
Two-dimensional diffusiophoretic colloidal banding: optimizing the spatial and temporal design of solute sinks and sources

Abstract
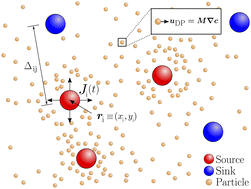
Diffusiophoresis refers to the phenomenon where colloidal particles move in response to solute concentration gradients. Existing studies on diffusiophoresis, both experimental and theoretical, primarily focus on the movement of colloidal particles in response to one-dimensional solute gradients. In this work, we numerically investigate the impact of two-dimensional solute gradients on the distribution of colloidal particles, i.e., colloidal banding, induced via diffusiophoresis. The solute gradients are generated by spatially arranged sources and sinks that emit/absorb a time-dependent solute molar rate. First we study a dipole system, i.e., one source and one sink, and discover that interdipole diffusion and molar rate decay timescales dictate colloidal banding. At timescales shorter than the interdipole diffusion timescale, we observe a rapid enhancement in particle enrichment around the source due to repulsion from the sink. However, at timescales longer than the interdipole diffusion timescale, the source and sink screen each other, leading to a slower enhancement. If the solute molar rate decays at the timescale of interdipole diffusion, an optimal separation distance is obtained such that particle enrichment is maximized. We find that the partition coefficient of solute at the interface between the source and bulk strongly impacts the optimal separation distance. Surprisingly, the diffusivity ratio of solute in the source and bulk has a much weaker impact on the optimal dipole separation distance. We also examine an octupole configuration, i.e., four sinks and four sources, arranged in a circle, and demonstrate that the geometric arrangement that maximizes enrichment depends on the radius of the circle. If the radius of the circle is small, it is preferred to have sources and sinks arranged in an alternating fashion. However, if the radius of the circle is large, a consecutive arrangement of sources and sinks is optimal. Our numerical framework introduces a novel method for spatially and temporally designing the banded structure of colloidal particles in two dimensions using diffusiophoresis and opens up new avenues in a field that has primarily focused on one-dimensional solute gradients.
- This article is part of the themed collections: Soft Matter Lectureship Winners and Soft Matter Emerging Investigators Series
 Please wait while we load your content...
Please wait while we load your content...

