Visible-light driven photocatalytic CO2 reduction promoted by organic photosensitizers and a Mn(I) catalyst†
Received
27th April 2023
, Accepted 12th June 2023
First published on 27th June 2023
Abstract
Photocatalytic systems for CO2 reduction can greatly benefit from the development of fully organic photoredox sensitizers, so as to move away from the use of rare elements. In this study, a series of organic molecules, displaying thermally activated delayed fluorescence (TADF) containing diphenylamine (4DPAIPN, 3DPAFIPN) or carbazole (5CzBN, 4CzIPN, 3CzClIPN) moieties as electron donating groups were systematically investigated as photoredox sensitizers for CO2 reduction, coupled with a Mn(I)-complex as the catalyst (Mn). All of the TADF dyes were reductively quenched by BIH in triethanolamine (TEOA)–N,N-dimethylacetoamide solutions. However, their photocatalytic performances were markedly different. 5CzBN, 4CzIPN, and 3CzClIPN did not work as photosensitizers in the studied photocatalytic system because of low absorbance in the visible region and/or low reducing power of their one-electron reduced species. On the other hand, TADF molecules possessing diphenylamine groups are characterized by stronger absorption in the visible region and their one-electron reduced species have stronger reducing power. In particular, 4DPAIPN proved to be the best performing photosensitizer when using a molar ratio of photosensitizer![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :catalyst = 1:1 and a 470 nm LED source, yielding TONCO+HCOOH > 650 and ΦCO+HCOOH = 22.8 ± 1.5%.
:catalyst = 1:1 and a 470 nm LED source, yielding TONCO+HCOOH > 650 and ΦCO+HCOOH = 22.8 ± 1.5%.
Introduction
The depletable nature of fossil fuels and the release of enormous amounts of CO2 into the atmosphere (about 34 Gt per year) upon their combustion have prompted mankind to devise artificial photosynthetic systems that are able to store sunlight into high-energy products known as solar fuels.1 The ultimate goal is therefore to produce fuels by using low-energy and widely available feedstock, such as water and carbon dioxide, and sunlight as the sole energy source.2,3 To reach this goal an artificial photosynthetic system should comprise a photosensitizer (PS) that efficiently absorbs visible light for initiating electron transfer, a catalyst (CAT) that is able to store electrons or holes and drive multielectronic redox processes, and a sacrificial electron donor (S) that is irreversibly oxidized (Fig. 1).4,5 This approach simplifies the process and facilitates the study of the kinetics of the relevant processes and of the photo- and chemical stability of the different components. In such systems, the photosensitizer should be characterized by strong visible-light absorption, photostability, a long lifetime of the involved excited state and appropriate redox potentials. On the other hand, a catalyst should be able to accumulate electrons, have a high selectivity towards CO2 reduction, and be chemically stable under the reaction conditions.
 |
| | Fig. 1 Schematic representation of the studied system. | |
Given these requirements, most systems capable of reducing CO2 traditionally rely on heavy-metal complexes (e.g. Ru and Ir) as photosensitizers,6,7 whereas a large-scale application should desirably be based on abundant and non-toxic compounds. For this reason, research has recently been focusing on the development of both photosensitizers8 and catalysts9–12 that are either based on abundant metals or consist of purely organic compounds. Recently, a copper(II) purpurin complex,13 and mononuclear and dinuclear copper(I) complexes14–20 have for instance been utilized as photosensitizers in order to achieve the photochemical reduction of CO2. Although Zn porphyrins have been used as photosensitizers for CO2 reduction, their efficiency and durability are relatively low, especially in systems coupled with abundant-metal complexes as catalysts.21,22 Attempts at using metal-free photosensitizers to drive the photoreduction of CO2 initially included the use of cyanoanthracene23 and p-terphenyl24–26 dyes, which have the disadvantage of a short excited-state lifetime and absorption maxima located in the UV region. More recently, organic photosensitizers with stronger visible light absorption such as purpurin derivatives27–29 or the ability to populate their long-lived triplet excited state like phenazine30,31 have been employed.
A long excited-state lifetime is an essential feature of a photosensitizer since it increases the efficiency of bimolecular interactions, such as reductive quenching by a sacrificial electron donor. This feature is commonly found in transition metal complexes, as in the case of [Ru(bpy)3]2+.32 Nonetheless, strategies have been devised to induce intersystem crossing (ISC) in organic chromophores33–35 like the synthesis of compounds that are characterized by highly twisted electron donating and electron accepting units.36–38 In this way, it is possible to minimize the energy gap (ΔE) between the lowest singlet excited state (S1) and the lowest triplet excited state (T1) promoting intersystem crossing (from S1 to T1) and reverse intersystem crossing (RISC) (from T1 to S1), and thus gives rise to thermally activated delayed fluorescence (TADF) (Fig. 2). On top of enabling the population of a long-lived excited state, TADF compounds allow the tuning of redox potentials thanks to the strong localization of their HOMO and LUMO orbitals.38 Despite an extensive use of this class of organic dyes in photoredox catalysis,39,40 only one organic TADF molecule, i.e., 4CzIPN (Chart 1), has been used as a photosensitizer for the reduction of CO2.41–45 In order to construct highly efficient and durable photocatalytic systems using organic TADF dyes, improvements are needed in terms of stronger and red-shifted absorption of visible light, stronger oxidation power in the excited state, and higher stability of the reduced state. For example, in the case of 4CzIPN, its molar absorption coefficient is negligible at wavelengths longer than 470 nm, and this implies that an excess of 4CzIPN compared to the amount of catalyst is used (typical molar ratios: 5–10:1).41–45 Moreover, addition of an excess of PS frequently induces its decomposition, owing to the accumulation of its reduced form in the reaction solution. Therefore, although the turnover number (TON) based on the used CAT is high (in the order of 103), TON based on the used PS is much lower (ca. 102) and the reported quantum yields of CO2 reduction are less than 5%.46
 |
| | Fig. 2 Jablonski diagram illustrating the relevant steps for TADF emission. | |
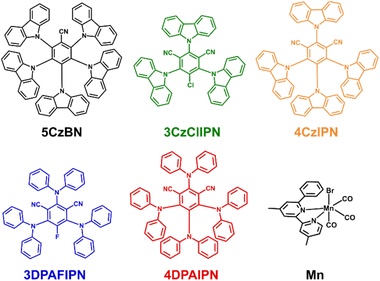 |
| | Chart 1 Structures and abbreviations of the organic TADF dyes and of the Mn(I) complex. | |
In this work, we investigate the ability of five organic TADF molecules to work as PSs in a three-component artificial photosynthetic system for photocatalytic CO2 reduction. These were coupled with a new earth-abundant Mn(I) catalyst46,47 (Chart 1), which has a sterically hindered phenyl group close to the central Mn for suppressing reductive dimerization,47,48 and with 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzimidazole (BIH)49 as the sacrificial electron donor to successfully develop an efficient and durable photocatalytic system without using heavy metals. Comparison of the photophysical and electrochemical properties of the chromophores allowed the rationalization of the different catalytic activities, which reach TONCO+HCOOH > 650 and ΦCO+HCOOH = 22.8 ± 1.5% in the case of 4DPAIPN.
Results and discussion
Chart 1 shows the investigated TADF photosensitizers together with the Mn(I) complex used as the catalyst, which has a phenyl group at the 6 position of the 4,4′-dimethyl-2,2′-bipyridine ligand in order to suppress dimerization of its reduced state.48
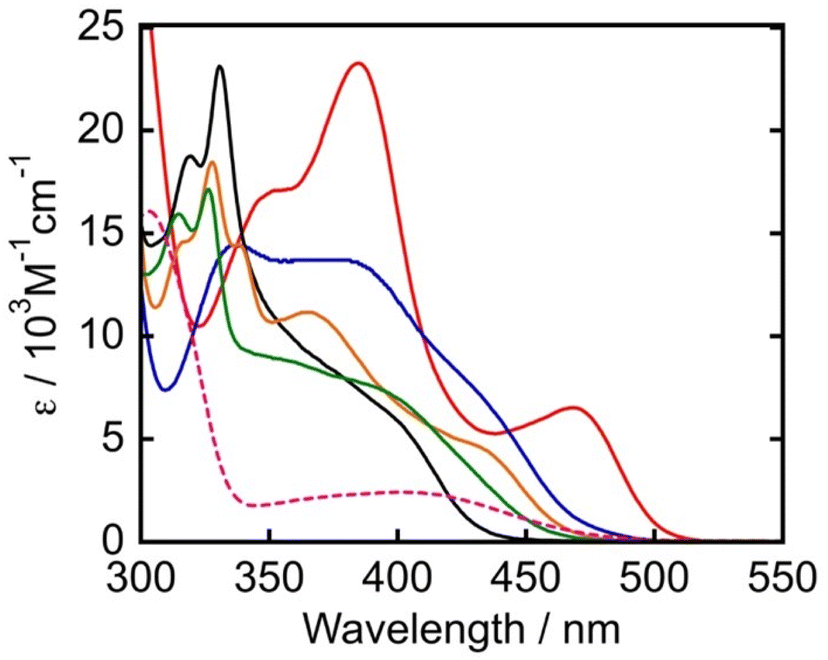
Fig. 3 shows the absorption spectra of the TADF chromophores measured in N,N-dimethylacetamide (DMA). All compounds present broad absorption bands in the visible region attributable to charge-transfer transitions from the electron donating diphenylamine (DPA) or carbazole (Cz) moieties to the electron accepting cyanobenzene unit. The absorption onsets were observed in the order of 4DPAIPN > 3DPAFIPN > 4CzIPN > 3CzClIPN > 5CzBN. The red-shifted absorption of 4DPAIPN and 3DPAFIPN compared to their carbazole analogues is attributed to the more electron-rich nature of DPA moieties compared to Cz moieties. This leads to a destabilization of the HOMO, resulting in smaller transition energies for visible-light absorption. In particular, 4DPAIPN is characterized by an absorption onset at 516 nm (ε = 100 M−1 cm−1). The photophysical properties of the PSs are summarized in Table 1.
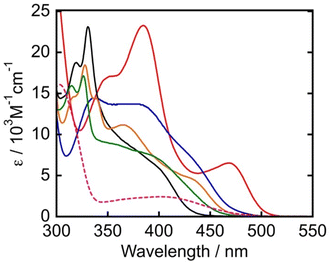 |
| | Fig. 3 UV-vis absorption spectra of PSs in DMA (4DPAIPN: red, 3DPAFIPN: blue, 5CzBN: black, 4CzIPN: orange, 3CzClIPN: green). The pink dashed line indicates the UV-vis absorption spectrum of Mn in DMA. | |
Table 1 Photophysical properties of PSs and Mn in DMAa
|
|
ε/103 M−1 cm−1 |
λ
abs/nm |
λ
em/nm |
τ
PF
/ns |
τ
TADF
/μs |
Φ
em
/% |
η
q
/% |
| (440 nm) |
(470 nm) |
(ε = 100) |
|
The estimated errors in the photophysical measurements are reported in the ESI.
Lifetime of prompt fluorescence.
Lifetime of TADF emission.
Emission quantum yield in an Ar atmosphere.
Quenching fraction of the emission calculated from ηq = 1 − I/I0; I: emission intensity in Ar-saturated DMA containing both TEOA (1.5 M) and BIH (0.1 M); I0: emission intensity in Ar-saturated DMA.
|
|
4DPAIPN
|
5.29 |
6.50 |
516 |
527 |
2.9 |
84 |
48.9 |
93 |
|
3DPAFIPN
|
5.93 |
1.11 |
498 |
536 |
3.6 |
43 |
16.0 |
93 |
|
5CzBN
|
0.31 |
0.05 |
454 |
515 |
24 |
10 |
33.9 |
98 |
|
4CzIPN
|
3.88 |
0.42 |
486 |
552 |
21 |
2.1 |
30.0 |
98 |
|
3CzClIPN
|
2.15 |
0.19 |
478 |
554 |
13 |
32 |
9.9 |
94 |
|
Mn
|
1.50 |
0.48 |
501 |
— |
— |
— |
— |
— |
All these organic PSs exhibit emission spectra in the visible region. From time-resolved emission measurements, all of these emission decays can be fitted with double exponential functions. The short components (in the range of ns) are attributed to prompt fluorescence, while the longer components (in the range of μs) are due to TADF (Table 1). For example, the TADF emission of 4DPAIPN displays a lifetime of 84 μs, which is long enough to function as a photosensitizer in bimolecular quenching processes. Interestingly, for all of the studied TADF dyes, strong emission quenching is observed in the presence of BIH (0.1 M)49 and triethanolamine (TEOA, 1.5 M) (quenching efficiencies ηq = 93–98%, Fig. S1†), which are the same conditions used in the photocatalytic reactions described below. These results indicate that the excited states of all of these organic compounds are quenched by BIH and/or TEOA. Therefore, these organic compounds can possibly work as photosensitizers to drive photocatalytic CO2 reduction.
Fig. 3 also shows the absorption spectrum of Mn in DMA. Since the onset of its absorption band is 501 nm, irradiation with visible light at λex ≤ 501 nm can potentially induce photodecomposition of Mn. Consequently, different reaction conditions (in terms of light sources and concentrations) were employed for testing the organic chromophores' ability of promoting photocatalytic CO2 reduction. In the first case, blue light LED peaked at 440 nm was used to irradiate a CO2-saturated DMA solution containing PS (250 μM), Mn (50 μM), BIH (0.1 M) and TEOA (1.5 M). Since Mn also absorbs light at 440 nm (ε = 1.50 × 103 M−1 cm−1), 5-times higher concentrations of PSs were used to limit the percentage of light absorbed by Mn and thus reduce its decomposition. For example, in the case of 3DPAFIPN (Fig. 4), after 20 h of irradiation, 53.8 μmol of CO and 13.0 μmol of HCOOH were produced along with 5.3 μmol of H2, which corresponds to turnover numbers based on 3DPAFIPN of TON3DPAFIPNCO = 108 and TON3DPAFIPNHCOOH = 26 and TON based on Mn of TONMnCO = 538 and TONMnHCOOH = 130. HCOOH formation stopped rapidly after approximately 3–5 h of irradiation, although CO kept on being produced even at longer irradiation times. The results of the photocatalytic reactions carried out under the same conditions as shown in Fig. 4 but using the other organic photosensitizers are summarized in Table 2. High photocatalytic activity was also observed when using 4DPAIPN as PS, but its performance was slightly lower under these reaction conditions compared to that observed when using 3DPAFIPN. On the other hand, utilization of the PSs possessing Cz moieties gave poor results in the photocatalytic reduction of CO2. Noteworthily, 4CzIPN, which was used as the PS for photocatalytic CO2 reduction in previous studies,41–45 did not show photocatalytic ability for CO2 reduction in our system. The lack of activity observed for 4CzIPN and 3CzClIPN can be explained by comparison between the first reduction potential of Mn (Ep(Mn/Mn˙−) = −1.83 V vs. Fc+/Fc) and the reduction potentials of 4CzIPN (E1/2(PS/PS˙−) = −1.72 V vs. Fc+/Fc) and 3CzClIPN (−1.61 V vs. Fc+/Fc) (Fig. 5 and Table 3). Indeed, these two reduction potentials are more positive than the first reduction wave of Mn, therefore the electron transfer processes from one-electron reduced 4CzIPN and 3CzClIPN to Mn are endothermic and cannot take place. In contrast, 4DPAIPN, 3DPAFIPN, and 5CzBN possess more negative reduction potentials compared to Mn, and this makes the reduction of the catalyst thermodynamically allowed.
 |
| | Fig. 4 Photocatalytic production of CO ( ), HCOOH ( ), HCOOH ( ), and H2 ( ), and H2 ( ) using 3DPAFIPN as a photosensitizer: CO2-saturated DMA solutions (2 mL) containing 3DPAFIPN (250 μM), Mn (50 μM), BIH (0.1 M), and TEOA (1.5 M) were irradiated at λex = 440 nm. ) using 3DPAFIPN as a photosensitizer: CO2-saturated DMA solutions (2 mL) containing 3DPAFIPN (250 μM), Mn (50 μM), BIH (0.1 M), and TEOA (1.5 M) were irradiated at λex = 440 nm. | |
Table 2 Photocatalytic reactions using PSs and the Mn(I) catalyst with a 5:1 ratioa
| Entry |
Product/μmol |
TONPS |
TONMn |
| CO |
HCOOH |
H2 |
CO |
HCOOH |
H2 |
CO |
HCOOH |
H2 |
|
CO2-saturated DMA solutions (2 mL) containing PS (250 μM), Mn (50 μM), BIH (0.1 M), and TEOA (1.5 M) were irradiated using LED light at λex = 440 nm for 20 h.
|
| 1 |
4DPAIPN
|
40.8 |
9.7 |
5.5 |
82 |
19 |
11 |
408 |
97 |
55 |
| 2 |
3DPAFIPN
|
53.8 |
13.0 |
5.3 |
108 |
26 |
11 |
538 |
130 |
53 |
| 3 |
5CzBN
|
3.1 |
1.1 |
0.3 |
6 |
2 |
<1 |
31 |
11 |
3 |
| 4 |
4CzIPN
|
0.4 |
<0.1 |
1.2 |
<1 |
<1 |
2 |
4 |
<1 |
11 |
| 5 |
3CzClIPN
|
0.4 |
<0.1 |
0.3 |
<1 |
<1 |
<1 |
4 |
<1 |
3 |
 |
| | Fig. 5 Cyclic voltammograms of (a) 4DPAIPN (red line), 3DPAFIPN (blue line), 5CzBN (black line), 4CzIPN (orange line), and 3CzClIPN (green line) measured in Ar-saturated DMA containing Et4NBF4 (0.1 M) as the supporting electrolyte with a Ag/AgNO3 (10 mM) reference electrode, and (b) of Mn in Ar-saturated (broken line) and CO2-saturated (solid line) solutions using the same electrolyte and reference electrode. The Fc+/Fc redox couple was also measured and taken as the standard. Scan rate was 0.2 V s−1. The dashed vertical line in (a) indicates the first reduction potential of Mn: Ep(Mn/Mn˙−) = −1.83 V. | |
Table 3 Electrochemical properties of PSs and CAT
| PS |
E
1/2/V vs. Fc+/Fc |
|
4DPAIPN
|
−2.08 |
|
3DPAFIPN
|
−1.94 |
|
5CzBN
|
−1.99 |
|
4CzIPN
|
−1.72 |
|
3CzClIPN
|
−1.61 |
| CAT |
E
p/V vs. Fc+/Fc |
|
Mn
|
−1.83 |
5CzBN exhibited much lower photocatalytic activity compared to 3DPAFIPN and 4DPAIPN because of its lower molar absorption coefficient at 440 nm (Table 1). In fact, under the tested experimental reaction conditions the absorbance of 5CzBN at 440 nm is even less compared to that of Mn (Fig. S2†). Consequently, photosensitive Mn is exposed to light and this leads to its decomposition and interruption of catalysis.
Given that 4DPAIPN and 3DPAFIPN have much stronger absorbances at 440 nm and suitable redox potentials, we focused only on these two PSs in order to further optimize the reaction conditions. First, the concentrations of PSs were decreased to 50 μM, so that the ratio of PS:Mn was 1:1 (entries 1 and 2 in Table 4). Under these conditions, the system using 4DPAIPN maintained good photocatalytic activity (TONCO = 357 and TONHCOOH = 102), while photocatalysis in the presence of 3DPAFIPN drastically decreased (TONCO = 133 and TONHCOOH = 55), suggesting that the stability of 3DPAFIPN is less compared to 4DPAIPN during the reaction. Second, a longer wavelength (λex = 470 nm) was employed for irradiating solutions containing PS:Mn = 1:1 (50 μM each, entries 3 and 4 in Table 4). In this case, the superior performance of 4DPAIPN compared to 3DPAFIPN is even more pronounced, as expected from the higher absorbance of 4DPAIPN (ε470 nm = 6.50 × 103 M−1 cm−1) compared to that of 3DPAFIPN (ε470 nm = 1.11 × 103 M−1 cm−1) at wavelengths longer that 450 nm. Fig. 6 shows the formation of CO2-reduction products over time using 50 μM of 4DPAIPN and Mn. In the initial stage, a larger amount of HCOOH was produced in comparison to that of CO, followed by a halt in HCOOH formation after 5 h of irradiation. In contrast, formation of CO continued even after 20 h of irradiation and CO becomes the major product. After 20 h of irradiation, 47.6 μmol of CO and 18.9 μmol of HCOOH were formed along with 2.9 μmol of H2, which corresponds to TONCO = 476, TONHCOOH = 189, and TONH2 = 29. These results are slightly larger than those obtained under irradiation at 440 nm because Mn has very little absorption at 470 nm, resulting in reduced photodegradation of the catalyst. The fact that formation of HCOOH stopped after 5 h of irradiation while CO continued forming even after 20 h suggests that different manganese complexes derived from Mn (and possibly Mn itself) form during the reaction. Since blue colour was not detected in the reaction solution during the photocatalytic reaction, the reductive dimer of the Mn complex did not probably contribute to the photocatalytic CO2 reduction.12,47,48 The reaction mechanism related to Mn and its derivatives in the photocatalytic reactions is currently under investigation in our laboratory.
Table 4 Photocatalytic CO2 reduction using an organic photosensitizer and Mn at 1:1 ratioa
| Entry |
Photosensitizer |
λ
ex/nm |
Product/μmol |
TONb |
| CO |
HCOOH |
H2 |
CO |
HCOOH |
H2 |
|
CO2-saturated DMA solutions (2 mL) containing PS (50 μM), Mn (50 μM), BIH (0.1 M), and TEOA (1.5 M) were irradiated using LED light at λex = 440 nm or 470 nm for 20 h.
In the case of entry 1, the same experiments were repeated three times and the results including the experimental errors are as follows: TONCO = 476 ± 20, TONHCOOH = 189 ± 15, TONH2 = 29 ± 10.
|
| 1 |
4DPAIPN
|
440 |
35.7 |
10.2 |
7.1 |
357 |
102 |
71 |
| 2 |
3DPAFIPN
|
13.3 |
5.5 |
1.0 |
133 |
55 |
10 |
| 3 |
4DPAIPN
|
470 |
47.6 |
18.9 |
2.9 |
476 |
189 |
29 |
| 4 |
3DPAFIPN
|
4.3 |
2.5 |
0.5 |
43 |
25 |
5 |
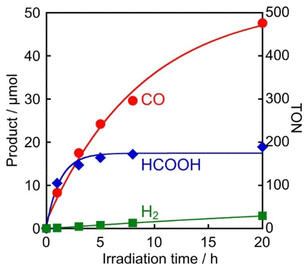 |
| | Fig. 6 Photocatalytic production of CO ( ), HCOOH ( ), HCOOH ( ), and H2 ( ), and H2 ( ) using 4DPAIPN as a photosensitizer: CO2-saturated DMA solutions (2 mL) containing 4DPAIPN (50 μM), Mn (50 μM), BIH (0.1 M), and TEOA (1.5 M) were irradiated at λex = 470 nm. ) using 4DPAIPN as a photosensitizer: CO2-saturated DMA solutions (2 mL) containing 4DPAIPN (50 μM), Mn (50 μM), BIH (0.1 M), and TEOA (1.5 M) were irradiated at λex = 470 nm. | |
In order to further characterize the system involving 4DPAIPN and Mn (1:1), the quantum yields (Φ) for CO2 reduction in the initial stage were determined using 480 nm monochromatic light with a light intensity of 5.0 × 10−9 Einstein per s. These were found to be ΦCO = 5.9 ± 0.2% and ΦHCOOH = 16.9 ± 1.3% from the linear production up to 9 h and 4 h of irradiation, respectively (Fig. 7 and S7†). This is the highest quantum yield of CO2 reduction using organic photosensitizers, to the best of our knowledge.
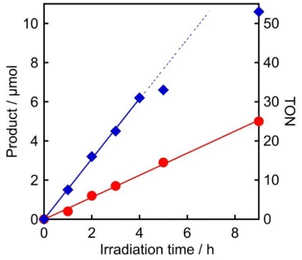 |
| | Fig. 7 Photocatalytic production of CO ( ) and HCOOH ( ) and HCOOH ( ) using 4DPAIPN: CO2-saturated DMA solutions (4 mL) containing 4DPAIPN (50 μM), Mn (50 μM), BIH (0.1 M), and TEOA (1.5 M) were irradiated at λex = 480 nm. Light intensity = 5.0 × 10−9 Einstein per s. ) using 4DPAIPN: CO2-saturated DMA solutions (4 mL) containing 4DPAIPN (50 μM), Mn (50 μM), BIH (0.1 M), and TEOA (1.5 M) were irradiated at λex = 480 nm. Light intensity = 5.0 × 10−9 Einstein per s. | |
Table 5 summarizes the control experiments for the photocatalytic reduction of CO2 using 4DPAIPN. In the absence of 4DPAIPN or Mn, no products were formed. Control experiments in the dark or using an Ar-saturated solution yielded no products of photocatalytic CO2 reduction, as well. Irradiation without BIH led to the formation of much smaller amounts of CO and HCOOH (TONCO = 7 and TONHCOOH = 4) compared to those with BIH (TONCO = 476 and TONHCOOH = 189) indicating that BIH mainly functioned as a sacrificial electron donor. In the absence of TEOA, the produced amounts of CO and HCOOH (TONCO = 68 and TONHCOOH = 24) also decreased. Therefore, the possible roles of TEOA should be as follows: (1) as a base, so as to deprotonate the one-electron oxidized BIH (BIH˙+) and prevent backward electron transfer from PS˙− to BIH˙+, (2) as a supporting ligand to capture CO2 into the manganese(I) complex to form carbonate ester species, i.e., Mn–CO2–OC2H4N(C2H4OH)2, which is a catalytic precursor for CO2 reduction,50 and (3) as a proton source. These results indicate that 4DPAIPN, Mn, and BIH worked as PS, CAT, and the sacrificial electron donor, respectively for photocatalytic reduction of CO2.
Table 5 Control experiments for photocatalytic CO2 reductiona
| Entry |
Absence |
Product/μmol (TON) |
| CO |
HCOOH |
H2 |
|
A CO2-saturated DMA solution (2 mL) containing 4DPAIPN (50 μM), Mn (50 μM), BIH (0.1 M), and TEOA (1.5 M) was irradiated using 470 nm light for 20 h.
Photocatalytic reactions were performed without 4DPAIPN, Mn, BIH, or TEOA, respectively.
The solution was placed in the dark for 20 h.
An Ar-saturated solution was used.
|
| 1 |
— |
47.6 (476) |
18.9 (189) |
2.9 |
| 2b |
4DPAIPN
|
<0.1 (<1) |
n.d. |
0.1 |
| 3b |
Mn
|
n.d. |
n.d. |
<0.1 |
| 4b |
BIH |
0.7 (7) |
0.4 (4) |
<0.1 |
| 5b |
TEOA |
6.8 (68) |
2.4 (24) |
1.5 |
| 6c |
Light irradiation |
n.d. |
n.d. |
n.d. |
| 7d |
CO2 |
n.d. |
n.d. |
0.7 |
In order to confirm that CO and HCOOH were produced via reduction of CO2, isotope labeling experiments using 13CO2 (99% 13C content) were performed. Fig. 8 displays the GC-MS chart of the gas phase after the photocatalytic reactions. When working in a 13CO2 atmosphere, a strong signal at m/z = 29 was observed at 5.5 min of retention time, which is attributed to 13CO, along with a weak signal at m/z = 28 attributable to 12CO (Fig. 8a). The ratio between these two signals is 97:3. When photocatalyzed under unlabeled CO2, on the other hand, almost only a signal at m/z = 28 was observed (Fig. 8b). For the identification of H13COOH and H12COOH, 1H and 13C NMR have been frequently used. In this photocatalytic system, however, NMR spectroscopy was not applicable probably due to the presence of paramagnetic manganese species. Therefore, HCOOH was extracted from the reaction solution with ethyl acetate and sulfuric acid aqueous solution, and was analyzed using GC-MS (Fig. 9).51 In the case of an unlabeled CO2 atmosphere, signals at m/z = 44, 45, 46 were observed at 8.0 min of retention time with the relative intensities of 0.18:0.71:1. Because the retention time and relative intensities of the signals were fairly similar to the reference sample of HCOOH solution, the observed signals are attributed to H12COOH (Fig. 9b). When photocatalyzed in a 13CO2 atmosphere, +1-shifted signals attributable to H13COOH were observed at m/z = 45, 46, 47 (Fig. 9a). The relative intensities of these signals were 0.21:0.74:1. These results clearly indicate that both CO and HCOOH were mostly produced from CO2.
 |
| | Fig. 8 GC-MS chromatograms of the gas phase after irradiation: a DMA (2 mL) solution containing 4DPAIPN (250 μM), Mn (250 μM), BIH (0.1 M), and TEOA (1.5 M) was irradiated at λex = 470 nm for 20 h under (a) 13CO2 or (b) unlabeled CO2 atmosphere. | |
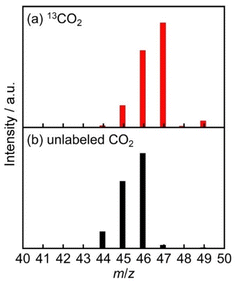 |
| | Fig. 9 GC-mass spectra of the liquid phase of the photocatalytic reaction at 8.0 min of retention time: a DMA (2 mL) solution containing 4DPAIPN (250 μM), Mn (250 μM), BIH (0.1 M), and TEOA (1.5 M) was irradiated at λex = 470 nm for 20 h under (a) 13CO2 or (b) unlabeled CO2 atmosphere. The produced HCOOH was extracted from the reaction solution with ethyl acetate and sulfuric acid aqueous solution (experimental details are shown in the ESI†). | |
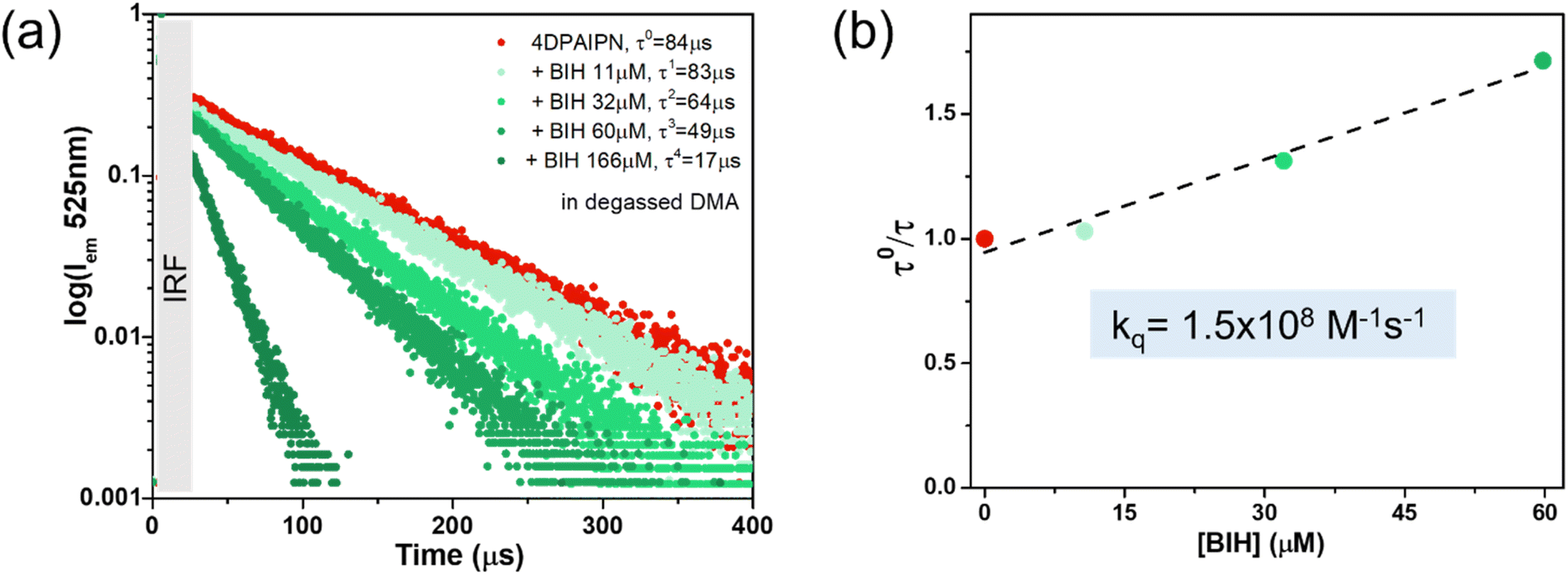
In order to study the mechanism of the reaction, Stern–Volmer analyses of 4DPAIPN using BIH and TEOA were performed. Since 4DPAIPN displays prompt fluorescence (PF) and TADF at the same wavelength (λem = 527 nm) with significantly different lifetimes (τPF = 2.9 ns and τTADF = 84 μs), Stern–Volmer plots using emission lifetimes were performed (Table 6). In the case of BIH, varying its concentration from the μM to mM range verified that both PF and TADF were efficiently quenched (Fig. S4† and 10) with large quenching constants, i.e., kq = 2.7 × 109 M−1 s−1 and 1.5 × 108 M−1 s−1, respectively.
Table 6 Quenching rate constants (kq) and quenching fractions in the presence of each quencher solely (ηq)
| Emission |
k
q/M−1 s−1 |
η
q in the presence of the quencher/% |
| BIH |
TEOA |
BIH (0.1 M) |
TEOA (1.5 M) |
| PF |
2.7 × 109 |
<107 |
44 |
∼0 |
| TADF |
1.5 × 108 |
3.2 × 104 |
>99 |
80 |
 |
| | Fig. 10 (a) Emission intensity decays (λex: 470 nm; λem: 525 nm) of 4DPAIPN upon addition of increasing amounts of BIH (up to 166 μM) in degassed DMA. Instrument response function has been grayed-out. (b) Corresponding Stern–Volmer plot. | |
On the other hand, 1.0 M of TEOA could not quench PF at all (Fig. S5†). Although TADF was quenched by TEOA, the rate constant (kq = 3.2 × 104 M−1 s−1) is much smaller compared to that of BIH (Fig. S6†). These results are further supported by thermodynamic calculations. In fact, the reported E00 transition energies of the singlet and triplet excited states of 4DPAIPN are 2.59 eV and 2.42 eV respectively,37 the reduction potentials of the singlet and triplet excited states of 4DPAIPN can be calculated as follows:
| | | E(1PS*/PS˙−) = E1/2(PS/PS˙−) + 1E00 = +0.51 V vs. Fc+/Fc | (1) |
| | | E(3PS*/PS˙−) = E1/2(PS/PS˙−) + 3E00 = +0.34 V vs. Fc+/Fc | (2) |
Since the oxidation potential of BIH is E(BIH˙+/BIH) = −0.09 V vs. Fc+/Fc,52 the electron transfer processes from BIH to the singlet and triplet excited states are highly exothermic and, given that E(1PS*/PS˙−) is more positive than E(3PS*/PS˙−), a higher quenching constant is expected for PF compared to TADF. In contrast, since E(TEOA˙+/TEOA) is ∼0.5 V vs. Fc+/Fc (0.80 V vs. SCE),53 reductive quenching of 4DPAIPN's T1 and S1 are both thermodynamically unfavorable or only slightly exergonic.
Furthermore, quenching fractions (ηq) can be calculated according to eqn (3):
| | | ηq = kqτ0[Q]/(1 + kqτ0[Q]) | (3) |
where
kq is the quenching constant,
τ0 is the emission lifetime in the absence of the quencher, and [Q] is the concentration of the quencher. Based on the quenching constant values obtained during Stern–Volmer experiments, when [BIH] = 0.1 M and in the absence of TEOA, the quenching efficiencies are
ηPFq = 44% and
ηTADFq > 99%. That is, BIH almost quantitatively quenches the longer-lived triplet excited state of
4DPAIPN (
i.e., TADF) and also about half of the singlet excited state that gives PF. In contrast, in the presence of only TEOA, no quenching of PF is possible even at [TEOA] = 1.5 M, whereas TADF is quenched with an efficiency of 80%. Nonetheless, under the reaction conditions employed in this work, TADF is quenched solely by BIH (>99%) because of the much faster quenching rate by BIH compared to that by TEOA (
kq[BIH] = 1.5 × 10
7 s
−1 and
kq[TEOA] = 4.8 × 10
4 s
−1). The pathway involving oxidative quenching of the excited state of
4DPAIPN by
Mn can be ruled out on the basis of its low concentration ([
Mn] = 50 μM). Moreover, the oxidation potential of the triplet excited state of
4DPAIPN calculated using
eqn (4) is almost equivalent to
Ep(
Mn/
Mn˙−) = −1.83 V, so oxidative quenching should be a slow process.
| | | E(PS˙+/3PS*) = E1/2(PS˙+/PS) − 3E00 = −1.87 V vs. Fc+/Fc | (4) |
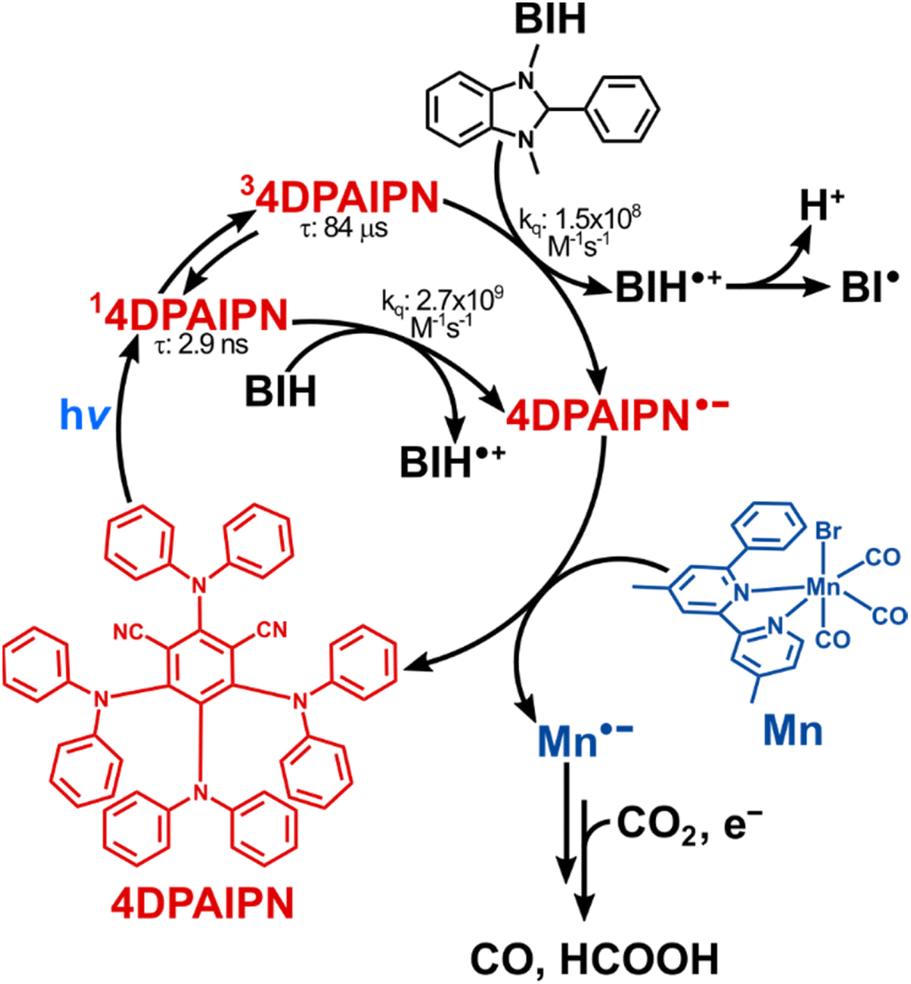
From these investigations, we can conclude that in the photocatalytic reaction using 4DPAIPN the long-lived excited state of 4DPAIPN is reductively quenched by BIH and the so-formed 4DPAIPN˙− reduces Mn. It is noteworthy that the control experiment without BIH (entry 4 in Table 5) produced only trace amounts of CO and HCOOH, even though 80% of TADF is quenched by TEOA in the absence of BIH as described above. The possible reasons are the following: (1) the oxidized product of TEOA might react with 4DPAIPN and/or Mn resulting in the loss of their function as a photosensitizer and/or catalyst; (2) as we previously reported, the slower quenching rate of the excited state might couple with more efficient back electron transfer in a solvated ion pair of [4DPAIPN˙−⋯TEOA˙+] resulting in a smaller quantum yield for the formation of 4DPAIPN˙−.52
On the basis of the experimental results described above, a hypothesis of the catalytic cycle that is active for the reduction of CO2 promoted by 4DPAIPN is reported in Fig. 11. The photocatalytic reduction is initiated by reductive quenching of both the singlet and triplet excited states of 4DPAIPN, mainly by BIH. The so-obtained 4DPAIPN˙− then efficiently donates one electron to Mn, thanks to its strong reduction power. The produced Mn˙− species might change its structure and react with CO2, although the exact mechanisms of these processes have not been clarified yet. Interesting mechanistic points including the interaction between the Mn complex and TEOA54,55 are currently under investigation in our laboratory. Finally, one more electron is supplied by BI˙ (produced by deprotonation of BIH˙+) and/or 4DPAIPN˙− to the reaction intermediate made from Mn˙− to give CO and HCOOH.
 |
| | Fig. 11 Proposed mechanism for CO2 reduction promoted by 4DPAIPN. | |
Conclusions
A series of fully organic TADF molecules with diphenylamine (4DPAIPN, 3DPAFIPN) or carbazole (5CzBN, 4CzIPN, 3CzClIPN) moieties as electron donating groups were systematically investigated as photoredox sensitizers for CO2 reduction coupled to a new Mn(I) molecular catalyst (Mn). All of the investigated TADF molecules were reductively quenched by BIH in DMA–TEOA solutions. Since the one-electron reduced species of 4CzIPN and 3CzClIPN cannot supply an electron to Mn owing to their low reductive powers, they cannot work as photosensitizers in the studied photocatalytic system. Similarly, 5CzBN cannot work as an efficient photosensitizer because of its low molar absorption coefficient in the visible region, even when using 440 nm light for irradiation. On the other hand, TADF molecules with diphenylamine groups are characterized by stronger absorption in the visible region and their one-electron reduced species have stronger reductive powers. In particular, 4DPAIPN proved to be the most efficient and durable photosensitizer in the photocatalytic reactions consisting in a molar ratio of photosensitizer:catalyst = 1:1 (TONCO+HCOOH > 650). The quantum yield for CO2 reduction was measured as 22.8 ± 1.5%, which, to the best of our knowledge, is the highest value among the reported photocatalytic systems for CO2-reduction using organic TADF molecules as photosensitizers. This demonstrated that a high molar absorption coefficient and red-shifted absorption, in addition to negative reduction potentials, are all beneficial for the reaction. Moreover, mechanistic investigation proved that a long excited state lifetime is essential in order to favour the efficient bimolecular quenching processes of the photosensitizer, and that BIH is the species mainly responsible for its reductive quenching. One limit to the overall reaction's quantum yield is the photosensitizer's internal conversion (S1 → S0) deactivation constant (in the order of 1 × 108 s−1) and prompt fluorescence, which lead to fast deactivation of the photosensitizer before it has been able to interact with the other species present in the mixture. Development of TADF photosensitizers with lower non-radiative and radiative deactivation from the singlet excited state will therefore be beneficial for the improvement of the reaction's efficiency.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
The work was supported by JSPS KAKENHI Grant Number JP22K19081, JP20H00396, and JP17H06440 in Scientific Research on Innovative Areas “Innovations for Light-Energy Conversion (I4LEC)”. Italian national projects (PRIN 2017 20174SYJAF, SURSUMCAT and PRIN2017 20172M3K5N, CHIRALAB) are acknowledged for financial support of this research. P. C., P. G. C., A. G., E. B., F. C., and S. P. also acknowledge the University of Bologna for financial support.
References
- N. Armaroli and V. Balzani, Angew. Chem., Int. Ed., 2007, 46, 52–66 CrossRef CAS PubMed
 .
.
- G. Segev, J. Kibsgaard, C. Hahn, Z. J. Xu, W. S. Cheng, T. G. Deutsch, C. Xiang, J. Z. Zhang, L. Hammarström, D. G. Nocera, A. Z. Weber, P. Agbo, T. Hisatomi, F. E. Osterloh, K. Domen, F. F. Abdi, S. Haussener, D. J. Miller, S. Ardo, P. C. McIntyre, T. Hannappel, S. Hu, H. Atwater, J. M. Gregoire, M. Z. Ertem, I. D. Sharp, K.-S. Choi, J. S. Lee, O. Ishitani, J. W. Ager, R. R. Prabhakar, A. T. Bell, S. W. Boettcher, K. Vincent, K. Takanabe, V. Artero, R. Napier, B. R. Cuenya, M. T. M. Koper, R. Van De Krol and F. Houle, J. Phys. D: Appl. Phys., 2022, 55, 323003 CrossRef .
- V. Balzani, G. Bergamini and P. Ceroni, Angew. Chem., Int. Ed., 2015, 54, 11320–11337 CrossRef CAS PubMed .
- Y. Yamazaki, H. Takeda and O. Ishitani, J. Photochem. Photobiol., C, 2015, 25, 106–137 CrossRef CAS .
-
Y. Tamaki, H. Takeda and O. Ishitani, Molecular Technology, Wiley, 2019, vol. 1, pp. 209–249 Search PubMed .
- A. M. Cancelliere, F. Puntoriero, S. Serroni, S. Campagna, Y. Tamaki, D. Saito and O. Ishitani, Chem. Sci., 2020, 11, 1556–1563 RSC .
- R. Konduri, H. Ye, F. M. MacDonnell, S. Serroni, S. Campagna and K. Rajeshwar, Angew. Chem., Int. Ed., 2002, 41, 3185–3187 CrossRef CAS PubMed .
- C.-F. Leung and T.-C. Lau, Energy Fuels, 2021, 35, 18888–18899 CrossRef CAS .
- K. E. Dalle, J. Warnan, J. J. Leung, B. Reuillard, I. S. Karmel and E. Reisner, Chem. Rev., 2019, 119, 2752–2875 CrossRef CAS PubMed .
- H. Takeda, C. Cometto, O. Ishitani and M. Robert, ACS Catal., 2017, 7, 70–88 CrossRef CAS .
- C. Steinlechner, A. F. Roesel, E. Oberem, A. Päpcke, N. Rockstroh, F. Gloaguen, S. Lochbrunner, R. Ludwig, A. Spannenberg, H. Junge, R. Francke and M. Beller, ACS Catal., 2019, 9, 2091–2100 CrossRef CAS .
- L. Rotundo, R. Gobetto and C. Nervi, Curr. Opin. Green Sustainable Chem., 2021, 31, 100509 CrossRef CAS .
- H. Yuan, B. Cheng, J. Lei, L. Jiang and Z. Han, Nat. Commun., 2021, 12, 1835 CrossRef CAS PubMed .
- A. Rosas-Hernández, C. Steinlechner, H. Junge and M. Beller, Green Chem., 2017, 19, 2356–2360 RSC .
- L. Gracia, L. Luci, C. Bruschi, L. Sambri, P. Weis, O. Fuhr and C. Bizzarri, Chem.–Eur. J., 2020, 26, 9929–9937 CrossRef CAS PubMed .
- Y. Sakaguchi, A. Call, M. Cibian, K. Yamauchi and K. Sakai, Chem. Commun., 2019, 55, 8552–8555 RSC .
- H. Takeda, K. Ohashi, A. Sekine and O. Ishitani, J. Am. Chem. Soc., 2016, 138, 4354–4357 CrossRef CAS PubMed .
- H. Takeda, Y. Monma, H. Sugiyama, H. Uekusa and O. Ishitani, Front. Chem., 2019, 7, 1–12 CrossRef PubMed .
- X. Zhang, M. Cibian, A. Call, K. Yamauchi and K. Sakai, ACS Catal., 2019, 9, 11263–11273 CrossRef CAS .
- H. Takeda, H. Kamiyama, K. Okamoto, M. Irimajiri, T. Mizutani, K. Koike, A. Sekine and O. Ishitani, J. Am. Chem. Soc., 2018, 140, 17241–17254 CrossRef CAS PubMed .
- C. Bizzarri, Eur. J. Org. Chem., 2022, e202200185 CAS .
- J. Shipp, S. Parker, S. Spall, S. L. Peralta-Arriaga, C. C. Robertson, D. Chekulaev, P. Portius, S. Turega, A. Buckley, R. Rothman and J. A. Weinstein, Inorg. Chem., 2022, 61, 13281–13292 CrossRef CAS PubMed .
- J. Bonin, M. Robert and M. Routier, J. Am. Chem. Soc., 2014, 136, 16768–16771 CrossRef CAS PubMed .
- S. Matsuoka, K. Yamamoto, C. Pac and S. Yanagida, Chem. Lett., 1991, 20, 2099–2100 CrossRef .
- S. Matsuoka, K. Yamamoto, T. Ogata, M. Kusaba, N. Nakashima, E. Fujita and S. Yanagida, J. Am. Chem. Soc., 1993, 115, 601–609 CrossRef CAS .
- T. Dhanasekaran, J. Grodkowski, P. Neta, P. Hambright and E. Fujita, J. Phys. Chem. A, 1999, 103, 7742–7748 CrossRef CAS .
- L. Chen, Y. Qin, G. Chen, M. Li, L. Cai, Y. Qiu, H. Fan, M. Robert and T.-C. Lau, Dalton Trans., 2019, 48, 9596–9602 RSC .
- H. Rao, J. Bonin and M. Robert, ChemSusChem, 2017, 10, 4447–4450 CrossRef CAS PubMed .
- Z. Guo, S. Cheng, C. Cometto, E. Anxolabéhère-Mallart, S.-M. Ng, C.-C. Ko, G. Liu, L. Chen, M. Robert and T.-C. Lau, J. Am. Chem. Soc., 2016, 138, 9413–9416 CrossRef CAS PubMed .
- H. Rao, C.-H. Lim, J. Bonin, G. M. Miyake and M. Robert, J. Am. Chem. Soc., 2018, 140, 17830–17834 CrossRef CAS PubMed .
- T. Ogata, Y. Yamamoto, Y. Wada, K. Murakoshi, M. Kusaba, N. Nakashima, A. Ishida, S. Takamuku and S. Yanagida, J. Phys. Chem., 1995, 99, 11916–11922 CrossRef CAS .
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von Zelewsky, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS .
- D. Sasikumar, A. T. John, J. Sunny and M. Hariharan, Chem. Soc. Rev., 2020, 49, 6122–6140 RSC .
- J. Zhao, W. Wu, J. Sun and S. Guo, Chem. Soc. Rev., 2013, 42, 5323 RSC .
- E. Bassan, A. Gualandi, P. G. Cozzi and P. Ceroni, Chem. Sci., 2021, 12, 6607–6628 RSC .
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed .
- V. K. Singh, C. Yu, S. Badgujar, Y. Kim, Y. Kwon, D. Kim, J. Lee, T. Akhter, G. Thangavel, L. S. Park, J. Lee, P. C. Nandajan, R. Wannemacher, B. Milián-Medina, L. Lüer, K. S. Kim, J. Gierschner and M. S. Kwon, Nat. Catal., 2018, 1, 794–804 CrossRef CAS .
- E. Speckmeier, T. G. Fischer and K. Zeitler, J. Am. Chem. Soc., 2018, 140, 15353–15365 CrossRef CAS PubMed .
- M. A. Bryden and E. Zysman-Colman, Chem. Soc. Rev., 2021, 50, 7587–7680 RSC .
- A. Gualandi, M. Anselmi, F. Calogero, S. Potenti, E. Bassan, P. Ceroni and P. G. Cozzi, Org. Biomol. Chem., 2021, 19, 3527–3550 RSC .
- Y. Wang, X.-W. Gao, J. Li and D. Chao, Chem. Commun., 2020, 56, 12170–12173 RSC .
- Y. Wang, T. Liu, L. Chen and D. Chao, Inorg. Chem., 2021, 60, 5590–5597 CrossRef CAS PubMed .
- Y. Wang, L. Chen, T. Liu and D. Chao, Dalton Trans., 2021, 50, 6273–6280 RSC .
- T. Liu, Y. Li, Y. Fang, Y. Fu, Y. Chen and D. Chao, Chem.–Asian J., 2022, 17, e2022008, DOI:10.1002/asia.202200846 .
- Y. Fang, T. Liu, L. Chen and D. Chao, Chem. Commun., 2022, 58, 7972–7975 RSC .
- Quantum yields were recalculated by using the following equations (see ref. 4) because reductants such as triethylamine used in ref. 39–43 are known to work as two-electron donor in one photosensitized cycle. Φ = (total number of products)/(number of photons).
- M. Bourrez, F. Molton, S. Chardon-Noblat and A. Deronzier, Angew. Chem., Int. Ed., 2011, 50, 9903–9906 CrossRef CAS PubMed .
- M. D. Sampson, A. D. Nguyen, K. A. Grice, C. E. Moore, A. L. Rheingold and C. P. Kubiak, J. Am. Chem. Soc., 2014, 136, 5460–5471 CrossRef CAS PubMed .
- Y. Pellegrin and F. Odobel, C. R. Chim., 2017, 20, 283–295 CrossRef CAS .
- H. Koizumi, H. Chiba, A. Sugihara, M. Iwamura, K. Nozaki and O. Ishitani, Chem. Sci., 2019, 10, 3080–3088 RSC .
- Y. Kuramochi, M. Kamiya and H. Ishida, Inorg. Chem., 2014, 53, 3326–3332 CrossRef CAS PubMed .
- K. Ozawa, Y. Tamaki, K. Kamogawa, K. Koike and O. Ishitani, J. Chem. Phys., 2020, 153, 154302 CrossRef CAS PubMed .
- K. Kalyanasundaram, J. Chem. Soc., Faraday Trans. 2, 1986, 82, 2401 RSC .
- T. Nakajima, Y. Tamaki, K. Ueno, E. Kato, T. Nishikawa, K. Ohkubo, Y. Yamazaki, T. Morimoto and O. Ishitani, J. Am. Chem. Soc., 2016, 138, 13818–13821 CrossRef CAS PubMed .
- T. Morimoto, T. Nakajima, S. Sawa, R. Nakanishi, D. Imori and O. Ishitani, J. Am. Chem. Soc., 2013, 135, 16825–16828 CrossRef CAS PubMed .
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Rei
Inoue
c,
David
Fabry
c,
Francesco
Calogero
ab,
Simone
Potenti
ab,
Rei
Inoue
c,
David
Fabry
c,
Francesco
Calogero
ab,
Simone
Potenti




 ), HCOOH (
), HCOOH ( ), and H2 (
), and H2 ( ) using 3DPAFIPN as a photosensitizer: CO2-saturated DMA solutions (2 mL) containing 3DPAFIPN (250 μM), Mn (50 μM), BIH (0.1 M), and TEOA (1.5 M) were irradiated at λex = 440 nm.
) using 3DPAFIPN as a photosensitizer: CO2-saturated DMA solutions (2 mL) containing 3DPAFIPN (250 μM), Mn (50 μM), BIH (0.1 M), and TEOA (1.5 M) were irradiated at λex = 440 nm.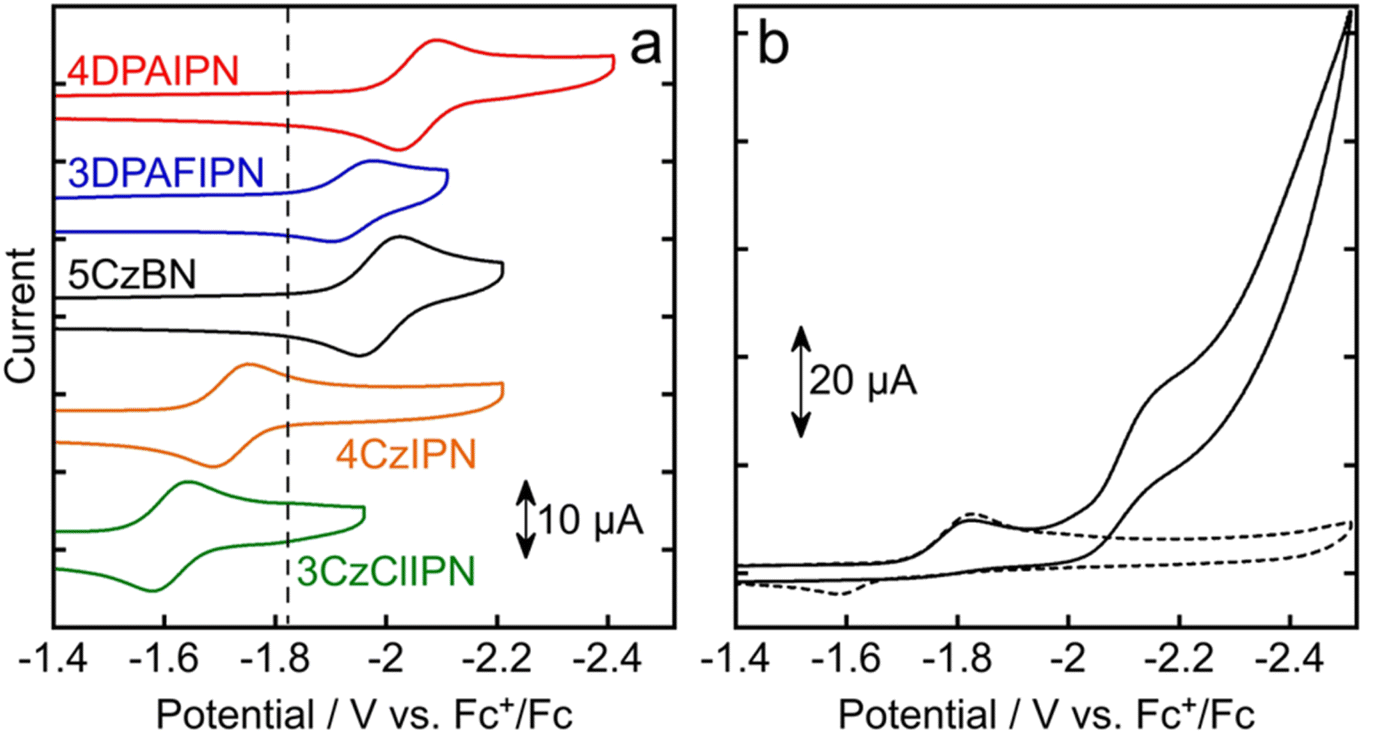

 ), HCOOH (
), HCOOH ( ), and H2 (
), and H2 ( ) using 4DPAIPN as a photosensitizer: CO2-saturated DMA solutions (2 mL) containing 4DPAIPN (50 μM), Mn (50 μM), BIH (0.1 M), and TEOA (1.5 M) were irradiated at λex = 470 nm.
) using 4DPAIPN as a photosensitizer: CO2-saturated DMA solutions (2 mL) containing 4DPAIPN (50 μM), Mn (50 μM), BIH (0.1 M), and TEOA (1.5 M) were irradiated at λex = 470 nm.
 ) and HCOOH (
) and HCOOH ( ) using 4DPAIPN: CO2-saturated DMA solutions (4 mL) containing 4DPAIPN (50 μM), Mn (50 μM), BIH (0.1 M), and TEOA (1.5 M) were irradiated at λex = 480 nm. Light intensity = 5.0 × 10−9 Einstein per s.
) using 4DPAIPN: CO2-saturated DMA solutions (4 mL) containing 4DPAIPN (50 μM), Mn (50 μM), BIH (0.1 M), and TEOA (1.5 M) were irradiated at λex = 480 nm. Light intensity = 5.0 × 10−9 Einstein per s.