
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Overcoming residual carbon-induced recombination in water-oxidation catalysis: combining a superior catalyst with low-carbon-content atomic layer deposition of SnO2 for improved catalysis†
Carly F.
Jewell
 a,
Ashwanth
Subramanian
b,
Won-Il
Lee
b,
Chang-Yong
Nam
*cb and
Richard G.
Finke
*a
a,
Ashwanth
Subramanian
b,
Won-Il
Lee
b,
Chang-Yong
Nam
*cb and
Richard G.
Finke
*a
aDepartment of Chemistry, Colorado State University, Fort Collins, Colorado 80523, USA. E-mail: Richard.Finke@colostate.edu; cynam@bnl.gov
bDepartment of Materials Science and Chemical Engineering, Stony Brook University, Stony Brook, New York, 11794, USA
cCenter for Functional Nanomaterials, Brookhaven National Laboratory, Upton, New York, New York 11973, USA
First published on 15th August 2023
Abstract
Previously an all earth-abundant dye-sensitized photoelectrochemical cell, composed of nanostructured SnO2 coated in perylene diimide dye (N,N′-bis(phosphonomethyl)-3,4,9,10-perylenediimide, PMPDI) plus photoelectrochemically deposited cobalt oxide (CoOx) catalyst, was shown to photoelectrochemically oxidize water. However, in that earlier work and device the addition of a known water-oxidation catalyst (WOCatalyst), CoOx, was found to reduce photocurrents, an effect that we termed an “anti-catalyst” effect to emphasize its unexpected, opposite-from-desired nature. The cause of the anti-catalyst effect was discovered to be residual carbon in the nanostructured SnO2 scaffolding necessary to increase surface area and photocurrents, hence actually residual-C-containing SnOxCy scaffolding. Herein, we examine two approaches to overcome this undesired anti-catalyst effect. First, we demonstrate photocurrent improvement with the addition of a more active, state-of-the-art, amorphous Li-IrOx, WOCatalyst. Second, we employ more phase-pure, planar SnO2 prepared by atomic layer deposition (ALD) and quantitatively compare, by optical absorption and double-layer capacitance measurements, the performance of ALD-SnO2vs. the nano-SnOxCy made in house following a literature recipe. The more phase-pure ALD-SnO2 yielded higher photocurrents based on either per dye or per electrochemically active surface area basis. Finally, in a third approach we combine both strategies for overcoming the anti-catalyst effect, specifically we use the best-in-class Li-IrOx catalyst and ALD-SnO2 to generate a best-performing device to date for this system. The results offer three successful approaches to overcome the anti-catalyst effect caused by residual-carbon-based recombination.
Introduction
Rising global demand for renewable energy has led to increased interest in and research on storable solar energy, particularly though the use of photocatalytic conversion of water into oxygen and hydrogen.1–5 A dye-sensitized photoelectrolysis cell (DS-PEC) designed to anodically split water generally requires a minimum of three components: a molecular light absorbing complex such as a dye, a semiconductor for charge transport, and a catalyst facilitating the oxidation of water.1,6,7 Organic dyes have drawn particular interest in recent years as light absorbers for solar-based water splitting as they are relatively cheap, can be made using all earth-abundant elements, and are synthetically highly tunable.1,8–10 The use of earth-abundant materials in water-oxidation catalysis (WOC) systems is desirable for sustainable, long-term use. Hence, significant work has been done researching WOC systems comprised of earth-abundant elements.11–14However, WOC is a complex kinetics system with many opportunities for inefficiency due to recombination. Prioritizing more plentiful elements over more efficient, but rarer-element, catalysts often introduces further inefficiencies. As a result, dye-sensitized water oxidation anodes made of all earth-abundant materials are less frequently reported, and those that have been reported are still not competitive with rare-element-based catalysts (Table 1). For example, a recently reported, all earth-abundant anode comprising a Prussian blue analog used with a Janus green B dye on rutile TiO2 resulted in photocurrents of 50 μA cm−2 at +0.8 V vs. NHE (normal hydrogen electrode), but exhibited charge recombination issues attributable to competitive charge recombination reactions and inefficient injection of the excited electrons.15
| Paper | Water oxidation catalyst | Dye | Scaffolding | Photocurrent | Light source | All earth-abundant system? |
|---|---|---|---|---|---|---|
| a Donor–π–acceptor dye. b Ru(bda)-(pyP)2 (bda is 2,2′-bipyridine-6,6′-dicarboxylate; pyP is (3-(pyridin-4-yloxy)propyl)phosphonic acid). c Ru-(pdc)(pic)3 with pyridine-2,6-dicarboxylic acid (pdc) as an anchoring group, pic = 4-picoline. d Donor–acceptor dye, 4-(diphenylamino)phenyl cyanoacrylic acid. e Ru4(μ-O)4(μ-OH)2(H2O)4(γSiW10O36)2]10−. f Bis-cationic perylenebisimides quantasomes interlocked with tetraethylene glycol. | ||||||
Finke and coworkers, 2017![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 12 12 |
CoOx | PMPDI | Nano-SnO2 | 20 μA cm−2 at +0.4 V vs. NHE, pH 7 | AM 1.5G | Yes |
| Karadas and coworkers, 202015 |
Prussian blue analog | Janus green B | Rutile TiO2 | 50 μA cm−2 at +0.8 V vs. NHE, pH 7 | 100 mW cm2, λ > 420 nm | Yes |
| Meyer and coworkers, 202232 |
CoF, Co4O4 cubic complex | D–π–Aa | TiO2 | 120 μA cm2 at +0.6 V vs. NHE), pH 7 | 100 mW cm2, λ > 400 nm | Yes |
| Bignozzi and coworkers, 201524 |
IrO2 | Perylene bisimide | WO3 | 70 μA cm−2 at +0.30 V vs. NHE, pH 3 | AM 1.5G, λ > 450 nm | No |
| Sun and coworkers, 201520 |
Ru-pdcc | D–Ad | TiO2 | 300 μA cm−2 at +0.36 V vs. NHE, pH 7 | 100 mW cm2, λ > 400 nm | No |
| Meyer and coworkers, 201730 |
Ru(bda)b | D–π–Aa | TiO2/Al2O3 core–shell | 400 μA cm−2 at +0.4 V vs. NHE, pH 4.8 | 100 mW cm2, λ > 400 nm | No |
| Bonchio and coworkers, 202231 |
Ru4POMe | PBI QS-TEGlockf | Inverse opal-ITO | 370 ± 30 μA cm−2 at +1.52 V vs. RHE, pH 7 | AM 1.5G, 850 mW cm2, λ > 450 nm | No |
Dye-sensitized water-oxidizing photoanodes using rare-metal catalyst are much more common in the literature and achieve much higher photocurrents. Many current, state-of-the-art water oxidation catalysts (WOCatalysts) are made of more expensive metals such as ruthenium16–21 and iridium.22–29 Anodes using Ru-based catalysts have achieved photocurrents on the order of 400 μA cm−2 at +0.4 V vs. NHE,30 370 ± 30 μA cm−2 at +1.52 V vs. RHE,31 and 300 μA cm−2 at +0.36 V vs. NHE.20 On the basis of the literature, a rare-element-based Ir catalyst was chosen for the research herein in order to optimize photocurrents while addressing the specific questions posed, vide infra.
Previously, our group developed a water-oxidation system consisting of all earth-abundant materials: nano-SnOxCy coated with a perylene diimide dye, N,N′-bis(phosphonomethyl)-3,4,9,10-perylenediimide (PMPDI), and CoOx as a WOCatalyst.12,13 Although the materials in this system are generally more environmentally friendly than typical Ru or Ir-based catalysts, the addition of the CoOx catalyst resulted in an, at the time unexpected, undesired decrease in photocurrent that we termed an “anti-catalyst” effect.12,13 Tracking down the origins of this “anti-catalyst” effect eventually led to the discover that this undesired behavior is attributable to carbon impurities in the SnO2, that is, to residual carbon in the nano-SnOxCy.33 Research by other groups found they, too, can alter the photoelectrochemistry of a SnO2 system by changing the carbon in the system.34 Interestingly, an increase in photocurrent density was observed in Sn–C systems, which was attributed to the formation of a heterostructure with decreased resistance,34 a reversal in trend from the observed effect in the system detailed in this paper.33
Herein, we pose and experimentally address the following five questions: (1) Can improving the WOCatalyst in the system overcome and outcompete such recombination pathways? That is, can a more active WOCatalyst impact the primary charge-transfer pathways during water oxidation by altering its kinetics, an important question with implications for optimizing the WOC in the present device and potentially other WOC devices moving forward. Note here that the cost of initially more expensive catalysts can prove to tend towards zero if their catalytic lifetimes tend toward “infinity”, that is reach high numbers of total turnovers of catalysis. Then, (2) what is the maximum current from such an improved device? Benchmarking that maximum current is important as one strives to prepare improved, future earth-abundant WOCs devices. Additionally, (3) can we utilize atomic-layer deposition (ALD) of planar SnO2 as an effective way33 to address the residual carbon in the nano-SnOxCy scaffolding, that is, as a way to reduce the residual C in the nano-SnOxCy, ideally sufficiently to produce a positive catalytic effect following CoOx addition?33 To this end, we will compare the performance of the lower-surface-area planar ALD-SnO2 device to its high-surface-area nano-SnOxCy counterpart by developing a way to correct for the very different surface areas of the two systems while answering the question: (4) is an intrinsic improvement in photocatalytic efficiency achieved in the planar system with its low-C-content SnO2?; (5) Finally, can we achieve and quantify a further improved WOC device by combining the best-performing ALD-SnO2 and a best-in-class WOCatalyst, Li-IrOx? Overall, this research strives to provide quantitative insights into the ways one can test new materials for complex WOC anodes en route to ensuring that the device components are not limited by catalyst choice. Our results also illustrate how and why testing both earth-abundant and precious metal catalysts can be used to address charge recombination issues and to set a benchmark for the maximum current available from a given device.
Experimental
Experimental conditions used herein are identical to those in our previously published work,11–13,33 unless otherwise noted. Key experimental details are provided below and in the ESI† as necessary to ensure that the present manuscript is largely self-contained and replicable by others.Materials
The following starting materials and solvents were used as received to generate buffer solutions: KOH (Fisher, Certified ACS grade, 98.5%, 1.5% water, 0.00028% Fe, 0.0008% Ni); KH2PO4 (Fisher, Certified ACS Grade, 99.3%, 0.0005% Fe); ethanol (Pharmco-Aaper, ACS grade); hydroquinone (Aldrich, >99%); NANOpure water (Barnstead NANOpure ultrapure water system, 18.0 MΩ). NANOpure water was used for all experiments.Synthesis of Li-IrOx
Using the synthesis procedure reported by Gao et al.,35 Li-IrOx was synthesized as detailed in Scheme 1. Briefly, 250 mg of iridium(III) chloride hydrate (Aldrich, reagent grade) was dissolved into 5 mL of water. Then, 5.0 g of LiOH·H2O (Sigma; 150 equivalents vs. Ir) was added. This solution was then heated to 90 °C with stirring in order to remove the water by evaporation. The solid remaining was then placed in a crucible and heated to 400 °C for 2 hours under air. After cooling, the resultant solid Li-IrOx product was rinsed three times with water and ethanol then dried at 60 °C overnight at 1 atm. The final Li-IrOx product was found to be comparable to the previously published material by X-ray photoelectron spectroscopy (XPS), detailed below and in the ESI.†35 | ||
| Scheme 1 Literature-based synthesis of Li-IrOx following Gao and coworkers experimental.35 | ||
Planar SnO2 preparation by ALD
As before,33 ultrathin conformal coatings of SnOx were carried out using the Cambridge Nanotech Savannah S100 ALD system (base pressure of ∼0.43 Torr) at a temperature of 85 °C. SnOx depositions were done using sequential exposures to tetrakis(dimethylamido)tin(IV) (TDMASn) (Strem Chemicals) (200 ms) and water vapor (15 ms) with an intermediate purge time of 10 s for both the precursors.Preparation of nano-SnOxCy/PMPDI anodes
Nano-SnOxCy was prepared as before using a modified literature procedure36 previously optimized for the present system.12,33 Briefly, 800 mg of white commercially available SnO2 nanopowder (SnO2, NanoArc©, Alpha Aesar, Lot Analysis: 99.6% SnO2, 47 m2 g−1 specific surface area by BET (Brunauer–Emmett–Teller) method, 18 nm average particle size, 6.95 g cm−3 density) was massed into a 20 mL scintillation vial to which a Teflon-coated magnetic stir bar was added. The powder was then wetted with 1.0 mL of glacial acetic acid (ACS grade, Mallinckrodt), stirred until fully mixed, and then placed into an ultrasonic water bath (Branson 2510) for 5 min. Next, 4.0 mL of water was added to the paste and the resulting mixture was stirred for 5 min on a stir plate. The paste was then sonicated twice using a QSonica Q125 ultrasonic liquid microprocessor (1/8′′ probe), which was held at 80% amplitude. To this mixture, 1.10 g of polyethyleneglycol bisphenol A epichlorohydrin copolymer (PEG-BAE) (Sigma, Mw = 15000–20000), which serves as a thickening agent, was added in individual small chunks while stirring. After all the PEG-BAE had been added and dissolved fully upon visual inspection, one drop (approximately 20 mg) of Triton X-100 (octyl phenoxy polyethoxyethanol, Sigma) was added using a plastic transfer pipet in order to increase adhesion of the paste to the glass substrate. This final paste was then stirred overnight and then refrigerated in a sealed vial. All synthetic and manufacturing details for PMPDI dye can be found in our previous publications.11,12 In order to manufacture the nano-SnO2 anodes, the previously described nano-SnO2 paste was doctor bladed onto fluorine-doped tin oxide (FTO)-coated glass using a 2-Scotch thickness and annealed under air for 1 hour at 450 °C.12 The PMPDI dye was loaded onto the FTO coated in either nano-SnO2 or ALD-SnO2 anodes for 24 hours at 95 °C from a saturated solution of fully protonated PMPDI in water.12
Deposition of Li-IrOx onto nano-SnOxCy/PMPDI anodes
Li-IrOx catalyst (2.5 mg), prepared as above, was dissolved into 1.0 mL of a 95:5 v/v ethanol: water mixture. Previous work using the Li-IrOx catalyst deposited the Li-IrOx using a “custom external-mixing two-fluid nozzle”.29In lieu of this unavailable custom-made device, a handheld nebulizer (Hudson RCI, Micro Mist) was used with air to spray droplets of the Li-IrOx solution onto the anode. Air pressure was set to the minimum value sufficient to nebulize the solution. Care was taken to ensure the mixture deposited uniformly onto the anode by using a modified nebulizer to narrow the size of the spray equal to the anode size. Further information on the nebulizing device can be found in ESI S3.† The resulting anode was then annealed at 120 °C for 10 min under air.
Deposition of CoOx
CoOx WOCatalyst was added to photoanodes by photoelectrochemical deposition.11,12 The FTO/SnO2/PMPDI anodes were placed into a solution of pH 7, 0.5 mM Co(NO3)2 and 0.1 M potassium phosphate buffer (KPi) and held at +0.2 V vs. Ag/AgCl under 1 sun illumination for 3 min.12 Anodes were then rinsed with water for 30 s and allowed to air dry. Note the same charge was observed to be passed on both planar and nanostructured SnO2 anodes.Photoelectrochemical testing
All photoelectrochemical experiments were done in a previously described11–13 custom two-compartment Pyrex cell. The custom cell has two compartments separated by a medium porosity glass frit: a working compartment (1 × 1.5 × 1.5 cm3, 5 mL) and an auxiliary compartment. A CH Instruments CHI-750D bipotentiostat, a Pt wire counter electrode, and a Ag/AgCl (3 M NaCl, +0.215 V vs. NHE) reference electrode were used for all experiments. The counter electrode was placed alone in the auxiliary compartment to separate the products from the working and counter electrodes. The reference electrode was positioned in the working compartment near the working electrode anode's surface in order to minimize resistance. The anode of interest was held with an alligator clip to the front wall of the working compartment with the undyed, nonconductive glass side pressed flat against the compartment wall nearest the incoming light. Both sides of the cell were filled with ca. 5 mL of electrolyte (pH 7, 0.1 M KPi buffer), an amount sufficient to cover the dyed film. The anode was illuminated using a 65 W xenon arc lamp (PTO model A1010), powered using an OLIS XL150 adjustable power supply. The light from the xenon arc lamp was passed through both a bandpass filter (315–710 nm, Thorlabs KG3, FGS900S) and an ultraviolet filter (400 nm long-pass, Thorlabs FGL400S) before reaching the anode. The power density of the light was adjusted to match the visible region of the airmass 1.5 global (AM 1.5G) spectrum.12 In experiments with transients (interrupted illumination) a manual shutter was used to block the light.12 Each anode was tested with a series of electrochemical experiments in the order detailed below: (1) the cell was allowed to short-circuit in the dark for 60 s to depopulate the electrons from SnO2 sub-bandgap states.12 (2) The open-circuit potential, Voc, of the anode was measured vs. the Ag/AgCl reference electrode for 90 s both in the dark and (3) again under illumination. (4) A photocurrent transient experiment in which the anode was held at +0.2 V vs. Ag/AgCl for 300 s with 30 s light/dark transients throughout was next conducted. (5) The anode was then allowed to short-circuit again in the dark for 60 s, and the Voc in both the (6) dark and (7) light were remeasured for 90 s each. (8) A current–voltage (i–V) photocurrent transient experiment was performed where the voltage was scanned from −0.2 V to +1.0 V vs. Ag/AgCl using a 10 mV s−1 scan rate and 5 s light transients. For each experiment and each set of conditions being tested, every experiment was reproduced a minimum of three times with three separate anodes prepared under identical conditions. All figures and values reported herein are representative examples of the photoactivity of the anode system indicated. Care was taken to ensure that the reported results are reproducible and are not due to a defective anode or otherwise irreproducible sample.XPS
XPS was done in house using a PE-5800 series Multi-Technique ESCA XPS system. An Al Kα monochromatic source operating at 350.0 W was used for all XPS experiments. High-resolution (HRES) scans were carried out for all elements present (carbon, lithium, oxygen, iridium) in the initial survey scan at a minimum of 3 spots across the sample surface for 30 min apiece. To fit the data, CASAXPS software was used to analyze the data. Quantification results for each element were tabulated using an average of each replicate scan. Consistent with both the literature method of XPS fitting and ensuring self-consistency across fits, HRES spectra were calibrated to a 285 eV aliphatic carbon peak.37,38 Fits were done using a minimum, Ockham's Razor type approach to ensure the data was not overfit (Fig. S1†).Oxygen detection
Oxygen yield was measured using the generator–collector (G–C) technique detailed previously.12,13,39–41 Photoanodes of interest, “generators”, were placed with conductive sides facing the conductive side of a FTO “collector”.12 The generator and collector were then separated by a Parafilm spacer. The G–C device was placed in the working compartment detailed above, with the generator side flush against the wall of the cell nearest the incoming light. The cell was filled with approximately 10 mL pH 7, 0.1 M KPi buffer, sufficient to fully cover the dyed portion of the anode, which was degassed with Ar for a minimum of 60 min to remove oxygen. Current was measured at both the generator and collector electrodes, which were held at +0.2 V and −0.65 V vs. Ag/AgCl, respectively. The current was collected for 300 s in the dark to obtain a background, 300 s in the light to generate oxygen, then 300 s in the dark to ensure all oxygen fully diffused between the two electrodes and that all current attributable to oxygen production was collected. Control experiments with FTO/CoOx anodes were used to determine the collection efficiency of the G–C device12 and used to calculate the O2 yield and faradaic efficiency of each photoanode.Results and discussion
As a test of our first specific question, of how a different WOCatalyst can impact primary charge transfer and possibly overcome recombination due to carbon defects,33 a known state-of-the-art WOCatalyst, amorphous Li-IrOx,29,35 was added our otherwise earth-abundant nano-SnOxCy/PMPDI DS-PEC water-oxidation catalysis system. The addition of the Li-IrOx allows us to continue to use the increased surface area provided by nanostructuring the device with nano-SnOxCy, but seeks to kinetically outcompete recombination attributed to carbon in the nano-SnOxCy as judged to start by an increase in photocurrent.33 Additionally, the highest photocurrent one can observe for the nano-SnOxCy/PMPDI/Li-IrOx anode sets a benchmark for photocurrents one can strive for via the future development of improved, ideally all earth-abundant, catalysts.Addition of Li-IrOx WOCatalyst
Li-IrOx was synthesized as detailed in the Experimental section and deposited onto the nano-SnOxCy/PMPDI, resulting in nano-SnOxCy/PMPDI/Li-IrOx anodes. The photoactivity of these anodes was then examined in pH 7 buffer with the addition of hydroquinone (H2Q). As before,11,12 H2Q was employed as a sacrificial reductant as it exhibits a kinetically facile, 2e− and 2H+, oxidation.1,12,21 Photocurrent transients were carried out on the nano-SnOxCy/PMPDI anodes with and without Li-IrOx catalyst in the presence of H2Q (Fig. 1). These results were also compared to our previously published and optimized nano-SnOxCy/PMPDI/CoOx12 to best understand how changing the catalyst impacts photoactivity and to attempt to answer the first question posed herein seeking to understand how changing the catalyst can impact the charge-transfer kinetics of the device.42 It is important to note here that catalyst loading was not optimized for the nano-SnOxCy/PMPDI/Li-IrOx anodes demonstrating again how much more effective the Li-IrOx is as a WOCatalyst compared to even the optimized CoOx. Optimizing catalyst loading is certainly also possible, but will only exaggerate (i.e., but not change the general trend of) the enhanced-current findings herein with the Li-IrOx catalyst.
 | ||
| Fig. 1 Photocurrent transients in the presence of 20 mM H2Q sacrificial reductant, with background dark current subtracted, at +0.2 V vs. Ag/AgCl in pH 7, 0.1 M KPi buffer with 30 s light/dark transients where blue is nano-SnOxCy/PMPDI/Li-IrOx, green is nano-SnOxCy/PMPDI, and purple is nano-SnOxCy/PMPDI/CoOx. Note here that each transient shown represents a minimum of three replicates of separately generated and tested anodes. | ||
Importantly, steady-state photocurrents increased 10% with the addition of Li-IrOx, the first time we have observed an increase in the nano-SnOxCycontaining anodes.12,13 This increase in photocurrent in a net improvement of 34% from the anode with the CoOx catalyst to the Li-IrOx catalyst. As reported previously, anodes with CoOx WOCatalyst addition yield a decrease in steady-state photocurrents of 24% upon catalyst addition.33 Hence, by changing the catalyst used with the nano-SnOxCy/PMPDI anodes from CoOx to Li-IrOx, the anti-catalyst effect is reversed and now the photocurrents increase to −1.2 mA cm−2 rather than decrease43 with catalyst addition. Two times the −1.2 mA cm−2 photocurrent observed for the Li-IrOx-containing system oxidizing H2Q, that is −2.4 mA cm−2, serves as an experimentally based, estimated theoretical maximum photocurrent achievable for water oxidation with this device. Also noteworthy is the significantly lower photocurrent decay over the course of the 30 s light transients for anodes with deposited Li-IrOx, suggestive of facile electron-delivery-kinetics protection of the oxidized PMPDI dye.
To ensure the effect of Li-IrOx addition in H2Q is reflective of the true WOC performance, anodes with and without catalyst were examined in pH 7, 0.1 M KPi buffer under actual WOC conditions (i.e., without any added sacrificial reductant), Fig. 2. As in the system with H2Q, the addition of the Li-IrOx catalyst to nano-SnOxCy/PMPDI anodes in the buffered water system again increases the system's photocurrent, contrary to what is observed for the nano-SnOxCy plus the less able CoOx catalyst anodes.12,13 With the addition of Li-IrOx, the steady-state photocurrent of anodes increases by 50% – an increase greater than seen for the anodes tested with H2Q. Upon reflection, this larger increase in photocurrent post catalyst addition makes sense: the 2 e−/2H+ reaction with H2Q is kinetically much more facile than the more difficult, 4 e−/4H+, oxidation of water. Hence, an inherently higher probability of recombination in the slower, more difficult, overall 4 e−/4H+, water oxidation reaction is expected, corresponding to a greater effect of overcoming such recombination on the H2O oxidation system vs. the H2Q, hydroquinone, system.
 | ||
| Fig. 2 Photocurrent transients for nano-SnOxCy/PMPDI (green) and nano-SnOxCy/PMPDI/Li-IrOx (blue) anodes in pH 7, 0.1 M KPi buffer. Scans were done from −0.2 to +1.0 V vs. Ag/AgCl with 5 s light dark transients. | ||
Understanding recombination with different WOCatalysts
To provide additional evidence that this increase in photocurrent corresponds with a decrease in recombination, the Voc of the anode under illumination was measured.12,13,44 In this system, the Voc can be directly related to recombination as the Voc signifies the quasi-Fermi energy of electrons (EF,n) in the nano-SnOxCy, with respect to the Ag/AgCl reference electrode potential.12,13,44,45 The Voc is determined by the steady-state charge-carrier concentration. This means that Voc is determined by the relative rates at which electrons are injected from the photoexcited PMDPI to SnO2vs. depopulated from the SnO2via recombination. We assume the deposition of catalyst will not significantly alter the rate of electron injection from the excited PMPDI to the SnO2 scaffolding. Thus, the Voc values of nano-SnOxCy/PMPDI anodes without catalyst, with CoOx added, and with Li-IrOx added were compared to gain additional evidence for recombination in the present systems, Fig. 3. | ||
| Fig. 3 V oc vs. Ag/AgCl reference electrode in a half-cell setup for nano-SnOxCy/PMPDI anodes with differing catalyst addition. Note, no catalyst is added to the far-left data point. In this system, Voc ≈ EF,n, the quasi-Fermi energy of electrons in the SnO2 sub-bandgap states.12,13 | ||
Notably, we found an average Voc of −0.07 ± 0.03 V vs. Ag/AgCl for nano-SnOxCy/PMPDI/Li-IrOx anodes, a measurable improvement over the 0.00 ± 0.02 V vs. Ag/AgCl Voc for anodes without the catalyst.12 The negative, −70 ± 30 mV shift in Voc of nano-SnOxCy/PMPDI/Li-IrOx anodes relative to nano-SnOxCy/PMPDI anodes suggests a ca. 15-fold decrease in the rates of recombination relative to injection12,44versus the system with the CoOx catalyst (see S4† for more details on how this 15-fold value was obtained). For comparison, the addition of CoOx catalyst yields a positive12Voc of +0.05 ± 0.02 V vs. Ag/AgCl, consistent with an (undesired) increase in recombination in that CoOx catalyst system.
Our first two of the five specific questions raised in the Introduction have been answered at this point: (1) yes, the kinetics of a WOC device can be altered by changing the WOCatalyst, such that undesirable kinetics pathways can be avoided through the use of a more effective catalyst; and (2) −1.2 mA cm−2 currents are achievable for H2Q catalysis in a nano-SnOxCy/PMPDI/Li-IrOx device.
Faradaic efficiencies for WOC
Often, photocurrent densities in DS-PECs are assumed to be entirely attributable to water oxidation catalysis. To avoid this unneeded assumption, we measured faradaic efficiencies for water oxidation catalysis with nano-SnOxCy/PMPDI/Li-IrOx anodes using the established Generator–Collector method.12,41 Faradaic efficiencies with Li-IrOx were found to be 37 ± 10%, interestingly the same within error as the 31 ± 7% for anodes with CoOx.12 Hence, evidence for the expected at least modest improvement in selectivity for water-oxidation catalysis when using the Li-IrOx WOCatalyst is not apparent within the stated error bars. However, the overall photocurrent is higher when using the Li-IrOx WOCatalyst, so that more O2 is in fact produced as expected when using Li-IrOx rather than CoOx as the WOCatalyst.Testing the amount of recombination as a function of the amount of carbon
Nanostructured SnOxCy anodes were made deliberately containing half as much PEG-BAE organic stabilizer to test the effects of altering the amount of carbon trap states. Previously, with CoOx, changing the amount of PEG-BAE added to nano-SnOxCy did change the amount of recombination observed.33 However, herein, in anodes with Li-IrOx catalyst, the degree of recombination in the halved PEG-BAE anodes as measured by Voc, was found to be the same within error, as the nano-SnOxCy anodes made by the original synthesis, respectively −0.05 ± 0.02 V vs. Ag/AgCl and −0.07 ± 0.03 V vs. Ag/AgCl. These unchanged Voc values within experimental error, despite halving the original many of C-containing stabilizer, suggests that Li-IrOx is able to outcompete and overcome all the C-impurity based recombination in the nano-SnOxCy. Previously, this “1/2 as much carbon” test, while using the slower CoOx catalyst, resulted in measurable differences in Voc, and by implication recombination, again consistent with and supportive of a kinetics protection against at least C-impurity-based recombination when using the superior Li-IrOx water-oxidation catalyst.28As a visual representation of the kinetics of our system, intended as a working hypothesis for going forward, a scheme showing charge-transfer pathways in the nano-SnOxCy/PMPDI/Li-IrOx system is shown in Scheme S1 of the ESI† for the interested reader.
A semi-quantitative comparison of the planar ALD-SnO2vs. the nanostructured SnOxCy systems: which is better?
To this point, we have shown that the employment of intrinsically superior WOCatalyst can outcompete the residual-carbon-related recombination, thus overcoming the anti-catalyst issue. In our previous report, we also have demonstrated that lowering carbon content in SnO2 using ALD can improve the WOC performance. Here, a semi-quantitative comparison of the photoactivity of the lower-carbon-content ALD-SnO2 planar system's performance compared to the higher-surface-area nano-SnOxCy system is carried out in order to quantitatively answer the question of “which system is fundamentally better at overcoming recombination attributed to residual carbon?”.46–50 Previously we have determined our synthesis of nano-SnOxCy to be approximately 11 ± 0.3 wt% carbon by XPS while the surface of the SnO2 deposited by ALD is 5 ± 3 wt% carbon, and approximately 3 ± 3 wt% carbon after sputtering.33Such a device comparison of the performance of the ALD-SnO2 system vs. the nano-SnOxCy anodes requires a correction for their ca. two-order of magnitude different (vide infra) surface areas that results in a much lower photocurrent in the planar ALD-SnO2 system. Additionally, different dye absorption in the ALD-SnO2vs. the nano-SnOxCy anode systems could be a factor that varies between the two anodes or catalyst deposition could change. Hence, we employed two methods that use a fundamentally different scientific basis to compare the two systems to mitigate assumptions that would significantly impact the conclusions, one based on (i) correcting the data for PMPDI absorbance, and a second method based on (ii) correcting for the electrochemically-active surface area.
Two methods for correcting for the different surface areas between the nano-SnOxCy system and the planar ALD-SnO2 systems
Because of the significantly larger amount of surface area present in the nano-SnOxCy system vs. the planar ALD-SnO2 system, considerably different dye optical absorption is observed. Indeed, saturation-dying of the planar and the nanostructured SnOxCy systems reveals a stark apparent difference between them, Fig. 4. In the first method, which corrects for the dye absorbance, Beer's Law was employed to calculate the ratio of dye on the planar ALD-SnO2/PMPDI anode vs. the nanostructured nano-SnOxCy/PMPDI anode. As before, an approximately monolayer of dye was assumed12,51 so as to obtain the PMPDI dye extinction coefficient used in the calculations. Using that extinction coefficient and Beer's Law on the optical absorption data, the ratio of dye absorbance at λmax, after correction for SnO2 absorbance, was found to be 1.0:94 for ALD-SnO2/PMPD vs. nano-SnOxCy/PMPDI. Additional calculations determining the number of PMPDI dye molecules on both nano-SnOxCy/PMPDI and ALD-SnO2/PMPDI anodes can be found in the ESI.†
 | ||
| Fig. 4 (a) Optical absorbance spectrum of PMPDI on SnOxCy (black) and PMPDI on ALD-SnO2 (red). Absorbance of bare SnO2 film was subtracted off. The insert shows a zoomed-in plot of the absorbance of PMPDI on ALD-SnO2. (b) Image of ALD-SnO2/PMPDI anode (left) and nano-SnOxCy/PMPDI anode (right), where only the bottom two thirds of the anode is dyed. | ||
Double-layer capacitance (Cd) was employed as a second means to differentiate and then compare the electrochemically active surface area of the two systems. By looking at the double-layer capacitance at a series of scan rates, a μF cm−2 scalar was determined (Fig. 5). This scalar was then used with the Cd of the nano-SnOxCy system to find the difference between the geometric and electrochemically-active surface area. This value can then be used with the geometric surface area of the nano-SnOxCy anode tested in that particular experiment to find the electrochemically-active surface area of that anode. This method assumes the double-layer capacitance response is the same for the nano-SnOxCy and ALD-SnO2. Using this determination, the ratio of electrochemically-active surface area for ALD-SnO2 anodes vs. nano-SnOxCy anodes was found to be 1.0:190.
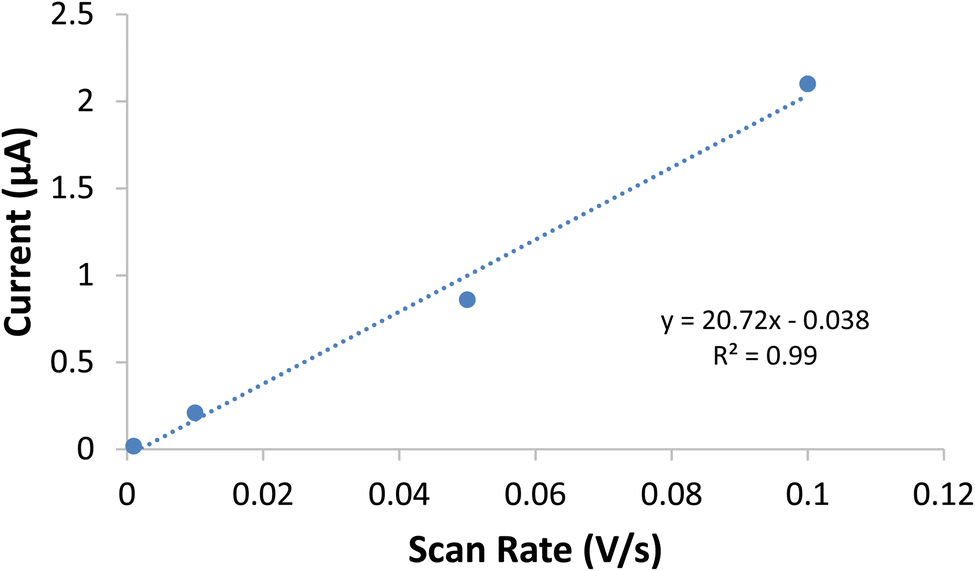 | ||
| Fig. 5 Plot of scan rate vs. capacitive current for the ALD-SnO2, where the slope of the line is directly proportional to the double layer capacitance. | ||
The different 1.0:94 and 1.0:190 values are, upon reflection, in reasonable (if not surprisingly good) agreement considering that the two methods measure a separate component of the system. The absorbance method looks at the PMPDI dye on dry anodes, while the capacitance method probes undyed anodes in solution (i.e., examines them prior to dyeing) while those anodes are undergoing electrochemistry. Given that the two techniques are measuring different aspects of the (different) systems under two distinct sets of conditions, the factor of two between the values is better than expected. In any event, the values are more than sufficient for the purposes of the present study. Additionally, so as to not artificially over-enhance/overcorrect the current (J) data for the ALD-SnO2 anodes, the corrections that follow deliberately emphasize the smaller, 1.0:94, value so as to understate the (corrected) current and implied conclusions. Moreover, control calculations are also provided in the ESI† using the other limit if the 1.0:190 correction to ensure that none of our conclusions change depending on which of the two possible surface-area corrections is used.
Results of comparing the Nano-SnOxCy system and the planar ALD-SnO2 systems
To compare the different anodes, the above correction was made on photocurrent transient plots with the addition of H2Q, Fig. 6. (The same data corrected using the higher electrochemically-active surface area value is provided as Fig. S3 of the ESI.†) The dashed lines represent a hypothetical nanostructured (high surface area) reduced-carbon version of the currently flat device. | ||
| Fig. 6 J−t transients in the presence of 20 mM H2Q sacrificial reductant, with background dark current subtracted, at +0.2 V vs. Ag/AgCl in pH 7, 0.1 M KPi buffer with 30 s light/dark transients where purple is nano-SnOxCy/PMPDI, red is nano-SnOxCy/PMPDI/CoOx, blue dashed is ALD-SnO2(5 nm)/PMPDI (X94), and green dashed is ALD-SnO2(5 nm)/PMPDI/CoOx (X94). Both planar (blue and green) anodes are corrected (X94) to match the dye absorbance of the nanostructured anodes. | ||
Several observations are readily apparent from examining Fig. 6: first, the corrected ALD-SnO2/PMPDI/CoOx anodes (green trace) demonstrate the highest photocurrent of all the systems—a conclusion even more exaggerated if the larger 1.0:190 correction is used instead (Fig. S3 of the ESI†). Second, the nano-SnOxCy/PMPDI/CoOx system is the worst performer (red trace). This conclusion, too, still holds with the larger correction (Fig. S3 of the ESI†). Third, the CoOx-free systems nano-SnOxCy/PMPDI and ALD-SnO2 (5 nm)/PMPDI perform essentially the same within experimental error (purple and blue curves, respectively). These results are consistent with and supportive of CoOx yielding enhanced recombination resulting in the observed “anti-catalyst” effect. (This conclusion, too, is unchanged with the higher correction, although with that correction the lower-C ALD-SnO2 (5 nm)/PMPDI system outperforms the greater C-containing nano-SnOxCy/PMPDI system. Evidence against diffusion limitations in the comparisons of the two systems are also presented in the ESI†). In other words, post scaling the ALD-SnO2 anodes' current density and comparing that directly to the nano-SnOxCy system's current density, the key finding is that the planar SnO2outperforms the nano-SnOxCyin terms of the current density. Meriting noting here, however, is that the high-surface-area nano-SnOxCy anodes do produce more oxygen on a per device basis than their planar counterparts. This finding answers question 4 listed in the Introduction, demonstrating that lower-carbon ALD-SnO2 produces anodes that outperform their nano-SnO2 anode counterpart systems, on both per amount of dye and per electrochemically-active surface area basis13 at potentials over +0.6 V vs. Ag/AgCl.
Perhaps most important are the steady-state photocurrents post the addition of CoOx catalyst: the ALD-SnO2 anodes show a ca. 44% increase in photocurrent in comparison to the nano-SnOxCy anodes that exhibit a ca. 24% decrease in photocurrent. This net 68% reversal of the “anti-catalyst” effect in anodes with less carbon impurity makes apparent that decreasing the amount of carbon by using ALD is a successful strategy en route to better performing anodes. This conclusion is further supported by the Voc values observed in the ALD-SnO2 system: ALD-SnO2 anodes coated in PMPDI were found to have a Voc of −0.03 ± 0.03 and to be the same within error, −0.03 ± 0.04, with the addition of the CoOx catalyst. This ALD-SnO2 strategy is of particular interest as it demonstrates a rare-element-free method of overcoming the issue of residual carbon dominated recombination without altering the WOCatalyst. With the ALD-SnO2(5 nm)/PMPDI/CoOx anodes, photocurrent densities are comparable to those seen in the nano-SnOxCy/PMPDI/Li-IrOx anodes on a per-dye or per-electrochemically-active-surface-area basis, without the use of iridium. Hence, if this level of photocurrent density can be achieved using a more carbon-free nano-SnO2 preparation, then in principle the use of a rare-element catalyst can be avoided while still achieving the higher photocurrent. It follows that studies aimed at the preparation of low-carbon, organic-stabilizer-free nano-SnO2 syntheses are, therefore, of considerable interest.
Finally, provided in Fig. S4 of the ESI,† is a comparison of photocurrents between the anodes described herein and the previously studied best performing nano-SnOxCy/PMPDI/PMPDI/AlOn (0.6 nm, 85 °C deposition)/CoOx anodes which added an additional alumina overlayer by ALD.13 When corrected for absorbance, the anodes made using planar SnO2 deposited by ALD were shown to have a higher photocurrent compared to the previous best nano-SnOxCy/PMPDI/AlOn (0.6 nm, 85 °C deposition)13 systems at potentials over +0.6 V vs. Ag/AgCl. In short, it is clear that using a more phase-pure ALD-SnO2 is a viable strategy to avoid the undesired recombination effects of residual carbon issue in metal-oxides such as nano-SnOxCy. In addition, designing a carbon-free nano-SnOxsynthesis that uses both aspects of the work herein—lowering the amount of carbon in the SnO2, while maintaining the high surface area—is a recommended direction of future research based on the findings in the present study.
Combining planar ALD-SnO2 and Li-IrOx catalyst in one, best-to-date, device
In order to study a best possible device in terms of its efficiency and address question 5 listed in the Introduction, the two methods detailed above were combined and tested together for water oxidation catalysis, specifically altering the WOCatalyst to the state-of-the-art Li-IrOx while also using lower-carbon ALD-SnO2. Photocurrent data for anodes in H2Q were measured for ALD-SnO2 anodes without catalyst, with CoOx, and with Li-IrOx (Fig. 7). | ||
| Fig. 7 J−t transients in the presence of 20 mM H2Q sacrificial reductant, with background dark current subtracted, at +0.2 V vs. Ag/AgCl in pH 7, 0.1 M KPi buffer with 30 s light/dark transients where blue is ALD-SnO2(5 nm)/PMPDI, red is ALD-SnO2(5 nm)/PMPDI/CoOx, and green is ALD-SnO2(5 nm)/PMPDI/Li-IrOx (green) anodes. | ||
Anodes with Li-IrOx catalyst again yield an increase in photocurrents of approximately 4-times with the WOCatalyst addition in the ALD-SnO2 system, resulting in significantly higher photocurrents than their counterparts with CoOx catalyst. Combining both methods of overcoming carbon-based recombination in the anode results in the most photoactive device by far on a per-dye or a per-electrochemically-active-surface-area basis, outperforming the ALD-SnO2/PMPDI/CoOx anodes, which themselves outperform their nano-SnOxCy counterparts. Similar, albeit significantly lower, results can be seen on ALD-SnO2 anode systems without H2Q with and without Li-IrOx, Fig. S5.†
The Voc was measured for the ALD-SnO2/PMPDI/Li-IrOx anodes in order to probe recombination in this combined system. The anodes exhibit a Voc of −0.09 ± 0.03 V vs. Ag/AgCl, the same value within error as both the nano-SnOxCy/PMPDI/Li-IrOx and the nano-SnOxCy/PMPDI/Li-IrOx anodes made with nano-SnOxCy containing half as much PEG-BAE organic stabilizer. These Voc values support our finding that the improved Li-IrOx catalyst by itself can overcome recombination in our devices attributable to residual carbon.
Lastly, the faradaic efficiency for water oxidation was also tested for the ALD-SnO2/PMPDI/Li-IrOx anodes resulting in an efficiency of 60 ± 11% for water oxidation. This faradaic efficiency is increased over the 37 ± 10% detailed above for nano-SnOxCy/PMPDI/Li-IrOx anodes. This increase in faradaic efficiency over the nano-SnOxCy/PMPDI/Li-IrOx anodes implies that more of the photocurrent generated is originating from oxygen generation, rather than peroxide formation or other side reactions. The increased faradaic efficiency is furthermore consistent with the increase we previously observed when comparing faradaic efficiencies for nano-SnOxCy/PMPDI/CoOx to that for ALD-SnO2/PMPDI/CoOx, 31 ± 7% and 45 ± 6%, respectively.12,33
Conclusions
The photoelectrochemical WOC research presented investigates the impact on photocurrent of changing the WOCatalyst, specifically the decrease in current due to carbon impurity-based recombination. We were able to answer the five specific questions posed at the beginning of the study: (1) yes, improving the WOCatalyst to the state-of-the-art Li-IrOx WOCatalyst that is the best-in-class alters the charge transfer kinetics to outcompete recombination pathways attributable to residual carbon. More specifically, the use of a Li-IrOx WOCatalyst yielded the desired increase in photocurrent upon catalyst addition, thereby overcoming the decrease in photocurrent and anti-catalyst effect seen with the slower, earth-abundant CoOx WOCatalyst. We also found (2) that the maximum steady-state current from such simple organic-dye-based devices, nano-SnOxCy/PMPDI/Li-IrOx produced −42 μA cm−2 at +0.4 V vs. Ag/AgCl for water oxidation (and −1.2 mA cm−2 photocurrent for H2Q catalysis). Additionally (3), dye adsorption and double-layer capacitance were studied and used to semi-quantitatively compare the two systems, from which we found the interesting result that (4) the simple, planar ALD-SnO2system is more efficient than the FTO/nano-SnOxCy/PMPDI/CoOx system, once the ≥ca. 102 different surface area of the two systems is taken into account. Lastly, (5) a device made using the combination of ALD-SnO2 and the superior Li-IrOx catalyst was shown to have the highest photoactivity on a per-dye or a per-electrochemically-active-surface-area basis, higher than both anodes made using only one of the two strategies described above. That achievement so noted, had we started with Li-IrOx as the WOCatalyst initially in 2017, we likely would have never discovered the role carbon plays when employing nano-SnOxCy, as we would have never observed the tell-tale anti-catalyst effect.Some needed directions of future research are apparent from the present studied. One is that the development of an earth-abundant WOCatalyst that is more selective for water oxidation's 4e− process over the 2e− peroxide reaction remains very desirable. Second, fast spectroscopic kinetic and mechanistic studies aimed at verifying or refuting the individual charge-transfer pathways proposed in Scheme S1† as a working hypothesis would also be most welcome. Finally, the development of an organic stabilizer-free, low-carbon nano-SnO2synthesis is a high priority that promises to be important to all areas in materials chemistry and science currently employing nano-SnOxCy. In short, DS-PECs for WOC and the areas they depend on continue to be interesting as well as promising areas with many directions of research meriting additional research.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Science Foundation under Grant No. 1664646. The authors thank the members of Prof. Finke's research group for reading draft versions of this manuscript and providing valuable feedback. We also thank the Analytical Research Core at Colorado State University for instrument access and assistance. This research used the Materials Synthesis and Characterization Facility of the Center for Functional Nanomaterials, which is a U.S. Department of Energy Office of Science User Facility, at Brookhaven National Laboratory under Contract No. DE-SC0012704.References
- J. T. Kirner and R. G. Finke, Water-Oxidation Photoanodes Using Organic Light- Harvesting Materials: A Review, J. Mater. Chem. A, 2017, 5, 19560–19592 RSC.
- A. J. Bard and M. A. Fox, Artificial Photosynthesis: Solar Splitting of Water to Hydrogen and Oxygen Water Splitting, Acc. Chem. Res., 1995, 28, 141–145 CrossRef CAS.
- R. E. Smalley, Future Global Energy Prosperity: The Terawatt Challenge, Materials Matters, 2004, 30, 412–417 Search PubMed.
- N. S. Lewis and D. G. Nocera, Powering the Planet: Chemical Challenges in Solar Energy Utilization, Proc. Natl. Acad. Sci. USA, 2006, 103, 15729–15735 CrossRef CAS PubMed.
- L. Steier and S. Holliday, A Bright Outlook on Organic Photoelectrochemical Cells for Water Splitting, J. Mater. Chem. A, 2018, 6, 21809–21826 RSC.
- D. Wang, Z. Xu, M. v. Sheridan, J. J. Concepcion, F. Li, T. Lian and T. J. Meyer, Photodriven Water Oxidation Initiated by a Surface Bound Chromophore-Donor-Catalyst Assembly, Chem. Sci., 2021, 12, 14441–14450 RSC.
- S. Yun, N. Vlachopoulos, A. Qurashi, S. Ahmad and A. Hagfeldt, Dye Sensitized Photoelectrolysis Cells, Chem. Soc. Rev., 2019, 48, 3705–3722 RSC.
- M. Bonchio, Z. Syrgiannis, M. Burian, N. Marino, E. Pizzolato, K. Dirian, F. Rigodanza, G. A. Volpato, G. la Ganga, N. Demitri, S. Berardi, H. Amenitsch, D. M. Guldi, S. Caramori, C. A. Bignozzi, A. Sartorel and M. Prato, Hierarchical Organization of Perylene Bisimides and Polyoxometalates for Photo-Assisted Water Oxidation, Nat. Chem., 2018, 11, 146–153 CrossRef.
- A. Mishra, M. K. R. Fischer and P. Büuerle, Metal-Free Organic Dyes for Dye-Sensitized Solar Cells: From Structure: Property Relationships to Design Rules, Angew. Chem., Int. Ed., 2009, 48, 2474–2499 CrossRef CAS PubMed.
- D. P. Hagberg, T. Marinado, K. M. Karlsson, K. Nonomura, P. Qin, G. Boschloo, T. Brinck, A. Hagfeldt and L. Sun, Tuning the HOMO and LUMO Energy Levels of Organic Chromophores for Dye Sensitized Solar Cells, J. Org. Chem., 2007, 72, 9550–9556 CrossRef CAS.
- J. T. Kirner, J. J. Stracke, B. A. Gregg and R. G. Finke, Visible-Light-Assisted Photoelectrochemical Water Oxidation by Thin Films of a Phosphonate-Functionalized Perylene Diimide Plus CoOx Cocatalyst, ACS Appl. Mater. Interfaces, 2014, 6, 13367–13377 CrossRef CAS.
- J. T. Kirner and R. G. Finke, Sensitization of Nanocrystalline Metal Oxides with a Phosphonate-Functionalized Perylene Diimide for Photoelectrochemical Water Oxidation with a CoOx Catalyst, ACS Appl. Mater. Interfaces, 2017, 9, 27625–27637 CrossRef CAS PubMed.
- C. F. Jewell, A. Subramanian, C.-Y. Nam and R. G. Finke, Ultrathin Alumina Passivation for Improved Photoelectrochemical Water Oxidation Catalysis of Tin Oxide Sensitized by a Phosphonate-Functionalized Perylene Diimide First without, and Then with, CoOy, Sustainable Energy Fuels, 2021, 5, 5257–5269 RSC.
- F. Liu, J. J. Concepcion, J. W. Jurss, T. Cardolaccia, J. L. Templeton and T. J. Meyer, Mechanisms of Water Oxidation from the Blue Dimer to Photosystem II, Inorg. Chem., 2008, 47, 1727–1752 CrossRef CAS PubMed.
- T. G. Ulusoy Ghobadi, A. Ghobadi, M. Buyuktemiz, E. A. Yildiz, D. Berna Yildiz, H. G. Yaglioglu, Y. Dede, E. Ozbay and F. Karadas, A Robust, Precious-Metal-Free Dye-Sensitized Photoanode for Water Oxidation: A Nanosecond-Long Excited-State Lifetime through a Prussian Blue Analogue, Angew. Chem., 2020, 132, 4111–4119 CrossRef.
- Z. Deng, H. W. Tseng, R. Zong, D. Wang and R. Thummel, Preparation and Study of a Family of Dinuclear Ru(II) Complexes That Catalyze the Decomposition of Water, Inorg. Chem., 2008, 47, 1835–1848 CrossRef CAS.
- Y. v. Geletii, C. Besson, Y. Hou, Q. Yin, D. G. Musaev, D. Quiñ, R. Cao, K. I. Hardcastle, A. Proust, P. Kö and C. L. Hill, Structural, Physicochemical, and Reactivity Properties of an All-Inorganic, Highly Active Tetraruthenium Homogeneous Catalyst for Water Oxidation, J. Am. Chem. Soc., 2009, 131, 17360–17370 CrossRef CAS PubMed.
- M. Yamamoto, L. Wang, F. Li, T. Fukushima, K. Tanaka, L. Sun and H. Imahori, Visible Light-Driven Water Oxidation Using a Covalently-Linked Molecular Catalyst–Sensitizer Dyad Assembled on a TiO2 Electrode, Chem. Sci., 2016, 7, 1430–1439 RSC.
- M. Yamamoto, Y. Nishizawa, P. Chá, F. Li, T. Rn Pascher, V. Sundströ, L. Sun and H. Imahori, Visible Light-Driven Water Oxidation with a Subporphyrin Sensitizer and a Water Oxidation Catalyst, Chem. Commun., 2016, 52, 13702–13705 RSC.
- F. Li, K. Fan, B. Xu, E. Gabrielsson, Q. Daniel, L. Li and L. Sun, Organic Dye-Sensitized Tandem Photoelectrochemical Cell for Light Driven Total Water Splitting, J. Am. Chem. Soc., 2015, 137, 9153–9159 CrossRef CAS.
- K.-R. Wee, B. D. Sherman, M. K. Brennaman, M. v. Sheridan, A. Nayak, L. Alibabaei and T. J. Meyer, An Aqueous, Organic Dye Derivatized SnO2/TiO2 Core/Shell Photoanode, J. Mater. Chem. A, 2016, 4, 2969–2975 RSC.
- G. F. Moore, J. D. Blakemore, R. L. Milot, J. F. Hull, H.-E. Song, L. Cai, C. A. Schmuttenmaer, R. H. Crabtree and G. W. Brudvig, A Visible Light Water-Splitting Cell with a Photoanode Formed by Codeposition of a High-Potential Porphyrin and an Iridium Water-Oxidation Catalyst, Energy Environ. Sci., 2011, 4, 2389–2392 RSC.
- J. R. Swierk, D. D. Méndez-Hernádez, N. S. Mccool, P. Liddell, Y. Terazono, I. Pahk, J. J. Tomlin, N. v Oster, T. A. Moore, A. L. Moore, D. Gust, T. E. Mallouk and D. D. Méndez-Hernández, Metal-Free Organic Sensitizers for Use in Water- Splitting Dye-Sensitized Photoelectrochemical Cells, Proc. Natl. Acad. Sci. USA, 2015, 112, 1681–1686 CrossRef CAS.
- F. Ronconi, Z. Syrgiannis, A. Bonasera, M. Prato, R. Argazzi, S. Caramori, V. Cristino and C. A. Bignozzi, Modification of Nanocrystalline WO3 with a Dicationic Perylene Bisimide: Applications to Molecular Level Solar Water Splitting, J. Am. Chem. Soc., 2015, 137, 4630–4633 CrossRef CAS PubMed.
- R. J. Kamire, K. L. Materna, W. L. Hoffeditz, B. T. Phelan, J. M. Thomsen, O. K. Farha, J. T. Hupp, G. W. Brudvig and M. R. Wasielewski, Photodriven Oxidation of Surface-Bound Iridium-Based Molecular Water-Oxidation Catalysts on Perylene-3,4-Dicarboximide-Sensitized TiO2 Electrodes Protected by an Al2O3 Layer, J. Phys. Chem. C, 2017, 121, 3752–3764 CrossRef CAS.
- M. Bledowski, L. Wang, A. Ramakrishnan, O. v. Khavryuchenko, V. D. Khavryuchenko, P. C. Ricci, J. Strunk, T. Cremer, C. Kolbeck and R. Beranek, Visible-Light Photocurrent Response of TiO2–Polyheptazine Hybrids: Evidence for Interfacial Charge-Transfer Absorption, Phys. Chem. Chem. Phys., 2011, 13, 21511–21519 RSC.
- Y. Liu, X. Liang, H. Chen, R. Gao, L. Shi, L. Yang and X. Zou, Iridium-Containing Water-Oxidation Catalysts in Acidic Electrolyte, Chin. J. Catal., 2021, 42, 1054–1077 CrossRef CAS.
- G. Hu, J. L. Troiano, U. T. Tayvah, L. S. Sharninghausen, S. B. Sinha, Y. Shopov, B. Q. Mercado, R. H. Crabtree and G. W. Brudvig, Accessing Molecular Dimeric Ir Water Oxidation Catalysts from Coordination Precursors, Inorg. Chem., 2021, 60, 14349–14356 CrossRef CAS PubMed.
- H. H. Cho, L. Yao, J. H. Yum, Y. Liu, F. Boudoire, R. A. Wells, N. Guijarro, A. Sekar and K. Sivula, A Semiconducting Polymer Bulk Heterojunction Photoanode for Solar Water Oxidation, Nat. Catal., 2021, 4, 431–438 CrossRef CAS.
- M. S. Eberhart, D. Wang, R. N. Sampaio, S. L. Marquard, B. Shan, M. K. Brennaman, G. J. Meyer, C. Dares and T. J. Meyer, Water Photo-Oxidation Initiated by Surface-Bound Organic Chromophores, J. Am. Chem. Soc., 2017, 139, 16248–16255 CrossRef CAS PubMed.
- T. Gobbato, F. Rigodanza, E. Benazzi, P. Costa, M. Garrido, A. Sartorel, M. Prato and M. Bonchio, Enhancing Oxygenic Photosynthesis by Cross-Linked Perylenebisimide “Quantasomes, J. Am. Chem. Soc., 2022, 144, 14021–14025 CrossRef CAS PubMed.
- Y. Zhu, D. Wang, W. Ni, G. G. Gurzadyan, L. Sun, T. J. Meyer and F. Li, Water Oxidation by a Noble Metal-Free Photoanode Modified with an Organic Dye and a Molecular Cobalt Catalyst, J. Mater. Chem. A, 2022, 10, 9121–9128 RSC.
- C. F. Jewell, A. Subramanian, C.-Y. Nam and R. G. Finke, Understanding the “Anti-Catalyst” Effect with Added CoOx Water Oxidation Catalyst in Dye-Sensitized Photoelectrolysis Cells: Carbon Impurities in Nanostructured SnO2 Are the Culprit, ACS Appl. Mater. Interfaces, 2022, 14, 25326–25336 CrossRef CAS PubMed.
- I. Neelakanta Reddy, B. Akkinepally, N. Siva Kumar, M. Asif, J. Shim and C. Bai, SnO2 Nanoparticles Anchored on Carbon Spheres for Enhanced Charge Generation and Potentiodynamic Effects, J. Electroanal. Chem., 2023, 937, 117411 CrossRef CAS.
- J. Gao, C.-Q. Xu, S.-F. Hung, W. Liu, W. Cai, Z. Zeng, C. Jia, H. Ming Chen, H. Xiao, J. Li, Y. Huang and B. Liu, Breaking Long-Range Order in Iridium Oxide by Alkali Ion for Efficient Water Oxidation, J. Am. Chem. Soc., 2019, 141, 3014–3023 CrossRef CAS PubMed.
- B. J. Brennan, A. C. Durrell, M. Koepf, R. H. Crabtree and G. W. Brudvig, Towards Multielectron Photocatalysis: A Porphyrin Array for Lateral Hole Transfer and Capture on a Metal Oxide Surface, Phys. Chem. Chem. Phys., 2015, 17, 12728–12734 RSC.
- S. Evans, Correction for the Effects of Adventitious Carbon Overlayers in Quantitative XPS Analysis, Surf. Interface Anal., 1997, 25, 924–930 CrossRef CAS.
- G. Greczynski and L. Hultman, X-Ray Photoelectron Spectroscopy: Towards Reliable Binding Energy Referencing, Prog. Mater. Sci., 2020, 107, 100591 CrossRef CAS.
- Y. Zhao, J. R. Swierk, J. D. Megiatto Jr, B. Sherman, W. J. Youngblood, D. Qin, D. M. Lentz, A. L. Moore, T. A. Moore, D. Gust, T. E. Mallouk and T. J. Meyer, Improving the Efficiency of Water Splitting in Dye-Sensitized Solar Cells by Using a Biomimetic Electron Transfer Mediator, Proc. Natl. Acad. Sci. USA, 2012, 109, 15612–15616 CrossRef CAS PubMed.
- D. L. Ashford, B. D. Sherman, R. A. Binstead, J. L. Templeton and T. J. Meyer, Electro-Assembly of a Chromophore-Catalyst Bilayer for Water Oxidation and Photocatalytic Water Splitting, Angew. Chem., Int. Ed., 2015, 54, 4778–4781 CrossRef CAS.
- B. D. Sherman, M. v Sheridan, C. J. Dares and T. J. Meyer, Two Electrode Collector−Generator Method for the Detection of Electrochemically or Photoelectrochemically Produced O2, Anal. Chem., 2016, 88, 7076–7082 CrossRef CAS PubMed.
- It is important to note here that a comparison of the catalysts based on the number of active sites or amount of catalyst deposited would be ideal, but is not yet possible as the amount of catalyst deposited, much less the number of active sites, is not known.
- One alternative hypothesis that needs to be noted is that because Li-IrOx is spray-deposited using a nebulizer and CoOx is photoelectrochemically deposited, possibly preferentially onto the electrically conductive carbon impurities, it is conceivable that electrodepositing Li-IrOx could result in further preferential deposition onto carbon impurities, but this is not experimentally feasible herein. Another alternative hypothesis is that spray depositing the Li-IrOx catalyst could passivate the carbon-based recombination sites.
- S. Ardo and G. J. Meyer, Photodriven Heterogeneous Charge Transfer with Transition-Metal Compounds Anchored to TiO2 Semiconductor Surfaces, Chem. Soc. Rev., 2009, 38, 115–164 RSC.
- J. R. Swierk, N. S. McCool, T. P. Saunders, G. D. Barber, M. E. Strayer, N. M. Vargas-Barbosa and T. E. Mallouk, Photovoltage Effects of Sintered IrO2 Nanoparticle Catalysts in Water-Splitting Dye-Sensitized Photoelectrochemical Cells, J. Phys. Chem. C, 2014, 118, 17046–17053 CrossRef CAS.
- H. Y. Kim, J. H. Nam, S. M. George, J. S. Park, B. K. Park, G. H. Kim, D. J. Jeon, T. M. Chung and J. H. Han, Phase-Controlled SnO2 and SnO Growth by Atomic Layer Deposition Using Bis(N-Ethoxy-2,2-Dimethyl Propanamido)Tin Precursor, Ceram. Int., 2019, 45, 5124–5132 CrossRef CAS.
- J. Sundqvist, J. Lu, M. Ottosson and A. Hårsta, Growth of SnO2 Thin Films by Atomic Layer Deposition and Chemical Vapour Deposition: A Comparative Study, Thin Solid Films, 2006, 514, 63–68 CrossRef CAS.
- M. N. Mullings, C. Hägglund and S. F. Bent, Tin Oxide Atomic Layer Deposition from Tetrakis(Dimethylamino)Tin and Water, J. Vac. Sci. Technol., A, 2013, 31, 061503 CrossRef.
- J. Heo, A. S. Hock and R. G. Gordon, Low Temperature Atomic Layer Deposition of Tin Oxide, Chem. Mater., 2010, 22, 4964–4973 CrossRef CAS.
- H. E. Cheng, D. C. Tian and K. C. Huang, Properties of SnO2 Films Grown by Atomic Layer Deposition, Procedia Eng., 2012, 36, 510–515 CrossRef CAS.
- I. Ichinose, H. Senzu and T. Kunitake, Stepwise Adsorption of Metal Alkoxides on Hydrolyzed Surfaces: A Surface Sol-Gel Process, Chem. Lett., 1996, 25, 831–832 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Details on the determination of the electrochemically-active surface area and the deposition of Li-IrOx. See DOI: https://doi.org/10.1039/d3se00222e |
| This journal is © The Royal Society of Chemistry 2023 |