
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Impact of impurities on water electrolysis: a review
Hans
Becker
 a,
James
Murawski
b,
Dipak V.
Shinde
a,
Ifan E. L.
Stephens
*b,
Gareth
Hinds
a and
Graham
Smith
*a
a,
James
Murawski
b,
Dipak V.
Shinde
a,
Ifan E. L.
Stephens
*b,
Gareth
Hinds
a and
Graham
Smith
*a
aNational Physical Laboratory, Teddington, Middlesex TW11 0LW, UK. E-mail: graham.smith@npl.co.uk
bDepartment of Materials, Imperial College London, Royal School of Mines, London SW7 2AZ, UK. E-mail: i.stephens@imperial.ac.uk
First published on 14th March 2023
Abstract
Low temperature water electrolysers such as Proton Exchange Membrane Water Electrolysers (PEMWEs), Alkaline Water Electrolysers (AWEs), and Anion Exchange Membrane Water Electrolysers (AEMWEs) are known to be sensitive to water quality, with a range of common impurities impacting performance, hydrogen quality and device lifetime. Purification of feed water adds to cost, operational complexity, and design limitations, while failure of purification equipment can lead to degradation of electrolyser materials and components. Increased robustness to impurities will offer a route to longer device lifetimes and reduced operating costs but understanding of the impact of impurities and associated degradation mechanisms is currently limited. This critical review offers, for the first time, a comprehensive overview of relevant impurities in operating electrolysers and their impact. Impurity sources, degradation mechanisms, characterisation techniques, water purification technologies and mitigation strategies are identified and discussed. The review generalises already reported mechanisms, proposes new mechanisms, and provides a framework for consideration of operational implications.
 Hans Becker | Hans Becker received his BSc and MSc in Chemical Engineering from Institut Teknologi Bandung and the Hong Kong University of Science and Technology, respectively. In 2018, he obtained his PhD in Electrochemistry from the Technical University of Denmark working on high temperature – Proton Exchange Membrane (PEM) fuel cells. Hans then moved to the National Physical Laboratory UK for 3 years as a Higher Research Scientist working on the metrology of PEM fuel cells and electrolysers. Hans is currently a Green Hydrogen Scientist at bp, leading the research of various water electrolyser technologies. |
 James Murawski | James Murawski received his MSci in Chemistry with industrial placement from the University of Southampton in 2018. He is currently pursuing a PhD under the supervision of Dr Ifan E. L. Stephens alongside industrial supervisors from Johnson Matthey and National Physical Laboratory. James' research focuses on benchmarking and improving understanding of oxygen evolution catalyst stability in both aqueous model systems and full cell testing for PEM water electrolyser applications. |
 Dipak V. Shinde | Dipak completed his PhD at Hanyang University (Seoul, South Korea). His doctoral research involved the synthesis and characterisation of metal oxide and chalcogenide nanostructures for use in solar cells and water splitting devices. His subsequent postdoctoral work included roles at POSTECH (Pohang, South Korea), working on perovskite solar cell fabrication, and at the Italian Institute of Technology (IIT, Genoa), where he worked to develop electrochemical experiments to investigate the hydrogen evolution and CO2 reduction reactions. Dipak's current work at NPL involves the development of new characterisation techniques for water electrolysers to understand their performance degradation. |
 Ifan E. L. Stephens | Ifan is a Reader in Electrochemistry at the Department of Materials, Imperial College London. After a PhD at the University of Cambridge, Ifan moved to the Department of Physics at the Technical University of Denmark as a postdoctoral researcher, then Assistant Professor and finally as an Associate Professor. In 2015, Massachusetts Institute of Technology awarded Ifan the Peabody Visiting Associate Professorship. Ifan has published 80 papers on topics including oxygen reduction, oxygen evolution, CO2 reduction and nitrogen reduction. Ifan co-founded HPNow, focused on H2O2 electrosynthesis. Ifan and colleagues were awarded the John Jeyes Award for their work on H2O2 production. |
 Gareth Hinds | Gareth is an NPL Fellow and Science Area Leader in the Electrochemistry Group at the National Physical Laboratory in Teddington, United Kingdom. His primary expertise is in the development of novel in situ diagnostic techniques and standard test methods for assessment of corrosion and material degradation in energy applications. Gareth is a fellow of the Royal Academy of Engineering and holds visiting academic positions at UCL, the University of Strathclyde and Harbin Institute of Technology. He is the author of over 200 publications and is currently President of the World Corrosion Organization and Vice President of the European Federation of Corrosion. |
 Graham Smith | Graham leads the UK's National Physical Laboratory's work on electrochemical hydrogen technologies and more broadly coordinates NPL's ‘Metrology for Clean Hydrogen Energy’ activities. His research focuses on developing high-quality measurements to support the global industrial and academic effort towards better electrochemical energy technologies. After an MSci and PhD at Imperial College London, Graham carried out post-doctoral research at the University of Southampton and the University of Cape Town. Prior to joining NPL, Graham worked as a researcher in the SINTEF (Norway). |
1 Introduction
1.1 Background
Low temperature water electrolysers are increasingly being deployed to produce green hydrogen worldwide.1 With applications in transport, heating and the chemical industry, the availability of cheap low carbon hydrogen will be crucial to maximise the use of renewable energy and reduce or eliminate net CO2 emissions.2–4 Electrolysers are currently being deployed at the MW scale5 but to make a substantial impact on climate change, cumulative deployments need to be on the TW scale. To facilitate this Hydrogen Europe's strategic innovation and research agenda has set targets for a reduction of the cost of hydrogen fuel from € 5–8 to <€ 3 kgH2−1 by 2030. This necessitates technology improvements. Thankfully there are still many opportunities to improve the understanding of the processes operating in electrolysers and use this understanding to improve electrolyser lifetime and performance.6–8 One little-studied area of practical importance is in understanding the impact of water impurities on the performance and durability of electrolysers.Electrolysers producing hydrogen require water to operate; approximately 45 kg of water is electrolysed to produce 5 kg (∼166 kW hHHV) of hydrogen, enough to fill the storage tank in a typical passenger fuel cell electric vehicle. In comparison, around 5 kg of fresh water is used in thermoelectric power plants to produce 1 kW h of energy,9 while the typical UK citizen has a domestic water consumption of 142 kg per day10 and a total daily water footprint estimated to be in excess of 4600 kg.11 Rather than quantity, the key challenge is the quality of the water.
It's well known that the water supplied to the electrolyser must be of high purity. Commercial electrolyser manufacturers typically specify a minimum required water supply quality in terms of conductivity, typically <1 μS cm−1 (>1 MΩ cm),12 and a total organic carbon (TOC) content. Academic research groups typically use ultra-pure, ASTM D1193-06 Type I water (resistivity 18.2 MΩ cm @ 25 °C, TOC < 50 μg L−1) supplied from a lab system, but the use of ion exchange resins or procedures to maintain resistivity in recirculating systems varies.13 The latest EU low temperature water electrolyser testing guidelines from the JRC14 call for ISO 3696 Grade 2 water, which has a conductivity of <1.0 μS cm−1 at the inlet and for PEM electrolysis also require the anode water recirculation loop to have ion exchange resins and a conductivity meter in the loop.
Deionisation can be a large burden on the cost of producing green hydrogen. In a recent analysis, deionised water purification comprised 22% of the total balance of plant (BoP) cost of a 1 MW PEMWE system and was relatively insensitive to both system size and annual production rate.15 Deionisation also incurs operational costs from energy use, water supply and maintenance. It is difficult to find reliable numbers for energy use associated with the production of high purity water but it is likely on the order of 5 kW h m−3,16,17 or 0.2 kW for 5 kg of H2. The energy demand for deionisation therefore likely represents less than 0.2% of the energy stored in the product hydrogen. As with any plant, the capacity influences the cost. For a 200 m3 per day throughput reverse osmosis (RO) plant, a capital expenditure (CAPEX) of $0.5 M and operating expenditure (OPEX) of ∼$3 m−3 has been estimated.18 The cost shifts to $2 M CAPEX and $2 m−3 OPEX when the capacity is increased to 1200 m3 per day. It should be noted that reports in the literature often assume the feed water of RO plants to either be seawater or brackish water. When the inlet of the RO plant is much cleaner, i.e., freshwater, the CAPEX and OPEX may be lower than these assumptions.
More subtly, the need for high purity water limits electrolyser design flexibility. For instance, in PEMWE systems, high purity feed water is typically deionised further using ion exchange resins as it recirculates through the system, maintaining purity during operation. The temperature sensitivity of these ion exchange membranes effectively limits the recirculation loop temperature and intolerance to ions requires use of particular materials in the stack and BoP. The conservatism in design caused by these issues increases the cost of green hydrogen.
There are a number of different mechanisms by which water impurities may conceivably impact the normal operation, performance, or lifetime of electrolysers. Many of these have not been directly observed experimentally in operating electrolysers, though there is evidence of their impact in analogous fuel cell systems or in ex situ experiments on relevant materials. Hydrogen Europe's Strategic Research and Innovation Agenda has set a 2030 target of 0.1–0.12% degradation per 1000 h of electrolyser operation.19 Therefore, even slow, and seemingly insignificant degradation processes can have a large impact on the viability of an electrolyser.
1.2 Structure and operation of low temperature electrolysers
There are three main categories of low temperature electrolyser, which are distinguished mainly by the type of electrolyte they use. The principles of operation of a PEMWE, AEMWE, and AWE are shown schematically in Fig. 1. | ||
| Fig. 1 Principles of operation of different low temperature water electrolyser technologies. | ||
PEMWEs are a semi-mature technology.6,7 They are typically operated with inlet water temperatures of around 60 °C and typical current densities of ∼2–4 A cm−2 resulting in acceptable cell voltages of 1.8 V and lifetimes of 6–9 years.22 Nonetheless, there is intensive research underway to increase lifetime, improve performance and decrease cost and these numbers are expected to improve. Operationally PEMWEs have a small volumetric footprint, are efficient, produce high purity hydrogen, can directly produce pressurised hydrogen and can rapidly respond to load variations which makes them well suited to handle the intermittency of renewable power.23
AEMWEs are of interest as they potentially retain many of the operational benefits of PEMWEs while avoiding the use of expensive and rare critical raw materials such as iridium and platinum. The latest AEMWEs in the academic literature tend to operate around 60 °C at 2 A cm−2 and ∼1.9 V vs. RHE.27,28 Like PEMWEs, they generally have fast response times, produce pure hydrogen, have a small volumetric footprint, and directly produce pressurised hydrogen. Current drawbacks include the comparatively slow rate of hydrogen evolution reactions under alkaline conditions29 and lifetime limitations (<4 years); with membrane, ionomer and catalyst developments needed in order to meet the long lifetimes required of commercial systems.30
AWEs are very mature systems and have been used industrially for almost a century, though the recent interest in electrolysis for energy applications is driving advances, for example zero-gap configuration cells.32 Typical current densities are 1 A cm−2 at 1.8 V, which results in larger footprints than membrane-based electrolysers. A 1 GW facility using PEMWE was estimated to have a footprint of 8–13 ha compared to AWE with 10–17 ha.33 The liquid electrolyte and high permeability of the porous separator to gases hinders operation at very low currents, which makes coupling with renewables more challenging and increases the difficulty of producing directly compressed gas. However, AWE systems have a low capital cost compared to PEMWE and are very robust, with examples operating for 30–40 years.34
1.3 Structure of the review
This review summarises existing published work on the behaviour of impurities inside low temperature electrolysers, highlights possible poisoning mechanisms based on ex situ and fuel cell experiments and identifies critical areas for future research. It also summarises techniques to measure the presence of impurities in situ, potential methods to improve tolerance to water impurities and strategies to recover performance after contamination events. Throughout, this paper draws upon a thorough review of the relevant electrolyser literature and is also informed by relevant results in fields such as proton exchange membrane fuel cells (PEMFCs) where the study of the impact of common impurities is more complete.Section 2 reviews the potential sources of impurity for the different electrolyser technologies, covering both exogenous impurities in the water or electrolyte and endogenous impurities arising from the degradation of stack and ancillary components. Section 3 addresses the impact of specific contaminants on PEMWEs, and Section 4 does the same for AWEs and AEMWEs. Section 5 provides an overview of the available characterisation techniques to measure the impact of impurities during in situ or ex situ measurements, together with a summary of commonly-used water purification technologies and a discussion of possible mitigation and recovery strategies. The review concludes with a summary and an outlook for the study of the impact of impurities in electrolysers.
2 Sources of impurities
The water supply process in an electrolyser system is fairly complicated, with the BoP conditioning the water both before the inlet and in the recirculation loop, Fig. 2. Specific commercial configurations are proprietary but the scheme in Fig. 2 presents an illustrative example for the case where water is supplied to both anode and cathode. Water is sourced, usually from a local water supply, and purified in a multi-step process, details of which are often proprietary to the system manufacturer. Water is then fed to the stack where it is electrolysed to produce hydrogen and oxygen; it's usual to operate liquid flows far in excess of that required to sustain the electrochemical reaction to ensure the stack operates at high efficiency and to control the temperature. The temperature of the feed to the electrolyser depends on the specific technology but inlet temperatures of 60 °C to 80 °C are typical. As the electrolyser technologies discussed here operate exothermally, the vast majority of the thermal energy is lost by heating the water. Liquid and product gases usually exit the stack together and undergo some separation process with the liquid being recirculated and the gases exiting the system. To achieve very high purity hydrogen gas (with <5 ppm of water35) there are further drying processes which are not considered here.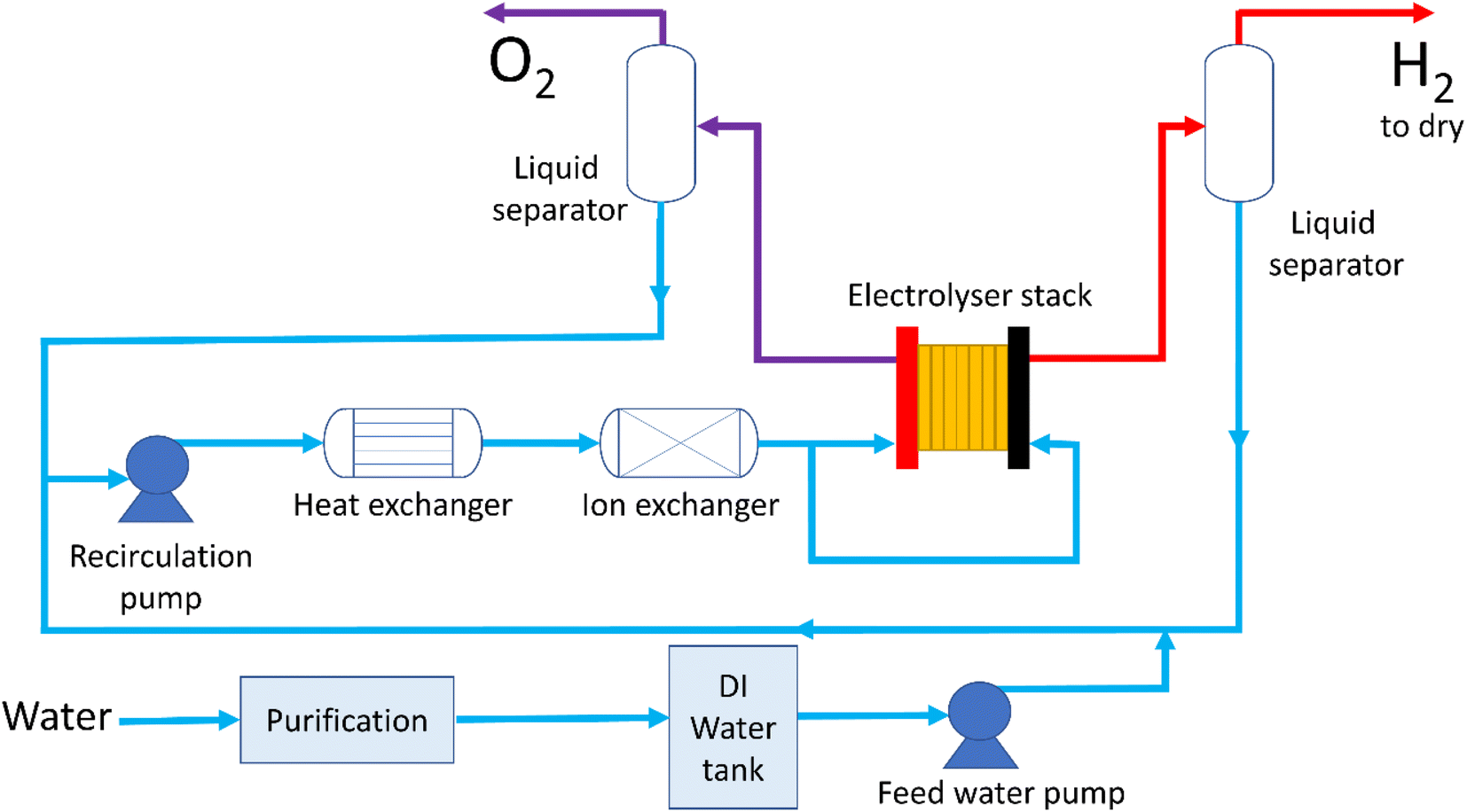 | ||
| Fig. 2 Schematic of typical water process flow for a PEMWE stack with water flowing to both anode and cathode. | ||
The recirculated water (or in the case of alkaline systems aqueous potassium hydroxide) must be cooled before supply back into the cell, though again the specific processes are usually proprietary. For PEMWE systems, ion exchange resins are used to remove contaminating anions and cations from the water supply and conductivity probes are commonly used to check for the build-up of ionic impurities. It is not possible to use ion exchange resins with alkaline electrolysers, due to the high concentration of potassium hydroxide. Behaviour of water in the stack is fairly intricate. In PEMWE systems it's common to feed only to the anode where the water is consumed by the electrochemical reaction so that the only water in the cathode outlet is that which crosses through the membrane owing to diffusion and electro-osmotic drag. The same mechanisms operate in alkaline cells with the added complexity that water is consumed on the cathode and produced at the anode stoichiometrically. In alkaline cells using a supporting electrolyte there is also the need to rebalance the concentration of the electrolyte, which will vary over time.36
Based on the process described above, it's possible to identify two different sources of impurities: exogenous and endogenous. We define exogenous impurities as those introduced by the feed water while endogenous impurities arise from degradation processes within the electrolyser and BoP itself. The following sections will discuss in detail the possible sources of impurities but with limited quantitative information available in the public domain from real electrolyser operation there remains much uncertainty.
2.1 Exogenous impurities
Water is an excellent solvent for cations and anions. Electrolysers typically require water that has been purified to a high resistivity, i.e., featuring a low concentration of charge-carrying ionic species. However, in reality, impurities may still enter an electrolyser. Table 1 compares three types of water and the concentration of ionic impurities: seawater, tap water, and ASTM Type II water (1 MΩ cm). Tap water and seawater are widely used feedstocks for water electrolysis, which need to be purified to the required specifications of ASTM Type II water before use.Compliant ASTM Type II water may still contain 5 μg L−1 Na+ and 5 μg L−1 Cl−, with the presence of other ions resulting in conductivity up to 1.0 μS cm−1. Crudely then, a 1 MW PEMWE stack operating at full capacity for 10 years may therefore be exposed to over 40 g of Na+, enough to ion exchange approximately one third of the membrane in such an electrolyser.† It is also important to note that, if not removed, impurities will become enriched; while water is consumed stoichiometrically in the electrolyser, the impurities it carries may not be. This process leads to a gradual increase in concentration of impurities in the cells and water recirculation loop, potentially by many orders of magnitude. Together, these issues justify the use of ion exchange resins in the recirculation loop of PEMWEs and clearly illustrate the possible implications of even a seemingly insignificant concentration of impurities. As mentioned previously, in-line ion exchange resins are not suitable for AWEs or AEMWEs operating with supporting electrolyte as the resins would quickly become saturated with the cations and anions from the electrolyte.
Beyond ions, other impurities may enter the electrolyser via the water supply. Organic impurities do not necessarily contribute to conductivity and the ASTM Type II water standard permits the presence of 50 μg L−1 TOC. Biological components are also present in many water sources, and care must be taken to periodically clean pure water reservoirs to prevent their growth. Dissolved gases such as nitrogen, oxygen, argon, carbon dioxide, etc. will also be present in concentrations from nmol L−1 to μmol L−1.39 While chemically inert gases are not likely to impact electrolyser performance, they may affect hydrogen quality, which is often required to meet the stringent quality standards.40 Oxygen and carbon dioxide, on the other hand, are not wholly inert, as discussed below.
While most impurities may be removed during purification, accidental water contamination due to equipment or maintenance failures must also be considered. A 2016 presentation by Proton Onsite (now NEL Hydrogen) showed that in the period 2006–2015, 65% of stack defects were caused by inadvertent customer contamination41 while errors in system design and maintenance may also lead to the introduction of impurities. Due to the cost associated with purification, and in many cases local scarcity of groundwater or potable water, there is growing academic interest in developing electrolysers capable of operating on lower quality water, or even on humidity from ambient air.42 Development towards cells able to run on seawater was the subject of a recent review.37 Generating hydrogen by the electrolysis of small organic compounds or their aqueous solutions is also of academic interest and has been the subject of recent reviews.43,44
2.2 Endogenous impurities
We classify any impurities arising from inside of the electrolyser system as endogenous. Operation of electrolysers for decades will lead to degradation of both the stack and BoP components, generating impurities in situ. Leaching from components45 and contamination from manufacturing, commissioning and maintenance may also result in impurities being introduced by the electrolyser itself.Much of the BoP, including tubes, tanks, gaskets, pumps, heat exchangers, sensors, filters, etc. are necessarily in intimate contact with the water supply to and from the electrolyser stack, which is both heated and saturated with either oxygen or hydrogen. Any corrosion products or leachants from BoP components are therefore able to enter the water supply. As there is little information available on the composition of water in operating commercial electrolysers it is difficult to quantify the likelihood and concentration of these impurities but there is literature evidence to suggest their presence at ppm concentrations.45 Twenty different components commonly used as sealants in PEMFCs were leach tested in 90 °C DI water and showed tens to hundreds ppm of leachant.45 There are reports of metals, even high-grade stainless steels, undergoing corrosion processes in ultrapure water with the expectation that ions are released,46 though we stress there is a paucity of good quality data under conditions directly applicable to electrolysers. Such corrosion processes may be accelerated when ions are present in the electrolyte, either due to the build-up of impurities from other sources or because of the potassium hydroxide in AWEs or supporting electrolyte in AEMWEs. An example of contamination in PEMFCs attributed to ions leaching from tubing was reported by Sun et al. The performance of a 9 cell stack operated for 7800 h was recovered almost completely by immersing the catalyst coated membrane (CCM) in 0.5 M sulphuric and subsequent washing in DI water47 with post mortem analysis of the CCM revealing copper, calcium, and iron within the membrane and the catalyst layer. Grigoriev et al. conducted a 5500 h PEMWE experiment consisting of on/off current cycles to simulate intermittent renewable energy operation, albeit with platinum as the anode.48 Unsurprisingly, the platinum was found to corrode and deposit within the membrane, with membrane thinning reported, but traces of silicon, potassium, and calcium contamination within the membrane were also reported. There were no traces of iron, nickel, or copper; therefore, it was concluded that impurities arose from degradation of water quality, with silicon suspected to leach out from a polypropylene tank into the water.
With the potential of the anode electrode routinely exceeding potentials of 1.5 V vs. RHE,49 and with parts of the cell either very highly acidic or basic, materials for components inside the stack must be carefully selected to avoid corrosion and strike a balance between cost, performance and lifetime. The typical route taken has been to choose materials with very high stability such as platinum-coated titanium for PTLs and BPPs in PEMWEs and nickel for components in AWEs. In AWEs, there is an absence of literature for nickel or iron-based bipolar plate degradation, and studies of these materials are often performed under the scope of catalysis research. PEMWE components, on the other hand, have been shown to introduce impurities even when expected to be stable.
When an uncoated titanium PTL was used at the anode for 1000 h, titanium corrosion was reported with titanium oxides found deposited at the cathode.50,51 This phenomenon took place during both constant current and current cycling operation. Similar titanium contamination was also reported when a titanium alloy with 1 wt% of iron was used as the anode PTL.52 After 4000 h of operation, up to 7 wt% titanium and 3.5 wt% of iron was detected in the cathode catalyst layer, sourced from migrated corroded titanium. The authors suggested that this must primarily stem from the cathode titanium PTL, where a sufficiently protective titanium dioxide layer does not form due to the very acidic and reductive environment. A half-cell experiment also showed that both titanium and iridium dissolved into the electrolyte solution when the operation mode was switched from OCV to 1 or 2 mA cm−2.53 Despite the relatively low current density and operation in a specially designed cell, this demonstrates that dissolution can occur during transition from open circuit to another current. Siracusano et al. also performed a 3500–5700 h durability test at 1 A cm−2 and 55 °C for a single cell PEMWE using CCMs manufactured via different hot-pressing protocols.54 They reported titanium and iron impurities present in the anode catalyst layer, while no impurities were detected at the cathode. The authors proposed that iron likely comes from stainless steel tubing degradation, while titanium was suspected to be corroded due to hydrogen fluoride formation. The hydrogen peroxide generated from oxygen crossover at the cathode may also trigger titanium dissolution, although the exact mechanism is not clear.55 There is a pressure to move from very stable materials such as titanium to less expensive materials. Though there is some scope to relax material requirements, for instance by recognising that not all parts of a PEMWE anode experience very high potentials,56 this results in an increased risk of components releasing ions and impacting the cell performance. Taken to the extreme, an uncoated stainless steel component was used as the PTL at the anode of a PEMWE57,58 and, unsurprisingly, extensive corrosion was observed within 15 h, and iron and nickel were detected at the anode, membrane and cathode.
Catalysts for electrochemical processes are chosen based on their stability, performance, and cost. With PEMWE catalysts for OER regularly showing an intrinsically inverse relationship between activity and stability,59 there is necessarily a trade-off. For instance, ruthenium oxides are well known to be excellent catalysts for OER in acid but iridium oxides (or a mixed oxide based on iridium and ruthenium) are more commonly chosen for their stability.59,60 Even iridium oxides are not completely stable, undergoing electrochemical dissolution at operational potentials,61 with possible heightened dissolution rates during transient operation as the oxidation state may change.62–65 PEMWE cathode catalysts are also susceptible to corrosion, as the cathode can be exposed to oxidising potentials >0.9 V vs. RHE during shutdown periods.49 Platinum black catalyst, for instance, was reported to lose 152 ng cm−2 of a 3 mg cm−2 loading over a 90 h OCV period.66 While negligible, corresponding to only 0.005% of total cathode loading, long periods of operation can easily accumulate these ions in the cathode water. Catalyst supports are less used within electrolyser research than in fuel cells but their increased development and use offers both another vector for impurities to be introduced and also a potential mitigation, with supported fuel cell catalysts routinely showing reduced sensitivity to impurities.67
While iridium (and/or ruthenium) dissolved during operation can redeposit at the anode, the dissolved species have a driving force to cross the membrane where they may be reduced by hydrogen. Deposition on the cathode catalyst may result in performance losses, and deposition inside the membrane as small particles can weaken the structural integrity of the membrane.68Post mortem investigation via scanning transmission electron microscopy X-ray energy dispersive spectroscopy (STEM-XEDS) of a low loading MEA (anode: 0.08 mgIr cm−2) tested for 4500 h @ 1.8 A cm−2 showed significant crossover of iridium as well as its distribution across the entire CCM, Fig. 3. In addition to the iridium crossover, dissolved platinum, thought to occur due to dissolution of smaller nanoparticles during start up and shutdown procedures, was demonstrated to redeposit in the membrane resulting in similar effects to iridium redeposition but to a lesser extent than the anode catalyst.
 | ||
| Fig. 3 Distribution of iridium and platinum across CCM after testing a low loading MEA (anode: 0.08 mgIr cm−2) for 4500 h @ 1.8 A cm−2. This figure has been adapted from ref. 68. | ||
The high pH present in AWEs and AEMWEs means that non-precious metals such as nickel are more stable to dissolution, as shown from their Pourbaix diagrams69 and therefore cobalt, nickel, and iron based catalysts are frequently used at both the anode and cathode and exhibit high stability.70,71 In contemporary commercial AWE systems, nickel-based catalysts are the standard25,31,72 with systems exhibiting stability for up to 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h.73 This degree of stability is not yet observed in AEMWE and there is as yet no standardised catalyst for this technology.74 One study of an AWE cell with a 316L stainless steel anode and a RANEY®–Ni cathode exhibited a negligible change in iron and nickel concentrations in the electrolyte after operation for 30 days at 0.3 A cm−2 in 6 M KOH at 75 °C.75 Another study using a specially devised ultraviolet-visible spectro-electrochemical cell reported that at potassium hydroxide concentrations of 0.1 M Ni(OH)2/NiOOH dissolves into the electrolyte at potentials of 1.2 V, 1.4 V, and 1.52 V vs. RHE. While the exact ionic species is uncertain, the absorption bands suggest that Ni2+ ions are the predominant species.76 PGM based catalysts have also been explored for AWEs and AEMWEs,77–79 although most research is limited to activity studies in 3-electrode setups.80–82
000 h.73 This degree of stability is not yet observed in AEMWE and there is as yet no standardised catalyst for this technology.74 One study of an AWE cell with a 316L stainless steel anode and a RANEY®–Ni cathode exhibited a negligible change in iron and nickel concentrations in the electrolyte after operation for 30 days at 0.3 A cm−2 in 6 M KOH at 75 °C.75 Another study using a specially devised ultraviolet-visible spectro-electrochemical cell reported that at potassium hydroxide concentrations of 0.1 M Ni(OH)2/NiOOH dissolves into the electrolyte at potentials of 1.2 V, 1.4 V, and 1.52 V vs. RHE. While the exact ionic species is uncertain, the absorption bands suggest that Ni2+ ions are the predominant species.76 PGM based catalysts have also been explored for AWEs and AEMWEs,77–79 although most research is limited to activity studies in 3-electrode setups.80–82
Despite improved PFSA membrane chemistry, membrane degradation as a result of hydroxy radical attack initiated by the Fenton reaction can occur and may release anions into the water.28 Degradation of the PFSA polymer releases SO42− from the end group and F− from the polymer main group and side chain as a by-product.83 Anion exchange membranes and ionomers are also subject to rapid degradation and may form various by-products.26,84 Indeed, the phenyl groups often found in hydrocarbon-based ionomers are known to adsorb strongly on the surface of AEMWE catalysts.30 In PEMFCs, additives such as ceria or manganese oxides that are sometimes added to membranes and catalyst layers to act as radical scavengers have been shown to be redeposited throughout fuel cell electrodes85,86 and to migrate into the exhaust water;87 similar behaviour may be expected with these materials in electrolysers.
Impurities may also originate from residual chemicals associated with the manufacture of components within the electrolyser system. In AWEs, potassium hydroxide is added to the water initially (note it is not consumed in the reaction so the system only requires a pure water supply) and bulk potassium hydroxide is routinely contaminated with ppm levels of impurities such as Cl−, PO43−, SO42− and a range of metals including aluminium, arsenic, calcium, copper, iron, sodium, lead, zinc, and silver ions.88 Residual solvents or salts can also be present from the manufacturing of catalysts and membranes. For instance, chloride-based precursors of platinum and iridium oxide nanoparticles can leave chlorine residues on the catalyst,89,90 while residual alcohol solvents such as methanol, ethanol, or propanol may result from the use of inks and slurries. Membranes also contain some residual impurities from the production process, which is why many research groups pre-clean purchased CCMs by immersing them in hot hydrogen peroxide and sulphuric acid before use. The break-in period for PEMFCs is frequently attributed to the in situ cleaning of catalyst layers and membranes91–93 and such break-in periods are also observed in PEMWEs.27
Finally, by-products from the electrolysis process and recombination reactions between hydrogen and oxygen may form highly reactive radical and peroxide species stoichiometrically.84,94 Though they have a low concentration and short lifetime they may eventually result in damage to components.
3 Impact of impurities in PEMWEs
3.1 Introduction
This section addresses the impact of endogenous and exogenous impurities at both the anode and cathode of PEMWEs. Impurities are classified into three main types: cationic, anionic, and organic (inert compounds are only briefly considered) with plausible mechanisms of operation and impact assessed as summarised in Fig. 4. For many classes of impurity there is limited quantitative information available for specific impurities, so here the discussion remains largely qualitative.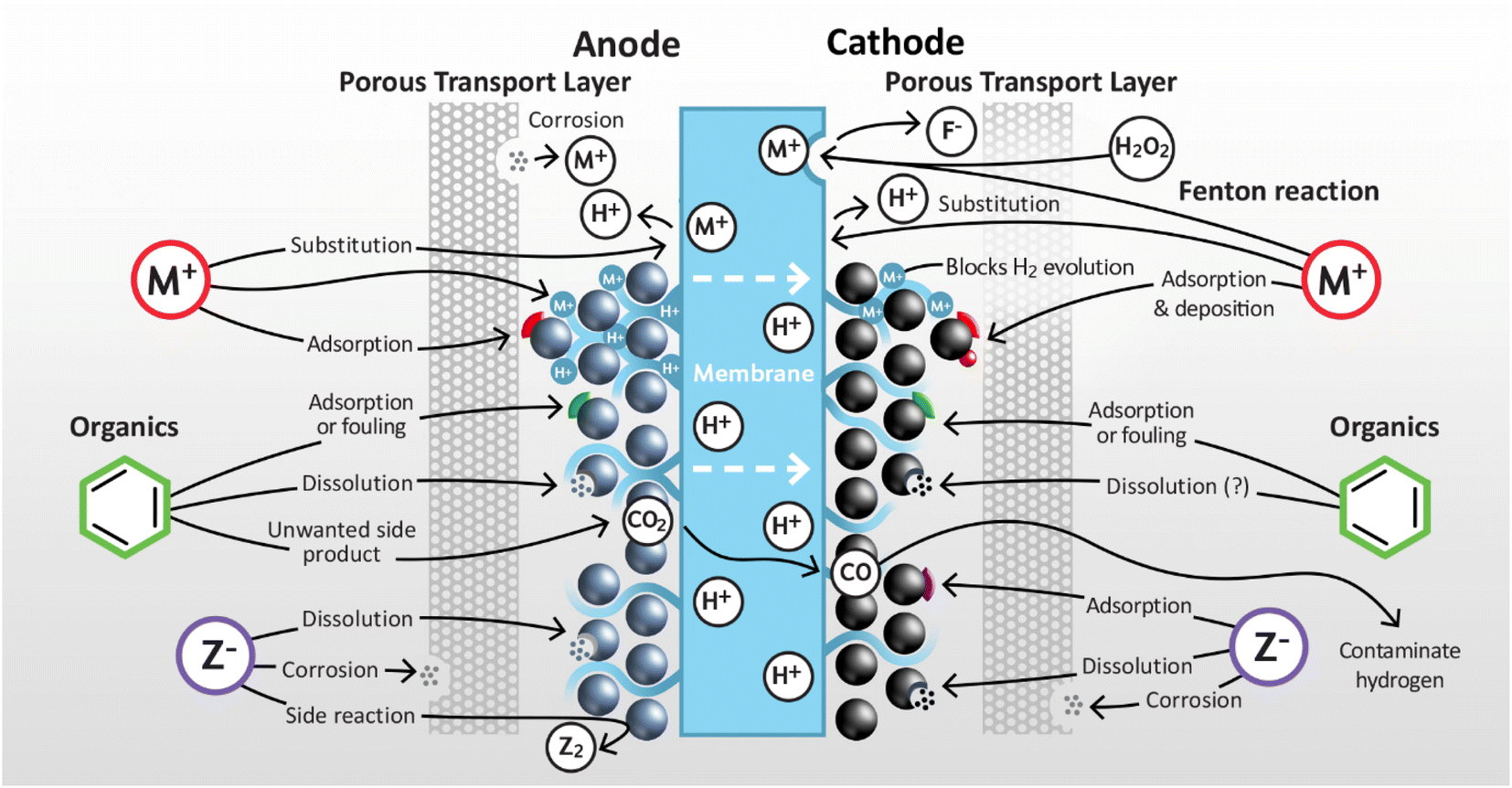 | ||
| Fig. 4 Schematic illustration of the impact of various types of impurities on PEMWEs. | ||
3.2 Cationic impurities
For PEMWEs, cations are by far the most problematic class of impurity owing to their ability to impact the catalyst, ionomer and membrane, and the number of metallic components in the system that present endogenous sources. Cationic impurities can conceivably degrade performance and lifetime via several mechanisms: substitution of available protons in the membrane and ionomer, adsorption and deposition, and the initiation of unfavourable reactions.3.2.1.1 Membrane. Cations degrade PEMWE performance primarily by lowering the membrane conductivity.95–104 PFSA-based membranes such as Nafion possesses a very high conductivity in the presence of water due to the hyper acidity of the SO3− end group (estimated pKa of ca. −6) that results in the formation of mobile H+ ions/protons.105 However, most cations have a higher affinity than protons for the SO3− end group,106 leading to proton displacement and a consequent reduction in the proton conductivity of the membrane. PFSA membranes in liquid water generally have a reported ionic conductivity in the range of 0.05–0.2 S cm−1,105 yet this value can decrease by 1–2 orders of magnitude when all of the protons are exchanged to metal cations.107
Not all impurity ions have the same impact; the situation is worse for higher valence ions which are preferentially absorbed, leading to higher concentrations in the membrane even at very low aqueous mole fractions (see Fig. 5).104,108–110 For example, Fe3+ has a Keq ∼5000 times higher than Na+. In some reports, at low concentrations (<10 ppm), absorption of Ni2+ and Cu2+ in the membrane was much higher than that of Fe3+; however, this was likely due to a relatively short membrane equilibrium time of 72 h.111 There is a threshold concentration of ions before a step change in conductivity can be observed. Ions such as Na+, Cu2+, Ni2+, and Fe3+ were only observed to yield a significant decrease of conductivity at concentrations >10 ppm in water. The effect of these impurities could be mitigated slightly when present at low pH. Upon contamination in 1 M H2SO4, cationic impurities such as Fe3+ or Cr2+ do not impose conductivity loss at concentrations below 200–300 ppm.112 On the other hand, contamination in distilled water results in conductivity loss at concentrations as low as 10 ppm.110
 | ||
| Fig. 5 Impact of immersion in cation solutions in distilled water for 48 h on PFSA conductivity. This figure has been adapted from ref. 110. | ||
Non-metallic cations, such as ammonium (NH4+), can also substitute protons within the PFSA membrane.113–115 As with metallic cations, the membrane conductivity in PEMFCs has been reported to decrease with an increased concentration of NH4+ absorbed by the membrane in both liquid phase113,114 and gas phase.114,115 As PEMWE membranes are typically fully equilibrated with liquid water, the effect of ammonium contamination may be less severe than that seen in PEMFCs and where the loss in conductivity was found to be lower at higher relative humidity. Note also, that in PEMFC, catalyst poisoning may dominate performance losses introduced by ammonia.116
Apart from conductivity loss, cationic impurities can also compromise membrane water transport properties and overall membrane stability. Na+ and Ca2+ adsorption has been reported to change the water balance by increasing the water transfer coefficient (tH2O) and decreasing water content.95,96 The tH2O for Nafion 115 increased from 2.5 to more than 10 as all the protons in the membrane were exchanged with Ca2+.117 Compromised water drag is problematic for gaseous applications such as fuel cells though the situation is less clear for electrolysers. Thermal stability is also affected by cations, with the onset temperature for thermal degradation of the SO3− group higher when Nafion® NR 211 membrane was contaminated with Ni2+, beginning at a Ni2+/H+ ratio of 0.2.118
3.2.1.2 Ionomer. PEMWE catalyst layers routinely contain an ion conducting phase, referred to here as ionomer, to increase the electrochemically active surface area119 – the area where reactant (water), ion conducting phase (protons) and electrically conducting phase (catalyst surface) are in intimate contact. PEMWE ionomer is typically also a PFSA and therefore suffers from the same issues as the membrane when exposed to cations. While loss of protons in the ionomer may increase the observed resistance of the cell, the catalyst layers are thin (typically <25 μm (ref. 120)) with ionomer layers on the catalyst surface many orders of magnitude thinner than this. The effective loss of electrochemically active surface area that results as proton transport is deactivated is therefore generally a larger issue. This is confirmed by studies that have introduced metallic impurities directly into the water feed of operating PEMWEs. In one case, the impact on a PEMWE cell of incorporating 0.05 M Na2SO4 into the anode and cathode water feeds was studied with an in situ reference electrode.121 Following contamination, the pH of the anode water decreased from pH 6 to pH 3 as the protons in the anode ionomer and membrane were substituted and formed sulphuric acid in the water. Intriguingly, the pH of the cathode water increased from pH 6 to pH 11 after 3 h of operation. The increase of pH at the cathode was attributed to Na+ migrating though the membrane and substituting for protons in the cathode catalyst layer, eventually resulting in a transition from the acidic HER to the alkaline HER, which releases OH−. Interestingly, the effect depends on which water supply is contaminated. When the anode supply was contaminated the cathode potential decreased from 0 V to −0.5 V vs. RHE, while a much smaller decrease was observed when the cathode water was contaminated (0 V to −0.1 V vs. RHE). It's unclear whether the presence of the impurity impacted the operation of the in situ reference electrode, which can be highly sensitive to their local environment.
Electromigration of cations causes the cathode ionomer to be more severely impacted than the anode ionomer, particularly when operating at high current densities.122–126Fig. 6 shows the effect when the anode of a PEMWE cell was poisoned with 0.001 M Na2SO4 at 0.5 A cm−2; the anode overpotential plateaued then decreased as the cathode pH spiked to 11.122 A similar potential peaking trend was found in a study injecting 0.005 M Fe2(SO4)3 with 0.01 M H2SO4 as the anode water feed of a PEMWE cell operated at 0.5 A cm−2.124 The phenomenon can be explained by cation electromigration to the cathode alleviating poisoning at the anode, while exacerbating it at the cathode. Post mortem Electron Probe Microanalysis (EPMA) showed cation accumulation in the cathode when the anode supply was poisoned by sodium105 and iron.124 Electrochemical Impedance Spectroscopy (EIS) also showed an increase in charge transfer resistance (Rct) rather than ohmic resistance (RΩ), indicating that the degradation mechanism is associated with loss of electrochemically active surface area. Damage to performance was also observed even at ppm levels of cationic impurities in water.125,126 A ∼0.2 V increase in cell voltage at 2 A cm−2 after 4 h was reported even with iron concentrations as low as 1 ppm, increasing to ∼0.9 V at 2 A cm−2 at 10 ppm,125 with EIS indicating that an increase in kinetic overpotential seemed to be the major reason for performance degradation.126
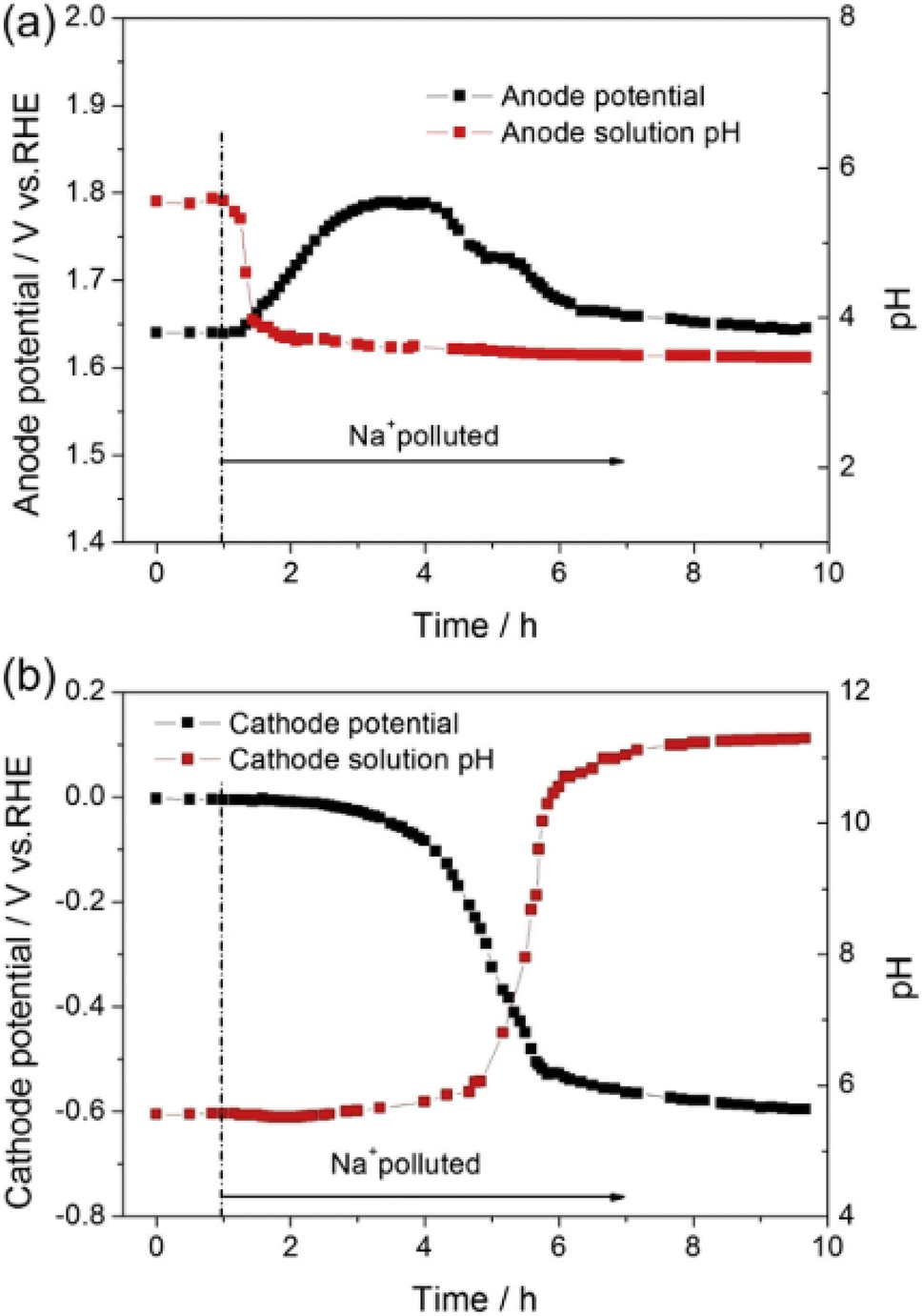 | ||
| Fig. 6 (a) Anode potential vs. time and anode water pH vs. time; (b) cathode potential vs. time and cathode water pH vs. time for a PEMWE cell poisoned with 1 mM Na+ at 60 °C, 500 mA cm−2 with flow rate of 1 ml min−1. This figure has been reproduced from ref. 122 with permission from Elsevier, Copyright 2013. | ||
While most studies point to accumulation of cations in the cathode catalyst layer as the primary degradation mechanism, the governing mechanism is largely controlled by the rate of ion migration (a function of current density) balanced by ion back-diffusion.123,127 A study of trace ion impurities in the cathode effluent of a PEM electrolyser for hydrogen peroxide production showed that the concentrations of titanium and iridium cations generated by corrosion processes in situ were dependent upon the operating current density.128 Gd3+ is a model trivalent cation used in neutron imaging studies due to its very high neutron cross-section, enabling good contrast resolution.123,127 Upon passage of electrical current at 1 A cm−2 in a single cell PEMWE poisoned with Gd3+, the ions accumulated in the cathode catalyst layer within ∼16 s. Once the current was switched off, accumulated Gd3+ back-diffused to the membrane within 200 s. This behaviour is identical to that reported for Fe3+,123 and is therefore likely to be expected for most cation poisoning. In case of Fe3+, voltage breakdowns at different current densities show that an increased RΩ dominates at <0.2 A cm−2 but that increased mass transport and kinetic overpotentials dominate at >1 A cm−2.123,127 At low current densities, the rate of ion migration can be countered by Fickian back-diffusion arising from the concentration gradient between cathode and anode. This prevents excessive accumulation in the cathode and thus RΩ degradation dominates at low current density. As the current density increases, back-diffusion is overpowered by electromigration, causing mass transport and kinetic overpotential to dominate degradation at high current density. Metallic cations are also much less mobile in the membrane meaning that they are not easily purged once present.
There are some reports where cations have been observed to deposit on the cathode during operation. A half-cell study of HER on platinum in the presence of 10−6 M CuSO4 exhibited visible copper deposition.129 The presence of CuSO4 led to an increase to the steady state overpotential of 300 mV, which was reached more rapidly at higher concentrations (<3 h at 5 × 10−4 M vs. >20 h at 10−6 M). Another study investigated the impact of 0.01 M NiSO4 in the feed water of a PEMWE cell operating with platinum on both cathode and anode.131 A decline in performance was observed when the cell voltage was held at 2.35 V (0.3 A cm−2 to ca. 0.15 A cm−2) attributed to membrane conductivity decrease. Bulk nickel deposition was also suspected to take place; however, this is unlikely to occur in state-of-the-art PEMWEs as the cathode potential rarely drops below −100 mV vs. RHE.
PGM ions have also been shown to deposit within the membrane, creating local hotspots that promote membrane degradation. Cycling the catalyst potential up to 1.0 V and 1.2 V vs. RHE during operation in PEMFCs results in platinum dissolution into the membrane, where it is subsequently chemically reduced by crossover hydrogen, forming characteristic metallic bands.132 Membrane thickness also affects deposition rate, as thinner membranes promote hydrogen crossover and exhibit more platinum bands within the membrane post-potential cycling.132 A PEMWE with 0.08 mg cm−2 iridium oxide anode and 0.3 mg cm−2 Pt/C operated for 4000 h at 1.8 A cm−2 showed iridium and platinum migration to the opposite electrodes,68 as shown in Fig. 7. The majority of iridium (46% of total iridium) migrated and was deposited in the cathode, while 28% remained inside the membrane. Platinum migration was less severe, with only 18% of total catalyst deposited inside the membrane. Significant loss of membrane mechanical integrity was also reported after the test, but the correlation with the deposited platinum/iridium remains unclear.
 | ||
| Fig. 7 Migration of Ir into the membrane (a, c) STEM images, (b, d) elemental mapping using STEM-EDS of non-degraded and degraded membranes (e and f) are high magnification images of Ir band while panel (g) shows elemental mapping of Ir for (f). This figure has been reproduced from ref. 68 with permission from Elsevier, Copyright 2020. | ||
Platinum deposition inside the membrane can have short-term beneficial side effects, as it can mitigate gas crossover, increasing hydrogen quality and ensuring that the flammability limit is not exceeded.133,134 Employing a thin platinum layer at 0.02 mg cm−2 loading between two Nafion 115 and 212 membranes was reported to reduce the concentration of hydrogen in oxygen from 0.2 vol% to <0.005 vol% at 1 A cm−2.133 Even when the cathode was operated at 30 bara, 0.06 mg cm−2 platinum loading in a thin Nafion 212 membrane could suppress hydrogen in oxygen from 3 vol% to <1 vol%.134 In all these studies, the reported downside is the notable increase of RΩ due to the imperfect interface between the recombination catalyst and the membrane.133,134 However, the long-term adverse effects remain unknown as all reported tests in the literature were performed over short timescales.
3.3 Anionic impurities
Unlike cations, anions cannot replace protons within the membrane and ionomer, and other mechanisms therefore operate. The most commonly highlighted problems are the initiation of side reactions, such as the chlorine evolution reaction that may lower hydrogen quality, and processes that accelerate the corrosion of metallic components. Literature reports predominantly feature investigations of chloride anions, and these therefore dominate the examples in this section, although it's likely that other anions have similar mechanisms of operation.| 2Cl− → Cl2 + 2e−, E° vs. SHE = 1.358 V | (1) |
Other halide anions, such as F−, Br−, and I−, can also be oxidised to their gaseous form with analogous evolution reactions:
| 2Br− → Br2(aq) + 2e−, E° vs. SHE = 1.087 V | (2) |
| 2F− → F2 + 2e−, E° vs. SHE = 2.866 V | (3) |
| 2I− → I2 + 2e−, E° vs. SHE = 0.535 V | (4) |
OER is more thermodynamically favourable than CER under acidic conditions relevant to PEMWEs142 as shown in Fig. 8, however during typical operation the PEMWE anode potential is higher than the CER onset potential. The CER is also more kinetically facile than the OER as the latter requires 4 electrons and is thought to have 3 intermediates, while CER only requires 2 electrons and has a single intermediate.143,144 As such the ratio of exchange current density for CER/OER on most PEMWE anode materials ranges from 103 to 107, and the evolution of chlorine is therefore much more favoured kinetically. For mixed oxide catalysts such as ruthenium dioxide, the selectivity for CER at E = 1.25 V (vs. Ag/AgCl) is >0.93 for compositions of ruthenium dioxide >20 wt%.145 In fact, virtually any catalyst that is active for OER is also highly active for CER.146 There is a growing trend of developing oxygen selective catalysts for use in saline conditions, but most of the work is conducted at alkaline pH.37
 | ||
| Fig. 8 Pourbaix diagram of the water-chloride electrolyte system. This figure has been adapted from ref. 148. | ||
A recent study of an iridium oxide catalyst on a rotating ring-disk electrode established that the OER and the CER reactions take place at different sites without competition;147 prior to this study, the conventional wisdom was that the two reactions occupy the same sites. The oxygen production rate remained constant even when the Cl− concentration was varied between 0–100 mM. The chlorine production rate increased linearly with Cl− concentration, with the CER selectivity approaching 100% at Cl− concentrations >50 mM, Fig. 9.
 | ||
| Fig. 9 Plot of OER and CER reaction rates as a function of [Cl−]. This figure has been adapted from ref. 147. | ||
Overall, the strength of halide ion adsorption follows the order I− > Br− > Cl−.160 The platinum cathode of a PEMWE typically encounters a negative potential, in the range of −0.1–0 V vs. RHE.49 Hence halide ions pose a risk by adsorbing on the catalyst surface and in the case of I− and Br− this may be irreversible at sufficiently high concentrations. HER on platinum in acid is, however, still a very fast reaction and so it stands to reason that minor anion contamination will not result in significant loss in performance. As a comparison, the hydrogen oxidation reaction (HOR) in PEMFCs only shows a major loss in performance when the catalyst is covered by 0.4–0.6 monolayer of CO.161,162
Adsorption of chloride on the platinum on carbon cathode catalyst of PEMFCs has also been reported to enhance hydrogen peroxide production,150,163 which as mentioned above is a necessary precursor in the Fenton reaction that attacks the PFSA backbone. Adsorption of Cl− accelerates the cleavage of the O–O bond, an important step in the hydrogen peroxide reduction reaction. Consequently, more hydrogen peroxide is released into the catalyst layer/membrane interface, promoting membrane thinning. In PEMWE cathodes, the low potential will likely hinder any hydrogen peroxide formation from taking place, though further study is needed to confirm this assumption.
Sulphate (SO42−) and bisulphate (HSO4−) anions are other by-products of membrane degradation. SO42− anions are well known to specifically adsorb onto platinum catalyst surfaces. SO42− adsorption is a function of potential and on Pt(111) peaks in the range 0.2–0.8 V vs. Ag/AgCl, with almost complete desorption at −0.2 V vs. Ag/AgCl, have been reported.164 At PEMWE cathode potentials (<0 V vs. RHE) and at the local acidic pH (from the Nafion ionomer), H+ adsorption overwhelms any SO42− adsorption.165 At the anode, SO42− at high concentrations (0.05 M H2SO4) have been shown to adsorb on Ir(111) and shift the onset of the OER to higher potentials.166 The lowest concentration of SO42− adsorption on iridium catalysts studied to date, to the best of our knowledge, is 0.05 M H2SO4.167 The concentration of SO42− from internal membrane degradation would likely fall in the ppb range, which is unlikely to pose a significant threat.
3.3.3.1 Cathode catalyst. Another degradation mechanism associated with chloride anions is their tendency to form chloroplatinic ligands that enhance platinum dissolution,168 a well-studied problem for platinum catalysts.169–177 The addition of just 2.8 μM of HCl to 1 M H2SO4 was reported to increase the dissolution rate of platinum by 2 orders of magnitude, even without any polarisation.168 Under constant applied potential, platinum dissolution in the absence of Cl− is slow; for example, potentiostatic testing of electrodes at 1.2 V vs. RHE in 0.5 M H2SO4 for 24 h did not exhibit any mass change as measured by electrochemical quartz crystal microbalance.178 However, platinum is much more prone to dissolution when the potential is cycled between low and high potential, particularly during cathodic scans.179,180 Potential cycling from 0.0–0.8 V vs. SHE was reported to induce dissolution of platinum even without any Cl− in the system, increasing substantially when the upper potential limit was 1.2 V; though we note the use of an Ag/AgCl reference electrode in the experiment, which could potentially leak trace amounts of Cl− into the electrolyte.173 Potential cycling of platinum takes place routinely in both PEMFCs and PEMWEs during start-up and shutdown.49,181 During shutdown of PEMWEs, the potential of the cathode may transition from <0.0 V vs. RHE to ca. 1.0 V vs. RHE due to crossover of oxygen from the anode. In the presence of Cl− the rate of platinum dissolution during potential cycling is higher, and this has been investigated as a function of Cl− concentration.174–176 The effect of ppm level Cl− concentration on carbon supported platinum was studied using ex situ quartz crystal microbalance. The combined effect of dissolution and adsorption reduced the platinum ECSA by 4–13% when Cl− concentration was varied between 500–2000 ppm.176 The corrosion potential of platinum was found to decrease from 1.23 V to as low as 0.82 V when 0.01 M of Cl− was present in the electrolyte.174
3.3.3.2 Anode catalyst. Unlike for platinum, there is a scarcity of studies on the impact of Cl− on the stability of iridium oxide anode catalysts. Iridium in perovskite form (Ba2BIrO6) cycled triangularly from 1.6–0 V vs. RHE showed enhanced iridium dissolution in the presence of 50 mM NaCl. The chloride was reported to exclusively affect noble metal dissolution in the perovskites, which occurred during potentiodynamic control and active OER/ClER.182 Ruthenium was also shown to have enhanced dissolution in the presence of Cl−.170 Platinum and ruthenium were cycled from 1.5–0.1 V vs. RHE in the presence of varying concentrations of Cl−,170 and it was found that in 0.1 M H+ + 0.01 M Cl−, the Ru dissolution was 11.8% after 120 cycles with dissolution increasing to almost 100% in 1 M Cl−. It should be noted that the oxide form of iridium and ruthenium is typically used at the anode, in which case the dissolution profile may be vastly different. For instance, a Dimensionally Stable Anode (DSA) that usually consists of iridium and ruthenium oxides is an industrial standard for the CER in the Chlor-Alkali Process and shows excellent stability in concentrated Cl− solutions.183
3.3.3.3 Porous transport layer/bipolar plate. Other components that can be attacked by anions are the PTL and BPP, which are typically manufactured from titanium, often with a platinum coating to minimise contact resistance. For platinum-coated components, the information presented above in the cathode section applies. For bare titanium components in oxygenated solution, an oxide layer forms at the surface of titanium and acts as a corrosion protection layer. This layer is even immune to the presence of Cl− at 3.5% concentration.184 However, anions such as F− are reported to be able to attack the TiO2 passive film, forming a soluble titanium-fluoride compound according to the following reaction:137,185
| TiO2 + 4H+(aq) + 6F−(aq) → TiF62− + 2H2O | (5) |
A threshold concentration of F− for titanium corrosion in the range 0.0005–0.02 M has been reported in 0.05 M H2SO4 and at 0.001 M F− in 1 M HClO4.185–187 Below the threshold concentration, the oxide film is partially damaged yet still protective. Above the threshold value, F− converts the compact film to a porous non-protective layer.185 Fluorine can also react with free protons to form HF, which is known to react with the surface oxide layer through the following reactions:
| Ti2O3 + 6HF → 2TiF3 + 3H2O | (6) |
| TiO2 + 4HF → TiF4 + 2H2O | (7) |
| TiO2 + 2HF → TiOF2 + H2O | (8) |
The rate of formation of HF depends on the proton concentration, i.e., the pH of the solution. In solutions of 200 ppm and 400 ppm of F−, corresponding to a pH of 4.0 and 4.3 respectively, it was found that the critical HF concentration before the corrosion current shifted was around 21 ppm.188 F− ions are a common impurity in PEMWE water as they are generated in situ when the membrane degrades; measurement of F− concentration is often used as an indicator of membrane degradation rate.138 F− has been largely overlooked as a potential corrodent in PEMWEs, and only a few studies have acknowledged its potential role in PTL and BPP degradation.54,137 However, at the typical pH of PEMWE water >1000 ppm of F− may be required exceed the critical concentration.188 When a PEMWE was operated at its normal conditions (60 °C, 2 A cm−2), <2 ppb of F− was detected at the cathode outlet.28 In order to trigger titanium corrosion, a commercial PEMWE with a correctly functioning water purification system would need to experience failure of both the ion exchange resin and the conductivity probe. Even then, it would take a very long time for the F− to accumulate to a concentration of 1000 ppm.
While chloride is typically the dominant ionic impurity in PEMWE feed water, the increase in concentration of any ions (anions or cations) can impact the water conductivity. In PEMWEs there is a distinct boundary between the highly ionically conducting catalyst layer and the very high resistance water circulating in the system. This means that the potential of electrolyser components such as the bipolar plate and large parts of a PTL are decoupled from that of the catalyst layer.56 If the conductivity of the water rises, the extent of this decoupling decreases and may lead to more rapid corrosion of electrolyser components. This highlights the possible damage that even chemically inert ions may facilitate when accumulated to high concentrations.
3.4 Organic & inert impurities
There are two main avenues by which electrolyser performance could be impeded by organic contaminants in PEMWE systems: (i) poisoning via adsorption of species on the catalyst surface decreasing electrochemical activity and (ii) enhancing catalyst dissolution.189 Non-ionic species are expected to have only a small impact on membrane and ionomer with PFSAs, well studied in analogous applications such as direct methanol fuel cells. In addition to directly poisoning the catalyst, the oxidation of organic molecules to carbon monoxide and carbon dioxide can also indirectly have a detrimental effect on the system. The ISO 14687 standard sets maximum permissible concentrations in hydrogen for automotive fuel cell use of 0.2 and 2 μmol mol−1 of carbon monoxide and carbon dioxide, respectively.40 Discussion in this section is restricted to the impact of organic impurities on the performance and stability of the catalyst layer; we are not aware of any studies on their impact on corrosion resistance of PTL and BPP components.
However, the kinetics of methanol oxidation are much less established on iridium or ruthenium oxide electrodes than for platinum containing electrodes. Typically, both ruthenium and iridium oxides require potentials >0.9 V vs. RHE to yield any measurable methanol oxidation activity.194,195 We anticipate that at typical PEMWE anode operating potentials (>1.5 V vs. RHE), there is sufficient driving force for the oxidation reaction to take place and trace impurities are unlikely to remain unoxidized. Indeed, certain types of water purification system use PEMWE-style devices to oxidise organics in wastewater.196,197 Using simulated contaminated water, Petrov et al. demonstrated that low concentrations of organic species are oxidised without significant effect on the performance of a PEMWE, utilising a Ru2IrTaOx anode catalyst and platinum cathode catalyst.196 The polarisation curve showed very little difference between pure water and pure water with 40 ppm TOC (a mixture of aromatic and non-aromatic organic alcohols and acids). In addition, the oxidation process was demonstrated to occur at high rates during OER with carbon dioxide detected using gas chromatography of the anode outlet gases. More challengingly, the formation of carbon monoxide and carbon dioxide via oxidation of organic molecules during operation introduces additional impurity into the PEMWE system. While this is likely not problematic at the anode, crossover to the cathode could prove detrimental as COads can easily form and block active sites of the platinum catalyst at cathode.198 At sufficiently low potentials (0.06 VRHE), carbon dioxide may also be reduced to COads, providing a further source of ECSA reduction.
Organic species containing certain functional groups, such as –OH and –CN, can form stable metal complexes with dissolved metal species.189 This effect can be further enhanced by chelating agents such as ethylenediaminetetraacetic acid (EDTA), which can enhance dissolution of many metals, including PGMs.199,200 The formation of these metal complexes could enhance dissolution of the catalyst by stabilising the dissolved metal species and preventing redeposition of the metal, leading to increased loss of catalyst performance. Nonetheless, the extent to which this phenomenon affects performance at both the small scale experimental (i.e., RDE) and the full system level (i.e., single cell/stack) is unknown. A recent study conducted by Kormányos et al. on fuel oxidizing PtRu/C catalysts demonstrates the effect of organic species on platinum and ruthenium dissolution rates, using online inductively coupled plasma mass spectrometry.201 In 0.1 M HClO4 the presence of different organic fuels (methanol, ethanol, and isopropyl alcohol) had no discernible effect on the onset potential for platinum or ruthenium dissolution. However, the presence of carbon monoxide did shift the dissolution potential to more negative values by 130–140 mV; we attribute this enhancement of dissolution in the presence of carbon monoxide to the roughening it can induce to precious metal catalysts,202,203 in turn exposing more under-coordinated surface sites, which are the most prone to dissolution.204 It is not clear either why this mechanism does not occur when operating on methanol as COads is a key intermediate or how this phenomenon may affect iridium oxide or platinum electrocatalysts.
Both platinum and ruthenium are found to dissolve when cycled to potentials of 1.2 V vs. RHE or higher, and this is enhanced in the presence of methanol, as shown in Fig. 10.205 We anticipate that oxide catalysts, such as iridium–ruthenium oxides or even perhaps iridium oxides, may also be subject to enhanced dissolution in the presence of methanol under PEMWE conditions.
 | ||
| Fig. 10 Results from online ICP-MS using a scanning flow cell indicating the effect of 0.05 M organic fuel contamination on catalyst dissolution. This figure has been reproduced from ref. 201 under CC-BY-NC-ND licence. | ||
3.5 Summary and outstanding research challenges
| Impurity | Source | Cell components | Membrane | Anode | Cathode |
|---|---|---|---|---|---|
| Na+, Mg2+, Ca2+ | Seawater, potable water | Lowers membrane conductivity95,96,104,106,107,131,159 | Lowers ionomer conductivity,207 catalyst poison47 | Lowers ionomer conductivity,47 decreases rate of HER57,123,126,127,129 | |
| Ti4+ | PTL corrosion | ||||
| Ni2+, Cu2+ | External contamination | ||||
| Fe2+, Fe3+ | Stainless steel tubing degradation | Lowers membrane conductivity, degrades membrane106,107,111,124,125,207,208 | |||
| Irn+ | Anode catalyst dissolution | ||||
| Cl− | Water purification failure | Enhances dissolution,170,182 catalyst poison, unwanted side product147,182,209 | |||
| F− | Water purification failure, membrane degradation | Corrodes Ti PTL and BPP54,137 | Enhances dissolution185–188 | ||
| Br−, I− | Water purification failure | Enhances dissolution,155,156 catalyst poison157–159 | |||
| SO42− | Water purification failure, membrane degradation | Catalyst poison166 | |||
| Organics | External contamination | Enhances dissolution,199,200,205 catalyst poison,198 unwanted side product194,195 | |||
| Solid particulates | External contamination | Blocks PTL pores and induces mass transport issues210 | |||
Anions are mainly sourced exogenously, except for F− and SO42−, which are products of membrane degradation. Anions are less damaging in comparison to cations as they cannot substitute protons and lower ionic conductivity. Most anions can be adsorbed on the catalyst surface, resulting in reduced activity. Of all anions, chlorides are the most widely studied as they are present at higher concentrations in seawater and tap water. Moreover, chlorides can be oxidised into chlorine at the PEMWE anode. This can lead to secondary damage to tubing and ancillary equipment, and contamination of product gases. In principle, other anions can be adsorbed on the catalyst but the low potential at the cathode (<0 V vs. RHE) and high potential at the anode (>1.5 V vs. RHE) usually prevent this issue.
Organics and inert impurities are the remaining two of the possible exogenous impurities in the PEMWE water. ‘Organic’ is a very broad classification and different functional groups can induce unique effects. Groups such as –OH and –CN may induce chelating effects and promote PGM dissolution but, most commonly, organics adsorb at the catalyst surface and block active sites. At the anode, organics such as methanol can be expected to be oxidised, but gaseous oxidation products can cross over to the cathode, lowering the hydrogen quality and poisoning the cathode catalyst. For inert impurities there is little research performed to date and the most feasible issue is solid particulates introduced by the BoP, leading to mass transport issues.
The impact of organic impurities is another topic that is barely touched upon in the literature. The primary industrial context for water-containing organics is the electrooxidation of wastewater, so the reported data are often focused more on the organic oxidation efficiency and less on cell performance. There is a clear economic benefit in using lower quality water as it would simplify the associated cost of water purification or reduce the energy required for hydrogen production via electrolysis. The underlying premise is that organics should not interfere with PEMWE performance as they would simply be oxidised. However, some concerns remain, such as the potential for crossover carbon monoxide and carbon dioxide to lower the quality of hydrogen and adsorb on the cathode catalyst. More detailed studies would help to verify these concerns and could aid the development of organic fed water electrolysis technology. Finally, the impact of solid and microbial impurities on PEMWE performance has yet to be assessed. In the event of rupture of the reverse osmosis membrane, these solid inert impurities could enter the water and disrupt the stack performance, but simple mitigation or recovery approaches may be possible.
4 Impact of impurities in AWEs and AEMWEs
4.1 Introduction
This section addresses the impact of endogenous and exogenous impurities at both the anode and cathode of AWEs and AEMWEs. As in the PEMWE section, impurities are classified into three main types: cationic, anionic, and organic, with plausible mechanisms of operation and impact assessed for both technologies simultaneously, as summarised in Fig. 11. For many classes of impurity there is limited quantitative information available, so the discussion again remains largely qualitative. | ||
| Fig. 11 Schematic summary of the impact of various types of impurity on AEMWEs (top) and AWEs (bottom). | ||
4.2 Cationic impurities
The main charge carriers in AWEs and AEMWEs are anions, particularly OH−. Therefore, cations do not pose a direct threat to the membrane, ionomer, and electrolyte as they do in PEMWEs. In fact, some of the cationic impurities that can commonly be found in water and potassium hydroxide electrolyte actually enhance the performance of some OER catalysts. The two main mechanisms by which cationic impurities can affect performance are adsorption on the catalyst and deposition in the solid phase.One of the mechanisms by which deposited iron improves the performance of Ni(OH)2/NiOOH electrocatalysts is by increasing the electrical conductivity by up to a factor of 30.214 However, the main reason behind the increased OER activity is apparently changes to the active site. Without any iron on the Ni(OH)2/NiOOH catalyst, γ-NiOOH was reported to be the most active phase for OER.214 Iron incorporation on Ni(OH)2 was observed to take place continuously in non-purified potassium hydroxide, with one experiment showing that the iron content increased to 23–26 at% of total metal content after 38 days of aging. The overpotential at 10 mA cm−2 was observed to reach a minimum at 12–17 at% iron after 5 days of aging. It was found that the deposited iron formed a NiFe layered double hydroxide structure,88 which is one of the most active non-precious metal OER catalysts in an alkaline environment reported to date.215 Intentionally adding iron salt such as FeO42− into the electrolyte was also reported to improve OER activity.216 When cycled from −0.14 V to 1.86 V vs. Hg/HgO using reagent-grade potassium hydroxide, an increase of OER current by a factor of 1.75 was observed after 150 cycles. Addition of K2FeO4 after the 150th cycle further increased the OER current by a factor of 1.1. Apart from iron, other metallic impurities can also play an active role in the performance of AWE systems. Fluorine-doped tin oxide on glass was used as an electrode to perform OER in a 0.5 M BK3O3 solution with metallic impurities. Initially no current was generated at 1.4 V vs. NHE, yet after prolonged anodization a rise of current was detected. The 17 nM concentration of nickel within the electrolyte was reported to be deposited and acted as an active catalyst. It was demonstrated that when amberlite resin was used to clean the electrolyte, the increase of current was smaller in magnitude.217
At the cathode, conflicting reports have been published on the impact of iron deposition on the performance of nickel catalysts. When a nickel wire was held at −1.5 V vs. Hg/HgO in 30% KOH with 0.5 ppm iron for several hours, the overpotential was observed to increase continuously.218Post mortem analysis revealed iron deposition and thus one of the causes of degradation was attributed to iron being a worse catalyst for HER than nickel. However, measurement of a Ni200 cathode (99.6% purity) showed contradictory results. The Ni200 cathode was tested at 37 °C, 250 mA cm−2 for 105 s in 30 wt% potassium hydroxide with the addition of either 0.03 ppm or 3 ppm iron. The cathode degraded at the same rate in both solutions until around 103 s, and the electrode in 3 ppm iron experienced a recovery to its original potential after 105 s. The electrode in 3 ppm iron was found to change colour to black and to have picked up iron impurities. The performance increase was then attributed to an increased active surface area.219
One of the possible reasons for the decrease in nickel performance during HER is hydride formation. In a specially designed permeation cell, nickel foil was secured between two compartments, with HER taking place in one compartment and the hydrogen diffusing through the metal to the other compartment, where a permeation current is recorded. In 30% potassium hydroxide, at charging currents of 25 mA and 50 mA, the hydrogen permeation current firstly increased and then dropped as the HER overpotential increased. This was attributed to the formation of beta nickel hydride on the surface, which acts as hydrogen diffusion barrier. The decrease in electroactivity was attributed to the d-band filling of nickel in beta nickel hydride.220 Nickel hydride was also observed on a RANEY® Ni–Al anode after operation at 300 mA cm−2 in 10% potassium hydroxide. A continuous increase in overpotential was observed and X-ray diffraction (XRD) revealed hydride formation. Post-HER, the electrode was annealed under argon at 1000 °C for 2 h, which significantly reduced the overpotential and removed nickel hydride.221 Iron deposition was reported to mitigate this hydride formation at the cathode. Nickel foil with 0.04 μm of sputter deposited iron on the surface was found to have a stable overpotential and stable hydrogen crossover current after 45 h in the permeation cell, in contrast to bare nickel.222 It has been shown using the permeation cell experiments that iron particles detached from the electrode form HFeO2− by corrosion, which can be reduced back to iron on the electrode surface at more negative potentials, as shown in the following reaction.223
| Fe + 2H2O → HFeO2− + 3H+ + 2e− | (9) |
This freshly deposited iron was proposed to increase the HER activity of iron-coated nickel cathodes. For nickel–iron cathodes, HER activity was reported to increase with iron content peaking at 90 wt% iron. Cathodic sweep experiments showed signs of reduction of Ni2+, Fe2+ and/or NiFe2O4 to nickel and iron, which in turn enlarged the active surface area. However, corrosion did take place on all nickel, iron, and Ni–Fe samples forming oxide layers. The cathodic potential was proposed to reduce these oxides into active intermediates such as adsorbed FeOH and adsorbed NiOH that further enhance HER activity.224
A similar increase of HER performance after iron or nickel deposition has been reported for silver, gold, copper225 and cobalt based catalysts.226 Improvement in HER activity of copper and ruthenium based catalysts due to leaching and deposition of titanium oxides from a titanium current collector has also been reported.80,227 Nickel hydroxides either introduced as an impurity in the electrolyte or deliberately deposited have also been shown to improve the catalytic activity of nickel, silver, copper and platinum based catalysts.228,229 The oxide and hydroxide-based impurities discussed above have been shown to improve the catalytic activity by optimizing the water dissociation step of HER. It is clear that any metallic impurities that are active for OER and HER reaction would likely not induce degradation upon deposition.
While some metallic impurities are beneficial for nickel-based cathodes, others can prove detrimental. Zn2+ is often present in low amounts in potassium hydroxide electrolyte and can also leach out from plastic components and gasket materials. Zn(NO3)2·6H2O at 0.067 mg mL−1, which is significantly higher than the trace ppm level found in potassium hydroxide, was added to the catholyte of a nickel/nickel electrolyser operated at 50 mA cm−2. The cell voltage increased from 2.6 V and reached a stable 3.1 V for the next 6 h of operation. Clear zinc dendrite deposits were identified post operation. Zinc was reduced at −1.43 V vs. Ag/AgCl/3 M KCl, while the cathode HER potential at 50 mA cm−2 was −1.55 V vs. Ag/AgCl/3 M KCl.230 Synthetic tap water was also reported to degrade the HER rate. The synthetic tap water consisted of mg L−1 levels of the following impurities: Na+, Ca2+, Mg2+, Al3+, Cl−, HCO3−, SO42−, and NO3−. Held at 0.25 A cm−2, the performance of the electrolyser degraded continuously during 55 h of operation, with the anode potential increasing from 1.8 V to 2.4 V vs. RHE. Interestingly, gas product analysis revealed no significant difference between using pure water and synthetic tap water. EDX of the nickel electrode surface showed the presence of carbon (39.82%), oxygen (15.39%), iron (28.73%), lead (4.78%), sodium (2.97%), chlorine (2.19%), potassium (3%), and zinc (1.7%).38 Most of these metallic impurities were not included when creating the synthetic tap water; therefore zinc, lead, and iron must have originated from salt impurities. Reports have shown that Ni–Fe215 and Ni–C231 are active catalysts for HER, so the most likely culprits for the deactivation are lead, zinc, and Cl−.
4.3 Anionic impurities
In both AWEs and AEMWEs OH− is usually the charge-carrying ion, though we note some AEMWEs use alternative or mixed anions. Potassium hydroxide at concentrations above 20 wt% is widely used as the circulated electrolyte in AWEs. In theory, AEMWEs may operate on pure water with the hydroxide in the anion exchange membrane transporting charge, however contemporary practice is to use a low concentration 0.1 M to 1 M supporting electrolyte to obtain higher performance and durability, with aqueous potassium hydroxide being the most common choice. The main mechanisms by which anionic impurities can impact AWE and AEMWE performance is through charge carrier substitution in the electrolyte and adsorption on the catalyst layer.4.3.1.1 Carbon dioxide. An impurity of concern in an alkaline environments is carbon dioxide. Carbon dioxide is present in ambient air at around 400 ppm and can readily react with potassium hydroxide to form bicarbonate (HCO3−) and carbonate (CO32−) anions according to the following reactions:
| OH− + CO2 ⇌ HCO3− | (10) |
| OH− + HCO3− ⇌ CO32− + H2O | (11) |
The radii of the hydrated forms of HCO3− and CO32− are 3.64 Å and 3.94 Å, respectively.232 The carbonates are larger in size than OH− at 3 Å, which in turn lowers their overall ionic mobility. Relative to K+, the mobilities of OH−, HCO3−, and CO32− are 2.71, 0.6, and 0.98, respectively.233 The presence of carbon dioxide lowers the overall cell performance of an AWE by reducing the effective electrolyte conductivity. The impact of carbon dioxide on the performance has been studied extensively in alkaline fuel cells (AFCs) which are necessarily exposed to ambient air in a way that electrolysers are not. A half-cell AFC study using 7.1 M potassium hydroxide and 5% carbon dioxide showed a linear increase of carbonate concentration during 3000 h of operation. Interestingly, this increased concentration of carbonate did not affect the performance of the electrode, as evidenced by similar performance degradation of control (pure H2 fed) and 5% CO2 containing H2 fed electrodes. The decrease of cell performance was mainly attributed to the decrease of electrochemical surface area of the catalyst over time.234 In another study using a microfluidic fuel cell, a critical ratio of hydroxide/carbonate to cause a 10% voltage decrease was proposed.235 In the case of 3 M potassium hydroxide, the critical potassium carbonate concentration was 0.75 M (reached after 1700 h operation) while for 5 M and 7 M potassium hydroxide it was 1.75 M (reached after 3900 h of operation). The absence of significant conductivity decline in ref. 234 was simply because the critical concentration of carbonate was not reached. Carbon dioxide contamination in AWEs should be much slower in sealed systems with water purification, so timescales for contamination should be longer.
In the case of AEMWEs, there have been various studies on anion exchange membrane fuel cells (AEMFCs) using similar AEMs.236–238 For three types of AEM with different anion forms (OH−, HCO3−, CO32−), the conductivity was found to decrease in the following order: OH− > HCO3− > CO32− with increased relative humidity and membrane hydration reported to remedy the conductivity decline.239 It is likely that the extent of carbon dioxide poisoning in AEMWEs, where water fully hydrates the membrane, is not as severe as that reported in AEMFCs; additionally, OH− is generated in situ by simply running alkaline water electrolysis reactions and this will gradually purge the carbonate as carbon dioxide.240 This has been demonstrated in AEMFCs when deliberately passing a low electrolysis current or operating at high current densities.241 In case of OER, the presence of carbonate was found to decrease the catalytic activity of LaNiO3 by formation of carbonates which altered the chemical environment of active sites.242 This may be generally applicable to other transition metal based OER catalysts.
Apart from decreasing the ionic conductivity and catalyst activity, carbon dioxide has been reported to affect membrane physicochemical properties such as membrane crystallinity and side-chain spacing.243 The reaction to form CO32− generates water, which then leaves the membrane; this shrinks the side-chain spacing and affects membrane crystallinity. However, a higher humidity (RH: 85%) was reported to reduce the extent of this polymer morphology change. Carbon dioxide contamination of the water tank was also a problem for a pure water operated AEMWE.244 Polarisation curves of the cell in quick succession showed rapid degradation while the HFR increased 3-fold from 0.6 Ω cm2 to 1.67 Ω cm2. This short-term degradation was not observed when measures to minimise carbon dioxide ingress, i.e., placing the whole setup inside a glovebox, were put in place, clearly illustrating the importance of effective gas-tight seals when operating AEMWEs with pure water.
| Cl− + 2OH− → OCl− + 2e−, E° = 0.89 V vs. SHE, pH = 14 | (12) |
The 480 mV overpotential window before the initiation of reaction (12) has routinely been exploited to facilitate operation of direct seawater electrolysis without generation of chlorine or OCl− at the anode.37 It is not within the scope of this article to explore in-depth the issue of seawater electrolysis; the reader is referred to other excellent reviews of the subject.37,142 However, most studies regarding the issue of chloride contamination have been in the context of seawater electrolysis applications.
NiFe layered double hydroxide (LDH) catalyst, a common AWE OER catalyst, was reported to be able to perform OER close to 100% faradaic efficiency in 0.5 M sodium chloride + 0.1 M potassium hydroxide.148 Keeping the pH at 13 with an overpotential of 480 mV, 10 mA cm−2 was achieved without any chlorine production. A 2 h stability test at 10 mA cm−2 under different pH showed degradation of NiFe LDH at pH 9.2 with and without sodium chloride. A sharp increase of potential from 1.7 V to 2.4 V vs. RHE was observed after 60 min of operation with sodium chloride. When both sodium chloride and potassium hydroxide were used, the cell potential remained below the 480 mV limit during 120 min of operation. The same catalyst was also proven to work well in a AEMWE configuration with a Pt/C cathode and Tokuyama A201 membrane.245 Three concentrations of potassium hydroxide (0.1 M, 0.5 M, and 1 M) were tested with and without addition of 0.5 M sodium chloride by setting the cell voltage at 1.6 V. All cells showed degradation over a 100 h period, except that in 0.1 M potassium hydroxide without sodium chloride. Scanning electron microscopy and impedance spectroscopy indicated no degradation of the electrode or membrane thickness with or without sodium chloride. The evidence suggested that the degradation was mainly due to chloride ions substituting hydroxyl ions and lowering conductivity. Interestingly, the assumption of a 480 mV window of operation is only valid from a thermodynamic perspective. Using a symmetrical feed of 0.5 M potassium hydroxide and sodium chloride at both cathode and anode, it took a cell voltage >2.4 V to initiate any OCl− formation using Ir-black and Ir/TiO2 as the anode catalyst. When NiFe LDH was used as the anode, prolonged operation at high current densities (0.2–0.7 A cm−2) corresponding to cell voltage of 2.0–2.4 V yielded a stable 94% faradaic efficiency, suggesting only a minor contribution from the CER. Iodometric analysis of the electrolyte showed no OCl− formation, demonstrating selectivity of NiFe LDH in the presence of Cl−.246
4.4 Organic & inert impurities
 | (13) |
 | (14) |
Various papers have pointed out that NiO(OH) is the required species for oxidation of organics, based on the fact that the oxidation potential coincides with the potential for NiO(OH) formation.252,253 One experiment showed that after cycling the potential (−0.9 V to 1.6 V vs. MMO) of Ni/C for 50 cycles, the magnitude of the methanol oxidation peak decreased by 65%, which was suggested to be as a result of nickel oxide formation. In commercial AWEs and AEMWEs with nickel-based catalysts, even though the potential of the anode would be high enough to initiate oxidation, it seems that catalyst passivation may prevent complete oxidation of the organics.
Another issue is the nature of the by-products of these organic fuels upon oxidation. One study of a Ni[salen] complex catalyst at different applied potentials showed that the oxidation reaction starts at 0.4 V vs. Ag/AgCl and generates a range of products. Methanol and glycerol are mainly oxidised into formate, while ethanol held at 0.6 V vs. Ag/AgCl forms acetate instead.254 In AWEs and AEMWEs, the anode potential will be much higher than this, in which case oxidation of the organics into carbon monoxide and carbon dioxide can be expected. Carbon dioxide poses significant challenges in alkaline electrolytes as discussed above. As with PEMWEs, both carbon dioxide and carbon monoxide may also contaminate the product hydrogen.
The right type of organic could also impose a chelating effect on the nickel catalysts. For instance, EDTA and [S,S]-ethylenediamine disuccinic acid ([S,S]-EDDS) have been used as chelating agents in metal recovery.255 Their ability to form stable metal complexes may cause or accelerate catalyst degradation. This should be given careful consideration should wastewater containing unknown organics be used as the water feed. For alkaline systems, the presence of isopropanol, methanol, ethanol and formic acid actually resulted in a stabilisation effect as it was noted that even up to the upper potential limit tested, 1.5 V vs. RHE, acetone was the primary product of oxidation and not carbon dioxide.201 The acetone is shown to stabilize the catalyst by irreversibly adsorbing on the surface, which prevents the dissolution of surface oxide layers during the reverse scan. The conclusion from this work suggests that the mechanism by which enhancement or stabilisation of catalysts occurs, via organic species, is very dependent on the specific surface adsorption and resulting oxidation pathway of these species. This indicates that studies on materials specifically relevant to OER in both acid and alkaline environments are necessary to determine whether organic species are having a significant effect on catalyst stability.
4.5 Summary and outstanding research challenges
| Impurity type | Source | Anode | Separator/membrane/conductivity | Cathode |
|---|---|---|---|---|
| Fe3+ | Electrolyte impurity | Adsorbs on catalyst improvement of OER activity88,211,214,216 | Improvement of HER activity222–224 increased surface area219 | |
| Ni2+ | Electrolyte impurity | Adsorbs on catalyst. Improvement of OER activity217 | Improvement of HER activity228,229 | |
| Al3+ | Electrolyte impurity | Adsorbs on catalyst. No adverse effect reported | No adverse effect reported | |
| Zn2+ | Gasket leaching out, electrolyte impurity | Catalyst poison212 | Forms dendrites at negative potentials, catalyst poison230 | |
| Cd2+, Pb2+ | External contamination | Catalyst poison212 | ||
| Ti3+/Ti4+ | Electrode leaching out | Improvement of HER activity80,227 | ||
| Na+ | Water impurity (if NaOH is not used) | No adverse effect reported | No adverse effect reported | |
| Ca2+, Mg2+ | Water impurity | Deposits Mg(OH)2 at high pH,31,258 may cause mass transport issues at electrodes | ||
| Cl− | Water impurity | Nickel corrosion, apart from special structures.247,248 | ||
| CO2 | Air contamination | Lowers conductivity,232,234,235,237–239,259 reduces side chain spacing and membrane crystallinity243 | Increases charge transfer resistance (AEMWE)259 | |
| Organics | Water impurity | Catalyst poison255 | Catalyst poison255 | |
| Inert impurities | Precipitates formed by reaction with electrolyte | Clogging of pores256,257 | ||
Since the charge carriers in AWEs and AEMWEs are anions, anionic impurities pose a significant threat. Carbon dioxide from ambient air can easily diffuse into the cell if there is a gasketing problem or a slightly loose connection within the stack. It forms HCO3− and CO32− anions that lower the cell ionic conductivity and performance considerably. Moreover, carbon dioxide has also been reported to change the membrane physical properties in AEMFCs. Halogen ions can also be introduced from impure feed water or malfunction of the water purification unit. Cl−, in particular, can trigger unwanted side reactions and generate OCl−. If the electrode structure is incompatible, it could also trigger corrosion of the electrode.
There have been many studies of organic impurities in three-electrode setups, but all of them are framed from an alkaline fuel cell or organic electrooxidation perspective. Organic impurities can be introduced when lower quality water is used as the water feed. It is expected that at the anode, the organics will be oxidised into various products depending on the applied potential. Side products such as formates and acetates have been reported. Other oxidation products, such as carbon dioxide and carbon monoxide, are likely but have not been explicitly reported. Gaseous and volatile products can contaminate the product hydrogen quality, leading to a requirement for additional purification processes.
On the anion side, optimisation of electrodes can lead to OER selective anode catalysts in the presence of chloride anions. This would be beneficial for direct seawater electrolysis but could also prove important if lower quality water were used to cut purification costs. A detailed study of the impact of carbon dioxide is also lacking for alkaline electrolysers, as most papers report results within the context of fuel cells. Fuel cells operate with air and have a higher concentration of carbon dioxide, while electrolysers would likely have a lower concentration. The concentration should be addressed in a more relevant range.
Finally, there is no organic impurity related study within the context of AWEs and AEMWEs. Many questions remain to be answered, including how electrooxidation could impact the performance of AWEs and AEMWEs and impacts on catalyst durability. Carbon dioxide, in principle, could be formed at high anode potentials and would lower the ionic conductivity of the electrolyte. There is still a steep learning curve to study whether organics could interfere with AWE and AEMWE operation in the long term.
5 Characterisation techniques, water purification technologies, and mitigation strategies
5.1 Characterisation techniques
A wide variety of analytical techniques are used in the testing of impurities and the effects on the materials involved for PEMWE, AEMWE and AWE. This testing can be split between half-cell investigations of small quantities of catalyst and single cell testing of complete systems. In addition to this, testing is split further between testing that can be done in situ (in a relevant environment), in operando (in an operating device) and ex situ (not in a relevant environment) with half-cell or full cell design being the limiting factor in the accessibility toward many currently available in situ and in operando characterisation techniques. This section provides a summary of the more commonly used techniques that could be used for relevant studies in the future.Rotating Disc Electrode (RDE) or half cell testing are the most basic types of in situ electrochemical tests. Their main use is to characterise the behaviour of materials such as benchmarking the activity and stability of catalysts or other cell components. In a typical experiment the material under investigation in immersed in a liquid electrolyte along with a counter electrode and a reference electrode. A potential is applied to the material and the resulting current is measured. The benefit of these experiments is the well-defined potential of the sample, and ability to work with a range of electrode types including thin films of very small amounts of catalysts. A major limitation of these types of experiments is mass transport to and from the electrodes. In RDE, the electrode is rotated to precisely control the diffusion layer at the electrode surface but this is not often sufficient; for example the OER may form microbubbles that prohibit high current density operation.260,261 New cell designs, such as the floating electrode, that facilitate gas evacuation262 alleviate mass transport effects. Half-cell testing often facilitates the use of other in situ characterisation tools and it is easy to introduce impurities to the liquid electrolyte.
Rotating Ring Disk Electrode (RRDE) allows for additional in situ electrochemical probing of the process and in particular the intermediates that are formed at the working electrode.150 While the material being investigated on the RDE is under current or potential control at the centre of a rotator an additional ring separated from the working electrode allows for a separate reaction to be probed by holding the ring at a different potential. This technique has been used to probe the quantity of chlorine being formed at an electrode, as well as demonstrating that oxygen evolution on iridium oxide is not suppressed by increasing Cl− concentration.182 The chloride reduction reaction and oxygen reduction reaction can be competing reactions at the ring, so it was found to be necessary to select a condition that allowed only chlorine reduction: 0.95 V vs. RHE at pH 0.88.147 The limitation of the RRDE technique is that it cannot distinguish the chemical identify of species at the ring electrode. Other analytical techniques are required to specifically identify the species and at what potential they are selective to certain reactions.
Single cell/full cell testing is carried out by operating a scaled down electrolyser typically monitoring the cell voltage under a range of conditions. A single cell from a stack may be used or more commonly a small ∼10's cm2 cell is assembled as a scaled down device.49,124 As all of the electrochemical components in an electrolyser are present and acting as they do in a real device, the results are highly representative of real systems. However, this means that deconvoluting the response to determine mechanisms is challenging and usually relies on complementary techniques such as carbon monoxide stripping to determine ECSA, linear sweep voltammetry to determine hydrogen crossover, and electrochemical impedance spectroscopy to determine cell resistance and kinetic parameters.
Stack/system testing is the most relevant type of electrochemical testing, as this uses a real device, or one identically configured. Testing at this scale is resource intensive, time consuming and as with single cell testing it is challenging to deconvolute underlying processes. However, it can highlight phenomena that are only present in real systems, such as impurity enrichment, BoP degradation or stack design choices that exacerbate the impact of impurities.47
Electrochemical Quartz Crystal Microbalance (EQCM) uses the high mass sensitivity of the resonant frequency of thin quartz crystal in an electrical field to measure changes in mass of a working electrode.263 These features allow EQCM to probe mass changes during in situ electrochemical half-cell experiments. This has allowed the investigation of the effects of impure potassium hydroxide on NiOOH films where iron contamination results in increased activity, but in particular the technique facilitated distinguishing between the effects of structure changes vs. iron incorporation that caused an increased mass of the electrode.88 A limitation of this technique is that it cannot distinguish between different sources of mass loss such as dissolution and particle detachment.264
Attenuated Total Reflectance – Fourier Transform Infrared Spectroscopy (ATR-FTIR) has been used to investigate degradation of the Nafion membrane in half cell tests.265 In particular, the ability of ATR-FTIR to investigate changing states of the functional groups within the membrane provides additional information about the impact of contaminants on membrane degradation that complements elemental mapping of contaminants. ATR-FTIR can also be used in half-cell tests on organic impurities and to reveal the by-products of oxidation of organics.254
Ultraviolet-visible spectrophotometery (UV-Vis) has been used for the quantification of the valence state of iron in cathode water by comparison of the Fe2+ signal recorded at 510 nm and using a standard concentration curve of prepared solutions to assess concentration.124 In addition, in situ UV-Vis can be useful for detecting reaction intermediates in oxides266 as well as establishing the effect of various contaminants on surface species.267
X-ray absorption spectroscopy (XAS): X-ray absorption near edge spectroscopy (XANES), and extended X-ray absorption fine structure (EXAFS) typically use a synchrotron to provide a X-ray beam to excite core electrons.268 These techniques have been used to investigate the in situ effects of anion absorption of Cl−, Br−, OH−, and HSO4− on platinum surfaces.151 While unlikely to be a commonly used technique for contaminant investigation in either PEMWE or AWE setups due to the requirement for a synchrotron facility as well as further limitations with cell design, it has proved insightful for fundamental studies of absorbed anionic species as well as demonstrating the capability of conducting in situ measurements.
Scanning Electron Microscopy (SEM)/Energy Dispersive X-ray Spectroscopy (EDX or EDS) involves coupling the detailed imaging of SEM with elemental mapping via EDS and is one of the common techniques used for post mortem analysis of CCMs. SEM provides different imaging modes with standard instruments usually offering both composition and topographical contrast and resolution on the sub μm scale. EDX has been used to particular effect both for the detection of cation contaminants and for analysing the amount of catalyst dissolved and where it deposits on the MEA.68 Typically EDX has a detection range of 0.1–100 wt% of the material,68 though the quantification of lighter elements such as oxygen and carbon is often unreliable, and the technique is not surface sensitive; as such it is better at detecting larger quantities of material deposited.
X-ray photoelectron spectroscopy (XPS) utilises the photoelectric effect, which involves firing X-rays at a sample and measuring the energy of emitted electrons, with each element giving a specific fingerprint signature.269 While in situ ambient pressure XPS has been used for the investigation of oxygen evolution materials267,270,271 it is more commonly used for ex situ characterisation of catalyst materials, where it is complementary to EDX with a high degree of sensitivity to the surface of materials and quantification of lighter elements is more precise.
Electron probe microanalysis (EPMA), like EDS, utilises an electron beam and recording of emitted X-rays.272 This provides spatial resolution on the sub-micrometer scale as well as the ability to detect concentration down to at least 100 ppm for most elements. This facilitates investigation of cross sections of CCMs for various cation contaminants such as Na+,122 and Fe3+.124
Online neutron and X-ray imaging techniques utilise either a neutron or X-ray beam directed towards the sample, and the subsequent absorption and scattering of the radiation. The technique has often been employed to elucidate water-bubble dynamics within the flow field of PEMWEs.273–276 Applying neutron scattering to impurity studies is not straightforward, as it requires the impurity to have a sufficiently contrasting neutron scattering cross-section to water and other components. Certain ions, such as Gd3+, fulfil this criteria by having an extremely high neutron scattering cross-section per atom, making them discernible.123
Nanoscale Secondary Ion Mass Spectrometry (Nano SIMS) is an imaging technique that analyses the atomic mass of species on a solid sample surface with a resolution of 50 nm and a detection range in the sub-ppm range. The technique works by sputtering the top monolayer sample via a primary ion beam and analysing the ionised atomic and molecular species with a mass spectrometer. Nano SIMS has been routinely used for biological applications, but to date it has not been employed for post mortem CCM analysis.277 This technique offers a better resolution and detection for trace impurities and could be exploited in future research.
Inductively Coupled Plasma Mass Spectrometry (ICP-MS) is a type of mass spectrometry that uses inductively coupled plasma to ionise samples, allowing trace element analysis of liquid solutions.278 In the case of PEMWE and AWE research this can allow for analysis of recirculated water125 or electrolyte for a range of contaminants. In addition, the technique can be used to monitor catalyst losses due to dissolution by analysing the quantity of dissolved catalyst in water/electrolyte in cell testing.201 This may be of limited use for MEA analysis of flow water for anode catalyst dissolution as a majority of the dissolved anode catalyst will travel and deposit either in the membrane or on the cathode.68 The detection limit of most elements in ICP-MS is also much lower than that of inductively coupled plasma optical emission spectroscopy (ICP-OES) with a lower limit of parts per trillion (ppt) for some elements/machines.
Scanning Flow Cell Inductively Coupled Mass Spectrometry (SFC ICP-MS) couples an electrochemical flow cell and ICP-MS allowing the composition of the electrolyte to be determined as a function of electrochemical properties. It allows in situ studies of both acid and alkali cell setups, good time resolution and potential dependent studies.279 The high potential resolution enables the study of catalyst dissolution during potential scanning, allowing clear identification of the sensitivity to potential as well as the effect of contaminants on the quantity of catalyst that dissolves at a given potential.
pH monitoring encompasses a wide variety of techniques that measure pH. The most common methods are electrochemical, monitoring the potential difference between two electrodes.280 It is routinely used to monitor the quality of the feed water and acidity/alkalinity of prepared electrolyte. Monitoring the pH of the anode and cathode water in an electrolyser has been used to investigate the mechanism operating when poisoning with Na+ occurs.121
Ion chromatography measures the concentration of ions by separating them based on interaction with a resin and eluent. It is a technique that is often used to characterise ionic products in the water phase, particularly from membrane degradation, and can reliably detect impurities to the ppb level.141,281 While often used ex situ, incorporation of the ion chromatograph in situ to record the measurement of F− ions during PEMWE operation has also been reported.28
Polarography is an electrochemical technique that can be used to measure the concentration of metal ions in an electrolyte and can be incorporated in-line with continuous logging of data. The working electrode is either a dropping mercury cathode282 or a hanging mercury drop electrode. Noble metal ions, such as platinum, can be measured in the ng L−1 range283 and it has recently been used to measure the dissolution of PEMWE cathode material at open circuit with a limit of detection of 2 ng L−1.66
Ion selective electrodes are an off-the-shelf solution for measuring the concentration of specific ions. They work in a similar way to pH electrodes; instead of a proton selective membrane, a specific ion selective membrane is used.284,285 For example, a F− ion sensitive electrode uses a LaF3 crystalline membrane that only allows F− to pass.286 These electrodes have been routinely employed for in situ and ex situ measurement of ions in PEMFCs, mainly for F−.87 Their limit of detection is at the ppm level, so ICP-MS is preferred when reliable measurement at the ppb level is needed. However, such electrodes have the advantages of being cheap, robust, and modular.
Water conductivity is typically measured as a resistance between two electrodes placed in a water container of accurately known dimensions.287 In very pure water the self-ionisation of water contributes a significant proportion of conducting ions. Temperature influences the viscosity and ion movement which in turn affects the measured conductivity.288 Conductivity meters are routinely employed for on-line conductivity measurements of feed water in PEMWEs.289
Total Organic Carbon (TOC) is the amount of carbon present in organic compounds and is generally used as a non-specific indicator of water quality.290 TOC analysers measure amount of carbon dioxide formed by catalytic oxidation of organic content. The carbon dioxide is measured either using a conductivity cell (if it's in the aqueous phase) or by a non-dispersive infrared cell. TOC measurements have recently been employed to measure MEA degradation in PEMWEs.289
Gas chromatography (GC) can be used to determine the quantity of evolved gas via the use of an analytical column where different gases are separated by their interaction with the stationary phase.291 This is particularly relevant for the study of both carbon dioxide and carbon monoxide as contaminant gases. The monitoring of these gases also allows for studying the effects of organic contaminant consumption at high potential operation where they are expected to undergo oxidation.196
Differential Electrochemical Mass Spectrometry (DEMS)292 allows for online detection of electrochemical products and intermediate species via a catalyst layer deposited onto a porous Teflon membrane supported on a glass or steel frit whose pores then lead to a mass spectrometer. The hydrophobic membrane prevents liquid from entering the pores but allows gaseous and volatile products to pass through. In addition, it can be used to semi-quantitatively determine the quantity of species produced. This technique has been applied to the study of Cl− contamination on carbon supported iridium oxides to determine at the ratio of evolved chlorine to oxygen in conjunction with RRDE experiments.147
On Chip Electrochemical Mass Spectrometry (EC-MS) is very similar to DEMS.293 This variant utilises a type of membrane chip that avoids the need for differential pumping, allowing faster time resolution and complete sampling of gaseous and volatile products/intermediates, providing a fully quantitative analysis. While it has not been used to date for any contaminant studies of OER catalysts, the orders of magnitude enhancement in sensitivity to molecules such as carbon dioxide and oxygen could make it highly useful to probe organic oxidation reactions.
5.2 Water purification technologies
Despite the critical role of water purity in the longevity of various low temperature electrolyser technologies, little has been openly published on the development of specialist water purification systems or techniques to recover performance loss due to poisoning incidents. The water purification requirements for PEMWE systems vary with manufacturer and installation, but in general most manufacturers stipulate a minimum feed water resistivity of 1 MΩ cm. AWE water purity is not often discussed in the literature, but a similar minimum value of 1 MΩ cm is to be expected as impurities such as Ca2+, Mg2+, and Cl− need to be removed.While fresh feed water is easily supplied at ambient temperature it is common in PEMWE systems to have purification in the recirculation loop fed from the outlet of the stack. A SAC resin can operate reliably above 100 °C. The bottleneck in ion exchange resin temperature stability is the reactivity of cationic headgroups in SBAs in the presence of hydroxide anions. As is very well known to researchers working with AEM materials, high temperatures facilitate degradation of these materials. Some commercial anion exchange resins can tolerate temperatures up to 70 °C but this may still be the most limiting component in PEMWE systems with respect to temperature, though we note that higher temperatures may compromise membrane mechanical integrity through creep deformation.7 Enhanced thermal stability may eventually be obtained by employing the techniques recently developed to improve the stability of AEM materials or potentially using non-hydroxide anions that would not demineralise the water, but which would also not adversely impact performance.
5.3 Mitigation and recovery strategies
In the event of a contamination event or impurity accumulation, it may be worthwhile to attempt to recover the performance of an electrolyser. This section will suggest a few possible avenues to recover from impurity contamination in PEMWEs, AWEs, and AEMWEs. To the best of our knowledge none of these are used in practice.Contamination of the ion conducting phase of PEMWEs, AWEs and AEMWEs is likely to be reversible. Metallic cation contamination of the PFSAs in PEMWEs may be removed by immersing in an acidic solution, such as 0.5–1.0 M H2SO4, to re-protonate the poisoned ionomer.124 A novel approach to re-protonate the ionomer in situ is by flowing carbon dioxide at moderate pressures through the anode water tank to form carbonic acid.123 Pressurising with carbon dioxide for 36 h was found to recover 95% cell voltage efficiency after poisoning by Fe2+ ions. In AWEs and AEMWEs using recirculating electrolyte an ion exchange resin cannot be placed inside the recirculation loop. Recovery from any contamination is therefore likely to require the replacement of the electrolyte. As with PEMWEs, anionic contamination of AEM and ionomer may be reversible. Light carbonate contamination of the membrane or ionomer is expected to be self-reversing with operation at high current densities as OH− is constantly generated at the cathode side241 and would eventually purge carbonate and bicarbonate from the system as carbon dioxide. Other soluble anionic impurities may be flushed with repeated changes of electrolyte. If insoluble metal oxide precipitates are formed, then these may be removed by purging the system with an acidic solution before the electrolyte is replaced, though this may damage electrode materials.
At the catalyst layer, the chemistry of specific impurities will require different methods to clean the catalyst. Many ionic impurities be removed by oxidation or repeated rinsing of the system. Adsorbed halide anions on any catalyst surfaces may be desorbed by either oxidising at very positive potentials (>1 V vs. RHE) to oxidise the halide or by holding the catalyst at reducing potentials to evolve copious volumes of hydrogen. These techniques may require the cell potentials to be reversed, all techniques which involve potential excursions for the electrode may have a detrimental impact on catalyst longevity, but it may be relevant when contamination is critical. Similarly, high potentials may resolve heavy contamination from organic compounds via oxidation. Also noteworthy here are attempts to produce catalysts which are selective to OER in the presence of chlorine.209 Metallic or insoluble salt impurities may require removal by flushing the system with a solution which adjusts the pH into a range where such impurities are soluble (for example an acidic solution to remove iron precipitates). The challenge in all cases is to remove the impurity while avoiding, as far as possible, damage to the electrolyser components. This will require careful calibration of conditions for the recovery step.
Impurities added to hydrogen as a result of contamination may be mitigated by hydrogen purification steps, and very low concentrations of trace impurities such as carbon dioxide and halides are tolerated in hydrogen fuel specifications, as discussed above. Radical formation may be mitigated using techniques applied in PEMFCs. The first method is to limit the crossover of hydrogen and oxygen by introducing a layer of recombination catalyst into the membrane or catalyst layer.133,299 Another method is to introduce a radical scavenger or peroxide decomposition catalyst inside the membrane to reduce the concentration of radicals that cause damage. Metal oxides such as titanium dioxide, zirconium dioxide, and cerium dioxide have proven their utility in suppressing the release of F− under PEMFC operation.300
5.4 Summary and outstanding research challenges
| Technique | Possible purpose | Detection limits |
|---|---|---|
| Rotating disc electrode | Impact of impurity on catalyst performance260,261 | Not applicable |
| Single cell testing | Impact of impurity on CCM level performance124 | Not applicable |
| Stack or system level testing | Impact of impurity on overall stack performance47 | Not applicable |
| Rotating ring disc electrode | Side product measurement147,182 | ppm (depending on molecule) |
| Electrochemical quartz crystal microbalance | Impact of change in mass due to impurity on electrode performance88 | ng cm−2 |
| Attenuated total reflectance-Fourier transform infrared spectroscopy | Detecting change in functional group structure to study impact of impurities on membrane degradation207 | Not applicable |
| Ultraviolet-visible spectrophotometry | Concentration of impurities267 | ppb to ppm (depending on molecule) |
| X-ray absorption spectroscopy | Structure of catalyst surface151 | ∼5% by weight (depends on the element) |
| X-ray photoelectron spectroscopy | Surface characterization of catalysts267,270,271 | 0.1–1 at.% |
| Inductively coupled plasma-mass spectroscopy | Liquid phase quantification of metallic elements68,278 | ppb |
| Scanning flow cell-inductively coupled mass spectrometry | Operando concentration detection279 | ppb |
| pH monitoring | Indication of impurity or side reaction taking place121 | Not applicable |
| Ion chromatography | Concentration measurement of ions28,141,281 | ppb |
| Polarography | Concentration measurement of a specific ion (e.g. Pt2+)66,282 | ppb |
| Ion selective electrodes | Concentration measurement of a specific ion (e.g. F−)301 | ppb to ppm |
| Water conductivity | Conductivity measurement of feed water210,287,288 | 1 μS cm−1 |
| Total organic carbon | Concentration of organic carbon210,290 | 5 mgC L−1 |
| Gas chromatography | Concentration measurement of a molecule196,291 | ppm |
| Differential electrochemical mass spectrometry | Qualitative concentration measurement of product and intermediate product209,292 | Semi-quantitative |
| On chip electrochemical mass spectrometry | Quantitative concentration measurement of product and intermediate product293 | pmol s−1 |
| Scanning electron microscopy-energy dispersive spectroscopy | Impurity distribution in CCM68 | >0.1 wt% |
| Electron probe microanalysis | Impurity distribution in CCM124,272 | ppm |
| Neutron/X-ray imaging | Operando distribution of impurity in the cell273–276 | Semi-quantitative |
| Nanoscale secondary ion mass spectrometry | Impurity distribution in CCM277 | ppb to ppm |
In the liquid phase, the most commonly applied technique is ICP-MS, which can detect metallic elements down to the ppb level. However, most experiments use ICP-MS ex situ, meaning the water or electrolyte is collected at certain time intervals and analysis is performed afterwards. SFC ICP-MS offers an operando solution by connecting the cell directly with an ICP-MS. When the desired impurity is in ionic form, ion chromatography and polarography can work well; concentrations down to the ppb level can be measured. When the concentration is slightly higher, pH electrodes and ion sensitive electrodes can provide a cheaper and more robust alternative for operando measurement of impurity in the water.
Online gas phase analysis is rarely performed to study impurities; however, gas chromatography is the most common analytical technique applied, with different detectors and columns able to measure a range of gas phase impurities down to the ppb level. In the half cell setting, DEMS and EC-MS can be employed to track rapid evolution of side products semi-quantitatively. The RRDE, in this case, can also be used to monitor gaseous impurities that are generated as side-products at the anode.
CCM characterisation is mostly performed post mortem, with the exception of online neutron imaging of impurities. Neutron imaging is a powerful operando tool that can show the distribution and poisoning behaviour of certain impurities within the CCM; however, this is usually limited to less relevant ions such as Gd3+ that have a sufficient neutron scattering cross-section. In post mortem testing the cell can be disassembled and individual components observed using SEM/EDX to map the distribution of impurities within the cell. EDX can only measure impurities at quite high concentrations, so for lower concentrations other techniques must be used. XPS and EPMA have been reported to be sensitive down to the ppm level while nano SIMS can go to ppb levels. Overall, the choice of technique depends on the nature and concentration of impurity present.
Water purification is a mature technology and there is little information available in the literature on the specific, proprietary systems implemented in commercial systems, but a combination of RO and ion exchange resin technologies is typical where high purity water is required.
The mitigation and recovery strategies presented are mostly theoretical processes that may be applied to recover electrolysers in the event of severe poisoning or when the stack feed water is known to be impure. Processes exist for recovering poisoning by most types of impurity at low concentrations, but the processes may themselves cause damage to the electrolyser.
A clear challenge for water purification is to further reduce the capital and operating costs for purification. Ion exchange resins stable to increased temperature may be critical to support higher temperature operation of PEMWEs. Existing mitigation and recovery strategies are largely theoretical and need to be demonstrated in real systems in order to properly evaluate their benefits, costs, and drawbacks.
6 Conclusions
As electrolyser developments continue and long-term, large-scale deployment ramps up, the limitations that impurities place on the economics, performance and durability of low temperature electrolysers will become clearer. To date this has been a little studied area; the authors hope that this review will stimulate consideration of this area by summarising the state of the art, highlighting illustrative work in adjacent fields, and flagging outstanding research questions.For PEMWEs, impurity testing at the cell level has only been performed explicitly for a handful of cations with others studied only in half-cell configurations or else their response inferred from results in analogous systems, such as PEMWEs. This is even more marked for anionic impurities, which have largely focused on Cl− in half-cell tests; even here it is unclear how damaging chlorine evolution is when Cl− is present at low concentrations for long time periods. The impact of simple organic molecules may be inferred from studies on alcohol-fed electrolysers, but more complex molecules are likely to have an impact on durability.
The AWEs have been routinely operated for long periods suggesting that they are, in practice, robust to trace impurities. However, the limited studies on impurity tolerance mean that the impact on more modern higher-performing devices is unclear, as is the potentially unnecessary degree of conservatism in the water quality used. The use of lower catalyst loadings and more complex interfaces in AEMWEs is likely to make them less robust to impurities than AWEs, but again there is little hard information. Studies of the impact of impurities on AEMWEs are hindered by their ongoing development, which means that there is not yet a clear consensus on materials, design, or operation.
General questions relevant to all device types include methods for recovery from poisoning events, the stability/solubility of BoP materials in ultra-pure water, and the development of methods to assess the long-term stability towards impurities over short-time scales.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the National Measurement System of the UK's Department of Business, Energy & Industrial Strategy. JM acknowledges the Engineering and Physical Sciences Research Council for an Industrial CASE Studentship EP/S513635/1 with contributions from National Physical Laboratory and Johnson Matthey.References
- M. Thema, F. Bauer and M. Sterner, Power-to-gas: electrolysis and methanation status review, Renewable Sustainable Energy Rev., 2019, 112, 775–787 CrossRef CAS.
- J. Rissman, C. Bataille, E. Masanet, N. Aden, W. R. Morrow, N. Zhou, N. Elliott, R. Dell, N. Heeren, B. Huckestein, J. Cresko, S. A. Miller, J. Roy, P. Fennell, B. Cremmins, T. Koch Blank, D. Hone, E. D. Williams, S. de la Rue du Can, B. Sisson, M. Williams, J. Katzenberger, D. Burtraw, G. Sethi, H. Ping, D. Danielson, H. Lu, T. Lorber, J. Dinkel and J. Helseth, Technologies and policies to decarbonize global industry: review and assessment of mitigation drivers through 2070, Appl. Energy, 2020, 266, 114848 CrossRef CAS.
- J. M. Thomas, P. P. Edwards, P. J. Dobson and G. P. Owen, Decarbonising energy: the developing international activity in hydrogen technologies and fuel cells, J. Energy Chem., 2020, 51, 405–415 CrossRef.
- Hydrogen Council, How hydrogen empowers the energy transition, https://hydrogencouncil.com/wp-content/uploads/2017/06/Hydrogen-Council-Vision-Document.pdf Search PubMed.
- A. Buttler and H. Spliethoff, Current status of water electrolysis for energy storage, grid balancing and sector coupling via power-to-gas and power-to-liquids: a review, Renewable Sustainable Energy Rev., 2018, 82, 2440–2454 CrossRef CAS.
- K. Ayers, N. Danilovic, R. Ouimet, M. Carmo, B. Pivovar and M. Bornstein, Perspectives on Low-Temperature Electrolysis and Potential for Renewable Hydrogen at Scale, Annu. Rev. Chem. Biomol. Eng., 2019, 10, 219–239 CrossRef CAS PubMed.
- K. Ayers, The potential of proton exchange membrane-based electrolysis technology, Curr. Opin. Electrochem., 2019, 18, 9–15 CrossRef CAS.
- U. Babic, M. Suermann, F. N. Büchi, L. Gubler and T. J. Schmidt, Critical Review—Identifying Critical Gaps for Polymer Electrolyte Water Electrolysis Development, J. Electrochem. Soc., 2017, 164, F387–F399 CrossRef CAS.
- E. A. Byers, J. W. Hall and J. M. Amezaga, Electricity generation and cooling water use: UK pathways to 2050, Glob. Environ. Change, 2014, 25, 16–30 CrossRef.
- At home with water, https://www.energysavingtrust.org.uk/sites/default/files/reports/AtHomewithWater%287%29.pdf Search PubMed.
- A. Chapagain and S. Orr, UK Water Footprint: the impact of the UK's food and fibre consumption on global water resources, https://wwfeu.awsassets.panda.org/downloads/wwf_uk_footprint.pdf Search PubMed.
- NEL Hydrogen, M series containerized PEM electrolyser, https://nelhydrogen.com/product/m-series-containerized/ Search PubMed.
- Standard Specification for Reagent Water, https://www.astm.org/d1193-99.html Search PubMed.
- G. Tsotridis and A. Pilenga, EU harmonized protocols for testing of low temperature water electrolysis, Publications Office of the EU, 2021 Search PubMed.
- A. Mayyas, M. Ruth, B. Pivovar, G. Bender, K. Wipke, A. Mayyas, M. Ruth, B. Pivovar, G. Bender and K. Wipke, Manufacturing Cost Analysis for Proton Exchange Membrane Water Electrolyzers, National Renewable Energy Laboratory, NREL/TP-6A, 65, 2019 Search PubMed.
- M. Qasim, M. Badrelzaman, N. N. Darwish, N. A. Darwish and N. Hilal, Reverse osmosis desalination: a state-of-the-art review, Desalination, 2019, 459, 59–104 CrossRef CAS.
- U. Caldera, D. Bogdanov, S. Afanasyeva and C. Breyer, Role of Seawater Desalination in the Management of an Integrated Water and 100% Renewable Energy Based Power Sector in Saudi Arabia, Water, 2017, 10, 3 CrossRef.
- A. K. Plappally and J. H. Lienhard, Costs for water supply, treatment, end-use and reclamation, Desalin. Water Treat., 2013, 51, 200–232 CrossRef.
- Fuel cells and Hydrogen Joint Undertaking (FCH 2 JU), Annual Work Plan and Budget, 2019, https://ec.europa.eu/research/participants/data/ref/h2020/other/wp/jtis/h2020-wp19-fch_en.pdf Search PubMed.
- M. Carmo, D. L. Fritz, J. Mergel and D. Stolten, A comprehensive review on PEM water electrolysis, Int. J. Hydrogen Energy, 2013, 38, 4901–4934 CrossRef CAS.
- M. Bernt, A. Hartig-Weiß, M. F. Tovini, H. A. El-Sayed, C. Schramm, J. Schröter, C. Gebauer and H. A. Gasteiger, Current Challenges in Catalyst Development for PEM Water Electrolyzers, Chem.-Ing.-Tech., 2020, 92, 31–39 CrossRef CAS.
- S. A. Grigoriev, V. N. Fateev, D. G. Bessarabov and P. Millet, Current status, research trends, and challenges in water electrolysis science and technology, Int. J. Hydrogen Energy, 2020, 45, 26036–26058 CrossRef CAS.
- M. Götz, J. Lefebvre, F. Mörs, A. McDaniel Koch, F. Graf, S. Bajohr, R. Reimert and T. Kolb, Renewable Power-to-Gas: a technological and economic review, Renewable Energy, 2016, 85, 1371–1390 CrossRef.
- H. A. Miller, K. Bouzek, J. Hnat, S. Loos, C. I. Bernäcker, T. Weißgärber, L. Röntzsch and J. Meier-Haack, Green hydrogen from anion exchange membrane water electrolysis: a review of recent developments in critical materials and operating conditions, Sustainable Energy Fuels, 2020, 4, 2114–2133 RSC.
- I. Vincent and D. Bessarabov, Low cost hydrogen production by anion exchange membrane electrolysis: a review, Renewable Sustainable Energy Rev., 2018, 81, 1690–1704 CrossRef CAS.
- D. Henkensmeier, M. Najibah, C. Harms, J. Žitka, J. Hnát and K. Bouzek, Overview: State-of-the Art Commercial Membranes for Anion Exchange Membrane Water Electrolysis, J. Electrochem. Energy Convers. Storage, 2021, 18, 024001 CrossRef CAS.
- G. Bender, M. Carmo, T. Smolinka, A. Gago, N. Danilovic, M. Mueller, F. Ganci, A. Fallisch, P. Lettenmeier, K. A. Friedrich, K. Ayers, B. Pivovar, J. Mergel and D. Stolten, Initial approaches in benchmarking and round robin testing for proton exchange membrane water electrolyzers, Int. J. Hydrogen Energy, 2019, 44, 9174–9187 CrossRef CAS.
- P. Marocco, K. Sundseth, T. Aarhaug, A. Lanzini, M. Santarelli, A. O. Barnett and M. Thomassen, Online measurements of fluoride ions in proton exchange membrane water electrolysis through ion chromatography, J. Power Sources, 2021, 483, 229179 CrossRef CAS.
- J. Durst, A. Siebel, C. Simon, F. Hasché, J. Herranz and H. A. Gasteiger, New insights into the electrochemical hydrogen oxidation and evolution reaction mechanism, Energy Environ. Sci., 2014, 7, 2255–2260 RSC.
- D. Li, A. R. Motz, C. Bae, C. Fujimoto, G. Yang, F.-Y. Zhang, K. E. Ayers and Y. S. Kim, Durability of anion exchange membrane water electrolyzers, Energy Environ. Sci., 2021, 14, 3393–3419 RSC.
- K. Zeng and D. Zhang, Recent progress in alkaline water electrolysis for hydrogen production and applications, Prog. Energy Combust. Sci., 2010, 36, 307–326 CrossRef CAS.
- R. Phillips and C. W. Dunnill, Zero gap alkaline electrolysis cell design for renewable energy storage as hydrogen gas, RSC Adv., 2016, 6, 100643–100651 RSC.
- IRENA, Green Hydrogen Cost Reduction: Scaling up Electrolysers to Meet the 1.5 °C Climate Goal, https://www.irena.org/-/media/Files/IRENA/Agency/Publication/2020/Dec/IRENA_Green_hydrogen_cost_2020.pdf Search PubMed.
- M. Bodner, A. Hofer and V. Hacker, H2 generation from alkaline electrolyzer, Wiley Interdiscip. Rev.: Energy Environ., 2015, 4, 365–381 CAS.
- T. Bacquart, A. Murugan, M. Carré, B. Gozlan, F. Auprêtre, F. Haloua and T. A. Aarhaug, Probability of Occurrence of ISO 14687-2 Contaminants in Hydrogen: Principles and Examples from Steam Methane Reforming and Electrolysis (Water and Chlor-alkali) Production Processes Model, Int. J. Hydrogen Energy, 2018, 43, 11872–11883 CrossRef CAS.
- J. Brauns and T. Turek, Alkaline Water Electrolysis Powered by Renewable Energy: A Review, Processes, 2020, 8, 248 CrossRef CAS.
- S. Dresp, F. Dionigi, M. Klingenhof and P. Strasser, Direct electrolytic splitting of seawater: opportunities and challenges, ACS Energy Lett., 2019, 4, 933–942 CrossRef CAS.
- Y. Petrov, J. P. Schosger, Z. Stoynov and F. De Bruijn, Hydrogen evolution on nickel electrode in synthetic tap water - alkaline solution, Int. J. Hydrogen Energy, 2011, 36, 12715–12724 CrossRef CAS.
- J. Colt, in Dissolved Gas Concentration in Water: Computation as Functions of Temperature, Salinity and Pressure, Elsevier, 2012, pp. 179–197 Search PubMed.
- Hydrogen fuel quality — Product specification (ISO 14687:2019(E)), https://www.iso.org/standard/69539.html Search PubMed.
- E. Anderson, in 2nd International Workshop on Durability and Degradation Issues in PEM Electrolysis Cells and its Components, 2016 Search PubMed.
- J. Guo, Y. Zhang, A. Zavabeti, K. Chen, Y. Guo, G. Hu, X. Fan and G. K. Li, Hydrogen production from the air, Nat. Commun., 2022, 13, 1–9 Search PubMed.
- H. K. Ju, S. Badwal and S. Giddey, A comprehensive review of carbon and hydrocarbon assisted water electrolysis for hydrogen production, Appl. Energy, 2018, 231, 502–533 CrossRef CAS.
- C. Coutanceau and S. Baranton, Electrochemical conversion of alcohols for hydrogen production: a short overview, Wiley Interdiscip. Rev.: Energy Environ., 2016, 5, 388–400 CAS.
- M. Opu, G. Bender, C. S. Macomber, J. W. Van Zee and H. N. Dinh, Understanding the Effects of PEMFC Contamination from Balance of Plant Assembly Aids Materials: In Situ Studies, J. Electrochem. Soc., 2015, 162, F1011–F1019 CrossRef CAS.
- X. Dong, R. G. Iacocca, B. L. Bustard and C. A. J. Kemp, Investigation of Stainless Steel Corrosion in Ultrahigh-Purity Water and Steam Systems by Surface Analytical Techniques, J. Mater. Eng. Perform., 2010, 19, 135–141 CrossRef CAS.
- S. Sun, Z. Shao, H. Yu, G. Li and B. Yi, Investigations on degradation of the long-term proton exchange membrane water electrolysis stack, J. Power Sources, 2014, 267, 515–520 CrossRef CAS.
- S. A. Grigoriev, K. A. Dzhus, D. G. Bessarabov and P. Millet, Failure of PEM water electrolysis cells: case study involving anode dissolution and membrane thinning, Int. J. Hydrogen Energy, 2014, 39, 20440–20446 CrossRef CAS.
- E. Brightman, J. Dodwell, N. Van Dijk and G. Hinds, In situ characterisation of PEM water electrolysers using a novel reference electrode, Electrochem. Commun., 2015, 52, 1–4 CrossRef CAS.
- C. Rakousky, U. Reimer, K. Wippermann, S. Kuhri, M. Carmo, W. Lueke and D. Stolten, Polymer electrolyte membrane water electrolysis: restraining degradation in the presence of fluctuating power, J. Power Sources, 2017, 342, 38–47 CrossRef CAS.
- C. Rakousky, U. Reimer, K. Wippermann, M. Carmo, W. Lueke and D. Stolten, An analysis of degradation phenomena in polymer electrolyte membrane water electrolysis, J. Power Sources, 2016, 326, 120–128 CrossRef CAS.
- S. A. Grigoriev, D. G. Bessarabov and A. S. Glukhov, On the contamination of membrane–electrode assembles of water electrolyzers with solid polymer electrolyte by the elements of titanium alloys, Russ. J. Electrochem., 2017, 53, 808–812 CrossRef CAS.
- S. Cherevko, T. Reier, A. R. Zeradjanin, Z. Pawolek, P. Strasser and K. J. J. Mayrhofer, Stability of nanostructured iridium oxide electrocatalysts during oxygen evolution reaction in acidic environment, Electrochem. Commun., 2014, 48, 81–85 CrossRef CAS.
- S. Siracusano, N. Van Dijk, R. Backhouse, L. Merlo, V. Baglio and A. S. Aricò, Degradation issues of PEM electrolysis MEAs, Renewable Energy, 2018, 123, 52–57 CrossRef CAS.
- C. Fonseca and M. Barbosa, Corrosion behaviour of titanium in biofluids containing H2O2 studied by electrochemical impedance spectroscopy, Corros. Sci., 2001, 43, 547–559 CrossRef CAS.
- H. Becker, L. Castanheira and G. Hinds, Local measurement of current collector potential in a polymer electrolyte membrane water electrolyser, J. Power Sources, 2020, 448, 227563 CrossRef CAS.
- J. Mo, S. Steen, Z. Kang, G. Yang, D. A. Taylor, Y. Li, T. J. Toops, M. P. Brady, S. T. Retterer, D. A. Cullen, J. B. Green and F.-Y. Zhang, Study on corrosion migrations within catalyst-coated membranes of proton exchange membrane electrolyzer cells, Int. J. Hydrogen Energy, 2017, 42, 27343–27349 CrossRef CAS.
- J. Mo, S. M. Steen, F. Y. Zhang, T. J. Toops, M. P. Brady and J. B. Green, Electrochemical investigation of stainless steel corrosion in a proton exchange membrane electrolyzer cell, Int. J. Hydrogen Energy, 2015, 40, 12506–12511 CrossRef CAS.
- T. Reier, M. Oezaslan and P. Strasser, Electrocatalytic Oxygen Evolution Reaction (OER) on Ru, Ir, and Pt Catalysts: A Comparative Study of Nanoparticles and Bulk Materials, ACS Catal., 2012, 2, 1765–1772 CrossRef CAS.
- K. E. Ayers, L. T. Dalton and E. B. Anderson, Efficient Generation of High Energy Density Fuel from Water, ECS Trans., 2019, 41, 27–38 CrossRef.
- C. Daiane Ferreira Da Silva, F. Claudel, V. Martin, R. Chattot, S. Abbou, K. Kumar, I. Jiménez-Morales, S. Cavaliere, D. Jones, J. Rozière, L. Solà-Hernandez, C. Beauger, M. Faustini, J. Peron, B. Gilles, T. Encinas, L. Piccolo, F. H. Barros De Lima, L. Dubau and F. Maillard, Oxygen Evolution Reaction Activity and Stability Benchmarks for Supported and Unsupported IrOx Electrocatalysts, ACS Catal., 2021, 11, 4107–4116 CrossRef CAS.
- M. Milosevic, J. Knöppel, K. Ehelebe, D. Abbas, D. Escalera López, S. Thiele and S. Cherevko, Catalyst Dissolution Analysis in PEM Water Electrolyzers during Intermittent Operation, ECS Meet. Abstr., 2022, 1369 CrossRef.
- P. J. Rheinländer and J. Durst, Transformation of the OER-Active IrOx Species under Transient Operation Conditions in PEM Water Electrolysis, J. Electrochem. Soc., 2021, 168, 024511 CrossRef.
- S. Cherevko, S. Geiger, O. Kasian, A. Mingers and K. J. J. Mayrhofer, Oxygen evolution activity and stability of iridium in acidic media. Part 1. – Metallic iridium, J. Electroanal. Chem., 2016, 773, 69–78 CrossRef CAS.
- S. Cherevko, S. Geiger, O. Kasian, A. Mingers and K. J. J. Mayrhofer, Oxygen evolution activity and stability of iridium in acidic media. Part 2. – Electrochemically grown hydrous iridium oxide, J. Electroanal. Chem., 2016, 774, 102–110 CrossRef CAS.
- J. Dodwell, M. Maier, J. Majasan, R. Jervis, L. Castanheira, P. Shearing, G. Hinds and D. J. L. Brett, Open-circuit dissolution of platinum from the cathode in polymer electrolyte membrane water electrolysers, J. Power Sources, 2021, 498, 229937 CrossRef CAS.
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Activity benchmarks and requirements for Pt, Pt-alloy, and non-Pt oxygen reduction catalysts for PEMFCs, Appl. Catal., B, 2005, 56, 9–35 CrossRef CAS.
- H. Yu, L. Bonville, J. Jankovic and R. Maric, Microscopic insights on the degradation of a PEM water electrolyzer with ultra-low catalyst loading, Appl. Catal., B, 2020, 260, 118194 CrossRef CAS.
- B. Beverskog and I. Puigdomenech, Revised Pourbaix diagrams for nickel at 25–300 °C, Corros. Sci., 1997, 39, 969–980 CrossRef CAS.
- Y. Yan, B. Y. Xia, B. Zhao and X. Wang, A review on noble-metal-free bifunctional heterogeneous catalysts for overall electrochemical water splitting, J. Mater. Chem. A, 2016, 4, 17587–17603 RSC.
- C. K. Kjartansdóttir, L. P. Nielsen and P. Møller, Development of durable and efficient electrodes for large-scale alkaline water electrolysis, Int. J. Hydrogen Energy, 2013, 38, 8221–8231 CrossRef.
- S. Kotrel and S. Brauninger, in Handbook of Heterogeneous Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2008 Search PubMed.
- M. B. I. Janjua and R. L. Le Roy, Electrocatalyst performance in industrial water electrolysers, Int. J. Hydrogen Energy, 1985, 10, 11–19 CrossRef CAS.
- D. Xu, M. B. Stevens, M. R. Cosby, S. Z. Oener, A. M. Smith, L. J. Enman, K. E. Ayers, C. B. Capuano, J. N. Renner, N. Danilovic, Y. Li, H. Wang, Q. Zhang and S. W. Boettcher, Earth-Abundant Oxygen Electrocatalysts for Alkaline Anion-Exchange-Membrane Water Electrolysis: Effects of Catalyst Conductivity and Comparison with Performance in Three-Electrode Cells, ACS Catal., 2019, 9, 7–15 CrossRef CAS.
- A. N. Colli, H. H. Girault and A. Battistel, Non-precious electrodes for practical alkaline water electrolysis, Materials, 2019, 12, 1–17 CrossRef PubMed.
- J.-M. Ye, D.-H. He, F. Li, Y.-L. Li and J.-B. He, Roles of soluble species in the alkaline oxygen evolution reaction on a nickel anode, Chem. Commun., 2018, 54, 10116–10119 RSC.
- J. J. Kaczur, H. Yang, Z. Liu, S. D. Sajjad and R. I. Masel, Carbon dioxide and water electrolysis using new alkaline stable anion membranes, Front. Chem., 2018, 6, 263 CrossRef PubMed.
- Y. Leng, G. Chen, A. J. Mendoza, T. B. Tighe, M. A. Hickner and C.-Y. Wang, Solid-State Water Electrolysis with an Alkaline Membrane, J. Am. Chem. Soc., 2012, 134, 9054–9057 CrossRef CAS PubMed.
- N. S. Luna, G. Correa-Perelmuter, G. I. Lacconi, L. A. Diaz and E. A. Franceschini, In operando activation of alkaline electrolyzer by ruthenium spontaneous deposition, J. Solid State Electrochem., 2021, 25, 1019–1027 CrossRef CAS.
- D. V. Shinde, T. M. Kokumai, J. Buha, M. Prato, L. De Trizio and L. Manna, A robust and highly active hydrogen evolution catalyst based on Ru nanocrystals supported on vertically oriented Cu nanoplates, J. Mater. Chem. A, 2020, 8, 10787–10795 RSC.
- J. Mahmood, F. Li, S. M. Jung, M. S. Okyay, I. Ahmad, S. J. Kim, N. Park, H. Y. Jeong and J. B. Baek, An efficient and pH-universal ruthenium-based catalyst for the hydrogen evolution reaction, Nat. Nanotechnol., 2017, 12, 441–446 CrossRef CAS PubMed.
- D. H. Kweon, M. S. Okyay, S.-J. Kim, J.-P. Jeon, H.-J. Noh, N. Park, J. Mahmood and J.-B. Baek, Ruthenium anchored on carbon nanotube electrocatalyst for hydrogen production with enhanced faradaic efficiency, Nat. Commun., 2020, 11, 1278 CrossRef CAS PubMed.
- C. Chen and T. F. Fuller, The effect of humidity on the degradation of Nafion® membrane, Polym. Degrad. Stab., 2009, 94, 1436–1447 CrossRef CAS.
- J. Parrondo, Z. Wang, M.-S. J. Jung and V. Ramani, Reactive oxygen species accelerate degradation of anion exchange membranes based on polyphenylene oxide in alkaline environments, Phys. Chem. Chem. Phys., 2016, 18, 19705–19712 RSC.
- C. Lim, A. S. Alavijeh, M. Lauritzen, J. Kolodziej, S. Knights and E. Kjeang, Fuel Cell Durability Enhancement with Cerium Oxide under Combined Chemical and Mechanical Membrane Degradation, ECS Electrochem. Lett., 2015, 4, F29–F31 CrossRef CAS.
- A. M. Baker, R. Mukundan, D. Spernjak, E. J. Judge, S. G. Advani, A. K. Prasad and R. L. Borup, Cerium Migration during PEM Fuel Cell Accelerated Stress Testing, J. Electrochem. Soc., 2016, 163, F1023–F1031 CrossRef CAS.
- M. Zatoń, B. Prélot, N. Donzel, J. Rozière and D. J. Jones, Migration of Ce and Mn Ions in PEMFC and Its Impact on PFSA Membrane Degradation, J. Electrochem. Soc., 2018, 165, F3281–F3289 CrossRef.
- S. Klaus, Y. Cai, M. W. Louie, L. Trotochaud and A. T. Bell, Effects of Fe electrolyte impurities on Ni(OH)2/NiOOH structure and oxygen evolution activity, J. Phys. Chem. C, 2015, 119, 7243–7254 CrossRef CAS.
- A. S. Aricò, S. Siracusano, N. Briguglio, V. Baglio, A. Di Blasi and V. Antonucci, Polymer electrolyte membrane water electrolysis: status of technologies and potential applications in combination with renewable power sources, J. Appl. Electrochem., 2013, 43, 107–118 CrossRef.
- K. Hengge, C. Heinzl, M. Perchthaler, D. Varley, T. Lochner and C. Scheu, Unraveling micro- and nanoscale degradation processes during operation of high-temperature polymer-electrolyte-membrane fuel cells, J. Power Sources, 2017, 364, 437–448 CrossRef CAS.
- S. S. Kocha and B. G. Pollet, Advances in Rapid and Effective Break-in/Conditioning/Recovery of Automotive PEMFC Stacks, Curr. Opin. Electrochem., 2021, 100843 Search PubMed.
- X. Yuan, S. Zhang, J. C. Sun and H. Wang, A review of accelerated conditioning for a polymer electrolyte membrane fuel cell, J. Power Sources, 2011, 196, 9097–9106 CrossRef CAS.
- D. Yang, Y. Lan, T. Chu, B. Li, P. Ming, C. Zhang and X. Zhou, Rapid activation of a full-length proton exchange membrane fuel cell stack with a novel intermittent oxygen starvation method, Energy, 2022, 260, 125154 CrossRef CAS.
- F. Fouda-Onana, M. Chandesris, V. Médeau, S. Chelghoum, D. Thoby and N. Guillet, Investigation on the degradation of MEAs for PEM water electrolysers part I: effects of testing conditions on MEA performances and membrane properties, Int. J. Hydrogen Energy, 2016, 41, 16627–16636 CrossRef CAS.
- T. Okada, N. Nakamura, M. Yuasa and I. Sekine, Ion and Water Transport Characteristics in Membranes for Polymer Electrolyte Fuel Cells Containing H+ and Ca2+ Cations, J. Electrochem. Soc., 1997, 144, 2744–2750 CrossRef CAS.
- T. Okada, S. Møller-Holst, O. Gorseth and S. Kjelstrup, Transport and equilibrium properties of Nafion® membranes with H+ and Na+ ions, J. Electroanal. Chem., 1998, 442, 137–145 CrossRef CAS.
- T. Okada, J. Dale, Y. Ayato, O. A. Asbjørnsen, M. Yuasa and I. Sekine, Unprecedented effect of impurity cations on the oxygen reduction kinetics at platinum electrodes covered with perfluorinated ionomer, Langmuir, 1999, 15, 8490–8496 CrossRef CAS.
- T. Okada, G. Xie, O. Gorseth, S. Kjelstrup, N. Nakamura and T. Arimura, Ion and water transport characteristics of Nafion membranes as electrolytes, Electrochim. Acta, 1998, 43, 3741–3747 CrossRef CAS.
- T. Okada, Y. Ayato, J. Dale, M. Yuasa, I. Sekine and O. Andreas Asbjørnsen, Oxygen reduction kinetics at platinum electrodes covered with perfluorinated ionomer in the presence of impurity cations Fe3+, Ni2+ and Cu2+, Phys. Chem. Chem. Phys., 2000, 2, 3255–3261 RSC.
- T. Okada, Y. Ayato, H. Satou, M. Yuasa and I. Sekine, The effect of impurity cations on the oxygen reduction kinetics at platinum electrodes covered with perfluorinated ionomer, J. Phys. Chem. B, 2001, 105, 6980–6986 CrossRef CAS.
- T. Okada, H. Satou, M. Okuno and M. Yuasa, Ion and water transport characteristics of perfluorosulfonate ionomer membranes with H+ and alkali metal cations, J. Phys. Chem. B, 2002, 106, 1267–1273 CrossRef CAS.
- T. Okada, H. Satou and M. Yuasa, Effects of Additives on Oxygen Reduction Kinetics at the Interface between Platinum and Perfluorinated Ionomer, Langmuir, 2003, 19, 2325–2332 CrossRef CAS.
- M. Saito, K. Hayamizu and T. Okada, Temperature dependence of ion and water transport in perfluorinated ionomer membranes for fuel cells, J. Phys. Chem. B, 2005, 109, 3112–3119 CrossRef CAS PubMed.
- T. Okada, Y. Ayato, M. Yuasa and I. Sekine, The effect of impurity cations on the transport characteristics of perfluorosulfonate ionomer membranes, J. Phys. Chem. B, 1999, 103, 3315–3322 CrossRef CAS.
- A. Kusoglu and A. Z. Weber, New Insights into Perfluorinated Sulfonic-Acid Ionomers, Chem. Rev., 2017, 117, 987–1104 CrossRef CAS PubMed.
- K. Hongsirikarn, J. G. Goodwin, S. Greenway and S. Creager, Effect of cations (Na+, Ca2+, Fe3+) on the conductivity of a Nafion membrane, J. Power Sources, 2010, 195, 7213–7220 CrossRef CAS.
- S. Shi, A. Z. Weber and A. Kusoglu, Structure-Transport Relationship of Perfluorosulfonic-Acid Membranes in Different Cationic Forms, Electrochim. Acta, 2016, 220, 517–528 CrossRef CAS.
- K. Hongsirikarn, J. G. Goodwin, S. Greenway and S. Creager, Effect of cations (Na+, Ca2+, Fe3+) on the conductivity of a Nafion membrane, J. Power Sources, 2010, 195, 7213–7220 CrossRef CAS.
- S. Logette, C. Eysseric, G. Pourcelly, A. Lindheimer and C. Gavach, Selective permeability of a perfluorosulphonic membrane to different valency cations. Ion-exchange isotherms and kinetic aspects, J. Membr. Sci., 1998, 144, 259–274 CrossRef CAS.
- M. J. Kelly, B. Egger, G. Fafilek, J. O. Besenhard, H. Kronberger and G. E. Nauer, Conductivity of polymer electrolyte membranes by impedance spectroscopy with microelectrodes, Solid State Ionics, 2005, 176, 2111–2114 CrossRef CAS.
- M. J. Kelly, G. Fafilek, J. O. Besenhard, H. Kronberger and G. E. Nauer, Contaminant absorption and conductivity in polymer electrolyte membranes, J. Power Sources, 2005, 145, 249–252 CrossRef CAS.
- H. Wang and J. A. Turner, The influence of metal ions on the conductivity of Nafion 112 in polymer electrolyte membrane fuel cell, J. Power Sources, 2008, 183, 576–580 CrossRef CAS.
- R. Halseid, P. J. S. Vie and R. Tunold, Influence of Ammonium on Conductivity and Water Content of Nafion 117 Membranes, J. Electrochem. Soc., 2004, 151, A381 CrossRef CAS.
- K. Hongsirikarn, J. G. Goodwin, S. Greenway and S. Creager, Influence of ammonia on the conductivity of Nafion membranes, J. Power Sources, 2010, 195, 30–38 CrossRef CAS.
- T. Lopes, D. S. Kim, Y. S. Kim and F. H. Garzon, Ionic Transport and Water Vapor Uptake of Ammonium Exchanged Perfluorosulfonic Acid Membranes, J. Electrochem. Soc., 2012, 159, B265–B269 CrossRef CAS.
- A. Verdaguer-Casadevall, P. Hernandez-Fernandez, I. E. L. Stephens, I. Chorkendorff and S. Dahl, The effect of ammonia upon the electrocatalysis of hydrogen oxidation and oxygen reduction on polycrystalline platinum, J. Power Sources, 2012, 220, 205–210 CrossRef CAS.
- T. Okada, Ion and Water Transport, Characteristics in Membranes for Polymer Electrolyte Fuel Cells Containing H+ and Ca2+ Cations, J. Electrochem. Soc., 1997, 144, 2744 CrossRef CAS.
- C. Bas, N. D. Albérola and L. Flandin, Effects of contaminant on thermal properties in perfluorinated sulfonic acid membranes, J. Membr. Sci., 2010, 363, 67–71 CrossRef CAS.
- W. Xu and K. Scott, The effects of ionomer content on PEM water electrolyser membrane electrode assembly performance, Int. J. Hydrogen Energy, 2010, 35, 12029–12037 CrossRef CAS.
- M. Bernt, A. Siebel and H. A. Gasteiger, Analysis of Voltage Losses in PEM Water Electrolyzers with Low Platinum Group Metal Loadings, J. Electrochem. Soc., 2018, 165, F305–F314 CrossRef CAS.
- L. Zhang, X. Jie, Z.-G. Shao, Z.-M. Zhou, G. Xiao and B. Yi, The influence of sodium ion on the solid polymer electrolyte water electrolysis, Int. J. Hydrogen Energy, 2012, 37, 1321–1325 CrossRef CAS.
- L. Zhang, X. Jie, Z.-G. Shao, X. Wang and B. Yi, The dynamic-state effects of sodium ion contamination on the solid polymer electrolyte water electrolysis, J. Power Sources, 2013, 241, 341–348 CrossRef CAS.
- U. Babic, M. Zlobinski, T. J. Schmidt, P. Boillat and L. Gubler, CO2− Assisted Regeneration of a Polymer Electrolyte Water Electrolyzer Contaminated with Metal Ion Impurities, J. Electrochem. Soc., 2019, 166, F610–F619 CrossRef CAS.
- X. Wang, L. Zhang, G. Li, G. Zhang, Z.-G. Shao and B. Yi, The influence of Ferric ion contamination on the solid polymer electrolyte water electrolysis performance, Electrochim. Acta, 2015, 158, 253–257 CrossRef CAS.
- N. Li, S. S. Araya and S. K. Kær, The effect of Fe3+ contamination in feed water on proton exchange membrane electrolyzer performance, Int. J. Hydrogen Energy, 2019, 44, 12952–12957 CrossRef CAS.
- N. Li, S. S. Araya and S. K. Kær, Long-term contamination effect of iron ions on cell performance degradation of proton exchange membrane water electrolyser, J. Power Sources, 2019, 434, 226755 CrossRef CAS.
- M. Zlobinski, U. Babic, M. Fikry, L. Gubler, T. J. Schmidt and P. Boillat, Dynamic Neutron Imaging and Modeling of Cationic Impurities in Polymer Electrolyte Water Electrolyzer, J. Electrochem. Soc., 2020, 167, 144509 CrossRef CAS.
- J.-P. B. Haraldsted, Z. Révay, R. Frydendal, A. Verdaguer-Casadevall, J. Rossmeisl, J. Kibsgaard and I. Chorkendorff, Trace anodic migration of iridium and titanium ions and subsequent cathodic selectivity degradation in acid electrolysis systems, Mater. Today Energy, 2019, 14, 100352 CrossRef.
- E. R. Kötz and S. Stucki, Ruthenium dioxide as a hydrogen-evolving electrode, J. Appl. Electrochem., 1987, 17, 1190–1197 CrossRef.
- D. M. Kolb, M. Przasnyski and H. Gerischer, Underpotential deposition of metals and work function differences, J. Electroanal. Chem. Interfacial Electrochem., 1974, 54, 25–38 CrossRef CAS.
- P. Millet, T. Alleau and R. Durand, Characterization of membrane-electrode assemblies for solid polymer electrolyte water electrolysis, J. Appl. Electrochem., 1993, 23, 322–331 CrossRef CAS.
- K. Yasuda, A. Taniguchi, T. Akita, T. Ioroi and Z. Siroma, Platinum dissolution and deposition in the polymer electrolyte membrane of a PEM fuel cell as studied by potential cycling, Phys. Chem. Chem. Phys., 2006, 8, 746–752 RSC.
- C. Klose, P. Trinke, T. Böhm, B. Bensmann, S. Vierrath, R. Hanke-Rauschenbach and S. Thiele, Membrane Interlayer with Pt Recombination Particles for Reduction of the Anodic Hydrogen Content in PEM Water Electrolysis, J. Electrochem. Soc., 2018, 165, F1271–F1277 CrossRef CAS.
- S. Garbe, E. Samulesson, T. J. Schmidt and L. Gubler, Comparison of Pt-Doped Membranes for Gas Crossover Suppression in Polymer Electrolyte Water Electrolysis, J. Electrochem. Soc., 2021, 168, 104502 CrossRef CAS.
- A. Collier, H. Wang, X. Zi Yuan, J. Zhang and D. P. Wilkinson, Degradation of polymer electrolyte membranes, Int. J. Hydrogen Energy, 2006, 31, 1838–1854 CrossRef CAS.
- V. O. Mittal, H. R. Kunz and J. M. Fenton, Membrane Degradation Mechanisms in PEMFCs, J. Electrochem. Soc., 2007, 154, B652 CrossRef CAS.
- Q. Feng, X. Z. Yuan, G. Liu, B. Wei, Z. Zhang, H. Li and H. Wang, A review of proton exchange membrane water electrolysis on degradation mechanisms and mitigation strategies, J. Power Sources, 2017, 366, 33–55 CrossRef CAS.
- M. Chandesris, V. Médeau, N. Guillet, S. Chelghoum, D. Thoby and F. Fouda-Onana, Membrane degradation in PEM water electrolyzer: Numerical modeling and experimental evidence of the influence of temperature and current density, Int. J. Hydrogen Energy, 2015, 40, 1353–1366 CrossRef CAS.
- S. Stucki, G. G. Scherer, S. Schlagowski and E. Fischer, PEM water electrolysers: Evidence for membrane failure in 100 kW demonstration plants, J. Appl. Electrochem., 1998, 28, 1041–1049 CrossRef CAS.
- P. S. Ruvinskiy, A. Bonnefont, C. Pham-Huu and E. R. Savinova, Using Ordered Carbon Nanomaterials for Shedding Light on the Mechanism of the Cathodic Oxygen Reduction Reaction, Langmuir, 2011, 27, 9018–9027 CrossRef CAS PubMed.
- S. H. Frensch, G. Serre, F. Fouda-Onana, H. C. Jensen, M. L. Christensen, S. S. Araya and S. K. Kær, Impact of iron and hydrogen peroxide on membrane degradation for polymer electrolyte membrane water electrolysis: computational and experimental investigation on fluoride emission, J. Power Sources, 2019, 420, 54–62 CrossRef CAS.
- W. Tong, M. Forster, F. Dionigi, S. Dresp, R. Sadeghi Erami, P. Strasser, A. J. Cowan and P. Farràs, Electrolysis of low-grade and saline surface water, Nat. Energy, 2020, 5, 367–377 CrossRef CAS.
- J. Bennett, Electrodes for generation of hydrogen and oxygen from seawater, Int. J. Hydrogen Energy, 1980, 5, 401–408 CrossRef CAS.
- H. A. Hansen, I. C. Man, F. Studt, F. Abild-Pedersen, T. Bligaard and J. Rossmeisl, Electrochemical chlorine evolution at rutile oxide (110) surfaces, Phys. Chem. Chem. Phys., 2010, 12, 283–290 RSC.
- Y. Takasu, T. Arikawa, S. Sunohara and K. Yahikozawa, Direct detection of competitively evolving chlorine and oxygen at anodes, J. Electroanal. Chem., 1993, 361, 279–281 CrossRef CAS.
- S. Trasatti, Electrocatalysis in the anodic evolution of oxygen and chlorine, Electrochim. Acta, 1984, 29, 1503–1512 CrossRef CAS.
- J. G. Vos and M. T. M. Koper, Measurement of competition between oxygen evolution and chlorine evolution using rotating ring-disk electrode voltammetry, J. Electroanal. Chem., 2018, 819, 260–268 CrossRef CAS.
- F. Dionigi, T. Reier, Z. Pawolek, M. Gliech and P. Strasser, Design Criteria, Operating Conditions, and Nickel-Iron Hydroxide Catalyst Materials for Selective Seawater Electrolysis, ChemSusChem, 2016, 9, 962–972 CrossRef CAS PubMed.
- M. C. Santos, D. W. Miwa and S. A. S. Machado, Study of anion adsorption on polycrystalline Pt by electrochemical quartz crystal microbalance, Electrochem. Commun., 2000, 2, 692–696 CrossRef CAS.
- T. J. Schmidt, U. A. Paulus, H. A. Gasteiger and R. J. Behm, The oxygen reduction reaction on a Pt/carbon fuel cell catalyst in the presence of chloride anions, J. Electroanal. Chem., 2001, 508, 41–47 CrossRef CAS.
- T. M. Arruda, B. Shyam, J. M. Ziegelbauer, S. Mukerjee and D. E. Ramaker, Investigation into the competitive and site-specific nature of anion adsorption on Pt using in situ X-ray absorption spectroscopy, J. Phys. Chem. C, 2008, 112, 18087–18097 CrossRef CAS.
- K. Saadi, P. Nanikashvili, Z. Tatus-Portnoy, S. Hardisty, V. Shokhen, M. Zysler and D. Zitoun, Crossover-tolerant coated platinum catalysts in hydrogen/bromine redox flow battery, J. Power Sources, 2019, 422, 84–91 CrossRef CAS.
- M. W. Breiter, Voltammetric study of halide ion adsorption on platinum in perchloric acid solutions, Electrochim. Acta, 1963, 8, 925–935 CrossRef.
- S. Ferro and A. De Battisti, The bromine electrode. Part I: adsorption phenomena at polycrystalline platinum electrodes, J. Appl. Electrochem., 2004, 34, 981–987 CrossRef CAS.
- N. M. Marković, C. A. Lucas, H. A. Gasteiger and P. N. Ross, Bromide adsorption on Pt(100): rotating ring-Pt(100) disk electrode and surface X-ray scattering measurements, Surf. Sci., 1996, 365, 229–240 CrossRef.
- G. N. Salaita, D. A. Stern, F. Lu, B. Helmut, D. G. Frank, B. C. Schardt, J. L. Stickney, M. P. Soriaga and A. T. Hubbard, Structure and Composition of a Platinum(111) Surface as a Function of pH and Electrode Potential in Aqueous Bromide Solutions, Langmuir, 1986, 2, 828–835 CrossRef CAS.
- M. Goor-Dar, N. Travitsky and E. Peled, Study of hydrogen redox reactions on platinum nanoparticles in concentrated HBr solutions, J. Power Sources, 2012, 197, 111–115 CrossRef CAS.
- Z. X. Shu and S. Bruckenstein, Iodine adsorption studies at platinum, J. Electroanal. Chem., 1991, 317, 263–277 CrossRef CAS.
- D. C. Johnson, A Study of the Adsorption and Desorption of Iodine and Iodide at Platinum Electrodes in 1.0M Sulfuric Acid, J. Electrochem. Soc., 1972, 119, 331 CrossRef CAS.
- V. S. Bagotzky, Y. B. Vassilyev, J. Weber and J. N. Pirtskhalava, Adsorption of anions on smooth platinum electrodes, J. Electroanal. Chem., 1970, 27, 31–46 CrossRef.
- Y. Matsuda, T. Shimizu and S. Mitsushima, Adsorption behavior of low concentration carbon monoxide on polymer electrolyte fuel cell anodes for automotive applications, J. Power Sources, 2016, 318, 1–8 CrossRef CAS.
- H. Becker, T. Bacquart, M. Perkins, N. Moore, J. Ihonen, G. Hinds and G. Smith, Operando characterisation of the impact of carbon monoxide on PEMFC performance using isotopic labelling and gas analysis, J. Power Sources Adv., 2020, 6, 100036 CrossRef.
- I. Katsounaros, J. C. Meier and K. J. J. Mayrhofer, The impact of chloride ions and the catalyst loading on the reduction of H2O2 on high-surface-area platinum catalysts, Electrochim. Acta, 2013, 110, 790–795 CrossRef CAS.
- A. Kolics and A. Wieckowski, Adsorption of bisulfate and sulfate anions on a Pt(111) electrode, J. Phys. Chem. B, 2001, 105, 2588–2595 CrossRef CAS.
- F. Gossenberger, F. Juarez and A. Groß, Sulfate, Bisulfate, and Hydrogen Co-adsorption on Pt(111) and Au(111) in an Electrochemical Environment, Front. Chem., 2020, 8, 1–11 CrossRef PubMed.
- A. Ganassin, V. Colic, J. Tymoczko, A. S. Bandarenka and W. Schuhmann, Non-covalent interactions in water electrolysis: influence on the activity of Pt(111) and iridium oxide catalysts in acidic media, Phys. Chem. Chem. Phys., 2015, 17, 8349–8355 RSC.
- J. A. Arminio-Ravelo, A. W. Jensen, K. D. Jensen, J. Quinson and M. Escudero-Escribano, Electrolyte Effects on the Electrocatalytic Performance of Iridium-Based Nanoparticles for Oxygen Evolution in Rotating Disc Electrodes, ChemPhysChem, 2019, 20, 2956–2963 CrossRef CAS PubMed.
- S. Mitsushima, Y. Koizumi, S. Uzuka and K. I. Ota, Dissolution of platinum in acidic media, Electrochim. Acta, 2008, 54, 455–460 CrossRef CAS.
- S. Geiger, S. Cherevko and K. J. J. Mayrhofer, Dissolution of Platinum in Presence of Chloride Traces, Electrochim. Acta, 2015, 179, 24–31 CrossRef CAS.
- S. Kanamura and M. Yagyu, Electrochemical dissolution of platinum and ruthenium from membrane electrode assemblies of polymer electrolyte fuel cells, Mater. Trans., 2016, 57, 1972–1976 CrossRef CAS.
- A. P. Yadav, A. Nishikata and T. Tsuru, Effect of halogen ions on platinum dissolution under potential cycling in 0.5 M H2SO4 solution, Electrochim. Acta, 2007, 52, 7444–7452 CrossRef CAS.
- K. Matsuoka, S. Sakamoto, K. Nakato, A. Hamada and Y. Itoh, Degradation of polymer electrolyte fuel cells under the existence of anion species, J. Power Sources, 2008, 179, 560–565 CrossRef CAS.
- Y. Sugawara, T. Okayasu, A. P. Yadav, A. Nishikata and T. Tsuru, Dissolution Mechanism of Platinum in Sulfuric Acid Solution, J. Electrochem. Soc., 2012, 159, F779–F786 CrossRef CAS.
- A. Pavlišič, P. Jovanovič, V. S. Šelih, M. Šala, N. Hodnik, S. Hočevar and M. Gaberšček, The influence of chloride impurities on Pt/C fuel cell catalyst corrosion, Chem. Commun., 2014, 50, 3732–3734 RSC.
- Z. Wang, E. Tada and A. Nishikata, In Situ Analysis of Chloride Effect on Platinum Dissolution by a Channel-Flow Multi-Electrode System, J. Electrochem. Soc., 2014, 161, F845–F849 CrossRef CAS.
- A. Lam, H. Li, S. Zhang, H. Wang, D. P. Wilkinson, S. Wessel and T. T. H. Cheng, Ex situ study of chloride contamination on carbon supported Pt catalyst, J. Power Sources, 2012, 205, 235–238 CrossRef CAS.
- S. Mitsushima, Y. Koizumi, S. Uzuka and K.-I. Ota, Dissolution of platinum in acidic media, Electrochim. Acta, 2008, 54, 455–460 CrossRef CAS.
- A. B. Ofstad, M. S. Thomassen, J. L. Gomez de la Fuente, F. Seland, S. Møller-Holst and S. Sunde, Assessment of Platinum Dissolution from a Pt/C Fuel Cell Catalyst: An Electrochemical Quartz Crystal Microbalance Study, J. Electrochem. Soc., 2010, 157, B621 CrossRef CAS.
- A. A. Topalov, S. Cherevko, A. R. Zeradjanin, J. C. Meier, I. Katsounaros and K. J. J. Mayrhofer, Towards a comprehensive understanding of platinum dissolution in acidic media, Chem. Sci., 2014, 5, 631 RSC.
- A. A. Topalov, I. Katsounaros, M. Auinger, S. Cherevko, J. C. Meier, S. O. Klemm and K. J. J. Mayrhofer, Dissolution of platinum: limits for the deployment of electrochemical energy conversion?, Angew. Chem., Int. Ed., 2012, 51, 12613–12615 CrossRef CAS PubMed.
- E. Brightman and G. Hinds, In situ mapping of potential transients during start-up and shut-down of a polymer electrolyte membrane fuel cell, J. Power Sources, 2014, 267, 160–170 CrossRef CAS.
- J. G. Vos, Z. Liu, F. D. Speck, N. Perini, W. Fu, S. Cherevko and M. T. M. Koper, Selectivity Trends between Oxygen Evolution and Chlorine Evolution on Iridium-Based Double Perovskites in Acidic Media, ACS Catal., 2019, 9, 8561–8574 CrossRef CAS.
- S. Trasatti, Electrocatalysis: understanding the success of DSA®, Electrochim. Acta, 2000, 45, 2377–2385 CrossRef CAS.
- N. T. Thomas and K. Nobe, The Electrochemical Behavior of Titanium: Effect of pH and Chloride Ions, J. Electrochem. Soc., 1969, 116, 1748–1751 CrossRef CAS.
- Z. B. Wang, H. X. Hu, C. B. Liu and Y. G. Zheng, The effect of fluoride ions on the corrosion behavior of pure titanium in 0.05 M sulfuric acid, Electrochim. Acta, 2014, 135, 526–535 CrossRef CAS.
- D.-S. Kong, The Influence of Fluoride on the Physicochemical Properties of Anodic Oxide Films Formed on Titanium Surfaces, Langmuir, 2008, 24, 5324–5331 CrossRef CAS PubMed.
- D.-S. Kong and Y.-Y. Feng, Electrochemical Anodic Dissolution Kinetics of Titanium in Fluoride-Containing Perchloric Acid Solutions at Open-Circuit Potentials, J. Electrochem. Soc., 2009, 156, C283 CrossRef CAS.
- M. Nakagawa, S. Matsuya, T. Shiraishi and M. Ohta, Effect of Fluoride Concentration and pH on Corrosion Behavior of Titanium for Dental Use, J. Dent. Res., 1999, 78, 1568–1572 CrossRef CAS PubMed.
- G. N. Martelli, R. Ornelas and G. Faita, Deactivation mechanisms of oxygen evolving anodes at high current densities, Electrochim. Acta, 1994, 39, 1551–1558 CrossRef CAS.
- S. S. Pethaiah, K. K. Sadasivuni and A. Jayakumar, Methanol Electrolysis for Hydrogen Production Using, Energies, 2020, 13, 5879 CrossRef CAS.
- G. Sasikumar, A. Muthumeenal, S. S. Pethaiah, N. Nachiappan and R. Balaji, Aqueous methanol electrolysis using proton conducting membrane for hydrogen production, Int. J. Hydrogen Energy, 2008, 33, 5905–5910 CrossRef CAS.
- S. P. Sethu, S. Gangadharan, S. H. Chan and U. Stimming, Development of a novel cost effective methanol electrolyzer stack with Pt-catalyzed membrane, J. Power Sources, 2014, 254, 161–167 CrossRef CAS.
- C. Lamy, T. Jaubert, S. Baranton and C. Coutanceau, Clean hydrogen generation through the electrocatalytic oxidation of ethanol in a Proton Exchange Membrane Electrolysis Cell (PEMEC): effect of the nature and structure of the catalytic anode, J. Power Sources, 2014, 245, 927–936 CrossRef CAS.
- R. Ortiz, O. P. Márquez, J. Márquez and C. Gutiérrez, Necessity of oxygenated surface species for the electrooxidation of methanol on iridium, J. Phys. Chem., 1996, 100, 8389–8396 CrossRef CAS.
- O. Sahin, H. Kivrak, M. Karaman and D. Atbas, The effect of iridium addition to platinum on the alcohol electrooxidation activity, Am. J. Mater. Sci. Eng., 2015, 3, 15–20 CAS.
- K. M. Petrov, L. M. Kaba, S. Srinivasan and A. J. Appleby, Water post-treatment without expendables – proton exchange membrane based electrolysis system, Int. J. Hydrogen Energy, 1993, 18, 377–382 CrossRef CAS.
- T. Muddemann, D. Haupt, M. Sievers and U. Kunz, Electrochemical Reactors for Wastewater Treatment, ChemBioEng Rev., 2019, 6, 142–156 CrossRef CAS.
- T. Smolinka, M. Heinen, Y. X. Chen, Z. Jusys, W. Lehnert and R. J. Behm, CO2 reduction on Pt electrocatalysts and its impact on H2 oxidation in CO2 containing fuel cell feed gas – a combined in situ infrared spectroscopy, mass spectrometry and fuel cell performance study, Electrochim. Acta, 2005, 50, 5189–5199 CrossRef CAS.
- S. Lustig, S. Zang, W. Beck and P. Schramel, Dissolution of metallic platinum as water soluble species by naturally occurring complexing agents, Mikrochim. Acta, 1998, 129, 189–194 CrossRef CAS.
- S. Zimmermann, C. M. Menzel, D. Stüben, H. Taraschewski and B. Sures, Lipid solubility of the platinum group metals Pt, Pd and Rh in dependence on the presence of complexing agents, Environ. Pollut., 2003, 124, 1–5 CrossRef CAS PubMed.
- A. Kormányos, F. D. Speck, K. J. J. Mayrhofer and S. Cherevko, Influence of Fuels and pH on the Dissolution Stability of Bifunctional PtRu/C Alloy Electrocatalysts, ACS Catal., 2020, 10, 10858–10870 CrossRef.
- D. N. McCarthy, C. E. Strebel, T. P. Johansson, A. den Dunnen, A. Nierhoff, J. H. Nielsen and I. Chorkendorff, Structural Modification of Platinum Model Systems under High Pressure CO Annealing, J. Phys. Chem. C, 2012, 116, 15353–15360 CrossRef CAS.
- S. Rudi, C. Cui, L. Gan and P. Strasser, Comparative Study of the Electrocatalytically Active Surface Areas (ECSAs) of Pt Alloy Nanoparticles Evaluated by Hupd and CO-stripping voltammetry, Electrocatalysis, 2014, 5, 408–418 CrossRef CAS.
- R. Jinnouchi, E. Toyoda, T. Hatanaka and Y. Morimoto, First Principles Calculations on Site-Dependent Dissolution Potentials of Supported and Unsupported Pt Particles, J. Phys. Chem. C, 2010, 114, 17557–17568 CrossRef CAS.
- P. Jovanovič, V. S. Šelih, M. Šala, S. Hočevar, F. Ruiz-Zepeda, N. Hodnik, M. Bele and M. Gaberšček, Potentiodynamic dissolution study of PtRu/C electrocatalyst in the presence of methanol, Electrochim. Acta, 2016, 211, 851–859 CrossRef.
- G. Wei, Y. Wang, C. Huang, Q. Gao, Z. Wang and L. Xu, The stability of MEA in SPE water electrolysis for hydrogen production, Int. J. Hydrogen Energy, 2010, 35, 3951–3957 CrossRef CAS.
- S. Xu, X. Wang, L. Zhang, S. Sun, G. Li, M. Zhang, Z. G. Shao and B. Zhu, The Fe3+ role in decreasing the activity of Nafion-bonded IrO2 catalyst for proton exchange membrane water electrolyser, Int. J. Hydrogen Energy, 2020, 45, 15041–15046 CrossRef CAS.
- H. Yu, L. Bonville, J. Jankovic and R. Maric, Microscopic insights on the degradation of a PEM water electrolyzer with ultra-low catalyst loading, Appl. Catal., B, 2020, 260, 118194 CrossRef CAS.
- J. G. Vos, T. A. Wezendonk, A. W. Jeremiasse and M. T. M. Koper, MnOx/IrOx as Selective Oxygen Evolution Electrocatalyst in Acidic Chloride Solution, J. Am. Chem. Soc., 2018, 140, 10270–10281 CrossRef CAS PubMed.
- G. Wei, Y. Wang, C. Huang, Q. Gao, Z. Wang and L. Xu, The stability of MEA in SPE water electrolysis for hydrogen production, Int. J. Hydrogen Energy, 2010, 35, 3951–3957 CrossRef CAS.
- D. A. Corrigan and S. P. Maheswari, Catalysis of the Oxygen Evolution Reaction By Trace Iron Impurities in Thin Film Nickel Oxide Electrodes., Electrochem. Soc. Ext. Abstr., 1985, 85–1, 934–935 Search PubMed.
- D. A. Corrigan and R. M. Bendert, Effect of Coprecipitated Metal Ions on the Electrochemistry of Nickel Hydroxide Thin Films: Cyclic Voltammetry in 1 M KOH, J. Electrochem. Soc., 1989, 136, 723–728 CrossRef CAS.
- L. J. Enman, M. S. Burke, A. S. Batchellor and S. W. Boettcher, Effects of Intentionally Incorporated Metal Cations on the Oxygen Evolution Electrocatalytic Activity of Nickel (Oxy)hydroxide in Alkaline Media, ACS Catal., 2016, 6, 2416–2423 CrossRef CAS.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, Nickel-Iron oxyhydroxide oxygen-evolution electrocatalysts: The role of intentional and incidental iron incorporation, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS PubMed.
- L. Trotochaud, J. K. Ranney, K. N. Williams and S. W. Boettcher, Solution-cast metal oxide thin film electrocatalysts for oxygen evolution, J. Am. Chem. Soc., 2012, 134, 17253–17261 CrossRef CAS PubMed.
- M. S. Ali Akbari, R. Bagheri, Z. Song and M. M. Najafpour, Oxygen-evolution reaction by nickel/nickel oxide interface in the presence of ferrate(VI), Sci. Rep., 2020, 10, 1–11 CrossRef PubMed.
- I. Roger and M. D. Symes, Efficient Electrocatalytic Water Oxidation at Neutral and High pH by Adventitious Nickel at Nanomolar Concentrations, J. Am. Chem. Soc., 2015, 137, 13980–13988 CrossRef CAS PubMed.
- J. Y. Huot and L. Brossard, Time dependence of the hydrogen discharge at 70°C on nickel cathodes, Int. J. Hydrogen Energy, 1987, 12, 821–830 CrossRef CAS.
- M. A. Riley, The Influence of Iron Deposition on the Voltage-Time Behavior of Nickel Cathodes in Alkaline Water Electrolysis, J. Electrochem. Soc., 1986, 133, 760 CrossRef CAS.
- H. E. G. Rommal, The Role of Absorbed Hydrogen on the Voltage-Time Behavior of Nickel Cathodes in Hydrogen Evolution, J. Electrochem. Soc., 1988, 135, 343 CrossRef CAS.
- D. M. Soares, Hydride Effect on the Kinetics of the Hydrogen Evolution Reaction on Nickel Cathodes in Alkaline Media, J. Electrochem. Soc., 1992, 139, 98 CrossRef CAS.
- A. E. Mauer, D. W. Kirk and S. J. Thorpe, The role of iron in the prevention of nickel electrode deactivation in alkaline electrolysis, Electrochim. Acta, 2007, 52, 3505–3509 CrossRef CAS.
- I. Flis-Kabulska and J. Flis, Hydrogen evolution and corrosion products on iron cathodes in hot alkaline solution, Int. J. Hydrogen Energy, 2014, 39, 3597–3605 CrossRef CAS.
- I. Flis-Kabulska and J. Flis, Electroactivity of Ni–Fe cathodes in alkaline water electrolysis and effect of corrosion, Corros. Sci., 2016, 112, 255–263 CrossRef CAS.
- G. I. Lacconi, H. M. Villullas and V. A. Macagno, The effect of metallic impurities on the hydrogen evolution reaction rate on group-Ib metals in alkaline solution, J. Appl. Electrochem., 1991, 21, 1027–1030 CrossRef CAS.
- J. Y. Huot and L. Brossard, In situ activation of cobalt cathodes in alkaline water electrolysis, J. Appl. Electrochem., 1988, 18, 815–822 CrossRef CAS.
- D. V. Shinde, Z. Dang, U. Petralanda, M. Palei, M. Wang, M. Prato, A. Cavalli, L. De Trizio and L. Manna, In Situ Dynamic Nanostructuring of the Cu-Ti Catalyst-Support System Promotes Hydrogen Evolution under Alkaline Conditions, ACS Appl. Mater. Interfaces, 2018, 10, 29583–29592 CrossRef CAS PubMed.
- R. Subbaraman, N. Danilovic, P. P. Lopes, D. Tripkovic, D. Strmcnik, V. R. Stamenkovic and N. M. Markovic, Origin of anomalous activities for electrocatalysts in alkaline electrolytes, J. Phys. Chem. C, 2012, 116, 22231–22237 CrossRef CAS.
- N. Danilovic, R. Subbaraman, D. Strmcnik, K. C. Chang, A. P. Paulikas, V. R. Stamenkovic and N. M. Markovic, Enhancing the alkaline hydrogen evolution reaction activity through the bifunctionality of Ni(OH)2/metal catalysts, Angew. Chem., Int. Ed., 2012, 51, 12495–12498 CrossRef CAS PubMed.
- S. Barwe, B. Mei, J. Masa, W. Schuhmann and E. Ventosa, Overcoming cathode poisoning from electrolyte impurities in alkaline electrolysis by means of self-healing electrocatalyst films, Nano Energy, 2018, 53, 763–768 CrossRef CAS.
- I. Flis-Kabulska, J. Flis, Y. Sun and T. Zakroczymski, Hydrogen evolution on plasma carburised nickel and effect of iron deposition from the electrolyte in alkaline water electrolysis, Electrochim. Acta, 2015, 167, 61–68 CrossRef CAS.
- A. M. Kiss, T. D. Myles, K. N. Grew, A. A. Peracchio, G. J. Nelson and W. K. S. Chiu, Carbonate and Bicarbonate Ion Transport in Alkaline Anion Exchange Membranes, J. Electrochem. Soc., 2013, 160, F994–F999 CrossRef CAS.
- J. R. Varcoe, P. Atanassov, D. R. Dekel, A. M. Herring, M. A. Hickner, P. A. Kohl, A. R. Kucernak, W. E. Mustain, K. Nijmeijer, K. Scott, T. Xu and L. Zhuang, Anion-exchange membranes in electrochemical energy systems, Energy Environ. Sci., 2014, 7, 3135–3191 RSC.
- E. Gülzow and M. Schulze, Long-term operation of AFC electrodes with CO2 containing gases, J. Power Sources, 2004, 127, 243–251 CrossRef.
- M. S. Naughton, F. R. Brushett and P. J. A. Kenis, Carbonate resilience of flowing electrolyte-based alkaline fuel cells, J. Power Sources, 2011, 196, 1762–1768 CrossRef CAS.
- J. Peng, A. L. Roy, S. G. Greenbaum and T. A. Zawodzinski, Effect of CO2 absorption on ion and water mobility in an anion exchange membrane, J. Power Sources, 2018, 380, 64–75 CrossRef CAS.
- K. N. Grew, X. Ren and D. Chu, Effect of CO 2 on the Alkaline Membrane Fuel Cell, ECS Trans., 2019, 41, 1979–1985 CrossRef.
- J. A. Vega, C. Chartier and W. E. Mustain, Effect of hydroxide and carbonate alkaline media on anion exchange membranes, J. Power Sources, 2010, 195, 7176–7180 CrossRef CAS.
- N. Ziv, A. N. Mondal, T. Weissbach, S. Holdcroft and D. R. Dekel, Effect of CO2 on the properties of anion exchange membranes for fuel cell applications, J. Membr. Sci., 2019, 586, 140–150 CrossRef CAS.
- N. Ziv and D. R. Dekel, A practical method for measuring the true hydroxide conductivity of anion exchange membranes, Electrochem. Commun., 2018, 88, 109–113 CrossRef CAS.
- M. Inaba, Y. Matsui, M. Saito, A. Tasaka, K. Fukuta, S. Watanabe and H. Yanagi, Effects of carbon dioxide on the performance of anion-exchange membrane fuel cells, Electrochemistry, 2011, 79, 322–325 CrossRef CAS.
- C. Baeumer, A. Y.-L. Liang, U. Trstenjak, Q. Lu, R. Waser, J. T. Mefford, F. Gunkel, S. Nemšák and W. C. Chueh, Carbonate formation lowers the electrocatalytic activity of perovskite oxides for water electrolysis, J. Mater. Chem. A, 2021, 9, 19940–19948 RSC.
- A. G. Divekar, A. C. Yang-Neyerlin, C. M. Antunes, D. J. Strasser, A. R. Motz, S. S. Seifert, X. Zuo, B. S. Pivovar and A. M. Herring, In-depth understanding of the CO2 limitation of air fed anion exchange membrane fuel cells, Sustainable Energy Fuels, 2020, 4, 1801–1811 RSC.
- J. Parrondo, C. G. Arges, M. Niedzwiecki, E. B. Anderson, K. E. Ayers and V. Ramani, Degradation of anion exchange membranes used for hydrogen production by ultrapure water electrolysis, RSC Adv., 2014, 4, 9875–9879 RSC.
- S. Dresp, F. Dionigi, S. Loos, J. Ferreira de Araujo, C. Spöri, M. Gliech, H. Dau and P. Strasser, Direct Electrolytic Splitting of Seawater: Activity, Selectivity, Degradation, and Recovery Studied from the Molecular Catalyst Structure to the Electrolyzer Cell Level, Adv. Energy Mater., 2018, 8, 1800338 CrossRef.
- S. Dresp, T. Ngo Thanh, M. Klingenhof, S. Brückner, P. Hauke and P. Strasser, Efficient direct seawater electrolysers using selective alkaline NiFe-LDH as OER catalyst in asymmetric electrolyte feeds, Energy Environ. Sci., 2020, 13, 1725–1729 RSC.
- G. Amikam, P. Nativ and Y. Gendel, Chlorine-free alkaline seawater electrolysis for hydrogen production, Int. J. Hydrogen Energy, 2018, 43, 6504–6514 CrossRef CAS.
- Y. Kuang, M. J. Kenney, Y. Meng, W. H. Hung, Y. Liu, J. E. Huang, R. Prasanna, P. Li, Y. Li, L. Wang, M. C. Lin, M. D. McGehee, X. Sun and H. Dai, Solar-driven, highly sustained splitting of seawater into hydrogen and oxygen fuels, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 6624–6629 CrossRef CAS PubMed.
- S. Maximovitch and G. Bronoel, Oxidation of methanol on nickel-zinc catalysts, Electrochim. Acta, 1981, 26, 1331–1338 CrossRef CAS.
- M. A. A. Rahim, R. M. A. Hameed and M. W. Khalil, Nickel as a catalyst for the electro-oxidation of methanol in alkaline medium, J. Power Sources, 2004, 134, 160–169 CrossRef.
- M. Fleischmann, K. Korinek and D. Pletcher, The oxidation of organic compounds at a nickel anode in alkaline solution, J. Electroanal. Chem., 1971, 31, 39–49 CrossRef CAS.
- A. Cuña, C. Reyes Plascencia, E. L. da Silva, J. Marcuzzo, S. Khan, N. Tancredi, M. R. Baldan and C. de Fraga Malfatti, Electrochemical and spectroelectrochemical analyses of hydrothermal carbon supported nickel electrocatalyst for ethanol electro-oxidation in alkaline medium, Appl. Catal., B, 2017, 202, 95–103 CrossRef.
- A. A. El-Shafei, Electrocatalytic oxidation of methanol at a nickel hydroxide/glassy carbon modified electrode in alkaline medium, J. Electroanal. Chem., 1999, 471, 89–95 CrossRef CAS.
- J. L. Bott-Neto, T. S. Martins, S. A. S. Machado and E. A. Ticianelli, Electrocatalytic Oxidation of Methanol, Ethanol, and Glycerol on Ni(OH)2 Nanoparticles Encapsulated with Poly[Ni(salen)] Film, ACS Appl. Mater. Interfaces, 2019, 11, 30810–30818 CrossRef CAS PubMed.
- G. Chauhan, K. K. Pant and K. D. P. Nigam, Extraction of Nickel from Spent Catalyst Using Biodegradable Chelating Agent EDDS, Ind. Eng. Chem. Res., 2012, 51, 10354–10363 CrossRef CAS.
- H. Schultz, G. Bauer, E. Schachl, F. Hagedorn and P. Schmittinger, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2000 Search PubMed.
- G. McLean, An assessment of alkaline fuel cell technology, Int. J. Hydrogen Energy, 2002, 27, 507–526 CrossRef CAS.
- J. Juodkazytė, B. Šebeka, I. Savickaja, M. Petrulevičienė, S. Butkutė, V. Jasulaitienė, A. Selskis and R. Ramanauskas, Electrolytic splitting of saline water: Durable nickel oxide anode for selective oxygen evolution, Int. J. Hydrogen Energy, 2019, 44, 5929–5939 CrossRef.
- J. Hnát, M. Paidar, J. Schauer and K. Bouzek, Polymer anion-selective membrane for electrolytic water splitting: The impact of a liquid electrolyte composition on the process parameters and long-term stability, Int. J. Hydrogen Energy, 2014, 39, 4779–4787 CrossRef.
- A. Hartig-Weiss, M. F. Tovini, H. A. Gasteiger and H. A. El-Sayed, OER Catalyst Durability Tests Using the Rotating Disk Electrode Technique: The Reason Why This Leads to Erroneous Conclusions, ACS Appl. Energy Mater., 2020, 3, 10323–10327 CrossRef CAS.
- H. A. El-Sayed, A. Weiß, L. F. Olbrich, G. P. Putro and H. A. Gasteiger, OER Catalyst Stability Investigation Using RDE Technique: A Stability Measure or an Artifact?, J. Electrochem. Soc., 2019, 166, F458–F464 CrossRef CAS.
- S. Martens, L. Asen, G. Ercolano, F. Dionigi, C. Zalitis, A. Hawkins, A. Martinez Bonastre, L. Seidl, A. C. Knoll, J. Sharman, P. Strasser, D. Jones and O. Schneider, A comparison of rotating disc electrode, floating electrode technique and membrane electrode assembly measurements for catalyst testing, J. Power Sources, 2018, 392, 274–284 CrossRef CAS.
- D. A. Buttry and M. D. Ward, Measurement of Interfacial Processes at Electrode Surfaces with the Electrochemical Quartz Crystal Microbalance, Chem. Rev., 1992, 92, 1355–1379 CrossRef CAS.
- R. Frydendal, E. A. Paoli, B. P. Knudsen, B. Wickman, P. Malacrida, I. E. L. Stephens and I. Chorkendorff, Benchmarking the Stability of Oxygen Evolution Reaction Catalysts: The Importance of Monitoring Mass Losses, ChemElectroChem, 2014, 1, 2075–2081 CrossRef CAS.
- S. Xu, X. Wang, L. Zhang, S. Sun, G. Li, M. Zhang, Z. G. Shao and B. Zhu, The Fe3+ role in decreasing the activity of Nafion-bonded IrO2 catalyst for proton exchange membrane water electrolyser, Int. J. Hydrogen Energy, 2020, 45, 15041–15046 CrossRef CAS.
- L. Francàs, S. Corby, S. Selim, D. Lee, C. A. Mesa, R. Godin, E. Pastor, I. E. L. Stephens, K.-S. Choi and J. R. Durrant, Spectroelectrochemical study of water oxidation on nickel and iron oxyhydroxide electrocatalysts, Nat. Commun., 2019, 10, 5208 CrossRef PubMed.
- K. Zhu, X. Zhu and W. Yang, Application of In Situ Techniques for the Characterization of NiFe-Based Oxygen Evolution Reaction (OER) Electrocatalysts, Angew. Chem., Int. Ed., 2019, 58, 1252–1265 CrossRef CAS PubMed.
- M. Wang, L. Árnadóttir, Z. J. Xu and Z. Feng, In Situ X-ray Absorption Spectroscopy Studies of Nanoscale Electrocatalysts, Nano-Micro Lett., 2019, 11, 47 CrossRef CAS PubMed.
- G. Greczynski and L. Hultman, X-ray photoelectron spectroscopy: Towards reliable binding energy referencing, Prog. Mater. Sci., 2020, 107, 100591 CrossRef CAS.
- M. Favaro, J. Yang, S. Nappini, E. Magnano, F. M. Toma, E. J. Crumlin, J. Yano and I. D. Sharp, Understanding the Oxygen Evolution Reaction Mechanism on CoOx using Operando Ambient-Pressure X-ray Photoelectron Spectroscopy, J. Am. Chem. Soc., 2017, 139, 8960–8970 CrossRef CAS PubMed.
- H. G. Sanchez Casalongue, M. L. Ng, S. Kaya, D. Friebel, H. Ogasawara and A. Nilsson, In Situ Observation of Surface Species on Iridium Oxide Nanoparticles during the Oxygen Evolution Reaction, Angew. Chem., Int. Ed., 2014, 53, 7169–7172 CrossRef CAS PubMed.
- R. Rinaldi and X. Llovet, Electron Probe Microanalysis: A Review of the Past, Present, and Future, Microsc. Microanal., 2015, 21, 1053–1069 CrossRef CAS PubMed.
- M. Maier, J. Dodwell, R. Ziesche, C. Tan, T. Heenan, J. Majasan, N. Kardjilov, H. Markötter, I. Manke, L. Castanheira, G. Hinds, P. R. Shearing and D. J. L. Brett, Mass transport in polymer electrolyte membrane water electrolyser liquid-gas diffusion layers: a combined neutron imaging and X-ray computed tomography study, J. Power Sources, 2020, 455, 227968 CrossRef CAS.
- C. H. Lee, R. Banerjee, N. Ge, J. K. Lee, B. Zhao, E. Baltic, J. M. LaManna, D. S. Hussey, D. L. Jacobson, R. Abouatallah, R. Wang and A. Bazylak, The effect of cathode nitrogen purging on cell performance and in operando neutron imaging of a polymer electrolyte membrane electrolyzer, Electrochim. Acta, 2018, 279, 91–98 CrossRef CAS.
- E. Leonard, A. D. Shum, S. Normile, D. C. Sabarirajan, D. G. Yared, X. Xiao and I. V. Zenyuk, Operando X-ray tomography and sub-second radiography for characterizing transport in polymer electrolyte membrane electrolyzer, Electrochim. Acta, 2018, 276, 424–433 CrossRef CAS.
- C. Lee, J. K. Lee, B. Zhao, K. F. Fahy, J. M. LaManna, E. Baltic, D. S. Hussey, D. L. Jacobson, V. P. Schulz and A. Bazylak, Temperature-dependent gas accumulation in polymer electrolyte membrane electrolyzer porous transport layers, J. Power Sources, 2020, 446, 227312 CrossRef CAS.
- J. Nuñez, R. Renslow, J. B. Cliff and C. R. Anderton, NanoSIMS for biological applications: current practices and analyses, Biointerphases, 2018, 13, 03B301 CrossRef PubMed.
- D. Beauchemin, Inductively coupled plasma mass spectrometry, Anal. Chem., 2008, 80, 4455–4486 CrossRef CAS PubMed.
- S. O. Klemm, A. A. Topalov, C. A. Laska and K. J. J. Mayrhofer, Coupling of a high throughput microelectrochemical cell with online multielemental trace analysis by ICP-MS, Electrochem. Commun., 2011, 13, 1533–1535 CrossRef CAS.
- W. Vonau and U. Guth, pH Monitoring: a review, J. Solid State Electrochem., 2006, 10, 746–752 CrossRef CAS.
- J. Zhang, B. A. Litteer, F. D. Coms and R. Makharia, Recoverable Performance Loss Due to Membrane Chemical Degradation in PEM Fuel Cells, J. Electrochem. Soc., 2012, 159, F287–F293 CrossRef CAS.
- M. Heyrovský, Ninety Years of Polarography, Chem. Rec., 2012, 12, 14–16 CrossRef PubMed.
- Z. Zhao and H. Freiser, Differential pulse polarographic determination of trace levels of platinum, Anal. Chem., 1986, 58, 1498–1501 CrossRef CAS.
- K. Štulík, in Encyclopedia of Analytical Science, Elsevier, 2005, pp. 493–498 Search PubMed.
- M. A. Arnold and M. E. Meyerhoff, Ion-selective electrodes, Anal. Chem., 1984, 56, 20–48 CrossRef.
- J. Thomas and H. J. Gluskoter, Determination of fluoride in coal with the fluoride ion-selective electrode, Anal. Chem., 1974, 46, 1321–1323 CrossRef CAS.
- R. G. Jones, Measurements of the electrical conductivity of water, IEE Proc.: Sci., Meas. Technol., 2002, 149, 320–322 CrossRef CAS.
- Does Temperature Affect pH?, https://techiescientist.com/does-temperature-affect-ph/ Search PubMed.
- G. Wei, Y. Wang, C. Huang, Q. Gao, Z. Wang and L. Xu, The stability of MEA in SPE water electrolysis for hydrogen production, Int. J. Hydrogen Energy, 2010, 35, 3951–3957 CrossRef CAS.
- I. Bisutti, I. Hilke and M. Raessler, Determination of total organic carbon – an overview of current methods, TrAC, Trends Anal. Chem., 2004, 23, 716–726 CrossRef CAS.
- I. A. Fowlis, Gas Chromatography: Analytical Chemistry by Open Learning, Wiley, 2nd edn, 1995 Search PubMed.
- H. Baltruschat, Differential electrochemical mass spectrometry, J. Am. Soc. Mass Spectrom., 2004, 15, 1693–1706 CrossRef CAS PubMed.
- D. B. Trimarco, S. B. Scott, A. H. Thilsted, J. Y. Pan, T. Pedersen, O. Hansen, I. Chorkendorff and P. C. K. Vesborg, Enabling real-time detection of electrochemical desorption phenomena with sub-monolayer sensitivity, Electrochim. Acta, 2018, 268, 520–530 CrossRef CAS.
- High PEM Project End Report, 2016 Search PubMed.
- Z. Yang, Y. Zhou, Z. Feng, X. Rui, T. Zhang and Z. Zhang, A review on reverse osmosis and nanofiltration membranes for water purification, Polymers, 2019, 11, 1–22 Search PubMed.
- D. Li and H. Wang, Recent developments in reverse osmosis desalination membranes, J. Mater. Chem., 2010, 20, 4551–4566 RSC.
- J. M. Gohil and A. K. Suresh, Chlorine attack on reverse osmosis membranes: Mechanisms and mitigation strategies, J. Membr. Sci., 2017, 541, 108–126 CrossRef CAS.
- M. Al-Abri, B. Al-Ghafri, T. Bora, S. Dobretsov, J. Dutta, S. Castelletto, L. Rosa and A. Boretti, Chlorination disadvantages and alternative routes for biofouling control in reverse osmosis desalination, npj Clean Water, 2019, 2, 2 CrossRef CAS.
- P. Trogadas, J. Parrondo, F. Mijangos and V. Ramani, Degradation mitigation in PEM fuel cells using metal nanoparticle additives, J. Mater. Chem., 2011, 21, 19381–19388 RSC.
- M. Zatoń, J. Rozière and D. J. Jones, Current understanding of chemical degradation mechanisms of perfluorosulfonic acid membranes and their mitigation strategies: a review, Sustainable Energy Fuels, 2017, 1, 409–438 RSC.
- M. Zatoń, B. Prélot, N. Donzel, J. Rozière and D. J. Jones, Migration of Ce and Mn Ions in PEMFC and Its Impact on PFSA Membrane Degradation, J. Electrochem. Soc., 2018, 165, F3281–F3289 CrossRef.
Footnote |
| † Assuming an electrolyser using Nafion-115 operating constantly at a current of 2 A cm−2 and a water stoichiometry of 1. |
| This journal is © The Royal Society of Chemistry 2023 |