
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Light-driven (cross-)dimerization of terpenes as a route to renewable C15–C30 crudes for fuel and lubricant oil applications†
Leandro
Cid Gomes
 a,
Anup
Rana
a,
Mathias
Berglund
b,
Per
Wiklund
*c and
Henrik
Ottosson
*a
a,
Anup
Rana
a,
Mathias
Berglund
b,
Per
Wiklund
*c and
Henrik
Ottosson
*a
aDepartment of Chemistry – Ångström Laboratory, Uppsala University, Box 523, Uppsala 751 20, Sweden. E-mail: henrik.ottosson@kemi.uu.se
bRISE Research Institutes of Sweden, Brinellgatan 4, Box 857, Borås 501 15, Sweden
cBiobase Sweden AB, Götlundagatan 3, Bandhagen, Stockholm 124 71, Sweden. E-mail: per@bioba.se
First published on 9th January 2023
Abstract
Non-fossil hydrocarbons are desirable for transport fuels and lubricant oils to reach a fossil carbon neutral economy. Herein, we show the production of such end-products from crude raw materials via the photosensitized dimerization of terpenes. Terpenes are hydrocarbons originating from renewable sources, such as forestry, industrial bio-waste and photosynthetically active microorganisms. Under irradiation at 365 nm, we observed high conversions of terpenes with conjugated diene segments into their dimers (e.g. 96.1 wt%, 12 h for α-phellandrene), and remarkable results were obtained using simulated and natural sunlight (90.8 wt% and 46.6 wt%, respectively, for α-phellandrene). We show that the lower reactivities of some isomeric monoterpenes could be overcome by a cross-photodimerization with α-phellandrene. We also utilized the cross-photodimerization approach to obtain C15 and C30 products, combining mixtures of isoprene, monoterpenes and sesquiterpenes. Hydrogenation of the terpene dimers gave materials with physical properties suitable as high energy density fuels and lubricant oils. Finally, our preliminary analysis based on recent literature points to the commercial viability of this route to produce fuels and lubricant oils, as well as to a potential for reduction of the environmental impact compared to fossil-based routes.
Introduction
According to the International Renewable Energy Agency (IRENA), liquid biofuels will represent 20% of the total final energy consumption of the transport sector in 2050.1 The need to develop alternative fuels applies in particular to the shipping and aviation sectors, for which IRENA considers advanced biofuels to be a key solution – i.e. biofuels from nonfood sources.2 At the same time, other fossil derived products such as solvents and lubricant oils must also be replaced to meet sustainability targets.3As an example of biomass from a nonedible source, terpenes are major components of essential oils from plants and they can also be extracted from conifer resins (Fig. 1).4 The main component in the leaf oil of Schinus mole is α-phellandrene 1 (45.7%), and the eucalyptus oil from some species (e.g. E. dives, especially the phellandrene variant) contains up to 80% of α-phellandrene.5–7 Other monoterpenes, such as pinenes, are obtained industrially as byproducts of paper pulping processes.8 β-Pinene can be converted to myrcene 2 through pyrolysis,8,9 while ocimene 3 can be obtained photochemically from α-pinene.10 Mono- and sesquiterpene syntheses by metabolically engineered organisms have also been the subject of extensive research.11–20 These routes utilize sugars derived from lignocellulosic biomass, or CO2 and water as feedstock, making terpenes potential candidates for sustainable syntheses of biofuels and lubricants.
 | ||
| Fig. 1 Terpenes that were used in this work and their possible sources: evergreen trees and plants (essential oils), industrial waste (turpentine), sugars derived from lignocellulosic biomass (converted into terpenes by microorganisms) and production by metabolically engineered photosynthetic microorganisms (cyanobacteria). | ||
The importance of terpenes, beside the fact that their use does not induce any food security threats, stems from their chemical similarity to fuel molecules derived from crude oil, making terpenes suitable as drop-in surrogate for fossil fuels.21 Indeed, hydrogenated monoterpenes themselves have been reported to have jet fuel properties,22,23 whilst their hydrogenated C20 dimers can be suitable for diesel-like or other high energy density fuels.24–29 Such reported methods of dimerization of monoterpenes are acid or metal catalyzed thermal reactions. The two monoterpenes α- and β-pinene have also been used in self-photosensitized cycloaddition with biomass-derived cyclic enones, yielding spiro-compounds suitable as jet fuels.30 All these reactions rely on the reactivity of isolated as well as conjugated π-bonds of monoterpenes.
Conjugated dienes have long been known to undergo triplet sensitized reactions promoted by organic photosensitizers.31,32 As such, the precursor molecule of terpenes, isoprene 4, undergoes triplet photosensitized dimerization to give [2 + 2], [4 + 2] and [4 + 4] photocycloaddition products under near-UV light.33 Since cycloalkanes are reported as high energy density combustion fuels,34 our group has recently reported on a fruitful route to solar jet fuels, where the photobiologically produced isoprene is subject to photosensitized dimerization.35 Herein, we apply the photochemical dimerization approach to mono- and sesquiterpenes bearing conjugated diene units, i.e. α-phellandrene 1, myrcene 2 and ocimene 3. The triplet photosensitized dimerization of 1–3 was reported earlier,36–38 yet, these reactions of 2 and 3 were carried out in dilute solutions in order to favor other intramolecular photoreactions in the triplet state – i.e. the internal cyclization of myrcene and the E/Z isomerization of ocimene. On the other hand, the earlier reported triplet sensitized dimerization of 1 by using naphthalene as photosensitizer was very inefficient (conversion of 12% after 24 h, Scheme 1A).38
 | ||
| Scheme 1 (A) Previous work by Baldwin and Nelson on the photosensitized dimerization of α-phellandrene using naphthalene as the photosensitizer. (B) Our present approach on the photosensitized dimerization of α-phellandrene and other terpenes as key step to produce high energy density fuels and base oils. NHOC (Net Heat of Combustion); KV40 (Kinematic Viscosity at 40 °C). | ||
Naphthalene could be an inefficient photosensitizer due to a large difference in the triplet energies of α-phellandrene and naphthalene. Matching of the triplet energies of the molecule to be sensitized and that of the photosensitizer is an important factor in photosensitized reactions.39 For conjugated dienes, the triplet energy is around 50 kcal mol−1 and an efficient triplet photosensitizer should have a relatively higher triplet energy to enable effective energy transfer to dienes.40 Conveniently, solar irradiation provides wavelengths down to the near-UV range, between 300–400 nm, which is equivalent to the energy of 71–95 kcal mol−1. Hence, it is possible to envision a photosensitizer that absorbs in the near-UV spectral region of sunlight and undergoes intersystem crossing, enabling the photosensitized dimerization of terpenes with conjugated diene moieties. Aryl ketones are good candidates, and extending the π-conjugation in aryl ketones or changing the substituents have been shown to promote a redshifted absorption.41,42 This is the case for some benzophenone derivatives, such as the 1,1-dinaphthylmethanone 8, which is the key photosensitizer in our studies (Scheme 1).
Thereby, we report a route to renewable hydrocarbon crudes from the photosensitized dimerization of monoterpenes, using near-UV light and, ultimately, by using simulated and natural sunlight (Scheme 1B). We find that α-phellandrene is especially reactive, and we utilize this in the cross-dimerization between α-phellandrene 1 and the less reactive monoterpenes and isoprene, giving, respectively C20 and C15 cross-dimers. We also describe the dimerization of sesquiterpenes present in ginger oil leading to C30 dimers. Finally, we present the physical properties of some of the hydrogenated crudes, finding that they are suitable as high energy density fuels and lubricant oils.
Experimental section
Materials
All chemicals – α-phellandrene (natural, 85% and synthetic, 95%), myrcene (93%), ocimene (94%), α-terpinene (97%), isoprene (99%), ginger oil (natural, containing α-zingiberene and β-sesquiphellandrene), xanthone (97%), thioxanthone (97%), benzophenone (99%) and 3-chloroperbenzoic acid (77%) – and reagent grade solvents were obtained from Sigma-Aldrich and were used as received, unless otherwise stated. Prior to use, the inhibitors p-tert-butylcatechol and butylated hydroxytoluene were removed from isoprene, myrcene and ocimene, by passing them through activated basic alumina. Prior to use, the ginger oil sample was passed through a column of silica gel and distilled under reduced pressure to obtain a C15-enriched fraction. The photosensitizer 8 was prepared as previously described.35 The synthesis of myrcene epoxide was performed by following the method reported by Hioki et al.43Dimerization and hydrogenation procedures
Characterization
Computational methods
All calculations were carried out using the Gaussian 16 program package,44 at the (U)M06-2X/6-311+G(d,p) level.45,46 Stationary points were characterized as minima through frequency calculations. The heats of combustion were calculated at the M06-2X/6-31+G(d,p) level using the method developed by Pahima et al.23Results and discussion
The initial studies and optimization of the photosensitized dimerization reactions at 365 nm light, and simulated and natural sunlight are presented. Further, the relative reactivities of monoterpenes are discussed, followed by the outcomes of the cross-dimerization between α-phellandrene and other terpenes, and the dimerization of sesquiterpenes in ginger oil. To conclude, the fuel and lubricant oil properties of selected hydrogenated crudes produced in this study are reported, together with a preliminary assessment of the sustainability and commercial viability of the process.Initial investigation and optimization of reactions
On the basis of our previous study,35 1,1-dinapthylmethanone 8 was used as photosensitizer. Additionally, screening with three other aromatic ketone derivatives (benzophenone, xanthone and thioxanthone) was performed for dimerization of myrcene in search of a better photosensitizer. However, 8 was proved to be the best choice (Table S1, ESI†). Among the four photosensitizers, thioxanthone has the highest molar absorption coefficient at 365 nm (Fig. S2A, ESI†), yet, it has a very low solubility in non-polar compounds such as the monoterpenes, which limits further optimization of the photosensitizer load. 1,1-Dinaphthylmethanone still has the second highest absorption coefficient at 365 nm, higher than those of xanthone and benzophenone (Fig. S2B and C, ESI†). It further has a triplet lifetime of 300 ns in toluene,41 which is long enough to partake in triplet energy transfer.Monoterpenes 1–5 (Fig. 1) were chosen based on their high abundance in nature,5,6,8 directly or after conversion from other monoterpenes,9 as well as their availability from metabolically engineered microorganisms, including cyanobacteria.11–13,20 Their adiabatic first triplet state energies (E(T1)) were calculated at the (U)M06-2X/6-311+G(d,p) level and found to be lower than 55.5 kcal mol−1, i.e. the E(T1) of 8 (Table 1 and Fig. S3, ESI†). The lower E(T1)'s may enable an efficient energy transfer from the photosensitizer to the monoterpenes.
| Entry | Monoterpene | E(T1) (kcal mol−1)a | Quartz test tube | CMS I | ||||
|---|---|---|---|---|---|---|---|---|
| 8b (mol%) | Time (h) | Dimersc (wt%) | 8b (mol%) | Time (h) | Dimersc (wt%) | |||
| a Adiabatic E(T1) calculated at the (U)M06-2X/6-311+G(d,p) level with thermal free energy corrections at 298 K. b 1,1-Dinaphthylmethanone 8 used as sensitizer. c Isolated yield of dimers – gravimetrically determined. d 262 g scale experiment using CMS II (260.6 g of dimers obtained). | ||||||||
| 1.1 | α-Phellandrene | 49.59 | 0.50 | 24 | 82 | 0.10 | 12 | 76 |
| 1.2 | α-Phellandrene | — | 0.50 | 12 | 77 | 0.20 | 6 | 63 |
| 1.3 | α-Phellandrene | — | — | — | — | 0.20 | 12 | 96 |
| 1.4 | α-Phellandrene | — | — | — | — | 0.20 | 24 | 99d |
| 2 | β-Myrcene | 52.09 | 0.50 | 48 | 44 | 0.50 | 48 | 59 |
| 3 | E-Ocimene | 47.42 | 0.50 | 48 | 10 | 0.50 | 48 | 16 |
| Z-Ocimene | 47.54 | |||||||
| 4 | α-Terpinene | 49.32 | 0.50 | 48 | 18 | — | — | — |
The selected monoterpenes were initially photo-irradiated with 8 for dimerization in quartz test tubes and at 365 nm. The formation of dimers is confirmed by the parent ion m/z 272 in the GC-MS analyses, which indicates a C20H32 molecular formula (Fig. S6, S14 and S20, ESI†). The chromatograms of α-phellandrene dimers reveal the presence of at least three major isomers, whereas there is a larger number of dimer isomers of myrcene and ocimene (Fig. 2). While ocimene and myrcene showed rather low conversions after 48 h of irradiation, α-phellandrene exhibited 82% conversion after 24 h (Table 1, entries 2–4). It is known that triplet 1,3-cyclohexadiene, which is a structural element of α-phellandrene, has a significantly longer lifetime than acyclic dienes – approximately two orders of magnitude longer.47 Therefore, triplet α-phellandrene has also a longer lifetime than myrcene and ocimene, and this leads to a higher concentration of the former, which impacts on the kinetics giving a more rapid reaction.
 | ||
| Fig. 2 Gas chromatograms of the dimers obtained from the photodimerization of three monoterpenes (1–3). Their respective mass spectra show [M+] = 272 m/z. | ||
Despite α-terpinene 5 having similar structure to α-phellandrene and suitable triplet energy, the reaction of 5 gave a much lower conversion to dimers (Table 1, entry 4). This finding can be correlated either to a preference for the reduction of 8 (H-transfer) leading to the aromatization of 5 and depletion of the photosensitizer, or to steric congestion at the termini of the diene moieties. The first explanation is supported by the observed increase of aromatic content in the reaction (signal at 7.1 ppm in the 1H NMR spectrum, Fig. S4, ESI†) and formation of the alcohol derived from 8 (Fig. S5, ESI†). A similar result was observed for the reaction of 5 with duroquinone.37 Since this side reaction causes photosensitizer depletion, the reactions with α-terpinene 5 were not further investigated. A similar reduction of 8 was not observed on the reaction with α-phellandrene (Fig. S6, ESI†). We also measured the UV-vis absorption spectra of the reaction mixtures of myrcene, ocimene and α-phellandrene before and after irradiation (Fig. S8†), and the optical activity of the photosensitizer is not reduced in the 365 nm region throughout the reactions. We also performed control experiments without photosensitizer and the irradiated samples did not show formation of dimers, supporting the triplet photosensitized dimerization process (Fig. S9, ESI†).
As the most reactive monomer, α-phellandrene was used to assess the effects of varied photosensitizer loading for the reaction in quartz tubes (Table S2, ESI†). Starting from 0.50 mol% the conversion to dimers dropped as the photosensitizer loading was decreased. However, the correlation was not linear and similar conversion was observed for three different loadings (0.125 and 0.250 mol%). Interestingly, even as low concentrations as 0.01 mol% enabled the dimerization (12.4 wt% conversion, 12 h). From those results, 0.125 mol% was indicated to be the optimal loading.
The photoreactions were next carried out in an alternative setup constructed by coiling a FEP tubing onto a glass condenser (Fig. S1A, ESI†). This setup, CMS I, provides a higher ratio of surface-area-to-volume exposed to light. We observed the expected increase of the conversion of monomers to dimers for the three monoterpenes 1–3 (Table 1, entries 1–3, CMS I column). The most remarkable change was noted for the dimerization of α-phellandrene 1. A complete conversion to dimers (12 h, 0.2 mol% and 96 wt%) was observed at much lower loading of 8 (0.2 mol%) compared to the reaction in quartz tube (12 h, 0.5 mol%, 77 wt%). The conversion of 63 wt% was observed after 6 h of photoirradiation with 0.2 mol% loading of 8. Unlike the results in quartz tube, decreasing the amount of 8 resulted in a lower conversion (76 wt% in 12 h, 0.1 mol% of 8), making the 0.2 mol% the optimal loading in the CMS I. These results show higher efficiency than the earlier reported naphthalene photosensitized dimerization, in which a conversion of only 12% was reached after 24 h.38 The conversion of myrcene 2 to dimers was increased to 59 wt%, while there was negligible change in the yield of ocimene 3 dimers. Following these results, we attempted to run dimerization of α-phellandrene in 260 g scale setup by using FEP tubing with larger internal diameter (CMS II, Fig. S1B, ESI,† internal diameter = 6.35 mm). A conversion of 99 wt% was observed after 24 h of photoirradiation (Table 1, entry 1.4, CMS I column). The longer reaction time could be attributed to the increased internal diameter of the FEP tubing used, which reduces the light penetration into the sample.
Based on the previously reported photodimerization of conjugated dienes in the presence of triplet photosensitizers,33 the dimers reported on Table 1 are expected to be cycloalkenes resulting from [2 + 2], [4 + 2] and [4 + 4] photocycloadditions (Fig. 3). Baldwin et al. suggested structure for the dimers of α-phellandrene in the photosensitization promoted by naphthalene, in which the main dimers should be a [2 + 2] cycloadduct between the methyl substituted double bonds (Fig. 3, structure 1a), derived from the most stable biradical intermediate (Fig. 3, 1BR).38 Meanwhile, the dimers from reactions of acyclic monoterpenes are more complex in nature and the structures 2a–3l (Fig. 3) are assumed based on the possible outcomes from the aforementioned photocycloadditions.
 | ||
| Fig. 3 Plausible structures of [2 + 2], [4 + 2] and [4 + 4] cycloadducts of α-phellandrene, myrcene and ocimene produced by the photosensitized dimerization. | ||
The myrcene dimers possibly consist of at least seven major isomers (Fig. 2), while for ocimene this distinction is less straightforward. The 1H NMR data of reported camphorene (product of the [4 + 2] cycloaddition, Fig. 3 structures 2g–2j) with myrcene dimers confirmed the presence of camphorene isomers in the mixture (Fig. S15, ESI†).24 Further structure assignments are challenging as the 1H NMR spectra for the mixture of dimers have several overlapping signals. However, the reasoning above strongly supports the notion that the product mixtures consist mainly of cycloalkene adducts.
Dimerization using simulated and natural sunlight
The photosensitizer 8 exhibits a broad absorption band within the range of 300–400 nm (Fig. S2B†),35,41 which might enable the dimerization to be carried out under simulated and natural sunlight. Therefore, mixtures of myrcene and α-phellandrene with 8 were irradiated under simulated sunlight with an intensity of 1 sun equivalent (AM 1.5 G) and using a flat coiled FEP tubing (CMS IV, Fig. S1D, ESI†). The amounts of 8 and the results are summarized on Table 2. The conversion of α-phellandrene to dimers was 90.8 wt% and achieved after 12 h which is comparable to the experiments using 365 nm light (96.1 wt%, 12 h, CMS I). The conversion of myrcene under solar simulated light was also comparable to the conversion observed under 365 nm irradiation (Table 2, entry 2).Later, a mixture of α-phellandrene and 8 in the CMS IV was irradiated with natural sunlight for 18 h (Uppsala, Sweden 59°51′09.5′′N 17°39′19.9′′E, approx. 30 m above sea level on September 19th and 20th, 2020), which led to 46.6 wt% conversion into dimers (Table 2, entry 3). Some important aspects must be considered before assessing the relevance of this last result. First, the average yearly global horizontal solar irradiance measured for Arlanda-Stockholm (35 km south of Uppsala) is 3319 MJ m−2 per year,48 which is lower than the average in several other places located in lower latitudes worldwide (Table S3, ESI†). Second, our experiment with sunlight was performed in mid-September, which accounts to an average solar irradiance that is half of the solar irradiance during summer months (Table S4, ESI†).48 Finally, the sky was not entirely clear during the second day of reaction. Therefore, given these non-optimal conditions, a conversion of 46.6 wt% is very promising, and one can expect higher conversions to α-phellandrene dimers during optimal annual season and in locations with higher solar irradiance.
We then evaluated to what extent the light intensity affects the photosensitized dimerization of α-phellandrene with 0.2 mol% of 8. We used again 365 nm light, setting a custom-made LED panel to three different intensities (6.5 mW cm−2, 4.0 mW cm−2 and 2.0 mW cm−2). The conversion of α-phellandrene into dimers versus time is plotted in Fig. 4A along with the linear fitting to determine their pseudo rate constants. We then plotted the rate constants versus the light intensity (Fig. 4B), which reveals the conversion rate dependency of light intensity to be 1.19 mmol per h mW−1 cm2. The distribution ratio of the C20-isomers did not vary with the different light intensities, as it can be seen on the 1H NMR spectra of the reaction mixtures (Fig. S23–S25, ESI†).
 | ||
| Fig. 4 Experiments with varying light intensities. (A) Conversion of α-phellandrene into dimers versus irradiation time, under irradiation at 365 nm in different intensities, 0.2 mol% of 1,1-dinaphthylmethanone. (B) Correlation between the light intensity and the conversion rate constant in the dimerization of α-phellandrene. | ||
Less reactive monoterpenes
Based on what is known from small cyclic and acyclic dienes,47 myrcene and ocimene have shorter triplet state lifetimes compared to α-phellandrene, due to the formers being acyclic conjugated dienes. Additionally, both myrcene and ocimene can undergo competitive side-reactions, a fact that may explain their modest reactivities. For instance, the triplet photosensitized internal cyclization of myrcene to 5,5-dimethyl-L-vinylbicyclo[2.1.1]hexane 9 (Fig. 5A) has been studied in solution, and the formation of this isomer was also observed in our experiments (18.3 wt%, Fig. S12, ESI†). As soon as the bicyclic structure 9 is formed, it can no longer partake in a photosensitized dimerization, because the reactive diene segment is lost and non-conjugated alkenes show higher triplet energies that are above the triplet energy of the photosensitizer 8. Indeed, the vertical triplet energy of 9 is 103 kcal mol−1 (calculated at the (U)M06-2X/6-311+G(d,p) level), much higher than that of 2 (76 kcal mol−1). The adiabatic triplet energy of 9 was not possible to calculate since the compound dissociates during the optimization. Therefore, the hypothesis was that the formation of 9 traps the myrcene that could have reacted in the photosensitized dimerization. In order to verify this hypothesis, we performed an epoxidation of myrcene to protect the isolated double bond and halt the internal cyclization of myrcene (Fig. 5A).43 The myrcene epoxide 10 was then mixed with the photosensitizer 8 and irradiated in the CMS I (0.50 mol%, 365 nm, 48 h). After the irradiation, the formation of an intramolecular cyclization product equivalent to 9 was not observed from the epoxide 10 (Fig. S19, ESI†). The isolated yield of dimers of 10 was 80.6 wt%, which is considerably higher than the 59.4 wt% (observed yield in the photodimerization of myrcene 2). The difference between these two outcomes is also close to the yield of 9. Furthermore, the unreacted amounts of myrcene and myrcene epoxide were very similar. These observations support the intramolecular cyclization to 9 as a limiting factor for the lower yield of myrcene dimers. | ||
| Fig. 5 (A) Myrcene and myrcene epoxide dimerization reactions with their respective isolated yields of dimers and recovered byproducts and reactants after distillation. (B) Ocimene dimerization with the isolated yields for dimers and the observed changes on the E/Z ratio for the recovered ocimene. The reactions were performed in CMS I. (C) Triplet biradical myrcene and ocimene structures. (D) Spin densities of myrcene and ocimene in their first triplet state, calculated at the (U)M06-2X/6-311+G(d,p) level. Note: the depicted structures in (A) and (B) are not all the dimers formed in the reaction. | ||
In the case of ocimene, a photosensitized E/Z-isomerization is also possible under the conditions described (Fig. 5B).31 Although in this case the resulting triplet structure is still reactive in the dimerization, the unimolecular intramolecular E/Z isomerization occurs faster than the encounter of one triplet excited ocimene molecule with another ocimene molecule, which would lead to dimers. The E/Z ratio determined by 1H NMR changed from 2.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.0 on the starting material to 3.0:1.0 for the ocimene recovered after irradiation. Therefore, the photosensitized E/Z isomerization is possibly a de-excitation method, forming the more stable isomer (E)-ocimene.
:1.0 on the starting material to 3.0:1.0 for the ocimene recovered after irradiation. Therefore, the photosensitized E/Z isomerization is possibly a de-excitation method, forming the more stable isomer (E)-ocimene.
Besides the highly competitive E/Z photoisomerization pathway, triplet excited ocimene is also more hindered than myrcene. In the triplet biradical intermediate of myrcene the radical character is always on a terminal carbon. In the case of ocimene, the radical character can reside either at the terminal carbon or at the internal carbon (Fig. 5C). Indeed, the calculated triplet state structures for ocimene indicate that the most stable biradical configuration occurs when the allyl radical is at the terminal position and the single radical is on in the internal carbon of the diene (Fig. 5D). This is similar to the case of the biradical intermediate 1BR in the α-phellandrene dimerization (Fig. 3), as in both cases the most stable biradical is the one with allyl radical having a stabilizing methyl group bonded to the allyl radical edge, where the molecular orbital has a non-zero coefficient (see SOMO of the allyl radical in Fig. 5C). Therefore, the non-allylic radical of triplet state ocimene is more hindered to react as it occurs from the internal carbon.
We further investigated how the quenching of the photosensitizer triplet energy varies among the three different monoterpenes. A series of emission spectra (phosphorescence) were measured at 77 K for samples containing 25 μM of 8 and increasing concentrations of one of the quenchers (α-phellandrene, myrcene and ocimene), using cyclohexane as the solvent. From the emission spectrum of 8 when no quencher has been added the band with highest intensity is at 560 nm (Fig. 6A) and the spectrum matches well with the spectrum reported by Rajagopal et al.41 We chose the band at 560 nm to follow the effect of adding increasing amounts of the monoterpenes. Despite that the setup for low temperature measurements creates more sources of errors, and the baseline varied from sample to sample, we were able to see the quenching promoted by the monoterpenes. As the concentration of the monoterpene increased, the intensity of the emission decreased, as seen in the Stern–Volmer plots for each of the monoterpenes (Fig. 6B). Using 300 ns as the triplet lifetime τ for 8, the Stern–Volmer plots for these quenching experiments allow us to determine the quenching rate constant kq for the different monoterpenes (Fig. 6B). Ocimene has a kq 10-fold higher than myrcene, while the kq of α-phellandrene is hundred times higher than that of ocimene. The differences in the quenching rate constants might also influence in the observed reactivities in the photosensitized dimerization. Yet, we use neat conditions of monoterpene in the reactions, i.e. high amounts of the quencher, which therefore makes the quenching of the photosensitizer, i.e. the energy transfer, very favorable in all cases.
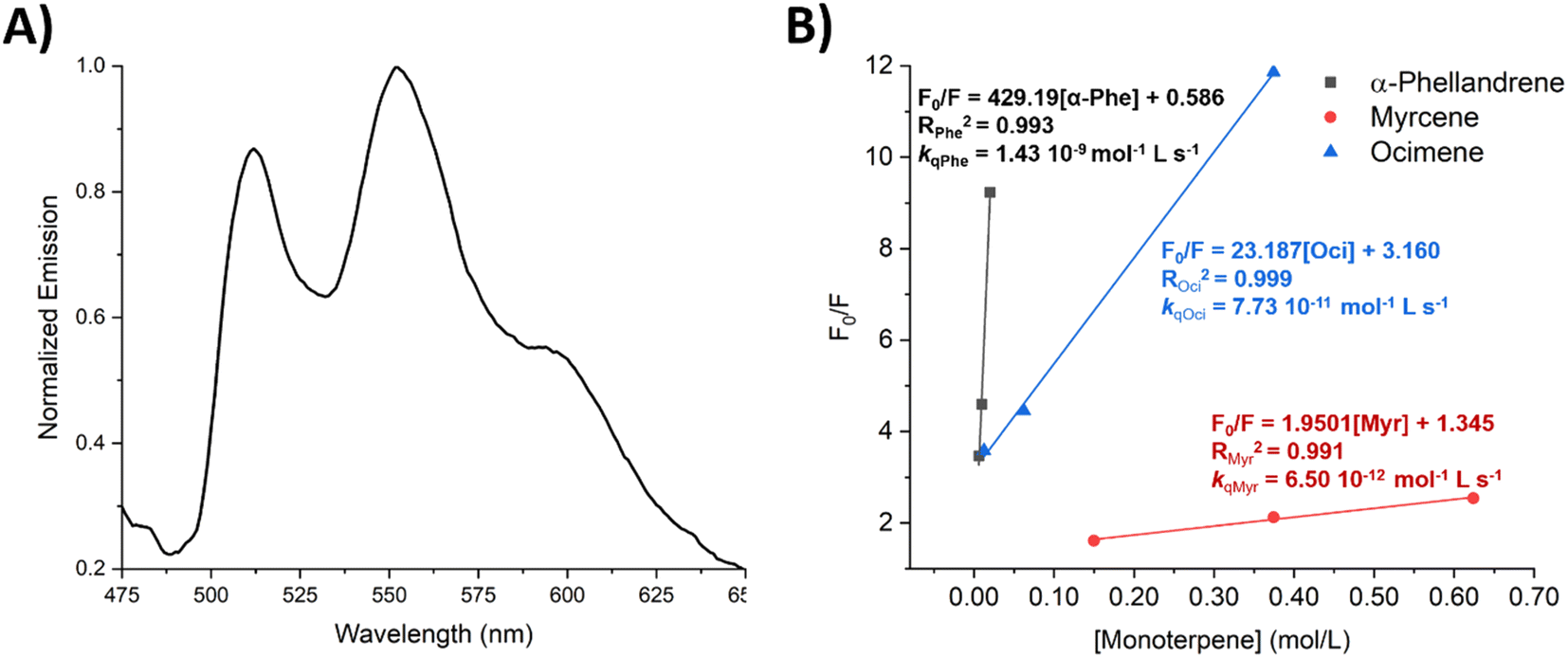 | ||
| Fig. 6 (A) Emission spectrum of 1,1-dinaphthylmethanone in cyclohexane, 25 μM, excitation at 338 nm. (B) Stern–Volmer plot for the quenching of 1,1-dinaphthylmethanone by the three monoterpenes (α-phellandrene, myrcene and ocimene). The origins of the axes were set to non-zero for better visualization. | ||
Cross-dimerization of monoterpenes
To improve the yield of ocimene and myrcene dimerization, we explored a cross-dimerization strategy between these less reactive compounds and α-phellandrene. Ocimene and myrcene were mixed with α-phellandrene in different molar ratios and with 0.25 mol% of photosensitizer 8. The results from the cross-dimerization are listed in Table 3. The individual conversions (column “Conversion (wt%)”, footnote b) express how much of the starting α-phellandrene, myrcene or ocimene ended up in the C20 mixture – as dimers to themselves or as cross-dimers formed with α-phellandrene.| Entry | Monoterpeneb | Time (h) | Conversion (wt%) | |||
|---|---|---|---|---|---|---|
| Myrcene or Ocimenec | α-Phellandrenec | Total | ||||
| a Reactions performed under 365 nm irradiation in the CMS I by using 8 as the photosensitizer (0.25 mol%). b Monoterpene amounts related to α-phellandrene (mol equivalent). c Individual conversions of α-phellandrene, myrcene or ocimene into dimers/cross-dimers, quantified by 1H NMR. | ||||||
| 1 | Myrcene | 1.00 eq. | 48 | 69.4 | 99.8 | 84.6 |
| 2 | Myrcene | 0.50 eq. | 48 | 91.0 | 98.2 | 95.8 |
| 3 | Myrcene | 0.50 eq. | 24 | 94.5 | 90.6 | 91.9 |
| 4 | Ocimene | 1.00 eq. | 48 | 68.7 | 65.7 | 67.2 |
| 5 | Ocimene | 0.50 eq. | 48 | 76.6 | 91.9 | 86.8 |
The best results were found when the 2:1 ratio of α-phellandrene:ocimene or α-phellandrene:myrcene was used. The yield of the cross-dimers was determined by the quantitative 1H NMR spectrum of the crude irradiated mixture by comparing with the unreacted 2 and 3. Ocimene showed almost a fivefold increase in its conversion to C20 hydrocarbons, reaching 76.6 wt% after 48 h (Table 3, entry 5). Yet, we observed that the individual conversion of α-phellandrene is reduced when ocimene is present (Table 3, entries 4 and 5) due to a dilution effect. The dilution effect is less present in the cross-dimerization with myrcene, since it is more reactive than ocimene, and therefore the chances that one triplet myrcene successfully attacks one α-phellandrene is higher than the chances of the attack by a triplet ocimene. Myrcene conversion exceeded 90 wt% already after 24 h (Table 3, entry 3). The improvement of the conversion of myrcene into C20 products is evident from the analysis of the 1H NMR spectrum of the crude irradiated mixture, in which protons corresponding to unreacted myrcene are barely visible due to its conversion into dimers and/or the intramolecular cyclized product 9 (Fig. S32, ESI†). The values from cross dimerization with α-phellandrene in Table 3 are plotted in Fig. 7 and compared to the dimerization of ocimene and myrcene (Table 1).
 | ||
| Fig. 7 Conversion to dimers of ocimene and myrcene in the cross-dimerization with α-phellandrene. Data from Table 3 compared to their conversions when neat conditions were used (Table 1). Labels 1.0 eq. and 0.5 eq. describe the amount of monoterpene used as starting material related to α-phellandrene (mol). | ||
The higher conversions of myrcene and ocimene also resulted from the long-lived triplet state of α-phellandrene – as discussed above, α-phellandrene being a cyclic conjugated diene has longer lifetime than acyclic dienes.47 Because triplet α-phellandrene has a longer lifetime, its concentration is higher when compared to the concentration of triplet myrcene or triplet ocimene. Therefore, it is the same principle as in ground state reactions: as the triplet reactant is in higher concentration, its reaction with a ground state myrcene or ocimene (and α-phellandrene) is more likely. On the other hand, as discussed above, the addition of a less reactive monoterpene to react with α-phellandrene can cause a dilution effect that decreases the dimerization of α-phellandrene. Interestingly, the cross-dimerization attempt using 1:1 ratio of α-phellandrene and myrcene did not significantly improve the conversion of myrcene to C20 products. However, the implicit dilution led to enhance the formation of 9 (30 wt%).
Route to C15 and C30 hydrocarbons
Following the cross-dimerization of α-phellandrene with other monoterpenes, we tested the reaction with isoprene and monoterpenes in order to obtain C15 hydrocarbons. The cross-dimerization with isoprene led mainly to the formation of C15 and C20 compounds (Fig. 8A). However, a minor amount of C10 hydrocarbons (dimers of isoprene) was also observed.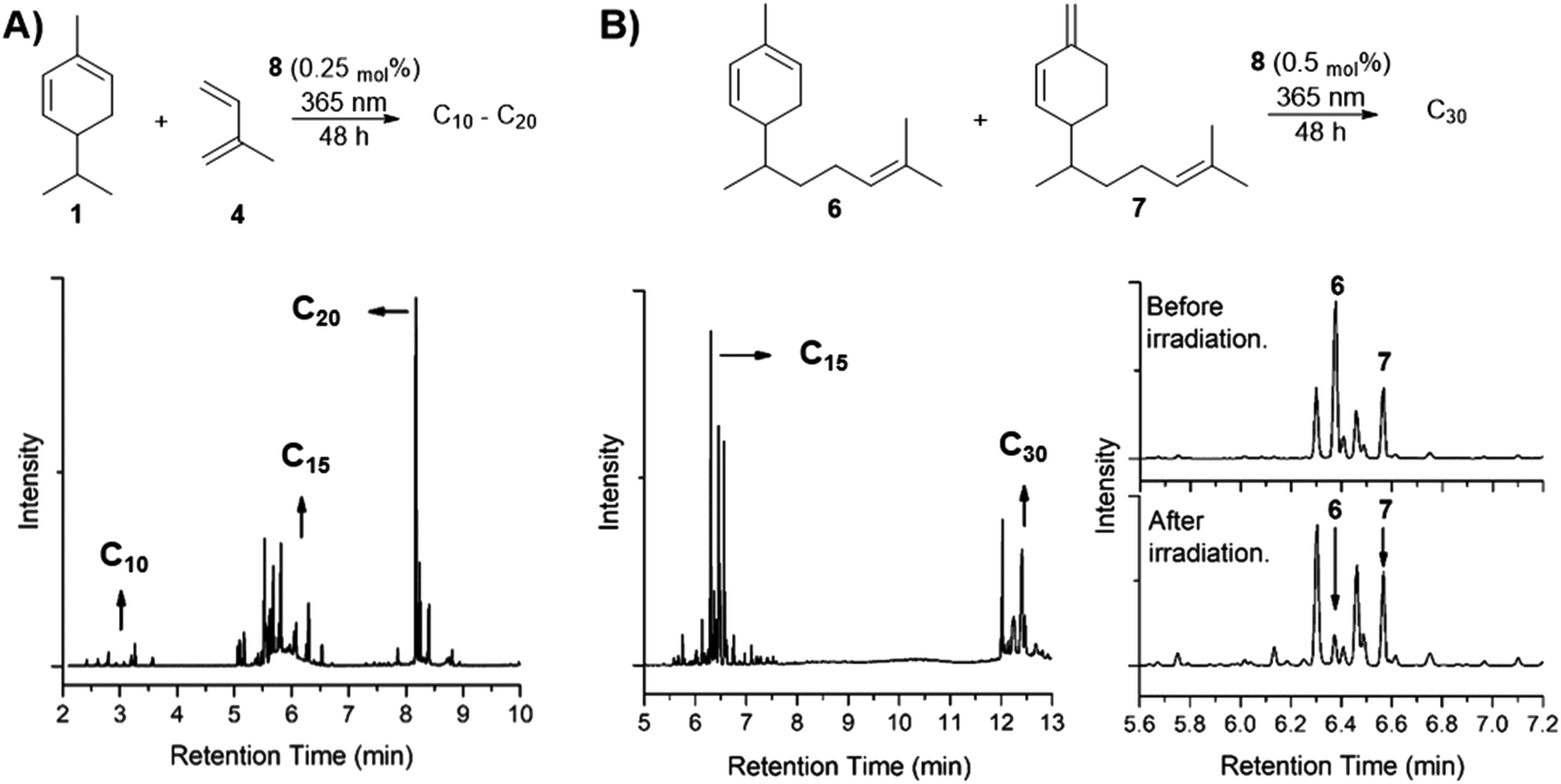 | ||
| Fig. 8 (A) GC trace of cross-dimerization between α-phellandrene and isoprene (1:1), where C10–C20 hydrocarbons can be observed. (B) GC trace of the ginger oil sample after irradiation under 365 nm, mixed with photosensitizer 8 (0.5 mol%) showing C30 dimers. Inset: chromatogram for the region of C15, before and after irradiation show that α-zingiberene peak (6) is reduced after irradiation, as well as 7, though to a lower extent. | ||
Conjugated dienes larger than monoterpenes are also available in nature. Examples of those fall in the continuation of the terpene homologue series (sesquiterpenes, diterpenes, etc.), and their dimerization could also provide hydrocarbon precursors to possible applications as lubricant oils. The observed reactivity of α-phellandrene makes endocyclic dienes the natural candidates for further investigations, such as the sesquiterpenes α-zingiberene 6 and β-sesquiphellandrene 7. These compounds are two of the main components present in ginger oil. Therefore, we irradiated a C15 fraction sample of ginger oil (87 g) mixed with photosensitizer 8 (0.25 mol%) in the CMS III, under 365 nm for 48 h. The viscosity of the crude mixture produced was visually higher, and almost complete conversion of α-zingiberene 6 to its dimers was observed by comparing the GC-MS trace of starting material and irradiated sample (Fig. 8B). The isolated yield of C30 dimers was 61 wt% (considering the amount of conjugated dienes in the starting material, determined by 1H NMR – see Fig S38, ESI†). Together with previous results reported herein, this is a further piece of evidence that endocyclic diene structures are a key feature for efficient photosensitized dimerization of hydrocarbon biomolecules towards renewable fuels and materials.
Fuel and lubricant related properties of selected C15 to C30 crudes
We computed the heats of combustion for the hydrogenation products of some of the compounds in Fig. 3 by following the procedure reported by Pahima et al.23 As expected, these compounds have high heats of combustion between 42.3 and 43.3 MJ kg−1, which are suitable for high energy density fuel applications (Table S5, ESI†). Interestingly, the dimers with cyclobutane and cyclooctane rings have higher NHOCs than dimers with cyclohexane rings. Hence, we synthesized hydrogenated samples of α-phellandrene dimers (HAPD), myrcene dimers (HMD), α-phellandrene-isoprene cross-dimers (HAPID) and ginger oil dimers (HGOD), and some physical properties relevant to fuels and lubricant oils were measured (Table 4).| Diesel fuels | |||||
|---|---|---|---|---|---|
| Property | HAPD | HMD | HAPID | Diesel D-2 (ref. 49) | Biodiesel49 |
| a LBO = lubricant base oil. | |||||
| Molecular formula | C20H36 | C20H40 | C20H36 C15H28 | — | — |
| Hydrogen content, % mass | 13.12 | 14.37 | 13.74 | 13 | 12 |
| Density at 25 °C, g mL−1 | 0.931 | 0.861 | 0.904 | 0.850 | 0.880 |
| Volumetric NHOC, MJ L−1 at 25 °C | 40.13 | 37.65 | 39.15 | 35.97 | 32.94 |
| Kinematic viscosity at 40 °C, mm2 s−1 | 41.1 | 12.7 | 6.45 | 1.3–4.1 | 4.0–6.0 |
| Pour point, °C | −21 | ≤45 | ≤45 | −35 to −15 | −5 to 10 |
| Lubricant and base oils | ||||||
|---|---|---|---|---|---|---|
| Property | HAPD | HMD | HGOD | C30 PAO mixture50 | LBO API group II 6 (ref. 51) | LBO from estolides52 |
| Molecular formula | C20H36 | C20H40 | C30H56 | — | — | — |
| Kinematic viscosity at 40 °C, mm2 s−1 | 41.1 | 12.7 | 303 | 15.23 | 41.5 | 102 to 519 |
| Kinematic viscosity at 100 °C, mm2 s−1 | 4.16 | 2.89 | 14.7 | 3.63 | 6.4 | — |
| Pour point, °C | −21 | ≤45 | −15 | −72 | −12 | −15 to −6 |
The crudes HMD, HAPD and HAPID have the experimental net heats of combustion (NHOC) higher than 43 MJ kg−1, which agrees with our computed values for some of the hydrogenated dimers (Table S5, ESI†). The volumetric NHOC values were higher than those for D2 diesel and biodiesel. Such values are desired for high energy density fuel applications.34 However, the obtained kinematic viscosities may restrict their use in such applications. While HAPD and HMD crudes have too high viscosities to be used stand-alone as diesel, they could be mixed in blends to afford the required viscosities.24,29 Yet, these blends could possibly be made entirely from non-fossil sources, using for example the C15 fraction produced in the cross-dimerization of α-phellandrene and isoprene. Indeed, adjustments of HAPID viscosity would be required to a lesser extent, as its viscosity is closer to that of D2 diesel (6.45 mm2 s−1, at 40 °C). Moreover, the volumetric NHOC of HAPID was 8.8% higher than the one for a typical D2 diesel. Alternatively, the C15 fraction in HAPID can be partially or entirely separated from the C20 fraction, affording a crude with higher proportion of C15 compounds, which would reduce the kinematic viscosity and likely approach the properties of hydrogenated sesquiterpenes, such as bisabolene.19 The pour point values found for the HAPID crudes were even lower than those for D2 diesel and biodiesel. Furthermore, earlier reports proposed to use C18–C20 hydrocarbons blends suitable for jet fuels.25,26,34,53 Therefore, this could also be an alternative end-use to the crudes HAPD and HMD.
The high viscosities of HAPD and HMD makes it possible to use these crudes for lubricant oil formulations. The kinematic viscosity at 40 °C (KV40) of HMD is 12.7 mm2 s−1, which is comparable to C30 poly alpha-olefins (POA) mixtures (15.23 mm2 s−1).50HAPD crudes have values for KV40 comparable to those of base oils belonging to API Group II 6 (41.5 mm2 s−1).51 The dimers derived from ginger oil (HGOD) have a very high KV40, and both HAPD and HGOD KV40 values are comparable to those of base oil synthesized from triesters and estolides derived from vegetable oils,52 showing a lower pour point in some cases. On the other hand, the viscosity index (VI) values were very low for both crudes HAPD, HMD and HGOD, as it was expected considering their cyclic structures. Yet, the use of viscosity index improvers or further refining for ring opening of some of the structures could increase the VI values.51 While the presence of rings negatively affects the VI, it should be considered that the cyclic structures also afford better solvency,51 therefore, the choice for keeping the rings could be interesting for applications where good solvency is required.
Outlines in GHG mitigation and economic feasibility
Our studies were carried out in a laboratory scale with maximum production rate at 11 g h−1 (Table 1, entry 1.4) of dimers under 365 nm light. Therefore, further work on scaling-up the process is required for a robust life cycle assessment (LCA) and techno-economic analysis (TEA). Yet, we briefly discuss the potential strengths and impacts of the photosensitized dimerization of terpenes.The non-edible evergreen trees of the Eucalyptus spp. correspond to 20 million hectares of forests worldwide.54 Among them, the E. dives (phellandrene variant) has 60–80% of α-phellandrene in its extracted oil.7 Assuming the highest oil yield of the E. dives to be similar to the one for E. globulus (900 kg ha−1 per year−1), the highest potential of global production of α-phellandrene would be 14 million tonnes per year. The actual yield would be lower since there is a large variation of constituents, oil yields, harvest windows, and other properties among different species.6 Because high α-phellandrene-content eucalyptus oils are used for inexpensive cleaning products,6,7 their use instead as a feedstock to lubricant oils and fuel production could be an added value to the eucalyptus oil market. On that regard, a recent work showed the economic potential of producing jet fuel from the leaf oil of Eucalyptus spp.55
Studies on the environmental impact of using eucalyptus tree to produce biomass for energy purpose have shown an opportunity to reduce GHG emissions up to 80% when compared to fossil fuels,56,57 especially due to the sequestration of CO2 by the crops offsets the emissions during the process, associated to their fast growth and short-rotation. The main source in these studies is the wood from the eucalyptus trees. Therefore, obtaining α-phellandrene from the leaf oil could be an additional route to valuable and sustainable goods. On the other hand, the high demand of land-use is a drawback, as well as the risk of threatening of biodiversity, as specific species that produce high amounts of α-phellandrene would be the optimal choice.
The α-phellandrene production from eucalyptus trees can also be a side route in the paper manufacturing, as well as it is for other terpenes. For instance, the pulp from paper manufacturing is a source to the bio-waste sulfate turpentine, which has a worldwide production of 330000 tonnes per year.8 The pyrolysis of β-pinene, one of the main components in the sulfate turpentine, produces myrcene,9 making turpentine a bio-waste source for myrcene production.
Alternatively, α-phellandrene and myrcene can also be produced by metabolically engineered cyanobacteria. Formighieri and Melis reported on the cyanobacterial production of α-phellandrene to reach titers of 0.15 mg g−1 dcw (dry cell weight),20 while titers of 0.33 mg of β-phellandrene g−1 dcw were achieved, over an incubation period of 48 h. Another strain reported on the same work produced mainly β-myrcene (62%) – but the overall amount of terpenes produced by that strain was low.
Recent works have explored the techno-economics and environmental impacts of the production of small hydrocarbons by cyanobacteria at industrial-scale.58–60 These studies reveal the possibility of reducing GHG emissions and they also indicate commercial viability. Their results depended heavily on the use of clean energy as input, optimal photobioreactors and improvements on the metabolic engineering of cyanobacteria in order to enhance cell productivity.58,59 The high volatility of small molecules enables an easier escape from the cell culture,59,60 but in the case of monoterpenes and larger molecules, more energy demanding methods may be required to extract molecules from culture such as the use of organic solvents, which might increase the environmental impact.17,20 The toxicity of monoterpenes to the cells represents another issue.61
As the sources examined and the ways to obtain them are considered to be renewable, we now reflect on the later steps to transform such feedstock into fuels and lubricant oils. Our approach uses low amounts of photosensitizer, and heating or cooling is not required during the dimerization step. Indeed, the main resource consumption would be electricity to power UV LEDs. While this could be provided by clean energy sources, such as photovoltaic cells or hydropower, it is also righteous to consider natural sunlight, as we have demonstrated the significant conversions obtained under simulated and natural sunlight. In this case, energy consumption would be much lower for the dimerization step. However, the land-use could be significant if one considers that the flat FEP setup (CMS IV) must be very wide to accommodate a multi-liter reaction mixture.
The steps after the dimerization are existing downstream processes in the oil industry – hydrogenation and distillation. Optimal and greener hydrogenation methods, with cheaper catalysts, and use of clean energy inputs should be considered to afford a final product that accounts for a reduced environmental impact as well as being commercially feasible.62
Conclusions
We have applied an established organic photochemical process to the conversion of key hydrocarbon biomolecules into renewable crudes for fuels and lubricant oils. By combining reactor design, selection of photosensitizer and use of reactive substrates, we have described the sunlight-promoted dimerization and cross-dimerization of terpenes to obtain C15–C30 hydrocarbon mixtures as potential precursors to drop-in diesel fuel and lubricant oil surrogates. Our preliminary assessment depicted the sustainability and viability of this process, which still requires further modification and scaling up in order to be submitted to a robust LCA and TEA in future.Making use of isoprene, monoterpenes and sesquiterpenes, a full sunlight-driven process to C10–C30 hydrocarbons would be possible, encompassing a diversity of sustainable liquid biofuels and lubricant oils produced.
Conflicts of interest
There are no conflicts of interests.Acknowledgements
We are grateful to Dr Wangchuk Rabten for design and construction of the flat tubing reactor, Dr William Siljebo for assistance in the construction of the LED panel, Dr João Rodrigues for discussions regarding production of terpenoids by microorganisms, Nathalie Proos Vedin and Dr Ouissam El Bakouri for assistance on the quantum chemical calculations, and Salauat R. Kiraev for assistance during the quenching experiments. Financial support from the Formas agency (grant 2017-00862) is greatly acknowledged. The computations were enabled by resources provided by the Swedish National Infrastructure for Computing (SNIC) at the National Supercomputer Center (NSC), Linköping, partially funded by the Swedish Research Council through grant agreement number 2018-05973.References
- International Renewable Energy Agency, IRENA (2019), Global Energy Transformation: A Roadmap to 2050, 2019 Search PubMed.
- International Renewable Energy Agency, Advanced biofuels what holds them back?, 2019 Search PubMed.
- R. Shah, M. Woydt and S. Zhang, The economic and environmental significance of sustainable lubricants, Lubricants, 2021, 9, 21 CrossRef CAS.
- A. E. Harman-Ware, in Chemical catalysts for biomass upgrading, Wiley-VCH, 2020, pp. 529–568 Search PubMed.
- K. H. C. Baser, M. Kürkçüoglu, B. Demirçakmak, N. Uülker and S. H. Beis, Composition of the essential oil of schinus molle l. grown in Turkey, J. Essent. Oil Res., 1997, 9, 693–696 CrossRef CAS.
- J. J. W. Coppen, Eucalyptus: the genus eucalyptus (medicinal and aromatic plants – industrial profiles), 2002 Search PubMed.
- B. E. J. Small, The Australian eucalyptus oil industry—an overview, Aust. For., 1981, 44, 170–177 CrossRef.
- A. Behr and L. Johnen, Myrcene as a natural base chemical in sustainable chemistry: a critical review, ChemSusChem, 2009, 2, 1072–1095 CrossRef CAS PubMed.
- R. L. Burwell, The mechanism of the pyrolyses of pinenes, J. Am. Chem. Soc., 1951, 73, 4461–4462 CrossRef CAS.
- G. Frank, Photosensitised reactions of α-pinene, J. Chem. Soc. B, 1968, 130–132 RSC.
- E. M. Kim, J. H. Eom, Y. Um, Y. Kim and H. M. Woo, Microbial synthesis of myrcene by metabolically engineered Escherichia coli, J. Agric. Food Chem., 2015, 63, 4606–4612 CrossRef CAS PubMed.
- B. Pattanaik and P. Lindberg, Terpenoids and their biosynthesis in cyanobacteria, Life, 2015, 5, 269–293 CrossRef CAS PubMed.
- N. Betterle and A. Melis, Photosynthetic generation of heterologous terpenoids in cyanobacteria, Biotechnol. Bioeng., 2019, 116, 2041–2051 CrossRef CAS PubMed.
- M. Gutensohn, T. T. H. Nguyen, R. D. McMahon, I. Kaplan, E. Pichersky and N. Dudareva, Metabolic engineering of monoterpene biosynthesis in tomato fruits via introduction of the non-canonical substrate neryl diphosphate, Metab. Eng., 2014, 24, 107–116 CrossRef CAS PubMed.
- C. Formighieri and A. Melis, Sustainable heterologous production of terpene hydrocarbons in cyanobacteria, Photosynth. Res., 2016, 130, 123–135 CrossRef CAS PubMed.
- A. L. Schilmiller, I. Schauvinhold, M. Larson, R. Xu, A. L. Charbonneau, A. Schmidt, C. Wilkerson, R. L. Last and E. Pichersky, Monoterpenes in the glandular trichomes of tomato are synthesized from a neryl diphosphate precursor rather than geranyl diphosphate, Proc. Natl. Acad. Sci. U.S.A., 2009, 106, 10865–10870 CrossRef CAS PubMed.
- J. S. Rodrigues and P. Lindberg, Metabolic engineering of synechocystis sp. PCC 6803 for improved bisabolene production, Metab. Eng. Commun., 2020, 12, e00159 CrossRef PubMed.
- R. Davidovich-Rikanati, E. Lewinsohn, E. Bar, Y. Iijima, E. Pichersky and Y. Sitrit, Overexpression of the lemon basil α-zingiberene synthase gene increases both mono- and sesquiterpene contents in tomato fruit, Plant J., 2008, 56, 228–238 CrossRef CAS PubMed.
- P. P. Peralta-Yahya, M. Ouellet, R. Chan, A. Mukhopadhyay, J. D. Keasling and T. S. Lee, Identification and microbial production of a terpene-based advanced biofuel, Nat. Commun., 2011, 2, 483 CrossRef PubMed.
- C. Formighieri and A. Melis, Cyanobacterial production of plant essential oils, Planta, 2018, 248, 933–946 CrossRef CAS PubMed.
- J. Filley, A. Miedaner, M. Ibrahim, M. R. Nimlos and D. M. Blake, Energetics of the 2 + 2 cyclization of limonene, J. Photochem. Photobiol., A, 2001, 139, 17–21 CrossRef CAS.
- J. D. Woodroffe and B. G. Harvey, High-performance, biobased, jet fuel blends containing hydrogenated monoterpenes and synthetic paraffinic kerosenes, Energy Fuels, 2020, 34, 5929–5937 CrossRef CAS.
- E. Pahima, S. Hoz, M. Ben-Tzion and D. T. Major, Computational design of biofuels from terpenes and terpenoids, Sustainable Energy Fuels, 2019, 3, 457–466 RSC.
- O. Staples, J. H. Leal, P. A. Cherry, C. S. McEnally, L. D. Pfefferle, T. A. Semelsberger, A. D. Sutton and C. M. Moore, Camphorane as a renewable diesel blendstock produced by cyclodimerization of myrcene, Energy Fuels, 2019, 33, 9956–9964 CrossRef.
- H. A. Meylemans, R. L. Quintana and B. G. Harvey, Efficient conversion of pure and mixed terpene feedstocks to high density fuels, Fuel, 2012, 97, 560–568 CrossRef CAS.
- B. G. Harvey, M. E. Wright and R. L. Quintana, High-density renewable fuels based on the selective dimerization of pinenes, Energy Fuels, 2010, 24, 267–273 CrossRef CAS.
- J. Xu, P. Zhu, X. Liu, Y. Hou, X. Yang, S. Shan, Y. Ma, D. Pan, B. Dong and Z. Guo, Preparation of high-density fuel through dimerization of β-pinene, Chem. Eng. Technol., 2020, 43, 2259–2265 CrossRef CAS.
- B. Yuan, Z. Wang, X. Yue, F. Yu, C. Xie and S. Yu, Biomass high energy density fuel transformed from α-pinene catalyzed by Brönsted-Lewis acidic heteropoly inorganic-organic salt, Renewable Energy, 2018, 123, 218–226 CrossRef CAS.
- J. D. Woodroffe and B. G. Harvey, A simple process for the dimerization and cross-coupling of isoprene and myrcene to high-performance jet and diesel blendstocks, Energy Fuels, 2022, 36, 2630–2638 CrossRef CAS.
- J. Xie, L. Pan, G. Nie, J. Xie, Y. Liu, C. Ma, X. Zhang and J. J. Zou, Photoinduced cycloaddition of biomass derivatives to obtain high-performance spiro-fuel, Green Chem., 2019, 21, 5886–5895 RSC.
- G. S. Hammond, P. A. Leermakers and N. J. Turro, Photosensitized cis-trans isomerization of the piperylenes, J. Am. Chem. Soc., 1961, 83, 2396–2397 CrossRef CAS.
- G. S. Hammond, N. J. Turro and A. Fischer, Photosensitized cycloaddition reactions, J. Am. Chem. Soc., 1961, 83, 4674–4675 CrossRef CAS.
- G. S. Hammond, N. J. Turro and R. S. H. Liu, Mechanisms of photochemical reactions in solution. XVI. Photosensitized dimerization of conjugated dienes, J. Org. Chem., 1963, 28, 3297–3303 CrossRef CAS.
- J. A. Muldoon and B. G. Harvey, Bio-based cycloalkanes: the missing link to high-performance sustainable jet fuels, ChemSusChem, 2020, 13, 5777–5807 CrossRef CAS PubMed.
- A. Rana, L. Cid Gomes, J. S. Rodrigues, D. M. M. Yacout, H. Arrou-Vignod, J. Sjölander, N. P. Vedin, O. el Bakouri, K. Stensjö and P. Lindblad, A combined photobiological-photochemical route to C10 cycloalkane jet fuels from carbon dioxide via isoprene, Green Chem., 2022, 24, 9602–9619 RSC.
- R. S. H. Liu and G. S. Hammond, Photosensitized internal addition of dienes to olefins, J. Am. Chem. Soc., 1967, 89, 4936–4944 CrossRef CAS.
- W. L. Dilling, Photochemical cycloaddition reactions of nonaromatic conjugated hydrocarbon dienes and polyenes, Chem. Rev., 1969, 69, 845–877 CrossRef CAS.
- J. E. Baldwin and J. P. Nelson, Cycloadditions. VI. Photosensitized dimerization of α-phellandrene, J. Org. Chem., 1966, 31, 336–338 CrossRef CAS.
- N. J. Turro, V. Ramamurthy and J. C. Scaiano, Principles of molecular photochemistry: an introduction, University Science Books, 2009 Search PubMed.
- P. Klán and J. Wirz, Photochemistry of organic compounds: from concepts to practice, John Wiley & Sons, 2009 Search PubMed.
- S. K. Rajagopal, K. Nagaraj, S. Deb, V. Bhat, D. Sasikumar, E. Sebastian and M. Hariharan, Extending the scope of the carbonyl facilitated triplet excited state towards visible light excitation, Phys. Chem. Chem. Phys., 2018, 20, 19120–19128 RSC.
- L. D. Elliott, S. Kayal, M. W. George and K. Booker-Milburn, Rational design of triplet sensitizers for the transfer of excited state photochemistry from UV to visible, J. Am. Chem. Soc., 2020, 142, 14947–14956 CrossRef CAS PubMed.
- H. Hioki, H. Ooi, M. Hamano, Y. Mimura, S. Yoshio, M. Kodama, S. Ohta, M. Yanai and S. Ikegami, Enantioselective total synthesis and absolute stereostructure of hippospongic acid A, Tetrahedron, 2001, 57, 1235–1246 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other function, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- R. A. Caldwell and M. Singh, Lifetimes of conjugated diene triplets, J. Am. Chem. Soc., 1982, 104, 6121–6122 CrossRef CAS.
- Energy Plus Weather Data Download – Stockholm Arlanda 024600 (IWEC), https://energyplus.net/weather-location/europe_wmo_region_6/SWE/SWE_Stockholm.Arlanda.024600_IWEC, accessed 8 October 2021.
- T. L. Alleman, R. L. McCormick, E. D. Christensen, G. Fioroni, K. Moriarty and J. Yanowitz, Biodiesel Handling and Use Guide, National Renewable Energy Lab, United States, 5th edn, 2016 Search PubMed.
- S. Shylesh, A. A. Gokhale, C. R. Ho and A. T. Bell, Novel strategies for the production of fuels, lubricants, and chemicals from biomass, Acc. Chem. Res., 2017, 50, 2589–2597 CrossRef CAS PubMed.
- Ã. K. Rudolphi, E. Kassfeldt and M. Torbacke, Lubricants: introduction to properties and performance, John Wiley & Sons, Incorporated, New York, United Kingdom, 2014 Search PubMed.
- B. Saha and D. Vlachos, Synthesis of (hemi) cellulosic lubricant base oils via catalytic coupling and deoxygenation pathways, Green Chem., 2021, 23, 4916–4930 RSC.
- C. F. Ryan, C. M. Moore, J. H. Leal, T. A. Semelsberger, J. K. Banh, J. Zhu, C. S. McEnally, L. D. Pfefferle and A. D. Sutton, Synthesis of aviation fuel from bio-derived isophorone, Sustainable Energy Fuels, 2020, 4, 1088–1092 RSC.
- T. H. Booth, Eucalypt plantations and climate change, For. Ecol. Manage., 2013, 301, 28–34 CrossRef.
- M. R. Davis, D. Kainer, G. A. Tuskan, M. H. Langholtz, C. M. Hellwinckel, M. Shedden and L. Eaton, Modeled economic potential for Eucalyptus spp. production for jet fuel additives in the United States, Biomass Bioenergy, 2020, 143, 105807 CrossRef CAS.
- B. Gabrielle, N. Nguyen The, P. Maupu and E. Vial, Life cycle assessment of eucalyptus short rotation coppices for bioenergy production in southern France, GCB Bioenergy, 2013, 5, 30–42 CrossRef.
- S. González-García, M. T. Moreira and G. Feijoo, Environmental aspects of eucalyptus based ethanol production and use, Sci. Total Environ., 2012, 438, 1–8 CrossRef PubMed.
- A. Nilsson, K. Shabestary, M. Brandão and E. P. Hudson, Environmental impacts and limitations of third-generation biobutanol: life cycle assessment of n-butanol produced by genetically engineered cyanobacteria, J. Ind. Ecol., 2020, 24, 205–216 CrossRef CAS.
- J. N. Markham, L. Tao, R. Davis, N. Voulis, L. T. Angenent, J. Ungerer and J. Yu, Techno-economic analysis of a conceptual biofuel production process from bioethylene produced by photosynthetic recombinant cyanobacteria, Green Chem., 2016, 18, 6266–6281 RSC.
- M. Amer, E. Z. Wojcik, C. Sun, R. Hoeven, R. Hoeven, J. M. X. Hughes, M. Faulkner, I. S. Yunus, S. Tait, L. O. Johannissen, S. J. O. Hardman, D. J. Heyes, G. Q. Chen, G. Q. Chen, M. H. Smith, P. R. Jones, H. S. Toogood, N. S. Scrutton, N. S. Scrutton and N. S. Scrutton, Low carbon strategies for sustainable bio-alkane gas production and renewable energy, Energy Environ. Sci., 2020, 13, 1818–1831 RSC.
- J. Keasling, H. Garcia Martin, T. S. Lee, A. Mukhopadhyay, S. W. Singer and E. Sundstrom, Microbial production of advanced biofuels, Nat. Rev. Microbiol., 2021, 19, 701–715 CrossRef CAS PubMed.
- N. R. Baral, O. Kavvada, D. Mendez-Perez, A. Mukhopadhyay, T. S. Lee, B. A. Simmons and C. D. Scown, Techno-economic analysis and life-cycle greenhouse gas mitigation cost of five routes to bio-jet fuel blendstocks, Energy Environ. Sci., 2019, 12, 807–824 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2se01370c |
| This journal is © The Royal Society of Chemistry 2023 |