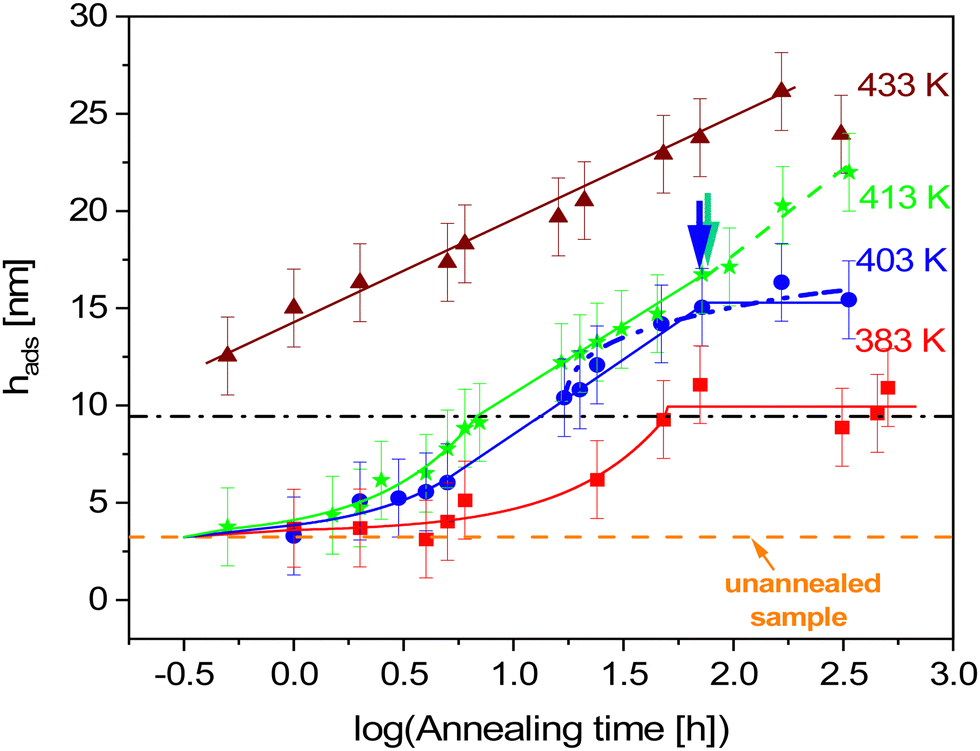 | ||
| Fig. 2 Time dependence of the thickness of the adsorbed layer at different annealing temperatures: red squares – 383 K (110 °C, Tg + 12 K), blue circles – 403 K (130 °C, Tg + 32 K), green asterisks – 413 K (140 °C, Tg + 42 K) and brown triangles – 433 K (160 °C, Tg + 62 K). The error bars are estimated from measurements of three different scratches. The orange dashed line represents the thickness of the unannealed sample. Colored solid lines are fits of linear and logarithmic growth to the data in the corresponding regions of the data. The dashed-dotted-dotted line is a fit of eqn (2c) to the data of the sample annealed at 413 K in the logarithmic growth regime. The green dashed line indicates the time dependence in the logarithmic growth regime at longest times for 413 K. | ||
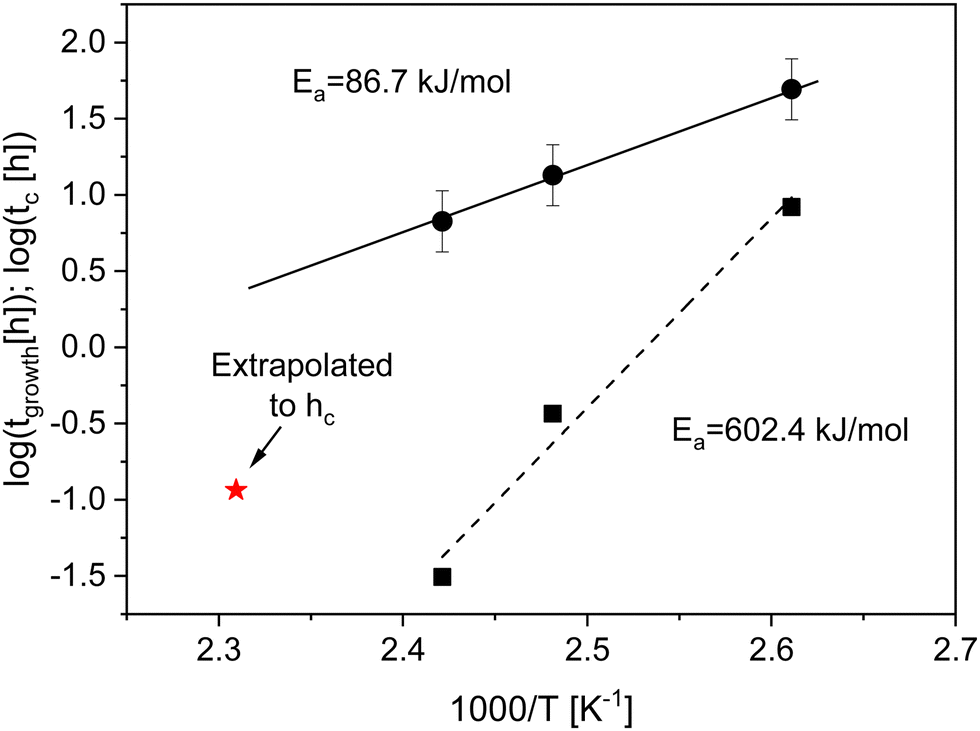 | ||
| Fig. 3 Time constant tgrowth (solid squares) and crossover time tc (solid circles) versus the reciprocal temperature in the Arrhenius diagram at temperatures of 383 K, 403 K and 413 K. The dashed line is the fit of the Arrhenius equation to tgrowth. The errors of tgrowth are smaller than the sizes of the symbols and given in the ESI.† The solid line is the fit of the Arrhenius equation to tc. The red asterisk is the extrapolated crossover time to the thickness of the adsorbed layer at an annealing temperature of 433 K (see text for more details). | ||
In a second approach the growth kinetics of the adsorbed layer of P2VP will be discussed in the frame of the two adsorption regimes proposed in ref. 11 and for other polymers discussed elsewhere36 which has a theoretical justification. As discussed above, for theoretical reasons, the growth kinetics of the adsorbed layer should follow time dependence with two different regimes:
| hads(t) = ht=0 + vt for t < tc | (2a) |
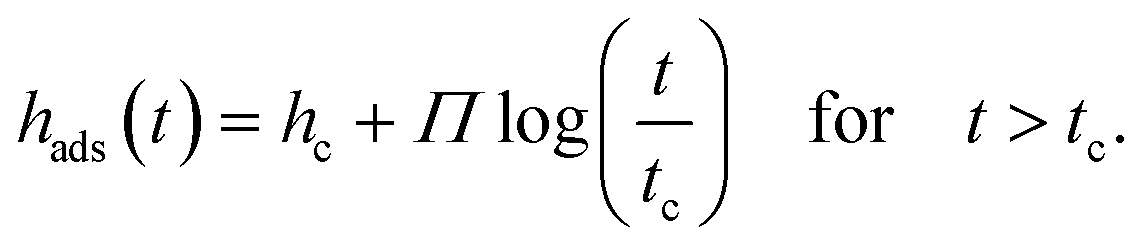 | (2b) |
In eqn (2), tc is the crossover time from the linear growth regime to the logarithmic one. ht=0 and hc are the thicknesses of the adsorbed layer at t = 0 and t = tc, respectively. v is the growth rate in the linear regime and Π is related to the growth rate in the logarithmic regime. The physical basis of these equations is discussed in detail in ref. 1. Eqn (2a) (the first mechanism) results from the direct pinning of the segments onto the substrate, whereas the second mechanism (eqn (2b)) is due to the growth of the adsorbed layer by diffusion of segments through the already adsorbed layer. This takes place at a cost of entropy. One must note that for soft matter systems transitions between the different regimes are not that sharp like that for crystals. For soft matter systems gradients are involved which is already discussed in the three-layer model for thin films. Eqn (2b) predicts unlimited growth of the adsorbed layer which is physically not possible. Therefore, an alternative approach was developed which considers this issue. These considerations result in43
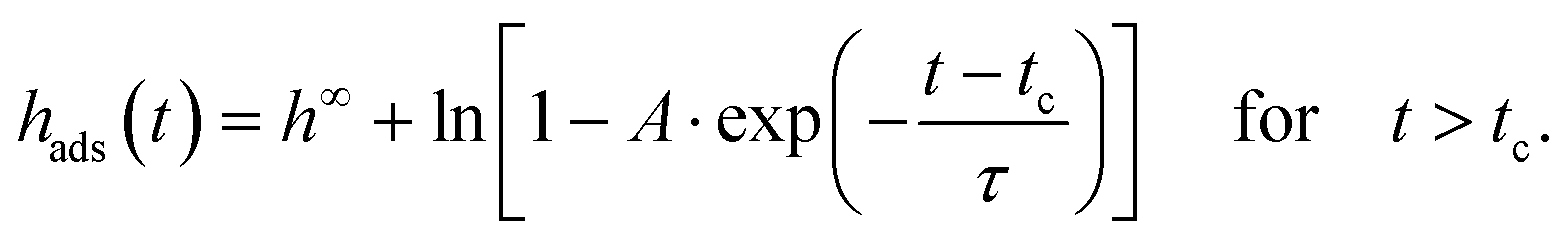 | (2c) |
At first glance, the estimated growth kinetics of the adsorbed layer of P2VP seems to be in agreement with the two regimes proposed in ref. 11 and for other polymers discussed elsewhere.36
At the lowest annealing temperature of 383 K (110 °C, Tg + 12 K) and for short times a linear growth regime of the adsorbed layer is observed. It is worth to note that also the power law approach to the growth kinetics at this temperature predicts a linear growth (see Fig. S2, ESI†). After an annealing time of ca. 40 h the thickness of the adsorbed layer becomes independent of time. The crossover time is estimated by a plot of the thickness of the adsorbed layer versus the linear time scale. The crossover time is taken as the time when the growth kinetics deviates from a linear one. A logarithmic growth regime of the adsorbed layer is not observed for this annealing temperature. This result is further proved in Fig. S2 in the ESI.† Likely the molecular mobility of the segments and chains is too low at this temperature to allow for a logarithmic growth step. Hence the segments cannot diffuse though the already adsorbed layer.
At the annealing temperature of 403 K (130 °C, Tg + 32 K) also a linear growth kinetics is observed up to an annealing time tc of 13.5 h. For longer times the linear growth regime is followed by a logarithmic one till a time of 73 h (indicated by a blue arrow in Fig. 2). The transition from the linear to the logarithmic growth becomes also clear in Fig. S6 (ESI†) where the thickness of the adsorbed layer is plotted versus time in the linear scale. This means that at this annealing temperature obviously the segments have enough molecular mobility to diffuse through the already formed tightly bound adsorbed layer. For annealing times longer than 73 h the thickness of the adsorbed layer becomes independent of time, showing a plateau. Thus, it is concluded that at an annealing temperature of 403 K and an annealing time of 73 h the thickness of the adsorbed layer reaches a maximum value and cannot increase further. Moreover, it was tried to fit eqn (2c) to the data. Fig. 2 shows that the fit of eqn (2c) did not work quite well.
When the annealing temperature is further increased to 413 K (140 °C, Tg + 42 K) again a linear growth regime is observed for short annealing times which changes to a logarithmic growth at a tc of 7.1 h. Surprisingly no change of the logarithmic growth to a plateau is observed for the covered annealing times. This is not expected because the annealing temperature is only slightly increased compared to the lower annealing temperature of 403 K, where a plateau is observed. Instead of a plateau, a turn-up of the growth kinetics is observed which is indicated by a green arrow in Fig. 2. This might imply that at this annealing temperature and time the structure of the adsorbed layer is changed, showing an eased diffusion of the segments through the already adsorbed ones.
At an annealing temperature of 433 K (160 °C, Tg + 62 K) only a logarithmic growth regime of the adsorbed layer is observed. No linear growth regime of the adsorbed layer is found. This means that the linear growth regime at this annealing temperature is probably too fast to be observed in the experimentally accessible time window. In general, compared to the other annealing temperatures, the adsorption kinetics measured for 433 K seems to be different.
At annealing temperatures of 383 K, 403 K and 413 K the thickness of the adsorbed layer hc at the crossover time seems to be independent of temperature having a value of ca. 9.5 nm. It should be noted that the error of hc is the same as that of the measured data which is ±2 nm. In the linear regime the adsorbed layer growth by a direct pinning of segments to the substrate. This process leads to a crowding at the substrate interface and the growth kinetics changes to the logarithmic one. The crowding at the interface is only due to the segment density at tc which should be only weakly dependent on temperature. An annealing temperature independent thickness of the adsorbed layer is also reported in the literature.11 Moreover, Fig. 2 shows further that there is a finite thickness of the absorbed layer at t = 0 of 3.3 nm. The segments forming that part of the adsorbed layer will be probably pinned to the surface by the spin coating process.
At annealing temperatures of 383 K, 403 K and 413 K the crossover time tc could be experimentally estimated as the time when the growth kinetics deviates from a linear one as suggested in the literature (see Fig. S2 and S3a, ESI†).11tc is plotted versus the annealing temperature in a kind of activation diagram (Fig. 3). The temperature dependence of tc for these data points follows a straight line which can be described using an Arrhenius equation. The fit of eqn (1) to the data yields an activation energy of ca. 86.7 kJ mol−1 which is a reasonable physical value. This value is also comparable with the data from the literature. For instance, for polystyrene an activation energy of 80 kJ mol−1 is found.1 It is worth to mention that the observed value for the activation energy for the crossover time corresponds to the activation energy found recently for an equilibration mechanism with a slow mode (SAP).44 This agreement might indicate that such a slow process might be responsible for the adsorption of polymer segments on the substrates. To confirm this assumption further investigations are necessary.
At an annealing temperature of 433 K only the logarithmic growth regime is observed in the experimental time window as discussed above. Therefore, the crossover time tc could not be directly extracted. As experimentally observed, the thickness of the adsorbed layer hc at the crossover time is approximately independent of the annealing temperature. To estimate a crossover time at an annealing temperature of 433 K the time dependence of the adsorption kinetics is extrapolated to hc. The value obtained by this procedure is added to Fig. 3. Obviously, the extrapolated data point does not fit to the Arrhenius-like dependence of tc defined by the lowest annealing temperatures. This might indicate that the growth kinetics of the adsorbed layer formed at an annealing temperature of 433 K is different from that at lower annealing temperatures.
This conclusion is further supported by Fig. 4, where the thickness of the adsorbed layer for an annealing time of 316.2 h is depicted versus the annealing temperature. The temperature dependence shows a transition from a low temperature behavior to a high temperature one at around 410 K. This means that the structure of the adsorbed layer depends on the annealing temperature. This can be discussed in the framework of weaker interactions of polymer segments at higher temperatures. This can be due to enthalpic and entropic effects. The latter contribution is discussed in ref. 20. Probably at higher annealing temperatures a more loosely adsorbed layer with a higher free volume is formed which eases the diffusion of segments. This might suggest that at higher temperatures the polymer segments can be desorbed from the substrate as discussed in ref. 20. The possible desorption of polymer segments can be also the reason that only a logarithmic growth regime is observed. Moreover, the upturn observed at an annealing temperature of 413 K in the logarithmic growth regime for longer annealing times might be also due to desorption effects. This seems to be realistic as the desorption of polymer segments depends not only on the annealing temperature but also on time.
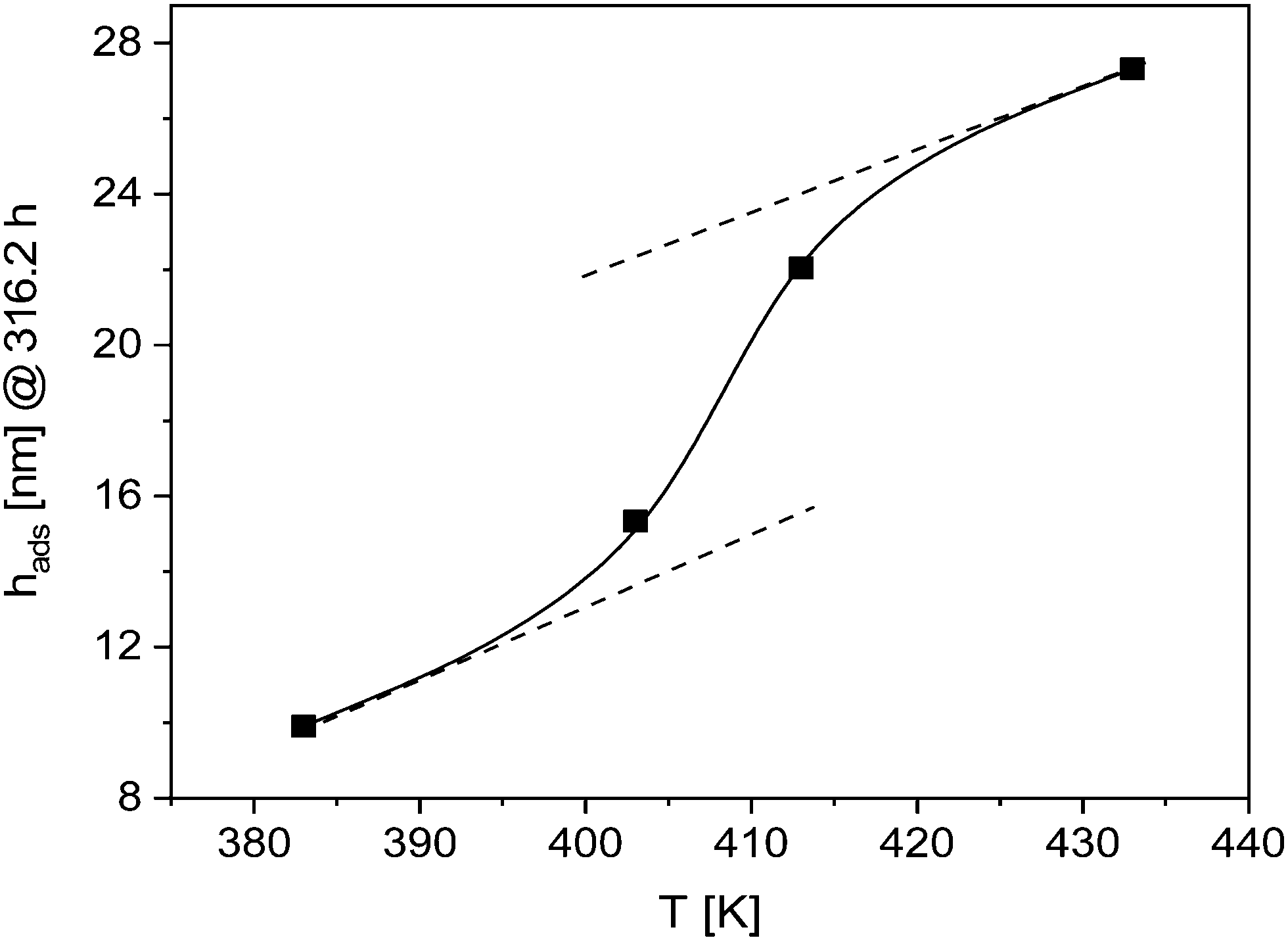 | ||
| Fig. 4 Thickness of the adsorbed layer formed for an annealing time of 316.2 h versus temperature. The solid line is a guide to the eyes. | ||
Fig. 3 and 4 indicate a change in the mechanism of growth kinetics. Besides a possible desorption of the polymer chains from the substrate also a change of the growth kinetics to a growth regime assisted by the dynamic glass transition (α-relaxation) might be considered. To prove this hypothesis further investigations are necessary specially to study the growth of the adsorbed layer at higher annealing temperatures. Such investigations are planned.
Fig. 5 depicts the growth rate in the linear regime versus inverse temperature. In the considered temperature range the data seem to follow Arrhenius dependence with an activation energy of approximately 85.3 kJ mol−1. This value corresponds to the activation energy estimated for the temperature dependence of the cross-over time tc. This seems to be consistent because reaching the cross-overtime is determined by the growth rate in the linear regime.
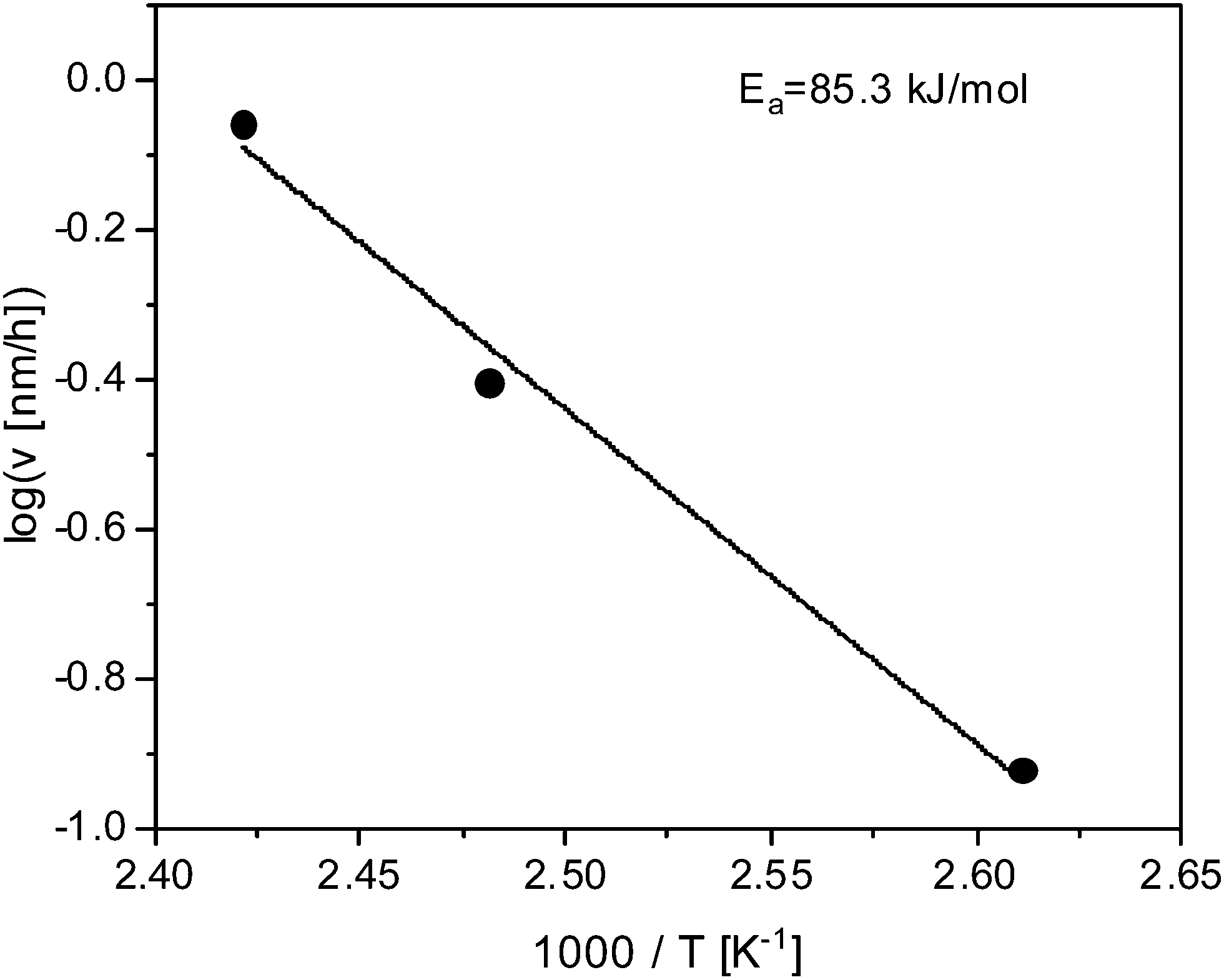 | ||
| Fig. 5 Growth rate in the linear regime versus inverse temperature. The solid line is a fit of the Arrhenius equation to the data. | ||
Finally, the influence of the leaching time is considered. All results discussed above were obtained for a leaching time of 30 min. To investigate the influence of the leaching time at an annealing temperature of 413 K and an annealing time of 3 days (72 h) different leaching times have been considered in Fig. 6. With increasing leaching time, the thickness of the adsorbed layer decreases slightly and continuously decreases although absolute values are in the range of the experimental errors. The thickness of the adsorbed layer reaches a plateau value at a leaching time of 24 h. The maximal difference in the thickness of the adsorbed layer for a leaching time of 30 min and 24 h is around 5 nm for the selected annealing conditions which is close to the experimental error. In further studies different annealing temperatures will be considered to investigate the influence of leaching time in more detail.
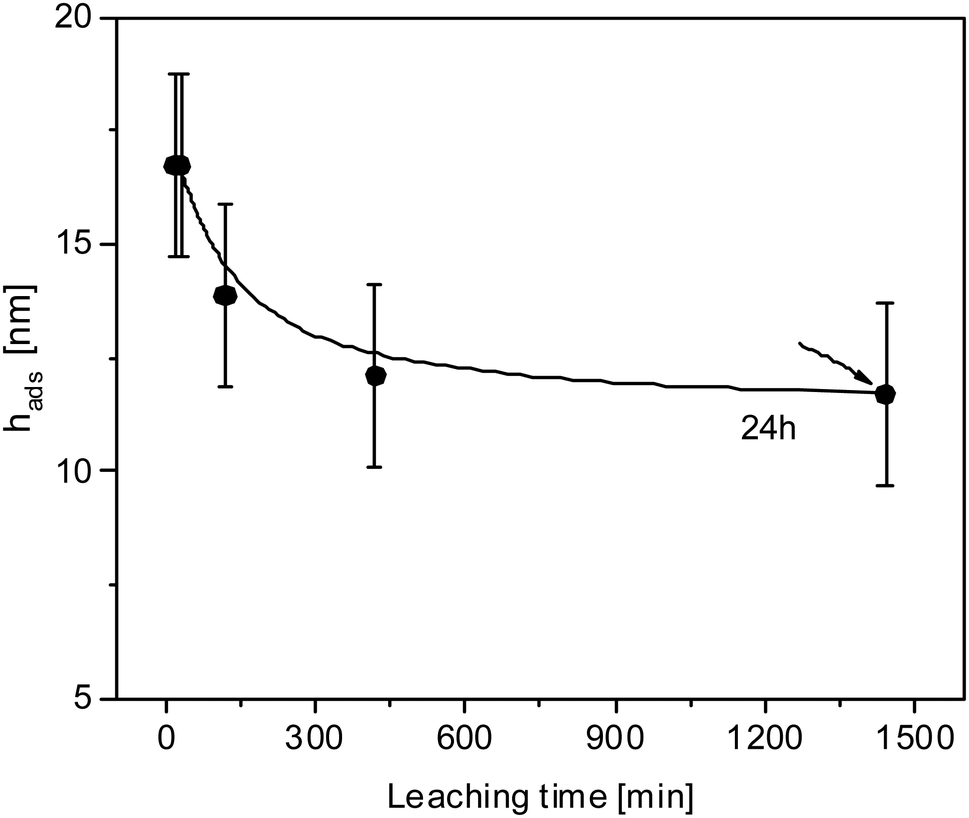 | ||
| Fig. 6 Thickness of the adsorbed layer versus the leaching time for samples annealed at 413 K for 3 days. The line is a guide to the eyes. | ||
Conclusions
The growth kinetics of the adsorbed layer of P2VP was investigated using a leaching approach where the thickness of the adsorbed layer was estimated via AFM measurements. In these experiments, the annealing temperature is varied in a broader range than previously reported in the literature.10 At the lowest annealing temperature, only a linear growth regime is observed followed by a plateau at longer time periods. This is reasoned considering that at low annealing temperatures, the molecular mobility of the segments is too low to allow for diffusion through the already adsorbed layer formed in the linear growth regime. At the next higher annealing temperature, linear and logarithmic growth regimes as well as a plateau for the longest annealing time periods are observed. As expected, for annealing times long enough at intermediate annealing temperatures, the growth of the adsorbed layer saturates, and no further adsorption or desorption of segments takes place. Upon increasing the annealing temperatures, the growth kinetics differs from that at low and intermediate temperatures. At an annealing temperature of 413 K, first a linear as well as logarithmic growth regime is observed. Second, the logarithmic growth kinetics shows an upturn to a faster increase of the growth of the adsorbed layer. At an annealing temperature of 433 K, only a logarithmic growth regime is observed. Moreover, the cross-over time estimated by extrapolation does not agree with the temperature dependence obtained for the lower annealing temperatures. From these results it can be concluded that the structure of the adsorbed layer depends strongly on the annealing temperature. As a possible molecular interpretation, desorption of segments from the substrate is discussed.Conflicts of interest
There are no conflicts of interest.Acknowledgements
The German Science Foundation DFG (Project number 124846229) is acknowledged for financial support to AS.References
- D. N. Simavilla, W. Huang, C. Housmans, M. Sferrazza and S. Napolitano, Taming the strength of interfacial interactions via nanoconfinement, ACS Cent. Sci., 2018, 4, 755 CrossRef PubMed.
- K. J. Johnson, E. Glynos, S.-D. Maroulas, S. Narayanan, G. Sakellariou and P. F. Green, Confinement Effects on Host Chain Dynamics in Polymer Nanocomposite Thin Films, Macromolecules, 2017, 50(18), 7241–7248 CrossRef CAS.
- S. Cheng, A. P. Holt, H. Wang, F. Fan, V. Bocharova, H. Martin, T. Etampawala, T. White, T. Saito, N. Kang, M. D. Dadmun, J. W. Mays and A. P. Sokolov, Unexpected Molecular Weight Effect in Polymer Nanocomposites, Phys. Rev. Lett., 2016, 116, 038302 CrossRef PubMed.
- D. N. Voylov, A. P. Holt, B. Doughty, V. Bocharova, H. M. Meyer, S. Cheng, H. Martin, M. Dadmun, A. Kisliuk and A. P. Sokolov, Unraveling the Molecular Weight Dependence of Interfacial Interactions in Poly(2-vinylpyridine)/Silica Nanocomposites, ACS Macro Lett., 2017, 6, 68–72 CrossRef CAS PubMed.
- E. Bailey, P. J. Griffin, M. Tyagi and K. I. Winey, Segmental Diffusion in Attractive Polymer Nanocomposites: A Quasi-Elastic Neutron Scattering Study, Macromolecules, 2019, 52, 669–678 CrossRef CAS.
- J. K. Bal, T. Beuvier, A. B. Unni, E. A. C. Panduro, G. Vignaud, N. Delorme, M. S. Chebil, Y. Grohens and A. Gibaud, Stability of polymer ultrathin films (< 7 nm) made by a top-down approach, ACS Nano, 2015, 9, 8184–8193 CrossRef CAS PubMed.
- S. Granick, Perspective: Kinetic and mechanical properties of adsorbed polymer layers, Eur. Phys. J. E: Soft Matter Biol. Phys., 2002, 9, 421 CrossRef CAS PubMed.
- B. O’Shaughnessy and D. Vavylonis, Non-equilibrium in adsorbed polymer layers, J. Phys.: Condens. Matter, 2005, 17, R63 CrossRef.
- J. N. Israelachvili, Intermolecular and surface forces, Academic Publisher, San Diego, 2011 Search PubMed.
- N. Jiang, J. Shang, X. Di, M. K. Endoh and T. Koga, Formation mechanism of high-density, flattened polymer nanolayers adsorbed on planar solids, Macromolecules, 2014, 47, 2682 CrossRef CAS.
- C. Housmans, M. Sferrazza and S. Napolitano, Kinetics of irreversible chain adsorption, Macromolecules, 2014, 47, 3390 CrossRef CAS.
- M. Braatz, L. I. Melendez, M. Sferrazza and S. Napolitano, Unexpected Impact of Irreversible Adsorption on Thermal Expansion: Adsorbed Layers Are Not That Dead, J. Chem. Phys., 2017, 146(20), 203304 CrossRef PubMed.
- F. D. Blum, G. Xu, M. Liang and C. G. Wade, Dynamics of poly(vinyl acetate) in bulk and on silica, Macromolecules, 1996, 29, 8740 CrossRef CAS.
- H. Mortazavian, C. J. Fennell and F. D. Blum, Structure of the interfacial region in adsorbed poly(vinyl acetate) on silica, Macromolecules, 2016, 49, 298 CrossRef CAS.
- E. U. Mapesa, N. Shahidi, F. Kremer, M. Doastakis and J. Sangoro, Interfacial dynamics in supported ultrathin polymer films – From the solid to the free interface, J. Phys. Chem. Lett., 2021, 12, 117–125, DOI:10.1021/acs.jpclett.0c03211.
- P. Gin, N. Jiang, C. Liang, T. Taniguchi, B. Akgun, S. K. Satija, M. K. Endoh and T. Koga, Revealed architectures of adsorbed polymer chains at solid-polymer melt interfaces, Phys. Rev. Lett., 2012, 109, 265501 CrossRef PubMed.
- M. Asada, N. Jiang, L. Sendogdular, P. Gin, Y. Wang, M. K. Endoh, T. Koga, M. Fukuto, D. Schultz, M. Lee, X. Li, J. Wang, M. Kikuchi and A. Takahara, Heterogeneous lamellar structures near the polymer/substrate interface, Macromolecules, 2012, 45, 7098 CrossRef CAS.
- N. Jiang, M. Endoh and T. Koga, in Structures and Dynamics of Adsorbed Polymer Nanolayers on Planar Solids, Non-equilibrium Phenomena in Confined Soft Matter, ed. S. Napolitano, Springer International, Switzerland, 2015, p. 129 Search PubMed.
- C. Rotella, S. Napolitano, S. Vandendriessche, V. K. Valev, T. Verbiest, M. Larkowska, S. Kucharski and M. Wübbenhorst, Adsorption Kinetics of Ultrathin Polymer Films in the Melt Probed by Dielectric Spectroscopy and Second-Harmonic Generation, Langmuir, 2011, 27(22), 13533–13538 CrossRef CAS PubMed.
- X. Monnier, S. Napolitano and D. Cangialosi, Direct observation of desorption of a melt of long polymer chains, Nat. Commun., 2020, 11, 4354 CrossRef CAS PubMed.
- K. Randazzo, M. Bartkiewicz, B. Graczykowski, D. Cangialosi, G. Fytas, B. Zuo and R. D. Priestley, Direct Visualization and Characterization of Interfacially Adsorbed Polymer atop Nanoparticles within Nanocomposites, Macromolecules, 2021, 54(21), 10224–10234 CrossRef CAS.
- S. Napolitano, E. Glynos and N. Tito, Glass transition of polymers in bulk, confined geometries, and near interfaces, Rep. Prog. Phys., 2017, 80, 036602 CrossRef PubMed.
- S. Madkour, P. Szymoniak, A. Hertwig, M. Heidari, R. Klitzing, S. von; Napolitano, M. Sferrazza and A. Schönhals, Decoupling of Dynamic and Thermal Glass Transition in Thin Films of a PVME/PS Blend, ACS Macro Lett., 2017, 6, 1156–1161 CrossRef CAS PubMed.
- S. Madkour, P. Szymoniak, J. Radnik and A. Schönhals, Unraveling the Dynamics of Nanoscopically Confined PVME in Thin Films of a Miscible PVME/PS Blend, ACS Appl. Mater. Interfaces, 2017, 9, 37289–37299 CrossRef CAS PubMed.
- S. Madkour, P. Szymoniak, M. Heidari, R. von Klitzing and A. Schönhals, Unveiling the Dynamics of Self-Assembled Layers of Thin Films of Poly(vinyl methyl ether) (PVME) by Nanosized Relaxation Spectroscopy, ACS Appl. Mater. Interfaces, 2017, 9, 7535–7546 CrossRef CAS PubMed.
- S. Kumar, K. K. Sriramoju, V. K. Aswal, V. Padmanabhan and G. Harikrishnan, Unraveling the Polymer Chain-Adsorbed Constrained Interfacial Region on an Atomistically Thin Carbon Sheet, J. Phys. Chem. B, 2019, 123, 2994–3001 CrossRef CAS PubMed.
- X. Li and X. Lu, Evolution of Irreversibly Adsorbed Layer Promotes Dewetting of Polystyrene Film on Sapphire, Macromolecules, 2018, 51, 6653–6660 CrossRef CAS.
- N. Jiang, M. Sen, M. K. Endoh, T. Koga, E. Langhammer, P. Bjöörn and M. Tsige, Thermal Properties and Segmental Dynamics of Polymer Melt Chains Adsorbed on Solid Surfaces, Langmuir, 2018, 34, 4199–4209 CrossRef CAS PubMed.
- M. Füllbrandt, P. J. Purohit and A. Schönhals, Combined FTIR and Dielectric Investigation of Poly(vinyl acetate) Adsorbed on Silica Particles, Macromolecules, 2013, 46, 4626–4632 CrossRef.
- N. Jiang, J. Cheung, Y. Guo, M. K. Endoh, T. Koga, G. Yuan and S. K. Satija, Stability of Adsorbed Polystyrene Nanolayers on Silicon Substrates, Macromol. Chem. Phys., 2018, 219, 1700326 CrossRef.
- D. N. Simavilla, A. Panagopoulou and S. Napolitano, Characterization of Adsorbed Polymer Layers: Preparation, Determination of the Adsorbed Amount and Investigation of the Kinetics of Irreversible Adsorption, Macromol. Chem. Phys., 2018, 219, 170030 Search PubMed.
- S. Madkour, M. Gawek, P. Penner, F. Paneff, X. Zhang, A. Gölzhäuser and A. Schönhals, Can Polymers be Irreversible Adsorbed on Carbon Nanomembranes? A Combined XPS, AFM, and Broadband Dielectric Spectroscopy Study, ACS Appl. Polym. Mater., 2022, 4, 8377–8385, DOI:10.1021/acsapm.2c01320.
- S. Madkour, M. Gawek, A. Hertwig and A. Schönhals, Do Interfacial Layers in Thin Films Act as an Independent Layer within Thin Films?, Macromolecules, 2021, 54, 509–519 CrossRef CAS.
- Q. Xu, N. Zhu, H. Fang, X. Wang, R. D. Priestley and B. Zuo, Decoupling Role of Film Thickness and Interfacial Effect on Polymer Thin Film Dynamics, ACS Macro Lett., 2021, 10(1), 1–8 CrossRef CAS PubMed.
- M. U. Rahman, Y. Xi, H. Li, F. Chen, D. Liu and J. Wie, Dynamics and Structure Formation of Confined Polymer Thin Films Supported on Solid Substrates, Polymers, 2021, 13(10), 1621 CrossRef CAS PubMed.
- S. Napolitano, Irreversible adsorption of polymer melts and nanoconfinement effects, Soft Matter, 2020, 16, 5348 RSC.
- M. F. Thees, J. A. McGuire and C. B. Roth, Review, and reproducibility of forming adsorbed layers from solvent washing of melt annealed films, Soft Matter, 2020, 16, 5366 RSC.
- M. Ishihara, T. Watanabe and T. Sasaki, Adsorption kinetics of polystyrene and poly(9-antracenyl methyl methacrylate) on SiO2 surface measured by chip nano-calorimetry, Polymers, 2022, 14, 605 CrossRef CAS PubMed.
- T. Orhan and J. Hacaloglu, Thermal degradation of poly(2-vinylpyridene), Polym. Degrad. Stab., 2013, 98, 356–360 CrossRef CAS.
- G. Reiter, Dewetting of thin polymer films, Phys. Rev. Lett., 1992, 68, 75–78 CrossRef CAS PubMed.
- A. Das, A. B. Dey, G. Manna, M. K. Sanyal and R. Mukherjee, Nanoparticle-mediated stabilization of a thin film bilayer, Macromolecules, 2022, 55, 1657–1668 CrossRef CAS.
- R. Mukherjee and A. Sharma, Instability. Self-organization and pattern formation in thin polymer films, Soft Matter, 2015, 11, 8717–8740 RSC.
- D. N. Simavilla, W. Huang, P. Vandestrick, J.-P. Ryckaert, M. Sferrazza and S. Napolitano, Mechanism of polymer adsorption onto solid substrates, ACS Macro Lett., 2017, 6, 975–979 CrossRef CAS PubMed.
- Z. Song, C. Rodríguez-Tinoco, A. Mathew and S. Napolitano, Fast equilibrium mechanisms in disordered materials mediated by slow liquid dynamics, Sci. Adv., 2022, 8, eabm7154 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sm00129f |
| This journal is © The Royal Society of Chemistry 2023 |