
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A manganese complex on a gas diffusion electrode for selective CO2 to CO reduction†
Catherine
Eagle
a,
Gaia
Neri
ab,
Verity L.
Piercy
ac,
Khadija
Younis
a,
Bhavin
Siritanaratkul
 a and
Alexander J.
Cowan
*a
a and
Alexander J.
Cowan
*a
aStephenson Institute for Renewable Energy and the Department of Chemistry, University of Liverpool, Liverpool, L69 7ZF, UK. E-mail: acowan@liverpool.ac.uk
bEnapter, Pisa, Tuscany, Italy
cNSG Pilkington, Latham, Ormskirk, UK
First published on 5th April 2023
Abstract
Manganese carbonyl complexes have been studied extensively in solution as low cost, selective electrocatalysts with a low overpotential for CO2 reduction but experiments are typically at low current densities. In this work, we examined their application in a gas diffusion electrode (GDE) flow cell and achieved partial current densities for CO, jCO of ∼14 mA cm−2 (−0.98 VRHE) with a Faradaic efficiency of >50%. Although we did observe a gradual decrease in activity for the [Mn(2,2′-bipyridine)(CO)3Br]/MWCNT (Mnbpy) GDE with a near neutral electrolyte over a 5 h experiment, it still achieves a higher initial partial current density for CO at a lower overpotential than a Ag nanoparticle benchmark electrode. Promisingly, initial studies of the Mnbpy GDE in a zero-gap electrolyser using a reverse biased bipolar membrane (BPM) achieved FE for CO of 70% at 50 mA cm−2, despite the acidic environment induced through directly contacting the membranes cation exchange layer. Overall this study demonstrates the potential of GDEs for CO2 reduction based on a catalyst using earth abundant elements.
Introduction
The electrocatalytic reduction of CO2 to useful products has been investigated extensively within the scientific community using heterogeneous metal catalysts such as Ag, Au and Cu.1 Molecular catalysts provide an opportunity to alter product selectivity due to the ability to tune electron density at the metal centre by altering ligands and functionality.2–5 One particular catalyst of note is a Re carbonyl complex ([Re(bpy)(CO)3Cl], bpy = 2,2′-bipyridine) which displayed good selectivity for CO.6 The related Mn derivative (Mnbpy = [Mn(bpy)(CO)3Br]) has since been shown to operate as a low-cost, low overpotential alternative when a suitable Brønsted acid is present,7 with selectivity equivalent to those achieved with Re (FECO = 85% at −1.32 VAg/AgCl).CO2 reduction using Mnbpy molecular catalysts has mainly been performed homogeneously in aprotic solvents with a low concentration of acid added. A small number of studies have shown that the Mn class of catalysts can operate in aqueous solvents either in solution or immobilised.8–14 Unfortunately, one of the major limitations of aqueous CO2 reduction is the low current densities achieved due to mass transport limitations of CO2 in aqueous electrolytes ([CO2]aq at 298 K, 1 atm = 33 mM).15 GDE architectures provide an opportunity to increase current densities and have been reported extensively,16 particularly with heterogeneous metal catalysts. The GDE allows a high concentration of CO2 to be maintained at the catalyst, even at high current densities.17,18 Recently a number of examples of molecular catalysts immobilised on GDEs for CO2 reduction have been reported.19,20
The only example of a Mnbpy derivative being used in a GDE cell structure that we are aware of used a modified bipyridine ligand with an aminophenyl functional group to enable covalent binding onto the carbon GDE.20 In these experiments the GDE was used with a flowed, near neutral pH, catholyte (0.2 M KHCO3 (aq)) at a range of potentials with good FE (FECO ∼60%) and a maximum jCO ∼0.7 mA cm−2 in variable potential experiments, although up to ∼6 mA cm−2 was reported when the same electrode was used for longer term electrolysis under apparently identical conditions.20 An alternative approach to covalent immobilisation that has been demonstrated is the simple co-deposition of Mnbpy on multi-walled carbon nanotubes (MWCNT) in a Nafion solution on a planar carbon electrode. In these studies, good partial current densities could be achieved (jCO ∼0.6 mA cm−2) even in an H-cell configuration.8,21 Building on these initial studies we report here the behaviour of Mnbpy/MWCNT GDEs prepared by a simple deposition method, and the behaviour of these electrodes in both a flow electrolyser (at near neutral pH) and in zero-gap configurations (both with high and low local pH at the cathode, Fig. 1).
 | ||
| Fig. 1 Schematic of the flow cell using an AEM and neutral electrolyte (a) and zero-gap electrolyser where either a reverse biased BPM or an AEM provide a low or high pH environment for the GDE cathode (b). The GDE structure with the catalyst/MWCNT coating shown in orange and expanded in the inset. | ||
The pH dependence of the Mnbpy GDE is of particular interest as carbonate formation and cross-over to the anode is a major loss pathway in the majority of GDE systems where anion exchange membranes (AEM) and a high local pH at the cathode are used. Carbonate cross-over leads to low CO2 utilisation yields which has been suggested to limit the commercial viability of many commonly studied low-temperature device architectures.22 The use of a reverse biased bipolar membrane (BPM) or cation exchange membrane (CEM) in a zero-gap architecture provides a low local pH, reducing HCO3−/CO32− formation and minimising cross-over of these species to the anode.23 However, an acidic environment can lead to high levels of H2 evolution and low FE for carbon products using conventional metal electrocatalysts which poses challenges for related systems.24–26 Molecular catalysts are of interest due to several demonstrating good tolerances to low pH, in particular, Mnbpy derivatives have been reported to operate in solution at pH ∼3.5.9 Here we find that the Mnbpy GDE is largely inactive for CO2 reduction at a high pH (in a zero-gap AEM cell) but at neutral (KHCO3 flow cell), and significantly at low pH (reverse biased BPM zero-gap cell), high FEs for CO production can be reached. Although device stabilities are limited at this point, this initial study shows that molecular catalysts based on earth abundant elements are of potential interest for use in CO2 electrolysers.
Experimental
Materials
Milli-Q water (18.2 MΩ) was used throughout. CO2 (1% CH4) and CO2 (CP grade) was purchased from BOC, potassium bicarbonate and potassium hydroxide from Sigma Aldrich (≥99.5%), Nafion 117 solution (∼5% in a mixture of water and aliphatic alcohols) from Sigma Aldrich, the Selemion anion exchange membrane (AEM) from Bellex International and Sustainion and Fumasep FBM from FuelCellStore. ELAT LT1400 (FuelCellStore) with a thickness 454 μm and a 5 wt% PTFE treatment was used as the GDE. RuO2 nanoparticles of size 5–10 nm were used as received (FuelCellStore) as were Ag nanoparticles with size < 150 nm (Sigma Aldrich). Mnbpy was synthesised according to a reported procedure.6Electrode preparation
Cathodes were prepared on ELAT LT1400 gas diffusion electrodes which were sprayed (back-side) with an additional 5 wt% PTFE (1 mL) layer and dried for 1 hour at 100 °C. Multi-walled carbon nanotubes (MWCNT, >98% carbon basis) were cleaned by sonication in 5% HCl for 30 min, filtered and dried in an oven. Catalyst inks were prepared by sonicating the cleaned MWCNT (2 mg cm−2) in ethanol (400 μL) and water (400 μL) for 1 hour, followed by the addition of Nafion 117 (2 μL), 60 wt% PTFE (9 μL) and Mnbpy (0.1, 1 or 4 mg cm−2) or Ag nanoparticles (4 mg cm−2). This solution was sonicated for 15 minutes and painted onto 1 cm2 active area (front-side) in 40 μL portions and dried overnight at 35 °C in the dark. Anodes were prepared using RuO2 (42 mg) in isopropyl alcohol (2.6 mL) and water (2.6 mL) which was sonicated for 1 hour, followed by the addition of Nafion 117 (420 μL) and further sonication for 1 hour. For flow cell experiments the titanium electrode surface of 10.5 cm2 was sprayed with the anode catalyst ink and dried at 100 °C for 1 hour prior to use. In the zero-gap structure the RuO2 was sprayed onto the carbon gas diffusion electrode held at 100 °C at a loading of 4 mg cm−2.Electrochemical measurements
All electrochemical measurements were performed using an Ivium Vertex potentiostat. Cyclic voltammograms (CVs) of the GDE were obtained by using the GDE as the working electrode, a platinum wire as the counter and a Ag/AgCl leak-free reference electrode. In flow cell experiments the electrolyte was 0.5 M KHCO3 (aq) and was pre-electrolysed (−0.1 mA) overnight with a titanium plate (working) and platinum wire (counter).27 The electrolyte was purged with either Ar or CO2 for 1 hour prior to each experiment. An ElectroCell Micro Flow Cell was used for all flow experiments (non-zero gap). The flow cell was composed of a titanium anode plate, a stainless-steel cathode plate, separated by PTFE electrolyte flow plates and rubber gaskets. The membrane (Selemion) was sandwiched between these plates to provide an assembly in which gas was flowed through the back-side of the cathode and electrolyte was flowed over the surface of the cathode and anode (separated by the membrane). CO2 (containing 1% CH4 as an internal calibrant) or pure CO2 was flowed at 20 mL min−1 through an Alicat mass flow controller. The catholyte and anolyte were pumped at 12 mL min−1 and 22 mL min−1 respectively and electrolysis begun once electrolyte had circulated through the tubing and dripped into the catholyte/anolyte beaker. The same supporting electrolyte concentrations were used for both the anolyte and catholyte. Data from the flow cell is reported with iR compensation (80%). The zero-gap electrolyser cell (Dioxide Materials) was composed of a serpentine anolyte chamber, a serpentine chamber for gas flow over the back of the cathode and two PTFE gaskets to separate the anode, membrane and cathode. Either Sustainion X37-50 (AEM, Dioxide Materials) or Fumasep (BPM, FuelCellStore) were sandwiched between the PTFE gaskets and pressed directly onto the cathode to provide an assembly where humidified CO2 was flowed over the backside of the cathode (20 mL min−1) and anolyte (1 M KOH (aq)) was flowed over the anode and chronopotentiometry was run in a two-electrode set-up. Gas chromatography was performed on an Agilent 6890N with a pulsed discharge detector using a N6 helium carrier gas, or using a Varian CP-4900 MicroGC with a Molsieve 5 Å column (10 m) with Ar carrier gas for H2 and CO detection by a thermal conductivity detector.Results and discussion
GDEs for CO2 reduction were prepared by deposition of a Mnbpy/MWCNT/Nafion mixture in ethanol/water onto the microporous layer of a commercial GDE. Scanning electron microscopy (SEM) and EDX-mapping show the structure of the electrode surface (Fig. S1†) and FTIR and XPS show the presence of Mnbpy on the electrode surface on the micron scale (Fig. S2–S4†). XPS analysis shows that the Mnbpy coated GDE has N 1s and Mn 2p peaks that are at 400.2 eV, 641.5 eV and 647.1 eV respectively. The binding energies of the immobilised Mnbpy are similar to Mnbpy powder (399.5 eV, 642.1 eV and 646.2 eV, Fig. S4†). We can conclude that the catalyst structure remains intact following deposition onto the GDE and that the interaction with the MWCNT has not substantially modified the electronic properties of the catalyst. Past studies of Mnbpy on carbon supports have shown that the catalyst loading can have a profound impact on operating potential and selectivity to carbon products.28 Therefore, electrodes were initially tested with variable Mnbpy loadings (0.1, 1, 4 mg cm−2) in a flow electrolyser with liquid catholyte at neutral pH (0.5 M KHCO3, pH ∼7.1 start, pH ∼8.6 end, Fig. S5†), Fig. 2. Chronopotentiometry experiments were recorded at 20 mA cm−2 for 90 minutes. A loading of 4 mg cm−2 resulted in the highest FECO (63%) and lowest cell potential (−0.95 VRHE). H2 (29% FE) and formate (6% FE) were also formed at lower levels, Fig. 2. | ||
| Fig. 2 FE and cathode potential for a Mnbpy CO2 reduction GDE in a flow cell with a 0.5 M KHCO3 catholyte with different catalyst loadings at 20 mA cm−2 (a) time-course FE data for 4 mg cm−2 Mnbpy GDE at 20 mA cm−2 (b). | ||
The FEs for formate and CO both decrease as the Mnbpy loading decreases and an increase in H2 evolution occurs. At the lower Mnbpy loadings we also found that a significant fraction of charge remains unaccounted for (total FE accounted for is 81.5% and 83.4% with 0.1 and 1 mg cm−2). One possible cause of the lower FE in these experiments is formate/formic acid transport across the AEM. We repeated the experiment using a Nafion membrane which minimises this crossover and it shows a constant 5% increase in the formate FE over the course of the reaction, indicating that some formate/formic acid crosses through the AEM to be oxidised at the anode and this may account for the lower FE when an AEM is used. The formation of formate from CO2 has been reported in several past studies, including on a Mnbpy catalyst derivatized with pyrene groups, which was immobilised on MWCNT in water.28 However, formate production has also been previously reported on MWCNT in the absence of a catalyst.29 Therefore we also prepared GDEs with only MWCNT and no Mnbpy. In experiments at 20 mA cm−2 the MWCNT electrodes generate appreciable levels of formate (∼10% FE) with H2 being the only other product detected, Fig. S6.† These control experiments indicate that the formate measured here is at least in part derived from the MWCNT and that CO is the primary CO2 reduction product produced by the Mnbpy catalyst.
Following an initial increase in FE for both CO production and H2 production over the first 25 minutes we find that the catalytic activity of the Mnbpy is relatively stable during 90 minutes of operation (Fig. 2b). Over 90 minutes we achieve a turnover number (TON) of 33 on the basis of the total Mnbpy deposited. (Note that this TON is a lower limit as the actual concentration of electrochemically active Mnbpy will be lower than the total deposited.) Control experiments in the absence of CO2 (with a N2 flow to the GDE) showed no significant CO formation demonstrating that the CO is produced as a result of the reduction of CO2 and not due to the breakdown of the Mnbpy catalyst (Fig. S7†). The increase in FE for H2 and CO production over the first 25 minutes correlates with an increase in overpotential, Fig. 2b. Although the electrolyser is flushed with CO2 for 15 minutes prior to experiments we believe that this initial onset is due to the reduction of trace O2 that is present within the porous GDEs.
As our initial studies identified that a 4 mg cm−2 loading of catalyst gave the highest activity, all further experiments described in the manuscript use GDEs prepared in this way. To examine the potential dependence of the electrochemical response of the GDE, four different potentials have been studied, Fig. 3. At −0.58 VRHE there is only a very low total current density (2.6 mA cm−2) and negligible CO production. CVs of the Mnbpy GDE immersed in a 0.5 M KHCO3 electrolyte, (Fig. S8†) show that at −0.58 VRHE there is a relatively small increase in current density under CO2vs. under Ar. Fig. S8† indicates that the catalytically active species, [Mn(bpy)(CO)3]−, is not formed until approximately −0.8 VRHE, in-line with past reports.7,8 Supporting the conclusion that [Mn(bpy)(CO)3]− is the catalytically active species, jCO increases to ∼0.5 mA cm−2 at −0.78 VRHE in the flow cell and at −0.98 VRHEjCO reaches a maximum of 13.7 ± 2.0 mA cm−2 (56 ± 8% FE). H2 (FE 30 ± 1%, 7.3 ± 0.2 mA cm−2) and formate (FE ∼11%, 2.7 mA cm−2) are also formed at −0.98 VRHE. We believe that this is amongst the highest CO partial current density to date for a Mn based CO2 reduction catalyst either in aqueous solution or on a GDE (see Table S1†). At more negative potentials (−1.18 VRHE) the hydrogen evolution reaction dominates and jCO actually decreases (7.7 ± 1.6 mA cm−2). Under operating conditions, we anticipated a local pH increase at the GDE due to the effects of CO2 reduction and hydrogen evolution. Reaction-diffusion simulations (see ESI for full details, Fig. S9†) of the local pH at the catalyst layer for varying current densities confirm that by 30 mA cm−2 there is a marked increase in local pH to 9.8. The pH behaviour of the Mnbpy GDE is discussed in more detail in the following section. Previous experimental and DFT studies7,14,30–33 have shown that CO2 binding to [Mn(bpy)(CO)3]− is proton assisted, therefore the predicted rise in pH rationalises the decrease in jCO at −1.18 VRHE.
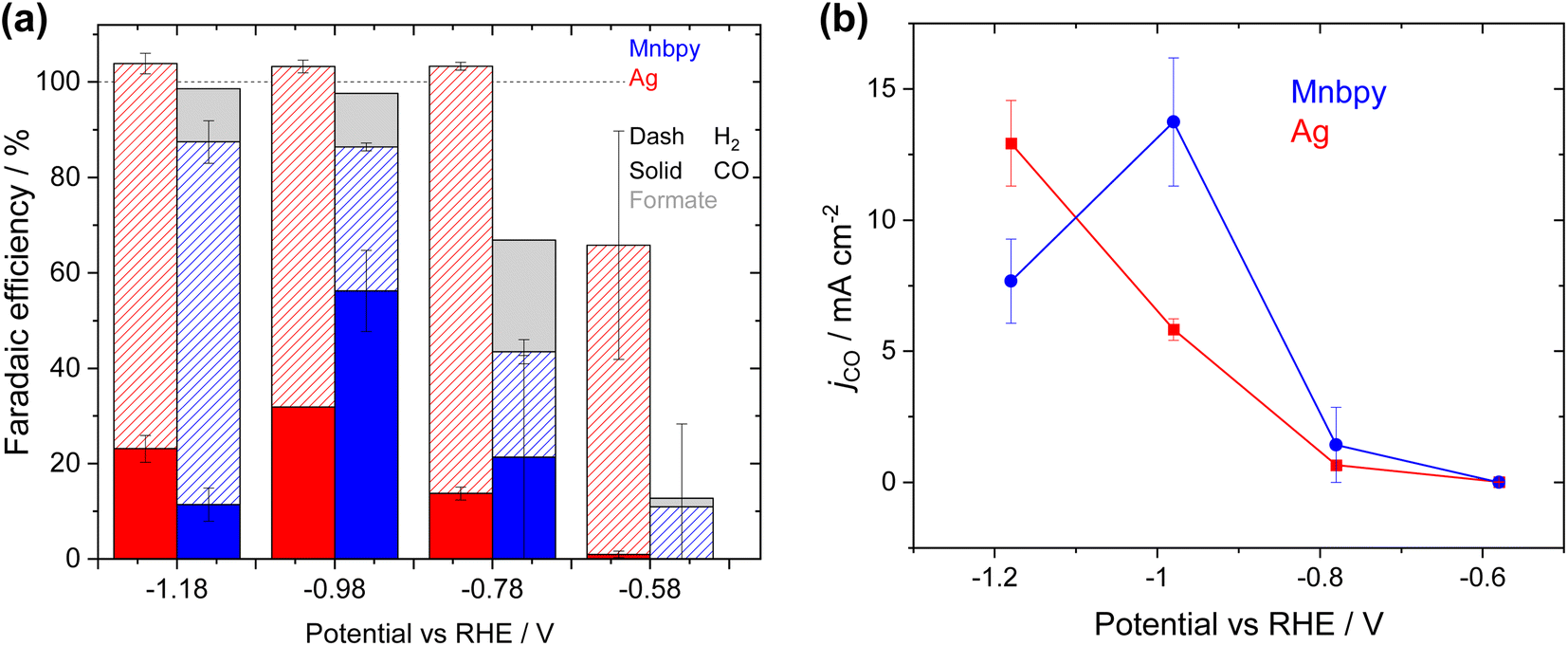 | ||
| Fig. 3 Mnbpy (blue) GDE and Ag (red) GDE FEs (a) and partial CO current densities (b) recorded over 60 min period when the GDE is used as the cathode in the 0.5 M KHCO3 flow cell for CO2 reduction. | ||
Fig. 4 shows the extended stability test of the Mnbpy GDE and the Ag GDE in the flow cell using a 0.5 M KHCO3 electrolyte (5 hours at −0.98 VRHE). The Mnbpy GDE initially shows a very high level of activity for CO2 reduction with FECO of 64% and jCO of 26 mA cm−2 within the first 30 minutes, but at >100 minutes we see activity slowly dropping and after 5 hours the FECO has decreased to <20% and the jCO ∼5 mA cm−2. One possible cause of the instability of the Mnbpy GDE is that it is operating at higher current densities which is known to accelerate GDE flooding,34–36 thought to be due to local pH changes and increased formation of KHCO3/K2CO3, but we rule this out here. Measurements of capacitance of a GDE before and after electrolysis have been shown to be an effective probe of flooding.37 Fig. S10† shows negligible differences in capacitance pre- and post-electrolysis (5 hours), furthermore there are no visible water droplets collecting in the cathode gas flow channels. Therefore, for the Mnbpy GDE we do not believe that electrode flooding is the cause of the decrease in activity.
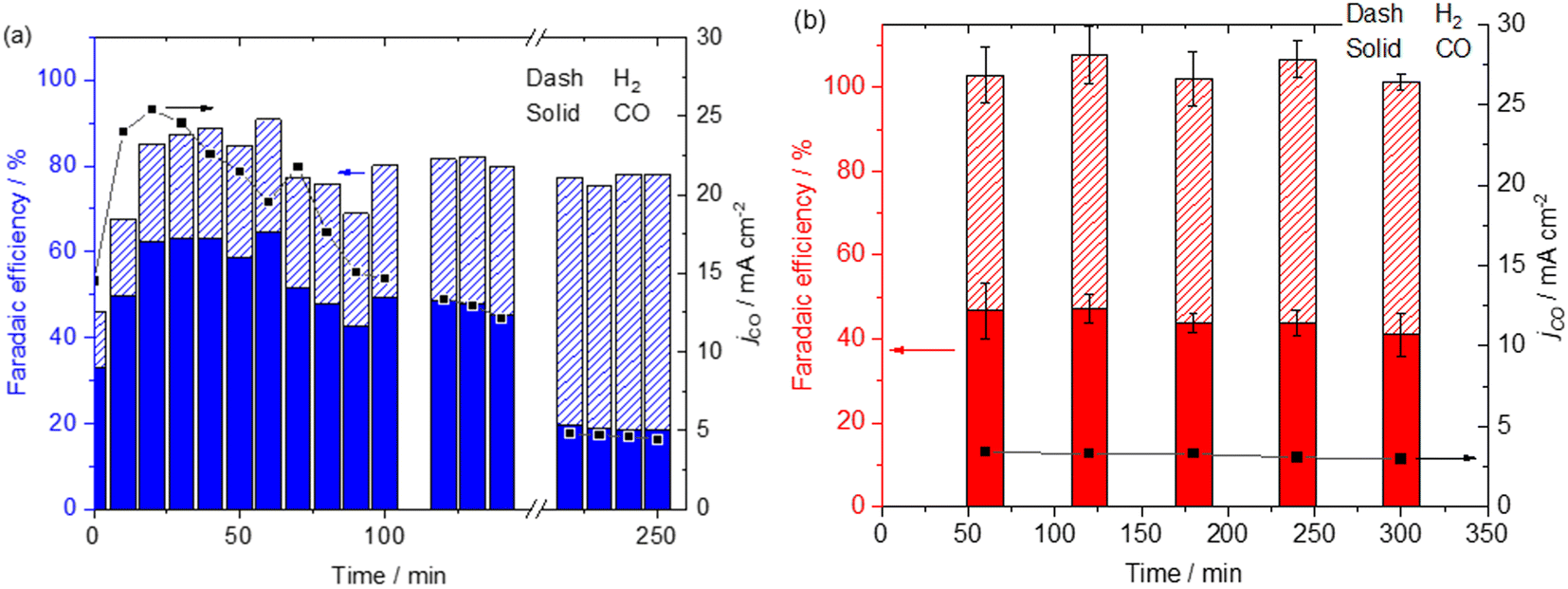 | ||
| Fig. 4 FE and partial current density for CO (jCO) of Mnbpy (a) and Ag (b) over 5 hour reaction operated in the flow cell. All experiments performed at −0.98 VRHE in 0.5 M KHCO3 (aq), RuO2 coated anode, Selemion membrane. The break in the data for the Mnbpy GDE is due to a GC failure at this time point. | ||
To assess the stability of the Mn catalyst SEM, EDX, FTIR and XPS studies were performed post-electrolysis (Fig. S1, S3 and S4†). These all show the presence of Mnbpy on the cathode surface with no evidence of catalyst degradation. However, analysis of post-electrolysis catholyte via UV/vis spectroscopy shows the presence of d–d transitions and the broad MLCT excited state of the Mnbpy complex8 (Fig. S11†), suggesting that Mn loss into the catholyte is the major activity loss mechanism. To assess whether the activity of the GDE could be recovered we performed experiments where the system was paused (30 minutes) to potentially re-adsorb the catalyst onto the surface of the GDE, however no recovery in jCO was achieved (Fig. S12†). As the Mnbpy is entering the liquid electrolyte of the flow cell it is important to test whether the activity is actually arising from the catalyst suspended in the electrolyte or from the catalyst supported on the GDE. When electrolysis was carried out with a GDE with only the MWCNT/polymer coating present in a 0.5 M KHCO3 catholyte containing deliberately added Mnbpy (∼0.1 mM), there was no CO detected during electrolysis, confirming that the activity only arises from catalyst immobilised on the GDE, Fig. S13†.19,38
To benchmark the activity of the Mnbpy GDE against a well-studied catalyst, we have prepared a conventional Ag nanoparticle (<150 nm) GDE and tested it in the same device architecture (0.5 M KHCO3 electrolyte), Fig. 3a and b. The Ag electrode shows similar behaviour to previous literature reports where jCO continues to increase significantly as the overpotential is increased with values of 5.8 ± 0.41 mA cm−2 and 12.9 ± 1.6 mA cm−2 at −0.98 VRHE and −1.18 VRHE.39,40 It is notable that at −0.98 VRHE the CO current density is significantly greater for the Mnbpy GDE (13.7 ± 2.0 mA cm−2) than the benchmark Ag catalyst at this potential (5.8 ± 0.41 mA cm−2). The achieved partial current densities using the Ag GDE are relatively low when compared to those reported in alkali electrolytes41 but they are in-line with past studies in KHCO3 electrolytes at low applied overpotentials42,43 and we anticipate that future optimisation of the formulation of the GDE may increase the activity of both the Mnbpy and the Ag electrodes.
Experiments in a flow cell using a near neutral electrolyte (KHCO3) showed that the Mnbpy GDE showed a good selectivity for CO2 reduction to CO, but that at higher current densities, which correlates to a higher local pH (Fig. S9†) there is a decrease in jCO. To further explore the pH dependence of the Mnbpy GDE we have also carried out a study of the Mnbpy GDE using zero-gap configurations. There is a high level of interest in the zero-gap architecture as a means to achieving higher current densities,44 making their development of particular interest. Furthermore, by directly contacting the GDE to the cation exchange layer of a BPM or to an AEM we are able to generate acidic or alkali environments for the Mnbpy catalyst.
When the Mnbpy GDE is directly contacted to an AEM the activity for CO2 reduction is very low, Fig. S14.† Even at low current densities (total cell current density 20 mA cm−2) jCO is low and it also drops rapidly from its initial value of 5 mA cm−2 (25% FE) after 10 minutes of electrolysis to ∼1 mA cm−2 (∼5% FE) at >45 minutes. The lack of activity for carbon dioxide reduction when the Mnbpy GDE is contacted directly to the AEM is rationalised by the high local pH. Mechanistic studies and calculations in organic solvents have shown that CO2 binding to [Mn(bpy)(CO)3]− is endergonic in the absence of a suitable Brønsted acid but that in the presence of an acid, [Mn(bpy)(CO)3(CO2H)] formation is exergonic.14,31,32,45 Spectroscopic studies have also directly correlated the presence of an intermediate on a low overpotential CO2 reduction pathway, [Mn(bpy)(CO)4]+, to the acid pKa.32 pH dependent studies of Mnbpy in water are limited but one past study9 of a water-soluble analogue of Mnbpy reported activity for CO production between pH 3.5–9 in line with the minimal CO2 reduction that occurred here in the zero-gap AEM device.
In contrast to the AEM system the reverse biased BPM zero-gap Mnbpy electrolyser (acidic environment for the GDE) is active for CO production, Fig. 5a. At 20 mA cm−2 the FE for CO is 61.6 ± 11.3% for CO, rising to 70.2 ± 7.2% for CO at 50 mA cm−2. At the highest current density tested (100 mA cm−2) we did observe a decrease in FE for CO to 30.6 ± 1.3% indicating that the Mnbpy catalyst may have reached its maximum turnover frequency. Experiments at higher current densities are complicated by the limited stability of the commercial BPM (Fumasep BPM) used here which is not recommended for use at >100 mA cm−2 for prolonged periods. A stability study of the zero-gap BPM system at 50 mA cm−2 (Fig. 5b), shows an initial drop in activity for CO production over the first 30 minutes but activity stabilises to ∼50% of its maximum for the remainder of the experiment. It is clear that the Mnbpy complex is able to operate in the acidic environment as anticipated.9 This is a significant result as it is recognised that operating the cathode at low pH overcomes the detrimental effects of CO32−/HCO3− formation that occurs in most reported devices22 and few studies have achieved good selectivity since H2 production often dominates in the acidic environment. The FE for CO at 50 mA cm−2 of the Mnbpy GDE exceeds that of our previously reported system using a Ni cyclam derived catalyst (48 ± 1%) in a reverse biased BPM cell (using the same Fumasep BPM)46 and for a benchmark Ag electrode (32 ± 0.5%) also at 50 mA cm−2 (Fig. S15†) and compares well to the highest reported to date for CO production using a reverse biased BPM.24–26,47 Further work is required to increase the achieved current densities and address the stabilities of these GDEs but these preliminary experiments indicate that Mnbpy is a promising electrocatalyst for use in electrolysers where there is an acidic or neutral environment at the cathode.
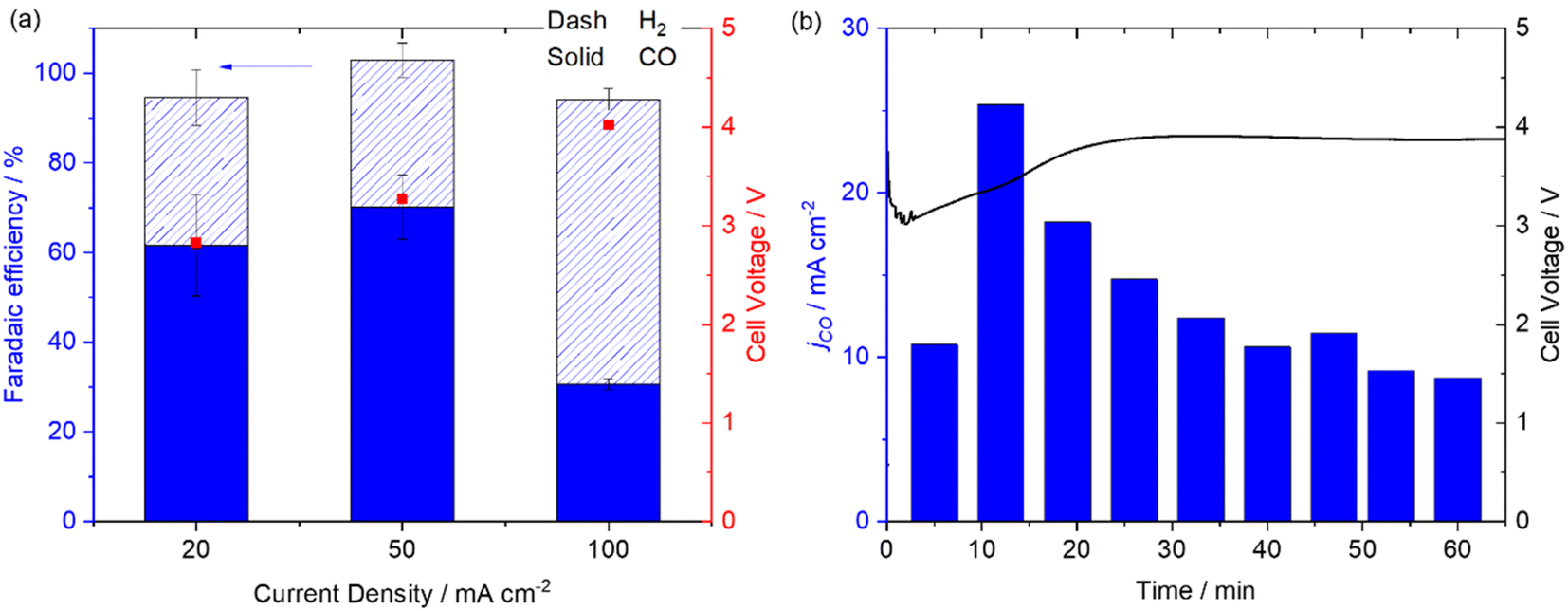 | ||
| Fig. 5 Mnbpy/MWCNT in a reverse biased BPM zero-gap electrolyser at −20, −50 and −100 mA cm−2 for ten minutes (a) and at −50 mA cm−2 for 1 hour (b). | ||
Conclusions
A GDE based on an earth abundant Mnbpy CO2 reduction catalyst requires a low overpotential to achieve higher partial current densities for CO than a GDE using a benchmark Ag catalyst in a flow electrolyser at a near neutral pH. The activity achieved here (jCO = 13.7 mA cm−2) at −0.98 VRHE under neutral pH conditions significantly exceeds that achieved in past studies with this class of catalysts but we do see a decrease in both partial current density for CO and selectivity towards CO2 reduction over a 5 hour period. This is proposed to be due to the loss of the catalyst from the GDE surface and future promising approaches to increasing stability in the flow electrolyser include optimisation of the electrode formulation and the potential modification of the Mn catalyst to decrease solubility and increase the strength of the interaction with the MWCNT. In a zero-gap configuration using an AEM we find that Mnbpy only shows low levels of activity. This observation is in-line with its known solution electrochemistry where protonation is critical in enabling initial CO2 binding. Promisingly, the Mnbpy GDE shows an excellent selectivity for CO when a reverse biased BPM configuration is used in preliminary studies. Such an approach is of particular interest as the BPM offers a way to overcome carbonate crossover losses but typically only low FEs for carbon products are achieved. Although operational stabilities of these GDEs is relatively low and significant improvements are needed, the work highlights the potential advantages of using selective molecular electrocatalysts such as Mnbpy in reverse biased BPM zero-gap electrolysers.Conflicts of interest
There are no conflicts to declare.Acknowledgements
CE thanks R&D Incubator, NSG Pilkington and the University of Liverpool for a PhD Studentship. This work was also supplied by UKRI-EPSRC through grants EP/V011863/1, EP/P034497/1 and EP/N01053/1. The X-ray photoelectron (XPS) data collection was performed at the EPSRC National Facility for XPS (“HarwellXPS”), operated by Cardiff University and UCL, under Contract No. PR16195.References
- Modern Aspects of Electrochemistry, ed. C. G. Vayenas, R. E. White and M. E. Gamboa-Aldeco, Springer New York, New York, NY, 2008, vol. 42 Search PubMed.
- J. P. Collin and J. P. Sauvage, Coord. Chem. Rev., 1989, 93, 245–268 CrossRef CAS.
- O. N. Efimov and V. V. Strelets, Coord. Chem. Rev., 1990, 99, 15–53 CrossRef CAS.
- M. R. Dubois and D. L. Dubois, Acc. Chem. Res., 2009, 42, 1974–1982 CrossRef.
- Z. Jiang, Z. Zhang, H. Li, Y. Tang, Y. Yuan, J. Zao, H. Zheng and Y. Liang, Adv. Energy Mater., 2023, 13(6), 2203603 CrossRef CAS.
- J. Hawecker, J.-M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1984, 328–330 RSC.
- M. Bourrez, F. Molton, S. Chardon-Noblat and A. Deronzier, Angew. Chem., Int. Ed., 2011, 50, 9903–9906 CrossRef CAS PubMed.
- J. J. Walsh, G. Neri, C. L. Smith and A. J. Cowan, Chem. Commun., 2014, 50, 12698–12701 RSC.
- J. J. Walsh, G. Neri, C. L. Smith and A. J. Cowan, Organometallics, 2019, 38, 1224–1229 CrossRef CAS.
- C. L. Smith, R. Clowes, R. S. Sprick, A. I. Cooper and A. J. Cowan, Sustainable Energy Fuels, 2019, 3, 2990–2994 RSC.
- T. E. Rosser, C. D. Windle and E. Reisner, Angew. Chem., Int. Ed., 2016, 55, 7388–7392 CrossRef CAS PubMed.
- C. Sun, L. Rotundo, C. Garino, L. Nencini, S. S. Yoon, R. Gobetto and C. Nervi, ChemPhysChem, 2017, 18, 3219–3229 CrossRef CAS PubMed.
- L. Rotundo, J. Filippi, R. Gobetto, H. A. Miller, R. Rocca, C. Nervi and F. Vizza, Chem. Commun., 2019, 55, 775–777 RSC.
- B. Siritanaratkul, C. Eagle and A. J. Cowan, Acc. Chem. Res., 2022, 55, 955–965 CrossRef CAS PubMed.
- R. Wiebe and V. L. Gaddy, Solubility of Carbon Dioxide in Water the Solubility of Carbon Dioxide in Water at Various Temperatures from 12 to 40° and at Pressures to 500 Atmospheres. Critical Phenomena*, 1940 Search PubMed.
- Y. Gu, J. Wei, X. Wu and X. Liu, Sci. Rep., 2021, 11, 11136 CrossRef CAS PubMed.
- C.-T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. G. D. Arquer, A. Kiani, J. P. Edwards, P. D. Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783–787 CrossRef CAS PubMed.
- F. P. G. de Arquer, C.-T. Dinh, A. Ozden, J. Wicks, C. McCallum, A. R. Kirmani, D.-H. Nam, C. Gabardo, A. Seifitokaldani, X. Wang, Y. C. Li, F. Li, J. Edwards, L. J. Richter, S. J. Thorpe, D. Sinton and E. H. Sargent, Science, 2020, 367, 661–666 CrossRef PubMed.
- S. Ren, D. Joulié, D. Salvatore, K. Torbensen, M. Wang, M. Robert and C. P. Berlinguette, Science, 2019, 365, 367–369 CrossRef CAS PubMed.
- J. Filippi, L. Rotundo, R. Gobetto, H. A. Miller, C. Nervi, A. Lavacchi and F. Vizza, Chem. Eng. J., 2021, 416, 147–160 CrossRef.
- J. J. Walsh, C. L. Smith, G. Neri, G. F. S. Whitehead, C. M. Robertson and A. J. Cowan, Faraday Discuss., 2015, 183, 147–160 RSC.
- J. A. Rabinowitz and M. W. Kanan, Nat. Commun., 2020, 11, 5231 CrossRef CAS PubMed.
- M. A. Blommaert, D. Aili, R. A. Tufa, Q. Li, W. A. Smith and D. A. Vermaas, ACS Energy Lett., 2021, 6, 2539–2548 CrossRef CAS PubMed.
- D. A. Salvatore, D. M. Weekes, J. He, K. E. Dettelbach, Y. C. Li, T. E. Mallouk and C. P. Berlinguette, ACS Energy Lett., 2018, 3, 149–154 CrossRef CAS.
- Y. C. Li, D. Zhou, Z. Yan, R. H. Gonçalves, D. A. Salvatore, C. P. Berlinguette and T. E. Mallouk, ACS Energy Lett., 2016, 1, 1149–1153 CrossRef CAS.
- Z. Yan, J. L. Hitt, Z. Zeng, M. A. Hickner and T. E. Mallouk, Nat. Chem., 2021, 13, 33–40 CrossRef CAS PubMed.
- Y. Hori, H. Konishi, T. Futamura, A. Murata, O. Koga, H. Sakurai and K. Oguma, Electrochim. Acta, 2005, 50, 5354–5369 CrossRef CAS.
- B. Reuillard, K. H. Ly, T. E. Rosser, M. F. Kuehnel, I. Zebger and E. Reisner, J. Am. Chem. Soc., 2017, 139, 14425–14435 CrossRef CAS PubMed.
- S. Pal, S. Narayanaru, B. Kundu, M. Sahoo, S. Bawari, D. K. Rao, S. K. Nayak, A. J. Pal and T. N. Narayanan, J. Phys. Chem. C, 2018, 122, 23385–23392 CrossRef CAS.
- C. Riplinger, M. D. Sampson, A. M. Ritzmann, C. P. Kubiak and E. A. Carter, J. Am. Chem. Soc., 2014, 136, 16285–16298 CrossRef CAS PubMed.
- Y. C. Lam, R. J. Nielsen, H. B. Gray and W. A. Goddard, ACS Catal., 2015, 5, 2521–2528 CrossRef CAS.
- G. Neri, J. J. Walsh, G. Teobaldi, P. M. Donaldson and A. J. Cowan, Nat. Catal., 2018, 1, 952–959 CrossRef CAS.
- D. C. Grills, M. Z. Ertem, M. McKinnon, K. T. Ngo and J. Rochford, Coord. Chem. Rev., 2018, 374, 173–217 CrossRef CAS.
- K. Yang, R. Kas, W. A. Smith and T. Burdyny, ACS Energy Lett., 2021, 6, 33–40 CrossRef CAS.
- Y. Wu, S. Garg, M. Li, M. N. Idros, Z. Li, R. Lin, J. Chen, G. Wang and T. E. Rufford, J. Power Sources, 2022, 522, 230998 CrossRef CAS.
- L. C. Weng, A. T. Bell and A. Z. Weber, Phys. Chem. Chem. Phys., 2018, 20, 16973–16984 RSC.
- M. E. Leonard, L. E. Clarke, A. Forner-Cuenca, S. M. Brown and F. R. Brushett, ChemSusChem, 2020, 13, 400–411 CrossRef CAS PubMed.
- Z. Zhang, L. Melo, R. P. Jansonius, F. Habibzadeh, E. R. Grant and C. P. Berlinguette, ACS Energy Lett., 2020, 5, 3101–3107 CrossRef CAS.
- C. Kim, H. S. Jeon, T. Eom, M. S. Jee, H. Kim, C. M. Friend, B. K. Min and Y. J. Hwang, J. Am. Chem. Soc., 2015, 137, 13844–13850 CrossRef CAS PubMed.
- T. Hatsukade, K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Phys. Chem. Chem. Phys., 2014, 16, 13814–13819 RSC.
- S. Ma, R. Luo, J. I. Gold, A. Z. Yu, B. Kim and P. J. A. Kenis, J. Mater. Chem. A, 2016, 4, 8573–8578 RSC.
- W. H. Cheng, M. H. Richter, I. Sullivan, D. M. Larson, C. Xiang, B. S. Brunschwig and H. A. Atwater, ACS Energy Lett., 2020, 470–476 CrossRef CAS.
- J. Lim, H. Lim, B. Kim, S. M. Kim, J.-B. Lee, K. R. Cho, H. Choi, S. Sultan, W. Choi, W. Kim and Y. Kwon, Electrochim. Acta, 2021, 395, 139190 CrossRef CAS.
- B. Endrődi, A. Samu, E. Kecsenovity, T. Halmágyi, D. Sebők and C. Janáky, Nat. Energy, 2021, 6, 439–448 CrossRef PubMed.
- C. Riplinger and E. A. Carter, ACS Catal., 2015, 5, 900–908 CrossRef CAS.
- B. Siritanaratkul, M. Forster, F. Greenwell, P. K. Sharma, E. H. Yu and A. J. Cowan, J. Am. Chem. Soc., 2022, 144, 7551–7556 CrossRef CAS PubMed.
- K. Yang, M. Li, S. Subramanian, M. A. Blommaert, W. A. Smith and T. Burdyny, ACS Energy Lett., 2021, 6, 4291–4298 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3se00236e |
| This journal is © The Royal Society of Chemistry 2023 |