
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInteractions in co-pyrolysis of Salicornia bigelovii and heavy fuel oil†
Jinan
Aljaziri
 *,
Ribhu
Gautam
* and
S. Mani
Sarathy
*,
Ribhu
Gautam
* and
S. Mani
Sarathy
Clean Combustion Research Center, Physical Science and Engineering Division, King Abdullah University of Science and Technology, Thuwal 23955-6900, Saudi Arabia. E-mail: jinan.aljaziri@kaust.edu.sa; ribhu.gautam@kaust.edu.sa
First published on 24th July 2023
Abstract
The interactions in the co-pyrolysis of biomass from Salicornia bigelovii (SB) with heavy fuel oil (HFO) were studied through an investigation of the yields, kinetics, and the quality of bio-oil and biochar produced. Co-pyrolysis experiments were performed at 550 °C in a quartz tube with three samples of different ratios of SB and HFO. The decomposition of the samples was investigated using a thermogravimetric analyzer (TGA). The effect of changing the geometry of the sample holder was also investigated in the TGA. The addition of HFO enhanced the volatilization of the sample which resulted in a decrease in char yield and an increase in gas yield. Mixing also increased reactivity and lowered the apparent activation energies in the last stage (227–500 °C) of the co-pyrolysis process. The amount of oxygenated compounds in the bio-oil collected from the mixtures was reduced. The selectivity towards aliphatic and aromatic hydrocarbons increased with an increased addition of HFO and the selectivity towards acids and nitrogen containing groups decreased. Co-pyrolysis of SB and HFO has the potential to produce better quality biofuels through an environmentally friendly process.
1. Introduction
Biomass is a promising clean energy source that has the potential to replace crude oil. The net CO2 emissions caused by burning biomass are theoretically significantly less than those produced from burning fossil fuels. This is due to the ability of biomass to reabsorb the emitted CO2 from the atmosphere as it grows during the photosynthesis process.1 Biomass usage for energy dates back to when burning wood was used for heating and cooking.2 Later on, other processes were developed to convert biomass into an energy crop including biochemical and thermochemical techniques. Conventional biomass used in such processes includes sugar, starches and vegetable oils. More recently, research has shifted to algae and cellulosic/lignocellulosic biomass species.2Pyrolysis is a single step thermochemical process that uses heat in the absence of O2 to decompose biomass into liquid (bio-oil), solid (biochar) and gaseous products.3 A variety of lignocellulosic-based biomass species have been explored for their potential to produce fuel and specialty chemicals.4 An interesting plant that is gaining more attention recently is Salicornia bigelovii (SB). SB is a plant that belongs to the halophyte species. Halophytes are salt and heat tolerant plants that can grow under harsh conditions.5 Therefore, it is possible to grow SB on marginal soils and irrigate with saline water. SB can be directly converted to fuel via pyrolysis and has been studied in different environments.6–8 The pyrolysis products of other halophytes such as Tamarix chinensis, Suaeda salsa, and Phragmites australis have also been explored.9,10
Pyrolysis bio-oil has potential in becoming a great candidate for energy generation. However, bio-oil still falls short when compared to fossil fuels due to the presence of heteroatoms, namely, N, O and S. Thus far, bio-oil has been used as a low-grade fuel,11 a source of value added chemicals,12 a binder13 and a functional carbon material.14,15 However, the use of bio-oil as transportation fuel is challenging due to its high content of oxygen and water, among other properties.16 This leads to issues of corrosion, chemical stability and low calorific value during the storage and use of bio-oil.17,18 Therefore, bio-oil processing is needed and multiple techniques have been developed in the past to treat bio-oil. Some examples of such techniques are: hydrotreating, hydrocracking, and solvent addition.19
A relatively newer technique to enhance the quality of bio-oil is co-pyrolysis, which is the simultaneous pyrolysis of two or more materials without requiring an additional reaction vessel, solvent, or catalyst, although a catalyst may be used to further improve the bio-oil quality.17,20 Co-pyrolysis of biomass with plastics has been extensively studied by numerous researchers.21–25 These studies have shown that co-pyrolysis of mixtures results in enhanced conversion of biomass to volatiles and gases resulting in lower char yields.22–24 The collected char from co-pyrolysis of pine cone with synthetic polymers had a higher calorific value compared to char from biomass pyrolysis alone.25 The bio-oil from co-pyrolysis of biomass and high-density polyethylene had 76% lower viscosity and lower water and oxygen content.26 Studies report that plastics contain large amounts of hydrogen and almost no oxygen, therefore could adjust the content of hydrogen, carbon, and oxygen in the bio-oil produced.25–27 Under co-pyrolysis, scientists speculate that the oxygen in the biomass reacts with the hydrogen and hydrogen radicals from plastics to produce H2O.28,29 This modifies the oxygen content and results in a more stable, less corrosive bio-oil.
Another rich source of hydrogen that can be used in biomass co-pyrolysis is heavy fuel oil (HFO). HFO is a black or brown viscous liquid with a tar-like consistency. It is a mixture of the residue of vacuum distillation and light distillate (cutter stock), that is difficult to burn because of its high concentration of complex hydrocarbons, known as asphaltene, that can cause fouling and corrosion in engines.30,31 HFO is mainly used in marine transportation, and although it is considered a low quality transportation fuel, 95% of marine engines use it due to its low cost.32 HFO, like plastics, is abundant in hydrogen and deficient in oxygen relative to biomass. Catalytic and non-catalytic gasification of HFO has been widely studied for the production of clean fuels.33 However, there are limited studies that have reported on the use of heavy fuels as feedstock or co-feedstock in pyrolysis, although petroleum/oily sludge has been used as a co-feed in biomass pyrolysis. The pyrolysis of mixtures of oily sludge with different biomass, such as sawdust, walnut shell, rice husk, and apricot shell, resulted in enhancements in heating value and H/C ratio.34,35 Moreover, bio-oil collected from co-pyrolysis of rice husk with oily sludge contained fewer oxygenates and a higher concentration of aromatics compared to bio-oil from biomass pyrolysis alone.36 Recently, the combustion behavior of SB pyrolysis bio-oil surrogates mixed with hydrocarbons was investigated, and better combustion behavior was reported.37 These studies demonstrate the potential advantages of using HFO in biomass co-pyrolysis.
The motivation behind this study is to explore SB and HFO co-pyrolysis as a possible bio-oil upgrading method. Different ratios of the two feedstock were used to study their interactions. SB, which is rich in nitrogen and oxygen, is expected to result in a significant amount of nitrogen- and oxygen-containing compounds during pyrolysis. The hydrogen-rich HFO can be a potential co-feed with SB to upgrade the pyrolysis vapors in situ. A tubular furnace pyrolyzer was used to study the effect of co-pyrolysis of SB and HFO on the liquid and solid products. The interactions were studied using a thermogravimetric analyzer (TGA). The effect of changing the geometry of the sample holder on the interactions was also studied in the TGA. The liquid product was then analyzed using a gas chromatography/mass spectrometer (GC/MS). In addition, a kinetic analysis of the process was investigated using the mass loss data obtained from the TGA measurements.
2. Materials and methods
2.1. Preparation and characterization
The SB samples were obtained as described in our previous study.7 SB plants were grown from seeds at the KAUST greenhouse for a period of approximately 6 months. The temperature was maintained at 28 °C during the day and 24 °C at night. The humidity was kept in the range of 50–60% during the growth period to ensure optimal growth conditions. All parts of the plant were utilized in this study. To reduce the high mineral content of the plant and avoid sodium volatilization during pyrolysis, a pretreatment washing method was employed.7 After drying and grinding, SB was placed in deionized water for 8 h at ∼50 °C in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 v/v ratio. The biomass was then filtered and dried overnight. The inherent sodium content of SB was 10.28 wt% before treatment and was reduced by almost 55% after the treatment.7 More than 86% of the sodium present in SB was retained in the biochar, thus sodium volatilization at the chosen pyrolysis temperature is not expected to be significant. The particle size of SB used in the experiments was 300 μm. The biochemical composition of SB is provided in Table 1.38 A typical Saudi Arabian heavy fuel oil sample was used, which contained 31 wt% diesel and 69 wt% vacuum residual oil. The SARA (Saturates, Aromatics, Resins, and Asphaltenes) of the HFO used is also available in Table 1.39
:20 v/v ratio. The biomass was then filtered and dried overnight. The inherent sodium content of SB was 10.28 wt% before treatment and was reduced by almost 55% after the treatment.7 More than 86% of the sodium present in SB was retained in the biochar, thus sodium volatilization at the chosen pyrolysis temperature is not expected to be significant. The particle size of SB used in the experiments was 300 μm. The biochemical composition of SB is provided in Table 1.38 A typical Saudi Arabian heavy fuel oil sample was used, which contained 31 wt% diesel and 69 wt% vacuum residual oil. The SARA (Saturates, Aromatics, Resins, and Asphaltenes) of the HFO used is also available in Table 1.39
| SB | HFO | ||
|---|---|---|---|
| Proximate analysis | (%mass) | Proximate analysis | (%mass) |
| a Reproduced with permission from Bañuelos et al.,38 Renewable Energy; published by Elsevier, 2018. b Reproduced with permission from Alabbad et al.,39 Journal of Thermal Analysis and Calorimetry; published by Springer Nature, 2023. | |||
| Moisture | 2.22 (0.4) | Moisture | 0.00 |
| Volatile matter | 84.12 (0.3) | Volatile matter | 89.66 (2.7) |
| Fixed carbon | 9.88 (0.6) | Fixed carbon | 9.78 (0.3) |
| Ash | 3.78 (0.5) | Ash | 1.24 (0.02) |
| Ultimate analysis | (%mass) | Ultimate analysis | (%mass) |
|---|---|---|---|
| C | 48.53 (1.04) | C | 83.21 (2.5) |
| H | 7.38 (0.2) | H | 10.19 (0.3) |
| N | 5.02 (0.2) | N | 0.12 (0.004) |
| O | 35.29 (1.76) | O | 1.37 (0.04) |
| S | <0.5 | S | 3.87 (0.1) |
Proximate and ultimate analyses of SB and HFO were performed and summarized in Table 1. The proximate analysis was performed following the ASTM E 1131-08 standard method using a TGA Q5000 (TA Instruments, USA).40 A sample mass of 5 mg was heated in the presence of N2 gas with a flowrate of 100 mL min−1. The temperature program employed started from room temperature to 105 °C at a heating rate of 20 °C min−1 and was held constant for five minutes. The temperature was then increased from 105 to 900 °C at 10 °C min−1. Once 900 °C was reached, the gas was switched to O2 at the same flowrate and held constant at 900 °C for 45 minutes. The elemental analysis was performed in a Thermo Flash 2000 (Thermo Fisher Scientific, U.S.A.) using a sample mass of 2–3 mg. Calibration of the instrument with 2,5-bis(5-tert-butyl-2-benzo-oxazol-2-yl) thiophene was performed to quantify the relative amounts of carbon, hydrogen, nitrogen and sulfur in the sample. The oxygen content of the sample was calculated by difference, which is commonly used for the elemental analysis of biomass species. Elemental analyses of biochar samples collected from co-pyrolysis experiments were also performed using the same procedure.
Mixtures of SB and HFO were prepared to study their interactions. The HFO content in the mixtures was 25%, 50% or 75%, with respect to the total dry mass of the sample. Pyrolysis experiments were performed on the individual components as well as the mixtures, resulting in a total of 5 samples: SB or 0% HFO, 25% HFO, 50% HFO, 75% HFO, and 100% HFO.
2.2. TGA experiments
The thermogravimetric analysis of the samples was performed using a TGA Q5000 (TA Instruments, USA). Approximately 5 mg of each sample was used in high-temperature platinum pans. The temperature ranged from ambience to 900 °C at a heating rate of 10 °C min−1 at atmospheric pressure. These experiments were performed under N2 conditions at a flow rate of 100 mL min−1.The thermogravimetric analyzer-Fourier transform infrared spectroscopy (TG-FTIR) experiments were performed using a Netzsch TG209 TGA coupled to a Bruker Tensor 27 FTIR. Approximately 5 mg of each sample was placed in open alumina crucibles. The experiments were conducted at atmospheric pressure with the same temperature range and heating rates as in the TGA experiments described above. The evolved vapors were carried to the FTIR via a transfer line made of Teflon tubes (800 mm (l) 4 mm (i.d.)), and were maintained at 230 °C to avoid the condensation of the vapors. The detector used was a Mercury–Cadmium–Telluride (MCT) IR detector. The FTIR spectra were collected in the wavenumber range of 650–4500 cm−1 at a scan rate of 2 cm−1 and are provided in the ESI.† Both TGA and TG-FTIR experiments were repeated to establish reproducibility, and negligible variations were observed.
The TGA mass loss profiles from the different sample holders were utilized to study the effect of the free space available above the sample on the mass loss data. A figure of the sample holders along with their dimensions can be found in ESI.†
2.3. Non-isothermal kinetic analysis
Thermogravimetric data was used to find the apparent activation energy for the pyrolysis of each sample. The general rate expression for solid-state decomposition is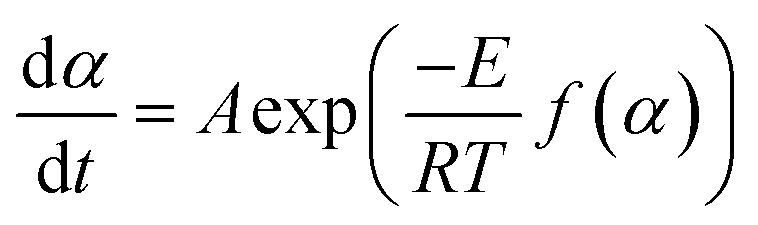 | (1) |
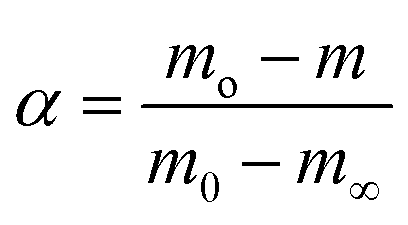 | (2) |
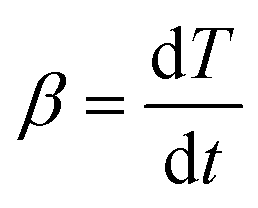 , the heating rate, is constant during the experiment.41–43 The following is obtained
, the heating rate, is constant during the experiment.41–43 The following is obtained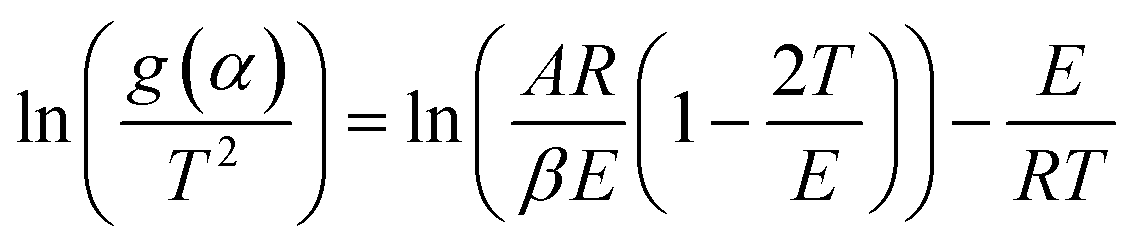 | (3) |
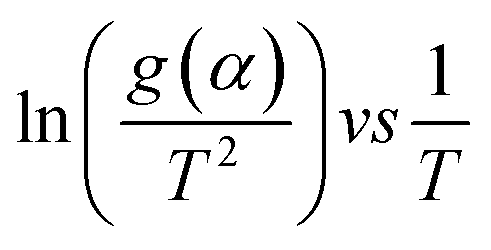 (eqn (3)). g(α) is evaluated using the normalized conversion and corresponding model function and temperature is recorded using a TGA. This will give a straight line with a slope equal to
(eqn (3)). g(α) is evaluated using the normalized conversion and corresponding model function and temperature is recorded using a TGA. This will give a straight line with a slope equal to  , from which the apparent activation energy can be evaluated.
, from which the apparent activation energy can be evaluated.
2.4. Experimental setup
Pyrolysis experiments were carried out in a customized tubular reactor. The reactor consisted of a quartz tube with stainless steel fittings attached at the ends for the gas inlet and outlet. The tube was placed in an electric furnace CY-O1200-50ICS (CY Scientific Instrument, China). The gas flow rate of N2 was maintained at 300 mL min−1. The reactor was purged with N2 prior to each experiment to ensure an inert environment. A sample of about 1.8 g was placed in a quartz boat sample holder and pushed into the center of the reactor tube once the desired temperature of 550 °C was reached. The temperature was also monitored by inserting a thermocouple into the reactor. In the mixtures, SB was placed at the bottom of the boat and the corresponding amount of HFO was placed above it, ensuring even distribution along the length of the quartz boat on the SB sample. A two-stage condenser was used at the outlet of the reactor to help condense the vapors and collect the liquid product. A schematic of the reactor setup can be found in ESI.† Each experiment was repeated at least three times non-consecutively to establish confidence in the data. The actual heating rate experienced by the sample was not determined in this study, however, other studies have shown that a biomass sample in this type of setup typically experiences a heating rate of about 100–200 °C min−1.45 This heating rate estimation will depend on the properties of the sample such as thermal conductivity, specific heat capacity and density. The gaseous product was not collected in this experiment, as the focus of the study was to understand the effect of interactions on the quality of the co-pyrolysis liquid bio-oil.2.5. Product characterization
The elemental analysis of the solid product (biochar) collected form SB, HFO and their mixtures was performed following the method presented in Section 2.1. The liquid product (bio-oil) collected from the pyrolysis experiments was analyzed in an Agilent 7890A gas chromatograph/mass spectrometer (GC/MS). The organic phase of the bio-oil was diluted with dichloromethane in equal amounts. An Agilent HP-5MS capillary column (60 m length × 0.25 mm diameter × 0.25 μm film thickness) was used for the separation. About 1 μL was injected into the column at an injection temperature of 250 °C. Ultra-pure helium (99.9995%) was used to carry the vapors through the column at a flow rate of 2 mL min−1 and a split ratio of 5:1. The oven temperature was programmed to increase at a rate of 10 °C min−1 from 35 to 320 °C. The oven was held at 320 °C for 5 min isothermally. The interface and detector temperatures were maintained at 300 °C and 250 °C, respectively. The electron ionization (EI) potential of the ion source was 70 eV and the scanning m/Z range was 35–700 Da. The National Institute of standards and technology (NIST) data library was used to identify the compounds present in the bio-oil based on the mass spectra. To understand the interactions between SB and HFO in the co-pyrolysis experiments, a deviation parameter (D) was introduced and was defined based on the selectivity as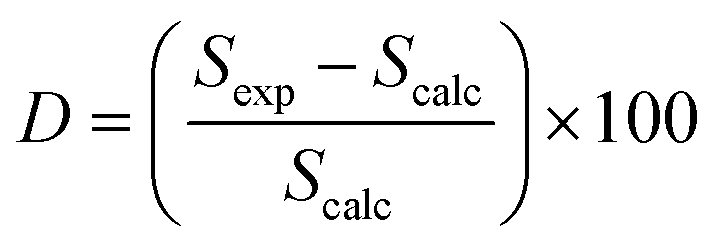 | (4) |
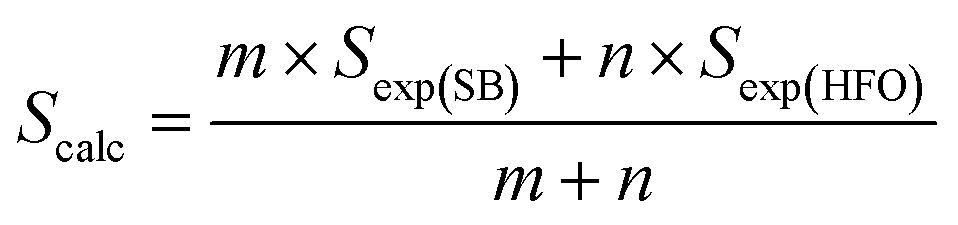 | (5) |
A positive D value represents an increase in selectivity of that group in the mixture ratios under study, which indicates synergistic interactions. A negative D value represents a decrease in the selectivity and indicates antagonistic interactions. A similar approach was used by Gautam and Vinu46 to study the interactions between binary and ternary mixtures of proteins, lipids and carbohydrates.
3. Results and discussion
3.1. Interactions between SB and HFO during co-pyrolysis in a TGA
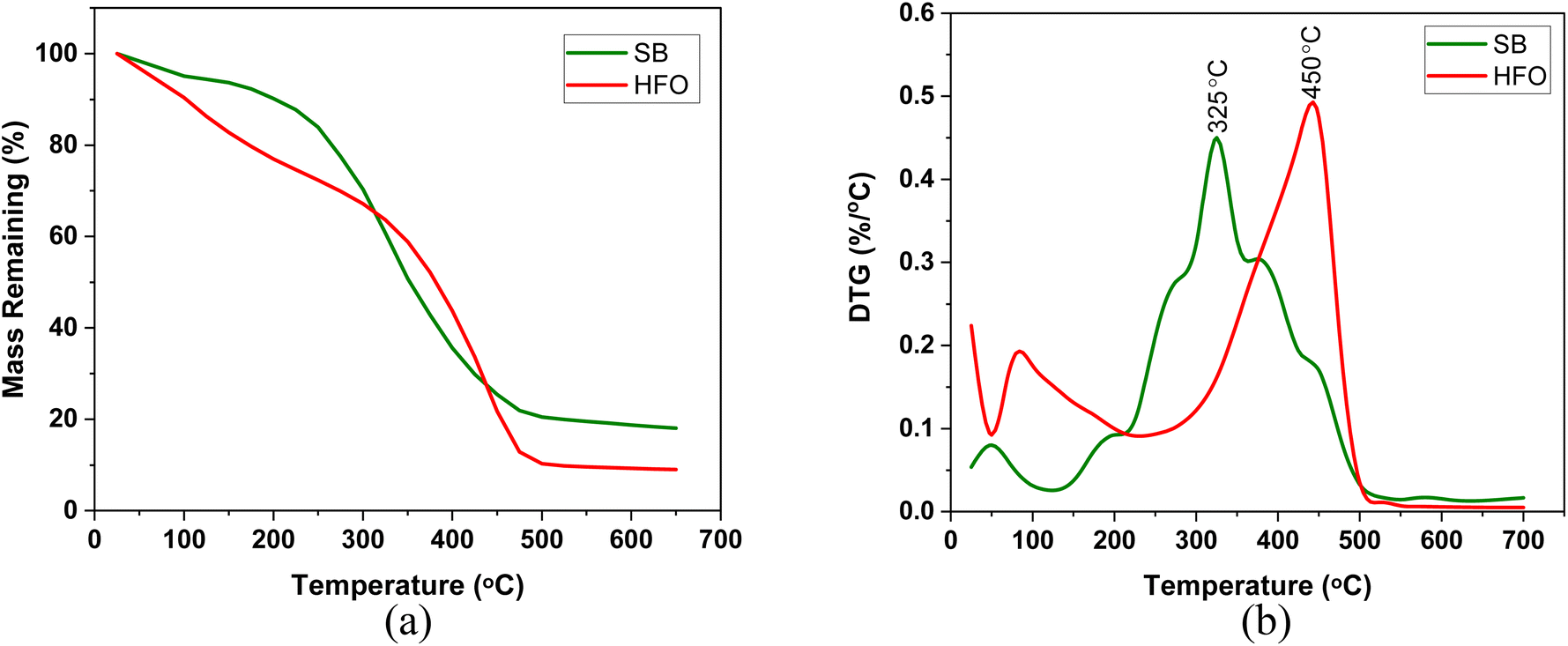 | ||
| Fig. 1 (a) TG and (b) DTG profiles of the individual components pyrolyzed in a high temperature platinum pan at 10 K min−1. | ||
The experimental and theoretical DTG profiles of the three mixtures are shown in Fig. 2. The theoretical values were calculated on a weighted average basis. Theoretical and experimental mass loss profiles overlapped, and no significant differences are observed. The DTG profiles of the mixtures are similar to those of individual SB and HFO, which is consistent with other studies on biomass co-pyrolysis.22,48 The Tgmax of the individual components, SB and HFO, are observed in the mixtures' DTG profiles as expected. A decrease in the maximum weight loss rate in the first stage and an increase in the second stage is observed when compared to the theoretical values. However, no significant interactions are observed in the DTG profiles. It should be noted that the pyrolysis interactions in a TGA do not capture any heat and mass transfer limitations due to the small sample size and low heating rate. Therefore, it does not provide an accurate representation of the interactions that would take place in a real size reactor.49 This is also supported by literature exploring the co-pyrolysis of cellulose with low density polyethylene and tobacco stalk with polymer.22,50 The pyrolysis process in a TGA is kinetically controlled and the TGA data collected was used for apparent kinetic analysis to understand the reactivity of SB and HFO mixtures, following the ICTAC Kinetics Committee recommendations.51 To further investigate the effect of mixing, other analyses such as TG-FTIR and GC/MS were also performed.
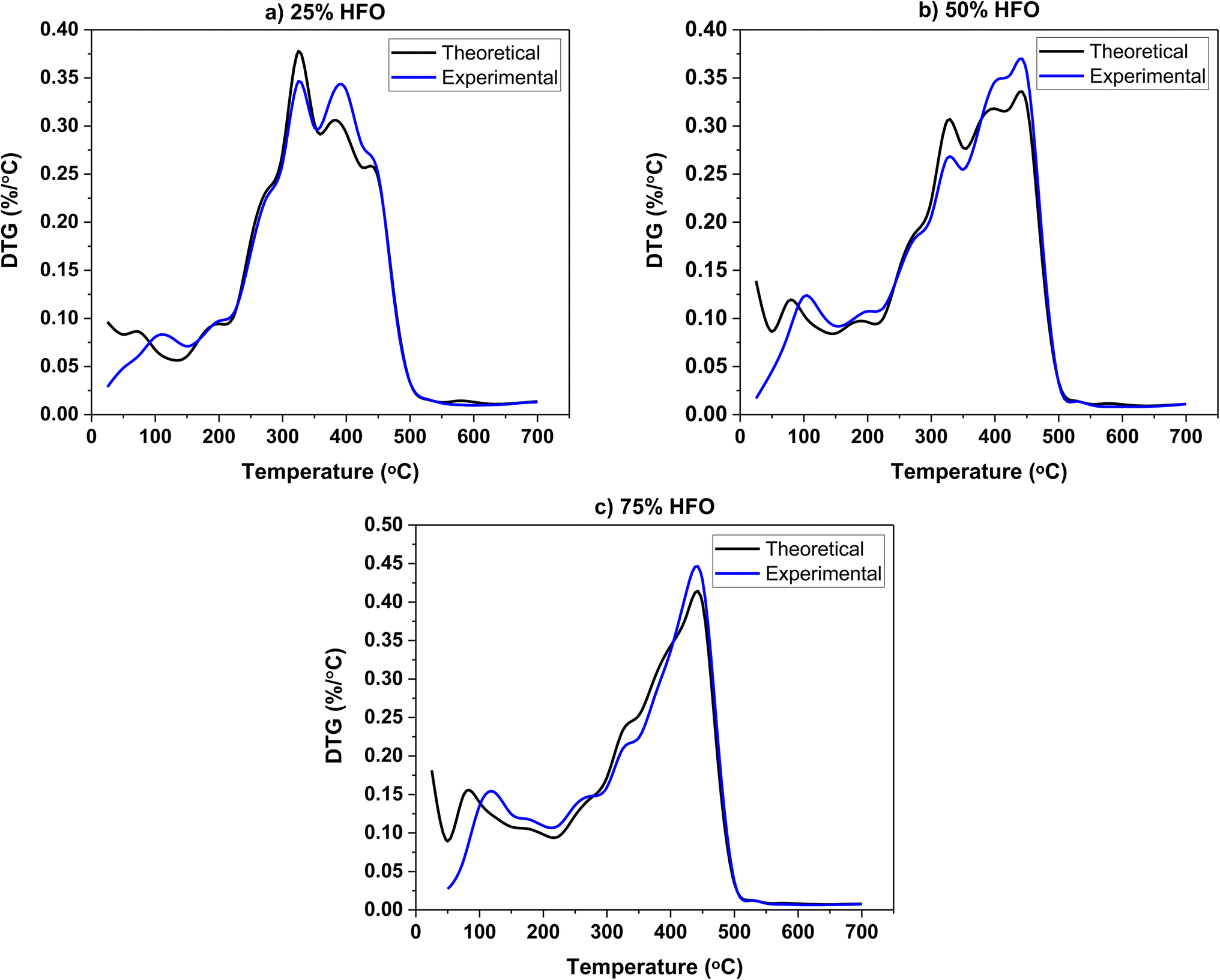 | ||
| Fig. 2 Theoretical and experimental DTG profiles of the three mixtures pyrolyzed in a high temperature pyrolysis pan (a) 25% HFO, (b) 50% HFO and (c) 75% HFO. | ||
A set of co-pyrolysis TG experiments were performed in an alumina crucible and a platinum pan (see ESI† for details). Studies have shown that the size and shape of the sample holder and the free space volume available can affect the mass loss profiles in TGA.52,53 To have uniformity in the measurements, the sample mass used in these experiments was 5 ± 0.5 mg. The two sample holders differ in the free space volume available above the sample. The alumina crucible offered larger free space volume compared to the pan. Fig. 3 shows the DTG curves obtained from the decomposition of SB and HFO in the alumina crucible and the platinum pan.
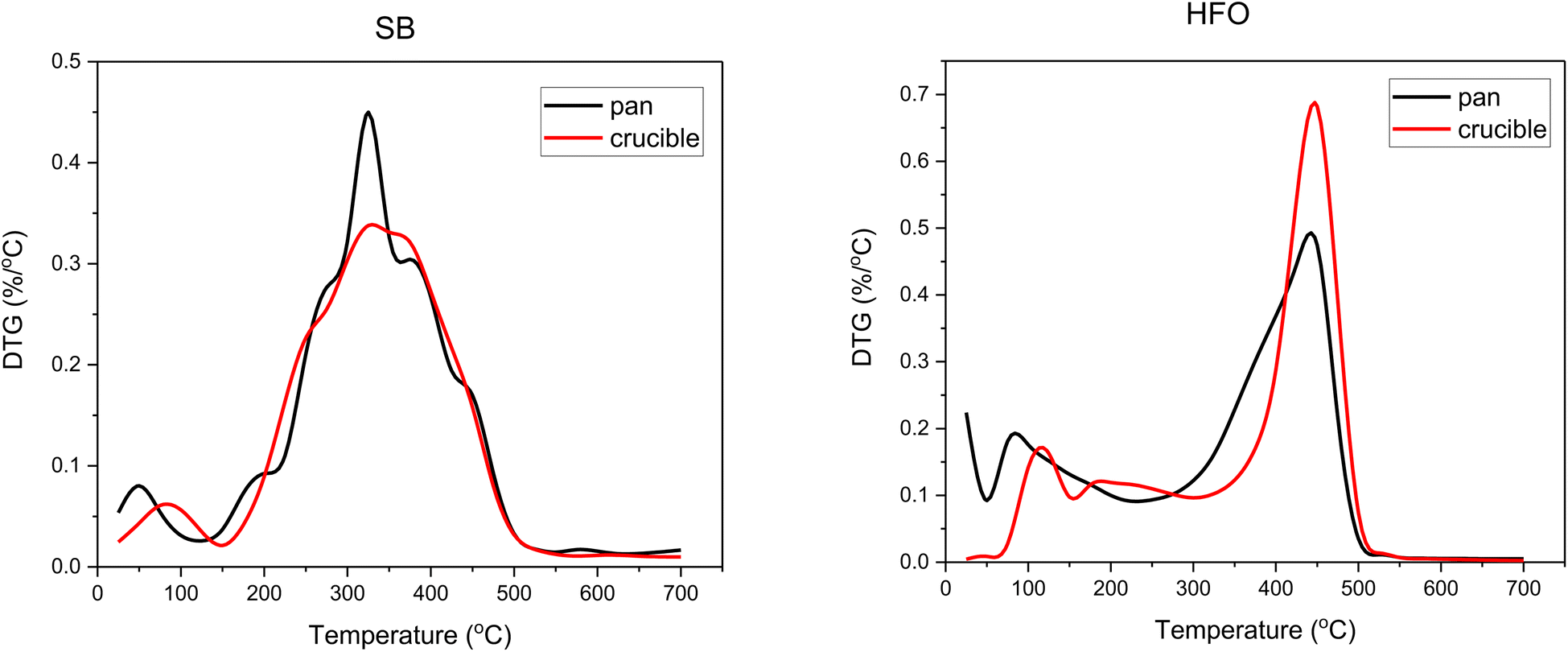 | ||
| Fig. 3 DTG curves of SB and HFO in alumina crucibles and platinum pans. | ||
The onset decomposition temperature of HFO shifted to higher temperatures using the alumina crucible. The temperature shifts from around 250 °C in the pan to 350 °C in the crucible. The peak temperature, however, remains the same. This could be explained by a delay in carrying out the vapors formed on top of the sample due to the difference in the height of the sample holder. In the pan, the purging gas can easily carry out the vapors, unlike the alumina crucible. Similar results have been observed by Shi et al. in which they have designed experiments to study the effect of the geometry of the crucible on thermal analysis.54 In SB the onset decomposition temperature has also slightly shifted to higher temperatures in the crucible. This is also due to delays in carrying out the pyrolysis vapors.
The peak temperature height was also affected by changing the sample holder. SB showed a higher peak height in the pan. This again could be due to the height of the walls. The free space available increases the diffusion pathway of the gas and hence results in a lower apparent rate. This was also observed by Nowak et al., as the free space available was altered by the addition of Al2O3 below the biomass sample.55 A lower rate was observed when the diffusion pathway through the free space was higher. In contrast, the opposite was observed in HFO. For HFO, a higher peak height was observed in the alumina crucible. This observation could be related to the concentration and rate of reaction. The initial concentration of the material at the decomposition temperature is lower in the pan compared to the crucible due to easier flashing of the lighter cut portion of HFO resulting in a lower apparent rate in the pan.
The DTG of the mixtures follow the characteristics observed in SB and HFO and any differences observed follow the same reasoning mentioned above. The DTG of mixtures can be found in ESI.†
The maximum weight loss of SB in the DTG profile was around 325 °C in both crucibles (Fig. 3). The evolution of gases around the peak temperature corresponds to cellulose, hemicellulose and protein decomposition.46 Around this temperature, depolymerization, cleavage of the glycosidic bond fragmentation, dehydration and rearrangement reactions occur. These reactions result in compounds such as furfural and benzene derivatives which evolve from cellulose decomposition. Whereas, hemicellulose decomposition results in furan derivatives, acetaldehyde and hydroxyacetaldehyde. Finally, protein decomposition results in compounds containing pyridine and indole as well as aldehydes and benzamine. Some of these compounds and reactions are represented in Fig. 7b. These reactions reduced as the composition of HFO increased in the mixture. Further discussion can be found in the TG-FTIR section in the light of product formation.
Fig. 4 depicts the major functional groups observed in the pyrolysis vapors of SB, HFO, and their mixtures. The major groups include CO2 (2350 cm−1), methyl C–H stretch (2935 cm−1), aromatic C–H in-plane-bend (906 cm−1) and aromatic C–H out-of-plane bend (690 cm−1). The maximum absorbance peak for CO2 was observed around 350 °C for all samples except 100% HFO. The intensity of the CO2 peak was highest for SB pyrolysis. A similar CO2 peak was observed upon pyrolysis of Tobacco stalk with a maximum at 350 °C.50 CO2 is a major gas produced during biomass pyrolysis due to the presence of oxygenated biomolecules, and it is also formed from the decarboxylation of carboxylic acids.57 The intensity of the CO2 peak decreased as more HFO was added to the mixture, until no CO2 was observed in the pyrolysis vapors of 100% HFO. This is expected, as HFO is primarily composed of carbon and hydrogen and lacks oxygen, which is required for the formation of CO2. It can be noted that CO2 peaked in the temperature range 340–345 °C for SB and SB/HFO mixtures. As the HFO composition increased, the experimental peak heights were 9.75% (25% HFO), 27.8% (50% HFO) and 46.67% (75% HFO) higher compared to theoretical peak heights indicating promotion of decarboxylation reactions under co-pyrolysis conditions.
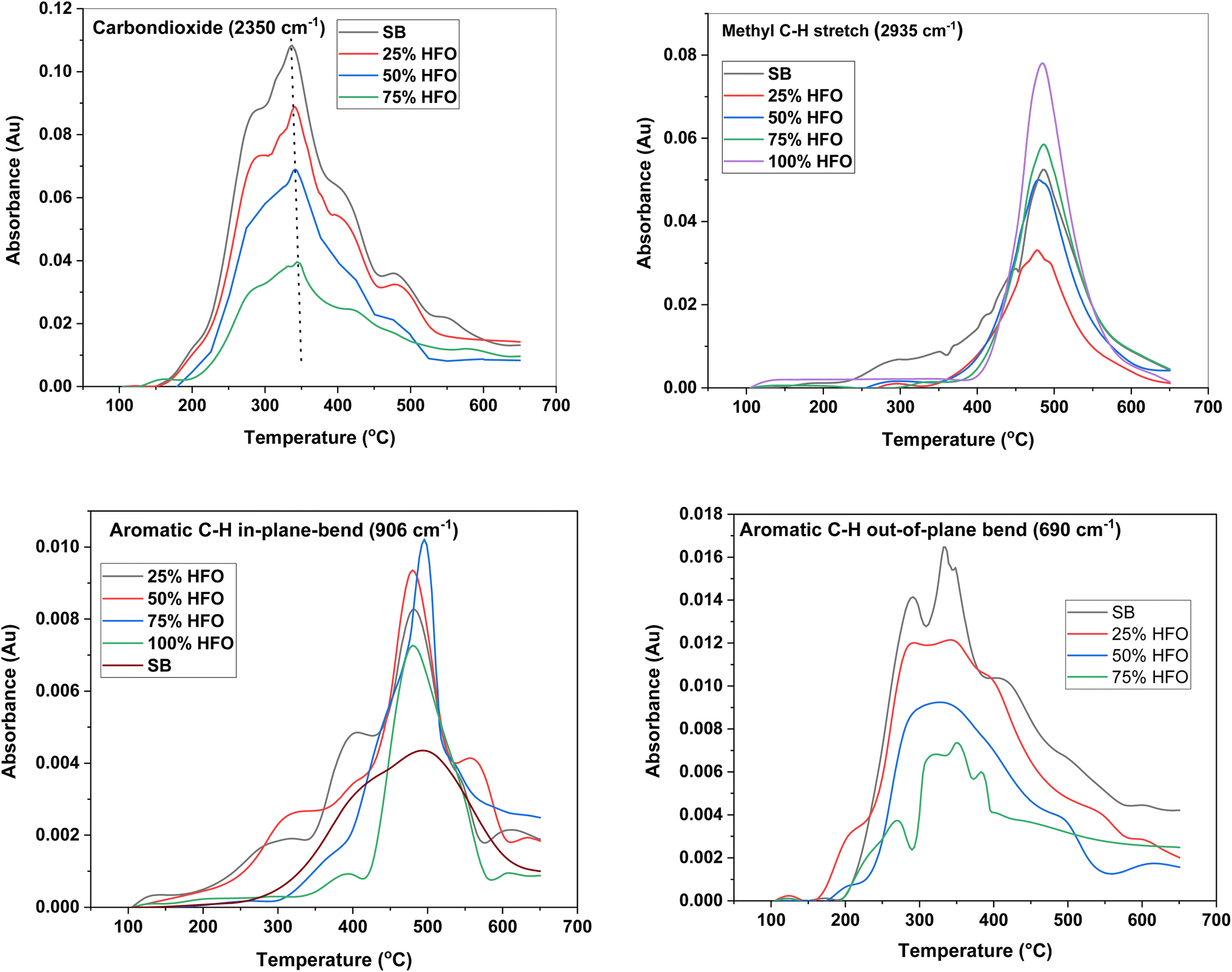 | ||
| Fig. 4 Temperature evolution of major function groups in the pyrolysis products from SB, HFO and their mixtures. | ||
HFO decomposes into different aliphatic and aromatic hydrocarbons under an inert environment,31 which is evident from the presence of peaks around 2935 cm−1, 906 cm−1 and 690 cm−1 in the FTIR spectra. These peaks correspond to the vibrations of aliphatic and aromatic hydrocarbons, respectively. Aliphatic hydrocarbons were produced from the pyrolysis of all five samples, with the highest intensity peak observed for HFO followed by the 75% HFO mixture. The gas generation of these hydrocarbons was concentrated between 400 and 650 °C for all samples except SB, which had a wider temperature range of 200–650 °C. This wider temperature range corresponds to the slow degradation of the biomass, as discussed earlier. Mixing of SB with HFO showed no effect on the evolution of aliphatic hydrocarbons as seen in Fig. 4. This suggests that the mixing of SB and HFO does not significantly alter the production of aliphatic hydrocarbons during co-pyrolysis.
Aromatic C–H in-plane bending, which is associated with the presence of polycyclic aromatic hydrocarbons (PAH) such as naphthalene,58 was present in the vapors produced from the pyrolysis of SB, HFO and their mixtures. PAH can form from phenol derivatives in lignin when hydroxyl and methoxy linkages are cleaved.59 The low intensity of this peak in SB can be attributed to the low content of lignin, which is around 2% by weight. HFO contains non-protonated aromatic carbons, which indicate the presence of fused rings.31 The mixtures exhibited a higher intensity peak than that from HFO alone, suggesting that the addition of SB helped in the extraction of the PAH present in HFO. This confirms the synergistic effects of mixing SB with HFO. The 75% HFO mixture resulted in the highest intensity peak, possibly due to the optimal balance of lignin and HFO content.
C–H out of plane bending, which is associated with the presence of monocyclic aromatic hydrocarbons (MAH), was not observed in HFO pyrolysis. The absence of MAH indicates the presence of more complex aromatics, such as polycyclic aromatic hydrocarbons (PAH). The intensity of the peak is highest for SB, which contains a higher percentage of MAH than HFO, and gradually decreases as the percentage of HFO added increases in the mixture until none is observed at 100% HFO.
3.2. Kinetic analysis
Eqn (3) was used to calculate the apparent activation energy (Eα) of the pyrolysis process for each sample. The apparent activation energy can be determined from the slope of the lines produced from plotting the logarithmic term, , against
, against 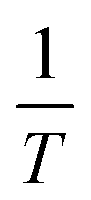 . The plot obtained from eqn (3) was split into three pyrolysis stages based on linearity. Multiple nucleation-growth kinetic models were tested in each stage to find the best fit model. The first stage of each sample was well represented by a model of the form, g(α) = −ln(1 − α), which describes the initial decomposition of the sample. The second and third stages were both sufficiently represented by a model of the form g(α) = [−ln(1 − α)]4, which describes the later stages of the decomposition process. Table 2 summarizes the Eα values for all samples calculated using these models.
. The plot obtained from eqn (3) was split into three pyrolysis stages based on linearity. Multiple nucleation-growth kinetic models were tested in each stage to find the best fit model. The first stage of each sample was well represented by a model of the form, g(α) = −ln(1 − α), which describes the initial decomposition of the sample. The second and third stages were both sufficiently represented by a model of the form g(α) = [−ln(1 − α)]4, which describes the later stages of the decomposition process. Table 2 summarizes the Eα values for all samples calculated using these models.
| Stage | Temperature (°C) | Conversion (α) | E a (kJ mol−1) | |
|---|---|---|---|---|
| 0% HFO | 1 | 25–62 | 0–0.03 | 39.18 |
| 2 | 62–227 | 0.03–0.14 | 35.98 | |
| 3 | 227–500 | 0.14–0.89 | 126.96 | |
| 25% HFO | 1 | 25–74 | 0–0.03 | 33.34 |
| 2 | 74–234 | 0.03–0.18 | 59.62 | |
| 3 | 234–500 | 0.18–0.93 | 125.03 | |
| 50% HFO | 1 | 25–106 | 0–0.06 | 33.56 |
| 2 | 106–286 | 0.06–0.28 | 52.72 | |
| 3 | 286–500 | 0.28–0.85 | 117.31 | |
| 75% HFO | 1 | 25–123 | 0–0.09 | 36.59 |
| 2 | 123–358 | 0.09–0.48 | 55.01 | |
| 3 | 358–500 | 0.48–0.96 | 191.62 | |
| 100% HFO | 1 | 25–73 | 0–0.05 | 51.15 |
| 2 | 73–356 | 0.05–0.46 | 43.10 | |
| 3 | 356–500 | 0.46–0.97 | 200.26 |
In TGA, the main objective is to perform measurements in a kinetically controlled regime. This is achieved using a low heating rate, small sample mass, and small particle size to ensure that the pyrolysis is kinetically controlled. The low heating rates also help to ensure that there is no (or minimal) gradient in the sample temperature and furnace temperature. In this study, the particle size of SB was 300 μm, sample mass was around 5 mg and heating rate was 10 °C min−1. The kinetic regime was verified by changing the sample mass and acquiring mass loss profiles. These overlapping mass loss profiles established the kinetically controlled regime for the performed TGA experiments. A detailed discussion can be found in literature.60–62 The main objective of investigating the non-isothermal kinetics in this study is to understand the shifts in different temperature regimes observed with mixing.
The thermal decomposition of the biomass, HFO and their mixtures followed a sigmoidal reaction regime. The kinetics of these reactions were well-captured by the nucleation-growth model. The kinetic functions used to evaluate the apparent activation energy represent both random nucleation and sequential growth mechanism. The corresponding equation leads to a sigmoidal reaction progression, as observed in many solid-state reactions, including this study. Solid-state reactions during pyrolysis commonly occur by the generation of nucleation sites followed by their geometric growth. It is worth noting that nucleation-growth models for solid-state reactions have been widely used in the last two decades to model the pyrolysis of organic materials.63
Decomposition occurs at higher temperatures, and therefore it is expected to observe higher Eα at higher temperatures. This is confirmed by the higher Eα values observed at stage 3 of all five samples compared to stages 1 and 2. HFO exhibited a higher Eα value compared to SB in all three stages. This can be attributed to its high thermal stability, which is due to the presence of asphaltenes that require more energy to decompose.
Biomass degradation takes longer, as most of the decomposition occurs in stage 3, where the conversion jumps from 0.14 to 0.82. However, HFO reaches almost 50% conversion in the second stage. The effect of this observation can also be extended to the mixtures, where higher percentages of HFO result in higher conversions at earlier stages corresponding to lower temperatures.
In stage 1, mixing resulted in slightly lower Eα values compared to the individual components. In stage 2, the mixtures showed slightly higher Eα values than SB and HFO individually, but the effect of mixing was not significant in these stages. The first stage of the mass loss corresponds to the release of moisture from the biomass and lighter components from the HFO, which is characterized by low Eα values. In stage 2, the decomposition of cellulose, hemicellulose and proteins begins, which is consistent with previous.46 In stage 3, the mixtures exhibited lower Eα values the individual components. Interestingly, Eα of HFO can be significantly reduced by about 42% by equal mixing with SB. This higher reactivity in the third stage could be attributed to biomass radicals potentially initiating reactions and enhancing the decomposition of HFO leading to lower Eα values. This finding is supported by previous studies that suggest biomass can act as a radical initiator in pyrolysis reactions.48
3.3. Interactions between SB and HFO during co-pyrolysis in tubular reactor
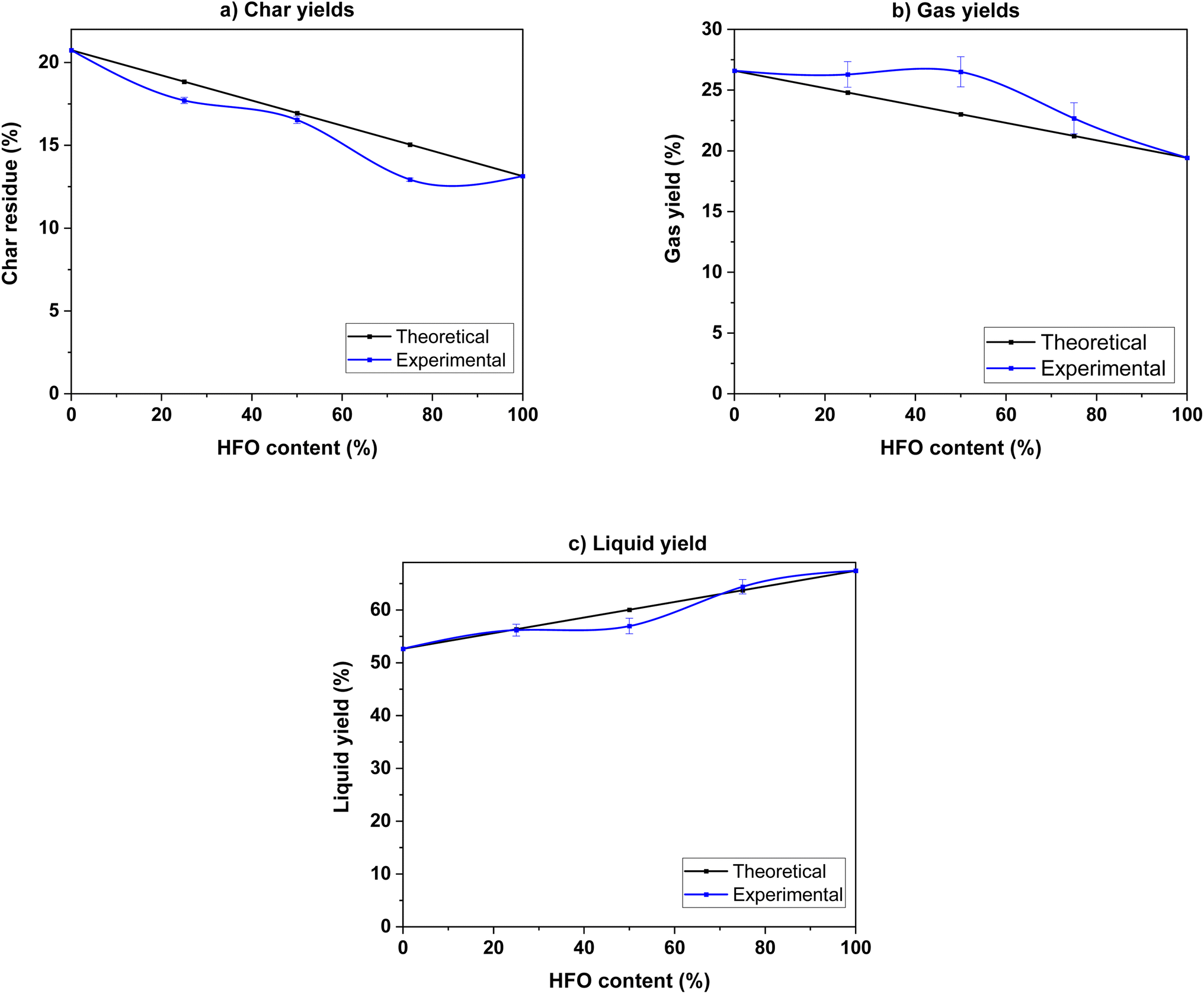 | ||
| Fig. 5 (a) Char, (b) gas and (c) liquid product yields of the pyrolysis of SB, HFO and their mixtures. | ||
To study the interactions during co-pyrolysis, the experimental yields were compared to the theoretical values calculated on a weighted average basis of the individual components. Synergistic interactions were observed in the gas and char yields. Mixing resulted in increased production of gas compared to the theoretical value, indicating a synergistic interaction. Similarly, the experimental char yields were lower than the theoretical values, which indicates that mixing enhanced devolatilization of the sample and allowed for a higher degree of conversion of the original feedstock. A hydrogen rich environment provided by HFO creates more radicals by transferring hydrogen to the biomass. These radicals initiate and promote scission and cracking reactions, which enhance char decomposition and increase gas yields. This phenomenon has been observed in previous studies.24,48,64
| Sample | C | H | N | S | O |
|---|---|---|---|---|---|
| SB | 61.23 (2.21) | 3.83 (0.02) | 6.98 (0.54) | n.d. | 27.96 (2.58) |
| 25% HFO | 66.78 (3.89) | 3.31 (0.01) | 5.52 (0.12) | 2.28 (0.13) | 22.11 (1.15) |
| 50% HFO | 75.15 (3.03) | 2.68 (0.05) | 3.82 (0.22) | 4.58 (0.15) | 13.77 (3.45) |
| 75% HFO | 79.52 (0.60) | 2.08 (0.17) | 2.48 (0.08) | 6.96 (0.87) | 8.96 (0.52) |
| 100% HFO | 86.94 (2.4) | 1.62 (0.04) | 0.92 (0.07) | 9.38 (0.09) | 1.14 (2.47) |
Carbon enrichment occurs via condensation and aromatization reactions. That involve the decarboxylation, dehydration, and dehydrogenation of the precursors.65 These reactions are accompanied by the reduction of nitrogen-containing compounds.66 The observed trend of increased carbon content in the solid residue accompanied by a decrease in H, N and O indicates that the addition of HFO might have enhanced these reactions.
Fig. 6 represents the selectivities of the major functional groups identified in the bio-oil from pyrolysis of SB, HFO and their mixtures along with the deviation in selectivities calculated using eqn (4). The compounds in the bio-oil can be linked to the components of SB and HFO.
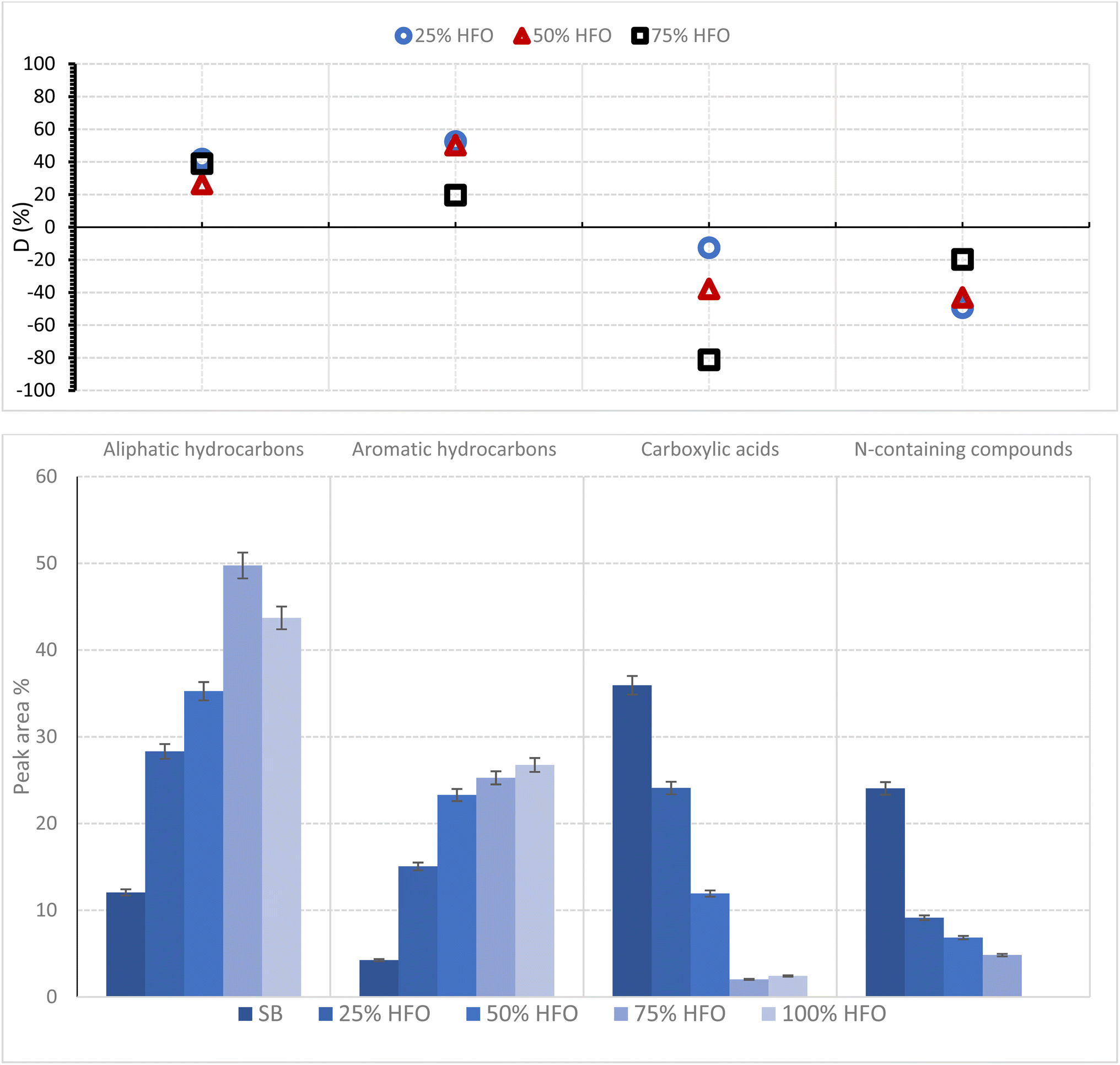 | ||
| Fig. 6 Distribution of compounds in the bio-oil from pyrolysis of SB, HFO and their mixtures. | ||
SB has typical characteristics of lignocellulosic biomass, which is ∼46 wt% cellulose, ∼15 wt% hemicellulose ∼2 wt% lignin.38 Lipids and protiens are also present in the biomass matrix. The major amino acids present are proline, phenylalanin, leucine, glutamic acid, tyrosine, histidine and aspartic acid.67,68 Lipids are represented by triglycerides in SB. HFO, on the other hand, is a mixture of polycyclic aromatic hydrocarbons containing sulfur, oxygen and nitrogen as heteroatoms. A surrogate has been used to represent HFO containing compounds such as naphthalene, benzothiophene, dibenzothiophene and quinoline.47Fig. 7 shows a representative composition of SB and HFO, as well as a broad overview of the reactions under pyrolysis and co-pyrolysis conditions.
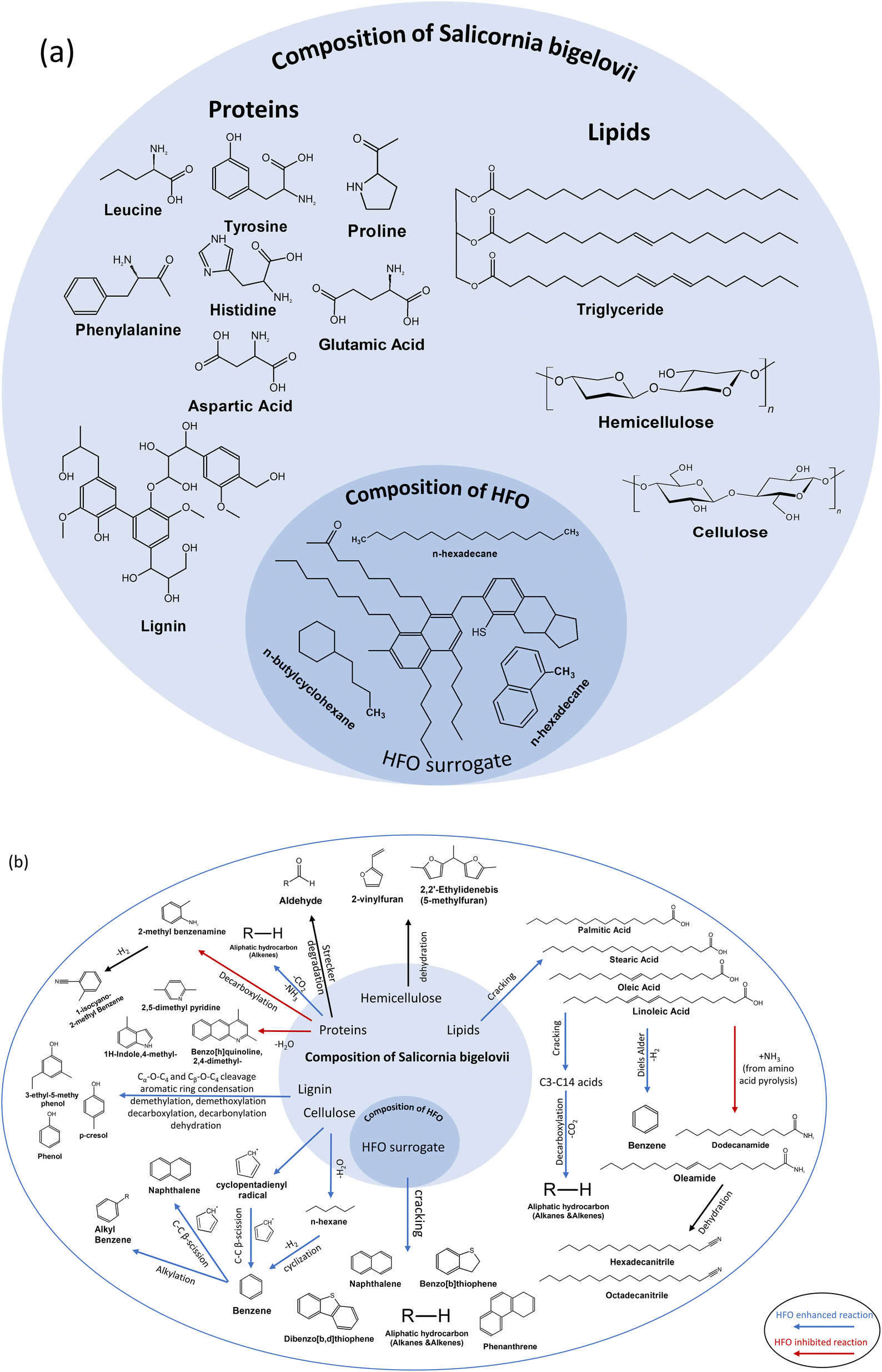 | ||
| Fig. 7 (a) Composition of SB and HFO. (b) Plausible reaction scheme for co-pyrolysis of SB and HFO. | ||
The addition of HFO resulted in a general increase in peak area for aliphatic and aromatic hydrocarbons in the mixture. The peak area % of aliphatic and aromatic hydrocarbons increased by 38% and 21%, respectively, when 75% HFO was added to SB. This increase was accompanied by a positive deviation in the selectivities for these two categories, indicating synergistic interactions. The positive deviation can be attributed to enhanced extraction of fatty acids assisted by pyrolysis and their subsequent decarboxylation in the presence of HFO, resulting in an increase in aliphatic hydrocarbons. Aromatic hydrocarbons, on the other hand, come from lignin dehydration, decarboxylation and demethylation reactions that are also enhanced by the addition of HFO. Furthermore, HFO cracking also contributes to the amount of aromatic hydrocarbons present in the bio-oil. The presence of HFO aids in the generation of more free radicals during co-pyrolysis, which facilitates cracking of C–C bonds and enhances the quality of oil products. The major hydrocarbons detected were eicosane and cetene, as well as benzene and naphthalene derivatives.
In contrast, carboxylic acids exhibit a decrease in peak area, and a negative deviation parameter was recorded as the percentage of HFO added increased. The area % of carboxylic acids from SB pyrolysis was 36%, which dropped to 2% in the 75% HFO mixture, with a deviation of about −81.2%. Cracking of lipids during the pyrolysis of SB results in the generation of fatty acids which are then consumed through decarboxylation reactions resulting in the formation of hydrocarbons and carbon dioxide. The decrease in area% of carboxylic acids confirmed that decarboxylation reactions were enhanced with the addition of HFO. The major acids found in the co-pyrolysis bio-oil were palmitic acid, stearic acid, oleic acid and linoleic acid.
N-containing groups exhibited a negative deviation parameter. This category includs amines/amides, N-aromatic compounds and nitriles. N-containing compounds come from protein cracking, as proteins are the major source of nitorgen in the mixture.46,69 Possible amino acid reaction pathways to produce a variety of N-conatining compounds are presented in Fig. 7b.70 For example, the benzene ring in tyrosine and phenylalanine and the five member ring with nitrogen in proline can undergo decarboxylation and dehydration reactions to form some of the major N-containing compounds present in the bio-oil, such as Indole and Imidazole. The major decomposition pathways of common amino acids in the protein structure of biomass during pyrolysis have been studied and correspond to those found in SB pyrolysis.71 Strecker degradation reactions of some proteins can form the aldehydes found in the bio-oil as seen in Fig. 7b. SB is known to be a protein-rich biomass as seen from the high N-content, which corresponded to a high area% of N-containing compounds in the bio-oil from SB.7,8,67,68 Lack of N-containing compounds in the 100% HFO sample asserts that only SB pyrolysis results in these species. Therefore, a decrease in the composition of this group is expected as more HFO is added. Mixing also contributed to the decrease, as evident in the negative deviation parameter. For example, the 25% HFO sample exhibited a deviation parameter of around −49.3%. The addition of HFO inhibited dehydration and decarboxylation of amino acids that would have allowed for the formation of N-containing compounds from the proteins present in the biomass. Examples of the compounds found in the bio-oil are amides such as oleamide and aromatics such as indole and 2,5-dimethylpyridine. Nitiriles were also present, such as hexadecanitrile, octadecanitrile and benzenepropanenitrile. Amides and nitriles were formed due to the reactions between the pyrolysis products from proteins and lipids.8,68
Other catagories represented only 1–3 area%, and therfore, were not included in the product analysis. However, a detailed list of products is provided in the SM. These catagories include esters such as methyl palmitate and oxalic acid (2-ehtyl hexyl) ester, alcohols such as palmityl and linoleyl alcohol, phenols such as p-cresol and finally furans such as 2-vinylfuran and 2,5-furandione. All of which were present in the bio-oil in trace amounts.
The primary pathways for biomass decomposition are cracking, depolymerization, rearrangement and char formation, with secondary reactions involving cracking and recombination also contributing to the process.75 Based on the GC/MS analysis of the bio-oil presented in the previous section, blue arrows indicate the reactions that were enhanced by the addition of HFO, while red arrows represent those that were inhibited.
During co-pyrolysis, the hydrocarbons generated from the cracking of HFO undergo chain-end and random scission, leading to the transfer of hydrogen to the anhydrosugars produced from cellulose pyrolysis and the formation carbonyl compounds such as aldehydes and ketones. The co-pyrolysis process promotes the conversion of aldehydes and ketones to hydrocarbons. Oxygenated compounds produced from cellulose pyrolysis act as acceptors, facilitating the scission of hydrocarbons into smaller alkanes and alkenes. As shown in Fig. 7, an increase in the biomass composition in the feedstock promotes the production of aromatic hydrocarbons via Diels–Alder reaction.76 Furan derivatives and other light oxygenates produced from biomass pyrolysis are consumed by the olefins produced from HFO pyrolysis. It is important to note that the interactions between the vapors from both the feedstocks during co-pyrolysis is known to be a complex phenomenon. If the biomass composition in the feedstock is too low or too high, it can significantly hinder the interactions between biomass and co-feed. This is why moderate compositions of HFO and biomass were chosen for this study.
A thorough analysis of gas and liquid samples would provide further insights into the interactions between biomass and HFO during co-pyrolysis, and this can be achieved by increasing the sample mass. The information from this study can be used, in part, for investigation of gasification of HFO with biomass as co-feed. Blends of pyrolysis bio-oil with heavy fuel oil have been shown to have good stability, reduced viscosity, and inhibition of polymerization.77 Although direct information on the properties of bio-oil obtained from pyrolysis of biomass with HFO is not available, studies elsewhere have compared the properties of bio-oil from co-pyrolysis of biomass with a hydrogen rich source (e.g., plastics) to that of bio-oil from biomass pyrolysis. For instance, bio-oil produced from the co-pyrolysis of rice straw with polyethylene waste exhibited lower viscosities and higher calorific values compared to bio-oil produced from biomass pyrolysis. The flash points of the bio-oil from mixtures were slightly higher than that of HFO and were comparable to that of diesel.78 Based on this, some enhancement in the fuel properties of the bio-oil from co-pyrolysis of SB and HFO is expected, suggesting their potential utilization for reduced emissions.
4. Conclusion
The effects of mixing SB biomass with HFO during pyrolysis at 550 °C were understood through experiments performed in a TGA and a tubular reactor. The free space available in the two sample holders used had some effect on the mass loss data obtained from the TGA. This was mainly due to the obstruction from the walls of the alumina crucibles in carrying out the vapors produced during pyrolysis. The results of the co-pyrolysis experiments show some synergistic effects between the two components. The addition of HFO resulted in increased extraction of PAH in the vapors produced during the pyrolysis. A kinetic analysis of the process revealed that the addition of HFO lowered the apparent activation energy in stage 3 (227–500 °C) of the co-pyrolysis process. Mixing also promoted cracking and volatilization hence increased gas yields by 7% and decreased char yields by 13% compared to the theoretical value of the 75% HFO mixture. A GC/MS analysis of the collected bio-oil showed an increase in selectivity towards aliphatic and aromatic hydrocarbons and a decrease towards carboxylic acids. The peak area% of aliphatic and aromatic hydrocarbons increased by 38% and 21%, respectively, and decreased by 34% for carboxylic acids when 75% HFO was added to the mixture. This study shows the benefits of co-pyrolysis of SB and HFO to produce valuable chemicals and possible upgrading of the fuel.Conflicts of interest
Authors have no competing financial interests to declare.Acknowledgements
This work was sponsored by King Abdullah University of Science and Technology (KAUST). The authors thank the Clean Combustion Research Center (CCRC) at KAUST for funding and allowing access to their experimental facilities.References
- J. Palmer and W. Carton, Carbon Removal as Carbon Revival? Bioenergy, Negative Emissions, and the Politics of Alternative Energy Futures, Front. Clim., 2021, 3, 60 CrossRef
.
-
R. Ruan, et al., Chapter 1 – Biofuels: Introduction, in Biofuels: Alternative Feedstocks and Conversion Processes for the Production of Liquid and Gaseous Biofuels, ed. A. Pandey, et al., Academic Press, 2nd edn, 2019, pp. 3–43 Search PubMed
- A. Demirbas and G. Arin, An Overview of Biomass Pyrolysis, Energy Sources, 2002, 24(5), 471–482 CrossRef CAS
-
A. A. Boateng, Energy crops—biomass resources and traits, in Pyrolysis of Biomass for Fuels and Chemicals, ed. A. A. Boateng, Academic Press, 2020, pp. 221–238 Search PubMed
- T. J. Flowers and T. D. Colmer, Salinity tolerance in halophytes*, New Phytol., 2008, 179(4), 945–963 CrossRef CAS PubMed
- Y. Makkawi,
et al., Assessment of the pyrolysis products from halophyte Salicornia bigelovii cultivated in a desert environment, Fuel, 2021, 290, 119518 CrossRef CAS
- A. Iaccarino, R. Gautam and S. M. Sarathy, Bio-oil and biochar production from halophyte biomass: effects of pre-treatment and temperature on Salicornia bigelovii pyrolysis, Sustainable Energy Fuels, 2021, 5(8), 2234–2248 RSC
- J. Aljaziri,
et al., On the effects of CO2 atmosphere in the pyrolysis of Salicornia bigelovii, Bioresour. Technol. Rep., 2022, 17, 100950 CrossRef CAS
- H. Xiao,
et al., Comparison of biochar properties from 5 kinds of halophyte produced by slow pyrolysis at 500 °C, Biochar, 2022, 4(1), 12 CrossRef CAS
- X. Dong,
et al., Characterization of halophyte biochar and its effects on water and salt contents in saline soil, Environ. Sci. Pollut. Res., 2022, 29(8), 11831–11842 CrossRef CAS
- S. Czernik and A. V. Bridgwater, Overview of Applications of Biomass Fast Pyrolysis Oil, Energy Fuels, 2004, 18(2), 590–598 CrossRef CAS
- Y. Jing,
et al., Catalytic Production of Value-Added Chemicals and Liquid Fuels from Lignocellulosic Biomass, Chem, 2019, 5(10), 2520–2546 CAS
-
Y. Lu, et al., Physical and Chemical Characterization of Bio-Pitch as a Potential Binder for Anode, in Light Metals 2019, Springer International Publishing, Cham, 2019 Search PubMed
- X. Hu and M. Gholizadeh, Progress of the applications of bio-oil, Renewable Sustainable Energy Rev., 2020, 134, 110124 CrossRef CAS
- X. Hu,
et al., High yields of solid carbonaceous materials from biomass, Green Chem., 2019, 21(5), 1128–1140 RSC
- B. B. Uzoejinwa,
et al., Co-pyrolysis of biomass and waste plastics as a thermochemical conversion technology for high-grade biofuel production: Recent progress and future directions elsewhere worldwide, Energy Convers. Manage., 2018, 163, 468–492 CrossRef CAS
- H. Hassan, J. K. Lim and B. H. Hameed, Recent progress on biomass co-pyrolysis conversion into high-quality bio-oil, Bioresour. Technol., 2016, 221, 645–655 CrossRef CAS
- F. Abnisa and W. M. A. Wan Daud, Optimization of fuel recovery through the stepwise co-pyrolysis of palm shell and scrap tire, Energy Convers. Manage., 2015, 99, 334–345 CrossRef
- S. Xiu and A. Shahbazi, Bio-oil production and upgrading research: A review, Renewable Sustainable Energy Rev., 2012, 16(7), 4406–4414 CrossRef CAS
- A. Alabdrabalnabi, R. Gautam and S. Mani Sarathy, Machine learning to predict biochar and bio-oil yields from co-pyrolysis of biomass and plastics, Fuel, 2022, 328, 125303 CrossRef CAS
- D. Phakedi, A. U. Ude and P. O. Oladijo, Co-pyrolysis of polymer waste and carbon-based matter as an alternative for waste management in the developing world, J. Anal. Appl. Pyrolysis, 2021, 155, 105077 CrossRef CAS
- S. D. Gunasee,
et al., Co-pyrolysis of LDPE and cellulose: Synergies during devolatilization and condensation, J. Anal. Appl. Pyrolysis, 2017, 126, 307–314 CrossRef CAS
- Ö. Çepelioğullar and A. E. Pütün, Products characterization study of a slow pyrolysis of biomass-plastic mixtures in a fixed-bed reactor, J. Anal. Appl. Pyrolysis, 2014, 110, 363–374 CrossRef
- K. G. Burra and A. K. Gupta, Kinetics of synergistic effects in co-pyrolysis of biomass with plastic wastes, Appl. Energy, 2018, 220, 408–418 CrossRef CAS
- M. Brebu,
et al., Co-pyrolysis of pine cone with synthetic polymers, Fuel, 2010, 89(8), 1911–1918 CrossRef CAS
- W. Chen,
et al., Co-pyrolysis of waste newspaper with high-density polyethylene: Synergistic effect and oil characterization, Energy Convers. Manage., 2016, 112, 41–48 CrossRef CAS
- S. Xiong,
et al., Study on the co-pyrolysis of high density polyethylene and potato blends using thermogravimetric analyzer and tubular furnace, J. Anal. Appl. Pyrolysis, 2015, 112, 66–73 CrossRef CAS
- B. Zhang,
et al., Production of aromatic hydrocarbons from catalytic co-pyrolysis of biomass and high density polyethylene: Analytical Py–GC/MS study, Fuel, 2015, 139, 622–628 CrossRef CAS
- Q. Bu,
et al., Microwave-assisted co-pyrolysis of microwave torrefied biomass with waste plastics using ZSM-5 as a catalyst for high quality bio-oil, J. Anal. Appl. Pyrolysis, 2018, 134, 536–543 CrossRef CAS
- L. Goldsworthy, Computational Fluid Dynamics Modelling of Residual Fuel Oil Combustion in the Context of Marine Diesel Engines, Int. J. Engine Res., 2006, 7(2), 181–199 CrossRef
- A. G. Abdul Jameel,
et al., Heavy fuel oil pyrolysis and combustion: Kinetics and evolved gases investigated by TGA-FTIR, J. Anal. Appl. Pyrolysis, 2017, 127, 183–195 CrossRef CAS
-
J. J. Corbett, Marine Transportation and Energy Use, in Encyclopedia of Energy, ed. C. J. Cleveland, Elsevier, New York, 2004, pp. 745–758 Search PubMed
- M. Jafarian,
et al., Emerging technologies for catalytic gasification of petroleum residue derived fuels for sustainable and cleaner fuel production—An overview, Energy Rep., 2023, 9, 3248–3272 CrossRef
- G. Hu,
et al., Investigation of waste biomass co-pyrolysis with petroleum sludge using a response surface methodology, J. Environ. Manage., 2017, 192, 234–242 CrossRef CAS PubMed
- S. Zhao,
et al., Dewatering and low-temperature pyrolysis of oily sludge in the presence of various agricultural biomasses, Environ. Technol., 2018, 39(21), 2715–2723 CrossRef CAS PubMed
- B. Lin, Q. Huang and Y. Chi, Co-pyrolysis of oily sludge and rice husk for improving pyrolysis oil quality, Fuel Process. Technol., 2018, 177, 275–282 CrossRef CAS
- R. Gautam,
et al., Combustion of Salicornia bigelovii Pyrolysis Bio-oil and Surrogate Mixtures: Experimental and Kinetic Study, Energy Fuels, 2023, 37(1), 385–400 CrossRef CAS
- J. A. Bañuelos,
et al., Production, characterization and evaluation of the energetic capability of bioethanol from Salicornia Bigelovii as a renewable energy source, Renewable Energy, 2018, 123, 125–134 CrossRef
- M. Alabbad,
et al., TG-DSC and TG-FTIR analysis of heavy fuel oil and vacuum residual oil pyrolysis and combustion: characterization, kinetics, and evolved gas analysis, J. Therm. Anal. Calorim., 2023, 148(5), 1875–1898 CrossRef CAS
-
ASTM-International, ASTM E1131-08 Standard Test Method for Compositional Analysis by Thermogravimetry, 2014 Search PubMed
- A. W. Coats and J. P. Redfern, Kinetic Parameters from Thermogravimetric Data, Nature, 1964, 201(4914), 68–69 CrossRef CAS
- M. Raza,
et al., Kinetic and thermodynamic analyses of date palm surface fibers pyrolysis using Coats-Redfern method, Renewable Energy, 2022, 183, 67–77 CrossRef CAS
- S. Ali,
et al., Investigation of kinetic decomposition characteristics of Malaysian wood species using Coats and Redfern (CR) method, Mater. Today: Proc., 2021, 42, 178–185 CrossRef CAS
- H. Jiang,
et al., Pyrolysis kinetics of phenol–formaldehyde resin by non-isothermal thermogravimetry, Carbon, 2010, 48(2), 352–358 CrossRef CAS
- W. Jerzak, M. Reinmöller and A. Magdziarz, Estimation of the heat required for intermediate pyrolysis of biomass, Clean Technol. Environ. Policy, 2022, 24(10), 3061–3075 CrossRef CAS
- R. Gautam and R. Vinu, Unraveling the interactions in fast co-pyrolysis of microalgae model compounds via pyrolysis-GC/MS and pyrolysis-FTIR techniques, React. Chem. Eng., 2019, 4(2), 278–297 RSC
- A. G. Abdul Jameel,
et al., Calculation of Average Molecular Parameters, Functional Groups, and a Surrogate Molecule for Heavy Fuel Oils Using 1H and 13C Nuclear Magnetic Resonance Spectroscopy, Energy Fuels, 2016, 30(5), 3894–3905 CrossRef CAS
- P. Lu,
et al., Synergistic effects on char and oil produced by the co-pyrolysis of pine wood, polyethylene and polyvinyl chloride, Fuel, 2018, 230, 359–367 CrossRef CAS
- Y. Long,
et al., Interactions among biomass components during co-pyrolysis in (macro)thermogravimetric analyzers, Korean J. Chem. Eng., 2016, 33(9), 2638–2643 CrossRef CAS
- R. Chen,
et al., Comparative study on synergistic effects in co-pyrolysis of tobacco stalk with polymer wastes: Thermal behavior, gas formation, and kinetics, Bioresour. Technol., 2019, 292, 121970 CrossRef PubMed
- S. Vyazovkin,
et al., ICTAC Kinetics Committee recommendations for collecting experimental thermal analysis data for kinetic computations, Thermochim. Acta, 2014, 590, 1–23 CrossRef CAS
- L. Shi,
et al., Reaction of volatiles from a coal and various organic compounds during co-pyrolysis in a TG-MS system. Part 1. Reaction of volatiles in the void space between particles, Fuel, 2018, 213, 37–47 CrossRef CAS
- L. Shi,
et al., Reaction of volatiles from a coal and various organic compounds during co-pyrolysis in a TG-MS system. Part 2. Reaction of volatiles in the free gas phase in crucibles, Fuel, 2018, 213, 22–36 CrossRef CAS
- L. Shi,
et al., Pyrolysis of coal in TGA: Extent of volatile condensation in crucible, Fuel Process. Technol., 2014, 121, 91–95 CrossRef CAS
- B. Nowak,
et al., Mass transfer limitation in thermogravimetry of biomass gasification, J. Therm. Anal. Calorim., 2013, 111(1), 183–192 CrossRef CAS
- G. Özsin and A. E. Pütün, TGA/MS/FT-IR study for kinetic evaluation and evolved gas analysis of a biomass/PVC co-pyrolysis process, Energy Convers. Manage., 2019, 182, 143–153 CrossRef
- C. Xu and J. Lancaster, Conversion of secondary pulp/paper sludge powder to liquid oil products for energy recovery by direct liquefaction in hot-compressed water, Water Res., 2008, 42(6), 1571–1582 CrossRef CAS
- M. Tommasini,
et al., Fingerprints of polycyclic aromatic hydrocarbons (PAHs) in infrared absorption spectroscopy, Spectrochim. Acta, Part A, 2016, 152, 134–148 CrossRef CAS PubMed
- D. K. Ojha, D. Viju and R. Vinu, Fast pyrolysis kinetics of lignocellulosic biomass of varying compositions, Energy Convers. Manage.: X, 2021, 10, 100071 CAS
- H. Bennadji,
et al., Effect of Particle Size on Low-Temperature Pyrolysis of Woody Biomass, Energy Fuels, 2014, 28(12), 7527–7537 CrossRef CAS
- J.-L. Dirion, C. Reverte and M. Cabassud, Kinetic parameter estimation from TGA: Optimal design of TGA experiments, Chem. Eng. Res. Des., 2008, 86(6), 618–625 CrossRef CAS
- G. Várhegyi, Aims and methods in non-isothermal reaction kinetics, J. Anal. Appl. Pyrolysis, 2007, 79(1), 278–288 CrossRef
- A. K. Burnham, X. Zhou and L. J. Broadbelt, Critical Review of the Global Chemical Kinetics of Cellulose Thermal Decomposition, Energy Fuels, 2015, 29(5), 2906–2918 CrossRef CAS
- E. Părpăriţă,
et al., TG/FT–IR/MS study on thermal decomposition of polypropylene/biomass composites, Polym. Degrad. Stab., 2014, 109, 13–20 CrossRef
- J. Zhang,
et al., Co-pyrolysis of sewage sludge and rice husk/bamboo sawdust for biochar with high aromaticity and low metal mobility, Environ. Res., 2020, 191, 110034 CrossRef CAS PubMed
- J. Jin,
et al., Cumulative effects of bamboo sawdust addition on pyrolysis of sewage sludge: Biochar properties and environmental risk from metals, Bioresour. Technol., 2017, 228, 218–226 CrossRef CAS PubMed
- R. El-Araby,
et al., Comparative evaluation of Salicornia bigelovii oil planted under different treatments, Bull. Natl. Res. Cent., 2020, 44(1), 133 CrossRef
- F. M. Attia,
et al., Nutrient composition and feeding value of Salicornia bigelovii torr meal in broiler diets, Anim. Feed Sci. Technol., 1997, 65(1), 257–263 CrossRef CAS
- R. Gautam and R. Vinu, Non-catalytic fast pyrolysis and catalytic fast pyrolysis of Nannochloropsis oculata using Co-Mo/γ-Al2O3 catalyst for valuable chemicals, Algal Res., 2018, 34, 12–24 CrossRef
-
S. C. Moldoveanu, Chapter 18 Pyrolysis of Amino Acids and Small Peptides, in Techniques and Instrumentation in Analytical Chemistry, ed, S. C. Moldoveanu, Elsevier, 2010, pp. 527–578 Search PubMed
- J. Yu, K. Maliutina and A. Tahmasebi, A review on the production of nitrogen-containing compounds from microalgal biomass via pyrolysis, Bioresour. Technol., 2018, 270, 689–701 CrossRef CAS PubMed
- D.-A. Lyra,
et al., Evaluation of Salicornia bigelovii Germplasm for Food Use in Egypt and the United Arab Emirates Based on Agronomic Traits and Nutritional Composition, Plants, 2022, 11, 2653 CrossRef CAS PubMed
- C. J. Mueller,
et al., Diesel Surrogate Fuels for Engine Testing and Chemical-Kinetic Modeling: Compositions and Properties, Energy Fuels, 2016, 30(2), 1445–1461 CrossRef CAS PubMed
- R. Gautam,
et al., On the products from the pyrolysis of heavy fuel and vacuum residue oil, J. Anal. Appl. Pyrolysis, 2023, 173, 106060 CrossRef CAS
- F.-X. Collard and J. Blin, A review on pyrolysis of biomass constituents: Mechanisms and composition of the products obtained from the conversion of cellulose, hemicelluloses and lignin, Renewable Sustainable Energy Rev., 2014, 38, 594–608 CrossRef CAS
- K. Ding,
et al., Improving hydrocarbon yield from catalytic fast co-pyrolysis of hemicellulose and plastic in the dual-catalyst bed of CaO and HZSM-5, Bioresour. Technol., 2018, 261, 86–92 CrossRef CAS PubMed
- M. D. Kass,
et al., Stability, Combustion, and Compatibility of High-Viscosity Heavy Fuel Oil Blends with a Fast Pyrolysis Bio-Oil, Energy Fuels, 2020, 34(7), 8403–8413 CrossRef CAS
- M. S. Hossain,
et al., Production of liquid fuel from co-pyrolysis of polythene waste and rice straw, Energy Procedia, 2019, 160, 116–122 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3se00063j |
| This journal is © The Royal Society of Chemistry 2023 |