
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Transient deformation and swelling of paper by aqueous co-solvent solutions†
C.-L.
Wong
a,
S.
Wang
a,
S.
Karimnejad
a,
M. G.
Wijburg
a,
H.
Mansouri
b and
A. A.
Darhuber
 *a
*a
aFluids & Flows Group, Department of Applied Physics, Eindhoven University of Technology, The Netherlands. E-mail: a.a.darhuber@tue.nl
bCanon Production Printing, Venlo, The Netherlands
First published on 16th January 2023
Abstract
Inkjet printing inks frequently contain polar liquids of low volatility such as glycerol or poly(ethylene glycols) in addition to the main solvent water. The deposition of these liquids on paper sheets induces swelling of the cellulose fibers, which leads to an overall, anisotropic deformation of the sheet. We characterized the corresponding strain components by means of a grid projection method and white light interferometry. For pure water, most of the hydroexpansion strain vanishes again after drying is complete. However, for aqueous solutions of non-volatile co-solvents, a large fraction of the deformation persists after the water has evaporated. Because swelling occurs only after liquid enters the cellulose fibers, monitoring the dynamics of expansion provides insight into the pore-fiber distribution of co-solvents. The corresponding timescales of pore-fiber transport strongly depend on the co-solvent concentration, as a sufficient quantity of water is needed to plasticize the fiber walls.
I. Introduction
The interaction of liquids and specifically water with paper is relevant to e.g. paper-making,1 food packaging2,3 and printing technology.4 In the field of inkjet printing, inks are often based on water as an environmentally friendly main solvent.5,6 Besides colorants and surfactants,7,8 they commonly contain so-called co-solvents – polar liquids such as glycerol or glycols - with a concentration up to 50 wt%.5 Their primary purpose is to serve as humectants for preventing inkjet nozzle clogging, but also as anti-cockle and anti-curl agents.5,9–12Generally, polar liquids cause cellulose fibers to swell, leading to an overall deformation of the paper sheet. If gradients of liquid content in the thickness direction of a paper sheet are the dominant driving force, the deformation is called curl.13–17 Douezan et al.18 and Reyssat et al.19 presented experiments of the curling dynamics of tracing paper deposited on a water surface. Moreover they formulated theoretical models for the bending induced by the differential expansion of paper fibers at different positions depending on the imbibition dynamics of the water in the thickness direction. Leppänen et al.14 accounted for the anisotropy of the elastic properties of paper as well as its heterogeneity. Lipponen et al.15 and Erkkilä et al.20 extended this model by accounting for the elasto-plastic material response of paper.
If the presence of in-plane gradients in liquid content is the dominant mechanism, the paper sheet can undergo a buckling instability.20,21 De Böck et al.22 presented a theoretical analysis of the buckling of paper sheets due to an inhomogeneous moisture distribution based on solutions of the Von Kármán equations. However, they disregarded the sensitive dependence of the elastic properties of paper on the moisture content, which implies that their results are restricted to the case of small excursions in moisture content. Lee et al. studied the bending and buckling of paper strips that were supported at their ends and moistened in the center by means of a capillary tube.23 Their numerical analysis accounted for the moisture-induced change in elastic properties.
In this manuscript, we studied the deformation of paper sheets after deposition of long lines of aqueous co-solvent solutions, after spray deposition or complete immersion and subsequent drying. We characterized the corresponding strain components in the cross machine direction (CD) and the thickness direction (TD) by means of a grid projection method,24 sheet length monitoring and white light interferometry (WLI). For pure water, most of the hydroexpansion strain vanishes again after drying is complete. However, for aqueous solutions of non-volatile co-solvents, a large fraction of the deformation persists after the water has evaporated. Interestingly, this persistent post-drying deformation amplitude first increases with co-solvent concentration up to about 50 wt%, beyond which it decreases. This non-monotonic behavior is attributed to a partial retardation of pore-fiber transport for low water concentrations, as water plays the role of a plasticizer of the cellulose fiber walls.6
II. Materials and methods
A. Materials and material properties
The co-solvents glycerol, ethylene glycol (EG), tri- and tetra ethylene glycol (TrEG and TEG) were purchased from Sigma-Aldrich and used as received. The molecular weight (MW), viscosity μcs, surface tension γcs and mass density ρcs of the pure co-solvents are listed in Table 1. Aqueous co-solvent solutions were prepared by mixing deionized water (Millipore Direct-Q3 R) and a pure co-solvent at a certain mass ratio in a glass bottle. Masses were measured with a Kern ALT 220-5DAM scale.| Co-solvent | Product# | M W | μ cs (mPa s) | γ cs (mN m−1) | ρ cs (kg m−3) |
|---|---|---|---|---|---|
| Glycerol | 449770 | 92.1 | 1206 | 63.5 | 1258 |
| Ethylene glycol (EG) | 324558 | 62.1 | 19.8 | 48.0 | 1110 |
| Triethylene glycol (TrEG) | 90390 | 150.2 | 49.0 | 45.5 | 1120 |
| Tetra thylene glycol (TEG) | 110175 | 194.2 | 58.3 | 44.0 | 1121 |
In some experiments we used aqueous solutions of the surfactants sodium dodecyl sulfate (SDS, anionic, MW 288, Sigma Aldrich, product number 436143) and Triton X-100 (TX-100, non-ionic, average MW 647, Sigma Aldrich, product number T9284). The critical micelle concentration (cmc) of SDS is 0.234 wt%, that of Triton X-100 is 0.019 wt%.
As substrates we used two uncoated paper types from different manufacturers that were both developed for inkjet printing applications.
• paper ‘A’ (DNS High-Speed Inkjet Natural Feel, Mondi, grammage g = 80 g m−2, thickness dsub = 104 μm),
• paper ‘B’ (Z-Plot 650, Ziegler, g = 90 g m−2, dsub = 116 μm, ash content 12%, surface pH 7.3).
The papers are stored in a sealed container until each experiment to reduce the effect of humidity fluctuations.
Nissan developed an empirical model for the dependence of Young's modulus Y of paper on its moisture content θw based on the effect of water on the number of inter-fiber hydrogen bonds. This model predicts a drastic reduction of Y as θw approaches the maximum holding capacity.25 Such a decrease of Y with increasing θw has been confirmed by many studies.23,26–30 Typically, the value of Y for fully wet paper is more than 20 times lower than that of dry paper.31
The elastic properties of machine-made paper are anisotropic due to the extensional deformation and tension in the so-called machine direction (MD). Typically the ratio of YMD/YCD ranges between 2 and 3, i.e. paper is more compliant in the cross-machine direction (CD).32–39 This behavior persists upon increasing the moisture content.26 Typical values of YCD are in the range of 1.4–4 GPa.26,35,37,40,41 The hygroexpansion coefficient is also strongly anisotropic. It is commonly larger in the CD than in the MD direction by a factor typically ranging from 2 to 5.38,39,42,43 The hygroexpansion coefficient in the thickness direction (TD) is typically a factor of 7 to 30 larger than in the plane of the paper sheet.44 The hydroexpansion coefficients for paper in contact with liquid-phase water have been less intensively studied. Larsson et al. spray-deposited water to increase the moisture content of magazine paper.45 For an increment Δθw ≈ 0.04 kg kg−1, they found a strain amplitude ΔεCD ≈ 0.14%. Figueiredo et al. measured the wet expansion of different paper types46 and found values in the range of ΔεCD ≈1.5–3%.
B. Liquid deposition and optical grid projection
Fig. 1(a) shows a sketch of the setup used for grid projection metrology, a corresponding photograph is available in Fig. 1 of the ESI.† A sheet of paper is suspended about 2 mm above the top surface of a motorized translation stage (Newport, model UTS 100CC). A motorized syringe pump (KD Scientific, model Legato 180) equipped with a gastight syringe (Hamilton, product number 1010, total volume 10 mL) supplies liquid onto the paper through a plastic tube (Hamilton, product number 90618). The tubing orifice was suspended approximately 0.5 mm above the paper surface. The paper is clamped on both sides by two metal plates that have an oblong opening with a width of wclamp = 40 mm. A grid projector (Advanced Illumination, model SL191-530IC) is mounted approximately 15 cm above the paper and projects gridlines under an angle of incidence of about θ = 40°. A CMOS camera (Thorlabs, model DCC3240M) mounted 35 cm above the paper sheet monitors the displacement and deformation of the projected gridlines. The lateral shift of the projected line corresponding to a certain vertical displacement of the paper sheet was calibrated using a precision labjack (Thorlabs, model number L490) and a digital displacement gauge (Mitutoyo, model number ID-H0530). | ||
| Fig. 1 (a) Sketch of the experimental setup. (b and c) Top-view photographs of wet zones deposited via substrate motion in the (b) machine direction (MD) and (c) cross direction (CD). | ||
Fig. 1(b and c) shows top-view photographs of wet zones deposited via substrate motion in the (b) machine direction (MD) and (c) cross direction (CD) of the paper. In both cases the expansion of the paper leads to a buckling instability with bulges oriented along the MD, because both the expansion coefficient and the compliance in the CD is significantly larger than in the MD. In all following experiments, the direction of substrate motion was oriented along the MD. The substrate speed and the volume flow rate of the syringe pump were set to Usub = 2 mm s−1 and 0.1 mL min−1, respectively, unless stated otherwise. The time delay between liquid deposition at the measurement location and the time corresponding to the first frame of the CMOS camera recording (which we denote as t = 0) is about Δt ≈ 25 ± 5 s. All experiments were conducted at room temperature and an ambient humidity of (40 ± 10)%.
C. Sheet length monitoring of square-shaped samples
The combination of line deposition and grid projection allows for convenient monitoring of the time-dependence of the swelling. However, it is restricted to co-solvent contents θcs (kg co-solvent per kg of dry paper) below 0.5 kg kg−1 due to rapid lateral imbibition. To reach larger values of θcs, we prepared square paper samples of width w = 25.4 mm with sides parallel to the MD and CD, respectively, and positioned them on flat polycarbonate plates. A known volume (typically 20–50 μl) of co-solvent solution was deposited homogeneously on the sample by means of a Hamilton digital syringe. In this fashion values of θcs up to about 1.0 kg kg−1 could be reached. The swelling causes length expansion, which was measured using an upright microscope (Olympus BX50), a manual translation stage (Newport) and a digital displacement indicator (Mitutoyo, product number ID-H530) with a resolution of 0.5 μm.D. Microbead displacement
We positioned a glass microsphere (approximate diameter 200 μm) onto a dry sheet of paper, placed it onto an upright microscope (Olympus, BX50) and focused onto it. Subsequently, EG or glycerol was deposited onto the paper, which caused it to swell in the thickness direction and displace the bead vertically. We measured the change in focus position by means of a z-stage (Kohzu, model number ZM10A-C3C) and a digital displacement indicator (Mitutoyo, product number ID-H530). Conformal contact of the paper strip with the underlying substrate was ensured by slightly bending it into a parabolic shape.E. Sample preparation for white-light interferometry
Samples were fabricated by depositing solution droplets on a circular paper sample with a diameter of 12.7 mm. The paper circle was placed on a flat, polished silica or silicon substrate. Samples in which the co-solvent is intended to partially remain within the inter-fiber pores were created with a high concentration of 60 wt%. Samples in which a pore-fiber equilibrium is required were prepared with low concentration solutions of 30 wt% and underwent a post-deposition rehydration treatment with water,6 since water accelerates the pore-fiber equilibration. The desired co-solvent content θcs was achieved by deposition of aqueous co-solvent solution droplets of different volumes. Care was taken to prevent excess liquid from spilling over the paper sample. After completion of every deposition and rehydration step, the samples were allowed to dry for at least 45 minutes.F. White-light interferometry (WLI)
We used a Bruker NPFLEX white light interferometer with a 5× objective lens and a 0.55× demagnification lens, resulting in a field of view of approximately 2 mm per frame. In order to measure a representative area of the paper samples, we used overlapping regions of interest that the NPFLEX software stitched together, see Fig. 2 for an example. The instrument only records a signal from coordinates where the local slope is smaller than the numerical aperture of the objective lens. For a rough sample such as a paper sheet, this results in a sparse data set. Therefore, we applied a running average over ±5 neighbouring pixels for visualization purposes. Moreover, we fit a plane to the substrate signal (the blue region in Fig. 2) and subtracted it, to eliminate any non-zero tilt of the substrate. In the white region in the center of Fig. 2, the z-coordinates of the paper surface were beyond the scan range, due to the presence of a cavity between the paper and the substrate. The height variations outside of the cavity region represented by green and yellow colors amount to approximately ±25 μm. Part of this variation is due to the intrinsic heterogeneity of the paper, another part may be caused by imperfect attachment to the substrate. In our data analysis, we average over 2D regions (indicated by red rectangles in Fig. 2), that are large enough to eliminate the effect of heterogeneity and that do not overlap with the region of non-conformal contact. | ||
| Fig. 2 Surface topography of a sample of paper A measured with white-light interferometry. The glycerol content was θcs = 0.9 kg kg−1, the substrate a silica wafer. The color bar denotes the surface topography in μm. Red rectangles denote regions for which the average expansion strain is determined. | ||
III. Experimental results
We quantify the hydroexpansion in terms of the strain components in the CD | (1) |
 | (2) |
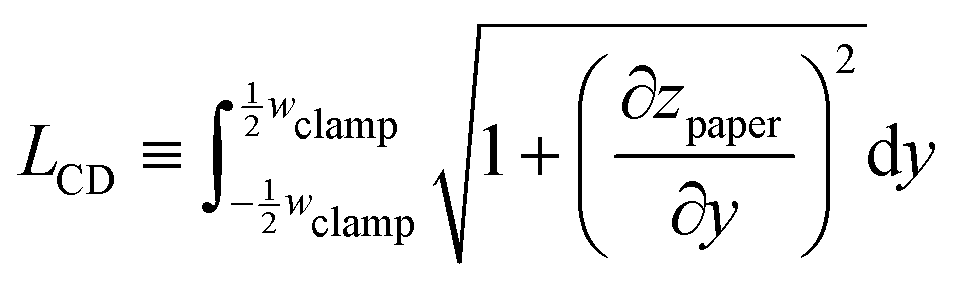 | (3) |
| Δz ≡ zpaper − zpaper,dry. | (4) |
We note that the division by wmax in eqn (2) corresponds to the notion that primarily the paper in the wet zone expands, whereas the paper that remains dry only deforms to maintain continuity of zpaper(y), but without concomitant change in length. Any hygroexpansion due to water evaporating from the wet zone and condensing on adjacent regions of dry paper is disregarded in this fashion. For the practical evaluation of eqn (3), we first smoothed the raw data using a running average, as otherwise the presence of noise will lead to an overestimation of  and thus of LCD.
and thus of LCD.
A. Expansion in the cross direction – grid projection
Fig. 3 shows the deformation amplitude Δz(y), the width of the wet zone w, and the expansion strain εCD as functions of time. The curve of w(t) terminates at t ≈ 15 min, due to the evaporation of the water. However, the curve of εCD(t) reaches a steady state only at t ≈ 21 min. This is because upon evaporation the water first depletes the micron-scale inter-fiber pores, which determine the optical appearance of paper.6 Most of the water in the fibers is still present and only after it has evaporated as well does εCD(t) reach a steady state. For pure water, 80–90% of the maximum hydroexpansion amplitude disappears after evaporation is complete. Fig. 3(a and c) shows that the paper sheet does not go back to a flat shape, but a small residual deformation remains, which we denote as persistent strain in Fig. 3(c).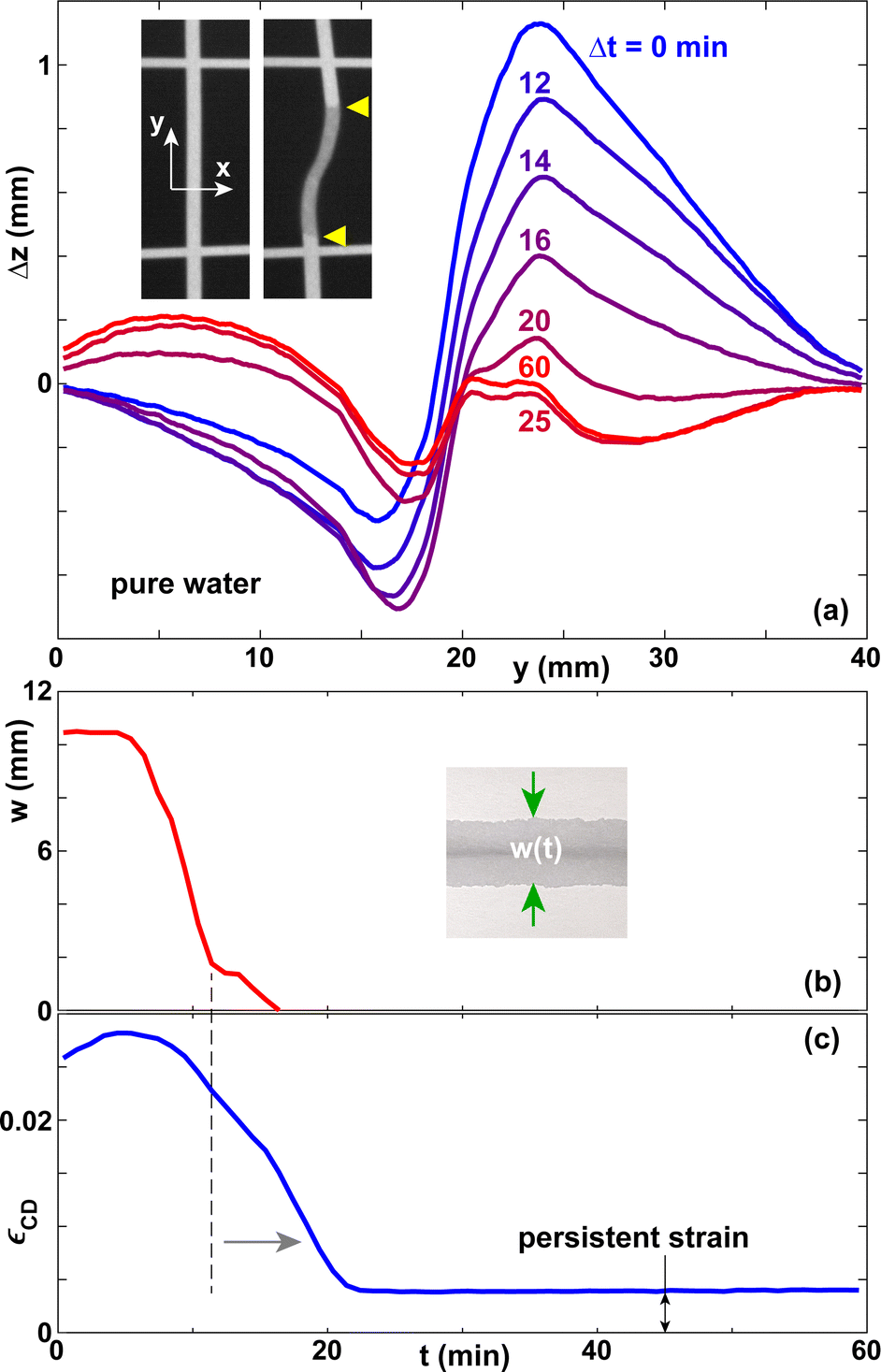 | ||
| Fig. 3 (a) Vertical displacement Δz(y) for different times Δt for lines of pure water deposited on paper A. The insets show the projected grid before and about 3 min after deposition. The yellow triangles illustrate the boundaries of the wet zone. The width of the images in the y-direction is about 20 mm. (b) Width of the wet zone w and (c) lateral expansion strain in the CD direction εCD as a function of time. | ||
Fig. 4 presents εCD for EG, TrEG and glycerol solutions of various initial concentrations c0. In contrast to pure water, the strain does not revert back to almost zero after the water has evaporated, but a large fraction of it persists. This is because the polar co-solvents studied are essentially non-volatile and induce a similar degree of swelling of the cellulose fibers as water, once they have penetrated the fiber walls.47
 | ||
| Fig. 4 Lateral expansion strain εCD in the cross direction induced by aqueous solutions of (a) EG, (b) TrEG and (c) glycerol as a function of time. Solid lines correspond to paper A, the dashed lines in (b) to paper B. | ||
For all co-solvents studied, εCD(t = 60 min) monotonically increases with increasing initial co-solvent concentration up to a critical value of about 50 wt%. For c0 exceeding this value, the persistent expansion strain εCD(t = 60 min) decreases. We attribute this effect to the lack of complete pore-fiber equilibration for high concentration solutions.6 EG has the smallest molecular weight of the set and thus suffers less from this effect than TrEG or glycerol. Moreover, the 60 wt% curve for EG has a noticeable upwards slope at t = 60 min, which is less pronounced or absent for TrEG and glycerol.
The dashed lines in Fig. 4(b) correspond to experiments with paper B, which generally results in lower values of εCD than paper A (solid lines) by about 15–30%. The bending rigidity48 of linear elastic plates scales as dsub3. The thickness of paper B (dsub = 116 μm) is approximately 11% larger than that of paper A (dsub = 104 μm). The above scaling relation thus would imply a reduction of the deformation amplitude for paper B by approximately 27% compared to paper A. While this is a reasonable agreement, we note that the different expansion amplitudes of the two paper types might also be affected by structural or compositional differences and not just their thicknesses.
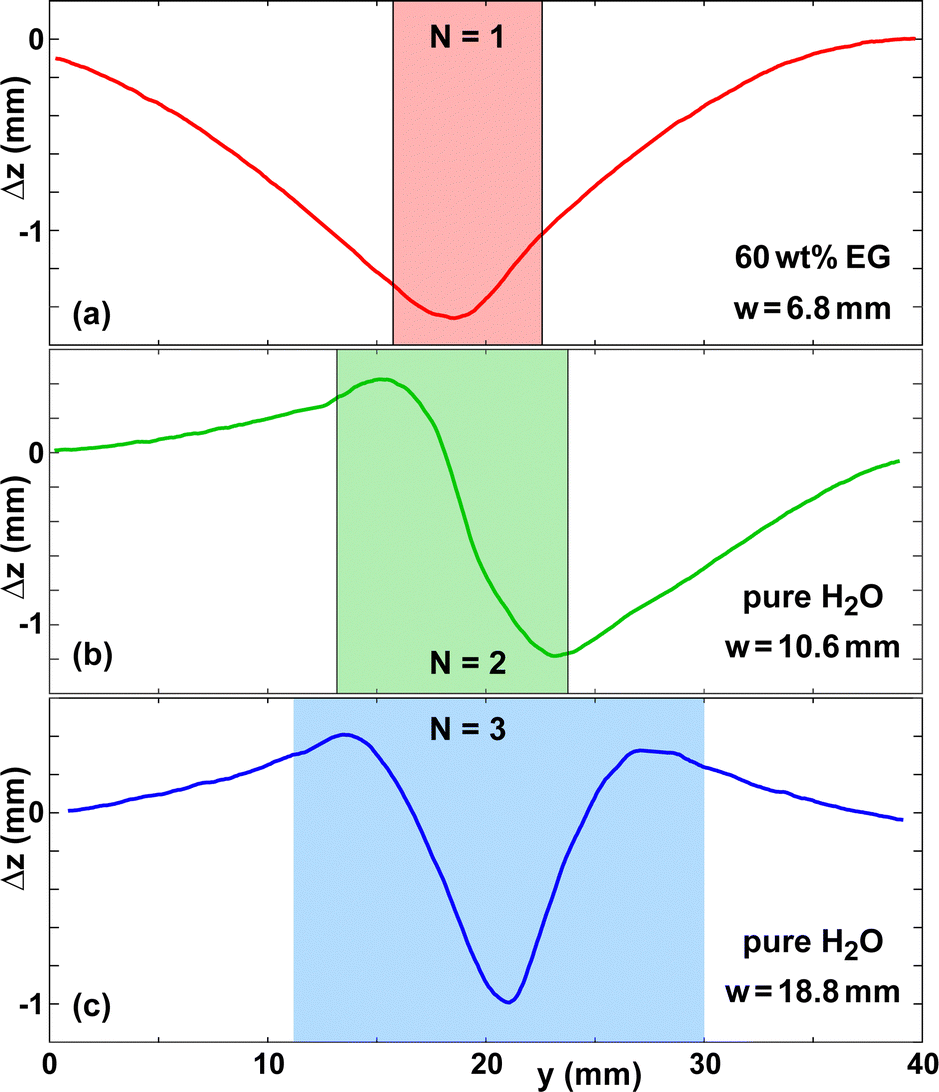 | ||
| Fig. 5 Examples of deformation profiles Δz(y) for (a) 60 wt% EG on paper B and (b and c) pure water on paper A. The substrate speed of (c) was Usub = 1 mm s−1 in contrast to all other experiments. | ||
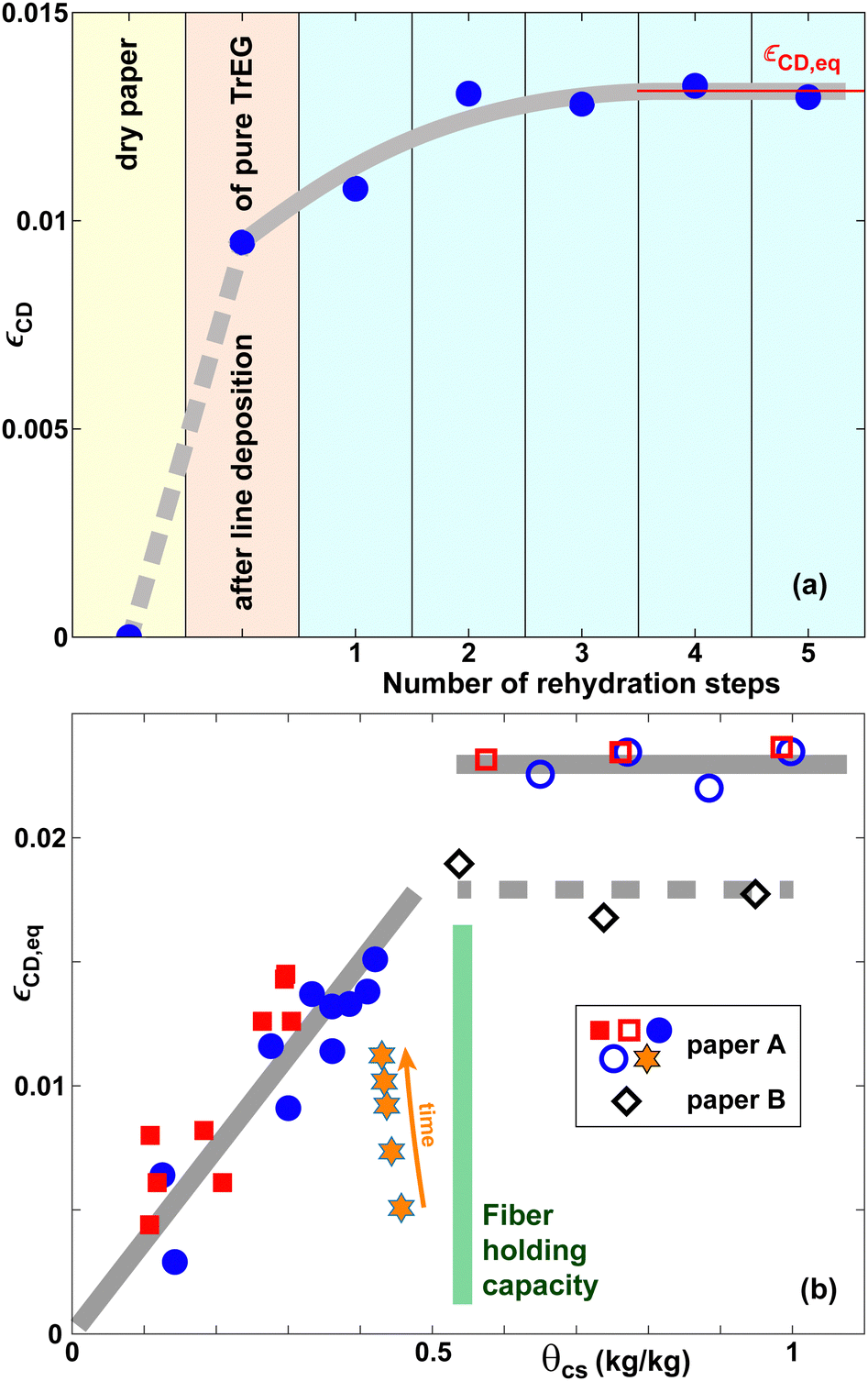 | ||
| Fig. 6 (a) Lateral expansion strain εCD in the CD as a function of the number of rehydration steps for pure TrEG. (b) Equilibrium lateral expansion strain εCD,eq in the cross direction (CD) as a function of θcs for EG (red squares) and TrEG (blue circles, diamonds, hexagrams). The gray lines in (a and b) are guides to the eye. Squares, circles and hexagrams refer to paper A, diamonds to paper B. The orange hexagrams represent selected timesteps of the c0 = 80 wt% curve in Fig. 4(b). | ||
The values of εCD,eq increase with θcs for θcs ≲ 0.5 kg kg−1 approximately linearly and then saturate. There is essentially no difference in εCD,eq for EG and TrEg to within experimental uncertainty. Our interpretation is that for small quantities of co-solvent, the co-solvent is essentially fully absorbed by the fibers until the maximum holding capacity of the fibers θHC,fibers ≈ 0.5 kg kg−1 is reached. For co-solvent quantities beyond this limit, a fraction of co-solvent resides in the inter-fiber pores, where it does not induce any further lateral expansion, however.
The open diamonds in Fig. 6(b) correspond to paper B, all other symbols to paper A. Paper B exhibits expansion strains that are approximately 20% smaller than for paper A, consistent with the behavior found in Fig. 4(b).
The hexagrams in Fig. 6(b) represent selected datapoints from the 80 wt% TrEG curve in Fig. 4(b). The expansion strain increases in time, but after 60 min it is still smaller than the equilibrium value corresponding to the same value of θcs. In contrast, the strain value for 50 wt% EG solutions in Fig. 4(a) after 1 hour is essentially equal to the equilibrium value.
B. Thickness increase – white-light interferometry
Fig. 7 shows the vertical expansion strain εTD measured as a function of co-solvent content θcs. Due to the tendency of paper to wrinkle, which prevents conformal contact with the substrate, we were unable to do reliable measurements for θcs below 0.5 kg kg−1. Filled symbols in Fig. 7 correspond to rehydrated samples and thus equilibrium co-solvent distributions. Open symbols in Fig. 7 represent measurements for non-rehydrated, as-deposited, non-equilibrium 60 wt% co-solvent distributions, which result in smaller swelling amplitudes, consistent with the behavior observed in Fig. 4. Semi-filled symbols refer to non-rehydrated, as-deposited, co-solvent distributions of glycerol (triangle) and EG (square) measured using microbead displacement. | ||
| Fig. 7 Vertical expansion strain εTD in the TD as a function of co-solvent content θcs for TEG (squares, circles) and glycerol (triangles). Solid squares and triangles refer to paper A and circles to paper B containing equilibrium co-solvent distributions after multiple rehydration treatments. (Half)open symbols refer to paper A and non-equilibrium, as-deposited 60 wt% co-solvent distributions without rehydration steps. | ||
Above θcs > 0.5 kg kg−1, εTD is only weakly dependent on θcs, consistent with the in-plane behavior shown in Fig. 6(b). It is evident that the strain amplitude in the TD is approximately a factor of 20 larger than in the CD. Cellulose fibers can expand easily in the TD, because they have been strongly compressed in this direction during the paper fabrication process. There is little difference between TEG and glycerol concerning the asymptotic value of εTD. There is a roughly 10% difference in the swelling amplitudes for TEG in papers A and B.
C. Expansion in the machine direction
In the MD, we observed an expansion strain of about 0.2% one hour after deposition of pure TrEG onto dry NF paper. After repeated water spray treatments, however, we observed a net shrinkage relative to the initial, dry state of approximately 0.2%. This is consistent with the hysteretic behavior observed for paper subject to cycling of the ambient relative humidity.39,50,51IV. Discussion
A. Mode selection
An interesting question pertains to the direction of surface deformation of the N = 1 deformation mode. There are three competing mechanisms at play. The first is gravity, which induces a downwards deformation. The second is the elastocapillary deformation of the paper sheet due to the interfacial tension and capillary pressure of the deposited droplet, which both lead to a deformation towards the underside of the paper sheet.52–55 This deformation mechanism is facilitated by the drastic decrease of the elastic modulus of paper with increasing moisture content. The gravitational force scales with ρliqVdropg, where g is the gravitational acceleration. In contrast, the surface tension force is proportional to the droplet footprint radius and scales as γliqVdrop1/3. Thus, for small droplet volumes, the influence of gravity becomes negligible compared to elastocapillary effects.The third mechanism is related to the existence of gradients of the moisture content or liquid content in the thickness direction of the paper sheet. Holmes et al.,17 Samy et al.56 and Sun et al.57 reported on experiments regarding the deposition of solvent droplets on polymer sheets. The solvent swells the top part of the polymer which induces a curvature corresponding to an upwards displacement. This is the same mechanism as responsible for the curl of paper sheets observed by Douezan et al.18 and Reyssat and Mahadevan.19
For co-solvent solutions with low concentration c0, the imbibition time to bridge the thickness of the paper sheet is short for the two paper types studied, such that vertical gradients in liquid content exist only for a brief time span. In contrast, the elastocapillary and gravitational force due to the presence of the droplet may act for a longer time. Consequently, we expect a bias towards a downwards displacement for low c0. For higher c0, vertical gradients persist for a longer time due to the higher viscosity, which may favor an upwards deformation for N = 1.
We performed a systematic study of the paper deformation after co-solvent deposition. However, we did not observe a clear trend concerning the direction of deformation, which may point at the influence of random heterogeneities in the paper structure.58–61
B. Pore-fiber distribution of co-solvents
Upon imbibition of liquid into a porous medium, the liquid fills first the largest pores and progressively enters the smaller ones owing to their lower permeability. Paper consists of cellulose fibers with typical intra-fiber pore sizes of approximately 1 to 100 nm and typical inter-fiber pore dimensions in the micron range. This implies that for an equilibrium distribution, characterized by a uniform capillary pressure, a liquid preferentially resides in the fibers, until they are saturated, i.e. completely filled. Further addition of liquid will also populate the micron-sized pores. The timescales relevant to this pore-fiber equilibration process, however, sensitively depend on the MW of the liquid, because the fiber walls act as a barrier. For swellable polymer membranes in contact with aqueous solutions, it is well known that the concentration of water has a large impact on the effective transport rate of large MW solutes through the membrane, because it plasticizes the polymer.62–64 We hypothesize that the same effect governs the pore-fiber transport dynamics of co-solvent solutions in paper.Stone and Scallan developed a solute exclusion technique for probing the accessibility of intra-fiber pores to molecules of increasing MW.65,66 Typical cut-off values of the solute MW are in the range 250–5000,67–69 therefore we conclude that the co-solvents we used can penetrate the fiber walls given enough time. However, without rehydration the co-solvents are unable to enter the fibres to the same extent in the time available between liquid deposition and completion of drying, compared to rehydrated samples. As only the liquid fraction in the fibers contributes to the expansion of the paper, the expansion strain provides information about the current pore-fiber distribution of the co-solvent. In ref. 6 we had studied the light transmittance I/I0 of co-solvent distributions in paper sheets. We found that light scattering is primarily sensitive to the co-solvent content in the inter-fiber pores. Therefore, monitoring εCD is a complementary technique for characterization of co-solvent distributions, as it is sensitive primarily to the co-solvent content in the intra-fiber pores. In the following subsection, the consistency of the results from strain and transmittance measurements is evaluated.
C. Quantification of pore-fiber distributions
Fig. 8(a) depicts the equilibrium transmittance I/I0 as a function of TEG content of paper A.6I/I0 is constant at low θcs ≲ 0.5, followed by an approximately linear increase in the range 0.5 ≲ θcs ≲ 1.1. In contrast, the equilibrium expansion strain εCD,eq is approximately a linear function of θcs for θcs ≲ 0.5, after which is reaches a plateau value [see Fig. 8(b)]. Thus, the equilibrium co-solvent distribution may be approximated as though the co-solvent resides exclusively in the intra-fiber pores at low θcs. When θHC,fibers is exceeded, the inter-fiber pores start to fill. This simplification is visualized in Fig. 8(c) in terms of the degrees of saturation S ≡ θcs/θcs,max of the intra- and inter-fiber pores. Here, θcs,max is the total holding capacity of the paper substrate.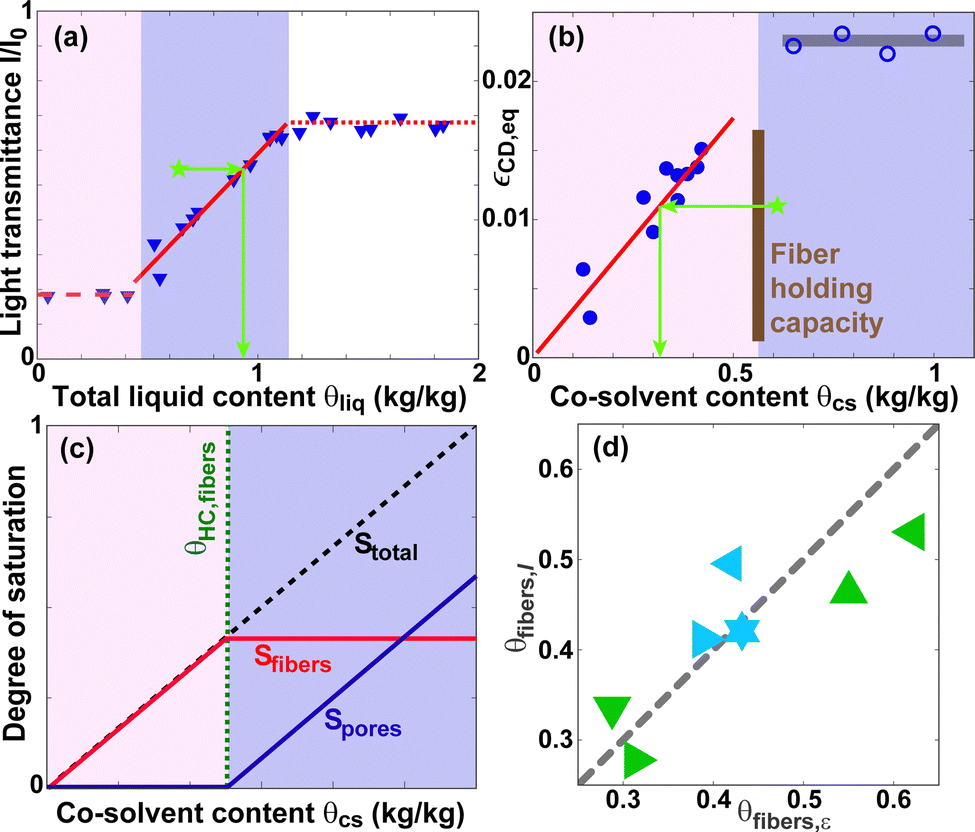 | ||
| Fig. 8 (a) Equilibrium light transmittance I/I0 data for TEG in paper A from ref. 6. The solid red line is a linear fit according to eqn (5). (b) Equilibrium expansion strain εCD,eq for TrEG in paper A. The red solid line is a linear fit according to eqn (6). (c) Simplified model of the equilibrium co-solvent distributions in the intra- and inter-fiber pores of paper. (d) The intra-fiber co-solvent content θfibers,I obtained from light transmittance data as a function of the intra-fiber content θfibers,ε determined from expansion strain data for the samples listed in Section IV of the ESI.† The dashed line corresponds to θfibers,I = θfibers,ε. | ||
The solid red line in Fig. 8(a) corresponds to the following linear fit of the relation between (I/I0)eq and θcs
| (I/I0)eq = 0.66θliq − 0.07 = 0.66θcs/0.95 − 0.07. | (5) |
| εCD,eq = 0.0347θcs. | (6) |
| θcs,fibers = εneq/0.0347. | (7) |
| θcs,pores = θcs,neq − θcs,fibers. | (8) |
| θcs,pores = 0.95[0.07 + (I/I0)neq]/0.66 − θHC,fibers | (9) |
| θcs,fibers = θcs,neq − θcs,pores. | (10) |
| Δθ = |θcs,fibers,ε − θcs,ibers,I| = |θcs,pores,I − θcs,pores,ε|. | (11) |
Consequently, the intra- and inter-fiber co-solvent content can be quantified consistently using either mechanical or optical data. Since the transmittance can be measured locally (i.e. with high position resolution), whereas the strain is an integral property of an entire sample, I/I0 provides more information.
D. Hysteresis and history dependence of paper properties
In this subsection, we would like to comment on the validity and consistency of our results, which is intrinsically limited by paper being a history-dependent material. The processes of coming into contact with liquid-phase water and subsequent drying induce irreversible changes not only in the fibrous network structure of the paper sheet but also in the internal conformation of the fibers.71 One related aspect is the breakage of inter-fiber hydrogen bonds upon water imbibition and the formation of new hydrogen bonds upon water evaporation. One manifestation thereof is the relaxation of the stress built into paper by its fabrication process upon water immersion, which accounts for the net shrinkage in the MD discussed in Section III.C. Changes of the internal fiber structure are related to e.g. hornification72–75 and microfibril coalescence.76The comparison of as-deposited samples with samples that have undergone multiple rehydration steps in Fig. 6 and 7 needs to be seen in the perspective of these phenomena. However, we believe that our method and our results are nevertheless valid and useful, because the irreversible and hysteretic strain amplitudes are typically much smaller than the expansion strain amplitudes we observe.77 An example is the persistent strain indicated in Fig. 3(c), which is approximately a factor of 10 smaller than the typical expansion strain amplitudes visible in Fig. 4 and 6.
V. Summary and conclusions
We have studied the anisotropic swelling of paper sheets after deposition of aqueous co-solvent solutions using four different experimental methods. Given the high elastic compliance of wet paper, optical grid projection has the advantage of being contactless. In contrast, in white-light interferometry, microbead displacement and sheet length monitoring, the paper is in contact with an underlying solid substrate.While the expansion in the cross machine direction (CD) was found to be on order of 2.5%, it amounts to approximately 40% in the thickness direction. Pure water gives rise to a persistent CD expansion strain of about 0.3% after drying, which rises up to 2.4% for solutions of non-volatile co-solvents that remain in the paper after the water has evaporated.
We found that the expansion strain not only depends on the quantity of co-solvent deposited, but also on the history of the sample, e.g. the initial co-solvent concentration in the solution. We ascribe this history dependence to the sensitivity of the co-solvent transport through the fiber walls to the concentration of water, which is needed to plasticize the fiber walls before molecules of larger molecular weight can pass through swiftly.
Co-solvents induce swelling primarily when they enter the intra-fiber pores, whereas they affect the optical light transmission primarily when they reside in the inter-fiber pores.6 Therefore, monitoring mechanical expansion and light transmission are two complementary methods for characterization of pore-fiber distributions of a co-solvent.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work is part of an Industrial Partnership Programme (i43-FIP) of the Foundation for Fundamental Research on Matter (FOM), which is part of the Netherlands Organisation for Scientific Research (NWO). This research programme is co-financed by Canon Production Printing, University of Twente, Eindhoven University of Technology, and the “Topconsortia voor Kennis en lnnovatie (TKI)” allowance from the Ministry of Economic Affairs. The authors thank Nicolae Tomozeiu, Herman Wijshoff and Louis Saes of Canon Production Printing for the fruitful cooperation.References
- Paper Products Physics and Technology, ed. M. Ek, G. Gellerstedt and G. Henriksson, de Gruyter, Berlin, 2009 Search PubMed.
- L. Söderhjelm and T. Sipiläinen-Malm, in Migration from Food Contact Materials, ed. L. L. Katan, Chapman & Hall, London, 1996, ch. 8 Search PubMed.
- J. Wang, M. Euring, K. Ostendorf and K. Zhang, J. Bioresour. Bioprod., 2022, 7, 1–13 CrossRef.
- Handbook of Print Media, ed. H. Kipphan, Springer, Berlin, 2001 Search PubMed.
- C. Schmid, in The Chemistry of Inkjet Inks, ed. S. Magdassi, World Scientific, Singapore, 2010, ch. 7 Search PubMed.
- M. G. Wijburg, S. Wang and A. A. Darhuber, Colloids Surf., A, 2023, 656, 130268 CrossRef CAS.
- G. Venditti, V. Murali and A. A. Darhuber, Colloids Surf., A, 2021, 634, 127832 CrossRef.
- G. Venditti, V. Murali and A. A. Darhuber, Langmuir, 2021, 37, 11726–11736 CrossRef CAS PubMed.
- J. R. Moffatt and H. P. Lauw, Formulation for Control of Paper Cockle in Thermal Inkjet Printing, US Pat., 5207824, 1992 Search PubMed.
- J. W.-P. Lin, Ink Jet Inks Comprising Anti-Curl Agents And Printing Processes, US Pat., 5997623, 1999 Search PubMed.
- G. W. Byers, R. Anderson and M. L. Choy, Cosolvents in Printing Fluids, US Pat., 2006/0065155A1, 2006 Search PubMed.
- K. M. Kosydar and R. P. Szajewski, Inkjet Printer With Inks Containing Polyoxygenated-Polyols, US Pat., 8936357B2, 2015 Search PubMed.
- W. Lu and L. A. Carlsson, Mech. Time-Depend. Mater., 2001, 5, 79–100 CrossRef CAS.
- T. Leppänen, J. Sorvari, A.-L. Erkkilä and J. Hämäläinen, Modell. Simul. Mater. Sci. Eng., 2005, 13, 841–850 CrossRef.
- P. Lipponen, T. Leppänen, J. Kouko and J. Hämäläinen, Int. J. Solids Struct., 2008, 45, 3596–3609 CrossRef.
- M.-L. Dano and J.-P. Bourque, Int. J. Solids Struct., 2009, 46, 1305–1316 CrossRef.
- D. P. Holmes, M. Roché, T. Sinha and H. A. Stone, Soft Matter, 2011, 7, 5188–5193 RSC.
- S. Douezan, M. Wyart, F. Brochard-Wyart and D. Cuvelier, Soft Matter, 2011, 7, 1506–1511 RSC.
- E. Reyssat and L. Mahadevan, Europhys. Lett., 2011, 93, 54001 CrossRef.
- A.-L. Erkkilä, T. Leppänen, J. Hämäläinen and T. Tuovinen, Int. J. Solids Struct., 2015, 62, 66–80 CrossRef.
- P. Broadbridge, G. R. Fulford, N. D. Fowkes, D. Y. C. Chan and C. Lassig, SIAM Rev., 1999, 41, 363–372 CrossRef.
- R. de Böck, A. van de Ven, L. Saes, J. Maubach and B. Koren, Int. J. Solids Struct., 2017, 128, 296–308 CrossRef.
- M. Lee, S. Kim, H.-Y. Kim and L. Mahadevan, Phys. Fluids, 2016, 28, 042101 CrossRef.
- S. S. Gorthi and P. Rastogi, Opt. Lasers Eng., 2010, 48, 133–140 CrossRef.
- A. Nissan, Trans. VIth Fund. Res. Symp., 1977, 609–629 Search PubMed.
- K. C. Yeh, J. M. Considine and J. C. Suhling, Proc. Tappi Int. Pap. Phys. Conf., 1991, 695–711 Search PubMed.
- S. Zauscher, D. F. Caulfield and A. H. Nissan, Tappi J., 1996, 79, 178–182 CAS.
- S. Zauscher, D. F. Caulfield and A. H. Nissan, Tappi J., 1997, 80, 214–223 CAS.
- I. H. Sahputra, A. Alexiadis and M. J. Adams, J. Polymer Sci. B: Polymer Phys., 2019, 57, 454–464 CrossRef CAS.
- B. Lin, J. Auernhammer, J.-L. Schäfer, T. Meckel, R. Stark, M. Biesalski and B.-X. Xu, Cellulose, 2022, 29, 1129–1148 CrossRef CAS.
- T. Vitale, MRS Proc., 1992, 267, 397–427 CrossRef CAS.
- D. Taylor and J. Craver, Trans. IIIrd Fund. Res. Symp., 1965, 852–872 Search PubMed.
- A. Jones, Tappi J., 1968, 51, 203–209 Search PubMed.
- G. Baum, C. Habeger and E. Fleischman, Trans. VIIth Fund. Res. Symp., 1981, 453–478 Search PubMed.
- K. Schulgasser, Fibre Sci. Technol., 1981, 15, 257–270 CrossRef.
- J. Lipponen, J. Pakarinen, J. Jääskeläinen and J. Grön, Paper & Timber, 2005, 87, 170–175 Search PubMed.
- T. Yokoyama and K. Nakai, Proc. SEM Ann. Conf. Exp. Appl. Mech., 2007, 3 CAS.
- E. Bosco, M. V. Bastawrous, R. H. Peerlings, J. P. Hoefnagels and M. G. Geers, Philos. Mag., 2015, 95, 3385–3401 CrossRef CAS.
- C. G. van der Sman, E. Bosco and R. H. Peerlings, Nord. Pulp Pap. Res. J., 2016, 31, 676–683 CrossRef CAS.
- Q. S. Xia, M. C. Boyce and D. M. Parks, Int. J. Solids Struct., 2002, 39, 4053–4071 CrossRef.
- P. Mäkelä and S. Östlund, Int. J. Solids Struct., 2003, 40, 5599–5620 CrossRef.
- T. Uesaka, J. Mat. Sci., 1994, 29, 2373–2377 CrossRef.
- C. Fellers, in Paper Products Physics and Technology, ed. M. Ek, G. Gellerstedt and G. Henriksson, de Gruyter, Berlin, 2009, ch. 4 Search PubMed.
- L. Salmén, C. Fellers and M. Htun, Trans. VIIIth Fund. Res. Symp., 1985, 511–527 Search PubMed.
- P. A. Larsson, M. Hoc and L. Wågberg, Nord. Pulp Pap. Res. J., 2009, 24, 371–380 CrossRef.
- A. Figueiredo, S. Magina, D. Evtuguin, E. Cardoso, F. João and P. Cruz, BioResources, 2016, 11, 2020–2029 CrossRef CAS.
- B. Philipp, H. Schleicher and W. Wagenknecht, J. Polym. Sci., Polym. Symp., 1973, 42, 1531–1543 CrossRef.
- S. Timoshenko and J. N. Goodier, Theory of Elasticity, McGraw-Hill, 1951 Search PubMed.
- I. Shepherd and H. Xiao, Colloids Surf., A, 1999, 157, 235–244 CrossRef CAS.
- T. Uesaka, I. Kodaka, S. Okushima and R. Fukuchi, Rheol. Acta, 1989, 28, 238–245 CrossRef CAS.
- I. Pulkkinen, J. Fiskari and V. Alopaeus, BioRes., 2009, 4, 126–141 CAS.
- R. Kern and P. Müller, Surf. Sci., 1992, 264, 467–494 CrossRef CAS.
- J. Olives, SIAM J. Appl. Math., 1996, 56, 480–493 CrossRef.
- V. Nair, I. Sharma and V. Shankar, Phys. Fluids, 2018, 30, 082114 CrossRef.
- T. Liu, Z. Liu, A. Jagota and C.-Y. Hui, J. Mech. Phys. Solids, 2020, 138, 103902 CrossRef.
- R. A. Samy, N. S. Satpathi and A. K. Sen, Europhys. Lett., 2020, 132, 44001 CrossRef CAS.
- W. Sun, A. Perez-Torres and F. Yang, Eur. Phys. J. E, 2020, 43, 49 CrossRef CAS PubMed.
- S. Jagannadh, H. Jordan and R. S. Brodkey, Chem. Eng. Sci., 1991, 46, 2997–3005 CrossRef CAS.
- B. Norman, in Paper Chemistry and Technology, ed. M. Ek, G. Gellerstedt and G. Henriksson, de Gruyter, Berlin, 2009, ch. 11 Search PubMed.
- B. Norman, in Paper Chemistry and Technology, ed. M. Ek, G. Gellerstedt and G. Henriksson, de Gruyter, Berlin, 2009, ch. 10 Search PubMed.
- K. T. Hu, M. Li and B. Hu, Adv. Mater. Processes Technol., 2012, 849–852 Search PubMed.
- J. S. Mackie and P. Meares, Proc. R. Soc. A: Math. Phys. Eng. Sci., 1955, 232, 498–509 CAS.
- H. Yasuda, L. D. Ikenberry and C. E. Lamaze, Makromol. Chem., 1969, 125, 108–118 CrossRef CAS.
- D. R. Paul, M. Garcin and W. E. Garmon, J. Appl. Polym. Sci., 1976, 20, 609–625 CrossRef CAS.
- J. E. Stone and A. M. Scallan, Cell. Chem. Technol., 1968, 2, 343–358 CAS.
- J. E. Stone, A. M. Scallan, E. Donefer and E. Ahlgren, Cellulases and Their Applications, ACS, 1969, ch. 13, pp. 219–241 Search PubMed.
- R. P. Neuman and L. P. Walker, Biotechnol. Bioeng., 1992, 40, 218–225 CrossRef CAS PubMed.
- J. K. Lin, M. R. Ladisch, J. A. Patterson and C. H. Noller, Biotechnol. Bioeng., 1987, 29, 976–981 CrossRef CAS PubMed.
- R. N. Ibbett, S. Kaenthong, D. A. S. Phillips and M. A. Wilding, J. Mat. Sci., 2007, 42, 6809–6818 CrossRef CAS.
- Physical properties of glycols, product data sheet, Dow Chemical Company Search PubMed.
- I. Déléris and J. Wallecan, Adv. Colloid Interface Sci., 2017, 246, 1–12 CrossRef PubMed.
- A. Scallan and G. Laivins, Trans. Xth Fund. Res. Symp., 1993, 1235–1260 Search PubMed.
- U. Weise, T. Maloney and H. Paulapuro, Cellulose, 1996, 3, 189–202 CrossRef.
- J. Diniz, H. Gil and J. Castro, Wood Sci. Technol., 2004, 37, 489–494 CrossRef.
- W. Mo, K. Chen, X. Yang, F. Kong, J. Liu and B. Li, Carbohydr. Polym., 2022, 289, 119434 CrossRef CAS PubMed.
- R. Pönni, T. Vuorinen and E. Kontturi, BioRes., 2012, 7, 6077–6108 Search PubMed.
- P. A. Larsson and L. Wågberg, Cellulose, 2008, 15, 515–525 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sm01388f |
| This journal is © The Royal Society of Chemistry 2023 |