
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On the understanding of bio-oil formation from the hydrothermal liquefaction of organosolv lignin isolated from softwood and hardwood sawdust†
Petter
Paulsen Thoresen
a,
Jonas
Fahrni
b,
Heiko
Lange
acd,
Jasmine
Hertzog
e,
Vincent
Carré
e,
Ming
Zhou
 f,
Anna
Trubetskaya
g,
Frédéric
Aubriet
e,
Jonas
Hedlund
f,
Tomas
Gustafsson
b,
Ulrika
Rova
a,
Paul
Christakopoulos
a and
Leonidas
Matsakas
*a
f,
Anna
Trubetskaya
g,
Frédéric
Aubriet
e,
Jonas
Hedlund
f,
Tomas
Gustafsson
b,
Ulrika
Rova
a,
Paul
Christakopoulos
a and
Leonidas
Matsakas
*a
aBiochemical Process Engineering, Division of Chemical Engineering, Department of Civil, Environmental and Natural Resources Engineering, Luleå University of Technology, 971-87, Sweden. E-mail: leonidas.matsakas@ltu.se; Tel: +46 (0) 920 493043
bRISE Processum AB, Department Biorefinery and Energy, Division of Bioeconomy and Health, Research Institute of Sweden, 981 22 Örnsköldsvik, Sweden
cDepartment of Earth and Environmental Sciences, University of Milano-Bicocca, Piazza della Scienza 1, 20126 Milan, Italy
dNBFC – National Biodiversity Future Center, 90133 Palermo, Italy
eUniversité de Lorraine, LCP-A2MC, 57000 Metz, France
fChemical Technology, Luleå University of Technology, 971 87 Luleå, Sweden
gDepartment of Bioscieces, Nord University, 7713 Steinkjer, Norway
First published on 4th October 2023
Abstract
Conversion of organosolv lignins isolated with and without an inorganic acid catalyst (H2SO4) from hard- and softwood (birch and spruce) into bio-oil through hydrothermal liquefaction has been investigated. Furthermore, fractions of the isolated bio-oils were catalytically deoxygenated to improve the bio-oil properties. As elucidated through NMR, both biomass source and extraction mode influence the bio-oil product distribution. Depending on whether the lignins carry a high content of native structures, or are depolymerized and subsequently condensed in the presence of sugar dehydration products, will dictate heavy oil (HO) and light oil (LO) distribution, and skew the HO product composition, which again will influence the requirements upon catalytical deoxygenation.
Introduction
Several issues are up for discussion when considering the persistent use and recovery of fossil fuels and petroleum. First, our economy and modern society are essentially founded on the idea that these resources are readily available.1 The abundance of non-renewable carbon is not completely known, its accessibility uncertain, so exactly when we will experience ‘peak oil’ and for how long these reserves will last, is questionable.2 The second discussion concerning the impact of fossil carbon on the climate is, to a larger extent, experiencing consensus,3 and the possible detrimental scenarios induced by extreme weather conditions include shortages of food, fresh drinking water, and outbreaks of diseases.4,5 Thus, even though the reserves of petroleum might, and likely will, sustain for several decades to come, the persistent usage of these will eventually be forced to a halt, and the global consequences upon on their depletion will be largely damaging for life as we know it today.A potential shift from traditional petroleum-based chemicals toward renewable ones is receiving increasing attention because of the aforementioned concerns.6 Especially the lignin fraction of lignocellulosic biomass is nowadays considered largely unexploited relative to its potential as the largest renewable source of aromatics.7 However, the main issue regarding achieving proper utilization of this vastly abundant component, moving toward integrating it as an industrial, aromatic building-block and mitigate toward lower global greenhouse gas emission, is to be established.8
To aid in overcoming this hurdle, the present work evaluates both an effective way of extracting this underutilized polymer, and its subsequent conversion into low molecular weight components in the form of bio-oil. This final bio-oil carries the potential of replacing important chemicals for the industry. For the initial lignin extraction process, organosolv fractionation was applied. This technology has over the recent years been well explored, with several recent papers reviewing the current state, and issues to be solved.9,10 Meanwhile, the key idea behind the process is to apply an extraction solution often consisting of water and organic solvents such as alcohols, organic acids or ketones,11 sometimes with the addition of a catalyst,12 which often has a profound effect on the chemistry of the obtained extract.13 Another important factor determining the chemistry of the obtained lignin isolate, is the type of raw material (hardwood, softwood or herbaceous raw material).14 While this extraction yields a product, which can be isolated to give lignin of high purity, the isolate will still be highly dispersed when considering its inherent composition and chemical structures. Thus, a subsequent treatment would be necessary to retrieve aromatic monomers with direct applications in the chemical industry. Hydrothermal liquefaction (HTL) is a way to achieve this.15 Application of this method for valorizing lignins into compounds of lower molecular weight is highly complex, both as a result of the initial lignin chemistry, but also because of the somewhat chaotic character of subcritical water where depolymerisation, decomposition, and recombination occur at the same time.16 For these reasons, the present work sets out to provide insight into how the initial lignin chemistry affects the distribution of the obtained bio-oil phases (volatiles (V), LO, HO, char (C)) but also the release of specific components seemingly originating from certain lignin motifs, or if specific motifs favors the formation of any product phase over the other. Finally, and for bringing the generated bio-oil fractions into suitable fuel components, catalytical upgrading will be performed. A recent work summarized the current state of catalytic deoxygenation of bio-mass derived fuel-precursors,17 and whereas the specific catalyst in combination with the precursors and its deoxygenation route varies, it essentially depends in the stability of the oxygen carrying group. This in general renders furan compounds harder to deoxygenate than phenols due to the integral role of the oxygen in the aromatic system.
Zeolites are the most prospective candidates for the upgrading of bio-oil attribute to their high hydrothermal stability, adjustable acidity, controllable pore structure, and shape selectivity. Especially, it has been noticed that ZSM-5 type zeolites have the perfect acidity and pore size for cracking and aromatization reactions, and improved accessibility to the acid sites can significantly increase the yielding of aromatics.18 However, catalyst suffers from exceedingly fast deactivation by coking.19 Some strategies have been proved to extend the life time of catalyst: (1) by the introduction of additional mesopores or reducing the crystal size, the diffusion path in zeolite can be reduced;20–22 (2) grow catalyst in fluoride medium to minimize the framework defects;23 (3) disperse zeolite by adding diluent for improve the heat transportation;24 and (4) co-feeding some molecules to increase the combined H/C ratio of the bio-oil.25
This study sets out to elucidate the factors influencing the properties of the end-fuel, starting from organosolv lignin extraction from both hard- and softwood raw materials. The primary lignins are characterized in detail (13C NMR and 1H–13C HSQC spectroscopy) and initially upgraded by hydrothermal liquefaction, with the characteristics of the obtained bio-oil fractions described (GC-MS and Fourier-transform ion cyclotron resonance mass spectrometry), before final catalytic upgrading is performed employing defect-free ultra-thin (35 nm) ZSM-5 catalyst. The catalyst was synthesized in fluoride medium and dispersed in 500 nm Stöber sol SiO2, as these can significantly increase the diffusion of hydrocarbons and the transportation of heat generated by the exothermic reaction, respectively. In addition, the bio-oil upon conversion compositionally renders them closer to petroleum. As such, the current work provides a novel start-to-end investigation of lignocellulosic valorization with in-depth characterization of the various intermediate stages evaluated in light of the native material structure.
Materials and methods
Lignin isolation
Birch (B) and spruce (S) lignins were obtained through organosolv (conditions given in Table 1) as previously described.26 In short, spruce and birch sawdust were treated at a solid-to-liquid ratio of 1-to-10 (dry weight biomass/volume solvent) in the presence or without acid catalyst (1% w/wbiomass). The solvent was a mixture of ethanol in water at a content of 60% v/v and 50% v/v for birch26 and spruce sawdust, respectively. Subsequent the organosolv treatment, the slurry was vacuum-filtered to separate the solids from the liquid fraction. Lignin solubility was then reduced by removing/evaporating the ethanol by rotary evaporation allowing its isolation as precipitate. Subsequently, the precipitate was isolated by centrifugation (14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm/29 416 g, at 4 °C for 15 min) and then freeze-dried to yield the final powdery product.
000 rpm/29 416 g, at 4 °C for 15 min) and then freeze-dried to yield the final powdery product.
| Code | Raw material | Temperature [°C] | Time [min] | EtOH [vol%] | Catalyst [H2SO4% w/wbiomass] |
|---|---|---|---|---|---|
| B | Birch sawdust | 200 | 15 | 60 | 0.0 |
| BA | Birch sawdust | 200 | 15 | 60 | 1.0 |
| S | Spruce sawdust | 200 | 30 | 50 | 0.0 |
| SA | Spruce sawdust | 200 | 30 | 50 | 1.0 |
Hydrothermal liquefaction (HTL)
An illustration of the set-up applied for the HTL is presented in Fig. 1. In short, it is a combination of three connected vessels. For trials performed in a semi-continuous fashion, the first reactor (semi-continuous feeding reactor in Fig. 1) is used as a feed tank where the starting feed is being stirred at room temperature. Next, when the feed liquid is pressurized, in this work by nitrogen at 55 bar, the feed is pushed into the pre-heated reactor (hydrothermal reactor in Fig. 1) by opening the valve for 0.5 seconds. All the valves in the hydrothermal system are pneumatic with on-off sensors to ensure safe operations. Initially, the pre-heated reactor contains 35 mL distilled water to ensure proper controller and thermometer function. When the desired treatment time is reached, the mixture is shot into a water-cooled cooling vessel (water-cooled vessel in Fig. 1) using the remaining steam and nitrogen pressure in the reactor. Heating from room temperature to 320 °C is achieved within 20 min, while cooling back to room temperature is done within 5 min. In the semi-continuous system, initial trials used a 10% suspension of lignin in water (B, BA, S, SA).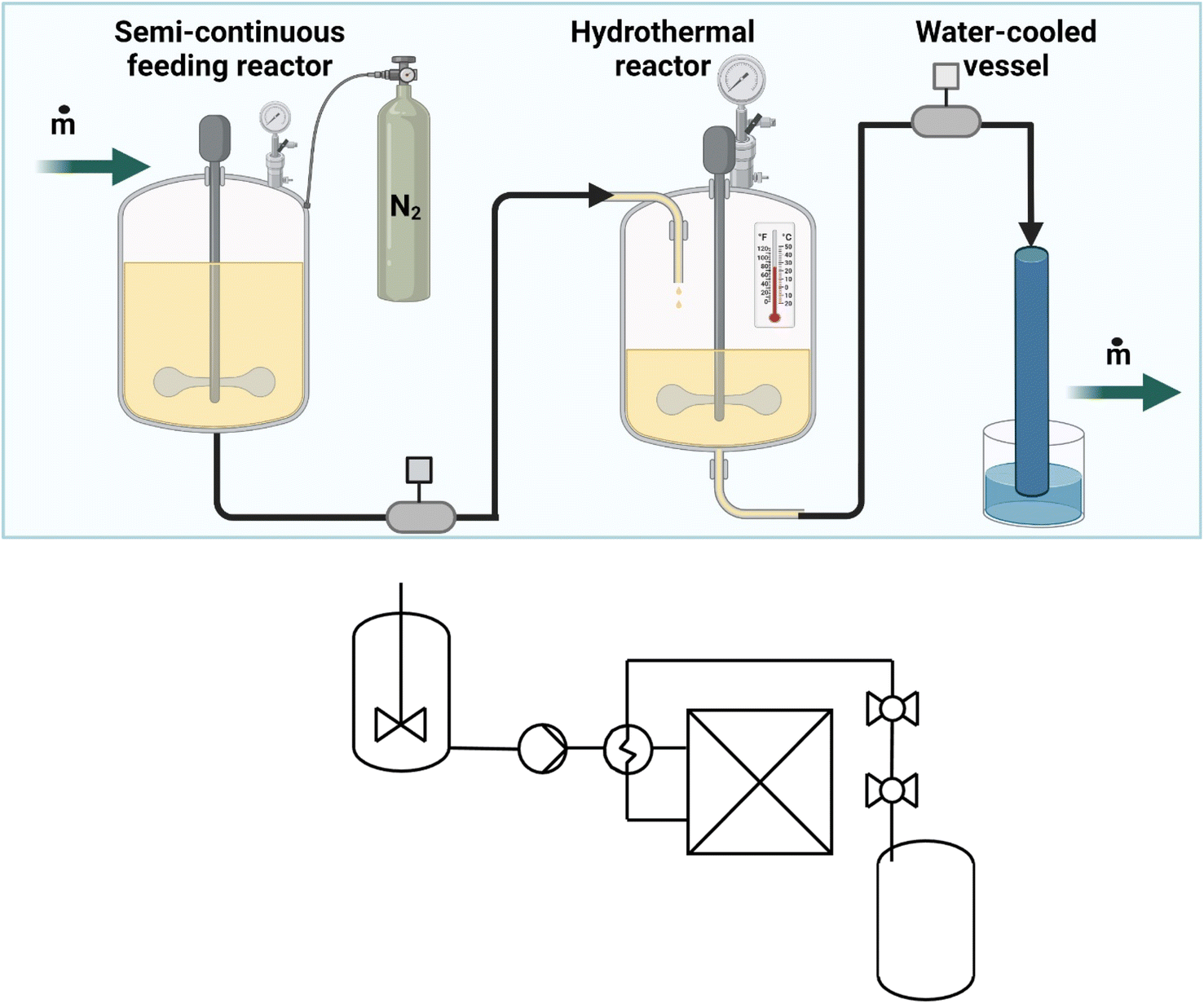 | ||
| Fig. 1 Top: The experimental set-up used for HTL of lignin with ball valves separating the pressurized zones. Bottom: PI&D of the continuous HTL including feeding tank, pump, heat exchanger, reactor and pressure release valves. | ||
The continuous reactor consists of a 5 L tank where the feed is stirred and pre-heated to 70 °C. Then with a dual-pumping system, the feed is fed into the reactor at 200 bar. Part of the feed stream is heated through a heat-exchanger system exchanging heat with the product stream, the remaining energy is obtained electrically until the desired temperature is obtained (PID controlled). At the end of the reaction time, the product is cooled down to 70 °C before being released through an alternating ball-valve system ensuring a pulse-flow operation. Overall reactor volume is 800 mL, and a flow-rate of 40 mL min−1 was applied. The residence time for the continuous system was 20 min. In the continuous system a 8.5% suspension of lignin in water was used.
The aqueous suspension in the crude reaction mixture was filtered over a 20 μm filter paper (Whatman 541). The filtrate was extracted twice with 50% of its volume of ethyl acetate to isolate the monomer rich light oil (LO). The remaining water phase was discarded. The HO and char were separated by washing the filter cake with 2-methyl-tetrahydrofuran (2-Me-THF). By removing the solvent, the HO is isolated. This fraction is the largest portion (see Fig. 3B) and is assumed to be comprised of depolymerized and deoxygenated species together with solvent-soluble unreacted lignin. An overview of the processing is presented in Fig. 2.
 | ||
| Fig. 2 An overall illustration of the workflow related to the HTL of lignin. | ||
Quantitative 13C nuclear magnetic resonance spectroscopy of the organosolv lignin
The lignin NMR analysis was performed as described previously.27,28 In short, for quantitative 13C NMR analysis, approx. 80 mg of lignin was dissolved in 500 μL DMSO-d6. We applied 50 μL (∼1.5 mg mL−1) of Cr(III) acetylacetonate in DMSO-d6 as a spin-relaxation agent and 50 μL (∼15 mg mL−1) of trioxane (92.92 ppm) in DMSO-d6 as an internal standard. Spectra were recorded on a Bruker 600 MHz AVANCE III spectrometer equipped with a 5 mm BBO broadband (1H/19F/2D) z-gradient cryo-probe and controlled with TopSpin 3.6.4 software at 30 °C, with a total of 20000–24000 scans. An inverse-gated proton decoupling pulse sequence was applied with a 90° pulse width, acquisition time of 1.2 s, and relaxation delay of 1.7 s. Data were processed with MestreNova Version 9.0.1 (Mestrelab Research S.L., Santiago de Campostela, Spain).
1H–13C heteronuclear single quantum coherence (HSQC) analysis of the organosolv lignins
The same sample prepared for the acquisition of the quantitative 13C NMR was used. HSQC spectra were obtained at 30 °C on a Bruker 600 MHz Avance III spectrometer (Bruker Biospin) controlled with Topspin 3.6.4 and equipped with a 5 mm BBO broadband (1H/19F/2D) z-gradient cryo-probe. The Bruker hsqcetgpsisp2.2 pulse program in DQD acquisition mode was applied, with NS = 64; TD = 2048 (F2) and 512 (F1); SQ = 12.9869 ppm (F2) and 164.9996 ppm (F1); O2 (F2) = 2601.36 Hz and O1 (F1) = 7799.05 Hz; D1 = 2 s; CNST2 1J(C–H) = 145; and acquisition time F2 channel = 197.0176 ms and F1 channel = 15.4164 ms. NMR data were processed with MestreNova.GC/MS-analysis of light oil
The LO were analysed by GC-MS to determine the amounts of monomers. Therefore, the samples were dissolved in acetone. Guaiacol, 4-methylguaiacol (creosol), 3-methoxycatechol and 2,6-dimethoxyphenol (syringol) were used as standards for quantification.Fourier-transform ion cyclotron resonance mass spectrometry (FT-ICR MS) of the heavy oil (HO)
The four heavy oils from liquefaction of birch or spruce lignin were prepared to a final concentration of 0.1 mg mL−1 in 90:10 (v/v) methanol/toluene (LC/MS grade).
Bio-oil mass spectra were acquired in positive-ion mode using atmospheric pressure photoionization (APPI) with a 7 T FT-ICR mass spectrometer (Solarix 2XR, Bruker Daltonics, Bremen, Germany). Ion source and instrument parameters were optimized via software FTMS-Control V2.3.0 (Bruker Daltonics). A 0.1 mg mL−1 sodium trifluoroacetate solution was used to externally calibrate the mass spectrometer, and to shim and gate the ICR detection cell. Mass spectra were acquired over a 193.5–2000 m/z range with a 4 megaword time-domain, and 300 scans were accumulated. Bio-oil samples were infused with a flow rate of 12 μL min−1. The APPI source conditions were set with drying gas temperature and flow rate at 200 °C and 2 L min−1, respectively, and the pressure of the nebulizer gas was 1.5 bar. The capillary voltage was 0.8 kV and the vaporizer was heated at 300 °C.
Software DataAnalysis 5.2 (Bruker, Daltonics) were used for data processing. Mass spectra were exported to peak list at a signal-to-noise ≥4. Internal calibration and peak list assignment were performed by Composer software (Sierra Analytics, Modesto, USA). Molecular formulae were assigned within a ±0.6 ppm mass error range, with the C, H, O, N, and S elements, and both radical cations and protonated ions were considered. This resulted to CH, CHO, CHN, CHON, and CHOS molecular series. The double bound equivalent (DBE) was calculated, as given by eqn (1), with nC, nH and nN being number of carbon, hydrogen, and nitrogen atoms, respectively. The DBE versus carbon number (#C) graphs give information on the aromaticity and unsaturation degree of a molecule.
 | (1) |
Catalytic upgrading of the light oil fraction
The zeolite ZSM-5 catalyst used here comprised 35 nm thick defect free crystals synthesized in fluoride medium as described previously.29 These defect free crystals are by far the thinnest reported, and it has been shown that these crystals displayed superior catalytic performance in terms of e.g. resistance to deactivation as compared with thicker ZSM-5 crystals and defective ZSM-5 crystals.29 In the present work, these crystals were supported on spherical Stöber SiO2 particles with a diameter of 500 nm in a ratio of 1:10. The mixture was pressed, crushed, and sieved to obtain a powder within the size range of 177 to 500 μm. Prior to use, the supported catalyst was dried at 110 °C for about 18 h.
For catalytic conversion of bio-oil, 1.7 g of the supported catalyst was loaded in a stainless-steel reactor with an inner diameter of 6 mm and a length of 140 mm. Quartz glass wool was used to keep the catalyst in the center of the reactor and graphite rings were used to seal the reactor. The bio-oil was mixed with methanol and water in a weight ratio of 60% bio-oil, 20% methanol, and 20% water or a ratio of 10% bio-oil, 30% methanol, and 60% water. A gas chromatograph (GC, Agilent 7890 B equipped with a capillary column CP-Sil PONA CB, 100 m × 250 μm × 0.5 μm and an FID detector) was connected online to the reactor.
The mixtures of bio-oil, methanol and water were fed at a weight hourly space velocity (WHSV) of 0.3 h−1 to the reactor at 360 °C, while the connection lines between the reactor and the GC were maintained at 220 °C. A stream of N2 at a flow rate of 9.5 mL min−1 was used as carrier gas. The reactor effluent was sampled every 70 minutes by the GC. The observed GC-peaks could be classified into the following groups of products according to their retention times: deoxygenated C5-products, e.g., light alkenes and alkanes (all peaks except for methanol and for dimethyl ether (DME) were observed at retention time less than 9.8 min); deoxygenated C6+ products, e.g., gasoline, (all peaks observed at retention time between 9.8 and 34 min); oxygenated products (all peaks observed with retention time greater than 34 min).
Results and discussion
Spruce sawdust
The lignins used in this work originate from spruce or birch sawdust, and while the specific mode of their extraction will affect their chemistry, they are also different from the start as they are isolated from soft- and hardwood, respectively.11Considering the weight distributions from HTL of spruce lignins (S and SA; Fig. 3), employing acid (SA) during the lignin extraction process does not largely affect the LO yield, while that of HO increases. Inevitably, a certain amount of low molecular weight species are still liberated yielding a similar LO yield; however, less volatiles and water solubles, i.e., substances lost with the waste water, are generated keeping more of the raw material in the HO fraction. To get an idea of the dynamics and what species are formed and potentially being either a precursor for either of the product fractions during the HTL, GC/MS-analysis was performed (Table 2).
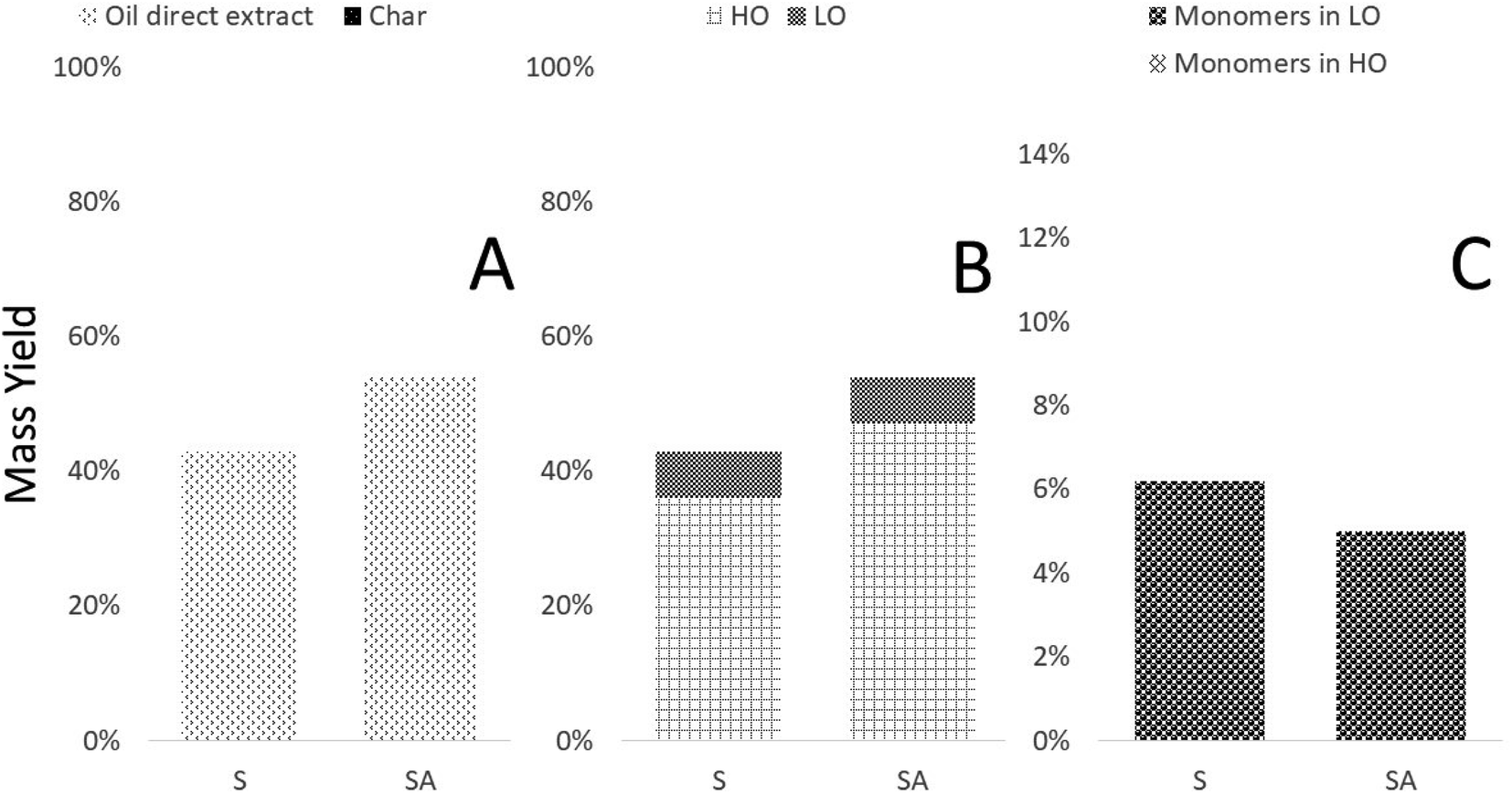 | ||
| Fig. 3 Weight distributions of the various fractions originating from spruce-based lignins, either extracted with (SA) or without acid (S): (A) weight distribution of the non-volatile fractions obtained; (B) division of oil direct extract into HO and LO fractions; (C) distribution of monomers in the LO and HO fractions. | ||
| Compounds | S [mg monomers per gLO] | SA [mg monomers per gLO] | B [mg monomers per gLO] | BA [mg monomers per gLO] |
|---|---|---|---|---|
| p-Cresol | 15.5 | 19.4 | ||
| Guaiacol | 274.2 | 212.9 | 60.1 | 66.3 |
| 4-Methylguaiacol | 113.3 | 73.7 | 24.1 | 45.1 |
| 3-Methoxycatechol | 35.1 | 89.3 | 57.4 | |
| 4-Ethylguaiachol | 48.4 | 34.6 | 21.4 | 96.7 |
| 4-Methylcatechol | 16 | 28.4 | ||
| 2,6-Dimethoxy phenol | 62.5 | 17.4 | 251 | 215.9 |
| 4-Propylguaiacol | 18.5 | 14.5 | 18 | 23.4 |
| 1,3,4-Dimethoxyphenol | 16.7 | 20.2 | ||
| Vanillin | 76 | 56.9 | 40.5 | |
| 5-Formylguaiacol | 21.8 | |||
| Phenol, 2,6-dimethoxy-4-methyl | 28.6 | 65.5 | 61.2 | |
| Acetoguaiacone | 35.8 | 26.4 | 15.7 | 22.4 |
| 2,6-Di-tert-butyl-p-cresol | 23.2 | 25.4 | ||
| 5-tert-Butylpyrogallol | 33.5 | |||
| Guaiacylacetone | 58.1 | 110.9 | 20.4 | 80.3 |
| 2-Butanone,4-(4-hydroxyphenyl) | 30.4 | |||
| Escaline | 18.3 | |||
| Syringaldehyde | 23.3 | |||
| Gallacetophenone-4′-methylether | 17.3 | |||
| Benzenepropanol, 4-hydroxy-3-methoxy- | 57.4 | 60.60 | 23 | |
| Syringaldehyde | 19.1 | 39.8 | 52.2 | |
| Gallacetophenone-4′-methylether | 17.1 | |||
| Phenol (unknown) | 24.4 | |||
| Acetosyringone | 28.7 | 56.2 | ||
| Syringylacetone | 42 | 121 | ||
| 5-(3-Hydroxypropyl)-2,3-dimethoxyphenol | 26.2 | 18.2 | ||
| Phenol (unknown) | 20.40 | |||
| Phenol (unknown) | 18.1 | |||
| Phenol (unknown) | 16.8 | 17.4 | ||
| Sum of quantified mass | 885.5 | 716 | 796 | 1143 |
| Monomers in fraction | 89% | 72% | 80% | 114% |
| Yield monomers in LO | 6.2% | 5.0% | 5.6% | 4.0% |
Furthermore, in order to potentially unravel any correlation between generated LO monomers with the initial spruce lignins, qualitative 1H–13C HSQC and quantitative 13C NMR analyses were performed on the two isolated spruce lignins. Quantization of the most common structures (Table 3) was achieved using the deviation of the HSQC data via quantitative 13C NMR.
| Motif (mode) | Shifts δ(1H) [ppm]/δ(13C) [ppm] | Spruce samplesa | Birch samplesb | ||
|---|---|---|---|---|---|
| S [mmol g−1] | SA [mmol g−1] | B [mmol g−1] | BA [mmol g−1] | ||
| a 200 °C; 30 min; 50 vol% EtOH. b 200 °C; 15 min; 60 vol% EtOH. c Overlapping signals. | |||||
| ArC-H | 125.5–100.0 | 14.65 | 16.06 | 10.87 | 22.19 |
| ArC-C | 125.5–140.0 | 7.15 | 7.72 | 10.87 | 10.55 |
| ArC-O-R | 140.0–160.0 | 13.26 | 14.73 | 12.73 | 13.08 |
| Ar(CH:CC:CO) |
42:20:38 |
42:20:38 |
32:32:37 |
48:23:29 |
|
| 13C–OMe (HSQC) | 54.5–56.5 | 6.28 (5.55) | 7.35 (6.59) | 11.30 (11.49) | 9.73 (9.98) |
| Sum aromatic carbons (per –OMe) | 35.05 (5.58) | 38.51 (5.24) | 34.47 (3.05) | 45.82 (4.71) | |
| Ketone | 206.50–207.10 | 0.50 | 0.42 | 0.00 | 0.04 |
| β-O-4′ to S | 4.88/72.04 | 0.03 | 0.03 | 0.36 | 0.10 |
| 4.13/85.91 | |||||
| 3.41/59.44 | |||||
| β-O-4′ to G | 4.90/71.34 | 0.01 | 0.02 | 0.19 | 0.03 |
| 4.21/84.59 | |||||
| 3.23/59.92 | |||||
| β–β′ | 4.67/84.98 | 0.01 | 0.01 | 0.06 | 0.01 |
| 3.06/53.50 | |||||
| 4.03/70.99 | |||||
| β-5′ | 5.43/87.00 | 0.02 | 0.02 | 0.19 | 0.08 |
| 3.48/52.99 | |||||
| 3.66/62.79 | |||||
| Xylopyranoside | 4.29/101.59 | 0.00 | 0.00 | 0.03 | 0.01 |
| 3.06/72.49 | |||||
| 3.26/73.94 | |||||
| 3.51/75.35 | |||||
| 3.36/62.50 | |||||
| Benzyl ether (LCC) | 4.59/80.92 | 0.00 | 0.01 | 0.04 | 0.01 |
| Guaiacyl (G6) | 6.25–7.20/117.0–122.75 | 2.21 | 2.59 | 0.78 | 1.90 |
| Ox. G2 | 7.40/110.48 | 0.53 | 0.77 | 0.14 | 0.44 |
| Guaiacyl (G2 + furan B C3) | G2: 6.80–7.13/108.0–113.5 | 3.80 | 3.68 | 0.87 | 3.02 |
| G2 + F3: 6.42–6.98/109.5–113.8 | |||||
| Furan B C4 | 7.36–7.68/121.5–125.0 | 1.40 | 1.31 | 0.06 | 1.22 |
| Syringyl (S2.6) | 6.20–7.00/101.8–104.4 | 0.07 | 0.07 | 2.34 | 1.39 |
| Furan A C3 | 6.15–6.75/104.4–107.4 | 0.12 | 0.18 | 0.69 | 2.99c |
| Furan A C4 | 6.90–7.45/105.2–108.2 | 0.11 | 0.18 | 0.65 | 1.58 |
| Sum furan rings | 1.50 | 1.48 | 0.70 | 2.80 | |
| Sum lignin rings | 2.28 | 2.66 | 3.11 | 3.29 | |
The general distribution of motifs in lignins originating from relevant crops has been summarized14 and will be applied as an initial foundation for the interpretation of the NMR data presented herein. In short, native lignins from softwoods are dominated by the guaiacyl monomer, derived from coniferyl alcohol. The interunit linkages vary, with the β-O-4′ linkage dominating (approx. 60%), while dibenzodioxocin (5–5′, 4-O-β′), phenylcoumaran (β-5′), and pinoresinol (β–β′) are usually present with contents of approx. 10, 10, and 5%, respectively. Apart from these more commonly occurring structures, spirodienone (β-1′) and biphenyl ether (4-O-5′) are present, albeit at significantly lower contents (approx. 1%). The analyses of the organosolv spruce lignins used for the HTL are in line with this general description, while indicating, however, that the standard motifs are drastically reduced in abundance. Analyses also indicate the presence of notable quantities of oxidized derivatives of typical interunit bonding motifs; condensation products are present.
The presented analytical data in form of GC-MS data and NMR data can serve to evaluate how the apparent structural changes in the raw-material, i.e., lignin modification, including cross-coupling both in the aliphatic but also in the aromatic domains, potentially incorporating sugar dehydration products in form of lignin–humin–hybrid structures,27 influences the dynamics and chemistry of the biomaterial during thermal treatment, thus determining final product formation.
Evaluating the results from Table 2, the monomers in the LO fraction with decreasing abundances as acid is used in the lignin extraction from spruce are guaiacol, 4-ethylguaiacol, and 4-methylguaiacol; this while the content of guaiacylacetone augments. The latter is understandable considering the data from the HSQC analysis in which G units with an oxidized side-chain are observed (Table 3). Importantly, liberated guaiacol units are in general decreasing abundancy in the LO fraction alongside a reduced overall monomer content, whereas the isolated HO fraction augments when moving from S to SA. For initially evaluating potential modification of the extracted lignins with respect to a putative lignin structure in planta during the initial organosolv process, the 13C NMR spectra where used to generate rough estimates for the content of distinct aromatic carbons and to compare against the methoxyl group content (Table 3). In general, the total content of aromatic carbon atoms is presenting a 6-to-1 ratio relative the methoxyl groups, which matches fairly well with what to expect for a spruce lignin. However, the ArC-H content appears low: for an ‘intact’ guaiacyl monomer one would expect ratios of CH:CC:CO being approx. 3:2:1, whereby the ratios found are approx. CH:CC:CO = 2:1:2. Worth to mention is that the exact distribution of ArC-C and ArC-O carbons might experience overlaps caused by other structural motifs generating peaks in their respective regions, however, the tertiary aromatic carbon content obtained from 13C NMR was found to match closely that obtained when integrating the HSQC spectra in the same regions, suggesting that tertiary carbons are reduced due to a higher content of quaternary aromatic carbons either linked to another carbon or oxygen. This trend appears to increase further as acid is employed in the extraction process, reducing still the number of aromatic carbons per measured methoxyl group. Interestingly, whereas the ratios of the various aromatic carbon types remain stable for the extraction treatments in absence (spruce lignin S) or presence of sulfuric acid (spruce lignin SA), the overall quantity increases for the latter alongside the oil-direct extract and amount of heavy oil (HO). Furthermore, the amount of monomers in the LO fraction decreases, suggesting that in fact the quantity and distribution of aromatic linkages influence how the oil fractions are distributed. Considering the composition of the LO fractions as elucidated through GC-MS, the monomers are largely of lignin origin, despite the fact that NMR analysis suggests the presence of furan moieties in the extracted lignins at significant contents (Table 3).
In order to evaluate the assigned lignin monomer and furan ring signals, the regions evaluated through 13C NMR where compared against the shifts assigned through HSQC (Fig. 4). A good agreement is found regarding the contents obtained through 13C NMR and their assigned HSQC shifts, using the quantification of the HSQCs on the basis of the quantitative 13C NMR as reported before. Interestingly, the fit especially improves for spruce derived lignins when assuming that the majority of the G units carries only two aromatic CH carbons, which would imply that the majority carries an aromatic substitution.
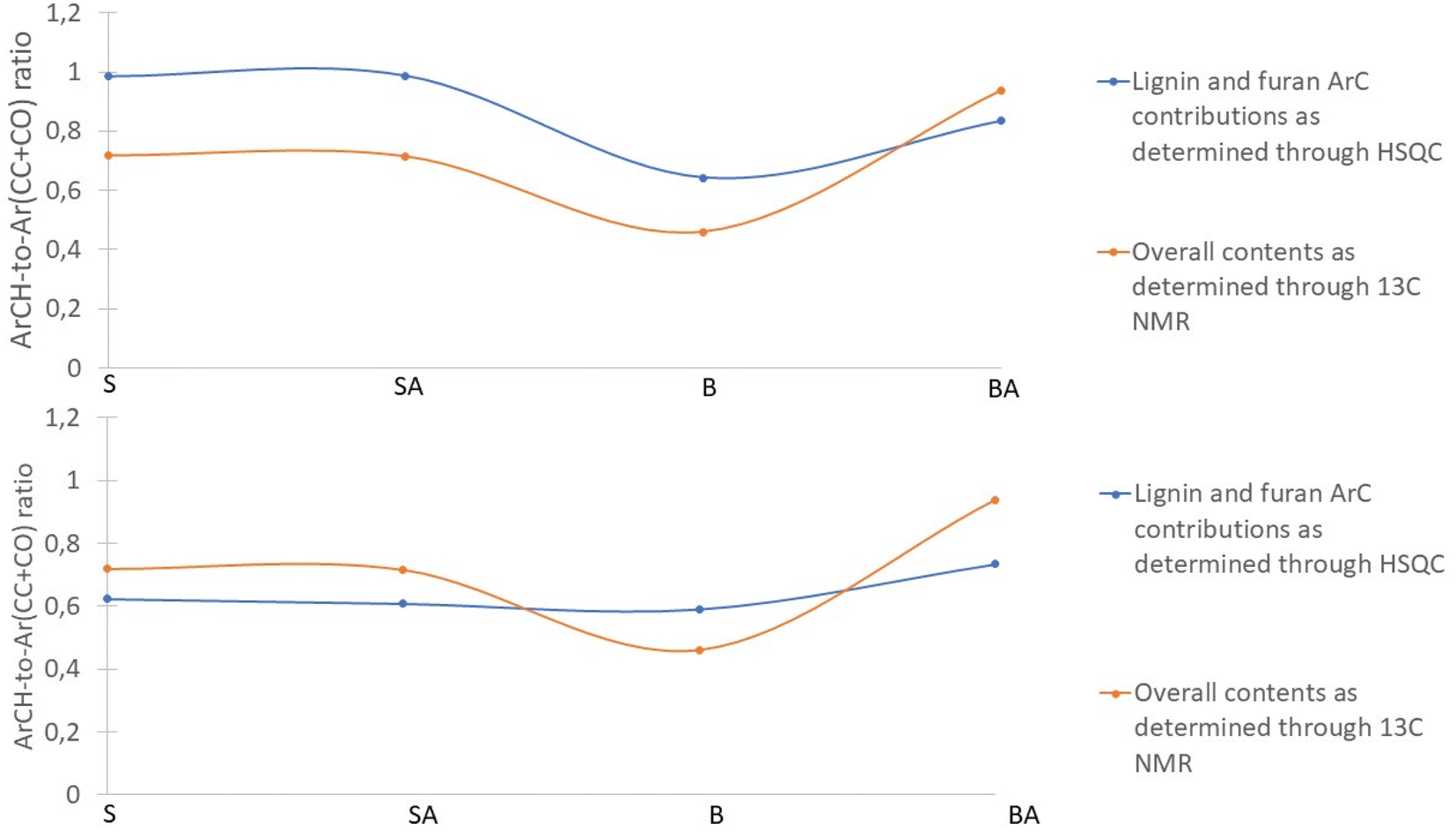 | ||
| Fig. 4 Top: Ratio of tertiary to quaternary aromatic carbons as measured through 13C shift ranges and specific 1H–13C assignments assuming that lignin monomers and furan rings are non-substituted. Bottom: As for the top figure except with the assumption that 1 of the 3 tertiary ring carbons of guaiacyl are substituted. | ||
For the spruce HO fractions, the results obtained by FT-ICR MS evidence an increase in CH species that originate on behalf of CHO compounds (Table 4), in the sample obtained under acidic conditions, i.e., SA. This does not contradict the NMR data where an increase in all aromatic carbon types (C–H, C–C, C–O) alongside the introduction of furans and aromatic substitution is observed. Fig. 5A also evidences the specificity of the spruce HO obtained under acidic conditions with the higher contribution of the CH species. For the CHO compounds, in both S and SA samples, the distribution is similar, although one can notice higher contribution of the oxygen-rich (>O9) compounds in the SA sample (Fig. 5).
| Sample | CH | CHO | CHN | CHNO | CHOS | Total |
|---|---|---|---|---|---|---|
| S | 251 (3.8%) | 6240 (95.5%) | 0 | 46 (0.7%) | 0 | 6537 |
| SA | 810 (10.6%) | 6597 (86.7%) | 8 (0.1%) | 127 (1.7%) | 64 (0.8%) | 7606 |
| B | 182 (2.5%) | 6975 (95.2%) | 5 (0.1%) | 53 (0.7%) | 112 (1.5%) | 7327 |
| BA | 631 (8.2%) | 6829 (89.2%) | 0 | 50 (0.7%) | 146 (1.9%) | 7656 |
 | ||
| Fig. 5 Oxygen-distribution of CH and CHO compounds detected in HO of (A) spruce organosolv lignins extracted with (SA) or without acid (S) and (B) birch organosolv lignins extracted with (BA) or without (B) acid. | ||
Similar observations are reported30 and were attributed to repolymerization phenomena. Such oxygen-rich compounds can also represent larger molecular weight lignin-components obtained, or formed, by acid catalysis. Regarding the pure hydrocarbon species, an insight was achieved by representing them according to their DBE vs. carbon number (Fig. 6). The differences between S and SA samples are noticeable with more CH compounds in the SA sample. These specific features are characterized by components that are of higher molecular weight and that display a higher number of unsaturated motifs (higher DBE) components. It is also noteworthy that from species with C > 30 and DBE > 8, there are some packets that differ by 5–6 carbon atoms and 1–3 DBE units. These CH compounds specifically observed in the SA sample can originate from high-molecular weight lignin species that underwent deoxygenation reactions under acidic conditions. These high-molecular weight compounds can be imagined to be similar to those observed in Fig. S2,† in the form of CHO compounds from non-acid catalyzed organosolv lignin comprising close to 60 carbon atoms. Similar graphic analyses were done using CHO compounds (Fig. S2†), which did not evidence particular influence of acid addition during organosolv fractionation on the distribution of these species, unlike in case of the CH species. These results are in line with the distributions shown in Fig. 5A. Thus, these compounds detected by FT-ICR MS derive from lignin oligomers and comprise between 10 and 70 carbon atoms, which explains O-distributions with up to 16 oxygen atoms.31 Relevant work performed which further aid the interpretation of the DBE vs. #C data suggest that, as acid is employed during the initial extraction treatment, the amount of saturated components augments within the produced HO fraction which potentially could be aliphatic segments linking aromatics.32
 | ||
| Fig. 6 DBE vs. carbon number graph of CH compounds detected by APPI FT-ICR MS of the four different HO obtained from spruce and birch lignins isolated in the absence (S, B) and presence (SA, BA) of acid-catalyst. | ||
At this point it is also worth mentioning the appearance of 1,3,4-dimethoxyphenol in the LO derived from SA, hence the acid extracted spruce lignin (Table 1). This component was not detected in the S-derived LO fraction. Other double methoxylated species appeared in the spruce-derived LO fractions and are in contrast to what one would initially expect to in fractions generated from softwoods. It seems also important to point out that the HO fractions of spruce, most noteworthy as well as the HO fractions from birch (vide infra) show different characteristics (DBE vs. #C) compared to that of polyaromatic hydrocarbons (PAHs).32,33 Instead, the DBE vs. #C slope suggest that aromatic groups carry increased contents of saturated carbon segments after HTL when acid is employed during the initial extraction.32 Liberation of such segments during HTL could certainly justify the appearance of pure CH components.
Birch sawdust
Moving from spruce to the birch extracts, Fig. 7 illustrates that use of acid during the lignin isolation prevents subsequent char formation, but also reduces the LO yield while instead increasing the formation of HO.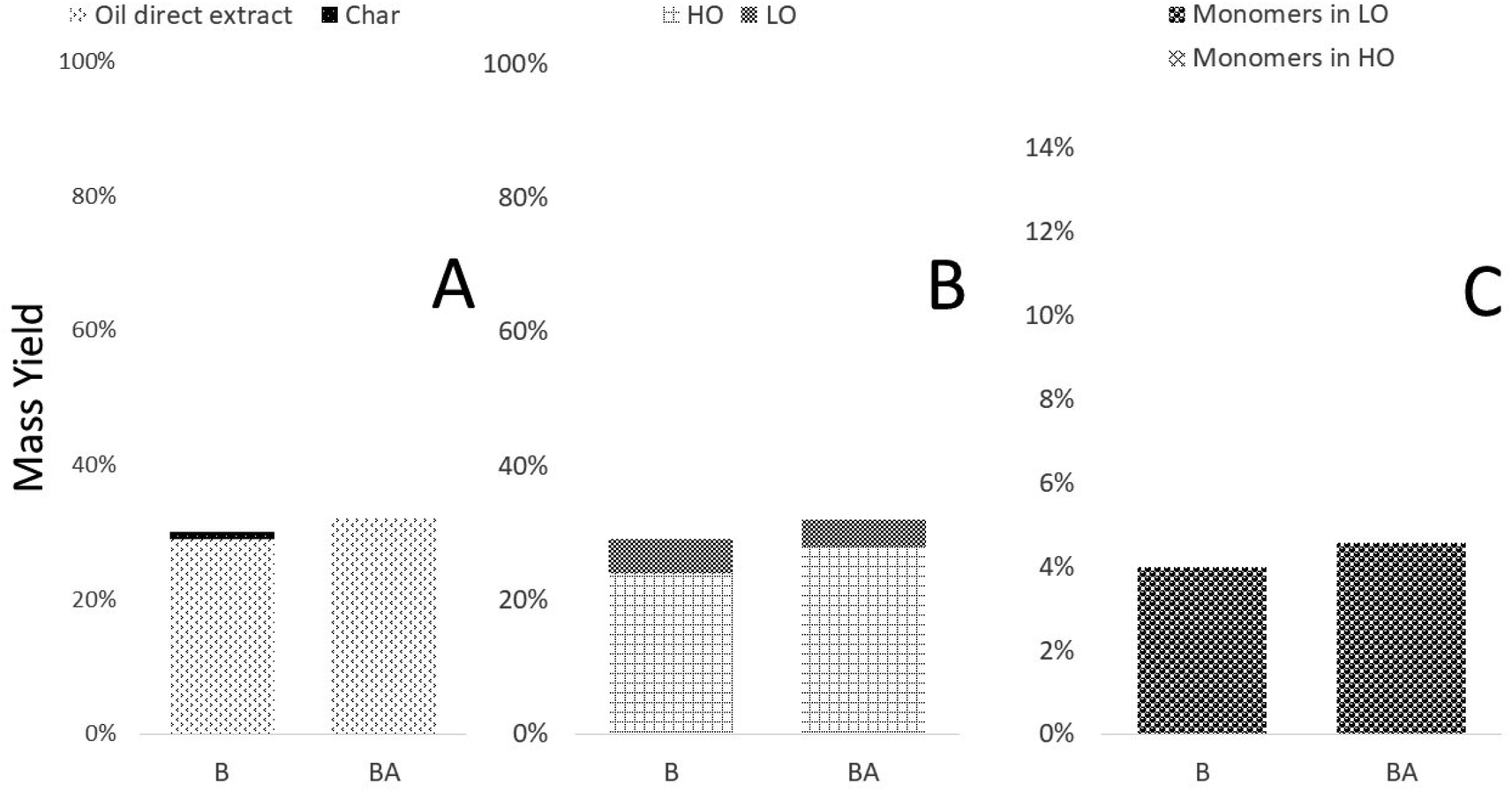 | ||
| Fig. 7 Weight distributions of various fractions originating from birch-based lignins, either extracted with (BA) or without acid (B): (A) weight distribution of the non-volatile fractions obtained; (B) division of oil direct extract into HO and LO fractions; (C) distribution of monomers in the LO and HO fractions. | ||
Considering the lignin motifs elucidated through NMR (Table 3), there is a significant difference between their contents depending on whether one employs acid or not. For example, the β-O-4′ linkage content to either G or S units decreases from 0.19 and 0.36 to 0.10 and 0.03 mmol g−1. Despite this, the overall changes in oil direct extract, char, HO and LO yield, are low compared to the spruce-derived products. When considering the distribution of aromatic carbons there is a large difference between B and BA. Especially the ArC-H content increases and is observed alongside a steep increase in the furan ring content (Table 2), which causes the sum of aromatic carbons per methoxyl to increase from the reasonable value of 3.05 to 4.71. This is not necessarily due to the introduction of furans considering that birch carries both guaiacyl and syringyl units and a value between 3 and 6 would be expected. Considering any aromatic substitutions (Fig. 4), the best fit for the aromatic carbon distributions, is found when not assuming the presence aromatic condensation of the G unit and in general correlate well with the measured contents of lignin monomers and furans. Compared to the S and SA-derived bio-oils, the direct oil extract is far lower than for the birch, further indicating that aromatic cross-linking could promote retention in the HO fraction.
Moving to the GC-MS-analyses of the LO fraction (Table 2), there is an increase in the guaiacol compounds, which would seem reasonable when recollecting the reduction of β-O-4′ to G linkages. This is in contrast to what was observed for the spruce lignin where they instead decreased. Expected to be exclusive for the hardwood derived bio-oil, also syringyl derivates, i.e., acetosyringone, syringylacetone, syringaldehyde, increase in content in LO when moving from B to BA and the same argument can be made from their β-O-4′ to S content. The differences between the BA fractions from softwood and the hardwood are also apparent looking at the total yields of monomers, even when considering an estimated error of 10% caused by the applied analyses, which is eventually biased by the fact that not all generated monomers are equally well ionizable: the total yields have been determined on the basis of the integrated area of the total ion chromatogram as obtained in the GC-MS analysis, causing in case of BA also a yield higher than 100%.
As with the spruce-derived HO fractions, the ones from birch show a comparable behavior when considering the molecular distribution obtained from FT-ICR of the two HO fractions (Table 4 and Fig. 5B). There is also a significant increase of the pure hydrocarbons contribution when lignin is extracted with acid catalyst, however, less CH formulae were observed for BA compared to the SA sample. Also, whereas less developed than SA at higher carbon numbers, the same is indicated in Fig. 6 where the DBE vs. carbon number graph obtained from the CH assignments of the BA sample also display signal for compounds with C > 30 and DBE > 8. As for samples based on spruce-lignin, CHO compounds did not seem to be impacted by addition of acid during organosolv as shown by Fig. 5B and S2.†
Process modification
Considering the resulting weight distributions (Fig. 8), the most obvious change introduced by making the process continuous is that the majority of the biooil product is found to be LO. Also, for the first time in the present work, spruce lignin is found to generate char, albeit to a negligible amount. Previous findings have found that the LO yield increases on behalf of HO upon elevated HTL severity,34 this while especially catechols have been found to be a precursor for bio-chars and active during repolymerization.35 A striking difference between SA and C-SA is the differences in guaiacol and catechol contents in the LO where the former is reduced by more than 50% per gram LO, whereas catechol and creosol appear for the first time in this work (Table 5), suggesting that the LO from C-SA is to a larger degree developed. Guaiacol is considered the precursor of catechol,36 with the latter tending to repolymerize into insoluble bio-char which makes the presented data reasonable. However, due to the lower thermal severity resulting from a more efficient heating, such a scenario must originate from the higher lignin loading applied during the continuous process.
 | ||
| Fig. 8 Weight distributions of various fractions originating from spruce when process configuration was altered to continuous (C-SA): (A) weight distribution of the non-volatile fractions obtained; (B) division of oil direct extract into HO and LO fractions; (C) distribution of monomers in the LO and HO fractions. | ||
| Compounds | C-SA [mg monomers per gLO] |
|---|---|
| Guaiacol | 106 |
| Catechol | 19 |
| Creosol | 21 |
| 3-Methoxycatechol | 3 |
| 4-Ethylguaiachol | 17 |
| 5-Formylguaiacol | 42 |
| Acetoguaiacone | 10 |
| 5-tert-Butylpyrogallol | 41 |
| Escaline | 11 |
| Syringaldehyde | 2 |
| Sum of quantified mass | 272 |
| Monomers in fraction | 27% |
 | ||
| Fig. 9 Conversion of biooil and deoxygenated C5- and deoxygenated C6+ yields (expressed as the percentage of the deoxygenated C5- and deoxygenated C6+ in the final product) for different feeds for the catalytic conversion of the light fraction of HTL oil, as a function of time on stream at 360 °C and a WHSV of 0.3 h−1 over the 35 nm thick defect free ZSM-5 catalyst supported on 500 nm Stöber SiO2 particles: (a) 60% bio-oil BA-LO, 20% methanol, and 20% water; (b) 10% bio-oil BA-LO, 30% methanol, and 60% water. | ||
Conclusion
Lignins extracted through organosolv processing, from spruce or birch, with or without sulfuric acid as acidic catalyst display varying chemical structures, which dictate how they behave upon formation of bio-oil during hydrothermal liquefaction. For spruce lignins, its innate ‘sensitive’ nature toward depolymerization and cross-coupling generate a bio-oil precursor, which upon HTL has the potential of generating an enlarged fraction of heavy oil (HO). The birch lignin experiences overall less modification, and despite similarities in terms of trends observed upon acid-catalyzed lignin extraction from birch when compared to spruce, the more ‘resistant’ nature of the hardwood lignin creates a starting-point where depolymerized traditional motifs refrain from undergoing further structural modifications that would give raise to an enhanced formation of the HO fraction as observed for the spruce-derived lignins. In addition, continuous processing was evaluated and found to primarily generate LO when SA was evaluated, i.e., a softwood lignin isolated using an acid-catalysed organosolv process. Finally, the LO fraction obtained from BA, i.e., lignin extracted using the acid-catalysed organosolv process, was catalytically converted to short-chained deoxygenated compounds with a conversion yield of more than 97%, prior to catalyst deactivation after approx. 18 h.Author contributions
PPT: investigation, data analysis, writing – original draft; JF: investigation, data analysis, writing – review & editing; HL: data analysis, supervision, writing – original draft; JH: investigation, data analysis, writing – review & editing; VC: investigation, data analysis, writing – review & editing; MZ: investigation, data analysis; AT: data analysis, writing – review & editing; FA: data analysis, supervision, writing – review & editing; JH: data analysis, methodology, supervision; TG: conceptualization, methodology, data analysis, supervision, writing – review & editing; UR: conceptualization, methodology, supervision, writing – review & editing; PC: conceptualization, methodology, supervision, writing – review & editing; LM: conceptualization, methodology, data analysis, supervision, writing – original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was part of the projects “Upgrading of organosolv lignin to jet fuel (GOLdJET FUEL)” and “Eco-efficient biorefinery for competitive production of green renewable shipping fuels (ECO-FORCE FUELS)” funded by the Swedish Energy Agency with reference numbers 2019-005832 and 2022-201046 respectively. Mattias Hedenström, Swedish NMR Centre (Umeå, Umeå University, VR RFI), João Figueira, Swedish NMR Centre (Umeå, Umeå University, Scilife Lab) and the NMR Core Facility (Swedish NMR Centre, SwedNMR, Umeå node), Umeå University are acknowledged for NMR support. FTICR MS equipment was funded by the European Regional Development Fund (FEDER), the general council of Moselle, Region Grand Est, Metz Metropole and the University of Lorraine (RESEX project).References
- I. v. Provornaya, I. v. Filimonova, V. Y. Nemov, A. v. Komarova and Y. A. Dzyuba, Energy Rep., 2020, 6, 514–522 CrossRef.
- U. Bardi, Energy Res. Soc. Sci., 2019, 48, 257–261 CrossRef.
- Climate Change 2022: Impacts, Adaptation and Vulnerability|Climate Change 2022: Impacts, Adaptation and Vulnerability, https://www.ipcc.ch/report/ar6/wg2/, accessed 10 May 2022 Search PubMed.
- J. Podesta and P. Ogden, Wash. Q., 2008, 31, 115–138 CrossRef.
- J. Gornall, R. Betts, E. Burke, R. Clark, J. Camp, K. Willett and A. Wiltshire, Philos. Trans. R. Soc., B, 2010, 365, 2973–2989 CrossRef PubMed.
- L. Dessbesell, M. Paleologou, M. Leitch, R. Pulkki and C. Xu, Renewable Sustainable Energy Rev., 2020, 123, 109768 CrossRef CAS.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- P. C. A. Bruijnincx and B. M. Weckhuysen, Nat. Chem., 2014, 6, 1035–1036 CrossRef CAS PubMed.
- D. Wei Kit Chin, S. Lim, Y. L. Pang and M. K. Lam, Biofuels, Bioprod. Biorefin., 2020, 14, 808–829 CrossRef CAS.
- P. P. Thoresen, L. Matsakas, U. Rova and P. Christakopoulos, Bioresour. Technol., 2020, 306, 123189 CrossRef CAS PubMed.
- C. Sun, G. Song, Z. Pan, M. Tu, M. Kharaziha, X. Zhang, P.-L. Show and F. Sun, Bioresour. Technol., 2023, 368, 128356 CrossRef CAS PubMed.
- W. J. J. Huijgen, A. T. Smit, J. H. Reith and H. den Uil, J. Chem. Technol. Biotechnol., 2011, 86, 1428–1438 CrossRef CAS.
- F. Hu, S. Jung and A. Ragauskas, Bioresour. Technol., 2012, 117, 7–12 CrossRef CAS PubMed.
- J. Ralph, C. Lapierre and W. Boerjan, Curr. Opin. Biotechnol., 2019, 56, 240–249 CrossRef CAS PubMed.
- A. R. K. Gollakota, N. Kishore and S. Gu, Renewable Sustainable Energy Rev., 2018, 81, 1378–1392 CrossRef.
- S. S. Toor, L. Rosendahl and A. Rudolf, Energy, 2011, 36, 2328–2342 CrossRef CAS.
- A. N. Kay Lup, F. Abnisa, W. M. A. W. Daud and M. K. Aroua, Appl. Catal., A, 2017, 541, 87–106 CrossRef CAS.
- A. J. Foster, J. Jae, Y.-T. Cheng, G. W. Huber and R. F. Lobo, Appl. Catal., A, 2012, 423–424, 154–161 CrossRef CAS.
- P. A. Alaba, Y. M. Sani, I. Y. Mohammed and W. M. A. Wan Daud, Rev. Chem. Eng., 2016, 32(1), 71–91 CAS.
- A. Veses, B. Puértolas, J. M. López, M. S. Callén, B. Solsona and T. García, ACS Sustain. Chem. Eng., 2016, 4, 1653–1660 CrossRef CAS.
- J. Hertzog, V. Carré, L. Jia, C. L. Mackay, L. Pinard, A. Dufour, O. Mašek and F. Aubriet, ACS Sustain. Chem. Eng., 2018, 6, 4717–4728 CrossRef CAS.
- L. Y. Jia, M. Raad, S. Hamieh, J. Toufaily, T. Hamieh, M. M. Bettahar, G. Mauviel, M. Tarrighi, L. Pinard and A. Dufour, Green Chem., 2017, 19, 5442–5459 RSC.
- Z. Qin, L. Lakiss, L. Tosheva, J.-P. Gilson, A. Vicente, C. Fernandez and V. Valtchev, Adv. Funct. Mater., 2014, 24, 257–264 CrossRef CAS.
- I. Yarulina, F. Kapteijn and J. Gascon, Catal. Sci. Technol., 2016, 6, 5320–5325 RSC.
- U. V. Mentzel and M. S. Holm, Appl. Catal., A, 2011, 396, 59–67 CrossRef CAS.
- M. Monção, K. Hrůzová, U. Rova, L. Matsakas and P. Christakopoulos, Molecules, 2021, 26, 6754 CrossRef PubMed.
- P. P. Thoresen, H. Lange, U. Rova, P. Christakopoulos and L. Matsakas, Int. J. Biol. Macromol., 2023, 233, 123471 CrossRef CAS PubMed.
- P. P. Thoresen, H. Lange, C. Crestini, U. Rova, L. Matsakas and P. Christakopoulos, ACS Omega, 2021, 6, 4374–4385 CrossRef PubMed.
- J. Hedlund, M. Zhou, A. Faisal, O. G. W. Öhrman, V. Finelli, M. Signorile, V. Crocellà and M. Grahn, J. Catal., 2022, 410, 320–332 CrossRef CAS.
- P. D. Kouris, X. Huang, X. Ouyang, D. J. G. P. van Osch, G. J. W. Cremers, M. D. Boot and E. J. M. Hensen, Catalysts, 2021, 11, 750 CrossRef CAS.
- E. Terrell, V. Carré, A. Dufour, F. Aubriet, Y. Le Brech and M. Garcia-Pérez, ChemSusChem, 2020, 13, 4428–4445 CrossRef CAS PubMed.
- R. Luo and W. Schrader, J. Hazard. Mater., 2021, 418, 126352 CrossRef CAS PubMed.
- A. T. Juarez-Facio, C. Castilla, C. Corbière, H. Lavanant, C. Afonso, C. Morin, N. Merlet-Machour, L. Chevalier, J.-M. Vaugeois, J. Yon and C. Monteil, J. Environ. Sci., 2022, 113, 104–117 CrossRef CAS PubMed.
- E. Miliotti, S. Dell'Orco, G. Lotti, A. Rizzo, L. Rosi and D. Chiaramonti, Energies, 2019, 12, 723 CrossRef CAS.
- S. Kang, X. Li, J. Fan and J. Chang, Renewable Sustainable Energy Rev., 2013, 27, 546–558 CrossRef CAS.
- J. Schuler, U. Hornung, N. Dahmen and J. Sauer, GCB Bioenergy, 2019, 11, 218–229 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3se00976a |
| This journal is © The Royal Society of Chemistry 2023 |