
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicrophase separation assisted reduction in the percolation threshold of MWCNT/block polymer composites†
Gokhan
Topcu
 *a,
David
Reinoso Arenas
b,
Tony
McNally
b and
C. Remzi
Becer
a
*a,
David
Reinoso Arenas
b,
Tony
McNally
b and
C. Remzi
Becer
a
aDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: Gokhan.Topcu@warwick.ac.uk; gt162@leicester.ac.uk
bInternational Institute for Nanocomposites Manufacturing (IINM), WMG, University of Warwick, Coventry, CV4 7AL, UK
First published on 7th January 2023
Abstract
Block copolymers continue to attract a great deal of interest since they allow the formation of microphase-separated domains, useful for nanopatterning/templating. Herein, we present the drastic effect of microphase separation of a diblock copolymer on the electrical properties of polymer nanocomposites. Microphase-separated poly(styrene-b-2-ethylhexyl acrylate) (P(St-b-EHA)) block copolymers having different block lengths were synthesized and utilized as templates for multi-walled carbon nanotubes (MWCNTs). The percolation threshold of the films decreased from 0.46 to 0.19 vol% with decreasing styrene phase fraction. More importantly, we observed a non-linear and unique reduction in percolation threshold with transforming the phase into lamellar structures.
Introduction
The incorporation of carbon nanotubes (CNTs) in polymeric materials to obtain enhanced electrical properties has been of significant interest from both the fundamental and engineering points of view for the last couple of decades. The improvement in electrical performance of the composites makes them potential candidates for various applications in the area of electronics, such as dielectrics for charge-storage capacitors,1 antistatic materials,2 electromagnetic shielding,3 piezoresistive sensors,4 and shape-memory polymer (SMP) materials.5,6 Nanocomposites of polymers and CNTs exhibit a sharp increase in their electrical conductivity with increasing nanofiller content above a percolation threshold that is described as a critical value to form an interconnected touching conductive network in 3D space.Additionally, the insulator-conductor transition of polymers by the incorporation of CNTs can be achieved at a low percolation threshold depending on the degree of CNT alignment and uniform spatial distribution of individual CNTs. Nevertheless, MWCNTs tend to form agglomerates and bundles in either liquid or solid media due to high electron delocalization induced by van der Waals interactions between nanotubes. Therefore, the vast majority of the studies have focused on reducing the percolation threshold via advanced chemical and manufacturing approaches.7,8 To eliminate CNT agglomeration, surface modification has been widely studied and can be grouped into two categories: covalent bonding of functional groups,9,10 non-covalent wrapping, or adsorption of mediating molecules onto the CNT surfaces.11–13 Covalent functionalization such as with acid treatment, esterification, or amidation reactions facilitates the separation of nanotube bundles into individual tubes or smaller bundles. However, the translational symmetry of the CNTs is disrupted by changing the hybridization of carbon atoms from sp2 to sp3, which leads to an interruption in electronic and transport properties. In contrast, the non-covalent functionalization approaches typically use π–π interactions to homogenize CNTs without creating defect sites on the CNT surface. Further approaches in attempts to achieve homogeneous CNT spatial distribution, such as double percolation, have been widely reported to reduce the percolation threshold by taking advantage of selective localization of CNTs in multi-component immiscible polymer systems.14 Compared to the homogenous distribution of CNTs in a single medium, this approach leads to two respective scenarios requiring co-continuous polymer morphologies; (i) the localization of fillers directly at the interface and (ii) the selective filling of only one of the phases. Even though the former case may be a theoretical ideal state to reach the lowest possible percolation threshold, to date it has s been difficult to achieve due to the very high aspect ratio of CNTs. Therefore, the latter scenario is considered more practical to govern particle distribution. In this sense, the double percolation concept has been extensively implemented for a wide variety of polymer systems including physical blends or block copolymers which enables the formation of conductive pathways with significantly reduced CNT content within the continuous phase.15–19 To tailor composite morphology, the utilisation of melt mixing and application of shear forces has been effectively adopted.14,20,21
Block copolymers offer remarkable high-ordered architectures with readily designed chemical structures; therefore, they are good candidates for templating CNTs. Upon appropriate conditions, polymer chains spontaneously self-assemble to form nanostructures in various geometry. From this perspective, the self-organization properties enable a possible route to the elimination of additional processing methods due to fabrication being relatively easy and reproducible. The thermodynamic driving force leading to the separation of the two (or more) chemically discrete blocks is the main reason for self-assembly on the nanoscale. The geometry, size, and symmetry of the structure are highly governed by the volume fraction of blocks, overall degree of polymerization, and incompatibility of the constituent monomers (Flory–Huggins parameter, χ). Recently, several studies of CNTs with block copolymer composites were reported, which examine the dispersion of CNTs and the electrical properties of composites.22–25 Staudinger et al. reported styrene–butadiene–styrene (SBS) triblock copolymer and MWCNT composites.22 They stated that shortened MWCNTs are more easily to be localised in one phase/interface but also tend to aggregate easier. Tzounis et al. showed that the CNTs can be aligned through the phase separation of SBS by applied shear force.24 On the other hand, Santos et al. presented MWCNT/SBS composites showing enhanced conductivity with a concentration of 0.1 vol%, which indicates block copolymers may enhance the percolation threshold.25 Nevertheless, the vast majority of recent studies only rely on the composites based on SBS, which are restricted in terms of phase control due to constant block fractions. A systematic investigation of block copolymers to enhance the electrical properties of CNT composites by controlling morphology hasn’t been attempted. Therefore, to further examine this electrical improvement, control over the microphase of block copolymers by using a precise chemical toolbox (i.e. controlled living radical polymerisation) is crucial which leads to the design of new types of polymers having better performance.
In this study, we enable a decrease in percolation threshold and a slight increase in electrical conductivity by controlling the microphase of diblock copolymers from one to another. As a template, P(St-b-EHA) diblock copolymers were synthesized in various block fractions to control MWCNT percolation. By tailoring the volume fraction and dimensions of the continuous phase, we examine the change in morphology from spherical to lamellar and AC conductivity of the nanocomposite. Our work suggests a generalizable strategy to improve the electrical properties of composites of block copolymers and CNTs having microphase separation and selective localization, thus, it may guide the way for the effective use of CNTs at optimum levels.
Experimental
Materials
Styrene (97%, Acros Organics) and 2-ethylhexyl acrylate (98%, Sigma-Aldrich) were purchased and purified by passing through basic aluminium oxide before usage as a monomer. BlocBuilder MA (Nitroxide initiator, Arkema) was supplied and used as received. All solvents including toluene, methanol, acetone, and chloroform used were in analytical grade. The MWCNTs (>98% carbon basis, Sigma-Aldrich, O.D. × L 6–13 nm × 2.5–20 μm) were used as a dispersion in chloroform. Before use, the conductive nanotubes were vigorously dispersed in an ultrasonic bath for 4 h.Synthesis of polystyrene macroinitiator
Polystyrene (PSt) macroinitiator (MI) was synthesized using NMP in an oil bath. Monomer (10 mmol), initiator (0.2 mmol), and toluene (2 ml) were mixed and loaded into a glass reactor. The mixture was degassed under nitrogen for 30 min. The reaction was maintained at 110 °C for various time scales for kinetic studies in different batches, and each was stopped at different time intervals. To use as MI, the reaction was stopped after 150 min to have more active radical end-groups that correspond to approximately 50% conversion according to kinetic studies. The reaction mixture was precipitated in cold methanol, dried under vacuum at room temperature for 8 h, and stored in a freezer.Synthesis of P(St-b-EHA) copolymers
The molar concentration of EHA is varied between 25 to 200 equivalent to MI. The monomer from (0.13 ml to 1 ml), MI (200 mg), and toluene (2 ml) were mixed and degassed under nitrogen for 30 min. Subsequently, the reaction was conducted at 110 °C in an oil bath for 4 h. All reaction mixtures were cooled down to end the reaction. The polymers were precipitated in excess cold methanol, filtered, and dried at 40 °C under a vacuum overnight.Preparation of composite films
All synthesized block copolymers (BCPs) were prepared as a solution (in chloroform, 20%) to fabricate as a film by using a spin coater. The desired concentration of MWCNTs in the composites was calculated and the MWCNT dispersion was diluted with chloroform to mix with the BCPs. The mixture was sonicated for 4 h to obtain surface-modified nanotubes. The surface cleaning procedure is as follows: placing an ultrasonic bath in a detergent solution for 15 min, rinsing with deionized water, washing with acetone, and finally drying under nitrogen. An aliquot of the polymer solution was either spun cast (250 μl, 1000 rpm, 1 min) or drop-cast (1 ml) onto glass slides. Afterwards, thin films were dried in ambient conditions overnight and then annealed in a sealed chamber filled with solvent chloroform vapour for 2 days.Characterization
GPC was utilised to determine molecular weight averages and polymer dispersity. GPC measurements were performed on an Agilent 390-LC system equipped with a PL-AS RT autosampler, 2 PL gel 5 μm mixed-C columns (300 × 7.5 mm), a PL gel 5 mm guard column (50 × 7.5 mm), and a differential refractive index (DRI). The system was eluted with THF containing 2% trimethylamine (TEA) at a flow rate of 1 mL min−1 and the DRI was calibrated with linear narrow poly(methyl methacrylate) standards. The molecular structure of polymers and reaction conversion was examined by NMR (Bruker Avance III HD 300 MHz instrument). The morphology of microphases was visualised by AFM (Bruker Dimension Icon instrument) in Quantitative Nanomechanical (QNM) in Air mode using silicon tip on nitride lever. Small-angle X-ray scattering (SAXS) measurements were performed on polymer films coated on Kapton tape by using a Xenocs Xeuss 2.0 instrument equipped with a micro-focus Cu_Ka source collimated with Scatterless slits. The scattering was measured using a Pilatus 300k detector with a pixel size of 0.172 mm2.Results and discussion
To achieve polymer templates, we systematically synthesized diblock copolymers of styrene and EHA by using Nitroxide-Mediated Polymerization in two steps, the first synthesis of a styrene macroinitiator, then the respective extension of the EHA block.26,27 Regarding the synthesis of the macroinitiator, the reaction was stopped at earlier stages to ensure high chain-end fidelity. The monomer conversion was approximately 50% and based on the kinetic studies (Fig. S1, ESI†) the polymerisation was stopped at 150 minutes. Therefore, the initial monomer amount was doubled compared to kinetic studies to obtain a poly(styrene) macroinitiator consisting of 50 units. Afterwards, P(St-b-EHA) block copolymers were synthesized by using the styrene macroinitiator and altering the molar concentration of EHA from 25 to 200 equivalents to MI. Therefore, this method enables having a controlled second block length, namely controlled volume fractions while keeping the first block length the same. The detailed monomer composition, molar mass and dispersity information regarding the obtained polymers are presented in Table 1.| Sample | n MI (mmol) | n EHA (mmol) | M n,GPC (g mol−1) | Đ | f St,theo | f St,exp |
|---|---|---|---|---|---|---|
| PSt MI | — | — | 7900 | 1.17 | — | — |
| P1 | 0.025 | 0.625 | 9700 | 1.24 | 0.62 | 0.81 |
| P2 | 0.05 | 1.25 | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 400 |
1.28 | 0.46 | 0.76 |
| P3 | 0.05 | 1.875 | 10600 |
1.32 | 0.36 | 0.74 |
| P4 | 0.05 | 2.5 | 10900 |
1.34 | 0.30 | 0.72 |
| P5 | 0.05 | 3.75 | 12200 |
1.41 | 0.22 | 0.64 |
| P6 | 0.05 | 5 | 14500 |
1.43 | 0.17 | 0.53 |
A systematic investigation of molecular weight was carried out by GPC. The GPC traces show a clear and gradual shift in lower retention time with increasing EHA fraction (Fig. 1a). Similarly, the 1H NMR SCspectra show a peak at 3.91 ppm arising from EHA and gradually increasing in intensity (Fig. 1b). This increase in the EHA fraction is slightly smaller than the theoretical value since the polymerization of the second block undergoes around 40% conversion. Therefore, the experimental styrene fraction is higher than the theoretical counterpart. Besides, increasing Mw with increasing EHA, inherently, the volume fraction of styrene (fSt) in the resulting block copolymer gradually decreases to 0.53 (Fig. 1c). To obtain thin films and examine microphase separation, polymer solutions in chloroform were spin-coated, thereafter, solvent-annealed (chloroform) for 2 days in a sealed chamber.
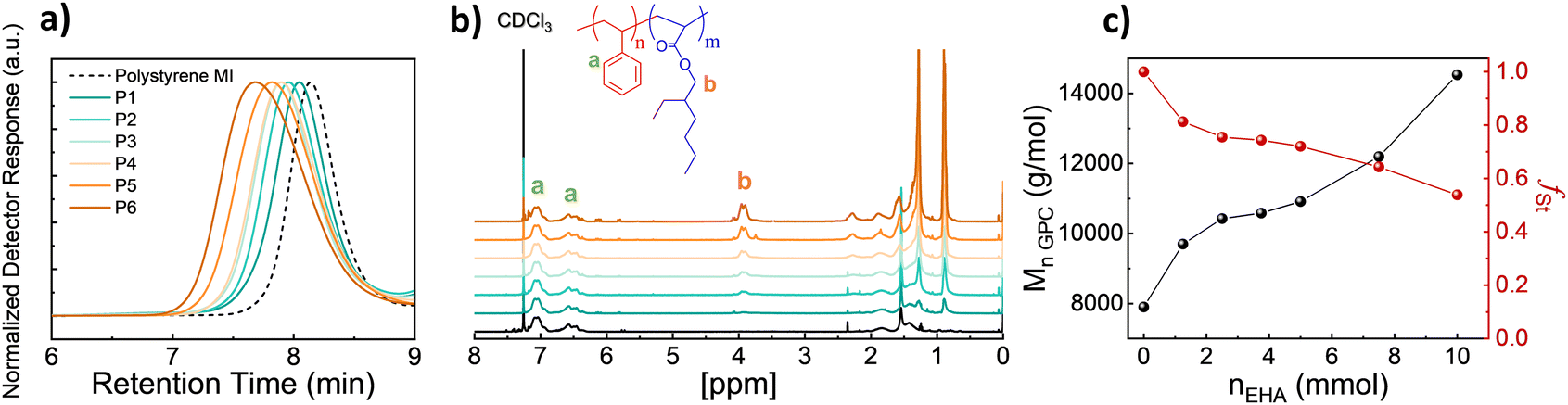 | ||
| Fig. 1 (a) GPC traces and (b) 1H NMR spectra of P(St-b-EHA) block copolymers with increasing EHA fraction. (c) Correlation between regent content (nEHA) and, Mn and experimental volume fraction of styrene (fSt), respectively. | ||
The implication of solvent vapour on polymer film provides a partial swelling that promotes the self-assembly of polymer chains.28–30 The microphase separation was examined by AFM, and the corresponding images for all block copolymers having various ratios are presented in Fig. 2a. The phase separation can be observed for the samples in different shapes according to volume fraction. The image of the block copolymers having the highest styrene volume fraction (fSt: 0.81) exhibits an amorphous and homogeneous structure without any phase separation. In the sample varying fSt between 0.75 and 0.72 fractions, the separation of two blocks can be identified ambiguously and EHA appears as small black spheres that refer to lower height and non-continuous fraction. For lower styrene fractions (fSt: 0.64 and 0.53), the phase separation distinctly appears in the lamellar shape.
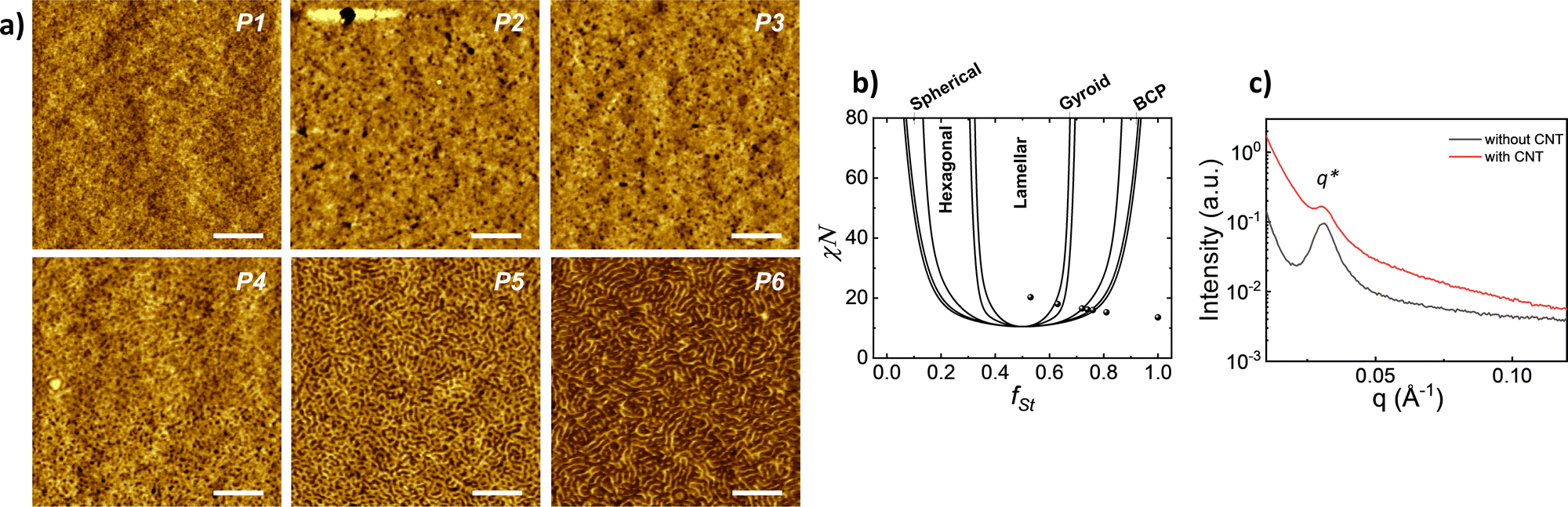 | ||
| Fig. 2 (a) AFM images of thin films of P(St-b-EHA) block copolymers having different block fraction after solvent annealing. The scale bar is 200 nm. (b) Self-Consistent Field Theory (SCFT) phase diagram of a diblock copolymer with theoretical phases labelled and corresponding points of synthesized P(St-b-EHA) block copolymers. (c) SAXS patterns of P6 film with/without CNT. | ||
According to previous experimental and theoretical studies,31,32 the equilibrium phase behaviour of diblock copolymers is governed by f and the segregation strength, χN, where N is the overall degree of polymerization. The energy of interactions between the two polymer blocks leads to microphase separation and the formation of various geometries. In order to predict the phase behaviour of block copolymers, Self-Consistent Field Theory (SCFT) is a practical and powerful tool that can help define critical transition points of χN as a function of f.33,34 At this critical transition (i.e. the order–disorder transition), χN can be given by:
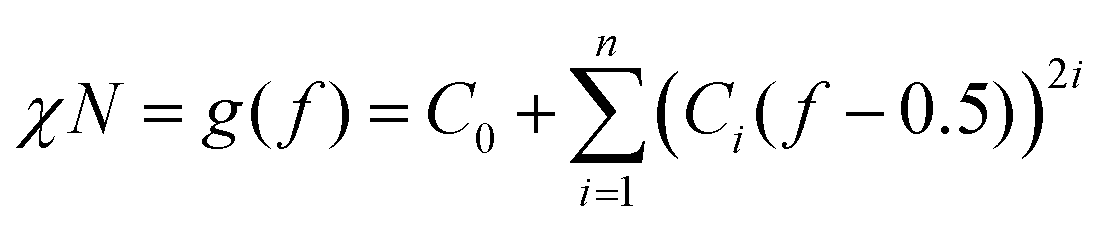 | (1) |
Both P(St-b-EHA)/MWCNT composite films were covered by conductive silver paste (1 × 1 cm2) and placed between 2 copper electrodes in the measurement cell. The AC conductivity of the composite films was recorded in the 1 Hz–1 MHz range. The conductivity of the PSt homopolymer composites with changing CNT content is presented in Fig. 3a. The conductivity increases with increasing CNT content and reaches 10−3 S m−1 at 2.5 vol%. The reason for the different behaviour is in part related to the increase in energy of free electrons with increasing frequency that can direct the electrons into conductivity for more insulating materials through the formation of an interconnected CNT network. Similarly, the conductivity of P6 composites, namely the lamellar-shaped counterparts, increases with increasing CNT content (Fig. 3b), however, this is more distinct and attains a higher (4 orders of magnitude higher) conductivity value of ≈0.13 S m−1. The relation between CNT content and conductivity for all polymer samples is presented in Fig. 3c. The plots show an increase in exponential decay as CNT content increases. This increase is attributed to the number of percolated interconnected CNT networks that have formed conductive pathways. After saturation, a further increase in CNT content is ineffective in increasing conductivity, new conductive paths are unable to form. For higher CNT contents (1.25 and 2.5% vol.), the conductivity is enhanced by 2 orders of magnitude in block copolymers having a lamellar phase. However, more importantly, very low percolation is obtained for P5 and P6, i.e., while fSt lowers.
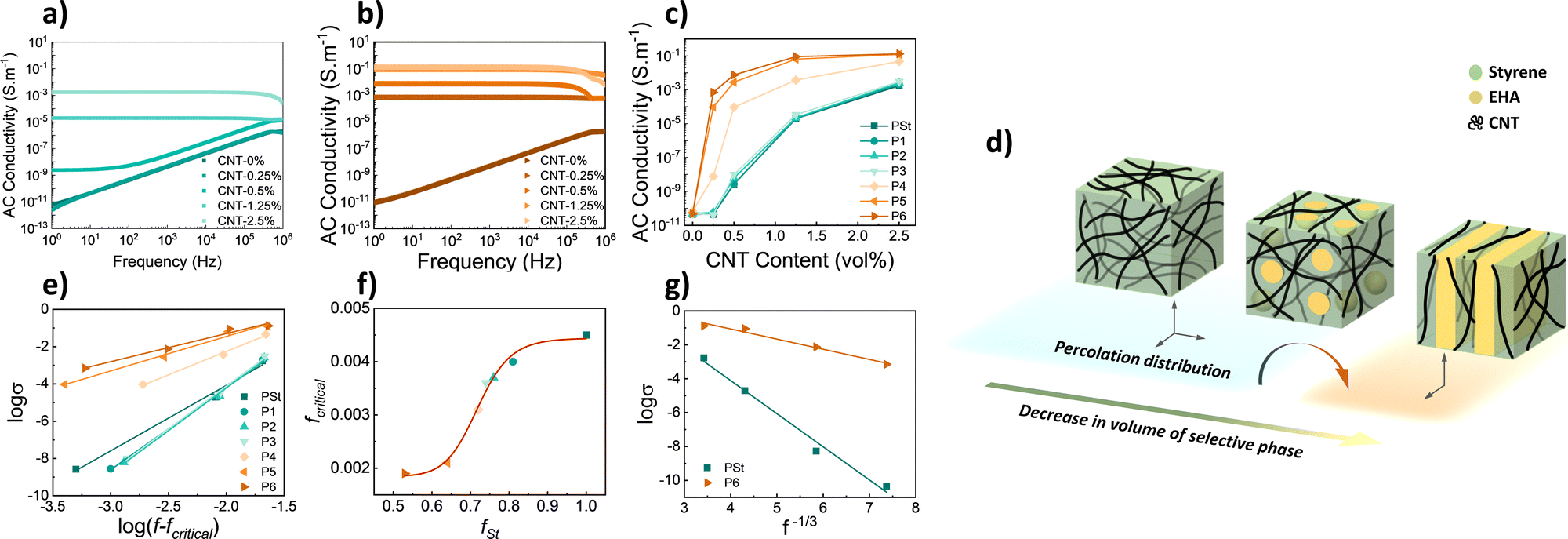 | ||
| Fig. 3 AC conductivity of (a) PS/MWCNT and (b) P(St-b-EHA)/MWCNT (P6) composite films between 1 Hz–1 MHz, (c) conductivity at 10 Hz for composites as a function of CNT content. (d) Schematic representation of possible CNT particle distribution forming percolation with changing phase-type. (e) Logarithmic plot of conductivity versus reduced CNT content, (f) relation between percolation threshold (fcritical) and fSt, and (g) relation between conductivity per unit volume styrene. | ||
It is known that the styrene block of copolymers will more readily physically bond to CNTs via π–π interaction.11,12,17,37 From that perspective, CNTs tend to localize in a continuous styrene phase, hence, a decrease in fSt provides a more dense localization of identical CNT content that in turn enables higher conductivity (Fig. 3d). To have a further understanding of the effect of the CNT localization on conductivity, various interpretations and approaches were performed. The relation between the composite conductivity (σ), CNT conductivity (σCNT), and CNT volume fraction fCNT can be given by a power law as below;
| σ = σCNT (fCNT − fcritical)t | (2) |
σ versus fCNT−1/3 was constructed (Fig. 3g), which yielded R2: 0.9942 and 0.9776, respectively. This linearity confirms the applicability of the theory for selective localization since there is a uniform dispersion of MWCNT in both cases. However, decreasing linearity as in P6 may hint that the CNTs in the composite are mildly selective to styrene and can also slightly involve the EHA phase.
Conclusions
In summary, P(St-b-EHA) copolymers were synthesized in various block length and their self-assembly characteristics were examined. The copolymers were used to obtain electrically conductive films by the inclusion of MWCNTs. By selectivity of styrene phase, MWCNTs were templated in microphase separation and their effect on electrical properties was studied. The distinct microphase separation properties of P(St-b-EHA) by changing the volume fraction of blocks were successfully visualized from disordered to ordered lamellar phase with decreasing styrene fraction. The AC conductivity measurement of P(St-b-EHA)/MWCNT composites showed that the phase separation results in higher conductivity values by using lower CNT content. Furthermore, the percolation threshold decreases with decreasing styrene fraction. This suggests selective localization of the CNTs in the continuous styrene phase, in other words, an increase of particles per unit styrene volume. Importantly and uniquely, the shape of block copolymer morphology has an additional effect on conductivity. The change in phase to a lamellar structure amplifies the conductivity of the composites as the CNTs are forced to align through-plane, i.e throughout the percolation direction. Using microphase separation as a strategy, more useful carbon-filled polymer conductors may be possible for use in microelectronics.Author contributions
G. Topcu – investigation, methodology, writing-original draft and conceptualization D. Reinoso Arenas – EIS measurements, editing manuscript T. McNally – review & editing manuscript C. R. Becer – conceptualization, writing – review and editing, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
G. Topcu acknowledges the Scientific and Technological Research Council of Turkey (TUBITAK) for supporting the Postdoctoral Research Fellowship (2219) ‘‘No. 1059B191900073’’.Notes and references
- Z. Wang, M. Zhu, Z. Pei, Q. Xue, H. Li, Y. Huang and C. Zhi, Mater. Sci. Eng., R, 2020, 139, 100520 CrossRef.
- R. Yadav, M. Tirumali, X. Wang, M. Naebe and B. Kandasubramanian, Def. Technol., 2020, 16, 107–118 CrossRef.
- S. N. Sultana, S. P. Pawar and U. Sundararaj, Ind. Eng. Chem. Res., 2019, 58, 11576–11584 CrossRef CAS.
- K. Ke, P. Pötschke, S. Gao and B. Voit, ACS Appl. Mater. Interfaces, 2017, 9, 5437–5446 CrossRef CAS PubMed.
- M. Behl, M. Y. Razzaq and A. Lendlein, Adv. Mater., 2010, 22, 3388–3410 CrossRef CAS PubMed.
- H. Meng and G. Li, Polymer, 2013, 54, 2199–2221 CrossRef CAS.
- V. D. Punetha, S. Rana, H. J. Yoo, A. Chaurasia, J. T. McLeskey Jr, M. S. Ramasamy, N. G. Sahoo and J. W. Cho, Prog. Polym. Sci., 2017, 67, 1–47 CrossRef CAS.
- S. W. Kim, T. Kim, Y. S. Kim, H. S. Choi, H. J. Lim, S. J. Yang and C. R. Park, Carbon, 2012, 50, 3–33 CrossRef CAS.
- J. Chen, L. Yan, W. Song and D. Xu, Composites, Part A, 2018, 114, 149–169 CrossRef CAS.
- S. Banerjee, T. Hemraj-Benny and S. S. Wong, Adv. Mater., 2005, 17, 17–29 CrossRef CAS.
- Y.-T. Liu, Z.-L. Zhang, W. Zhao, X.-M. Xie and X.-Y. Ye, Carbon, 2009, 47, 1883–1885 CrossRef CAS.
- Y.-T. Liu, W. Zhao, Z.-Y. Huang, Y.-F. Gao and X.-M. Xie, Carbon, 2006, 44, 1613–1616 CrossRef CAS.
- Z. Liu, S. M. Tabakman, Z. Chen and H. Dai, Nat. Protoc., 2009, 4, 1372–1381 CrossRef CAS PubMed.
- X.-d Qi, J.-h Yang, N. Zhang, T. Huang, Z.-w Zhou, I. Kühnert, P. Pötschke and Y. Wang, Prog. Polym. Sci., 2021, 123, 101471 CrossRef CAS.
- X. Zhao, J. Zhao, J.-P. Cao, D. Wang, G.-H. Hu, F. Chen and Z.-M. Dang, Mater. Des., 2014, 56, 807–815 CrossRef CAS.
- D. Wu, Y. Zhang, M. Zhang and W. Yu, Biomacromolecules, 2009, 10, 417–424 CrossRef CAS PubMed.
- F. Wode, L. Tzounis, M. Kirsten, M. Constantinou, P. Georgopanos, S. Rangou, N. E. Zafeiropoulos, A. Avgeropoulos and M. Stamm, Polymer, 2012, 53, 4438–4447 CrossRef CAS.
- N. K. Shrivastava, S. Suin, S. Maiti and B. Khatua, RSC Adv., 2014, 4, 24584–24593 RSC.
- Q. Li, J. Sun, J. Wang, G.-X. Chen, F. Li and Y. Zhang, RSC Adv., 2012, 2, 6637–6644 RSC.
- A. Göldel, G. Kasaliwal and P. Pötschke, Macromol. Rapid Commun., 2009, 30, 423–429 CrossRef PubMed.
- J.-F. Gao, D.-X. Yan, B. Yuan, H.-D. Huang and Z.-M. Li, Compos. Sci. Technol., 2010, 70, 1973–1979 CrossRef CAS.
- U. Staudinger, A. Janke, C. Steinbach, U. Reuter, M. Ganß and O. Voigt, Polymers, 2022, 14, 2715 CrossRef CAS PubMed.
- U. Staudinger, B. K. Satapathy and D. Jehnichen, Polymers, 2019, 11, 1831 CrossRef CAS PubMed.
- L. Tzounis, S. Pegel, N. E. Zafeiropoulos, A. Avgeropoulos, A. S. Paipetis and M. Stamm, Polymer, 2017, 131, 1–9 CrossRef CAS.
- J. P. Ferreira Santos, G. H. Franca Melo, A. M. Gonçalves, J. A. Eiras and R. E. Suman Bretas, J. Appl. Polym. Sci., 2018, 135, 46650 CrossRef.
- C. R. Becer, R. M. Paulus, R. Hoogenboom and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6202–6213 CrossRef CAS.
- C. R. Becer, C. Haensch, S. Hoeppener and U. S. Schubert, Small, 2007, 3, 220–225 CrossRef CAS PubMed.
- S. H. Kim, M. J. Misner, T. Xu, M. Kimura and T. P. Russell, Adv. Mater., 2004, 16, 226–231 CrossRef CAS.
- C. Jin, B. C. Olsen, E. J. Luber and J. M. Buriak, Chem. Mater., 2017, 29, 176–188 CrossRef CAS.
- G. Topcu, D. R. Arenas, S. Huband, T. McNally and C. R. Becer, J. Mater. Chem. C, 2022, 10, 9356–9363 RSC.
- F. S. Bates and G. H. Fredrickson, Annu. Rev. Phys. Chem., 1990, 41, 525–557 CrossRef CAS PubMed.
- E. W. Cochran, C. J. Garcia-Cervera and G. H. Fredrickson, Macromolecules, 2006, 39, 2449–2451 CrossRef CAS.
- M. W. Matsen and M. Schick, Phys. Rev. Lett., 1994, 72, 2660 CrossRef CAS PubMed.
- W. S. Loo, G. K. Sethi, A. A. Teran, M. D. Galluzzo, J. A. Maslyn, H. J. Oh, K. I. Mongcopa and N. P. Balsara, Macromolecules, 2019, 52, 5590–5601 CrossRef CAS.
- L. Leibler, Macromolecules, 1980, 13, 1602–1617 CrossRef CAS.
- W. Zha, C. D. Han, D. H. Lee, S. H. Han, J. K. Kim, J. H. Kang and C. Park, Macromolecules, 2007, 40, 2109–2119 CrossRef CAS.
- M. M. Arras, B. He and K. D. Jandt, Polymer, 2017, 127, 15–27 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sm01277d |
| This journal is © The Royal Society of Chemistry 2023 |