
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A novel biorefinery concept based on marginally used halophyte biomass†
Maxwel
Monção
a,
Petter Paulsen
Thoresen
a,
Tobias
Wretborn
a,
Heiko
Lange
abc,
Ulrika
Rova
a,
Paul
Christakopoulos
 a and
Leonidas
Matsakas
*a
a and
Leonidas
Matsakas
*a
aBiochemical Process Engineering, Division of Chemical Engineering, Department of Civil, Environmental and Natural Resources Engineering, Luleå University of Technology, SE-971 87 Luleå, Sweden. E-mail: leonidas.matsakas@ltu.se; Tel: +46 (0) 920 493043
bDepartment of Earth and Environmental Sciences, University of Milano-Bicocca, Piazza della Scienza 1, 20126 Milan, Italy
cNBFC – National Biodiversity Future Center, 90133 Palermo, Italy
First published on 30th June 2023
Abstract
Halophytes have major potential in biorefinery as these salt tolerant crops have prospects as an alternative biomass to meet energy demands and provide value-added products with reduced effects in terms of food security and environmental damage when compared to other crops. In this study, we investigated the effects of organosolv pretreatment process parameters on the fractionation of residual fibers from pressed Salicornia ramosissima and how it affects the fractions of cellulose, lignin, and hemicelluloses. Pretreated pulps contained as high as 48.95% w/w cellulose, a 2.9-fold increase from the untreated fibers. The delignification of pulp was as high as 75.01% and hemicellulose removal reached 96.38%. The hemicellulose fractions contained as high as 78.49% oligomers and we identified up to 30.4% linear xylooligosaccharides in the composition. The majority of the fragments of hemicelluloses had molecular weights lower than 1000 Da. Isolated lignin samples had in most cases very low sugar and ash contamination with a reduced molecular weight. The typical G-, S-, and H-type aromatic units were detected in the lignin, together with β-O-4′, β-5′, β-β′, and dibenzodioxocine links. The results suggest a novel applicability of S. ramosissima in a biorefinery context with fractionation deriving building blocks for value added products.
Introduction
Population growth, the expansion of cities, and an increased concern for the environment have induced a new urgency to the quest for renewable alternatives to fossil fuels.1,2 Droughts as well as exorbitant oil and gas prices have contributed to an energy crisis in several countries.3–5Biomass offers an alternative to coal and oil as a source of value-added products.6,7 Owing to the complexity of lignocellulosic biomass, fractionation is an indispensable tool for separating cellulose, hemicellulose, and lignin streams.8,9 The different fractions obtained can be directed towards downstream applications, such as the manufacturing of chemicals, biofuels for transport, textiles, and pharmaceuticals or nutraceuticals.6,10
During organosolv fractionation, biomass is typically treated at temperatures between 170 and 220 °C in a solution containing water and organic solvent, which enables both physical (temperature) and chemical (solvent) interactions.11,12 Organosolv fractionation is a very efficient means of producing high-purity cellulose, sulfur-free lignin residues, and liquor containing hemicelluloses hydrolyzed into oligomers, monomers, furans, and organic acids.13–15 Organosolv has been studied and applied to various lignocellulosic materials, including hardwoods, softwoods, and grasses as feedstock at both laboratory and industrial scales.16–18
The global increase in population results in a greater demand for arable land and urbanization, which may hinder the cultivation of plants destined for the production of lubricants, resins, clothing, and energy. To avoid taking away precious land from food production, plants aimed for non-food uses should be cultivated in terrain that is not immediately suitable for agriculture, such as coastal regions or drylands under desertification, whose salt content is too high for most crops to grow. Halophytes represent an excellent alternative for such regions,19 owing to their tolerance of up to 1 M NaCl.20 Highly saline soils may be either domesticated with progressively more tolerant halophytes or they can be cultivated with species already exhibiting an elevated salt tolerance.21
Halophytes have been associated with the production of fuel, charcoal, groundcover, feed, and food, as well as intercropped cultures and heavy metal remediation.22–24Salicornia ramosissima, which belongs to a widespread genus of halophytes, grows well at 110–200 mM NaCl, is tolerant also towards UV radiation and acidity, and is easy to cultivate.25,26 Research on the characterization of the plant has been carried out and the potential application of bioactive extracts in nutraceuticals, biopharmaceuticals, and cosmetics27 given the presence of phenolic acids, flavonols, organic acids, proteins, carbohydrates, and lipids.28,29 Lately, the research of S. ramosissima applications has been carried out on food applications.30–32 Other applications of S. ramosissima have been recently studied including as a source of cosmetics,33 kitchen salt,34 antioxidants35 and biogas.26
In this study, we have evaluated the application of S. ramosissima residual fibers, a by-product of the juicing process applied for the extraction of bioactive compounds. To determine the potential of this plant residue in a biorefinery context, parameters related to fractionation and downstream processing of samples have been assessed, and structures and components found in the lignin and hemicellulose fractions have been characterized.
Experimental
Feedstock
S. ramosissima was cultivated in sandy soil in Portugal and irrigated with water containing 12–13 g L−1 NaCl. Samples were harvested after 26 weeks of cultivation. Biomass was fractionated with a lab-scale single-auger juicer, yielding 66.7% juice and 33.3% fibers. Untreated dejuiced fibers included 16.64% w/w cellulose, 22.42% w/w hemicelluloses, 15.86% w/w lignin, 32.53% w/w extractives, and 0.78% w/w ashes (total ashes including those in the extractives amounted to 13.21%). Semi-quantitative elemental analysis identified sodium (6.30% w/w), potassium (0.78% w/w), magnesium (0.26% w/w), phosphorus (0.26% w/w), calcium (0.24% w/w), sulfur (0.17% w/w), and bromine (0.04% w/w) as the main inorganic elements.Organosolv pretreatment
The fibers were fractionated in a rotating multidigester organosolv reactor (Haato, Vantaa, Finland) composed of six vessels with 2.5 L capacity each. The biomass load for each condition was 90 g, with a solvent ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 w/v. The solvent was an ethanol:water solution at either 40%:60% v/v or 60%:40% v/v. After heating up to the selected temperature, the reaction was maintained at that temperature for a defined time (Table 1). Next, the reactor was cooled down to 40 °C and the material was vacuum-filtered to separate the pulp from the process liquor. Insoluble fibers (pulp) were washed with the same solvent as in the previous step, dried at room temperature, and collected in plastic bottles until further use. The liquor phase was processed in a rotary evaporator (Heidolph, Schwabach, Germany) to evaporate the ethanol, followed by centrifugation at 10000×g for 10 min to precipitate lignin. The latter was then freeze-dried (Telstar, Terrassa, Spain) and stored in plastic bottles until further use. The supernatant liquid (hemicellulose fraction) was collected and stored in plastic bottles at 4 °C.
:10 w/v. The solvent was an ethanol:water solution at either 40%:60% v/v or 60%:40% v/v. After heating up to the selected temperature, the reaction was maintained at that temperature for a defined time (Table 1). Next, the reactor was cooled down to 40 °C and the material was vacuum-filtered to separate the pulp from the process liquor. Insoluble fibers (pulp) were washed with the same solvent as in the previous step, dried at room temperature, and collected in plastic bottles until further use. The liquor phase was processed in a rotary evaporator (Heidolph, Schwabach, Germany) to evaporate the ethanol, followed by centrifugation at 10000×g for 10 min to precipitate lignin. The latter was then freeze-dried (Telstar, Terrassa, Spain) and stored in plastic bottles until further use. The supernatant liquid (hemicellulose fraction) was collected and stored in plastic bottles at 4 °C.
| Code | Temperature (°C) | Time (min) | Ethanol content (% v/v) |
|---|---|---|---|
| 0A4 | 200 | 15 | 40 |
| 0A6 | 200 | 15 | 60 |
| 0B4 | 200 | 30 | 40 |
| 0B6 | 200 | 30 | 60 |
| 0C4 | 200 | 45 | 40 |
| 0C6 | 200 | 45 | 60 |
| 1A4 | 180 | 15 | 40 |
| 1A6 | 180 | 15 | 60 |
| 1B4 | 180 | 30 | 40 |
| 1B6 | 180 | 30 | 60 |
| 1C4 | 180 | 45 | 40 |
| 1C6 | 180 | 45 | 60 |
| 1D4 | 180 | 60 | 40 |
| 1D6 | 180 | 60 | 60 |
| 2B4 | 160 | 30 | 40 |
| 2B6 | 160 | 30 | 60 |
Analytical methods
Extractives were isolated following the National Renewable Energy Laboratory NREL/TP 510 42619 protocol using a Soxhlet apparatus,36 first with water and then with ethanol, and finally with chloroform:methanol (2:1, v/v). The solvents were evaporated in a rotary evaporator, and the fractions were quantified and stored. The cellulose, hemicellulose, and lignin composition of untreated biomass and pretreated fractions was analyzed according to the NREL/TP 510 42618 protocol.37 The sugar content was quantified by high-performance anion exchange chromatography (HPAEC) (Thermo Scientific, Waltham, MA, USA) using a CarboPac PA-1 column (4 × 250 mm; Dionex™, Thermo Scientific) with a pulsed amperometric detector equipped with a gold electrode. Neutral sugars in hemicellulose monomers, linear cellooligosaccharies, and xylooligosaccharides were quantified directly, whereas oligomers were first hydrolyzed to monomers using 4% w/w aqueous H2SO4 according to the NREL/TP-510-42623 protocol.38 The samples were eluted with a sodium acetate gradient generated with buffer A (100 mm NaOH) and buffer B (1 m NaOAc in 100 mm NaOH). The gradient consisted of 0–30% buffer B for 25 min, followed by a wash step with 30–100% buffer B for 5 min, and equilibration with buffer A for 9 min at a flow rate of 1 mL min−1. For uronic acid analysis, the HPAEC apparatus was equipped with a PA-20 column (3 × 150 mm; Dionex™) maintained at 30 °C. The samples were eluted with buffer A (de-ionized water), buffer B (200 mm NaOH), and buffer C (100 mm NaOAc in 100 mm NaOH) according to the following gradient: 0–18 min, isocratic step (98.8% A and 1.2% B); 18–20 min, 1.2–50% B; 20–30 min, 50% A and 50% B; 30.1–46 min, 100% C; 46.1–50 min, 100% B; and 50.1–60 min, 98.8% A and 1.2% B at 0.4 mL min−1. The inorganic ash content was determined gravimetrically by ashing the samples at 550 °C for 3 h, with a temperature increase of 1 °C/min. The moisture content was determined gravimetrically after drying the samples at 95 °C overnight until a constant weight was attained.
Enzymatic saccharification
Enzymatic saccharification of pretreated biomass was performed in duplicate in 2 mL microcentrifuge tubes with 3% w/v pretreated biomass at a volume of 1 mL. Saccharification was evaluated using the commercial cellulase enzyme solution Cellic® CTec2 (Novozymes A/S, Bagsværd, Denmark) at an enzyme load of 20 FPU per gsolids in 50 mM citrate buffer (pH 5). The samples were incubated at 50 °C for 72 h in a thermomixer at 900 rpm, with aliquots checked every 24 h. After incubation, the enzyme solution was inactivated by placing the tubes in a water bath at 95 °C for 5 min and then centrifuging at 12000×g for 10 min at room temperature. The supernatant was filtered through a 0.22 μm filter (Sartorius, Göttingen, Germany) and the sugars were quantified by HPAEC with pulsed amperometric detection.
Liquid chromatography-mass spectrometry (LC-MS)
For LC-MS, 1 μL samples were purified prior to analysis on a graphite C18 ZipTip column (Merck Millipore, Burlington, MA, USA). The column was activated with 3 × 50 μL of a solution containing 90% v/v acetonitrile and 0.5% v/v formic acid, washed with 3 × 50 μL of 0.5% v/v formic acid, loaded with the sample, washed with 3 × 50 μL of a wash solution, and eluted with 3 × 50 μL of a glycan elution solution composed of 25% acetonitrile and 0.5% v/v formic acid. For reverse-phase chromatography, a column (10 cm × 250 μm) with 5 μm porous graphite particles (Hypercarb; Thermo-Hypersil, Runcorn, UK) was used. The samples were eluted with an acetonitrile gradient composed of buffer A (10 mm ammonium bicarbonate) and buffer B (10 mm ammonium bicarbonate in 80% acetonitrile). The gradient (0–45% buffer B) was eluted for 46 min, followed by a wash step with 100% buffer B and equilibration with buffer A for 24 min. The samples were analyzed on a linear ion trap mass spectrometer (LTQ XL; Thermo Electron, San José, CA, USA) in both negative and positive ion modes, using an Ion Max (Thermo Scientific) standard electrospray ionization source equipped with a stainless-steel needle kept at −3.5 kV. Compressed air was used as the nebulizer gas. The heated capillary was kept at 270 °C, and the capillary voltage was −50 kV. A full scan (m/z = 380–2000, two microscans, maximum 100 ms, and target value of 30000) was performed, followed by data-dependent MS2 scans (two microscans, maximum 100 ms, and target value of 10000) with a normalized collision energy of 35%, isolation window of 2.5 units, activation q = 0.25, and activation time of 30 ms. The threshold for MS2 was set to 300 counts. Data were acquired and processed with Xcalibur software (Version 2.0.7; Thermo Scientific). Spectra were interpreted manually using the GlycoWorkbench glycan analysis tool (https://www.eurocarbdb.org/applications/ms-tools).
Phosphorus-31 nuclear magnetic resonance (31P NMR)
Approx. 30 mg of lignin was accurately weighted and dissolved in 400 μL of anhydrous CDCl3/pyridine solution (1:1.6 (v/v)). 100 μL of a standard solution of N-hydroxy-5-norbornene-2,3-dicarboxylic acid imide (e-HNDI) (0.1 M in anhydrous CDCl3/pyridine solution) containing Cr(III) acetylacetonate as the relaxation agent (ca. 5 mg mL−1 was added. The phosphitylating reagent 2-chloro-4,4,5,5-tetramethyl-1,2,3-dioxiphosphospholane (100 μL) was added to the solution, which was then stirred for 2 h at room temperature. Spectra were recorded on a Bruker AVANCE 400 MHz equipped with a 5 mm double resonance broadband BBI inverse probe at 25 °C with a total of 160 scans. All chemical shifts reported are relative to the reaction product of water with Cl-TMDP, which gives a sharp signal in pyridine/CDCl3 at 132.2 ppm. Data were processed and quantified with MestreNova Version 9.0.1.Carbon-13 nuclear magnetic resonance (13C NMR)
For quantitative 13C NMR analysis, approx. 80 mg of lignin was dissolved in 500 μL DMSO-d6. We applied 50 μL (∼1.5 mg mL−1) of Cr(III) acetylacetonate in DMSO-d6 as a spin-relaxation agent and 50 μL (∼15 mg mL−1) of trioxane (92.92 ppm) in DMSO-d6 as an internal standard. Spectra were recorded on a Bruker 600 MHz AVANCE III spectrometer equipped with a 5 mm BBO broadband (1H/19F/2D) z-gradient cryo-probe and controlled with TopSpin 3.6.4 software at 30 °C, with a total of 20000–24000 scans. An inverse-gated proton decoupling pulse sequence was applied with a 90° pulse width, acquisition time of 1.2 s, and relaxation delay of 1.7 s. Data were processed with MestreNova Version 9.0.1 (Mestrelab Research S.L., Santiago de Campostela, Spain).
1H–13C heteronuclear single quantum coherence (HSQC)
For 1H–13C HSQC analysis, the same sample as prepared for the acquisition of the quantitative 13C NMR was used. Spectra were recorded on a Bruker 600 MHz AVANCE III spectrometer equipped with a 5 mm BBO broadband (1H/19F/2D) z-gradient cryo-probe and controlled with TopSpin 3.6.4 software at 30 °C. The Bruker hsqcetgpsisp2.2 pulse program in DQD acquisition mode was applied with NS = 64, TD = 2048 (F2) and 512 (F1), SQ = 12.9869 ppm (F2) and 164.9996 ppm (F1), O2 (F2) = 2601.36 Hz and O1 (F1) = 7799.05 Hz, D1 = 2 s, CNST2 1J (C–H) = 145, and acquisition times of 197.0176 ms (F2) and 15.4164 ms (F1). Data were processed with MestreNova Version 9.0.1.Size exclusion chromatography of lignins
To determine the molecular weight of lignin, we performed gel permeation chromatography (GPC). The samples were pretreated via acetobromination adopting the standard literature procedure39 by adding 0.9 mL glacial acetic acid and 0.1 mL acetyl bromide to 5 mg of lignin powder, followed by stirring for 2 h at room temperature in closed amber glass vials. The stirred solution was subsequently transferred to a round flask for the evaporation of the solvent in a rotary evaporator (Heidolph) at 50 °C and 50 mbar. Two wash steps were applied with 1 mL tetrahydrofuran (THF) followed by solvent evaporation. The material was dissolved in 1 mL THF and then filtered through 0.22 μm hydrophobic filters (Sartorius). Finally, the samples were analyzed by high-performance liquid chromatography (PerkinElmer, Waltham, MA, USA) using a Styragel® HR 4E column (Waters, Milford, MA, USA) and a UV detector (set to λ = 280 nm) at 40 °C with a flow rate of 0.6 mL min−1 and THF as the mobile phase. Calibration was carried out using polystyrene standards of 500–90000 Da (Sigma-Aldrich, St. Louis, MO, USA). The numbers were rounded up to 100 s due to resolution of the method.
Size exclusion chromatography of hemicelluloses
GPC was used to determine the molecular weight range of hemicelluloses in S. ramosissima. Samples were filtered through 0.22 μm hydrophilic filters (Sartorius) and analyzed by high-performance liquid chromatography using a refractive index detector and a series of two Ultrahydrogel 250 and 120 columns (Waters) operated at 60 °C, with deionized water as the mobile phase, and a flow rate of 0.6 mL min−1. Calibration was carried out using cellobiose (MW 342 Da; Sigma-Aldrich) and dextran (MW 1000, 5000, 12500, 25000, 50000, and 80000 Da; Sigma-Aldrich).
Results and discussion
Pulp
Organosolv pretreatment of dejuiced fibers from S. ramosissima was performed in 16 different combinations of temperature (160, 180, and 200 °C), duration (15, 30, 45, and 60 min), and solvent composition (40% and 60% v/v ethanol) (Table 1). Table 2 summarizes the composition of cellulose, hemicelluloses, lignin, ashes, and extractives following organosolv pretreatment.| Codes | Pulp recovery (% w/w) | Cellulose (% w/w) (% recovery) | Hemicelluloses (% w/w) (% recovery) | Lignin (% w/w) (% recovery) | Ashes (% w/w) | Extractives (% w/w) |
|---|---|---|---|---|---|---|
| a Codes: 0 – pretreatment at 200 °C; 1 – pretreatment at 180 °C; 2 – pretreatment at 160 °C; A – pretreatment for 15 min; B – pretreatment for 30 min; C – pretreatment for 45 min; D – pretreatment for 60 min; 4–40% v/v ethanol content; 6–60% v/v ethanol content. | ||||||
| 0A4 | 36.33 | 40.79 (89.06) | 11.14 (18.04) | 19.11 (43.76) | 2.26 | 14.16 |
| 0A6 | 33.80 | 37.95 (77.08) | 6.77 (10.20) | 17.91 (38.17) | 2.73 | 25.63 |
| 0B4 | 37.01 | 37.57 (83.54) | 2.19 (3.62) | 13.36 (31.17) | 2.21 | 32.16 |
| 0B6 | 33.91 | 45.32 (92.38) | 6.52 (9.86) | 15.17 (32.44) | 2.34 | 21.20 |
| 0C4 | 35.91 | 38.16 (68.62) | 3.35 (4.47) | 16.90 (31.89) | 2.12 | 29.05 |
| 0C6 | 30.35 | 48.95 (89.27) | 7.22 (9.78) | 13.06 (24.99) | 2.01 | 29.53 |
| 1A4 | 38.01 | 35.65 (81.45) | 8.49 (14.40) | 19.20 (46.01) | 2.30 | 24.54 |
| 1A6 | 39.08 | 36.60 (85.96) | 12.96 (22.60) | 15.79 (38.90) | 3.08 | 20.72 |
| 1B4 | 37.34 | 35.01 (78.55) | 7.56 (12.59) | 14.43 (33.97) | 2.30 | 32.06 |
| 1B6 | 37.32 | 36.54 (81.95) | 7.64 (12.72) | 15.89 (37.39) | 3.19 | 20.77 |
| 1C4 | 34.83 | 47.72 (99.89) | 6.78 (10.54) | 21.50 (47.21) | 1.85 | 13.82 |
| 1C6 | 34.78 | 38.73 (80.94) | 9.22 (14.30) | 18.62 (40.82) | 2.79 | 10.50 |
| 1D4 | 37.47 | 36.85 (83.00) | 8.33 (13.92) | 15.23 (35.99) | 2.07 | 26.84 |
| 1D6 | 36.64 | 38.51 (84.81) | 10.38 (16.96) | 13.66 (31.56) | 3.20 | 22.66 |
| 2B4 | 45.25 | 28.32 (77.02) | 12.76 (25.75) | 22.28 (63.58) | 1.96 | 16.29 |
| 2B6 | 49.64 | 27.83 (83.02) | 18.53 (41.03) | 21.06 (65.91) | 2.51 | 16.21 |
The cellulose content reached 48.95% w/w in samples pretreated at 180 °C for 45 min with 40% v/v ethanol, and its recovery ranged from 68.6% to 99.9%, for single treatment. Cellulose content was generally higher in samples pretreated at 200 °C or for longer times (45 and 60 min) at 180 °C. Except for pretreatments at 160 °C and the shortest pretreatment at 180 °C with 60% v/v ethanol, the recovery of hemicellulose from pulp was <20%. Delignification was >52.8% w/w except from the pretreatments at 160 °C. At 200 °C, hemicellulose recovery was generally higher at 40% v/v ethanol, while delignification increased with longer pretreatment time (61.8% to 75.0%) if 60% ethanol was provided. Besides pretreatments performed for 30 min at 180 and 200 °C, the lignin content in pretreated pulp was higher with 40% v/v ethanol. In our previous study with the same genus Salicornia dolichostachya, the cellulose content after organosolv pretreament was also higher in the sample pretreated at 200 °C, reaching 51.3% w/w, although it should be mentioned that the cellulose content in the untreated pulp was higher (25.56% w/w)40 compared to the cellulose content of the untreated pulps of S. ramosissima (16.64% w/w). As such the fold increase of cellulose with the S. ramosissima species was higher compared to the fold increase with S. dolichostachya.
The extractives obtained from pretreated pulp ranged from 10.5% to 32.2% w/w. The recovery of fibers was higher in the mildest pretreatments performed at 160 °C (45.3% and 49.6%), which coincided also with lower hemicellulose removal and delignification, thus indicating poor efficiency of these fractionation conditions in S. ramosissima.
Hemicelluloses
Table 3 lists the oligomer ratio for the sugars recovered in the hemicellulose fractions, as well as the yield of different monomers and oligomers. The yield of hemicellulosic sugars was higher following mild pretreatment at 180 °C for 15 min with 40% v/v ethanol (8.29 g per 100 gbiomass, which includes 2.38 g per 100 gbiomass monomers and 5.91 g per 100 gbiomass oligomers) and at 160 °C for 30 min with 40% v/v ethanol. The highest amount of total hemicellulose oligomers (6.23 g per 100 gbiomass), of which the majority were xylan and arabinan (3.62 and 2.01 g per 100 gbiomass, respectively), as well as nearly the highest ratio (76.35%) was achieved with pretreatment at 160 °C for 30 min with 40% v/v ethanol. Under these and all other conditions, 60% v/v ethanol yielded a higher oligomer ratio than 40% v/v ethanol. Only pretreatment at 200 °C for 45 min with 40% v/v ethanol yielded more monomers than oligomers, indicating that harsh conditions were required for hemicellulose recovery. Whereas monosaccharides can be directly applied for fermentation or as building blocks, hemicellulose oligomers are suitable as prebiotics, in regulating type II diabetes and cholesterol levels or as immunostimulants.41–43| Codes | Oligomers (%) | Monomers (g per 100 gbiomass) | Oligomers (g per 100 gbiomass) | Total cellulose sugars (g per 100 gbiomass) | Total hemicellulose sugars (g per 100 gbiomass) | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Glc | Rha | Ara | Man | Xyl | GalA | GlcA | Total | Glc | Rha | Ara | Man | Xyl | GalA | GlcA | Total | ||||
| a Codes: 0 – pretreatment at 200 °C; 1 – pretreatment at 180 °C; 2 – pretreatment at 160 °C; A – pretreatment for 15 min; B – pretreatment for 30 min; C – pretreatment for 45 min; D – pretreatment for 60 min; 4–40% v/v ethanol content; 6–60% v/v ethanol content. Glc, glucose; Rha, rhamnose; Ara, arabinose; Man, mannose; Xyl, xylose; GalA, galacturonic acid; GlcA, glucuronic acid. | |||||||||||||||||||
| 0A4 | 54.29% | 0.07 | 0.02 | 0.41 | 0.18 | 0.60 | 0.00 | 0.00 | 1.28 | 0.25 | 0.05 | 0.00 | 0.00 | 1.22 | 0.00 | 0.00 | 1.52 | 0.32 | 2.48 |
| 0A6 | 65.11% | 0.00 | 0.04 | 0.42 | 0.16 | 0.64 | 0.01 | 0.00 | 1.27 | 0.00 | 0.12 | 0.13 | 0.00 | 2.12 | 0.00 | 0.00 | 2.37 | 0.00 | 3.64 |
| 0B4 | 56.80% | 0.06 | 0.01 | 0.27 | 0.10 | 0.27 | 0.00 | 0.02 | 0.73 | 0.17 | 0.03 | 0.00 | 0.14 | 0.62 | 0.00 | 0.00 | 0.96 | 0.23 | 1.46 |
| 0B6 | 70.76% | 0.00 | 0.02 | 0.30 | 0.08 | 0.38 | 0.00 | 0.03 | 0.81 | 0.10 | 0.09 | 0.06 | 0.12 | 1.59 | 0.00 | 0.00 | 1.96 | 0.10 | 2.67 |
| 0C4 | 36.67% | 0.04 | 0.00 | 0.28 | 0.10 | 0.13 | 0.00 | 0.02 | 0.57 | 0.10 | 0.01 | 0.00 | 0.08 | 0.14 | 0.00 | 0.00 | 0.33 | 0.14 | 0.76 |
| 0C6 | 63.77% | 0.02 | 0.01 | 0.21 | 0.06 | 0.16 | 0.00 | 0.04 | 0.50 | 0.13 | 0.04 | 0.00 | 0.18 | 0.53 | 0.00 | 0.00 | 0.88 | 0.15 | 1.23 |
| 1A4 | 68.88% | 0.29 | 0.16 | 1.39 | 0.08 | 0.72 | 0.03 | 0.00 | 2.67 | 0.00 | 0.33 | 0.44 | 0.01 | 5.13 | 0.00 | 0.00 | 5.91 | 0.29 | 8.29 |
| 1A6 | 64.83% | 0.00 | 0.09 | 0.78 | 0.13 | 0.45 | 0.01 | 0.01 | 1.47 | 0.25 | 0.12 | 0.28 | 0.00 | 2.04 | 0.00 | 0.02 | 2.71 | 0.25 | 3.93 |
| 1B4 | 70.02% | 0.00 | 0.09 | 0.70 | 0.15 | 0.68 | 0.01 | 0.01 | 1.64 | 1.14 | 0.19 | 0.37 | 0.00 | 2.13 | 0.00 | 0.00 | 3.83 | 1.14 | 4.33 |
| 1B6 | 71.21% | 0.08 | 0.08 | 0.75 | 0.13 | 0.39 | 0.03 | 0.04 | 1.50 | 1.90 | 0.31 | 0.61 | 0.00 | 0.88 | 0.00 | 0.01 | 3.71 | 1.98 | 3.23 |
| 1C4 | 64.53% | 0.00 | 0.08 | 0.69 | 0.20 | 0.88 | 0.01 | 0.02 | 1.88 | 0.00 | 0.16 | 0.21 | 0.00 | 3.05 | 0.00 | 0.00 | 3.42 | 0.00 | 5.30 |
| 1C6 | 63.34% | 0.00 | 0.06 | 0.55 | 0.14 | 0.50 | 0.00 | 0.00 | 1.25 | 0.23 | 0.14 | 0.00 | 0.00 | 1.78 | 0.01 | 0.00 | 2.16 | 0.23 | 3.18 |
| 1D4 | 59.45% | 0.00 | 0.07 | 0.56 | 0.23 | 1.02 | 0.01 | 0.02 | 1.91 | 0.09 | 0.13 | 0.09 | 0.06 | 2.42 | 0.00 | 0.01 | 2.80 | 0.09 | 4.62 |
| 1D6 | 58.33% | 0.00 | 0.06 | 0.62 | 0.17 | 0.59 | 0.00 | 0.01 | 1.45 | 0.33 | 0.19 | 0.00 | 0.00 | 1.50 | 0.01 | 0.00 | 2.03 | 0.33 | 3.15 |
| 2B4 | 76.35% | 0.00 | 0.11 | 0.89 | 0.06 | 0.76 | 0.09 | 0.02 | 1.93 | 0.00 | 0.58 | 2.01 | 0.01 | 3.62 | 0.01 | 0.00 | 6.23 | 0.00 | 8.16 |
| 2B6 | 78.49% | 0.00 | 0.05 | 0.41 | 0.08 | 0.32 | 0.04 | 0.01 | 0.91 | 0.00 | 0.48 | 0.67 | 0.00 | 2.16 | 0.01 | 0.00 | 3.32 | 0.00 | 4.23 |
In 11 out of 16 pretreatments, arabinose was the main sugar monomer, whereas xylan was the main oligomer for all but one of the pretreatments. In arabinoxylan hemicelluloses, the arabinan present in the side chain is more susceptible to pretreatment, explaining its abundance among monomers and that of xylan among oligomers. Unlike cellulose, which has a partly crystalline structure,44 hemicellulose is less thermostable and is prone to the formation of thermally unstable anhydrosugars, such as arabinosan and xylosan.45 The mildest pretreatments at 160 °C presented the highest amounts of arabinan (2.01 and 0.67 g per 100 gbiomass), thus corroborating this tendency.
At 200 °C, the hemicellulose yield was higher in samples pretreated with 60% than 40% v/v ethanol. This trend was reversed at the other temperatures tested, demonstrating the importance of evaluating different combinations of parameters during organosolv pretreatment. Werner and collaborators studied the thermal decomposition of hemicelluloses and suggested that phenolic compounds cross-linked with arabinoxylan improved stability at higher temperatures.45 Numerous phenolic compounds have been reported in S. ramosissima, and their solubility tends to improve at higher ethanol ratios in the mixture.46,47
Table 4 lists the total values for different linear xylooligosaccharides (XOS), as well as some cello-oligosaccharides (COS) recovered from organosolv pretreatment of S. ramosissima. XOS and COS were found mostly as dimers, although some samples presented similar amounts of XOS2 and XOS3. Generally, in samples treated at 180 °C, XOS3 corresponded to at least 60% of the XOS2 content. Analogously, 60% v/v ethanol during organosolv pretreatment seemed to favor a higher XOS3/XOS2 ratio. Linear XOS4 forms were also detected, albeit at low concentrations. Notably, short pretreatments at 180 °C seemed to favor linear XOS4, whereas those at 160 °C prevented their formation. Finally, no linear XOS6 was detected in any sample. The highest amount of total linear XOS with a degree of polymerization (dp) of 2–6 was recorded following pretreatment at 180 °C for 15 min with 40% v/v ethanol, and was generally higher at 180 °C. Milessi and collaborators (2021) pretreated sugarcane bagasse with three different methods: alkaline, hydrothermal, and organosolv and they concluded that the hydrothermal and organosolv pretreatments were the best suitable for the production of XOS additionally suggesting that the organosolv pretreatment as best suitable given the lowest xylose formation.48
| Codes | Cello-oligosaccharides (g per 100 gbiomass) | Xylooligosaccharides (g per 100 gbiomass) | Other soluble oligosaccharides (g per 100 gbiomass) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| COS2 | COS4 | XOS2 | XOS3 | XOS4 | XOS6 | Total | XOS (2–6)/hemicellulose oligosaccharides | ||
| a Codes: 0 – pretreatment at 200 °C; 1 – pretreatment at 180 °C; 2 – pretreatment at 160 °C; A – pretreatment of 15 min; B – pretreatment for 30 min; C – pretreatment for 45 min; D – pretreatment for 60 min; 4–40% v/v ethanol content; 6–60% v/v ethanol content. COS2: 1,4-beta-D-cellobiose; COS3: 1,4-beta-D-cellotriose; XOS2: 1,4-beta-D-xylobiose; XOS3: 1,4-beta-D-xylotriose; XOS4: 1,4-beta-D-xylotetraose; XOS6: 1,4-beta-D-xylohexaose. Other soluble oligosaccharides: total mass of oligomer derived sugars excluding COS (dp 2,4) and XOS (dp 2–6). | |||||||||
| 0A4 | 0.04 | 0.00 | 0.31 | 0.11 | 0.04 | 0.00 | 0.46 | 30.26% | 1.02 |
| 0A6 | 0.05 | 0.00 | 0.31 | 0.20 | 0.02 | 0.00 | 0.53 | 22.36% | 1.79 |
| 0B4 | 0.02 | 0.00 | 0.12 | 0.03 | 0.02 | 0.00 | 0.17 | 17.71% | 0.77 |
| 0B6 | 0.02 | 0.00 | 0.20 | 0.09 | 0.04 | 0.00 | 0.33 | 16.84% | 1.61 |
| 0C4 | 0.03 | 0.00 | 0.03 | 0.01 | 0.01 | 0.00 | 0.05 | 15.15% | 0.25 |
| 0C6 | 0.01 | 0.00 | 0.08 | 0.02 | 0.02 | 0.00 | 0.12 | 13.64% | 0.75 |
| 1A4 | 0.09 | 0.00 | 0.61 | 0.39 | 0.07 | 0.00 | 1.07 | 18.10% | 4.75 |
| 1A6 | 0.04 | 0.00 | 0.17 | 0.16 | 0.06 | 0.00 | 0.39 | 14.39% | 2.28 |
| 1B4 | 0.07 | 0.00 | 0.50 | 0.31 | 0.05 | 0.00 | 0.86 | 22.45% | 2.90 |
| 1B6 | 0.06 | 0.00 | 0.26 | 0.21 | 0.06 | 0.00 | 0.53 | 14.29% | 3.12 |
| 1C4 | 0.08 | 0.00 | 0.52 | 0.37 | 0.05 | 0.00 | 0.94 | 27.49% | 2.40 |
| 1C6 | 0.06 | 0.00 | 0.23 | 0.22 | 0.03 | 0.00 | 0.48 | 22.22% | 1.62 |
| 1D4 | 0.06 | 0.00 | 0.51 | 0.31 | 0.03 | 0.00 | 0.85 | 30.36% | 1.89 |
| 1D6 | 0.07 | 0.00 | 0.30 | 0.27 | 0.03 | 0.00 | 0.60 | 29.56% | 1.36 |
| 2B4 | 0.02 | 0.00 | 0.15 | 0.05 | 0.00 | 0.00 | 0.20 | 3.21% | 6.01 |
| 2B6 | 0.02 | 0.00 | 0.07 | 0.02 | 0.00 | 0.00 | 0.09 | 2.71% | 3.21 |
The ratio of linear XOS (dp 2–6)/total hemicellulose oligomers varied between 2.71% (160 °C, 30 min, and 60% v/v ethanol) and 30.36% (180 °C, 60 min, and 60% v/v ethanol). Such a range indicates the presence of other soluble oligosaccharides in the samples, such as XOS with dp >6, pentoses such as arabinose (which was also detected), glucose, branched XOS, and XOS bound to alduronic acids.49,50
Spectra recorded during GPC were carefully interpreted, as the refractive index detector employed for the measurements could recognize also degradation compounds, residual ethanol, and salts. Such compounds are not expected to interact with the column in the same way the carbohydrates do, and are therefore likely to elute at lower elution volumes, which would correspond to higher molecular weight fractions. By taking into account the expected dp of soluble oligosaccharides, a cut-off of 1500 Da,51 corresponding to approximately 11 pentose (i.e., xylose) residues and 15.8 mL elution volume was chosen. The lowest cut-off was set at 150 Da, equivalent to that of a single xylose and an elution volume of 18.1 mL. To improve peak recognition and comparison between samples, GPC plots encompassed elution volumes of 15–19.5 mL, instead of the full range (0–36 mL). This strategy allowed the exclusion of peaks originating from other compounds or impurities.
Based on the chosen cut-off values, the signal in the ranges up to 500 Da, 500–1000 Da, and 1000–1500 Da was calculated (Table 5). Most of the signal was accounted for by MWs < 1000 Da. Only two of the samples (180 °C with 40% v/v ethanol for 15 or 30 min) had lower than 82% of the sugars at the range of MW < 1000 Da. A shift towards lower molecular weight with increasing treatment duration but the same temperature and ethanol content was observed. When evaluating the effect of treatment temperature, a shift towards lower molecular weights was observed with increasing temperature, indicating that higher temperatures promoted extensive hemicellulose depolymerization. This is evident in the chromatograms after 17.5 mL, whereby pretreatments at 200 °C result in fewer and broader peaks than observed for treatment products obtained at 180 and 160 °C. Finally, the ethanol content had a profound effect on the molecular weight of hemicellulose, with 60% v/v ethanol resulting in significantly more small hemicellulose molecules (<500 Da), except for treatments at 180 °C for 30 min.
| Codes | 1000 Da < MW < 1500 Da (%) | 500 Da < MW < 1000 Da (%) | MW < 500 Da (%) |
|---|---|---|---|
| a Codes: 0 – pretreatment at 200 °C; 1 – pretreatment at 180 °C; 2 – pretreatment at 160 °C; A – pretreatment for 15 min; B – pretreatment for 30 min; C – pretreatment for 45 min; D – pretreatment for 60 min; 4–40% v/v ethanol content; 6–60% v/v ethanol content. | |||
| 0A4 | 14.32% | 57.75% | 27.93% |
| 0A6 | 9.02% | 35.42% | 55.56% |
| 0B4 | 11.66% | 46.36% | 41.98% |
| 0B6 | 8.04% | 27.34% | 64.62% |
| 0C4 | 14.59% | 49.74% | 35.67% |
| 0C6 | 8.96% | 26.07% | 64.97% |
| 1A4 | 40.64% | 35.90% | 23.46% |
| 1A6 | 15.23% | 40.11% | 44.66% |
| 1B4 | 27.38% | 38.13% | 34.49% |
| 1B6 | 15.68% | 47.59% | 36.73% |
| 1C4 | 17.35% | 52.77% | 29.88% |
| 1C6 | 9.26% | 33.57% | 57.17% |
| 1D4 | 13.84% | 64.83% | 21.33% |
| 1D6 | 9.14% | 37.94% | 52.92% |
| 2B4 | 15.90% | 57.00% | 27.10% |
| 2B6 | 10.24% | 37.11% | 52.66% |
At 180 °C and 40% v/v ethanol, hemicellulose depolymerization led to an increase from 59.36% to 86.16% in the amount of molecules with MW < 1000 Da, when time increased from 15 to 60 min. Instead, at 60% v/v ethanol, the yield was above 84% at all times. For both ethanol contents, the total ratio of molecules with MW < 1000 Da was higher at 160 °C (84.1% and 89.8%) than at 180 °C (72.6% and 84.3%). This difference can be explained by the mildest conditions being less effective in fractionating biomass and thus causing slower release of hemicelluloses from the pulp.52
Based on the observed recovery of oligomers from hemicellulose, the mildest pretreatments were selected for LC-MS analysis. The specific LC-MS set-up employed could analyze glycans spanning 2–15 residues, but not monosaccharides. Moreover, MS could not differentiate between pentoses or hexoses, such as xylose and arabinose, because of identical mass. The samples were analyzed in their native, non-reduced form.
Fig. 1 reports the base peak chromatogram of samples pretreated at 160 °C for 30 min with 40% v/v ethanol analyzed in negative ion mode. The major peak at m/z 309 was assigned to a disaccharide consisting of a hexuronic acid linked to a pentose minus 16. The glycan was detected in positive ion mode as [M + NH4]+. This finding is important as biologically active XOS such as alduronic acids can influence prebiotic activity. Furthermore, hexuronic acid was suspected of forming a deoxyhexuronic acid (deoxyHexA). Mishra and collaborators (2013) studied the mass profiling of oligosaccharides of Salicornia brachiata using MALDI TOF–TOF MS analysis where they described signals for hexuronic acids and deoxy-pentoses.53 The formation of deoxyHexA during organosolv treatment is not commonly reported, likely due to more advanced analytical instrumentation being required to identify such compounds. However, deoxyHexA has been reported during organosolv treatment of biomass under alkaline conditions.54 Since alkaline organosolv treatment was not applied in this case, the elevated salt content of Salicornia pulp might be sufficient to induce deoxyHexA formation. Another peak at m/z 369 was assigned to a disaccharide consisting of a hexose with a methylated hexuronic acid. Moreover, strong signals were assigned to a pentose disaccharide, pentose trioses, and a C6 disaccharide. Finally, weaker signals that might correspond to acetylated C5 connected to C6 were also identified, along with acetylated hexuronic acids.
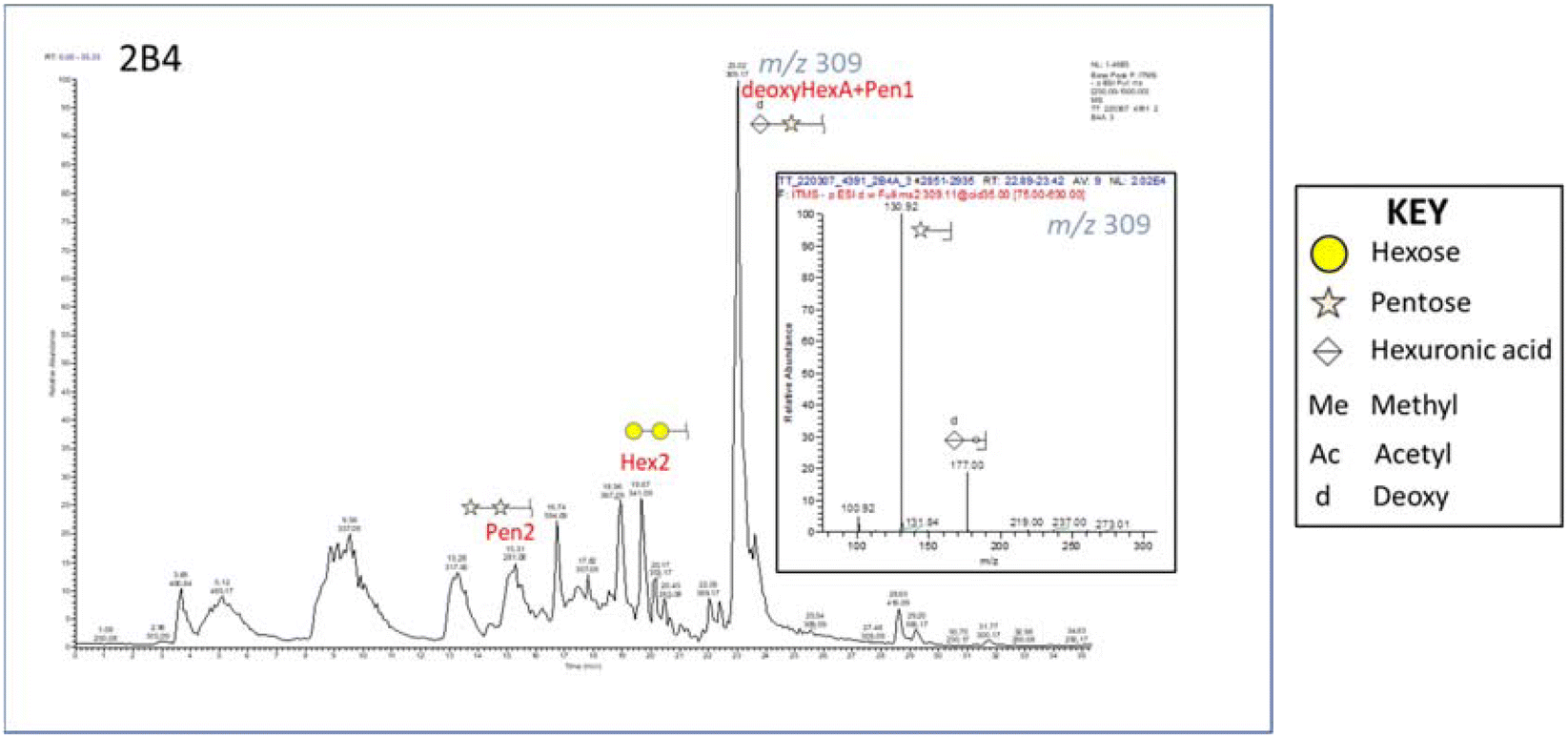 | ||
| Fig. 1 Base peak chromatogram of S. ramosissima pretreated at 160 °C for 30 min with 40% ethanol (2B4); the MS2 insert shows the major glycan at m/z 309 identified as a disaccharide consisting of a hexuronic acid linked to a pentose minus 16. | ||
A base peak chromatogram similar to the one above was obtained also for samples pretreated at 180 °C for 30 min with 60% v/v ethanol (Fig. S1†). In this case, a few early components, a major disaccharide at m/z = 367, as well as a later eluting disaccharide at 17 min (m/z = 353) were noted. The peak at m/z 353 pointed to a major fragment ion indicative of hexose (m/z = 161), whereas that at m/z = 367 indicated the presence of hexuronic acid. The major fragment ion at m/z = 191 in both components was assigned to acetylated pentose. A peak at m/z = 309 was also detected, although its abundance was almost 10 times lower than in the sample pretreated at 160 °C for 30 min with 40% v/v ethanol. The opposite was observed for the peak at m/z = 369. Finally, a peak pointing to a C6 disaccharide was detected, along with peaks corresponding to C5 tetraoses.
The sample pretreated at 160 °C for 30 min with 60% v/v ethanol presented a strong signal at m/z = 501 and 563, which corresponded to C6 trisaccharides and C5 tetraoses, respectively (Fig. S2†). No peak at m/z = 309 was detected in this sample. Peaks at m/z = 353 indicated the presence of acetylated pentoses connected to a C6 sugar. The peak at m/z = 367 was 1.7 times more abundant than in a similar sample pretreated at 180 °C. Also the peaks at m/z = 353 and = 367 exhibited a very strong signal, indicating an abundance of acetylated disaccharides. Other peaks suggested the presence of disaccharides composed of hexoses or one hexose and one pentose. Finally, a relatively abundant peak that could correspond to a tetraose consisting of three pentoses and one hexose was detected in this sample but to a much lesser extent in others.
The remaining samples, which included pretreatments at 180 °C for 15 or 30 min with 40% v/v ethanol or 15 min with 60% ethanol, contained the same glycans as described above, although at a varying relative abundance (Fig. S3†). Specifically, the sample pretreated at 180 °C for 30 min with 40% v/v ethanol was characterized by a strong peak at m/z = 353, followed by one at m/z = 369 (previously assigned to a hexose with a methylated hexuronic acid) and others corresponding to pentose disaccharides, tetraoses, hexose disaccharides, and trisaccharides. Finally, the peak at m/z = 309 was equally abundant as in the equivalent sample pretreated with 60% v/v ethanol. For the sample pretreated at 180 °C for 15 min with 40% v/v ethanol, the most abundant peak was at m/z = 367, which was not detected at 30 min, and was comparable to the one noted at 160 °C. A strong peak was detected again at m/z = 353, although it was 35–40% more abundant than in the sample pretreated at 160 °C for 30 min with 40% v/v ethanol and slightly higher than with 60% v/v ethanol. Again, a rather large peak was detected at m/z = 369, whereas the one at m/z = 563 (indicating C5 tetraose) was the most abundant across all tested samples. A peak assigned to a tetraose consisting of three pentoses and one hexose was also detected in this sample, although it was not as evident. Finally, in the sample pretreated at 180 °C for 15 min with 60% v/v ethanol, the strongest peaks were at m/z = 367 and 353, with the former similar to the one detected in the sample pretreated with 40% v/v ethanol and the latter approximately 43% more abundant. Additional peaks were assigned to C6 disaccharides, C5 tetraose, and tetraose consisting of three C5 and one C6 sugars, although the abundance was lower than in other samples. The same applied to a peak at m/z = 369. Overall, LC-MS analysis demonstrated that different hexurono-XOS/alduronic acids were the main branched oligosaccharides. Importantly, these acidic oligosaccharides exhibit various biological activities.55–57
The quantification of sugar degradation compounds and total phenolics in the hemicellulose fractions is reported in Table 6. Acetic acid originates from the acetylation of sugars, whereas furfural, hydroxymethylfurfural (HMF), levulinic acid, and formic acid derive from the degradation of sugars, thereby affecting their yields.58 Acetic acid presented the highest concentration, ranging from 0.636 to 4.766 g per 100 gbiomass. The second most common degradation compound was formic acid, which ranged from 0.046 to 0.493 g per 100 gbiomass, at the lowest and highest treatment temperatures, respectively. HMF varied between 0.037 and 0.356 g per 100 gbiomass, and furfural varied between 0.004 and 0.286 g per 100 gbiomass. Levulinic acid was detected only in the hemicellulose fraction pretreated at 200 °C for 15 min and 40% v/v ethanol, and even then at negligible levels (0.004 g per 100 gbiomass). Finally, total phenolic compounds ranged from 0.046 to 0.103 g per 100 gbiomass, with 40% v/v ethanol giving better results than 60% v/v ethanol, particularly at 180 °C for 15 min.
| Code | Acetic acid (g per 100 gbiomass) | Formic acid (g per 100 gbiomass) | HMF (g per 100 gbiomass) | Levulinic acid (g per 100 gbiomass) | Furfural (g per 100 gbiomass) | Phenolics (g per 100 gbiomass) | Total (g per 100 gbiomass) |
|---|---|---|---|---|---|---|---|
| a Codes: 0 – pretreatment at 200 °C; 1 – pretreatment at 180 °C; 2 – pretreatment at 160 °C; A – pretreatment for 15 min; B – pretreatment for 30 min; C – pretreatment for 45 min; D – pretreatment for 60 min; 4–40% v/v ethanol content; 6–60% v/v ethanol content. HMF, hydroxymethylfurfural. | |||||||
| 0A4 | 4.766 | 0.493 | 0.269 | 0.004 | 0.245 | 0.081 | 5.857 |
| 0A6 | 4.259 | 0.400 | 0.302 | 0.000 | 0.125 | 0.060 | 5.146 |
| 0B4 | 3.451 | 0.368 | 0.315 | 0.000 | 0.161 | 0.081 | 4.377 |
| 0B6 | 4.222 | 0.410 | 0.282 | 0.000 | 0.132 | 0.058 | 5.104 |
| 0C4 | 3.926 | 0.366 | 0.356 | 0.000 | 0.202 | 0.076 | 4.927 |
| 0C6 | 3.420 | 0.296 | 0.264 | 0.000 | 0.065 | 0.046 | 4.091 |
| 1A4 | 3.818 | 0.289 | 0.238 | 0.000 | 0.277 | 0.103 | 4.726 |
| 1A6 | 2.022 | 0.181 | 0.156 | 0.000 | 0.033 | 0.047 | 2.439 |
| 1B4 | 2.494 | 0.209 | 0.141 | 0.000 | 0.193 | 0.090 | 3.127 |
| 1B6 | 2.463 | 0.215 | 0.181 | 0.000 | 0.128 | 0.053 | 3.040 |
| 1C4 | 3.447 | 0.314 | 0.258 | 0.000 | 0.198 | 0.076 | 4.293 |
| 1C6 | 2.607 | 0.243 | 0.183 | 0.000 | 0.045 | 0.038 | 3.116 |
| 1D4 | 4.488 | 0.399 | 0.279 | 0.000 | 0.286 | 0.090 | 5.541 |
| 1D6 | 3.216 | 0.273 | 0.246 | 0.000 | 0.179 | 0.056 | 3.970 |
| 2B4 | 1.553 | 0.117 | 0.149 | 0.000 | 0.015 | 0.088 | 1.923 |
| 2B6 | 0.636 | 0.046 | 0.037 | 0.000 | 0.004 | 0.053 | 0.776 |
Except for pretreatment at 200 °C for 30 min, all other conditions yielded more degradation compounds at 40% than 60% v/v ethanol. Overall, samples pretreated at 160 °C led to lower yields with both 40% v/v ethanol (1.923 g per 100 gbiomass) and 60% v/v ethanol (0.776 g per 100 gbiomass).
To determine the potential of removing part of the degradation compound (if needed), an activated carbon treatment was applied and evaluated (Table 7). Activated carbon treatment was previously shown by our group not to affect the concentration of sugars.59 In general, furans (HMF and furfural) declined by more than 73%, with furfural dropping by more than 93% in all samples and even disappearing entirely in three of them. The highest reduction exhibited by HMF was 99.23% in the sample pretreated at 160 °C for 30 min with 40% v/v ethanol. In contrast, the loss of organic acids was not as dramatic, with acetic acid declining by 32.62% to 55.32%, and formic acid declining by 3.12% to 36.87%. The presence of acetic acid along with oligosaccharides might be beneficial in feed formulations. Prebiotic oligosaccharides (glucomannan) supplemented with acetic acid was shown to be effective at decreasing the shedding of Salmonella enteritidis in chicken feces, as well as in the re-isolation of S. enteritidis from the liver, spleen, and cecum.60 Additionally, it allowed chickens to attain the highest final body weight but the lowest feed conversion rate (gfeed/ggain in body weight), making it a promising strategy in feed trials.
| Code | Acetic acid | Formic acid | HMF | Furfural | ||||
|---|---|---|---|---|---|---|---|---|
| (g per 100 gbiomass) | Reduction | (g per 100 gbiomass) | Reduction | (g per 100 gbiomass) | Reduction | (g per 100 gbiomass) | Reduction | |
| a Representation of the codes: 0 – pretreatment at 200 °C; 1 – pretreatment at 180 °C; 2 – pretreatment at 160 °C; A – pretreatment for 15 min; B – pretreatment for 30 min; C – pretreatment for 45 min; D – pretreatment for 60 min; 4–40% v/v ethanol content; 6–60% v/v ethanol content. HMF, hydroxymethylfurfural. | ||||||||
| 0C4 | 1.939 | 50.61% | 0.245 | 33.18% | 0.028 | 92.04% | 0.010 | 95.31% |
| 0C6 | 1.905 | 44.30% | 0.209 | 29.36% | 0.046 | 82.49% | 0.005 | 92.93% |
| 1A4 | 2.279 | 40.31% | 0.228 | 21.29% | 0.023 | 90.42% | 0.012 | 95.75% |
| 1A6 | 0.982 | 51.44% | 0.128 | 29.29% | 0.009 | 94.25% | 0.000 | 100% |
| 1B4 | 1.114 | 55.32% | 0.146 | 29.87% | 0.002 | 98.39% | 0.001 | 99.65% |
| 1B6 | 1.629 | 33.87% | 0.180 | 16.09% | 0.042 | 76.94% | 0.008 | 93.62% |
| 1C4 | 2.242 | 34.97% | 0.252 | 19.83% | 0.012 | 95.29% | 0.005 | 97.45% |
| 1C6 | 1.757 | 32.62% | 0.196 | 19.60% | 0.050 | 72.92% | 0.002 | 94.83% |
| 1D4 | 2.113 | 52.91% | 0.252 | 36.87% | 0.015 | 94.49% | 0.005 | 98.24% |
| 1D6 | 1.735 | 46.03% | 0.184 | 32.63% | 0.035 | 85.77% | 0.009 | 94.84% |
| 2B4 | 0.730 | 53.01% | 0.112 | 4.16% | 0.001 | 99.23% | 0.000 | 100% |
| 2B6 | 0.391 | 38.62% | 0.056 | 3.12% | 0.001 | 98.58% | 0.000 | 100% |
Lignin
Compositional analysis of the lignin fraction obtained after organosolv pretreatment of S. ramosissima is summarized in Table 8. Undesired sugars were less abundant (<1%) following pretreatment at 200 °C, reaching instead the highest value (10.3%) in a pretreatment at only 160 °C. As stated previously (Table 2), delignification was less pronounced at the lower temperature, along with a duration of 15 min and 40% v/v ethanol. These results fit well the results of our previous study on the optimization on organosolv pretreatment of S. dolichostachya, where delignification was also temperature dependent with values ranging from 15.3% at 160 °C pretreatment to 56.4% in the pretreatment performed at 200 °C and longer pretreatment of 45 minutes achieved higher delignification (60.7%) when compared to lower pretreatment time (46.1%).40| Code | Delignification (%) | Cellulose (% w/w) | Hemicellulose (% w/w) | Sugars (%) | Ashes (%) | M n | M w | DI |
|---|---|---|---|---|---|---|---|---|
| a Codes: 0 – pretreatment at 200 °C; 1 – pretreatment at 180 °C; 2 – pretreatment at 160 °C; A – pretreatment for 15 min; B – pretreatment for 30 min; C – pretreatment for 45 min; D – pretreatment for 60 min; 4–40% v/v ethanol content; 6–60% v/v ethanol content. Mn, number average molecular weight; Mw, weight average molecular weight; DI, dispersity index (Mw/Mn). | ||||||||
| 0A4 | 56.24 | 1.58 | 0.68 | 2.25 | 1.16 | 600 | 1100 | 1.83 |
| 0A6 | 61.83 | 0.86 | 1.13 | 1.99 | 1.15 | 800 | 2000 | 2.50 |
| 0B4 | 68.83 | 0.29 | 0.34 | 0.63 | 2.01 | 500 | 900 | 1.80 |
| 0B6 | 67.56 | 0.15 | 0.66 | 0.82 | 2.29 | 800 | 2300 | 2.88 |
| 0C4 | 68.11 | 0.61 | 0.37 | 0.97 | 0.41 | 600 | 1000 | 1.67 |
| 0C6 | 75.01 | 0.12 | 0.35 | 0.48 | 0.49 | 700 | 1700 | 2.43 |
| 1A4 | 53.99 | 0.43 | 4.88 | 5.31 | 2.70 | 600 | 1100 | 1.83 |
| 1A6 | 61.1 | 0.62 | 7.12 | 7.73 | 2.99 | 600 | 1100 | 1.83 |
| 1B4 | 66.03 | 0.34 | 4.03 | 4.37 | 2.86 | 600 | 1000 | 1.67 |
| 1B6 | 62.61 | 0.54 | 6.87 | 7.41 | 4.17 | 900 | 2400 | 2.67 |
| 1C4 | 52.79 | 0.48 | 1.54 | 2.02 | 1.52 | 600 | 1000 | 1.67 |
| 1C6 | 59.18 | 0.4 | 1.24 | 1.64 | 0.04 | 800 | 2100 | 2.63 |
| 1D4 | 64.01 | 1.16 | 1.21 | 2.37 | 1.28 | 600 | 1000 | 1.67 |
| 1D6 | 68.44 | 1.34 | 1.76 | 3.11 | 1.98 | 800 | 2100 | 2.63 |
| 2B4 | 36.42 | 0.85 | 6.45 | 7.3 | 4.52 | 600 | 1200 | 2.00 |
| 2B6 | 34.09 | 1.08 | 9.64 | 10.71 | 9.00 | 600 | 1200 | 2.00 |
The number average molecular weight of lignin ranged from 500 to 900 Da, while the weight average was between 900 and 2400 Da. Moreover, 60% v/v ethanol generated lignin with the highest molecular weight in all but the mildest conditions (180 °C for 15 min and 160 °C pretreatments). The highest weight average (>2000 Da) was recorded in samples pretreated with 60% v/v ethanol at 200 °C and 180 °C, except for the 60 min pretreatment at 200 °C and the 15 min pretreatment at 180 °C.
To better understand the structure of lignin, we analyzed the samples by 31P NMR (Table 9). The aliphatic hydroxyl content exhibited a similar trend to sugar contamination (Table 8), with higher values for pretreatments at 160 °C or short incubations at 180 °C. Except for the latter, aliphatic hydroxyls were more abundant with 60% v/v ethanol. The sample pretreated at 160 °C for 30 min with 60% v/v ethanol yielded the most aliphatic hydroxyls (3.66 mmol g−1) and abundant acidic hydroxyls (0.67 mmol g−1), but the least aromatic hydroxyls. This finding thus corresponds to the finding of the higher sugar contents when applying the lower treatment temperature of 160 °C. As reported for grasses, free phenols of syringyl (S)-, guaiacyl (G)-, and para-hydroxyphenyl (H)-type were detected within the lignin fractions.61 The aromatic and acidic hydroxyls in lignin obtained following pretreatment at 200 °C increased continuously with increasing treatment duration at 40% v/v ethanol, but only between 15 and 30 min at 60% v/v ethanol. Samples pretreated at 160 °C displayed the lowest amount of aromatic hydroxyls.
| Code | Aliphatic OH (mmol g−1) | Aromatic OH (mmol g−1) | Aliphatic OH/aromatic OH | Acidic OH (mmol g−1) | |||
|---|---|---|---|---|---|---|---|
| Condensed | G-type | H-type | Total | ||||
| a Codes: 0 – pretreatment at 200 °C; 1 – pretreatment at 180 °C; 2 – pretreatment at 160 °C; A – pretreatment for 15 min; B – pretreatment for 30 min; C – pretreatment for 45 min; D – pretreatment for 60 min; 4–40% v/v ethanol content; 6–60% v/v ethanol content. | |||||||
| 0A4 | 1.50 | 1.41 | 0.74 | 0.33 | 2.49 | 0.60 | 0.59 |
| 0A6 | 1.98 | 1.26 | 0.69 | 0.30 | 2.26 | 0.88 | 0.57 |
| 0B4 | 1.46 | 1.43 | 0.76 | 0.35 | 2.54 | 0.57 | 0.61 |
| 0B6 | 1.86 | 1.55 | 0.84 | 0.37 | 2.77 | 0.67 | 0.59 |
| 0C4 | 1.68 | 1.87 | 0.98 | 0.46 | 3.31 | 0.51 | 0.77 |
| 0C6 | 1.73 | 1.53 | 0.82 | 0.37 | 2.72 | 0.64 | 0.57 |
| 1A4 | 2.53 | 0.93 | 0.54 | 0.22 | 1.69 | 1.50 | 0.45 |
| 1A6 | 2.46 | 0.85 | 0.54 | 0.22 | 1.61 | 1.53 | 0.49 |
| 1B4 | 2.75 | 1.34 | 0.76 | 0.30 | 2.40 | 1.15 | 0.61 |
| 1B6 | 2.54 | 0.88 | 0.55 | 0.22 | 1.65 | 1.54 | 0.53 |
| 1C4 | 1.80 | 0.93 | 0.53 | 0.22 | 1.68 | 1.07 | 0.51 |
| 1C6 | 1.84 | 1.04 | 0.59 | 0.24 | 1.87 | 0.98 | 0.47 |
| 1D4 | 1.61 | 1.23 | 0.66 | 0.27 | 2.16 | 0.75 | 0.52 |
| 1D6 | 2.29 | 1.23 | 0.72 | 0.30 | 2.24 | 1.02 | 0.53 |
| 2B4 | 2.60 | 0.69 | 0.50 | 0.23 | 1.42 | 1.83 | 0.52 |
| 2B6 | 3.66 | 0.56 | 0.50 | 0.23 | 1.29 | 2.84 | 0.67 |
The structure of the extracted lignin was elucidated in additional detail through quantitative 13C NMR and 1H–13C HSQC as described previously62 (Fig. 2 and 3, S4–S19†). The resulting motifs and concentrations (mmol g−1) are reported in Table 10. The traditional interunit motifs found in lignin, such as β-O-4′, β-5′, β-β′, and dibenzodioxocine, were readily detected. For the latter, the C–H shifts for all side-chain carbons (CHα, CHβ, and CHγ) were distinguished, and quantified via correlation with the quantitative 13C NMR using the cross-peaks at δH/δC 4.90/71.26 and 4.22/84.54 respectively for CHα and CHβ (linked to G/H), as well as 4.88/72.04 and 4.13/85.88, respectively (linked to S). The β-5′ structure was quantified on the basis of CHα and CHβ shifts appearing at δH/δC 5.43/87.06 and 3.48/52.98, respectively. Resinol (β-β′) was identified based on CHα, CHβ, and CHγ shifts at δH/δC 4.62/85.15, 3.06/53.52, and 4.78/71.0, respectively, and quantified using only the CHα and CHβ signals.63 The dibenzodioxocine motif was quantified based on CHα and CHβ shifts at δH/δC 4.57/80.09 and 4.01/86.67.64 The most common G, S, and H aromatic units in lignin were located and quantified using CH2 and CH6 shifts at δH/δC 6.92/110.11 and 6.77/118.38,63 CH2,6 shift at δH/δC 6.61/103.38,63 and CH2,6 shift at δH/δC 7.23/128.28,65 respectively.
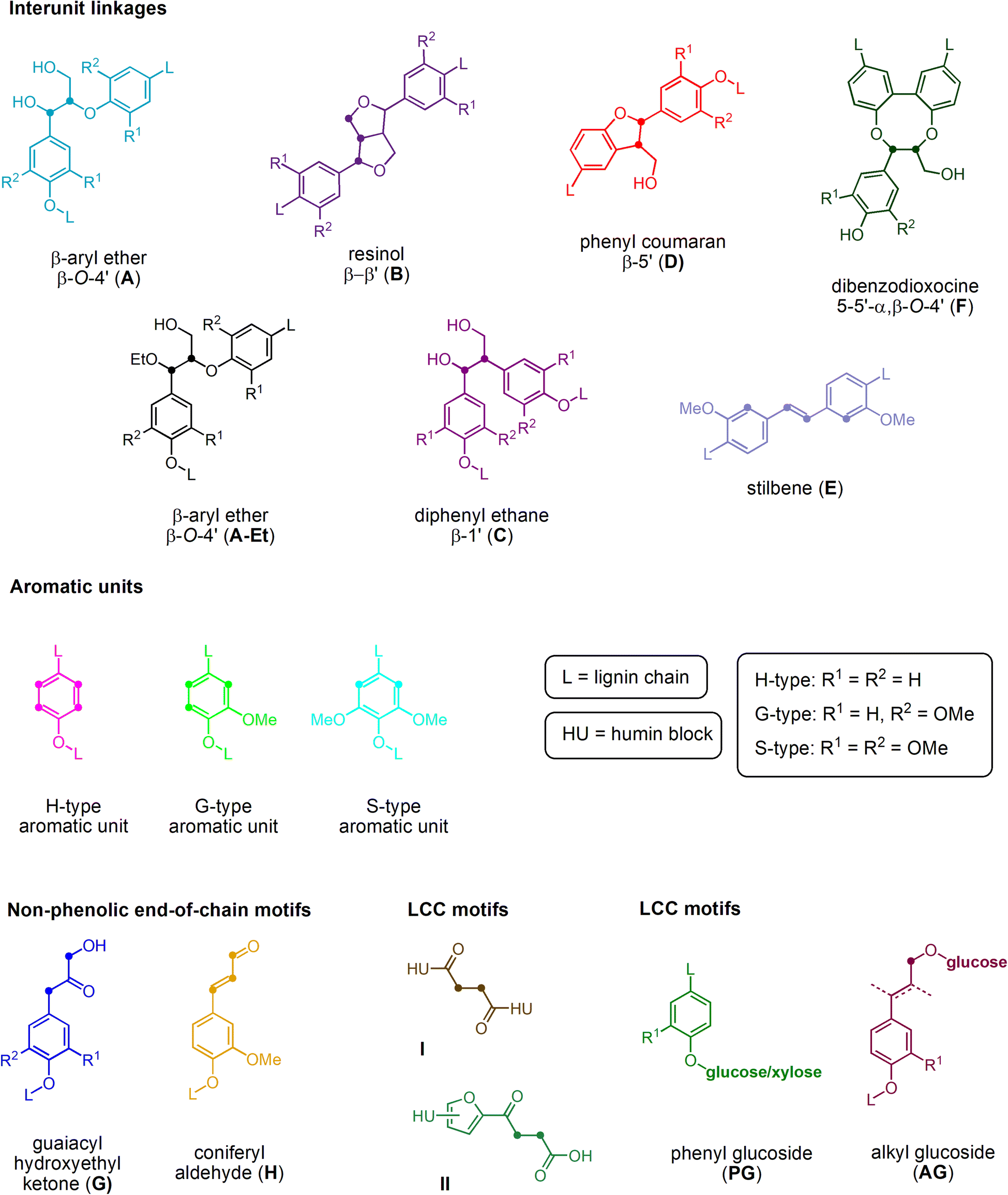 | ||
| Fig. 2 Structural representation of key functional motifs identified in the HSQC analysis of lignins isolated from S. ramosissima samples. | ||
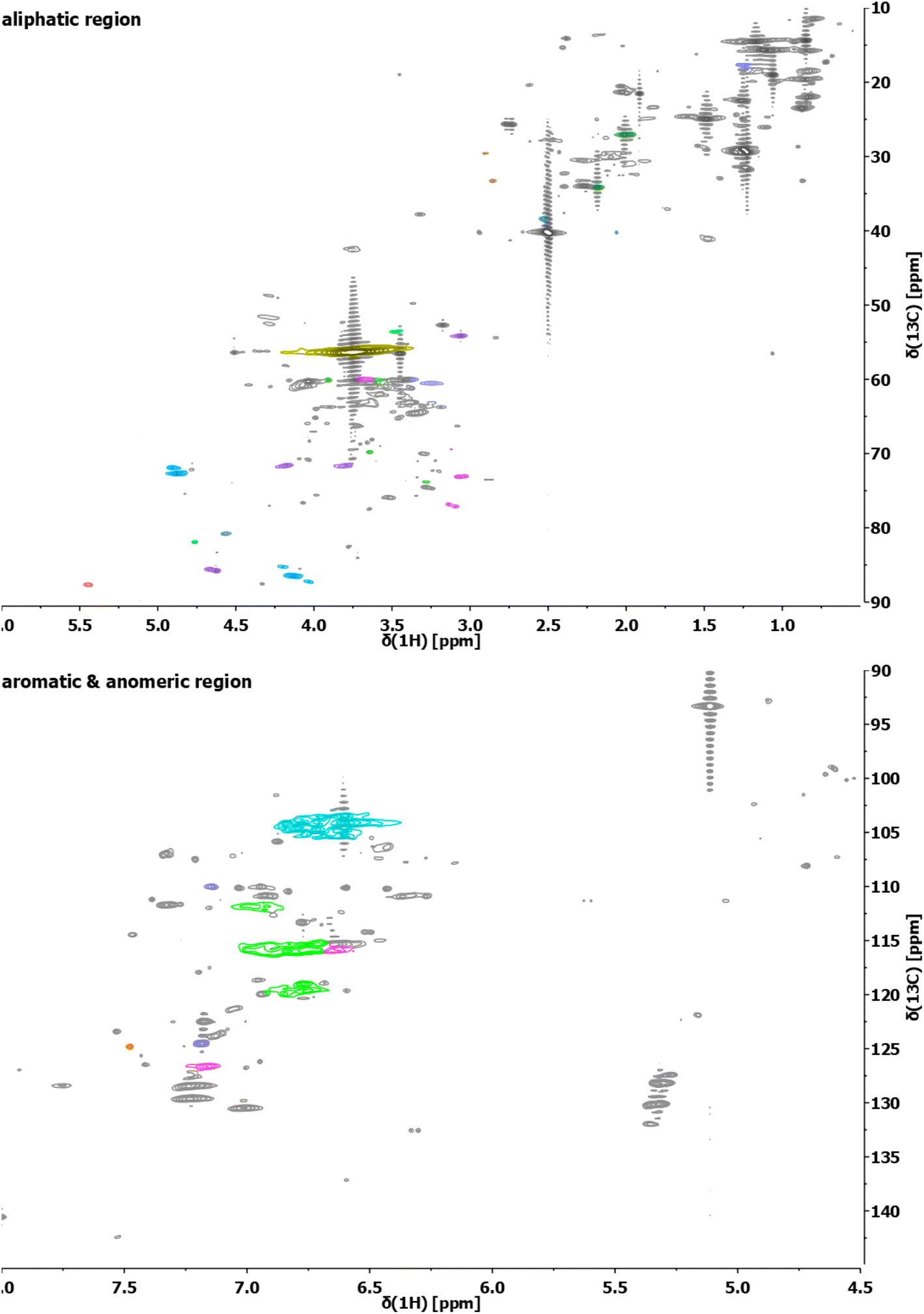 | ||
| Fig. 3 Exemplary HSQC analyses of the aliphatic region (top) and aromatic/anomeric region (bottom) of the lignin sample obtained from S. ramosissima samples pretreated at 160 °C for 30 min with 60% v/v ethanol (2B6). Colors refer to the colors used for the structural representation in Fig. 2. For complete spectra of this and other samples, refer to the ESI.† | ||
| Motif/structure | 0A4a | 0A6a | 0B4a | 0B6a | 0C4a | 0C6a | 1A4a | 1A6a | 1B4a | 1B6a | 1C4a | 1C6a | 1D4a | 1D6a | 2B4a | 2B6a |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Codes: 0 – pretreatment at 200 °C; 1 – pretreatment at 180 °C; 2 – pretreatment at 160 °C; A – pretreatment for 15 min; B – pretreatment for 30 min; C – pretreatment for 45 min; D – pretreatment for 60 min; 4–40% v/v ethanol content; 6–60% v/v ethanol content. | ||||||||||||||||
| β-O-4′ (to S) (CHα., CHβ) (A) | 0.23 | 0.24 | 0.14 | 0.16 | 0.10 | 0.15 | 0.57 | 0.46 | 0.41 | 0.44 | 0.27 | 0.31 | 0.35 | 0.35 | 0.40 | 0.26 |
| β-O-4′ (to G/H) (CHα., CHβ) (A) | 0.17 | 0.16 | 0.11 | 0.12 | 0.09 | 0.13 | 0.35 | 0.25 | 0.24 | 0.27 | 0.19 | 0.20 | 0.23 | 0.24 | 0.25 | 0.13 |
| β-β′ (CHα. CHβ) (B) | 0.32 | 0.30 | 0.27 | 0.23 | 0.29 | 0.31 | 0.49 | 0.45 | 0.40 | 0.43 | 0.29 | 0.33 | 0.41 | 0.35 | 0.34 | 0.24 |
| β-5′ (CHα, CHβ) (D) | 0.11 | 0.10 | 0.08 | 0.07 | 0.09 | 0.09 | 0.15 | 0.11 | 0.13 | 0.12 | 0.09 | 0.10 | 0.13 | 0.11 | 0.09 | 0.06 |
| Dibenzodioxocine (CHα, CHβ) (F) | 0.07 | 0.08 | 0.05 | 0.05 | 0.03 | 0.05 | 0.18 | 0.14 | 0.14 | 0.14 | 0.09 | 0.11 | 0.11 | 0.11 | 0.13 | 0.07 |
| S (C2,6-H) | 12.4 | 7.92 | 6.86 | 4.78 | 7.92 | 7.94 | 9.03 | 9.62 | 9.37 | 10.8 | 9.96 | 8.71 | 11.8 | 12.7 | 7.22 | 4.72 |
| G (C2–H and C6–H) | 10.6 | 7.15 | 7.56 | 7.22 | 9.23 | 10.5 | 10.0 | 9.83 | 11.0 | 8.81 | 6.10 | 10.2 | 9.07 | 8.60 | 5.61 | 3.51 |
| H (C2,6-H) | 3.82 | 2.05 | 1.75 | 2.20 | 2.84 | 3.48 | 4.03 | 4.01 | 4.01 | 3.89 | 2.52 | 3.86 | 2.70 | 3.31 | 3.04 | 2.33 |
| (1-> 4)-β-D-Xylp (CH1.2.3.4.5) | 0.01 | 0.04 | 0.01 | 0.02 | 0.00 | 0.00 | 0.24 | 0.28 | 0.15 | 0.24 | 0.09 | 0.06 | 0.06 | 0.12 | 0.11 | 0.13 |
| PG | 0.00 | 0.01 | 0.00 | 0.01 | 0.00 | 0.00 | 0.04 | 0.10 | 0.02 | 0.07 | 0.01 | 0.01 | 0.01 | 0.02 | 0.03 | 0.09 |
The identity of the signals denoting the H-unit has been a source of contention.66 Specifically, while the area surrounding the CH2,6 shift is somewhat populated, the shift appearing at δH/δC 6.69/115.06 should result as the overlapping signal from both the G5 and H3,5 groups, and correspond in intensity to the sum of G2 and H2,6. In fact, signals originating from non-lignin structures such as 1 → 4 linked β-D-xylopyranose have been located in the spectra at δH/δC 4.24/97.38 (C1–H),67 3.02/72.40 (C2–H), 3.28/74.02 (C3–H), 3.51/75.46 (C4–H), and 3.30/62.90 (C5–H).68 In addition, signals for lignin-carbohydrate complexes appear close to the anomeric region at δH/δC 4.72/100.88, attributed to phenyl glucoside linkages.67 A comparison of the abundances of β-5′, β-β′, dibenzodioxocine, and β-O-4′ interunit motifs with the sum of standard, i.e., unaltered, lignin G-, S-, and H-type aromatic units across the various treatments indicates that the ratios vary according to treatment times, indicating that the structures vary according to the treatment conditions. The lignin was altered to contain, according to current understanding,69 more condensed structures and/or structures incorporating sugars in the form of humins. Cross peaks are eventually identifiable that indicate humin-lignin interactions (Fig. 2 and 3, S4–S19†).
Mass balance
To evaluate the recovery of cellulose, hemicellulose, and lignin in the streams obtained from organosolv pretreatment, the total recovery of each compound was examined. As a result of a thermal pretreatment of the biomass, considerable losses exist due to degradation of compounds and this has been described in many studies.52,70–73 As shown in Fig. 4, most of the recovered cellulose was in the pretreated pulp fraction. The lowest cellulose recovery was obtained following pretreatment at 200 °C for 45 min with 40% v/v ethanol (0C4). The highest recovery and, therefore, best fractionation of cellulose were attained with the same ethanol content but with longer pretreatments at slightly lower 180 °C (1C4 and 1D4). Recovery of cellulose in the lignin fraction accounted for only a minor part (up to 1.3%) of the recovered cellulose. The highest values were observed with the shortest pretreatments (15 min) at 200 °C, which can be explained by their lower delignification values (Table 2). Conversely, at 180 °C, more cellulose was recovered from the longest pretreatment (60 min), which benefitted cellulose hydrolysis.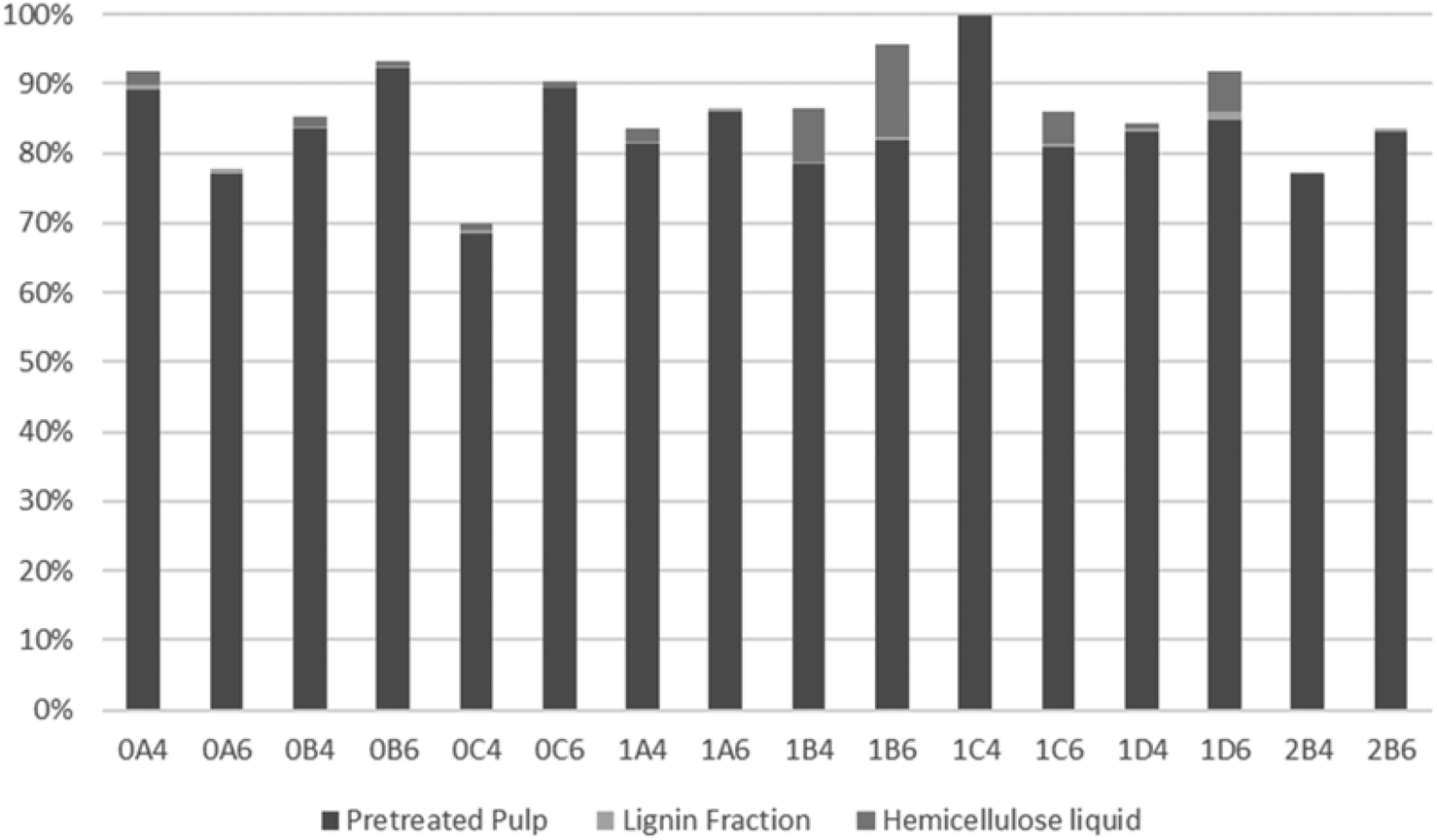 | ||
| Fig. 4 Cellulose distribution in the different S. ramosissima fractions. | ||
As shown in Fig. 5, lignin recovery was highest following pretreatment with 60% v/v ethanol, whereby the recovered content surpassed the initial amount of lignin. The increased lignin recovery can be explained by the formation of pseudo-lignin, an aromatic oligomeric or polymeric structure derived from furfural and HMF known to precipitate together with the Klason lignin, and/or covalently bound humin–lignin hybrids.69,74 In turn, both of these compounds are derived from sugar degradation.74 As shown on Table 6, the furfural content was higher following pretreatment with 40% v/v ethanol, while HMF was higher in six out of eight pretreatments. We suggest that ethanol composition affects the speed of sugar depolymerization, the consequent formation of furans, and conversion to pseudo-lignin and/or covalently bound humin–lignin hybrids. The latter can either solubilize in the liquor and appear in the lignin fraction, or become deposited and repolymerized within the solid fraction. Therefore, delignification ought to be considered also in the mildest pretreatments. Except for pretreatments at 160 °C and the shortest pretreatment at 180 °C with 40% v/v ethanol, the highest lignin recovery as lignin fraction was 77.9%.
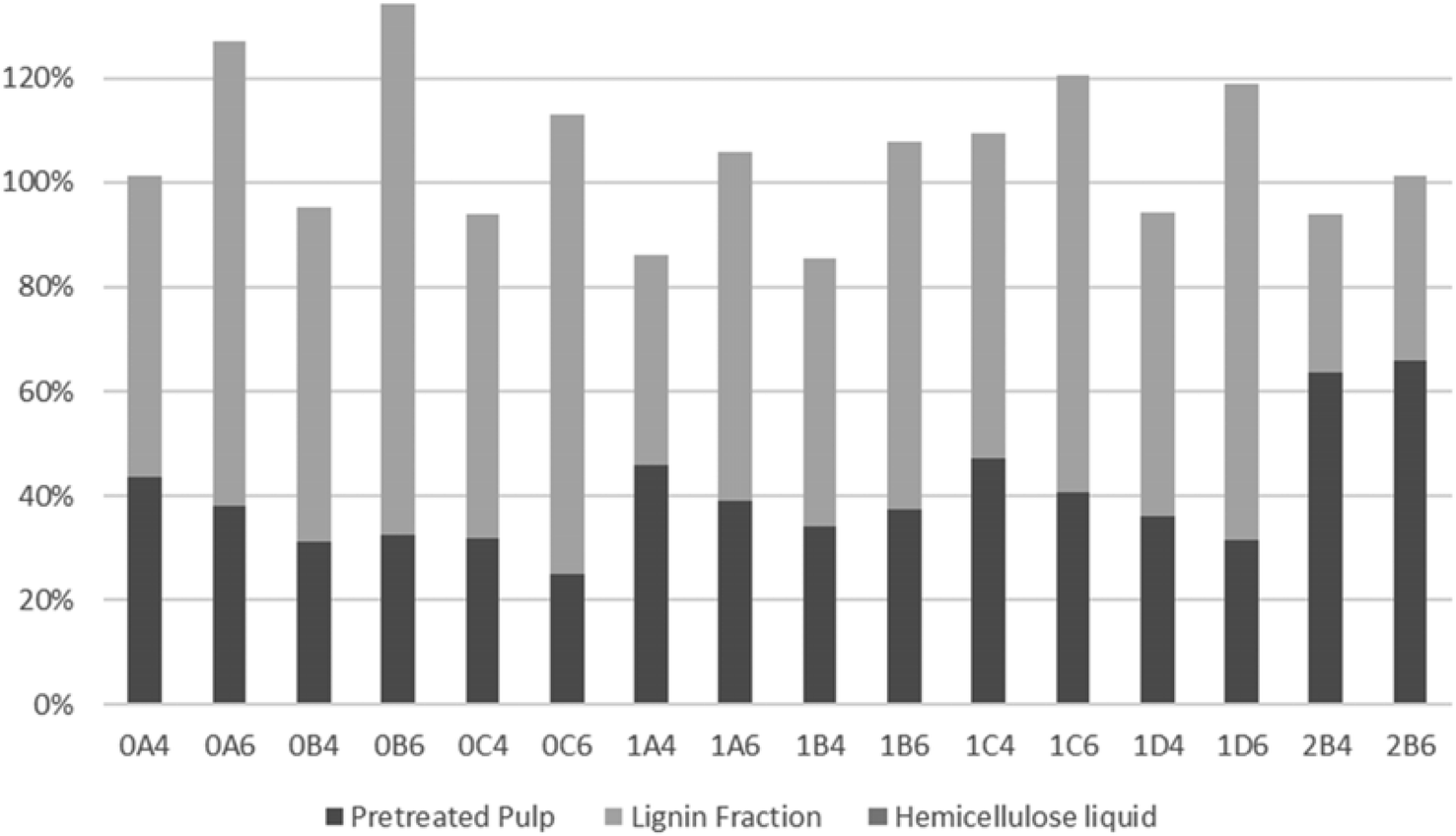 | ||
| Fig. 5 Lignin distribution in the different S. ramosissima fractions. | ||
The recovery of hemicellulosic sugars is shown in Fig. 6. The time effect is noticeable for pretreatment at both 200 and 180 °C, with significant reduction in hemicellulose recovery with increasing organosolv treatment duration. The majority of hemicellulose-derived sugars were present in the liquid fraction for most treatments. The lowest hemicellulose recoveries were obtained upon pretreatment at 200 °C with 40% v/v ethanol for 30 or 45 min (which were also the only conditions when the recovery was lower in 40% v/v ethanol). The amount of sugar degradation products (levulinic and formic acids plus furans) was temperature-dependent, with larger amounts attained at a higher temperature. The current results, whereby a lower recovery correlates with a more pronounced sugar degradation, corroborate this trend.
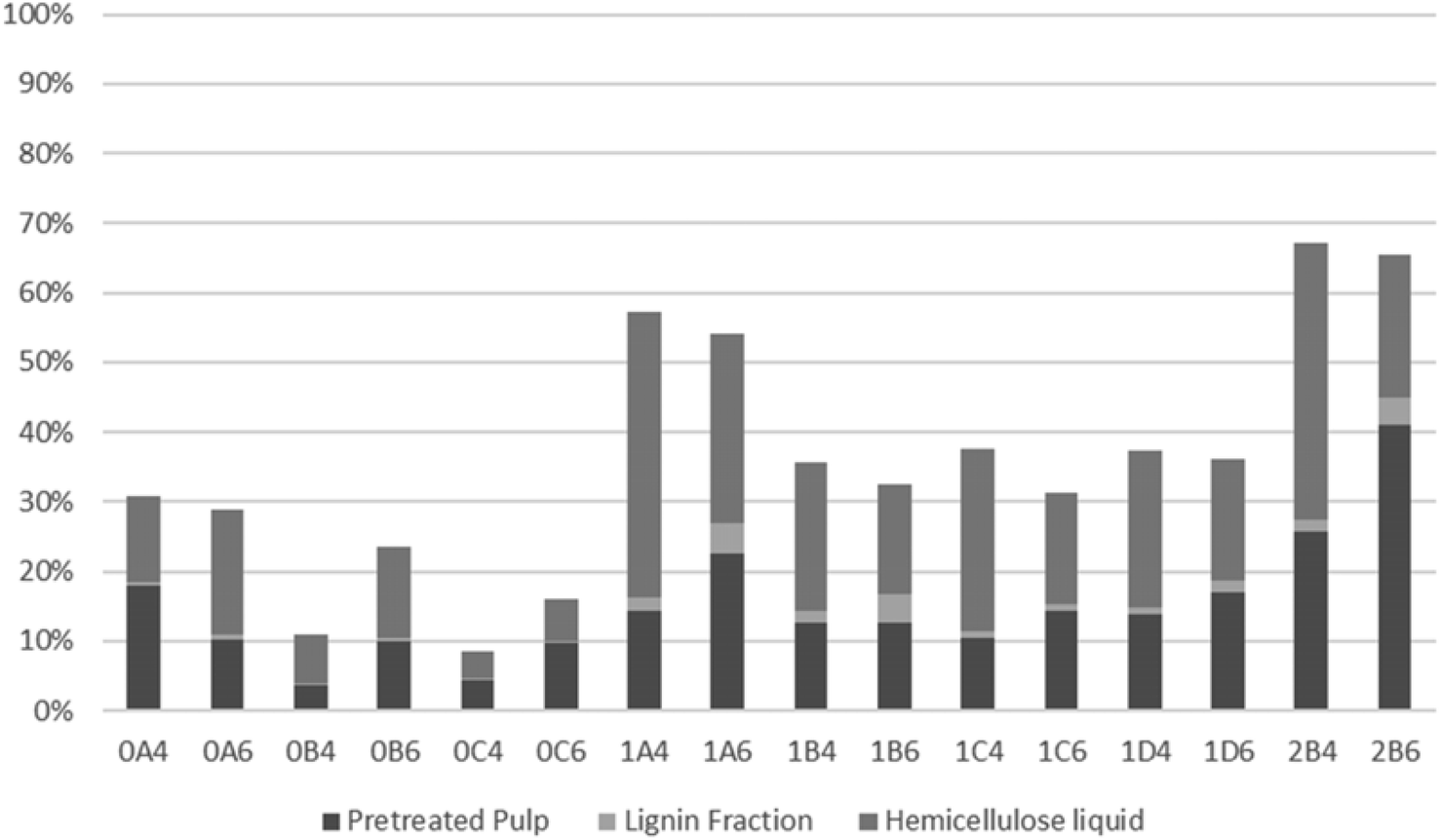 | ||
| Fig. 6 Hemicellulose distribution in the different S. ramosissima fractions. | ||
Enzymatic saccharification
The saccharification of pretreated pulps was performed to assess the viability of applications of the material in microbial cultivation for fermentation, and the results are presented in Fig. 7. The higher results of saccharification of 81% were achieved on the pretreatments at lower temperature that have lower amounts of degradation compounds that can inhibit the enzymatic activity. On the pulps produced in the pretreatments at 200 °C, higher saccharification (48.9%) was achieved when the pretreatment was performed for 15 minutes with 60% v/v ethanol content and besides the pretreatments of 15 minutes, the saccharification was under 30%. Shorter pretreatments at 180 °C in addition to pretreatments at 200 °C presented higher yields of saccharification and at this temperature, the advantage of a higher ethanol content is also shown. These results show that cellulose is readily hydrolyzed by enzymes to glucose and as such can serve as a feedstock for different bioprocesses.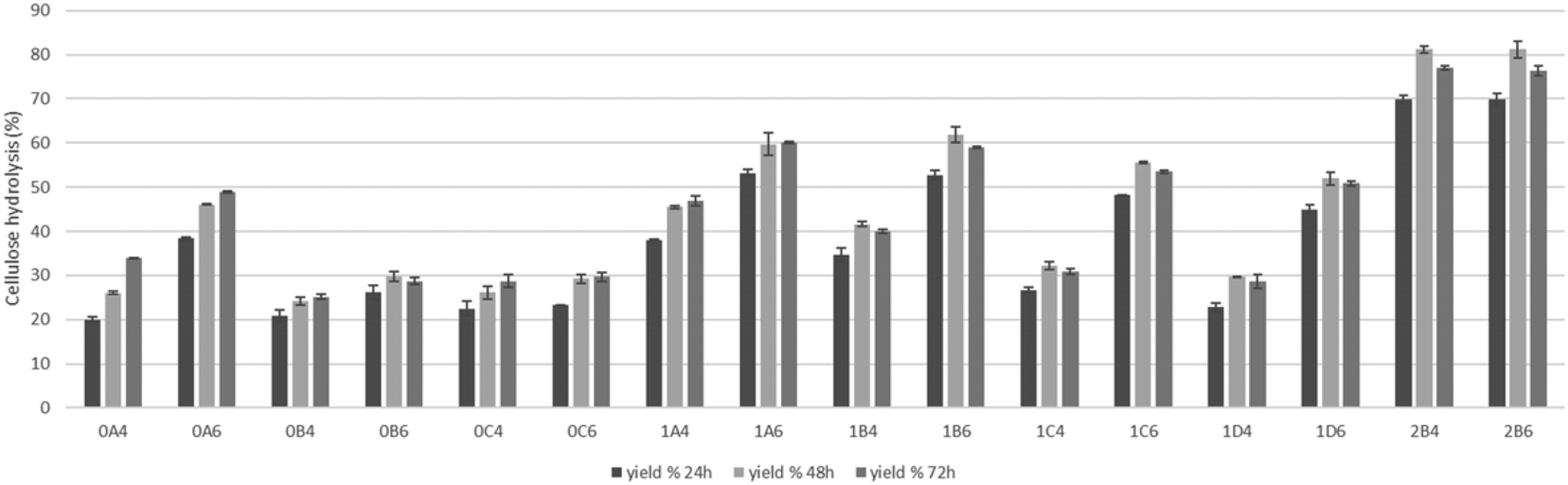 | ||
| Fig. 7 Cellulose enzymatic hydrolysis yields of Salicornia ramosissima pretreated biomasses. | ||
Conclusion
The present study demonstrates the applicability of residual dejuiced fibers of Salicornia ramosissima in a biorefinery context. Organosolv pretreatment was applied under 16 different conditions and detailed analysis of the streams was performed to elucidate the composition of the streams produced. With the exception of three mildest pretreatments, the removal of hemicelluloses was higher than 80% and delignification was as high as 68.8%. The mildest pretreatments on the other hand yielded higher hemicellulose fractions as well as an increased ratio of oligomers in the samples. The main branched oligosaccharides were hexurono-XOS/alduronic acids. Lignin fractions had low sugar and ash contamination and small molecular weight, characteristics desired for further applications. Overall our results suggest the potential of applying residual fibers of S. ramosissima in a biorefinery approach.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement no. 862834. Mattias Hedenström, Swedish NMR Centre (Umeå, Umeå University, VR RFI), João Figueira, Swedish NMR Centre (Umeå, Umeå University, Scilife Lab) and the NMR Core Facility (Swedish NMR Centre, SwedNMR, Umeå node), Umeå University are acknowledged for NMR support. Laura Hulkko (AAU Energy, Aalborg University) and Tanmay Chaturvedi (AAU Energy, Aalborg University) are acknowledged for the supply of the de-juiced fibers and technical assistance.References
- S. J. Malode, K. K. Prabhu, R. J. Mascarenhas, N. P. Shetti and T. M. Aminabhavi, Energy Convers. Manage.: X, 2021, 10, 100070 CAS.
- T. G. Ambaye, M. Vaccari, A. Bonilla-Petriciolet, S. Prasad, E. D. van Hullebusch and S. Rtimi, J. Environ. Manage., 2021, 290, 112627 CrossRef CAS PubMed.
- B. H. Kreps, Am. J. Sociol., 2020, 79, 695–717 CrossRef.
- Md. Washim Akram, Md. Arman Arefin and A. Nusrat, Energy Syst., 2022, 13, 749–787 CrossRef.
- Energy Crisis, World Bank, 2022.
- T. Werpy and G. Petersen, Top Value Added Chemicals from Biomass: Volume I – Results of Screening for Potential Candidates from Sugars and Synthesis Gas, Golden, CO (United States), 2004 Search PubMed.
- M. H. To, H. Wang, Y. Miao, G. Kaur, S. L. K. W. Roelants and C. S. K. Lin, Bioresour. Technol., 2023, 379, 128993 CrossRef CAS PubMed.
- M. Pauly and K. Keegstra, Curr. Opin. Plant Biol., 2010, 13, 304–311 CrossRef.
- T. Rodrigues Mota, D. Matias de Oliveira, R. Marchiosi, O. Ferrarese-Filho and W. Dantas dos Santos, AIMS Bioeng., 2018, 5, 63–77 Search PubMed.
- S. Varjani, W. Yan, A. Priya, F. Xin and C. S. K. Lin, Curr. Opin. Green Sustainable Chem., 2023, 41, 100806 CrossRef CAS.
- N. Brosse, M. H. Hussin and A. A. Rahim, in Biorefineries, ed. K. Wagemann and N. Tippkötter, Springer, Cham, 2017, pp. 153–176 Search PubMed.
- X. Zhao, K. Cheng and D. Liu, Appl. Microbiol. Biotechnol., 2009, 82, 815–827 CrossRef CAS PubMed.
- F. Xu, C. F. Liu, Z. C. Geng, J. X. Sun, R. C. Sun, B. H. Hei, L. Lin, S. B. Wu and J. Je, Polym. Degrad. Stab., 2006, 91, 1880–1886 CrossRef CAS.
- A. Johansson, O. Aaltonen and P. Ylinen, Biomass, 1987, 13, 45–65 CrossRef CAS.
- T. J. McDonough, in TAPPI Solvent Pulping Seminar, Boston, MA, 1992, p. 17 Search PubMed.
- J. H. Lora and S. Aziz, Tappi J., 1985, 68, 94–97 CAS.
- M. Akgul and H. Kirci, J. Environ. Biol., 2009, 735–740 CAS.
- A. Trubetskaya, H. Lange, B. Wittgens, A. Brunsvik, C. Crestini, U. Rova, P. Christakopoulos, J. J. Leahy and L. Matsakas, Processes, 2020, 8, 860 CrossRef CAS.
- A. Hameed and M. Ajmal Khan, Karachi Univ. J. Sci., 2011, 39, 40–44 Search PubMed.
- E. P. Glenn, J. J. Brown and E. Blumwald, CRC Crit. Rev. Plant Sci., 1999, 18, 227–255 CrossRef.
- J. M. Cheeseman, New Phytol., 2015, 206, 557–570 CrossRef.
- I. Sanjosé, F. Navarro-Roldán, Y. Montero, S. Ramírez-Acosta, F. J. Jiménez-Nieva, M. D. Infante-Izquierdo, A. Polo-Ávila and A. F. Muñoz-Rodríguez, Diversity, 2022, 14, 452 CrossRef.
- A. Castagna, G. Mariottini, M. Gabriele, V. Longo, A. Souid, X. Dauvergne, C. Magné, G. Foggi, G. Conte, M. Santin and A. Ranieri, Horticulturae, 2022, 8, 828 CrossRef.
- M. Lopes, M. J. Roque, C. Cavaleiro and F. Ramos, J. Food Compos. Anal., 2021, 104, 104135 CrossRef CAS.
- D. Ferreira, V. M. S. Isca, P. Leal, A. M. L. Seca, H. Silva, M. de Lourdes Pereira, A. M. S. Silva and D. C. G. A. Pinto, Arabian J. Chem., 2018, 11, 70–80 CrossRef CAS.
- A. Cayenne, A. E. Turcios, M. H. Thomsen, R. M. Rocha, J. Papenbrock and H. Uellendahl, Fermentation, 2022, 8, 189 CrossRef CAS.
- L. S. S. Hulkko, R. M. Rocha, R. Trentin, M. Fredsgaard, T. Chaturvedi, L. Custódio and M. H. Thomsen, Plants, 2023, 12, 1251 CrossRef CAS PubMed.
- L. S. S. Hulkko, A. E. Turcios, S. Kohnen, T. Chaturvedi, J. Papenbrock and M. H. Thomsen, Sci. Rep., 2022, 12, 20507 CrossRef CAS.
- A. M. Silva, J. P. Lago, D. Pinto, M. M. Moreira, C. Grosso, V. Cruz Fernandes, C. Delerue-Matos and F. Rodrigues, Appl. Sci., 2021, 11, 4744 CrossRef CAS.
- M. Lopes, C. Cavaleiro and F. Ramos, Compr. Rev. Food Sci. Food Saf., 2017, 16, 1056–1071 CrossRef CAS PubMed.
- S. C. Oliveira-Alves, F. Andrade, I. Prazeres, A. B. Silva, J. Capelo, B. Duarte, I. Caçador, J. Coelho, A. T. Serra and M. R. Bronze, Antioxidants, 2021, 10, 1312 CrossRef CAS PubMed.
- P. García-Rodríguez, F. Ma, C. del Río, M. Romero-Bernal, A. M. Najar, M. de la L. Cádiz-Gurrea, F. J. Leyva-Jimenez, L. Ramiro, P. Menéndez-Valladares, S. Pérez-Sánchez, A. Segura-Carretero and J. Montaner, Nutrients, 2022, 14, 5077 CrossRef PubMed.
- R. Giordano, G. E. Aliotta, A. S. Johannesen, D. Voetmann-Jensen, F. H. Laustsen, L. A. Andersen, A. Rezai, M. Fredsgaard, S. lo Vecchio, L. Arendt-Nielsen, M. H. Thomsen and A. Stensballe, Pharmaceuticals, 2022, 15, 150 CrossRef.
- A. R. Lima, N. L. Cristofoli, K. Filippidis, L. Barreira and M. C. Vieira, J. Food Process Eng., 2022, 45, e14154 CAS.
- M. R. Loizzo and R. Tundis, Antioxidants, 2022, 11, 797 CrossRef CAS PubMed.
- A. Sluiter, R. Ruiz, C. Scarlata, J. Sluiter and D. Templeton, Determination of Extractives in Biomass, Golden, CO, 2005 Search PubMed.
- A. Sluiter, B. Hames, R. Ruiz, C. Scarlata, J. Sluiter, D. Templeton and D. Crocker, Determination of Structural Carbohydrates and Lignin in Biomass: Laboratory Analytical Procedure (LAP), (Revised July 2011), 2008 Search PubMed.
- A. Sluiter, B. Hames, R. Ruiz, C. Scarlata, J. Sluiter and D. Templeton, Determination of Sugars, Byproducts, and Degradation Products in Liquid Fraction Process Samples, National Renewable Energy Laboratory, 2006, vol. 11, pp. 65–71 Search PubMed.
- J. Peng, F. Lu and J. Ralph, J. Agric. Food Chem., 1998, 46, 553–560 CrossRef CAS PubMed.
- M. Monção, T. Wretborn, U. Rova, L. Matsakas and P. Christakopoulos, RSC Adv., 2022, 12, 28599–28607 RSC.
- M. J. González-Muñoz, R. Alvarez, V. Santos and J. C. Parajó, Wood Sci. Technol., 2012, 46, 271–285 CrossRef.
- Z. Chen, S. Li, Y. Fu, C. Li, D. Chen and H. Chen, J. Funct. Foods, 2019, 54, 536–551 CrossRef CAS.
- Z. X. Lu, K. Z. Walker, J. G. Muir and K. O'Dea, Eur. J. Clin. Nutr., 2004, 58, 621–628 CrossRef CAS PubMed.
- Y. Takahashi and H. Matsunaga, Macromolecules, 1991, 24, 3968–3969 CrossRef CAS.
- K. Werner, L. Pommer and M. Broström, J. Anal. Appl. Pyrolysis, 2014, 110, 130–137 CrossRef CAS.
- S. Catena, N. Rakotomanomana, P. Zunin, R. Boggia, F. Turrini and F. Chemat, Ultrason. Sonochem., 2020, 68, 105231 CrossRef CAS PubMed.
- A. M. Silva, J. P. Lago, D. Pinto, M. M. Moreira, C. Grosso, V. Cruz Fernandes, C. Delerue-Matos and F. Rodrigues, Appl. Sci., 2021, 11, 4744 CrossRef CAS.
- T. S. Milessi, F. A. S. Corradini, J. V. M. Marçal, T. O. Baldez, W. Kopp, R. C. Giordano and R. L. C. Giordano, Ind. Crops Prod., 2021, 172, 114056 CrossRef CAS.
- M. S. Izydorczyk and C. G. Biliaderis, Carbohydr. Polym., 1992, 17, 237–247 CrossRef CAS.
- T. E. Timell, in Advances in Carbohydrate Chemistry, 1965, vol. 20, pp. 409–483 Search PubMed.
- S. Wichienchot, P. Thammarutwasik, A. Jongjareonrak, W. Chansuwan, P. Hmadhlu, T. Hongpattarakere, A. Itharat and B. Ooraikul, Songklanakarin J. Sci. Technol., 2011, 33, 517–523 CAS.
- C. S. Goh, H. T. Tan, K. T. Lee and N. Brosse, Biomass Bioenergy, 2011, 35, 4025–4033 CrossRef CAS.
- A. Mishra, M. Joshi and B. Jha, Carbohydr. Polym., 2013, 92, 1942–1945 CrossRef CAS PubMed.
- A. Shatalov, Carbohydr. Polym., 2002, 49, 331–336 CrossRef CAS.
- M. Nacos, P. Katapodis, C. Pappas, D. Daferera, P. Tarantilis, P. Christakopoulos and M. Polissiou, Carbohydr. Polym., 2006, 66, 126–134 CrossRef CAS.
- P. Christakopoulos, P. Katapodis, E. Kalogeris, D. Kekos, B. J. Macris, H. Stamatis and H. Skaltsa, Int. J. Biol. Macromol., 2003, 31, 171–175 CrossRef CAS PubMed.
- P. Katapodis, A. Kavarnou, S. Kintzios, E. Pistola, D. Kekos, B. J. Macris and P. Christakopoulos, Biotechnol. Lett., 2002, 24, 1413–1416 CrossRef CAS.
- S. Larsson, E. Palmqvist, B. Hahn-Hägerdal, C. Tengborg, K. Stenberg, G. Zacchi and N.-O. Nilvebrant, Enzyme Microb. Technol., 1999, 24, 151–159 CrossRef CAS.
- L. Matsakas, K. Novak, J. Enman, P. Christakopoulos and U. Rova, Bioresour. Technol., 2017, 242, 287–294 CrossRef CAS PubMed.
- Y. A. Attia, H. F. Ellakany, A. E. Abd El-Hamid, F. Bovera and S. A. Ghazaly, Arch. für Geflugelkunde, 2012, 76, 239–245 CAS.
- G. Gellerstedt and G. Henriksson, in Monomers, Polymers and Composites from Renewable Resources, Elsevier, 2008, pp. 201–224 Search PubMed.
- P. Paulsen Thoresen, H. Lange, C. Crestini, U. Rova, L. Matsakas and P. Christakopoulos, ACS Omega, 2021, 6, 4374–4385 CrossRef CAS PubMed.
- C. Cabral Almada, A. Kazachenko, P. Fongarland, D. da Silva Perez, B. N. Kuznetsov and L. Djakovitch, Biomass Convers. Biorefin., 2022, 12, 3795–3808 CrossRef CAS.
- R. H. Narron, H. Chang, H. Jameel and S. Park, ACS Sustainable Chem. Eng., 2017, 5, 10763–10771 CrossRef CAS.
- S. Constant, H. L. J. Wienk, A. E. Frissen, P. de Peinder, R. Boelens, D. S. van Es, R. J. H. Grisel, B. M. Weckhuysen, W. J. J. Huijgen, R. J. A. Gosselink and P. C. A. Bruijnincx, Green Chem., 2016, 18, 2651–2665 RSC.
- H. Kim, D. Padmakshan, Y. Li, J. Rencoret, R. D. Hatfield and J. Ralph, Biomacromolecules, 2017, 18, 4184–4195 CrossRef CAS PubMed.
- T.-Q. Yuan, S.-N. Sun, F. Xu and R.-C. Sun, J. Agric. Food Chem., 2011, 59, 10604–10614 CrossRef CAS.
- S. Bhagia, Y. Pu, B. R. Evans, B. H. Davison and A. J. Ragauskas, Bioresour. Technol., 2018, 269, 567–570 CrossRef CAS PubMed.
- P. P. Thoresen, H. Lange, U. Rova, P. Christakopoulos and L. Matsakas, Int. J. Biol. Macromol., 2023, 233, 123471 CrossRef CAS PubMed.
- A. De Santi, M. V. Galkin, C. W. Lahive, P. J. Deuss and K. Barta, ChemSusChem, 2020, 13, 4468–4477 CrossRef CAS PubMed.
- D.-E. Kim and X. Pan, Ind. Eng. Chem. Res., 2010, 49, 12156–12163 CrossRef CAS.
- L. F. Del Rio, R. P. Chandra and J. N. Saddler, Appl. Biochem. Biotechnol., 2010, 161, 1–21 CrossRef CAS PubMed.
- Z. Zhang, M. D. Harrison, D. W. Rackemann, W. O. S. Doherty and I. M. O'Hara, Green Chem., 2016, 18, 360–381 RSC.
- S. D. Shinde, X. Meng, R. Kumar and A. J. Ragauskas, Green Chem., 2018, 20, 2192–2205 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3se00458a |
| This journal is © The Royal Society of Chemistry 2023 |