
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Impact of granular inclusions on the phase behavior of colloidal gels
Yankai
Li
 a,
John R.
Royer
b,
Jin
Sun
a and
Christopher
Ness
*a
a,
John R.
Royer
b,
Jin
Sun
a and
Christopher
Ness
*a
aSchool of Engineering, The University of Edinburgh, King's Buildings, Edinburgh EH9 3FG, UK. E-mail: chris.ness@ed.ac.uk
bSUPA, School of Physics and Astronomy, The University of Edinburgh, King's Buildings, Edinburgh EH9 3FD, UK
First published on 26th January 2023
Abstract
Colloidal gels formed from small attractive particles are commonly used in formulations to keep larger components in suspension. Despite extensive work characterising unfilled gels, little is known about how the larger inclusions alter the phase behavior and microstructure of the colloidal system. Here we use numerical simulations to examine how larger ‘granular’ particles can alter the gel transition phase boundaries. We find two distinct regimes depending on both the filler size and native gel structure: a ‘passive’ regime where the filler fits into already-present voids, giving little change in the transition, and an ‘active’ regime where the filler no longer fits in these voids and instead perturbs the native structure. In this second regime the phase boundary is controlled by an effective colloidal volume fraction given by the available free volume.
1 Introduction
Dispersions of attractive colloids can form solid-like gels characterized by a system-spanning network of arrested particles.1–3 These colloidal gels are ubiquitous, encountered in disparate industries ranging from food and personal care products to building materials and catholyte slurries.4–7 While there has been considerable progress in understanding the formation, structure and rheology of ‘model’ colloidal gels formed from (nearly) uniformly sized spheres,8–11 most practical gels are more complex. In particular, colloidal gels frequently serve as a carrier for larger, non-Brownian ‘granular’ components (typical size ≳10 μm). In such composites, the gel often acts as a rheology modifier to prevent sedimentation. In some applications the gel network itself may be desired, for example catholyte slurries for battery manufacturing rely on a conductive carbon black gel to provide connectivity between the active Li-ion ‘grains’.It is thus critical to understand how granular inclusions alter the colloidal gel phase. Recent work examining the influence of inclusions on gel rheology suggests they have a significant impact,12,13 even introducing new phenomena such as rheological bi-stability in these filled systems.14 This previous work has largely focused on systems deep into the gel state, so it remains unclear how granular inclusions alter the gel transition and phase behavior (but see Jiang and Seto15).
For uniformly-sized colloidal spheres, the gelation phase boundaries depend on the colloid concentration and attraction strength, and there has been extensive work mapping these boundaries in a variety of systems.11,16–25 In depletion gels there is good agreement between gelation and the gas–liquid spinodal boundary,26 though there remains some debate concerning the generality of this agreement and the relative roles of percolation and clustering at the gel transition.23–25,27
The inclusion of larger grains introduces additional control parameters which can potentially alter these phase boundaries. For simple ‘hard’ grains interacting solely through their excluded volume, their influence will be set by the filler concentration and the size ratio rL/rS between the large (L) grains and the smaller (S) colloids. These granular inclusions reduce the free volume available to the colloids relative to the total volume, but the interplay between the inclusions and gel structure is non-trivial, as colloidal gels can be heterogeneous on length scales ≫rS.3,28,29
Here we characterise the influence of hard granular inclusions on the colloidal gel phase boundaries using numerical simulations, where the particle sizes, interactions and volume fractions can all be precisely varied. We find that the relative sizes of the inclusions and the void spaces present in the unfilled gels is the key parameter governing the phase behavior of the filled systems.
2 Methods
We simulate the trajectories of 104 colloidal and a smaller number of granular spheres in a periodic box (volume V), using LAMMPS.30 Motion is governed by the Langevin equation, which for particle i reads midUi/dt =FHi + FBi + FPi, with mi = (4/3)πri3ρ and Ui the particle mass and velocity respectively (we assume density ρ matching between all particle species and the solvent). The hydrodynamic force FHi= −6πηri(Ui − U∞i) captures Stokes drag on a sphere of radius ri, with U∞i the background fluid velocity at the sphere centre (generally set to 0 except under oscillatory shear).Brownian forces are generated as  , where Δt is the timestep, kT the thermal energy and the elements of the vector R are drawn from a Gaussian distribution with zero-mean and no time correlation. The characteristic diffusive timescale for a particle with radius ri is thus 6πηri3/kT. To avoid crystallisation in the small colloids, we use a binary size mixture with radii rS and 1.4rS (mixed equally by number), while the larger grains are monodisperse with radius rL varying from 8rS to 24rS. Since the diffusion time scales as ri3, for the larger grains it is >500 times longer than the colloidal timescale τB = 6πηrS3/kT, so that even though Brownian forces are applied uniformly to all particles the larger grains are effectively non-Brownian.
, where Δt is the timestep, kT the thermal energy and the elements of the vector R are drawn from a Gaussian distribution with zero-mean and no time correlation. The characteristic diffusive timescale for a particle with radius ri is thus 6πηri3/kT. To avoid crystallisation in the small colloids, we use a binary size mixture with radii rS and 1.4rS (mixed equally by number), while the larger grains are monodisperse with radius rL varying from 8rS to 24rS. Since the diffusion time scales as ri3, for the larger grains it is >500 times longer than the colloidal timescale τB = 6πηrS3/kT, so that even though Brownian forces are applied uniformly to all particles the larger grains are effectively non-Brownian.
Colloids (labeled i and j) at a distance r and surface-to-surface separation δij = r − (ri + rj) interact via a Morse potential, giving a pairwise force  with nij the center-to-center unit vector. This potential gives finite-ranged attraction, and repulsion for overlapping particles (δij < 0). The interaction length scale is set as κij−1 = (ri + rj)/200 to give short-ranged attraction. We evaluate the force when δij < 0.03(ri + rj) (following conventional practice, see e.g. Sciortino et al.31), beyond which the attractive force is <1% of its maximum value. We defined contacts using the same threshold of δij, having verified that a more stringent criteria does not affect any of the conclusions we draw. The depth of the attractive potential ε is varied between kT and 20kT, with a variable step size to refine our estimates of the gelation point. Colloid–granular and granular–granular contact forces are modeled as linear springs Fpij = −knδijnij, with a stiffness kn set sufficiently large (knrS2/kT = 5 × 104) to approximate hard-sphere interactions. The timestep Δt is set to 10−4, substantially smaller than the Brownian time τB, the contact time
with nij the center-to-center unit vector. This potential gives finite-ranged attraction, and repulsion for overlapping particles (δij < 0). The interaction length scale is set as κij−1 = (ri + rj)/200 to give short-ranged attraction. We evaluate the force when δij < 0.03(ri + rj) (following conventional practice, see e.g. Sciortino et al.31), beyond which the attractive force is <1% of its maximum value. We defined contacts using the same threshold of δij, having verified that a more stringent criteria does not affect any of the conclusions we draw. The depth of the attractive potential ε is varied between kT and 20kT, with a variable step size to refine our estimates of the gelation point. Colloid–granular and granular–granular contact forces are modeled as linear springs Fpij = −knδijnij, with a stiffness kn set sufficiently large (knrS2/kT = 5 × 104) to approximate hard-sphere interactions. The timestep Δt is set to 10−4, substantially smaller than the Brownian time τB, the contact time  and the inertial relaxation time τi = rS2ρ/η (with η sufficiently large that τi ≪ τB).
and the inertial relaxation time τi = rS2ρ/η (with η sufficiently large that τi ≪ τB).
We characterise the mechanical response of steady-state structures by applying an oscillatory shear through the liquid streaming velocity as U∞i(t,yi) = yiγ0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sinωt and turning off the Brownian forces. We remain in the low amplitude regime by setting γ0 = 0.005, and set ω such that inertia plays no role (in practice this is achieved when ωτi ≤ 0.005). The bulk shear stress is computed as
sinωt and turning off the Brownian forces. We remain in the low amplitude regime by setting γ0 = 0.005, and set ω such that inertia plays no role (in practice this is achieved when ωτi ≤ 0.005). The bulk shear stress is computed as  (with the sum being over all interacting pairs), and the viscoelastic moduli G′ and G′′ are computed from the Fourier transform of σxy averaged over 50 shear cycles. All results for both the elastic moduli and Liso are averages from at least 6 independent realisations with randomized granular and colloidal initial positions. We simulated a larger system of 105 colloids with rL/rS = 24 inclusions and confirmed that its results are consistent with the 104 system both in terms of Liso and in the distribution of isostatic cluster sizes.
(with the sum being over all interacting pairs), and the viscoelastic moduli G′ and G′′ are computed from the Fourier transform of σxy averaged over 50 shear cycles. All results for both the elastic moduli and Liso are averages from at least 6 independent realisations with randomized granular and colloidal initial positions. We simulated a larger system of 105 colloids with rL/rS = 24 inclusions and confirmed that its results are consistent with the 104 system both in terms of Liso and in the distribution of isostatic cluster sizes.
3 Results and discussion
The system initially equilibrates at a prescribed volume fraction in the absence of colloidal attraction, after which the interaction potential with depth ε/kT is turned on and the system evolves for up to 104τB to reach a steady state. To map the phase behavior of these systems, we vary ε/kT along with the colloidal and granular volume fractions, ϕS = Vs/V and ϕL = VL/V. These volume fractions are defined as the volume occupied by the colloids (grains), Vs (VL), relative to the total volume of the cubic simulation box, so that increasing ϕL at fixed ϕS decreases the free volume available to the colloids.3.1 Isostaticity percolation and mechanical response
To explore how large granular inclusions alter the colloidal microstructure, we characterize networks of isostatic particles, defined as colloids with N ≥ 6 contacts. This follows from the Maxwell criteria for stability in a system with pairwise central forces, requiring that constraints balance the degrees of freedom.32 While this isostaticity criterion is typically considered globally in the context of granular packings,33 it has been suggested that networks of locally isostatic particles control the rigidity of colloidal gels.9,34 Specifically, recent experimental work suggests the gel transition coincides with the formation of a percolating network of isostatic particles.35We define contacts among attractive colloids as pairs (radii ri and rj) within a separation 0.03(ri + rj). To characterise the distance from isostaticity percolation, we first remove colloids with N < 6 contacts and then identify clusters of isostatic particles. The isostatic length, Liso, gives the mean length of the largest isostatic cluster in all three spatial dimensions relative to the box size (Fig. 1(A)), so that Liso = 1 corresponds to isotropic isostaticity percolation (in principle Liso may exceed unity once the periodic box is unwrapped, in which case we limit it to this value).
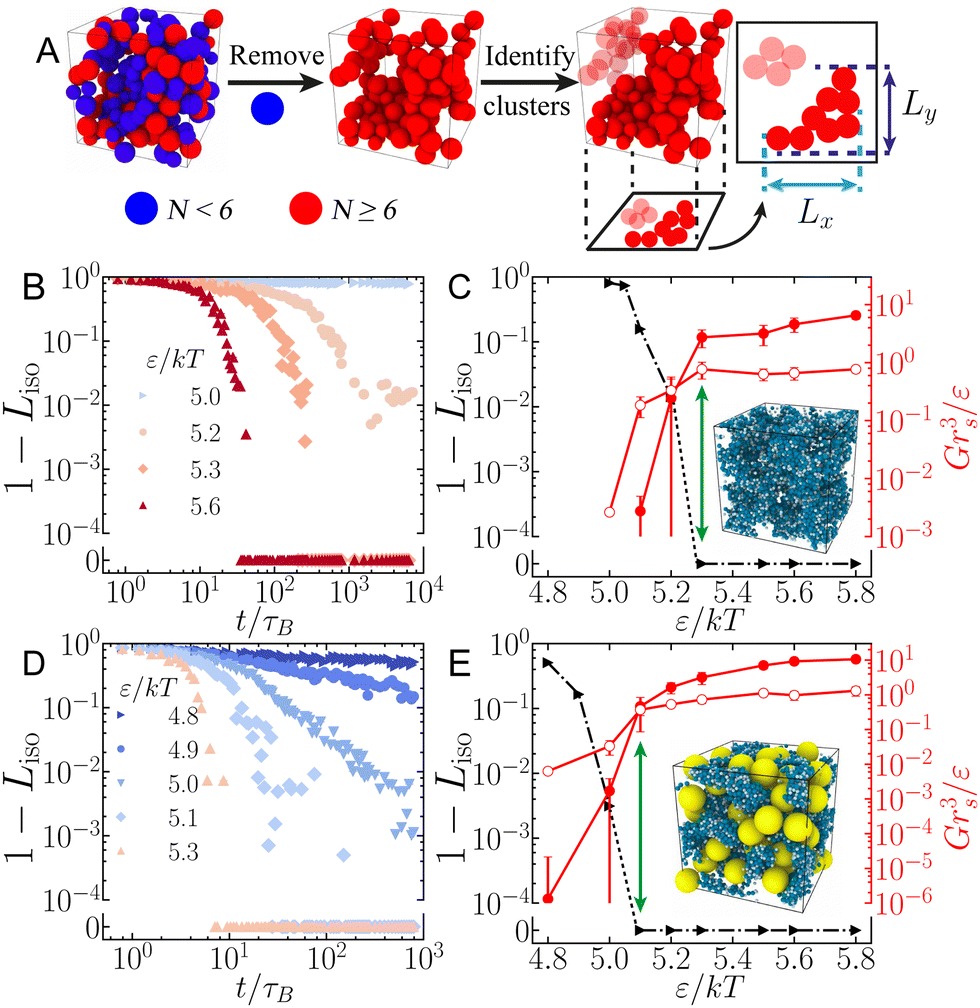 | ||
Fig. 1 Isostaticity percolation and its relation to the mechanical response. (A) Procedure for determining Liso. Left to right: Isostatic colloids with N ≥ 6 contacts (red) are isolated and contacting clusters identified. The isostatic length is computed from the span of the largest cluster, (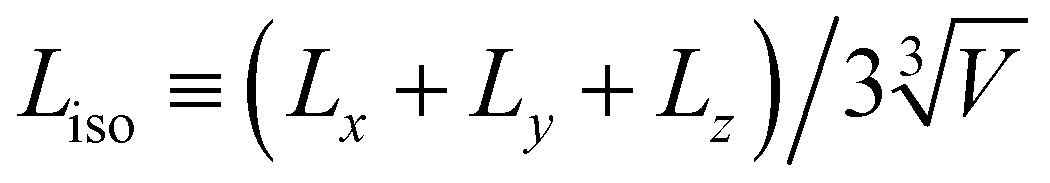 ). (B) Time evolution of 1 − Liso for an unfilled system (ϕS = 0.2, ϕL = 0) at varying attraction ε/kT. (C) Steady-state 1 − Lssiso (black, left axis) and viscoelastic moduli G′ and G′′ (red filled and open symbols, respectively, right axis) verses ε/kT. Green arrow highlights the coincidence between the crossover G′ = G′′ and 1 − Lssiso → 0. Inset: Rendering of a steady-state (t/τB > 103) gel state for ε/kT = 5.6 with ϕS = 0.2, ϕL = 0. (D) and (E) reproduce (B) and (C), respectively, for a filled system with ϕS = 0.20, ϕL = 0.3. Inset in (E) shows a rendering with granular inclusions (yellow) at ε/kT = 5.6. Error bars in (C) and (E) indicate the standard deviation from 6 realisations. Dotted lines in (C) and (E) (and later figures) are used to indicate data crossing the broken y-axis. ). (B) Time evolution of 1 − Liso for an unfilled system (ϕS = 0.2, ϕL = 0) at varying attraction ε/kT. (C) Steady-state 1 − Lssiso (black, left axis) and viscoelastic moduli G′ and G′′ (red filled and open symbols, respectively, right axis) verses ε/kT. Green arrow highlights the coincidence between the crossover G′ = G′′ and 1 − Lssiso → 0. Inset: Rendering of a steady-state (t/τB > 103) gel state for ε/kT = 5.6 with ϕS = 0.2, ϕL = 0. (D) and (E) reproduce (B) and (C), respectively, for a filled system with ϕS = 0.20, ϕL = 0.3. Inset in (E) shows a rendering with granular inclusions (yellow) at ε/kT = 5.6. Error bars in (C) and (E) indicate the standard deviation from 6 realisations. Dotted lines in (C) and (E) (and later figures) are used to indicate data crossing the broken y-axis. | ||
In pure colloidal suspensions (ϕL = 0), the colloids are initially well-dispersed, with Liso ≈ 0 (so that 1 − Liso ≈ 1). Turning on the attraction ε at time t = 0 causes clusters to form and grow, reflected in an increase in Liso(t) with time (Fig. 1(B)). As time progresses this initial growth slows and the isostatic length approaches a plateau at some steady-state value Lssiso at long times (t ≳ 103–104τB). The growth of Liso(t) depends on the attraction strength, with strongly attractive colloids rapidly reaching isostaticity percolation at Lssiso = 1 (1 −Lssiso = 0) while with weaker attraction Liso instead appears to plateau at some steady-state value Lssiso < 1, short of the percolation threshold. Reaching steady states in Liso(t) gets progressively slower as we approach the critical interaction energy ε*, and truncating our time series at 103–104τB limits our resolution in ε* to ±0.1kT. This limitation will not affect the outcomes of our work, and future studies aiming to obtain ε* with greater precision will require substantially larger particle numbers and run times.
We apply small amplitude oscillatory shear γ(t) = γ0sinωt to extract the viscoelastic moduli G′ and G′′ for the structures obtained at the end of each time series. We find a transition from liquid-like states (G′ < G′′) to solid-like states (G′ > G′′) as the attraction strength ε is increased (Fig. 1(C)), indicating the emergence of a solid-like gel. The crossover point where G′ = G′′ occurs as Lssiso approaches unity at interaction energy ε* indicating that the gel transition coincides with isostaticity percolation in agreement with Tsurusawa et al.35 We verified this agreement holds for ϕS ≤ 0.4, and that our phase diagram reported below is consistent with that obtained purely via rheology.
3.2 Role of granular inclusions
The addition of granular inclusions gives qualitatively similar behavior in both the evolution of Liso with time and the mechanical response of the steady state structures, Fig. 1(D) and (E). We again find good agreement between isostaticity percolation (Lssiso = 1) and the emergence of mechanical rigidity (G′ = G′′), indicated by green arrows in Fig. 1(C) and (E). Comparing these transition points for filled (rL = 8rS, ϕL = 0.3) and unfilled systems both at ϕS = 0.2, we find that the inclusions aid gelation with a reduced ε* in the filled system.To understand this shift in the gelation point, we examine how granular inclusions alter the structure and distribution of the isostatic colloidal particles. In a pure colloidal system below the gel transition, ε = 5.1kT = 0.94ε* and ϕS = 0.2, there are numerous disjoint clusters of isostatic particles (Fig. 2(A)) and the system remains well below the isostatic percolation threshold. The inclusion of the larger grains increases the number of isostatic colloidal particles, enabling them to instead form a large connected network which percolates across the sample for ϕL = 0.4 (Fig. 2(B)).
 | ||
| Fig. 2 Contrasting effects of granular inclusions. Shown are snapshots of steady-state (t = 103–104τB) configurations, with ϕS = 0.2 and ε = 0.94ε0* for (A) ϕL = 0 and; (B) ϕL = 0.4. Only isostatic colloids are rendered, with the largest cluster shown in red. Larger grains are shown (grey) in (B). (C) and (D) 1 − Lssiso(ϕL) at varying ε for (C) ϕS = 0.2 and (D) ϕS = 0.1. Values for ε given relative to the gelation point in the unfilled system, ε0*, with ε0* = 5.3kT for ϕS = 0.2 and ε0* = 6.1kT for ϕS = 0.1. Dashed squares (a) and (b) indicate states rendered in (A) and (B), respectively. (E) and (F) probability distribution of the number of contacts per particle as a function of ϕL, for (E) ϕS = 0.2, ε = 5.1kT and (F) ϕS = 0.07, ε = 6.6kT. Both cases cross the phase boundary as ϕL is increased. | ||
This observation is reflected in the microstructural statistics: for fixed ϕS = 0.2, we find that Lssiso of an initially liquid system increases dramatically with increasing ϕL (Fig. 2(C)), mirrored by a shift to larger N in the probability distribution of particle contact numbers (Fig. 2(E)). Defining ε0* ≡ ε*(ϕL = 0) the critical interaction energy in the unfilled system, granular inclusions can take a system initially quite far from the gelation point (ε = 0.91ε0* and Lssiso ≈ 0) nearly up to the transition point Lssiso ≈ 0.98 as ϕL increases up to ϕL = 0.4. Increasing the interaction energy to ε/ε0* = 0.96, the volume of granular filler needed to drive isostaticity percolation decreases, so that the gelation boundary ε* where Lssiso reaches unity continuously shifts to lower values with increasing ϕL.
Since the free volume available to the colloids decreases with increasing ϕL, one might expect this described increase in the number of isostatic particles, and hence an increase in Lssiso in the filled system. Furthermore, the gel phase boundary for unfilled systems, ε0*(ϕS), is a decreasing function of ϕS, at least at low to moderate concentrations.16–25 This suggests the possibility of capturing the shifting gel point with granular inclusions simply in terms of the reduced free volume available to the colloids. In this picture, one would expect the filler to have more pronounced effect at lower colloid concentrations, where the curve ε0*(ϕS) is steepest. However, simulations with ϕS = 0.1 (and lower) instead show the opposite, with the inclusion of large grains giving only a modest increase in Lssiso (Fig. 2(D)), having little to no effect on P(N) (Fig. 2(F)), and generating only minor shifts in the gelation phase boundary. This unexpected result demonstrates that a universal picture in which the gel state is defined entirely by an effective colloidal volume fraction does not apply.
3.3 Fillers and voids
To understand the reduced filler impact at low ϕS, we look at how the microstructure of the colloidal phase varies with concentration. Specifically, we focus on the size distribution of the empty voids between the colloids at (or close to) the gel transition.36 We compute this distribution by dividing the simulation volume into cubic cells (length rS) and then finding the largest possible sphere (radius rv) centered in each cell that avoids intersecting a colloidal particle (so that larger grains are treated as empty voids). Normalised histograms of these local void radii give the void size distribution P(rv).For an unfilled colloidal system with ϕS = 0.2 close to the gel transition (ε = 5.3kT ≈ ε0*), this distribution is nearly flat up to rv ≈ 3rS and then falls off rapidly as rv increases further (Fig. 3(A)) with voids larger than rv ≈ 6rS exceedingly rare. While precisely characterizing the rare-event tails in P(rv) would require significant computational effort, we can define an effective maximum void size P(rmaxv) = 10−4, as voids larger then this are effectively absent in our observed configurations. Adding larger granular inclusions, with rL = 8rS > rmaxv, perturbs the colloidal microstructure and shifts the shoulder in P(rv) to higher radii ≈rL, reflecting the voids created by the large grains.
 | ||
| Fig. 3 Void-volumes in filled and unfilled systems near the gel boundary. Normalized colloidal void size distribution P(rv) for (A) ϕS = 0.2 (ε = 5.3kT ≈ ε0*) and (B) ϕS = 0.1 (ε = 6.3kT ≈ ε0*) at varying ϕL. The large particle size is rL = 8rS (highlighted by black dashed vertical lines), with the exception of the red filled symbols in (B) where rL = 16rS (highlighted by the red dashed vertical line). (C) 1 − Lssiso(ϕL) for ε = 5.8kT (red circles) and 5.5kT (blue triangles), with open symbols for rL = 8rS and filled symbols for rL = 16rS. Schematic cartoon in (D) presents a simplified picture to understand the influence (or lack thereof) of the granular filler on the gel structure, with large grains either fitting into ‘natural’ voids in the gel, leaving the colloidal microstructure unaffected (black arrow), or forcing larger voids, which in turn distorts and compresses the colloidal phase (red arrows). | ||
However, reducing the colloid concentration to ϕS = 0.1 gives a significantly wider distribution of void sizes in the unfilled system, with the shoulder in P(rv) now around rv = 8rS and rmaxv ≈ 12rS (Fig. 3(B)). This indicates there are ‘pre-existing’ voids which can accommodate the larger rL = 8rS grains without forcing a significant change in the colloidal microstructure. Indeed, in contrast to the large shift seen for ϕS = 0.2, here increasing ϕL has only a minor impact on the shape of P(rv). This suggests a picture where dilute gels with rv > rL can effectively form around the immobile granular inclusions, forming a network of colloid–colloid contacts that is essentially indistinguishable from the unfilled case. With no change in this network, Liso is unaffected by the granular inclusions and there is little shift in the gel phase boundary. We would thus expect that increasing rL above the characteristic void size would result in a more pronounced filler-effect on Lssiso and the gelation point in these more dilute gels.
We find this is indeed the case when rL is increased from 8rS to 16rS. These larger grains now notably shift P(rv) to higher values (compare open and filled symbols in Fig. 3(B)), and we also now find a clear increase in Lssiso(ϕL) (Fig. 3(C)), similar to the impact of the rL = 8rS grains in the ϕS = 0.2 system with smaller voids. This supports a simplified picture of the interplay between the granular inclusions and attractive colloids (Fig. 3(D)); grains with rL < rv have little impact on the gel structure or transition point as they easily fit into the gel voids, while grains with rL > rv distort and compress colloidal structures, increasing Lssiso and reducing the attraction needed to form the gel.
3.4 Phase diagram
Having detailed the influence of granular inclusions at two specific ϕS, we now map out the gel phase boundaries over a range of ϕS and ϕL. Having demonstrated good agreement between the rigidity onset and isostaticity percolation, we use Lssiso to classify states as it provides a more direct microstructural measure. Carrying out a series of simulations at varying ε for given ϕS and ϕL, we define the transition point ε* as the point at which Lssiso = Liso* = 1. The dimensions of our simulation box are such that we cannot reach values of 0.9999 < Lssiso < 1, which again places a limitation on the precision with which we can determine ε*. For the range of ϕS explored here (up to ϕS = 0.4), we find the isostatic percolation boundary ε*(ϕS) monotonically decreases with increasing ϕS (Fig. 4 inset). For fixed rL = 8rS, increasing ϕL at fixed ϕS generally shifts this boundary to lower attraction strength. | ||
| Fig. 4 Isostatic percolation boundaries ε* varying ϕS and ϕL. Inset: Critical attractive energy ε*(ϕS) for varying ϕL (indicated in main panel legend) for rL = 8rS. Main panel: Open symbols show same data as inset, now plotted against ϕeff = ϕS/(1 − ϕL). Solid symbols show results for larger grains and smaller ϕS, with rL = 16rS for ϕS = 0.1 and rL = 24rS for ϕS = 0.07 so that rL > rmaxv in both cases. Error bars on ε* computed using error bars on Lssiso to obtain upper and lower bounds for the point the isostatic length reaches the threshold Lssiso = 1 and are shown only in the inset for clarity. | ||
Instead plotting these isostatic percolation boundaries as a function of an effective volume fraction ϕeff ≡ ϕS/(1 − ϕL), giving the volume fraction of the small colloids relative to the free volume excluding the large grains (1 − ϕL)V, we find reasonable collapse for ϕeff ≳ 0.2 (Fig. 4 main panel). This suggests that the filler-induced shifts in the phase boundary can be understood solely through the reduction in free volume available to small colloids, so that adding larger grains is effectively equivalent to shrinking the box volume. However, for ϕS ≲ 0.1 we find this collapse breaks down, with points at higher ϕL lying clearly above the ϕL = 0 boundary. This is consistent with the behavior seen in Fig. 2(D), where the granular inclusions only have a minor effect on Liso for ϕS = 0.1 compared to the significant enhancement seen at a higher ϕS = 0.2.
Increasing the size of the large particles to ensure rL > rmaxv, in this case setting rL = 16rS for ϕS = 0.1 and rL = 24rS for ϕS = 0.07, we find that this collapse can be recovered (filled symbols in Fig. 4). We thus see that when the granular inclusions are significantly larger than the typical voids in the unfilled gel, so that they force a notable change in the gel microstructure, the influence of the voids can be captured by the effective free volume available to the small colloids. With smaller grains this effect is diminished, with the phase boundary instead largely independent of the filler concentration.
4 Conclusions
Using Langevin dynamics simulations, we have mapped out the influence of larger granular inclusions on isostaticity percolation and the gel transition in suspensions of smaller attractive colloids. Varying the volume fractions of both species, we demonstrated two distinct regimes: (i) a ‘passive void-filling’ regime, where the granular inclusions can fit into already-present voids within the gel, so that the microstructure is effectively unchanged and the gel transition governed almost solely by ϕS and (ii) an ‘active void-enhancing’ regime where the granular inclusions perturb the gel structure by forcing larger voids and the gel transition is governed by an effective volume fraction ϕeff = ϕS/(1 − ϕL). These two limiting regimes are differentiated by the size ratio of the larger grains rL and the shape of the gel void size distribution P(rv), so that anticipating the impact of the granular filler requires detailed characterization of the unfilled gel structure.To obtain these insights it has been necessary to omit more detailed aspects of the physics from our simulation model. Importantly, we are operating under density matched conditions so that gravity may be neglected. Gravity plays a key role in filled gels (indeed in applications the role of the gelled phase is often to stabilise the grains against sedimentation) and in unfilled ones alike,37 and will be an important aspect of future research that builds on the results presented here. We have also omitted full hydrodynamics38 from our model, noting that they likely play an important role under dynamic conditions but are less important in determining the static gel structure.39,40
There is relatively little experimental work examining the phase behavior of filled colloidal gels, though a recent study using a battery electrode slurry (a carbon black gel with ≈10 μm granular inclusions) found little change with addition of the granular particles.7 Given the very low gel point (occurring at ϕS ≈ 0.02), it is plausible that the carbon black gel contains sufficiently large voids to place this system in regime (i), though detailed characterisation of the gel structure would be required to confirm this. Our results should be particularly relevant to battery slurry formulation and electrode fabrication, where particle connectivity is key for performance,6 providing a road map to match the native gel structure and filler properties to tune the electrode microstructure.
Author contributions
YL, JR, JS and CN planned the research; YL carried out the research; YL, JR and CN wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
YL is funded by the China Scholarship Council (CSC) and by the University of Edinburgh through a School of Engineering studentship. C. N. acknowledges support from the Royal Academy of Engineering under the Research Fellowship scheme. The work was supported by the UK Engineering and Physical Sciences Research Council under grant EP/N025318/1. For the purpose of open access, the authors have applied a Creative Commons Attribution (CC BY) licence to any Author Accepted Manuscript version arising from this submission. The data used to generate the figures in this article is available via Edinburgh DataShare at https://doi.org/10.7488/ds/3796.Notes and references
- V. Trappe and P. Sandkühler, Curr. Opin. Colloid Interface Sci., 2004, 8, 494–500 CrossRef CAS.
- E. Zaccarelli, J. Phys.: Condens. Matter, 2007, 19, 323101 CrossRef.
- C. P. Royall, M. A. Faers, S. L. Fussell and J. E. Hallett, J. Phys.: Condens. Matter, 2021, 33, 453002 CrossRef CAS PubMed.
- K. Y. Cho, Y. I. Kwon, J. R. Youn and Y. S. Song, Mater. Res. Bull., 2013, 48, 2922–2926 CrossRef CAS.
- T.-S. Wei, F. Y. Fan, A. Helal, K. C. Smith, G. H. McKinley, Y.-M. Chiang and J. A. Lewis, Adv. Energy Mater., 2015, 5, 1500535 CrossRef.
- W. B. Hawley and J. Li, J. Energy Storage, 2019, 25, 100862 CrossRef.
- S. L. Morelly, M. H. Tang and N. J. Alvarez, Polymers, 2017, 9, 461 CrossRef PubMed.
- V. Trappe, V. Prasad, L. Cipelletti, P. N. Segre and D. A. Weitz, Nature, 2001, 411, 772–775 CrossRef CAS PubMed.
- L. C. Hsiao, R. S. Newman, S. C. Glotzer and M. J. Solomon, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 16029–16034 CrossRef CAS.
- G. Wang, A. M. Fiore and J. W. Swan, J. Rheol., 2019, 63, 229–245 CrossRef CAS.
- J. Rouwhorst, P. Schall, C. Ness, T. Blijdenstein and A. Zaccone, Phys. Rev. E, 2020, 102, 022602 CrossRef CAS.
- C. Ferreiro-Córdova, G. Foffi, O. Pitois, C. Guidolin, M. Schneider and A. Salonen, Soft Matter, 2022, 18, 2842–2850 RSC.
- C. Ferreiro-Cordova, E. Del Gado, G. Foffi and M. Bouzid, Soft Matter, 2020, 16, 4414–4421 RSC.
- Y. Jiang, S. Makino, J. R. Royer and W. C. Poon, Phys. Rev. Lett., 2022, 128, 248002 CrossRef CAS PubMed.
- Y. Jiang and R. Seto, 2022, preprint, arXiv:2211.12978 DOI:10.48550/arXiv.2211.12978.
- M. C. Grant and W. B. Russel, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1993, 47, 2606–2614 CrossRef CAS PubMed.
- H. Verduin and J. K. Dhont, J. Colloid Interface Sci., 1995, 172, 425–437 CrossRef CAS.
- W. C. K. Poon, A. D. Pirie and P. N. Pusey, Faraday Discuss., 1995, 101, 65–76 RSC.
- P. N. Segrè, V. Prasad, A. B. Schofield and D. A. Weitz, Phys. Rev. Lett., 2001, 86, 6042–6045 CrossRef PubMed.
- S. A. Shah, Y.-L. Chen, K. S. Schweizer and C. F. Zukoski, J. Chem. Phys., 2003, 119, 8747–8760 CrossRef CAS.
- H. Sedgwick, K. Kroy, A. Salonen, M. B. Robertson, S. U. Egelhaaf and W. C. K. Poon, Eur. Phys. J. E: Soft Matter Biol. Phys., 2005, 16, 77–80 CrossRef CAS PubMed.
- P. J. Lu, E. Zaccarelli, F. Ciulla, A. B. Schofield, F. Sciortino and D. A. Weitz, Nature, 2008, 453, 499–503 CrossRef CAS.
- A. P. Eberle, N. J. Wagner and R. Castaneda-Priego, Phys. Rev. Lett., 2011, 106, 105704 CrossRef PubMed.
- M. E. Helgeson, Y. Gao, S. E. Moran, J. Lee, M. Godfrin, A. Tripathi, A. Bose and P. S. Doyle, Soft Matter, 2014, 10, 3122–3133 RSC.
- K. A. Whitaker, Z. Varga, L. C. Hsiao, M. J. Solomon, J. W. Swan and E. M. Furst, Nat. Commun., 2019, 10, 2237 CrossRef PubMed.
- R. Tuinier and H. N. Lekkerkerker, Colloids and the Depletion Interaction, Springer, Netherlands, 2011 Search PubMed.
- S. Zhang, L. Zhang, M. Bouzid, D. Z. Rocklin, E. Del Gado and X. Mao, Phys. Rev. Lett., 2019, 123, 058001 CrossRef CAS PubMed.
- A. Fierro, E. D. Gado, A. de Candia and A. Coniglio, J. Stat. Mech.: Theory Exp., 2008, 2008, L04002 Search PubMed.
- A. Zaccone, H. Wu and E. Del Gado, Phys. Rev. Lett., 2009, 103, 208301 CrossRef PubMed.
- A. P. Thompson, H. M. Aktulga, R. Berger, D. S. Bolintineanu, W. M. Brown, P. S. Crozier, P. J. in’t Veld, A. Kohlmeyer, S. G. Moore, T. D. Nguyen, R. Shan, M. J. Stevens, J. Tranchida, C. Trott and S. J. Plimpton, Comput. Phys. Commun., 2022, 271, 108171 CrossRef CAS.
- F. Sciortino, S. Mossa, E. Zaccarelli and P. Tartaglia, Phys. Rev. Lett., 2004, 93, 055701 CrossRef.
- J. C. Maxwell, London, Edinburgh Dublin Philos. Mag. J. Sci., 1864, 27, 294–299 CrossRef.
- M. van Hecke, J. Phys.: Condens. Matter, 2009, 22, 033101 CrossRef PubMed.
- G. Wang, A. M. Fiore and J. W. Swan, J. Rheol., 2019, 63, 229–245 CrossRef CAS.
- H. Tsurusawa, M. Leocmach, J. Russo and H. Tanaka, Sci. Adv., 2019, 5, eaav6090 CrossRef PubMed.
- N. Koumakis, E. Moghimi, R. Besseling, W. C. Poon, J. F. Brady and G. Petekidis, Soft Matter, 2015, 11, 4640–4648 RSC.
- R. Harich, T. Blythe, M. Hermes, E. Zaccarelli, A. Sederman, L. F. Gladden and W. C. Poon, Soft Matter, 2016, 12, 4300–4308 RSC.
- Z. Varga, G. Wang and J. Swan, Soft Matter, 2015, 11, 9009–9019 RSC.
- C. Ness and A. Zaccone, Ind. Eng. Chem. Res., 2017, 56, 3726–3732 CrossRef CAS.
- J. De Graaf, W. C. Poon, M. J. Haughey and M. Hermes, Soft Matter, 2019, 15, 10–16 RSC.
| This journal is © The Royal Society of Chemistry 2023 |