
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The application of the hierarchical approach for the construction of foldameric peptide self-assembled nanostructures†
Monika
Szefczyk
 *a,
Natalia
Szulc
bc,
Marlena
Gąsior-Głogowska
b,
Dominika
Bystranowska
d,
Andrzej
Żak
ef,
Andrzej
Sikora
g,
Oliwia
Polańska
b,
Andrzej
Ożyhar
d and
Łukasz
Berlicki
a
*a,
Natalia
Szulc
bc,
Marlena
Gąsior-Głogowska
b,
Dominika
Bystranowska
d,
Andrzej
Żak
ef,
Andrzej
Sikora
g,
Oliwia
Polańska
b,
Andrzej
Ożyhar
d and
Łukasz
Berlicki
a
aDepartment of Bioorganic Chemistry, Faculty of Chemistry, Wrocław University of Science and Technology, Wybrzeże Wyspiańskiego 27, 50-370 Wrocław, Poland. E-mail: monika.szefczyk@pwr.edu.pl
bDepartment of Biomedical Engineering, Faculty of Fundamental Problems of Technology, Wrocław University of Science and Technology, Wybrzeże Wyspiańskiego 27, 50-370 Wrocław, Poland
cDepartment of Physics and Biophysics, Wrocław University of Environmental and Life Sciences, Norwida 25, 50-375 Wrocław, Poland
dDepartment of Biochemistry, Molecular Biology and Biotechnology, Faculty of Chemistry, Wrocław University of Science and Technology, Wybrzeże Wyspiańskiego 27, 50-370 Wroclaw, Poland
eElectron Microscopy Laboratory, Faculty of Mechanical Engineering, Wrocław University of Science and Technology, Wybrzeże Wyspiańskiego 27, 50-370 Wrocław, Poland
fAdvanced Materials Engineering and Modelling Group, Faculty of Chemistry, Wrocław University of Science and Technology, Wybrzeże Wyspiańskiego 27, 50-370 Wrocław, Poland
gFaculty of Electronics, Photonics and Microsystems, Wrocław University of Science and Technology, Wybrzeże Wyspiańskiego 27, 50-370 Wrocław, Poland
First published on 9th May 2023
Abstract
In this paper, we show that a hierarchical approach for the construction of nanofibrils based on α,β-peptide foldamers is a rational method for the design of novel self-assembled nanomaterials based on peptides. Incorporation of a trans-(1S,2S)-2-aminocyclopentanecarboxylic acid residue into the outer positions of the model coiled-coil peptide led to the formation of helical foldamers, which was determined by circular dichroism (CD) and vibrational spectroscopy. The oligomerization state of the obtained peptides in water was established by analytical ultracentrifugation (AUC). The thioflavin T assay and Congo red methods showed that the obtained α,β-peptides possess a strong tendency to aggregate, leading to the formation of self-assembled nanostructures, which were assessed by microscopic techniques. The location of the β-amino acid in the heptad repeat of the coiled-coil structure proved to have an influence on the secondary structure of the obtained peptides and on the morphology of the self-assembled nanostructures.
Introduction
Compared to other polymeric materials, peptide-based materials possess many advantages, such as biocompatibility, easily modifiable properties, broad possibility of functionalization, and natural propensity for self-aggregation.1 As we have shown in previous studies,2 the individual features of the studied peptide sequences could be attributed to the achievement of a particular level of organization, which makes the design process rational. This so-called hierarchical approach offers the possibility of constructing complex mesoscopic size architectures in the process of self-assembly starting with the engineering of the peptide sequence. A well-designed amino acid sequence can propagate, producing a variety of secondary structures (i.e., helices, sheets, and turns), which can form 3D architectures (i.e., fibrils, tapes, and nanotubes).3 This approach enables control of the pathway that leads from the peptide building block to peptide-based smart materials showing specific functions.4 The main challenge in the construction of novel functional peptide-based biomaterials is to design constituent peptide monomers that employ these unique secondary structural conformations to establish complementary surfaces that interact noncovalently in a reproducible manner to create the desired nanostructures with favorable properties.5 Furthermore, if nanostructures are obtained by self-assembly, it is important to control the process itself to avoid defects in the structures and to design features that can be introduced to direct the process, for example, by externally applied stimuli.6 Sequence–structure–function dependency, the importance of noncovalent interactions, and other aspects in protein folding,7 assembly,8 and catalysis9 have been intensively studied. However, conclusions and rules have been developed mainly for proteins and peptides consisting of α-amino acids, raising the question of whether our understanding is specific to α-peptides or is universal for peptides containing noncanonical amino acids.10 To address this question, it is crucial to extend the studies to artificial, nonbiological systems, which can assure both testing our understanding of biological structures and developing new building blocks and molecular frameworks with applications in medicine,11 diagnostic agents,12 pharmaceuticals,13 novel materials,14 and others. Peptide foldamers containing nonnatural amino acids are perfect candidates for testing rules of folding and providing diverse building blocks for the construction of nanostructures15 because of the creation of the vast number of new structures and possibility of changing the identity of the backbone.Previously, we proposed an approach for the rational construction of helix-containing nanofibers using the modification of the outer positions of the coiled-coil structure with helix-promoting trans-aminocyclopentanecarboxylic acid (trans-ACPC).2 We have shown that the methodology based on α → β replacement and the incorporation of a constrained β-amino acid residue, apart from the formation of a structure analogous to the original, induces the development of larger nanometer-size structures. Trans-ACPC is a good candidate for the α → β residue substitution, since it is predisposed to adopt the local conformation required for α-helix mimicry16 and it has been shown to promote helical folding.17 However, previously published crystal structures supported by the CD data showed that an alpha/beta-backbone may have a different helix signature from a pure alpha-backbone.16,18 Additionally, the limited backbone modification using beta-residues can provide the stabilization of secondary, tertiary and quaternary structures without significantly altering native structure or function of peptides.18 Additionally, the introduction of β-amino acids can provide proteolytic stability,19 may exert some constraints on the dynamical properties of the peptide chain through increasing the conformational stability of the forming helices, and afford new properties of emerging α,β-peptides. In this work, trans-ACPC residues were placed in the outer positions of the CC structure so they could interact with each other to form a hydrophobic ‘zipper’ and improve the self-assembly properties of the peptides to form nanofibrils. Taking into account the above, we endeavor to design α,β-peptides that mimic the coiled-coil helicity and in the same time show self-association behavior and benefit from the structural preorganization imparted by the cyclic β-residues.
This methodology was probed in two sets of peptides with oligomerization states different from the oligomerization states in our previous studies.2 The secondary structure of the obtained peptides was evaluated using circular dichroism (CD) and vibrational spectroscopy. The oligomerization state of the obtained peptides was established by analytical ultracentrifugation (AUC). The aggregation behavior of the obtained peptides was followed by a thioflavin T (ThT) test and Congo red staining. The final nanostructures were characterized using atomic force microscopy (AFM) and transmission electron microscopy (TEM). Comparison of the results for three sets of peptides gave a broader view of the hierarchical approach and revealed some very interesting features for specific groups of peptides and structural polymorphism. Moreover, positioning of the β-amino acid in the heptad repeat of the coiled-coil structure was demonstrated to influence the secondary structure of the studied peptides and the morphology of the self-assembled nanostructures.
Results and discussion
Design and synthesis
The coiled-coil is a structural protein motif in which two to seven α-helices are coiled together to form a left-handed supercoil. The helices possess a characteristic seven-residue sequence repeat, commonly denoted with letters a to g, where the polar (p) and hydrophobic (h) residues are situated in a particular order, usually with the (hpphppp)n repeat pattern. The proper distribution of polar and hydrophobic residues along the helix ensures exceptional stability of the coiled-coil structure.20 Following the general principles that govern the design of coiled-coil motifs, it has been shown to be possible to construct peptides with a desired oligomerization state.21 However, the final oligomerization state of the designed peptide depends on the length of the peptides (number of heptad repeats) and the medium in which the peptide is dissolved.22 With these considerations in mind, we endeavored to construct two different sets of coiled-coil peptides with three heptad repeats to be model peptides 1 and 2, respectively (Fig. 1A), based on the previously published dimeric and tetrameric coiled-coil peptides.23 In our research, we shortened the sequence by one heptad repeat, applied α → cyclic β residue replacement in the outer positions of model coiled-coil peptides and then performed detailed studies. Therefore, for peptide 1, isoleucine and asparagine were incorporated at position a and leucine at position d, and for peptide 2, leucine was introduced into position a and isoleucine at position d. Lysine and glutamic acid were incorporated at positions e and g, respectively, as they can ensure additional stabilization of the structure by the formation of salt bridges. Alanine was introduced at positions b and c because it is a small helix-favoring amino acid that prevents undesirable influences of side-chain interactions. Glutamine, a charge-neutral polar amino acid, or tryptophan, a UV chromophore, was chosen to occupy the most external position f. Additionally, both ends of the peptide were capped with a glycine residue, and the sequence was appended with a two-residue sequence outside the helical region, in reference to peptides we previously described.2 Furthermore, the N-terminus was acetylated to reduce the net charge.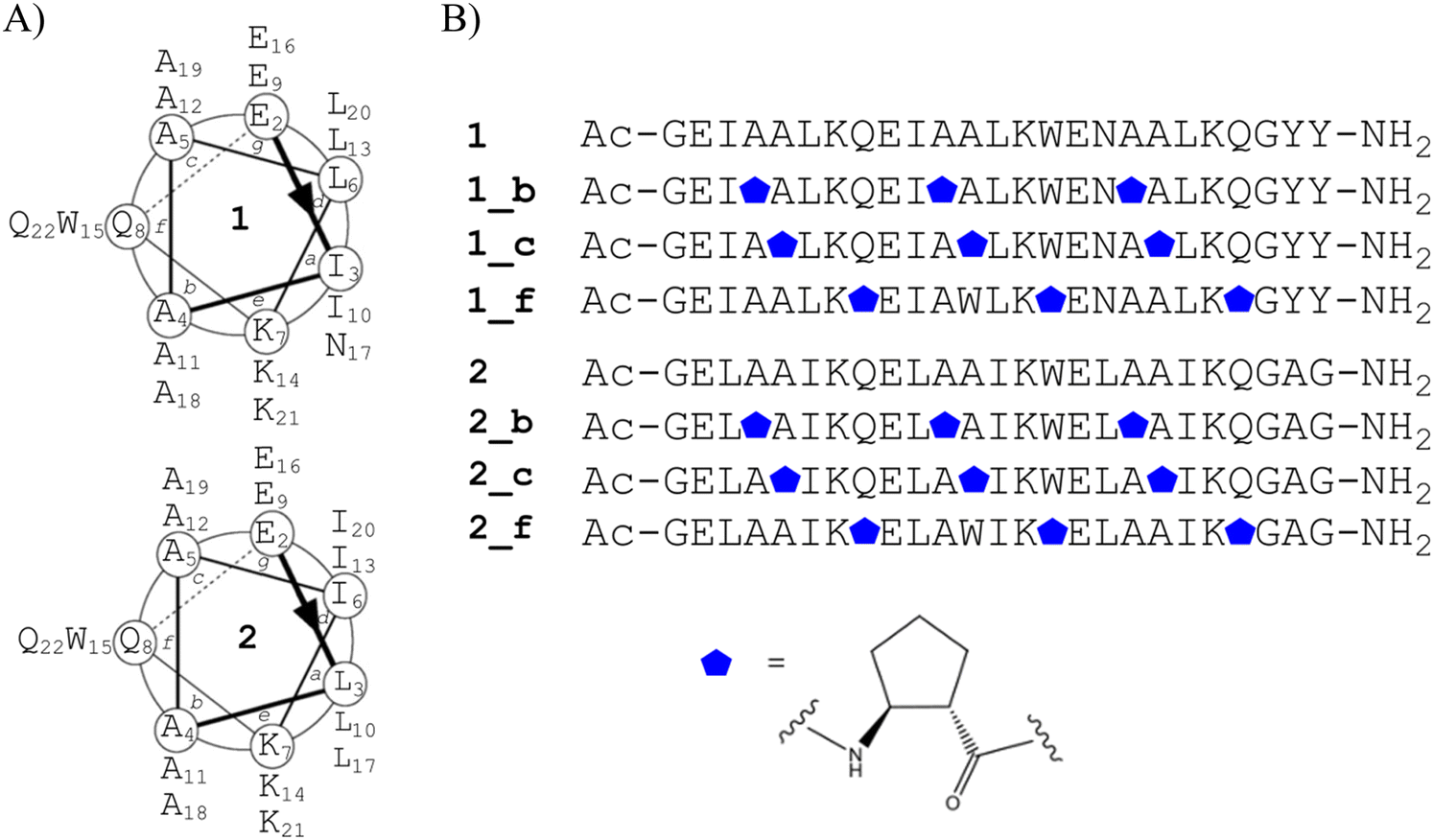 | ||
| Fig. 1 (A) Helical wheel representing the model coiled-coil peptides 1 and 2. (B) Sequences of the designed peptides, where the blue pentagon denotes the trans-aminocyclopentanecarboxylic acid (trans-ACPC) residue. | ||
In the next design step, the sequences of model peptides 1 and 2 were subjected to further modifications. A single α → cyclic β residue replacement was performed in the outer positions (b, c and f) in every heptad repeat of model peptides 1 and 2 with trans-2-aminocyclopentanecarboxylic acid (trans-ACPC), providing three α,β-peptides in this group of peptides (Fig. 1B).
Subsequently, the designed peptides were synthesized using solid-phase peptide synthesis (SPPS) and purified with high-performance liquid chromatography (HPLC). Next, the influence of this structurally constrained residue on the conformational behavior and stability of the helical structures as well as on nanostructure formation was evaluated.
Secondary structure evaluation
Circular dichroism (CD) spectroscopy was used to elucidate the secondary structure of the synthesized peptides in water (pH = 7) (Fig. 2). The model peptides 1 (Fig. 2A) and 2 (Fig. 2B) possess two minima at λ = 208 and 222 nm with the ratio of R(θ222/θ208) = 1, suggested to be typical for interacting α-helices.24 In most of the studied cases, the introduction of the trans-β residue in the chosen positions of the coiled-coil structure did not change the location of the minima compared to the model peptides but influenced the R(θ222/θ208) and mean residue ellipticity values, which is typical for helical α,β-peptides.25 However, in the CD spectra of 1_c and 2_c, shifts of the minimum position from 208 to 211 and 206 nm, respectively, can be observed. Therefore, we can conclude that peptide 1_c possesses only a partially folded conformation with a high contribution from β-sheet structure, since the described peptide was only partially folded, showed a low Cotton effect, and was very prone to aggregation under the study conditions. On the other hand, 2_c possesses a significantly distinct helical characteristics from the alpha-like helix present in the model coiled-coils. Also previous studies, including CD data with multiple crystal structures, showed that an alpha/beta combination backbone can have a different helix signature than a pure alpha backbone.16,18 We observed similar spectral disturbances in our previous studies for the trimeric peptide with trans-ACPC in the b position.2 Calculated mean residue ellipticity values (eqn (S4), ESI†) are generally lower for peptides based on 1 structure than those based on 2 coiled-coil. Thermal unfolding curves at 208 nm (Fig. 3A and B) were used to determine the conformational stability and calculate the melting temperatures (Fig. 3C and eqn (S5), ESI†). Model peptide 2 has a high melting temperature (79.5 °C) that can be associated with a stable and well-packed structure typical of coiled-coils.26 The introduction of the β-residue in position f resulted in a slight decrease in the melting temperature for peptide 2_f (72.4 °C). However, a more significant change in structure stability compared to model peptide 2 can be observed for peptides 2_b and 2_c, although their melting temperatures are still high (approximately 70 °C). Model peptide 1 proved to have a less stable structure than model peptide 2, and further α → β residue replacement in 1 resulted in peptides with even lower melting temperatures (Fig. 3C). The melting temperatures for peptides 1_c and 2_c are lower than melting temperatures for the other peptides in each group, which also confirms the disruption of their structures. Aggregation resulting from the high content of beta structures in some cases makes it impossible to determine the melting point, as was the case with the previously described trimeric peptide functionalized in the b position.2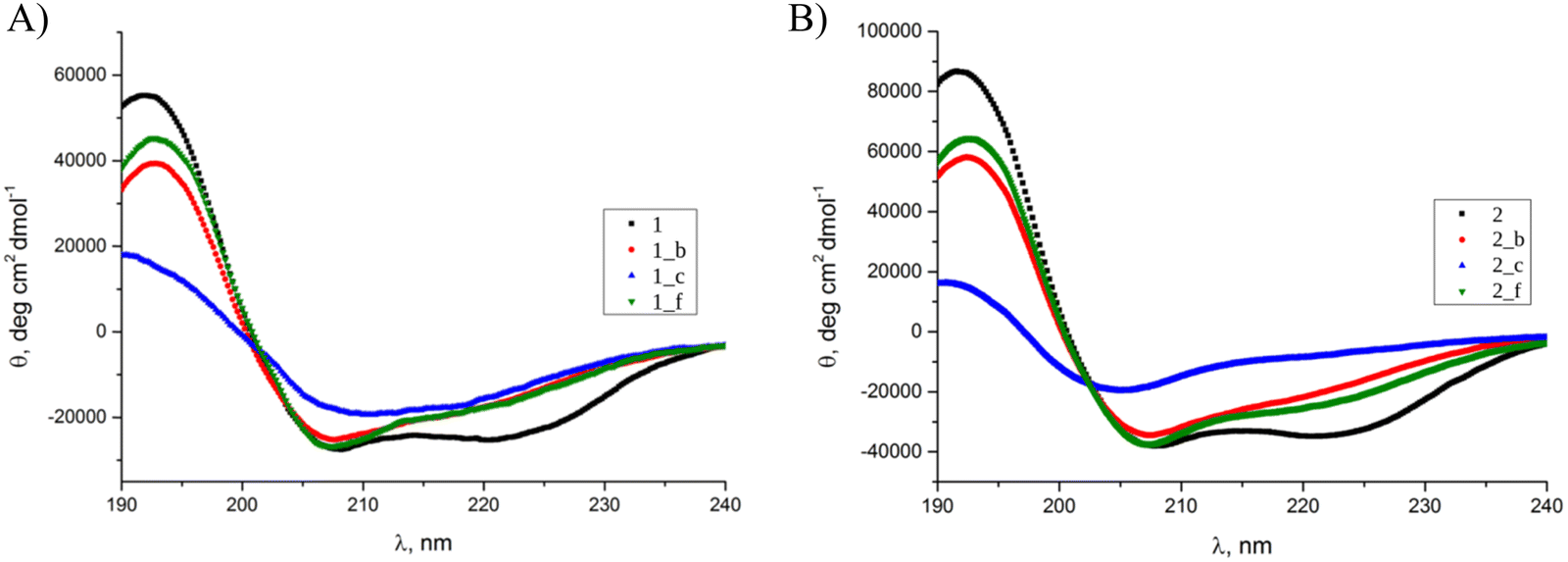 | ||
| Fig. 2 CD spectra of (A) 1, 1_b, 1_c, 1_f, and (B) 2, 2_b, 2_c, 2_f peptides in water. Cpep = 80 μM, pH = 7, T = 20 °C. | ||
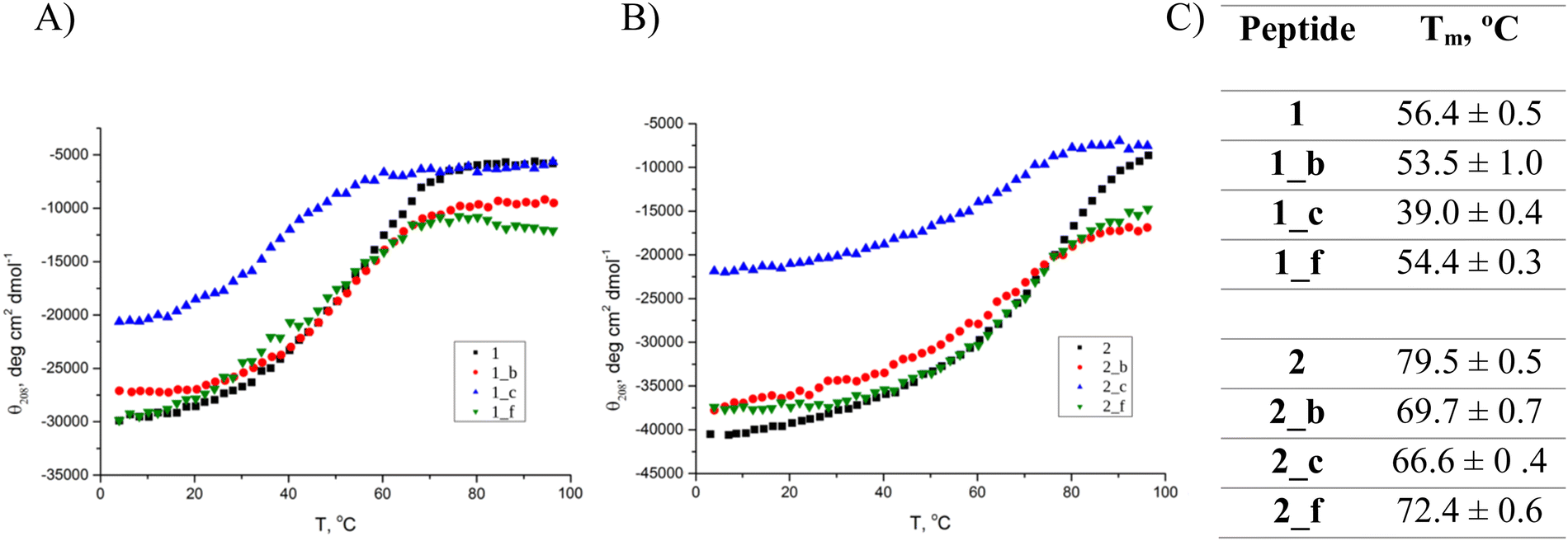 | ||
| Fig. 3 Thermal unfolding curves followed by CD signal at 208 nm of (A) 1, 1_b, 1_c, 1_f, and (B) 2, 2_b, 2_c, 2_f peptides in water. (C) The melting temperatures of the studied peptides were calculated based on thermal measurements at 208 nm in water. Cpep = 80 μM, pH = 7. | ||
The model coiled-coil peptides and the peptides modified with trans-ACPC were then subjected to sedimentation velocity analytical ultracentrifugation (SV-AUC) to provide new data on sample composition and collect information on the molecular weights and sedimentation coefficients of the observed species as well as on the effect of unnatural amino acid incorporation on sample heterogeneity. The continuous sedimentation coefficient distributions c(s) from the SV-AUC experiments revealed (Fig. S6 and Table 1, ESI†) that either the shape or the oligomerization state of the 1 and 2 peptides were changed in the presence of trans-ACPC, but in a different way for each of the peptides. Both 1 (Fig. S6A, ESI†) and 2 (Fig. S6B, ESI†) peptides generally had a single, narrow peak (at 0.672–0.689 S and 0.742–0.801 S for peptides 1 and 2, respectively), showing that there was only one predominant species present at the concentrations used and that there were no significant higher-order components, i.e., oligomers or aggregates. In these cases, the value of the sedimentation coefficients of the main peaks corresponded to the mass of the dimer. The sample of 80 μM–peptide 2 also contains a small portion (10%) of some lower molecular weight species that could result from its degradation. Additionally, a trace amount of higher molecular weight oligomers was observed at 1.993 S (9–10 mers, 5%), but only for a 100 μM–solution of peptide 2. At this concentration, a small fraction of monomers (4%) was also detected.
| Peptide | c (μM) | rmsd | s 20,w | f/f0 | MWapp (Da) | Oligomerization state |
|---|---|---|---|---|---|---|
| Numbers in brackets indicate the percentage of each fraction and are given considering 100% for the sum of the main indicated types of sedimenting species. rmsd – root-mean-square deviation; s20,w – sedimentation coefficient under the standard conditions (i.e., water, 20 °C); f/f0 – frictional ratio (the ratio of the actual frictional coefficient to the ratio for an anhydrous sphere with equal volume); MWapp – apparent molecular weight derived from SV-AUC experiments. The results collected for 1_f and 2_f should be treated with caution due to partial peptide precipitation. | ||||||
| 1 | 50 | 0.004919 | 0.672 | 1.40 | 4946 | Dimer (100%) |
| 80 | 0.005064 | 0.689 | 1.28 | 4469 | Dimer (100%) | |
| 100 | 0.005644 | 0.685 | 1.22 | 4273 | Dimer (100%) | |
| 1_c | 50 | 0.005914 | 0.473 | 1.54 | 3497 | Monomer (100%) |
| 80 | 0.005373 | 0.480 | 1.36 | 2957 | Monomer (100%) | |
| 100 | 0.005689 | 0.492 | 1.33 | 2947 | Monomer (100%) | |
| 1_f | 50 | 0.005048 | 0.646 | 1.48 | 5565 | Dimer (100%) |
| 80 | 0.005109 | 0.677 | 1.35 | 5210 | Dimer (100%) | |
| 100 | 0.005703 | 0.802 | 1.21 | 5687 | Dimer (100%) | |
| 2 | 50 | 0.005568 | 0.801 | 1.15 | 5209 | Dimer (100%) |
| 80 | 00.5486 | 0.235 | 1.17 | 850 | —(10%) | |
| 0.788 | 5216 | Dimer (90%) | ||||
| 100 | 0.009217 | 0.387 | 1.32 | 2149 | Monomer (4%) | |
| 0.742 | 5696 | Dimer (91%) | ||||
| 1.993 | 25096 | Higher aggregates (5%) | ||||
| 2_c | 50 | 0.004492 | 0.780 | 1.20 | 5762 | Dimer (100%) |
| 80 | 0.004920 | 0.588 | 1.20 | 3740 | Monomer (29%) | |
| 1.10 | 9584 | Trimer (71%) | ||||
| 100 | 00.5544 | 0.469 | 1.20 | 2672 | Monomer (20%) | |
| 1.04 | 8750 | Trimer (80%) | ||||
| 2_f | 50 | 0.006200 | 1.03 | 1.48 | 12.7 | Higher aggregates (100%) |
| 80 | 0.006322 | 0.92 | 1.25 | 8.34 | Trimer/tetramer (72%) | |
| 2.51 | 37.4 | Higher aggregates (28%) | ||||
| 100 | 0.006319 | 0.86 | 1.34 | 8.39 | Trimer/tetramer (54%) | |
| 2.09 | 31.9 | Higher aggregates (46%) | ||||
The results obtained for peptide 1_c revealed that the peptide existed exclusively in the monomeric state in solution (Fig. S6C, ESI†). In contrast, the analysis performed for peptide 2_c turned out to be more complex (Fig. S6D, ESI†). At the lowest peptide concentration one broad peak at 0.780 S was observed, which would correspond either to a conformation of the dimer or a mixture of different oligomeric forms that undergo fast and reversible association. As the concentration of the peptide 2_c increased the data revealed two significant species, with the principal component (at 1.04–1.10 S, 71–80%) being consistent in size with that expected for the trimer (Table 1). Its contribution increased slightly as the concentration of the peptide increases, which indicates that peptide 2_c exists in solution as a trimer in rapid equilibrium with smaller oligomeric forms. The second component (0.469–0.588 S, 20–29%) seemed to reflect the monomeric state of the peptide.
Peptides 1_f and 2_f appeared to be not as well soluble in water as the rest of all analyzed peptides. However, the c(s) distribution obtained for the solution of peptide 1_f (Fig. S6E, ESI†) similarly to 1 contains a single peak, corresponding to a dimer. This likely indicates that the incorporation of trans-ACPC did not significantly affected the oligomerization state of peptide 1_f compared to its unmodified version (Table 1). For peptide 2_f we observed an overall increase in the population of its conformations and the formation of higher-molecular-weight oligomers (Fig. S6F, ESI†). The amount of the oligomeric species (12-mers, 46%, Table 1) increased significantly with a 2-fold increase in protein concentration indicating that peptide 2_f is prone to undergo a concentration-dependent change in the oligomerization state.
The SV-AUC experiments revealed that not only the oligomerization state of the designed peptides may change after the trans-ACPC incorporation but also their shape. According to the collected data, peptide 1 turned out to be slightly more compact than peptide 2 (frictional ratio – f/f0 of 1.22 and 1.32 for 100 μM peptides, respectively). Moreover, the obtained results showed a concentration-dependent change in the f/f0 values: a decrease observed for peptide 1, and an increase – for peptide 2, indicating some conformational changes of the coiled-coils to a more compact (peptide 1) and more extended and/or asymmetric structure (for 2). Similar tendency, as for peptide 1 was observed for its modified version, 1_f. Simultaneously, no change in the f/f0 value was observed for peptide 2_c. Due to the high inaccuracy in the determined MW values for 1_b and 2_b, the data were not included in Table 1.
In summary, based on the hydrodynamic properties determined by SV-AUC, we can conclude that peptides 1, 1_c, 1_f, 2, 2_c, and 2_f exist in solution as moderately extended molecules with the propensity for oligomerization, at least up to a peptide concentration of 100 μM. Dimeric is the prevailing oligomerization state for most of the studied peptides. Although we based the design of peptide 2 on the previously published tetrameric peptide,23 the shortening of the sequence and performing experiments in water instead of buffer resulted in oligomerization of the resulting peptide 2 that was lower than expected.
The attenuated total reflectance-Fourier transform infrared (ATR-FTIR) and Fourier transform-Raman (FT-Raman) techniques were used for further studies on the secondary structure of peptides and for the evaluation of structural changes after 30 days of incubation at 37 °C. In the IR spectrum, trans-ACPC possesses a characteristic band located at 1691 cm−1.2 Therefore, the presence of the β-residue in the peptide sequence affects the area of the band with a maximum of 1675 cm−1 characteristic for the α-peptides. We observed that the introduction of trans-ACPC to the sequence caused notable changes in the spectral properties of the amide I band compared to the model peptides containing only α-residues. In particular, a significant widening was observed and an increase in full width at half maximum (FWHM), which is a consequence of the α → β residue substitution. However, significant changes in peak positions after the introduction of trans-ACPC can be directly attributed to specific alterations in structures, since they are not present in all spectra of α,β-peptides. A detailed analysis of the obtained spectra is given below. Additionally, a deconvolution process of the amide I band region (1705–1495 cm−1) provided more detailed information about the structural composition of the studied peptides. The raw and normalized ATR-FTIR and FT-Raman data after dissolving the peptides and after incubation can be found in the ESI† (Fig. S7–S13). The ATR-FTIR results showed that almost all the obtained peptides possess a helical structure due to the presence of a main characteristic absorption peak at approximately 1655–1652 cm−1 (Fig. 4A and C). In turn, for peptide 1_c, the maximum of the amide I band was located at 1629 cm−1, which in the literature is assigned to the β-cross structure,27,28 where β-sheets are stacked within the structure so that their axes are adjusted perpendicular to the stacking direction.29 This spectral signature is characteristic of amyloids, which are known to form long rigid fibrils.30,31 The distinctive structural characteristics of peptide 1_c can be explained by the structural transition from an α-helix to a β-sheet, which has also been observed in other studies of coiled-coil peptides.32 The variations in the helical packing of amino acid chains caused slight differences in the position of the local maxima of the amide I band in the spectra of the studied peptides. The shift toward higher wavelength values can be assigned to a looser packing of amino acids in the helices.2,33 Furthermore, the observed variations in the position of the amide I band in the ATR-FTIR spectra of the studied peptides (Fig. 4A and C) may be caused by the different accessibility of the carbonyl group to water molecules and thus the hydration level. Hydrogen bonds in coiled-coil peptides are bifurcated and involve both water molecules and neighboring double-stranded coiled-coils or their dimers.34 Fourth derivative spectra showed that the dominant component at ∼1651 cm−1 corresponding to the α-helix structure consists of three subcomponents at 1660, 1650 and 1640 cm−1. These findings are consistent with the findings of Heimburg et al., who observed characteristic triplets (1652 cm−1, 1638 cm−1 and 1626 cm−1 assigned to the A, E1, and E2 modes, respectively) instead of a dominant single peak (1650 cm−1) in the amide I′ band region for dimeric coiled-coil proteins, e.g., tropomyosin.35 The feature observed at ca. 1665 cm−1 is attributed to helix formation. Bands observed in the ranges from 1628 to 1610 cm−1 are typically assigned to β-sheet structures and intermolecular aggregates, respectively (Fig. 4).27 The presence of a β-sheet component was also observed in the case of cross-α peptides such as PSMα3.36 The percentage area of this subband clearly distinguishes coiled-coil oligomers from fibrils. The presence of both amide features arising from helical and intermolecular aggregates confirmed that α,β-peptide structures can form fibrils.2
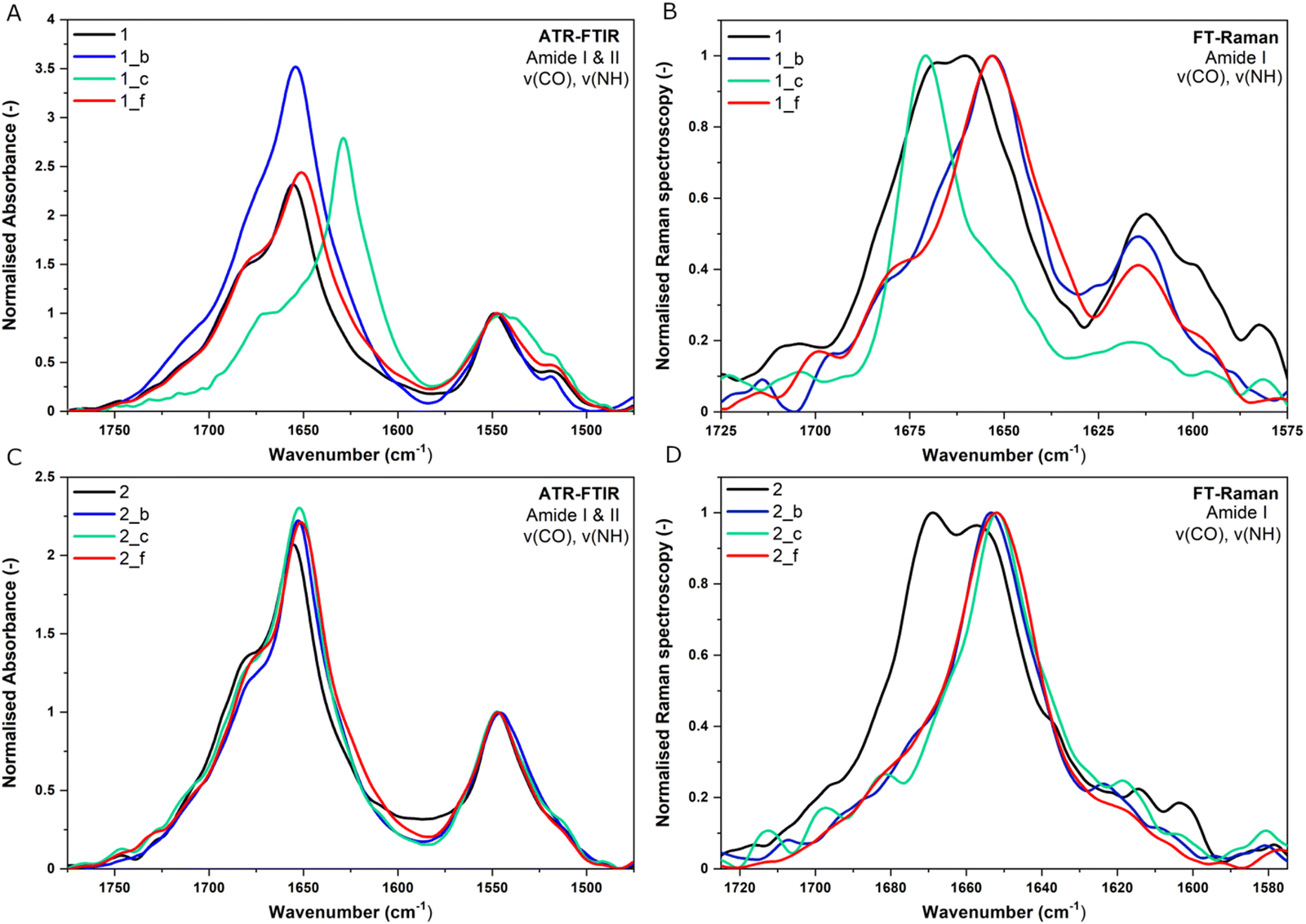 | ||
| Fig. 4 (A and C) Normalized ATR-FTIR spectra of the studied peptides in the wavenumber range of 1775–1475 cm−1 (amide I and II). (B and D) Normalized FT-Raman spectra of the studied peptides in the wavenumber range of 1725–1575 cm−1 (amide I). All spectra were registered directly on the day of dissolving. Cpep = 320 μM. | ||
In previous research, we noticed the structural complexity of the obtained peptides,2 so in this paper, we propose a detailed deconvolution of amide I in the region from 1750 to 1600 cm−1. Changes in the contribution of each structure to the studied peptides immediately after dissolving and then after 30 days of incubation can be observed on the pie chart (Fig. 5). There are three main structural contributions: (i) α-helices in the case of model peptides or disturbed helices in the case of peptides containing β-amino acids, (ii) coiled helices, and (iii) intermolecular aggregates/fibrils. The distinction between “helices” and “coiled helices” was based on the work of Heimburg et al.,35 where the authors showed that the coiled-coils display the position of the amide I band in the IR spectra different from the classical α-helix. It is a consequence of the different pitch value of the α-helix (α3.5) and helices in the CCs (α3.63). Most of the studied peptides possess a dominant helical conformation after dissolving in water, apart from peptide 1_c, which contains mainly intermolecular aggregates/fibrils. However, peptide 2_c contains the highest percentage of helical structures originating from coiled helices.37 The maximum amide I band at 1656 cm−1 is typically observed for collagen type I.38 The subbands in the range of 1745–1710 cm−1 origin from the side chains of amino acids, including trans-APPC amino acids (ATR-FTIR spectrum of trans published here2). After incubation, an increase in fibril content is visible for almost all peptides.
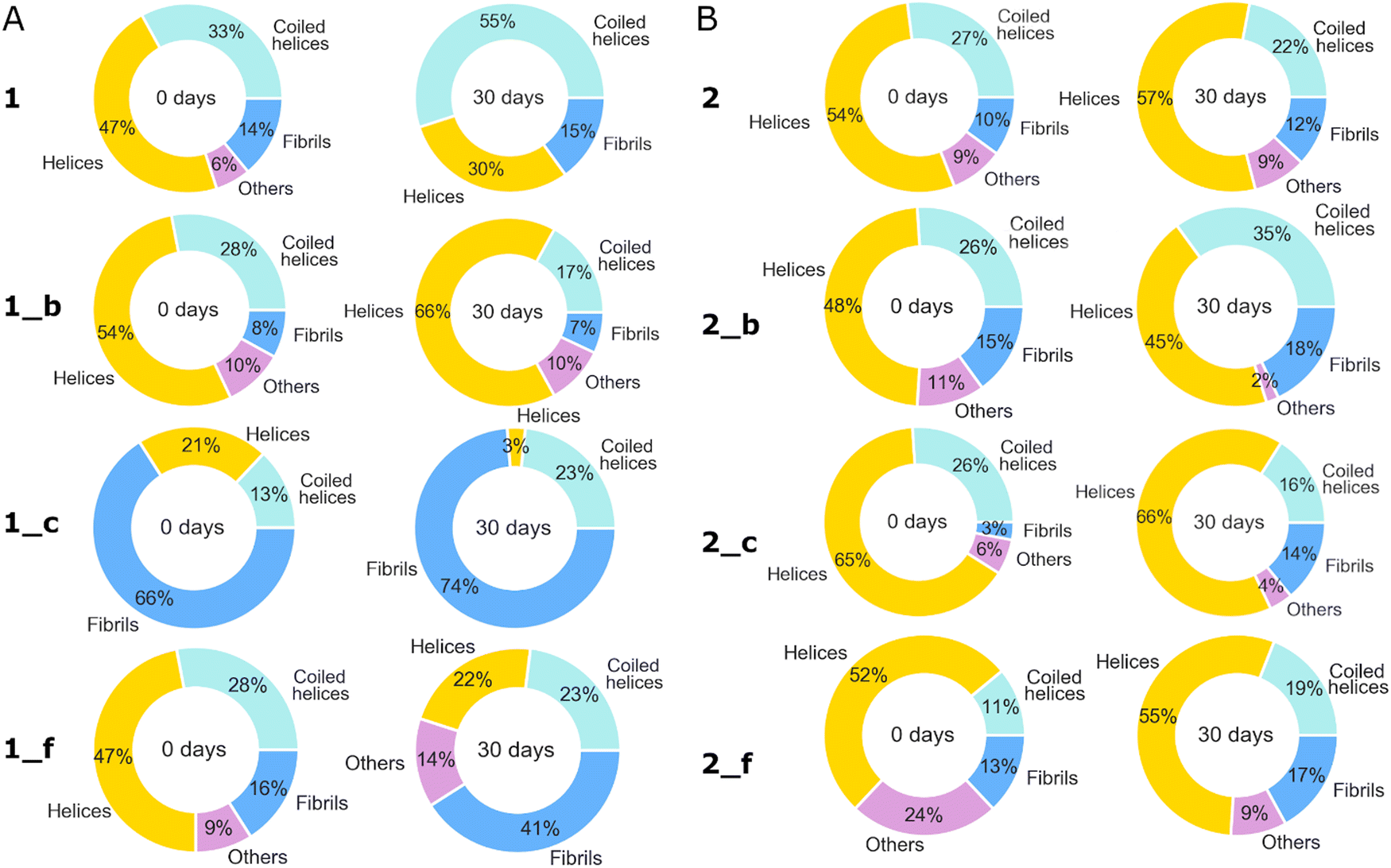 | ||
| Fig. 5 The percentage contribution of the amide I band subcomponents to the total area from 1750 to 1600 cm−1. Data based on deconvolution of ATR-FTIR spectra of (A) 1, 1_b, 1_c, 1_f and (B) 2, 2_b, 2_c, 2_f peptide samples on the day of dissolving (left column) and after 30 days of incubation at 37 °C (right column). Cpep = 320 μM. | ||
The FT-Raman results confirmed the characteristics typical for ordered helical structures for peptides 2_b, 2_c, 2_f, 1_b, and 1_f, due to the presence of the intensive narrow peak at approximately 1653 cm−1 (Fig. 4B and D), additionally showing distinguished 1_c features by the presence of a local maximum at ca. 1672 cm−1 assigned to β-sheet structures.39 Interestingly, FT-Raman analysis indicated a more complex structural composition for peptides 1 and 2 than we observed with CD and ATR-FTIR methods. Both peptides possess a broad amide I band with two local maxima at 1668 cm−1 (corresponding to β-sheet structures), and the second at ca. 1660–1665 cm−1 (representative of a helix or unordered structure), and the maximum of the amide III band located at 1335 cm−1 (indicating the presence of β-sheet structures).40 The results obtained with ATR-FTIR and FT-Raman methods are in concordance with CD studies regarding predominant helical conformation of the obtained peptides (except for the 1_c peptide) and higher conformational stability/helix-packing of the 2 group peptides compared to 1 group peptides. The distinct features of peptides 1_c and 2_c were also observed for peptides with trans-ACPC in the b position in our previous work.2
In summary, the CD and vibrational spectroscopy results that were obtained allowed us to evaluate the secondary structure composition of the synthesized peptides. Model peptides 1 and 2 show typical spectral characteristics for the coiled-coil structure with the addition of other structures (Fig. 2, 4 and 5). The introduction of the trans-ACPC residue in the model sequences influenced the secondary structural characteristics of the emerging α,β-peptide foldamers, namely, the secondary structure composition (Fig. 2, 4 and 5), stability (Fig. 3), and helix-packing (Fig. 4). The strongest impact can be observed for the α → β replacement in c position of the model peptides. We noticed similar spectral changes for the peptide with trans-ACPC in the b position of the model peptide from other sets of peptides.2 Based on the described results, we can conclude that peptides 1_c and 1_b (from our previous work2) exhibit a dominant β-sheet composition/aggregate content, and peptide 2_c seems to possess a disturbed helical structure, leading to the conclusion that, however, all positions b, c, and f are located outside the main coiled-coil core, the f position is the most available for modification without coiled-coil structure disturbance. However, positions b and c are located between long-chain amino acids (Q and K/E), which can create steric hindrance and direct the α → β replacement to create some disturbed helical structure and additional formation of the β-structures. Moreover, after 30 days of incubation, almost all the studied peptides except 1_c and 1_f preserved a dominant helical conformation; however, in most cases, an increase in fibril content was observed (Fig. 5).
Nanostructure studies
To identify the formation of nanostructures of the studied peptides, atomic force microscopy (AFM) and transmission electron microscopy (TEM) were applied. These two techniques were performed in various time steps to follow the self-assembly process. The AFM measurements showed that most of the studied peptides form fibrillar structures directly after dissolving with an average height of ca. 3.05 nm and an average diameter of 0.75 nm. The frequency distributions of the peptide height observed by AFM are presented in Fig. S14 (ESI†). The distinct features of peptides 1_c and 2_c are grooves that are located at equal intervals along the length of the fibril (Fig. 6). After 30 days of incubation, the average diameter of the fibrils increased to approximately 19 nm, and the length increased to several micrometers. TEM micrographs clearly indicate the presence of fibers that are formed by the lateral association of a few smaller fibrils. Interestingly, this lateral association for peptides 1_c, 1_f, 2, and 2_f occurred in the yarn-like structure (zoomed circles in Fig. 6). Moreover, branching effects can be observed for 1_c, 1_f, and 2_f in freshly dissolved samples and for 2 and 2_c after incubation. Furthermore, peptides 1_c and 1_f seemed to be more flexible because of the presence of bend fibrils. The 1_b and 2_b samples exhibit different morphologies, where the presence of globular structures with an average diameter of ca. 6.4 nm can be observed. The spherical/globular/oligomeric aggregates successively formed larger clusters, which after 30 days of incubation reached an average length of ca. 2 μm (Fig. S15, ESI†). In previous research on coiled-coiled-based peptides, we did not observe aggregation of the model peptide after two weeks of incubation.2 In the present work, we extended the incubation time to 30 days, and some aggregate formation for model peptides 1 and 2 was noticed. However, the self-association patterns of peptide 1 and 2 were different from the self-association patterns of other studied peptides; for instance, the aggregate morphology of peptide 1 after dissolving was the same as the aggregate morphology of samples 1_b and 2_b (globular structure formation), yet it changed to fibrils with irregular structural diameters ranging from 38.2 nm to 120.8 nm after 30 days of incubation. | ||
Fig. 6 Representative AFM images with the cross-section profiles measured after dissolving the studied peptides (left column). Electron micrographs of the studied peptides after 30 days of incubation at 37 °C (right column, magnification of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000). Inset: High magnification image of the yarn-like structure. Cpep = 1.6 μM (AFM) and 160 μM (TEM). 000). Inset: High magnification image of the yarn-like structure. Cpep = 1.6 μM (AFM) and 160 μM (TEM). | ||
A more irregular nanostructures morphology (with grooves or globular aggregates) is typical for peptides with trans-ACPC introduced in the b and c positions of the coiled-coil structure. Based on our previous work, we can conclude that the f position of the coiled-coil is the most prone to form homogenous secondary and regular higher-order structures.
Congo red (CR) staining was used as a supporting qualitative method for the characterization of the formed nanostructures. Although this method is used mainly for the identification of amyloid fibrils,41 CR also binds to collagen structures by forming hydrogen bonds with the side chains of the collagen triple helix42 and to oligomers and protofibrils to a lesser extent.43 In Fig. S16 (ESI†), the results of the dye binding to the studied peptide after two months of incubation at 37 °C are depicted. In the case of model peptide 1, amorphous aggregates as well as regular fibers, located mainly at the edges of the coffee ring in the drop, can be observed. In contrast, model peptide 2 binds CR very poorly. The peptides functionalized in the c or f position of the coiled-coil (1_c, 1_f, 2_c, and 2_f) formed unbranched fibrils that reached more than 100 μm in length. In the case of 1_c, fibrils are distributed over the entire surface of the sample. For the 1_f peptide, fibrils are arranged parallel to each other at the edges of the droplet on the slide. Similar effects were observed for the 2_c and 2_f samples. Interestingly, not all stained structures exhibit birefringence under polarized light, which is characteristic of amyloids in tissues.36,44 For peptides 1_b and 2_b, it was difficult to observe formed structures, presumably because shorter fibrils (a few μm long) may not be visible at the 200× magnification used during imaging.45 All the bright structures seen in the background of the samples that do not resemble fibrils are likely to be smaller structures that bind to CR dye. In summary, under the studied conditions, the longest and well-defined fibrils were formed by the peptides containing trans-ACPC in the c or f positions of the coiled-coil (1_c, 1_f, 2_c, and 2_f). These results are in compliance with high-resolution microscopy methods. However, we have not observed significant differences upon CR binding to helix-reach (1_b, 2_c) vs. beta-rich (1_c) peptides, probably because of the low resolution of the method.
The thioflavin T (ThT) fluorescence assay was performed as a supporting quantitative method for the characterization of the aggregation process. The studied peptides show different aggregation kinetics, as can be observed in Fig. S17 (ESI†). Because part of the results obtained cannot be fitted by the sigmoidal curve, the plateau region of each curve (from 5 to 12 hour) was selected, and the mean values of relative fluorescence (Table S18, ESI†) were calculated to compare different peptides. Under the conditions studied, aggregation kinetics with sigmoid curves were observed for samples 1_f and 2_f. Although the lag phase is not present, an elongation phase and a plateau that correspond to fibrillar structures can be observed for both peptides. Additionally, the oligomer/protofibril assemblies are formed faster in the 2_f sample, judging by a steeper curve slope than in the case of the 1_f sample. Additionally, the 2_f peptide had the highest relative fluorescence value. The fluorescence curves for peptides 1, 2, 2_b, and 2_c have some fluctuation, but the main trend is horizontally linear, and in consequence, these peptides possess the lowest fluorescence values. The curves of the 1_c and 2_b peptides start with a short but steep growth, followed by a stabilization and relatively flat plot.
The results obtained with AFM and TEM methods, supported by Congo red and ThT assays, demonstrate that the peptides obtained possess a diverse self-assembly propensity to form different nanostructures. There are no design features in the primary sequence of the studied peptides to promote the formation of “sticky-ended” nanofibers46 or “lock washer” nanotubes.47 Therefore, the increased self-aggregation propensity of some of the studied peptides (Fig. 6 and Fig. S16, S17, ESI†) compared to model peptides 1 and 2 is a consequence of the introduction of the trans-ACPC residues, which can form additional hydrophobic interactions between helices forming “cyclopentyl zipper”, similar to the “cyclohexyl zipper”-like motif observed in the ACHC reach peptides.48 Blunt-ended coiled-coils are formed above a minimum concentration and associate with one another in an offset pair of coiled-coils, which serves as a nucleation point for fiber formation.46 Therefore, the observed globular objects (Fig. 6) are a part of the fibrillogenesis process, not targeted structures, since they are observed mainly during the early stage of research, usually just after dissolving. A few coiled-coil bundles may aggregate to higher-order spherical structures due to shielding effects of hydrophobic sides in coiled-coil peptides.49 Another key parameter controlling fiber formation often is concentration.44,47 Increasing the time and/or concentration of the peptide leads to solutions containing both nanofibers and some number of globular aggregates and then mainly fibrillar nanostructures. Interestingly, peptides that form both β-sheets50,51 and α-helices52,53 exhibited similar fiber morphologies. That is why we observed similar nanostructure morphologies regardless of the secondary composition of the obtained peptides. However, peptides with trans-ACPC residue in the position b showed clearly different emerging structures compared to other peptides (Fig. 6). We observed a similar distinct association pattern for some specific peptides in our previous research.2 Moreover, helical peptides have been shown to convert to β-sheet nanofibers over time or under some conditions54–56 and from β-sheets to α-helices.57 Fibers can be created from both helices oriented parallel to the fiber axis37 or oriented perpendicular to the fiber axis58 since the presence of trans-ACPC residues allows nanofiber propagation in the axial direction (alternative for the “sticky-end” and “lock washer” strategies).
Conclusions
In conclusion, we have successfully applied the hierarchical approach for the construction of nanofibrils based on α,β-peptide The positions of the coiled-coil structure proved to be unequally prone to α → β residue replacement and may lead to the formation of α,β-peptides containing beta-sheets (1_c), different helix characteristics (2_c) or distinct morphology (1_b and 2_b) by creating a template for additional interactions, as can be inferred from the general picture given by the various techniques. AUC measurements showed that the introduction of trans-ACPC residues into the outer positions of the model coiled-coil peptides led to the formation of mainly dimeric foldamers with predominant helical structure and high thermal stability for the group 2 of peptides, as shown by CD data. Vibrational spectroscopy studies exposed a polymorphism of the obtained structures. The thioflavin T assay and Congo red methods showed that the α,β-peptides obtained possess a strong tendency to aggregate. The introduction of trans-ACPC residues into a sequence generally enhances the self-association process, leading to the formation of long, well-defined nanofibrils evaluated by microscopic techniques. Well-designed extended structures with identical, repeated, and regularly spaced subunits with the propensity to associate into high-order assemblies are the future of synthetic biology, nanomaterial engineering, drug delivery systems, and other fields. Peptides are designable and can be characterized in all respects, from sequence to structure, function, and interactions, making them perfect candidates for nanostructure building blocks.Experimental
Peptide synthesis
All commercially available reagents and solvents were purchased from Sigma-Aldrich, Merck or Lipopharm.pl, and used without further purification. Fmoc-(1S,2S)-2-aminocyclopentanecarboxylic acid (trans-ACPC) was purchased from Synnovator, Inc. Described peptides were obtained with an automated solid-phase peptide synthesizer (Biotage® Initiator + AlastraTM) using H-Rink amide ChemMatrix resin 35–100 mesh particle size (loading: 0.59 mmol g−1). Fmoc deprotection was achieved with 20% piperidine in DMF for 3 + 10 min. A double-coupling procedure was performed using 0.5 M solution of N,N′-diisopropylcarbodiimide (DIC) and 0.5 M solution of Oxyma Pure Novabiochem® in DMF, for α-amino acids 2 × 15 min and for β-amino acids 30 min at 75 °C. Acetylation reaction was performed using NMP/DIPEA/acetic anhydride (80:15:5) mixture at room temp. Cleavage of the peptides from the resin was carried out with the mixture of TFA/TIS/H2O (95:2.5:2.5) and 3 h of shaking. The crude peptide was precipitated with ice-cold Et2O and centrifuged (8 000 rpm, 10 min, 4 °C). Peptides were purified using preparative HPLC (Knauer AZURA ASM 2.1 L) with a C18 column (Thermo Scientific, Hypersil Gold 12 μm, 250 mm × 20 mm) with water/acetonitrile (0.05% TFA) eluent system.2
Analytical high-performance liquid chromatography (HPLC)
Analytical HPLC (UltiMate 3000 LC System Dionex) was performed using Kinetex 5 μm EVO C18 100 A 150 × 4.6 mm column. Program (eluent A: 0.05% TFA in H2O, eluent B: 0.05% TFA in acetonitrile, flow 0.9 mL min−1): A: t = 0 min, 90% A; t = 30 min, 10% A.2Mass spectrometry (MS)
Peptides were studied by WATERS LCT Premier XE System consisting of high resolution mass spectrometer with a time of flight (TOF) using electrospray ionization (ESI).2Concentrations
Due to the limitations of the method and/or device, measurements were made at the different concentrations, which may influence the aggregation process. However, we operate within the range of 50–320 μM and we use the same stock solutions for different measurements by adjusting the concentration to the method if possible. The concentrations used are indicated in the Experimental section and in the description of the tables and figures.pH adjustment
The peptides were dissolved in water and the pH was adjusted to 7 by adding 0.1 M NaOH solution.Circular dichroism (CD)
CD spectra were recorded on JASCO J-1500 at 20 °C between 240 and 190 nm in water with following parameters: 0.2 nm resolution, 1.0 nm bandwidth, 20 mdeg sensitivity, 0.25 s response, 50 nm min−1 scanning speed, 5 scans, 0.02 cm cuvette path length. The CD spectra of the solvents alone were recorded and subtracted from the raw data. The peptides were dissolved in water and pH was adjusted to 7 by adding 0.1 M NaOH solution. Typically, the samples were prepared by dilution of peptides stock solution to obtain peptide concentration of 80 μM. The concentration of the solution for the CD measurements was determined by measuring the UV absorbance at 280 nm and calculated using the Beer–Lambert law. The CD intensity is given as mean residue ellipticity (θ [deg × cm2 × dmol−1]) calculated using eqn (S4) (ESI†).2Temperature unfolding measurements using CD
To examine the thermal unfolding of the peptides, stock solutions were diluted to 80 μM in water (pH = 7) and the measurement was performed at 208 nm during the temperature increase from 4 to 98 °C in increments of 2 °C. Ellipticity measurements were recorded with 1 mm path length cuvette, others parameters remained unchanged.2 The melting temperatures of studied peptides were determined by nonlinear fitting to the equation described previously (eqn (S7), ESI†).59Sedimentation-velocity analytical ultracentrifugation (SV-AUC)
SV-AUC was conducted on a Beckmann Coulter Proteome-Lab XL-I ultracentrifuge (software version 6.0) equipped with a 4-position An-60Ti rotor. Peptides’ samples (50–100 μM) were prepared in water directly before measurements. Their concentrations were determined spectrophotometrically from absorbance values at 280 nm using the extinction coefficients of ε = 8480 M−1 × cm−1 for peptides 1, 1_c and 1_f, and ε = 5500 M−1 × cm−1 for peptides 2, 2_c and 2_f. All samples were subjected to centrifugation into 2-channel carbon-epoxy cells with 12 mm optical pathlengths at 20 °C and 50000 rpm using a step size of 0.003 cm, a delay time of 0 s. The absorbance across the cell was measured at 280 nm. The peptides’ partial specific volumes (0.75395, 0.75942, 0.76822, 0.76901, 0.77708 and 0.78578 mL g−1 for 1, 1_c, 1_f, 2, 2_c and 2_f, respectively), solvent density (0.99823 g mL−1) and viscosity (1.002 mPa × s) were calculated with SEDNTERP.60 Partial specific volumes of the peptides were estimated and corrected for the presence of trans-ACPC, N-terminal acetyl and C-terminal amine moieties using published molar increment values of chemical groups.61 Time-corrected data62 were analyzed with SEDFIT software (version 16.1c) using the built-in continuous sedimentation coefficient distribution model, c(s). Maximum-entropy regularization of the c(s) models was set to a confidence level of 0.68.63,64 The fits were calculated alternately using simplex and Marquardt–Levenberg algorithms. The best-fit c(s) distribution model was judged based on minimization of the root-mean-square deviation of the fit (rmsd), and also on the shape of the residuals plot. The obtained sedimentation coefficients are standardized s20,w values based on the solute and solvent properties. Data, fits, residuals, and c(s) distributions were plotted using the Gussi interface (version 1.4.2).65
Fourier-transform infrared (FTIR) spectroscopy
All spectra were collected using Nicolet 6700 FTIR spectrometer (Thermo Scientific, USA) with ATR accessory and heated diamond top-plate (PIKE Technologies), continuously purged with dry air. Each sample of 10 μL of peptide aqueous solution was dropped directly on the diamond surface and allowed to dry out. Spectra were obtained in the range of 3600–400 cm−1. For each spectrum, 512 interferograms was co-added with 4 cm−1 resolution at constant temperature 22 °C [71.6 F]. Directly before sampling, the background spectrum of diamond/air was recorded as a reference (512 scans, 4 cm−1). We used 320 μM peptide concentration, which was essential to obtain a good signal-to-noise ratio. The spectra were analyzed using OriginPro 2020b (OriginLab Corporation, USA). The analysis included: baseline correction, smoothing using the Savitzky–Golay filter (polynomial order 2, window size 19).66 Spectra were normalized to the absorbance of Amide II band. The raw and normalized data after dissolving of the peptides and after incubation can be found in the ESI† (Fig. S7–S10 and S13).2Fourier-transform Raman spectroscopy
Raman spectra were carried out using a Nicolet NXR 9650 FT-Raman spectrometer with MicroStage extension equipped with Nd:YVO4 laser (1064 nm, 500 mW) as an excitation source and InGaAs detector. A drop of 10 μL of each sample was deposited on the gold surface and dried under laser irradiation. All FT-Raman spectra were acquired in the range of 3700–0 cm−1 with 4 cm−1 resolution by averaging 1024 scans. The spectra were analyzed using OriginPro 2020b (OriginLab Corporation, USA). The analysis included baseline correction, smoothing using the Savitzky-Golay filter (polynomial order 2, window size 35).66 Spectra were normalized to the intensity of the band at 1450 cm−1 arising from deformation vibration of methylene (CH2) groups. Spectra were deconvoluted into subcomponents using the Lorentz function based on the second derivative spectra. The raw and normalized data can be found in the ESI† (Fig. S11 and S12).2Atomic force microscopy (AFM)
AFM images were collected using a MultiModeV atomic force microscope with a NanoScopeV controller (Bruker/formerly Veeco/, Santa Barbara, CA, USA). The topography was measured in TappingMode™ (TM) in ambient conditions. The semicontact silicon probes, RTESPW (Bruker Metrology, Santa Barbara, CA, USA), with following parameters: spring constant in range 20–80 N m−1, a resonance frequency of 264–342 kHz, a nominal tip radius of curvature smaller than 10 nm and a cantilever length of 115–135 μm, were used. The volume of 10 μL of the 1.6 μM peptide solution in water was applied to freshly cleaved mica sheet and after 10 seconds of adsorption the sheet was rinsed a few times with deionized water. Measurements conducted directly after 10 seconds of adsorption, on the mica surface, in the RT temperature. We scanned the area of: 2 μm × 2 μm and 5 μm × 5 μm, which resulted in 512 × 512 pixels images. The images were analyzed and processed using NanoScope Analysis 1.9 software (Bruker Corporation). We used a Gaussian third-order flattening filter. The average height of the nanostructures was established by performing a statistical analysis of the AFM images. The normal distribution is difficult to see in a small sample. Artifacts, such as curvature distortion (blowing effects), were considered during the morphological analysis.Transmission electron microscopy (TEM)
160 μM peptide water solutions were incubated for 30 days at 37 °C. Then, the 4 μL drop of each peptide solution was deposited onto glow discharged carbon on copper grid (Agar S160). An excess of the material was blotted after 1 minute of adhesion and 2% uranyl acetate was applied for 1 minute and then blotted. The samples were allowed to dry under normal conditions for at least 1 hour. TEM micrographs were collected using a Hitachi H-800 with 150 kV accelerating voltage.Congo red staining
The peptides were dissolved in water in the concentration of 1 mg mL−1 (Cpep ≅ 320 μM) and incubated in 37 °C for two months. A drop of peptide aliquot (15 μL) was fixed on a glass slide for 24 hours and then stained with Congo red (CR) dye dissolved in ethanol with 0.1 M NaOH. The CR working solution was centrifuged before staining the samples. Next, the samples were rinsed with ethanol and dried out in room temperature. Birefringence was determined with an ECLIPSE 50i microscope (Nikon, Japan). The images were preprocessed with a Darktable software to increase images’ contrast.Thioflavin T (ThT) fluorescence assay
Kinetic measurements were carried out in a 96-well plate on a CLARIOstar Plus, BMG LABTECH, at 37 °C, using wavelengths of 440 nm and 480 nm, for ThT excitation and emission respectively. Additionally, the plate was shaken for 30 s every 5 min during 12 hours of measurements. Final concentrations were 50 μM of ThT and 320 μM of peptide.2Abbreviations
| ACPC | Trans-(1S,2S)-2-aminocyclopentanecarboxylic acid |
| AFM | Atomic force microscopy |
| CD | Circular dichroism |
| FTIR | Fourier transform infrared spectroscopy |
| HPLC | High-performance liquid chromatography |
| MS | Mass spectrometry |
| TEM | Transmission electron microscopy |
| ThT | Thioflavin T fluorescence assay |
Conflicts of interest
Authors declare no potential conflict of interest.Acknowledgements
The work was financially supported by the National Science Centre, Poland, Grant No. 2017/26/D/ST5/00341 (to M. S.).References
- E. De Santis and M. G. Ryadnov, Peptide Self-Assembly for Nanomaterials: The Old New Kid on the Block, Chem. Soc. Rev., 2015, 44, 8288–8300 RSC.
- M. Szefczyk, N. Szulc, M. Gasior-Głogowska, A. Modrak-Wojcik, A. Bzowska, W. Majstrzyk, M. Taube, M. Kozak, T. Gotszalk, E. Rudzinska-Szostak and L. Berlicki, Hierarchical Approach for the Rational Construction of Helix-Containing Nanofibrils Using α,β-Peptides, Nanoscale, 2021, 13, 4000–4015 RSC.
- E. Gatto, C. Toniolo and M. Venanzi, Peptide Self-Assembled Nanostructures: From Models to Therapeutic Peptides, Nanomaterials, 2022, 12, 466 CrossRef CAS PubMed.
- B. B. Gerbelli, S. V. Vassiliades, J. E. U. Rojas, J. N. B. D. Pelin, R. S. N. Mancini, W. S. G. Pereira, A. M. Aguilar, M. Venanzi, F. Cavalieri, F. Giuntini and W. A. Alves, Hierarchical Self-Assembly of Peptides and its Applications in Bionanotechnology. Macromol, Chem. Phys., 2019, 220, 1900085 Search PubMed.
- K. Kulkarni, N. Habila, M. P. Del Borgo and M.-I. Aguilar, Novel Materials From the Supramolecular Self-Assembly of Short Helical β3-Peptide Foldamers, Front. Chem., 2019, 7, 70 CrossRef CAS.
- R. V. Ulijn and A. M. Smith, Designing Peptide Based Nanomaterials, Chem. Soc. Rev., 2008, 37, 664–675 RSC.
- K. A. Dill and J. L. Maccallum, The Protein-Folding Problem, 50 Years On, Science, 2012, 338, 1042–1046 CrossRef CAS.
- S. Lesne, M. T. Koh, L. Kotilinek, R. Kayed, C. G. Glabe, A. Yang, M. Gallagher and K. H. Ashe, A Specific Amyloid-β Protein Assembly in the Brain Impairs Memory, Nature, 2006, 440, 352–357 CrossRef CAS PubMed.
- G. L. Holliday, D. E. Almonacid, J. B. Mitchell and J. M. Thornton, The Chemistry of Protein Catalysis, J. Mol. Biol., 2007, 372, 1261–1277 CrossRef CAS PubMed.
- C. M. Goodman, S. Choi, S. Shandler and W. F. DeGrado, Foldamers as Versatile Frameworks for the Design and Evolution of Function, Nat. Chem. Biol., 2007, 3, 252–262 CrossRef CAS PubMed.
- J. Fraczyk, W. Lipinski, A. Chaberska, J. Wasko, K. Rozniakowski, Z. J. Kaminski, M. Bogun, Z. Draczynski, E. Menaszek, E. Stodolak-Zych, M. Kaminska and B. Kolesinska, Search for Fibrous Aggregates Potentially Useful in Regenerative Medicine Formed under Physiological Conditions by Self-Assembling Short Peptides Containing Two Identical Aromatic Amino Acid Residues, Molecules, 2018, 23, 568 CrossRef PubMed.
- G.-B. Qi, Y.-J. Gao, L. Wang and H. Wang, Self-Assembled Peptide-Based Nanomaterials for Biomedical Imaging and Therapy, Adv. Mater., 2018, 30, 1703444 CrossRef.
- M. Cao, S. Lu, N. Wang, H. Xu, H. Cox, R. Li, T. Waigh, Y. Han, Y. Wang and J. R. Lu, Enzyme-Triggered Morphological Transition of Peptide Nanostructures for Tumor-Targeted Drug Delivery and Enhanced Cancer Therapy, ACS Appl. Mater. Interfaces, 2019, 8, 16357–16366 CrossRef PubMed.
- C. Gong, S. Sun, Y. Zhang, L. Sun, Z. Su, A. Wu and G. Wei, Hierarchical Nanomaterials via Biomolecular Self-Assembly and Bioinspiration for Energy and Environmental Applications, Nanoscale, 2019, 11, 4147–4182 RSC.
- M. Szefczyk, Peptide Foldamer-Based Self-Assembled Nanostructures Containing Cyclic Beta-Amino Acids, Nanoscale, 2021, 13, 11325–11333 RSC.
- J. L. Price, W. S. Horne and S. H. Gellman, Structural Consequences of β-Amino Acid Preorganization in a Self-Assembling α/β-Peptide: Fundamental Studies of Foldameric Helix Bundles, J. Am. Chem. Soc., 2010, 132, 12378–12387 CrossRef CAS.
- J. L. Price, W. S. Horne and S. H. Gellman, Discrete Heterogeneous Quaternary Structure Formed by α/β-Peptide Foldamers and α-Peptides, J. Am. Chem. Soc., 2007, 129, 6376–6377 CrossRef CAS.
- W. S. Horne, J. L. Price and S. H. Gellman, Interplay among Side Chain Sequence, Backbone Composition, and Residue Rigidification in Polypeptide Folding and Assembly, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9151–9156 CrossRef CAS PubMed.
- D. Seebach, A. K. Beck and D. J. Bierbaum, The World of β- and γ-Peptides Comprised of Homologated Proteinogenic Amino Acids and Other Components, Chem. Biodiversity, 2004, 1, 1111–1239 CrossRef CAS PubMed.
- E. H. C. Bromley and K. J. Channon, Alpha-Helical Peptide Assemblies Giving New Function to Designed Structures, Prog. Mol. Biol. Transl. Sci., 2011, 103, 231–275 CAS.
- F. Lapenta, J. Aupic, Z. Strmsek and R. Jerala, Coiled Coil Protein Origami: from Modular Design Principles towards Biotechnological Applications, Chem. Soc. Rev., 2018, 47, 3530–3542 RSC.
- P. B. Harbury, T. Zhang, P. S. Kim and T. A. Alber, A Switch between Two-, Three-, and Four-Stranded Coiled Coils in GCN4 Leucine Zipper Mutants, Science, 1993, 262, 1401–1407 CrossRef CAS.
- J. M. Fletcher, A. L. Boyle, M. Bruning, G. J. Bartlett, T. L. Vincent, N. R. Zaccai, C. T. Armstrong, E. H. Bromley, P. J. Booth, R. L. Brady, A. R. Thomson and D. N. Woolfson, A Basis Set of de Novo Coiled-Coil Peptide Oligomers for Rational Protein Design and Synthetic Biology, ACS Synth. Biol., 2012, 1, 240–250 CrossRef CAS.
- S. A. Potekhin, T. N. Melnik, V. Popov, N. F. Lanina, A. A. Vazina, P. Rigler, A. S. Verdini, G. Corradin and A. V. Kajava, De Novo Design of Fibrils Made of Short Alpha-Helical Coiled Coil Peptides, Chem. Biol., 2001, 8, 1025–1032 CrossRef CAS PubMed.
- M. Szefczyk, E. Weglarz-Tomczak, P. Fortuna, A. Krzyszton, E. Rudzinska-Szostak and L. Berlicki, Controlling the Helix Handedness of ααβ-Peptide Foldamers through Sequence Shifting, Angew. Chem., Int. Ed., 2017, 56, 2087–2091 CrossRef CAS PubMed.
- J. M. Mason and K. M. Arndt, Coiled Coil Domains: Stability, Specificity, and Biological Implications, ChemBioChem, 2004, 5, 170–176 CrossRef CAS.
- K. Baginska, J. Makowska, W. Wiczk, F. Kasprzykowski and L. Chmurzyński, Conformational Studies of Alanine-Rich Peptide Using CD and FTIR Spectroscopy, J. Pept. Sci., 2008, 14, 283–289 CrossRef CAS PubMed.
- S. A. Tatulian, in FTIR Analysis of Proteins and Protein–Membrane Interactions, Lipid–Protein Interactions. Methods in Molecular Biology, ed. J. Kleinschmidt, Humana, New York, NY, 2019, vol. 2003 Search PubMed.
- R. Bücker, C. Seuring, C. Cazey, K. Veith, M. García-Alai, K. Grünewald and M. Landau, The Cryo-EM Structures of Two Amphibian Antimicrobial Cross-β Amyloid Fibrils, Nat. Commun., 2022, 13, 4356 CrossRef.
- R. Sarroukh, E. Goormaghtigh, J.-M. Ruysschaert and V. Raussens, ATR-FTIR: A “Rejuvenated” Tool to Investigate Amyloid Proteins, Biochim. Biophys. Acta, Biomembr., 1828, 2328–2338 Search PubMed.
- M. E. Gasior-Glogowska, N. Szulc and M. Szefczyk, in Challenges in Experimental Methods, Computer Simulations of Aggregation of Proteins and Peptides. Methods in Molecular Biology, ed. M. S. Li, A. Kloczkowski, M. Cieplak, M. Kouza, Humana, New York, NY, 2022, vol. 2340, pp. 281–307 Search PubMed.
- E. K. Roberts, K. M. Wong, E. J. Lee, M. M. Le, D. M. Patel and A. K. Paravastu, Post-assembly α-Helix to β-Sheet Structural Transformation Within SAF-p1/p2a Peptide Nanofibers, Soft Matter, 2018, 14, 8986–8996 RSC.
- V. Serrano, W. Liu and S. Franzen, An Infrared Spectroscopic Study of the Conformational Transition of Elastin-Like Polypeptides, Biophys. J., 2007, 93, 2429–2435 CrossRef CAS PubMed.
- T. Vajda and A. Perczel, The Clear and Dark Sides of Water: Influence on the Coiled Coil Folding Domain, Biomol. Concepts, 2016, 7, 189–195 CrossRef CAS PubMed.
- T. Heimburg, J. Schunemann, K. Weber and N. Geisler, FTIR-Spectroscopy of Multistranded Coiled Coil Proteins, Biochem., 1999, 38, 12727–12734 CrossRef CAS.
- O. M. Cracchiolo, D. N. Edun, V. M. Betti, J. M. Goldberg and A. L. Serrano, Cross-α/β Polymorphism of PSMα3 Fibrils, Proc. Natl. Acad. Sci. U. S. A., 2022, 11, e2114923119 CrossRef.
- L. Schefer, J. Adamcik, M. Dienera and R. Mezzenga, Supramolecular Chiral Self-Assembly and Supercoiling Behavior of Carrageenans at Varying Salt Conditions, Nanoscale, 2015, 7, 16182–16188 RSC.
- B. de Campos Vidal and M. L. S. Mello, Collagen Type I Amide I Band Infrared Spectroscopy, Micron, 2011, 42, 283–289 CrossRef.
- J. D. Flynn, R. P. McGlinchey, R. L. Walker III and J. C. Lee, Structural Features of α-Synuclein Amyloid Fibrils Revealed by Raman Spectroscopy, J. Biol. Chem., 2018, 293, 767–776 CrossRef CAS PubMed.
- A. Rygula, K. Majzner, K. M. Marzec, A. Kaczor, M. Pilarczyk and M. Baranska, Raman Spectroscopy of Proteins: a Review, J. Raman Spectrosc., 2013, 44, 1061–1076 CrossRef CAS.
- A. Espargaro, S. Llabres, S. J. Saupe, C. Curutchet, F. J. Luque and R. Sabate, On the Binding of Congo Red to Amyloid Fibrils, Angew. Chem., Int. Ed., 2020, 59, 8104–8107 CrossRef CAS PubMed.
- M. Bely and J. Makovitzky, Sensitivity and Specificity of Congo Red Staining According to Romhanyi. Comparison with Puchtler's or Bennhold's Methods, Acta Histochem., 2006, 108, 175–180 CrossRef PubMed.
- K. A. Bruggink, M. Muller, H. B. Kuiperij and M. M. Verbeek, Methods for Analysis of Amyloid-β Aggregates, J. Alzheimer's Dis., 2012, 28, 735–758 CAS.
- M. R. Nilsson, Techniques to Study Amyloid Fibril Formation in vitro, Methods, 2004, 34, 151–160 CrossRef CAS.
- C. C. vandenAkker, M. F. M. Engel, K. P. Velikov, M. Bonn and G. H. Koenderink, Morphology and Persistence Length of Amyloid Fibrils Are Correlated to Peptide Molecular Structure, J. Am. Chem. Soc., 2011, 133, 18030–18033 CrossRef CAS.
- M. J. Pandya, G. M. Spooner, M. Sunde, J. R. Thorpe, A. Rodger and D. N. Woolfson, Sticky-End Assembly of a Designed Peptide Fiber Provides Insight into Protein Fibrillogenesis, Biochemistry, 2000, 39, 8728–8734 CrossRef CAS.
- C. Xu, R. Liu, A. K. Mehta, R. C. Guerrero-Ferreira, E. R. Wright, S. Dunin-Horkawicz, K. Morris, L. C. Serpell, X. Zuo, J. S. Wall and V. P. Conticello, Rational Design of Helical Nanotubes from Self-Assembly of Coiled-Coil Lock Washers, J. Am. Chem. Soc., 2013, 135, 15565–15578 CrossRef CAS.
- W. C. Pomerantz, V. M. Yuwono, R. Drake, J. D. Hartgerink, N. L. Abbott and S. H. Gellman, Lyotropic Liquid Crystals Formed from ACHC-Rich β-Peptides, J. Am. Chem. Soc., 2011, 133, 13604–13613 CrossRef CAS.
- A. Lomander, W. Hwang and S. Zhang, Hierarchical Self-Assembly of a Coiled-Coil Peptide into Fractal Structure, Nano Lett., 2005, 5, 1255–1260 CrossRef CAS.
- H. Dong, S. E. Paramonov, L. Aulisa, E. L. Bakota and J. D. Hartgerink, Self-Assembly of Multidomain Peptides: Balancing Molecular Frustration Controls Conformation and Nanostructure, J. Am. Chem. Soc., 2007, 129, 12468–12472 CrossRef CAS PubMed.
- M. S. Lamm, K. Rajagopal, J. P. Schneider and D. J. Pochan, Laminated Morphology of Nontwisting β-Sheet Fibrils Constructed via Peptide Self-Assembly, J. Am. Chem. Soc., 2005, 127, 16692–16700 CrossRef CAS PubMed.
- E. Tayeb-Fligelman, O. Tabachnikov, A. Moshe, O. Goldshmidt-Tran, M. R. Sawaya, N. Coquelle, J.-P. Colletier and M. Landau, The Cytotoxic Staphylococcus aureus PSMα3 Reveals a Cross-α Amyloid-Like Fibril, Science, 2017, 355, 831–833 CrossRef PubMed.
- S. Q. Zhang, H. Huang, J. Yang, H. T. Kratochvil, M. Lolicato, Y. Liu, X. Shu, L. Liu and W. F. DeGrado, Designed Peptides that Assemble into Cross-α Amyloid-Like Structures, Nat. Chem. Biol., 2018, 14, 870–875 CrossRef CAS PubMed.
- H. Dong and J. D. Hartgerink, Short Homodimeric and Heterodimeric Coiled Coils, Biomacromolecules, 2006, 7, 691–695 CrossRef CAS.
- H. Dong and J. D. Hartgerink, Role of Hydrophobic Clusters in the Stability of α-Helical Coiled Coils and Their Conversion to Amyloid-like β-Sheets, Biomacromolecules, 2007, 8, 617–623 CrossRef CAS.
- K. Cieslik-Boczula, Alpha-Helix to Beta-Sheet Transition in Long-Chain Poly-l-Lysine: Formation of Alpha-Helical Fibrils by Poly-l-Lysine, Biochimie, 2017, 137, 106–114 CrossRef CAS PubMed.
- R. Xing, C. Yuan, S. Li, J. Song, J. Li and X. Yan, Charge-Induced Secondary Structure Transformation of Amyloid-Derived Dipeptide Assemblies from β-Sheet to α-Helix, Chem. Int. Ed., 2018, 57, 1537–1542 CrossRef CAS PubMed.
- K. L. Lazar, H. Miller-Auer, G. S. Getz, J. P. R. O. Orgel and S. C. Meredith, Helix-Turn-Helix Peptides That Form α-Helical Fibrils: Turn Sequences Drive Fibril Structure, Biochemistry, 2005, 44, 12681–12689 CrossRef CAS PubMed.
- D. F. Kreitler, D. E. Mortenson, K. T. Forest and S. H. Gellman, Effects of Single α-to-β Residue Replacements on Structure and Stability in a Small Protein: Insights from Quasiracemic Crystallization, J. Am. Chem. Soc., 2016, 138, 6498–6505 CrossRef CAS PubMed.
- T. M. Laue, B. Shah, T. M. Ridgeway and S. L. Pelletier, in Computer-aided Interpretation of Analytical Sedimentation Data for Proteins, Analytical Ultracentrifugation in Biochemistry and Polymer Science, ed. S. E. Harding, J. C. Hortong, A. J. Rowe, Royal Society of Chemistry, Cambridge, UK, 1992, pp. 90–125 Search PubMed.
- H. Durchschlag and P. Zipper, Calculation of Partial Specific Volumes and Other Volumetric Properties of Small Molecules and Polymers, J. Appl. Crystallogr., 1997, 30, 803–807, DOI:10.1107/S0021889897003348.
- H. Zhao, R. Ghirlando, G. Piszczek, U. Curth, C. A. Brautigam and P. Schuck, Recorded Scan Times Can Limit the Accuracy of Sedimentation Coefficients in Analytical Ultracentrifugation, Anal. Biochem., 2013, 437, 104–108 CrossRef CAS PubMed.
- P. Schuck, Sedimentation Analysis of Noninteracting and Self-Associating Solutes Using Numerical Solutions to the Lamm Equation, Biophys. J., 1998, 75, 1503–1512 CrossRef CAS PubMed.
- P. Schuck, Size-distribution Analysis of Macromolecules by Sedimentation Velocity Ultracentrifugation and Lamm Equation Modeling, Biophys. J., 2000, 78, 1606–1619 CrossRef CAS.
- C. A. Brautigam, Calculations and Publication-Quality Illustrations for Analytical Ultracentrifugation Data, Methods in Enzymology, Elsevier Inc, 1st edn, 2015, vol. 562, pp. 109–133 Search PubMed.
- A. Savitzky and M. J. E. Golay, Smoothing and Differentiation of Data by Simplified Least Squares Procedures, Anal. Chem., 1964, 36, 1627–1639 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sm00005b |
| This journal is © The Royal Society of Chemistry 2023 |