
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTarget-triggered CRISPR–Cas13a autocatalysis-driven amplification strategy for one-step detection of circadian clock gene†
Zhiyuan
Feng‡
,
Yi
Xue‡
,
Yangfang
Yun‡
,
Zheng
Liu
and
Jingjing
Zhang
 *
*
State Key Laboratory of Analytical Chemistry for Life Science, School of Chemistry and Chemical Engineering, Chemistry and Biomedicine Innovation Center (ChemBIC), Nanjing University, Nanjing 210023, China. E-mail: jing15209791@nju.edu.cn
First published on 19th April 2023
Abstract
The precise and sensitive detection of circadian clock genes is crucial for understanding their roles in regulating biological processes and their implications in various diseases. Here, we present a CRISPR/Cas13a-powered autocatalytic cleavage circuit (CRISPR-ACC) for one-step detection of the BMAL1 mRNA, a key component of the circadian clock. The CRISPR-ACC system utilizes a three-stranded RNA probe for Cas13a-mediated collateral cleavage and autocatalysis-driven amplification, resulting in attomolar sensitivity and single-base mismatch discrimination. We demonstrate the efficacy of CRISPR-ACC in detecting BMAL1 from cell extracts, with results that strongly correlate with RT-PCR. Our method provides a simple, rapid, and highly specific approach for evaluating circadian clock genes, with potential applications in circadian-related disease diagnosis. The CRISPR-ACC system can be readily adapted to detect other clock genes, highlighting its versatility and promise for advancing our understanding of circadian rhythms.
Introduction
Circadian rhythms are endogenous biological rhythms that enable organisms to adapt to the daily environmental changes.1,2 These rhythms are controlled by a complex network of molecular feedback loops, composed of clock genes and their protein products.3,4 Among clock genes, BMAL1 (brain and muscle ARNT-like protein 1) has been extensively studied for its involvement in cancers.5,6 For instance, BMAL1 is a transcription factor that regulates the expression of many downstream genes involved in cell proliferation, apoptosis, and metabolism.7 Recent studies have shown that the expression of BMAL1 is dysregulated in various cancers, including breast, prostate, liver, and colon cancer.8 Moreover, BMAL1 has been found to modulate the response of cancer cells to chemotherapy and radiation therapy, highlighting the importance of BMAL1 regulation in cancer diagnosis and treatment.9,10 Therefore, the accurate detection of BMAL1 in cancer cells may provide valuable insights into the molecular mechanisms underlying cancer development and progression, as well as potential targets for cancer therapy. Currently, BMAL1 mRNA was mainly identified based on polymerase chain reaction (PCR),11,12 northern blotting,13 microarray,14 and RNA sequencing.15 Despite some success, most of these methods still suffer from some drawbacks because of the tradeoffs between sensitivity, turnaround time, usability and costs. Therefore, it is essential to establish a new method for rapid, convenient, highly sensitive and selective detection of BMAL1 mRNA with the ultimate goal of understanding its biological functions.Recent years have witnessed an explosive development of CRISPR (clustered regularly interspaced short palindromic repeats) based nucleic acids detection platforms with the assistance of different CRISPR-associated (Cas) effectors.16,17 Among them, Cas13a is a CRISPR RNA (crRNA)-guided RNA-targeting effector and can be activated to engage in collateral cleavage of nearby non-targeted RNA after specifically recognition of its target RNA.18 The collateral cleavage ability of Cas13a has been widely used to cleave fluorophore-quencher labelled RNA probes for amplified fluorescence signals in optical detection.19,20 However, the sensitivity of this method is limited, typically in the femtomolar range, which is insufficient for detecting low abundance targets such as BMAL1. To improve detection sensitivity, Cas13a has been combined with various nucleic acid-based signal amplification strategies.21–25 However, these integrated systems may suffer from the time-consuming procedures, requirement of specialized equipment and careful probe design, as well as pre-amplification of RNA. Autocatalysis, a process vital to biochemistry and mimicking physiological mechanisms, has recently been utilized in nucleic acid-based circuits for detecting diverse biomarkers.26,27 Its sigmoidal reaction profile enables efficient signal amplification, rendering it a desirable choice for high-sensitivity detection. For instance, Shi et al. developed a CRISPR–Cas12a autocatalysis-driven feedback amplification network that could achieve attomolar DNA detection sensitivity.28 Inspired by this precedent work, we speculate that autocatalysis can be an ideal strategy to develop ultrasensitive sensors for BMAL1 mRNA assisted by Cas13a yet has rarely been explored.
With these considerations in mind, we present here the first report of CRISPR–Cas13a-powered autocatalytic cleavage circuit (CRISPR-ACC) for ultrasensitive and specific detection of BMAL1 mRNA. In our CRISPR-ACC, a three-stranded RNA probe (TSRP) that contains two uracil-rich consecutive regions for Cas13a-mediated collateral cleavage was designed, and applied as the fluorescence reporter assisted by the FAM-BHQ1 fluorophore-quencher pair. By targeting a 20 nt conserved region of BMAL1 mRNA, we demonstrated that CRISPR-ACC can be precisely implemented within one-step at physiological temperature, and achieved attomolar RNA detection sensitivity yet without any pre-amplification steps. Moreover, CRISPR-ACC was able to recognize single-base mismatches, even the mismatches are located distal to the 3′-protospacer-flank site (PFS). These specific features were further highlighted in evaluating BMAL1 levels in various cancer cells. By changing the sequence of targeting region of the Cas13a–crRNA, this CRISPR-ACC strategy could be readily applied for detection of other clock genes, exhibiting promising potential in circadian clock-associated disease diagnosis.
Materials and methods
Reagents and materials
All the DNA and RNA oligonucleotides (Table S1†) were obtained from Sangon Biotech Co. Ltd. (Shanghai, China) and purified by High-Performance Liquid Chromatography (HPLC). 2× TBE-Urea loading buffer (C506046) was obtained from Sangon Biotech Co. Ltd. (Shanghai, China). Reverse Transcription Kit (RR047A) and one-step TB Green RT-PCR Kit (RR420A) were purchased from Takara Biomedical Technology (Beijing, China). Monarch RNA Cleanup Kit (50 μg, T2040S) and RNA gel loading dye for non-denaturing polyacrylamide gels (6×, B7024S) were obtained from New England Biolabs (Beijing, China). Gel red nucleic acid stain was obtained from Vazyme (Nanjing, China). All solutions were prepared with Millipore water (18.25 MΩ cm−1). Nuclease free water was obtained from Servicebio (Wuhan, China). LbuCas13a and crRNA were expressed and purified as described in the ESI† (Section S1).The following buffers were used in the experiments. 1× TBE buffer consisted of 89 mM Tris-boric, 2 mM EDTA. 10× Cas13a reaction buffer contained 200 mM Tris-HCl (pH 7.5), 1.0 M NaCl, and 20 mM MgCl2. Protein storage buffer was composed of 20 mM Tris-HCl (pH 7.5), 300 mM NaCl, and 1 mM DTT.
Preparation of three-stranded RNA probe (TSRP)
The TSRP was prepared by mixing 6 μL of 3′-BHQ-1 labelled inhibitor RNA-1 (i1-RNA, 10 μM), 6 μL of inhibitor RNA-2 (i2-RNA, 10 μM), and 5 μL of 5′-FAM labelled RNA intermediate (10 μM) in 50 μL of 1× Cas13a reaction buffer. The reaction mixture was annealed by heating to 90 °C for 1 min followed by a 0.1 °C s−1 cooling ramp to 4 °C to allow complete hybridization. Finally, the resulting TSRP solution (1.0 μM) was stored at 4 °C in the dark until further use.Assembly of LbuCas13a/crRNA ribonucleoprotein (RNP) complex
LbuCas13a/crRNA1 (RNP1) and LbuCas13a/crRNA2 (RNP2) were prepared by respectively incubating LbuCas13a (1 μM) with crRNA1 (1 μM) and crRNA1 (1 μM) in 1× Cas13a reaction buffer at 37 °C for 30 min. The pre-assembled RNP complex was stored at 4 °C before use.Evaluation of the analytical performance of BMAL1 sensor
In a typical CRISPR-ACC assay, 5 μL of BMAL1 mRNA at various concentrations was mixed with 5 μL of TSRP (1.0 μM), 20 μL of RNP1 (50 nM), and 20 μL of RNP2 (50 nM). For real-time fluorescence monitoring of the assay, the resulting solution was added into a PCR tube and the fluorescence signal was collected using a LightCycler 96 Real-Time PCR system (Roche) at 37 °C for 10 min. For end-point fluorescence detection, the mixture solution was incubated at 37 °C for 30 min, and the fluorescent signal of the resulting solution was measured using a F-320 (TIANJIN GANGDONG, China). The excitation wavelength was 488 nm, and the fluorescence spectra were collected from 500 nm to 650 nm. For selectivity analysis, a series of BMAL1 mRNA with single-base mutation at different positions was tested following the same procedure described above.Gel electrophoresis studies
The purified LbuCas13a was characterized using 10% SDS-PAGE in SDS-PAGE running buffer at 200 V for 30 min, while the purified crRNA was analyzed using 10% Urea-PAGE in 1× TBE buffer at 13 mA for 30 min. Low MW DNA ladder(25–500 bp) was used as the marker. All nucleic acid gels were stained with GelRed Nucleic Acid Gel Stain, and visualized by UV shadowing using 3500 digital gel image analysis system (Tanon, China). The TSRP assembly (FAM labelled) was characterized using 12% native PAGE, and visualized by blue shadowing.Cell culture
All cell lines were obtained from the Cell Bank of Type Culture Collection of Chinese Academy of Sciences (Shanghai, China). The B16F10 and 4T1 cell lines were cultured in RPMI-1640 medium complemented with 10% fetal bovine serum (FBS), 100 U mL−1 penicillin, and 100 μg mL−1 streptomycin in a humidified incubator containing 5% CO2 at 37 °C. In contrast, the RAW264.7 cell line was cultured in DMEM as described above.Total RNA extraction and RT-PCR analysis
The above three cell lines were seeded in 6-well plates and grown until 60–70% confluency was reached. After that, the cells were collected through digestion and centrifugation. Then, the total RNA was extracted using the Trizol and UNlQ-10 column total RNA isolation kit (Sangon) according to the manufacturer's instructions. Total RNA concentration was determined spectroscopically using the Nanodrop, and 1 μg of total RNA was reverse transcribed using PrimeScript™ RT reagent Kit. The resulting cDNA samples were further amplified using real-time PCR with the primers indicated in Table S2† and a TB Green® Premix Ex Taq™. All RT-PCR tests were performed on a LightCycler® 96 System (Roche), and all results were quantified using the comparative threshold cycle (Cq) method.Results and discussion
Working Principle of CRISPR–Cas13a-powered autocatalytic cleavage circuit (CRISPR-ACC) for BMAL1 detection
As illustrated in Scheme 1, the CRISPR-ACC system is composed of three modules: 1) a Cas13a from Leptotrichia buccalis (LbuCas13a) that acts as a RNA-targeting Cas effector, without the requirement of a protospacer adjacent motif (PAM) in the target for recognition, 2) a three-stranded RNA probe (TSRP) as the substrate for LbuCas13a, which was constructed by hybridizing a 41 nt RNA intermediate with two inhibitor RNAs (i1-RNA and i2-RNA) respectively containing seven consecutive poly-U regions, and 3) two crRNAs (crRNA1 and crRNA2) containing the specific 20 nt RNA region at the 3′ end, targeting to the conserved region of BMAL1 mRNA and the above RNA intermediate, respectively. The TSRP is designed so that the crRNA2-recognition region within the RNA intermediate was blocked synergistically by i1-RNA and i2-RNA, preventing it from binding to the Cas13a–crRNA2 complex. In the presence of BMAL1 target, the Cas13a–crRNA1 complex recognizes BMAL1 and thereby activates the trans-cleavage activity of the Cas13a–crRNA1 complex. In addition, LbuCas13a preferentially cleaves single-stranded RNA rather than double-stranded RNA, and more importantly, at uracil flanking ribose-phosphodiester bond. Such a Cas13a-mediated cleavage significantly lowered the melting temperature of the RNA intermediate and the inhibitor RNAs (<37 °C), thus shifting the hybridization equilibrium of RNAs in TSRP to release the uncaged RNA intermediate. Subsequently, the decaged RNA intermediate binds to and activates the Cas13a–crRNA2 complex, which could cleave another TSRP to generate a new uncaged RNA intermediate. The newly liberated RNA intermediate could hybridize with another Cas13a–crRNA2 to trigger the next ACC cycle. When fluorophore-quencher labelled TSRP was used, the above target-triggered ACC process would promote the generation of amplified fluorescence signal through the accumulation of uncaged fluorophore-labelled RNA, forming a basis of autocatalysis-driven amplification detection of target mRNA.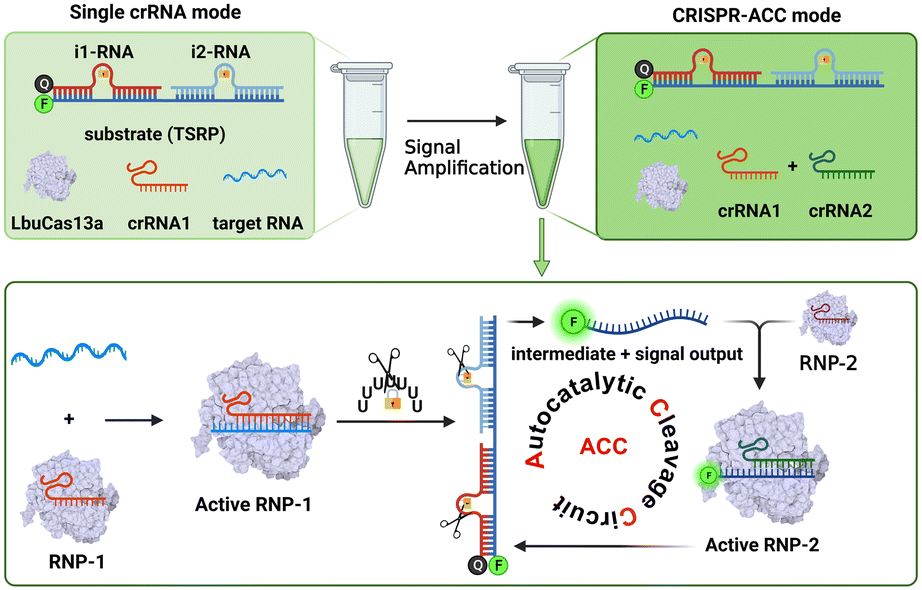 | ||
| Scheme 1 Schematic diagram of the construction of an autocatalytic cleavage circuit (ACC) platform for RNA detection using CRISPR–Cas13a. | ||
Design and characterization of LbuCas13a and crRNA
Compared to other Cas effectors, LbuCas13a functions by recognizing a non-G 3′-PFS in the target RNA, rather than requiring a protospacer adjacent motif (PAM) in the target. In the case of our study on BMAL1 mRNA, the PFS was designed to be rC, thereby ensuring the high cleavage activity of LbuCas13a. Firstly, the LbuCas13a with a molecular weight of approximately 140 kDa was expressed in Escherichia coli, following the previous protocol with some modifications.29 After purification with the nickel-nitrilotriacetic acid (Ni-NTA) affinity chromatography, the purity was verified by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) (Fig. S1†). Then, the specific crRNA1 targeting a 20 nt conserved region of BMAL1 mRNA (coding domain sequences region) was designed and transcribed in vitro. Similarly, a 51 nt crRNA2 targeting the specific sequence region in the RNA intermediate between position34 and 51 (full-length numbering, 5′ to 3′) was obtained. The purity and size of these two crRNAs was characterized using denaturing Urea-PAGE (Fig. S2†).Design and characterization of the three-stranded RNA probe (TSRP)
Unlike traditional Cas13a-based detection using a single-stranded RNA (ssRNA) fluorescent reporter, the performance of our CRISPR-ACC system relies on the effective target-triggered activation of autocatalytic cleavage circuit assisted by crRNA1/crRNA2. In this context, the TSRP should possess two functions: (1) effectively blocks the RNA intermediate binding to crRNA2, which is essential to minimize the nonspecific background signal; (2) effectively releases the RNA intermediate in response to the BMAL1 target, which is essential to maximize the signal output. In addition, LbuCas13a was reported to recognize ssRNA rather than dsRNA, and its collateral cleavage activity displayed highly base preference with an order of U > G > A = C.22 Based on the above considerations, two inhibitor RNAs were employed to lock the RNA intermediate. As shown in Fig. 1A, the BHQ-1 labelled i1-RNA contains a 21 bp sequence complementary to the 1–21th bases of the FAM-labelled RNA intermediate (full-length numbering, 5′ to 3′), while i2-RNA contains a 20 bp sequence complementary to the remaining 22–41 bases at the 3′ end. Meanwhile, both i1-RNA and i2-RNA was embedded with seven consecutive uracil ribonucleotides to form a loop-like structure, serving as the primary Cas13a cleavage site. The calculated melting temperature of i1-RNA and i2-RNA with the RNA intermediate is 53.0 and 55.0 °C, respectively, resulting in a stable TSRP in its close state. The assembly of TSRP was first verified by native PAGE (Fig. S3†), where a well-defined band with slower electrophoretic mobility (lane 4) was observed compared to the control samples (lane 1 to lane 3).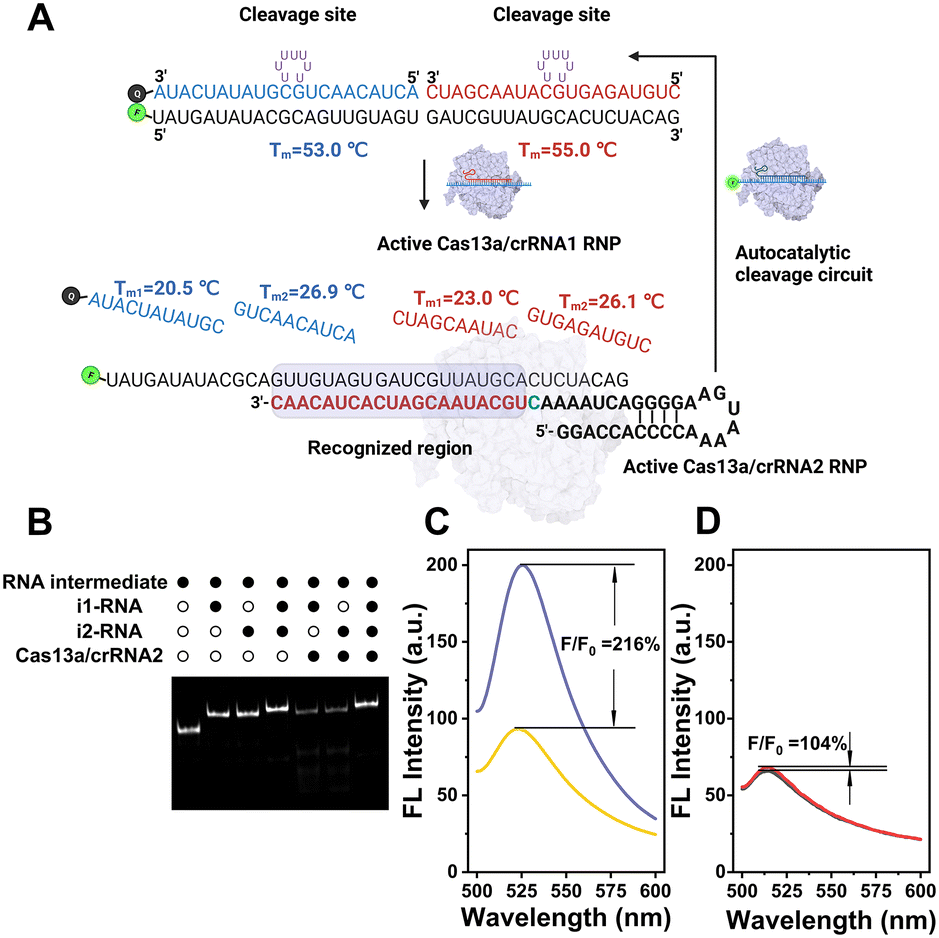 | ||
| Fig. 1 (A) Schematic of the CRISPR-ACC system for BMAL1 mRNA detection. (B) Native PAGE (12%) analysis showing that the TSRP (200 nM) is stable against Cas13a–crRNA2 in the absence of target RNA. The RNA intermediate is labelled by a 6-carboxyfluorescein at 5′ end. The symbols “solid circle” and “hollow circle”, indicate the presence and absence of the reagent, respectively. Comparison of fluorescence enhancement to BMAL1 (1 pM) using the CRISPR-ACC signal amplification system (C) and the single crRNA-based method (D). | ||
After confirming the successful assembly of TSRP, we asked whether it is stable against Cas13a–crRNA2 in the absence of target RNA. To visualize the RNA products, FAM-labelled RNA intermediate was used to construct the TSRP, and then analyzed using native PAGE (Fig. 1B). After annealing the RNA intermediate with i1-RNA or i2-RNA, a single band with a larger molecular weight than the RNA intermediate (lane 1) appeared in lane 2 and lane 3, respectively, indicating the formation of RNA duplex. Moreover, upon the addition of Cas13a–crRNA2 complex, the RNA duplex band in both lane 5 and lane 6 became evidently lighter, and concurrently several consecutive bands with higher electrophoretic mobility, assigned to cleaved RNA fragments, was observed. This result indicated a low blocking efficacy to the RNA intermediate in the presence of single inhibitor RNA. In contrast, no new band emerged for treating TSRP with Cas13a–crRNA2 complex (lane 7) compared to the control TSRP band (lane 4), indicating a high tolerance of TSRP against binding to Cas13a–crRNA2. Taken together, these findings suggest successful construction of highly effective caged TSRP through the introduction of two inhibitor RNAs.
Feasibility of CRISPR-ACC for fluorescent detection of BMAL1
Next, we attempt to evaluate the capability of signal enhancement through CRISPR-ACC coupled with TSRP. Using BMAL1 mRNA as a model, we initially performed a fluorescence assay to verify the collateral cleavage activity of the LbuCas13a/crRNA1 complex and its specific recognition. As shown in Fig. S4,† the TSRP (curve d) displayed a very weak fluorescence signal at 520 nm, indicating an effective fluorescence quenching due to the close proximity between FAM and BHQ-1 in the intact TSRP. Moreover, in the absence of either BMAL1 (curve b) or crRNA1 (curve c), no significant changes in fluorescence signal was observed. In contrast, upon the addition of 1 nM BMAL1, a dramatic increase of fluorescent signal was obtained (curve a). This result was attributed to the spatial separation of the fluorophore from the quencher upon the Cas13a-mediated TSRP cleavage.To evaluate the fluorescence signal amplification provided by the crRNA2-assisted ACC reaction, we compared the fluorescence responses of CRISPR–Cas13a assay for BMAL1 at the picomolar level with and without the crRNA2. As shown in Fig. 1D, in the presence of only crRNA1, a 4% increase in the signal was observed for 1 pM BMAL1 using CRISPR–Cas13a assay. However, when crRNA1 and crRNA2 were applied together (Fig. 1C), the fluorescence signal increased 116% over background. This result indicated that the crRNA2-assisted ACC amplification strategy could dramatically improve the signal-to-noise ratio and result in more sensitive detection of BMAL1.
Sensitivity of CRISPR-ACC for BMAL1 detection
To obtain the best performance of the CRISPR-ACC sensor for detecting BMAL1, we further optimized various experimental parameters, including the ratio between inhibitor RNAs and the RNA intermediate, and the concentrations of LbuCas13a and TSRP. As shown in Fig. S5A,† when the ratio between inhibitor RNAs and the RNA intermediate is 1.2, the maximum S/N ratio is obtained. This result may be attributed to the slightly degradation of inhibitor RNAs during the reaction.30 Similarly, the optimal concentration of LbuCas13a and TSRP was obtained as 20 nM (Fig. S5B†) and 200 nM (Fig. S5C†), respectively.Under optimized experimental conditions, we performed a series of fluorescence assays for quantitative detection of BMAL1 in Tris buffer. Firstly, various concentrations of BMAL1 (ranging from 10 fM to 1 nM) was tested by a traditional Cas13a-based assay using the 6 nt ssRNA reporter (5′-FAM-UUUUUU-BHQ1-3′) as the substrate. As shown in Fig. 2A, the fluorescence signal at 520 nm increased with increasing BMAL1 concentrations from 10 fM to 1 nM, and a limit of detection (LOD) of 2.9 fM was obtained based on a 3σb/slope (Fig. 2D), where σb is the standard deviation of three blank samples. Secondly, we conducted similar tests and tried to detect BMAL1 using an updated Cas13a-based assay by introducing the TSRP as the substrate for Cas13a–crRNA1 (Fig. 2B). We found that this updated system exhibited a narrow dynamic range from 1 pM to 1 nM (Fig. 2E), and showed a decreased sensitivity (LOD, 0.7 pM) compared to the above traditional assay. This observation may be due to slow dissociation of BHQ-1/FAM labelled RNA duplex compared to ssRNA, without the assistance of crRNA2. Finally, the BMAL1 was quantified using CRISPR-ACC system (Fig. 2C), and the end-point fluorescence signal (30 min) is positively correlated with the concentration of BMAL1. Moreover, a linear dynamic range of 100 aM to 1 nM as well as a LOD of 4.6 aM was obtained, which is approximately 630-fold and 1.5 × 105-fold lower than the above traditional and updated Cas13a-based assay, respectively. The linear range and LOD of the above three fluorescent detection methods are listed in Table S3.† In addition, a comparison between the recently developed CRISPR RNA detection methods and CRISPR-ACC is summarized in Table S4.† Taken together, these findings further highlighted the vital role of the ACC amplifier in improving the overall Cas13a-based RNA sensing performance.
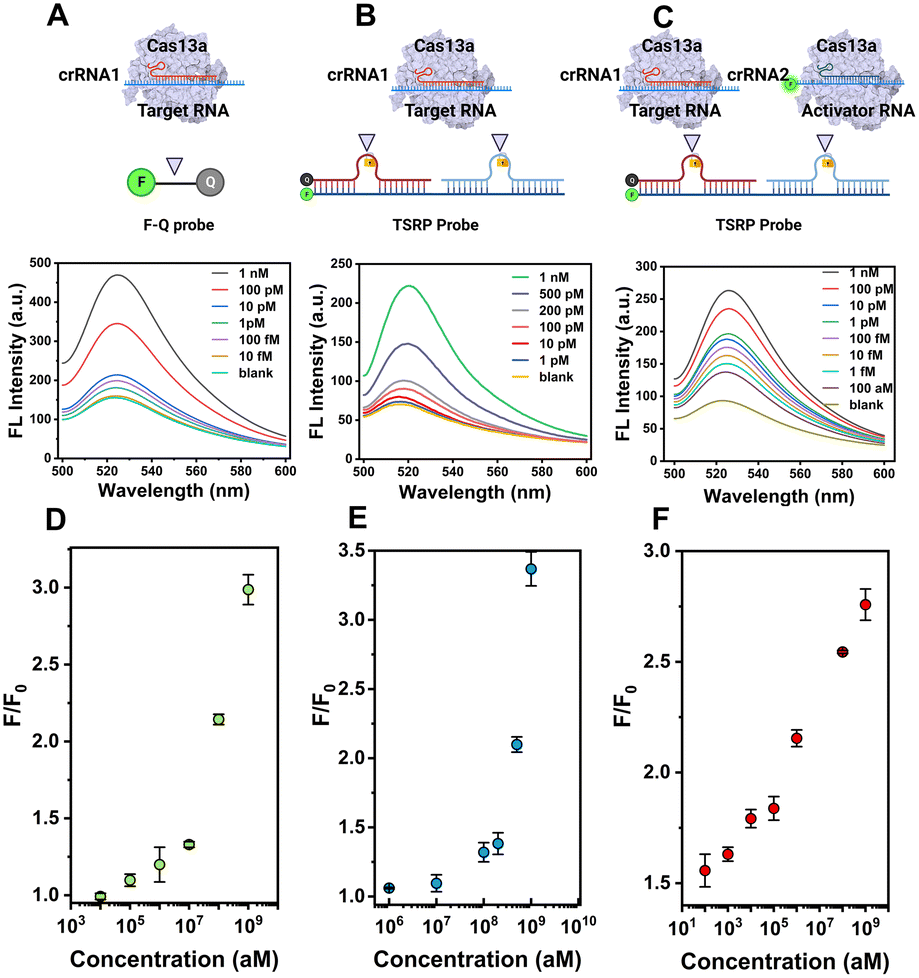 | ||
| Fig. 2 CRISPR-ACC system enables highly sensitive detection of RNA. Schematic and fluorescence spectra of the traditional Cas13a-based assay (A), the updated Cas13a-based assay (B), and the CRISPR-ACC system (C) responding to different concentrations of BMAL1 mRNA as indicated. The concentration of CRISPR/crRNA1 and CRISPR/crRNA2 is 20 nM, while the 6 nt ssRNA reporter (F-Q probe) and TSRP probe is 200 nM. (D), (E), and (F) represents the corresponding dynamic detection range for (A), (B), and (C), respectively. The data represents the mean value ± SD (n = 3). | ||
Specificity of CRISPR-ACC for BMAL1 detection
Encouraged by the high sensitivity of CRISPR-ACC, we further challenged its capacity of discrimination toward single-nucleotide mutation (SM) in BMAL1 mRNA. To this aim, we introduced ten SMs along the BMAL1 sequence, starting from its 5′ end, which led to mismatches at various positions in the crRNA1-target duplex (Fig. 3A). Significant differences in the normalized ΔF value were observed between wild-type BMAL1 and BMAL1 containing SMs at the same concentration (1.0 nM). To better evaluate the discrimination ability, the fluorescence enhancement ratio (FER, defined as ΔFWT/ΔFSM) was calculated as the discrimination factor. As shown in Fig. 3B, compared with the WT BMAL1(Perfect), the CRISPR-ACC system displayed the highest discrimination ability to the SMs located at position 1 (SM-1), with maximum FER value of 32.4. Moreover, a medium discrimination ability (10.0 ≤ FER ≤ 20.0) was obtained between WT BMAL1 and SM-5/SM-7/SM-8/SM-9/SM-10, indicating a good specificity of the CRISPR-ACC system in the discrimination of PFS-distal SMs. However, for SM-2, SM-3, SM-4 and SM-6, our CRISPR-ACC system only showed acceptable discrimination ability (1.0 ≤ FER < 5.0). The CRISPR system exhibits significant position-dependent specificity, which is determined by the varying tolerance of the spacer sequence to base mismatches, particularly in the seed region (8 to 14 nucleotides near the PAM sequence). Mismatches in the seed region can significantly affect the activity of the Cas9 protein.31 Additionally, it has been previously reported that Cas12a's crRNA is less sensitive to single-base mismatches when there are 4 to 6 nucleotides away from the PAM region at the 3′ end while maintaining high nuclease activity which similar to Cas 9 system.32 Such position-dependence in discrimination ability of SMs was also observed in other CRISPR–Cas-based sensors,28,33 because the SMs location relative to the PFS or PAM may affect the stability and specificity of the crRNA-target RNA complex.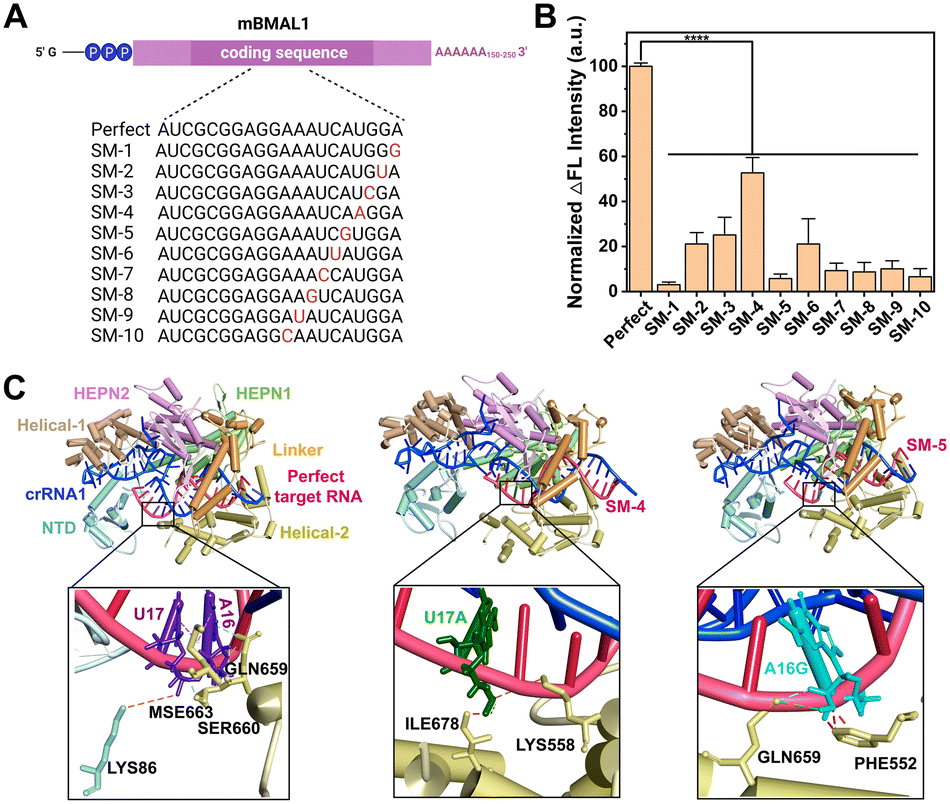 | ||
| Fig. 3 CRISPR-ACC system for single-nucleotide mutation (SM) detection. (A) Sequence information of the perfect target RNA and a series of RNA SMs, with the mutated site indicated in red. (B) Normalized fluorescence signal of RNA SMs and perfect target RNA. Each RNA has a concentration of 1.0 nM. Statistical significance: ****P < 0. 0001. The data represents the mean value ± SD (n = 3). (C) The 3D structures of LbuCas13a–crRNA1–BMAL1 (Perfect/SM-4/SM-5) ternary complex and the molecular interactions between BMAL1 and Cas13a. The orange, pink, green, and red dashed lines indicate electrostatic, hydrophobic, hydrogen bonding interactions, and spatial resistance, respectively. | ||
To explain the above position-dependent specificity, we further performed the computational modeling studies. Previous work confirmed that LbuCas13a displayed a bilobed architecture containing an α-helical recognition (REC) lobe and a nuclease (NUC) lobe. The REC lobe anchors the repeat region of the crRNA1, while the NUC lobe contains a channel where the guide-target RNA duplex is bound.34 In this scenario, we attempt to study the conformational change in the crRNA1 and LbuCas13a on ternary complex formation upon the recognition of WT or mutated BMAL1. In our study, the 51 nt crRNA sequence contained a 31 nt repeat region, similar to the previous report, and a 20 nt BMAL1 recognition region (Fig. 1A). Recent studies revealed that interactions between the crRNA and LbuCas13a play critical roles in the overall RNA cleavage, while the target RNA mainly interacts with the Helical-2, Linker, and HEPN1 domains through the sugar-phosphate backbone of nucleotides.33,34 Under these considerations, the target RNA SM-4, SM-5, and Perfect BMAL1 were chosen as the model, we first predicted the 3D binary structures of crRNA1-WT/mutated BMAL1 (Perfect/SM-4, SM-5) and optimized their structures by 10 ns MD simulation (Fig. S6A†). Next, we performed docking studies to investigate the conformational change in the crRNA upon binding with WT and mutated BMAL1. The ternary structures of the LbuCas13a–crRNA1-target RNA complex were shown in Fig. 3C, in which the WT sites were shown as purple stick mode and two mutation sites were highlighted in green (SM-4) within the WT BMAL1 contact the side chains of LYS86 within the NTD domain, and MSE663, SER660 and GLN659 within the Helical-2, domain (Fig. 3C). In contrast, the U17A and A16G mutations make a steric hindrance with ILE678 and PHE552, respectively (Fig. 3C). Thus, the introduction of SM-4 (U17A) and SM-5 (A16G) disrupts the interactions between crRNA and the NTD/Helical-2 domains in Cas13a. This is because the mutation causes a change in the complementary pairing of the target RNA with the BMAL1 recognition region of crRNA1. Furthermore, since the mutation sites of target RNA are adjacent to the middle of the repeat region and BMAL1 recognition region of crRNA1, the mutation resulted in a large change in the conformation of the repeat region of crRNA1 (Fig. S6B†), which may affect the binding mode of crRNA1-target RNA to Cas13a and reduce the cleavage activity of Cas13a.
Evaluation of BMAL1 levels in different cancer cells using CRISPR-ACC
Given the highly sensitive and selective detection of BMAL1, the CRISPR-ACC system was further applied to evaluate the BMAL1 levels in different cancer cells. Briefly, we initially prepared a series of samples containing different amounts of total RNA extracted from the B16F10 cells, which were subsequently analysed using the CRISPR-ACC system (Fig. 4A). As expected, the fluorescence signal at 520 nm increased with increasing total RNA amounts from 5 to 200 ng (Fig. 4B), and BMAL1 in 5 ng total RNA could be identified (inset). Moreover, using RAW264.7 cells as the control, we compared the relative BMAL1 levels extracted from the same concentration of B16F10 and 4T1 cells using both CRISPR-ACC system and RT-qPCR. As shown in Fig. 4C, the BMAL1 level in B16F10 cells is 0.5 ± 0.1 folds lower than that of RAW264.7 cells, whereas 4T1 cells showed a 1.8 ± 0.1 folds increase in BMAL1 level. These results match closely with the average values of 0.2 and 1.7 measured by RT-qPCR (Fig. S7 and Table S5†). Overall, these observations not only verify the feasibility of our CRISPR-ACC system for the detection of BMAL1 in real samples, but also demonstrate the good accuracy of our assay.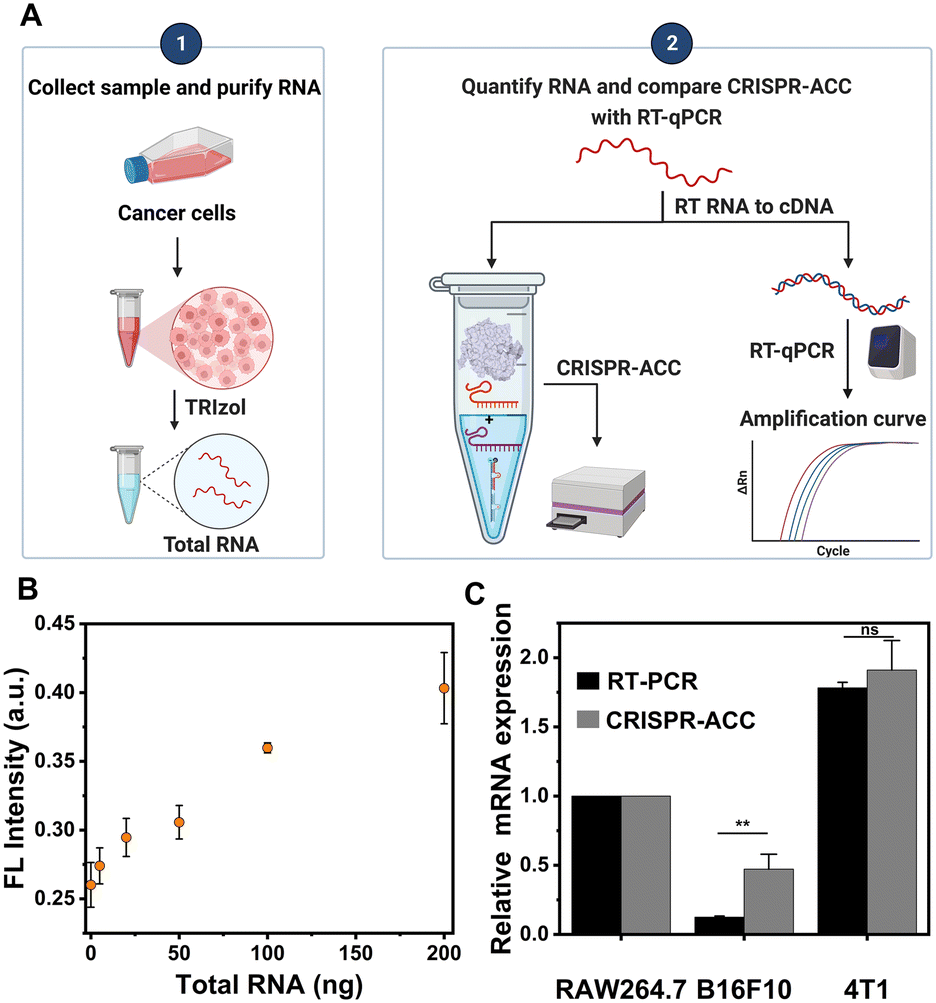 | ||
| Fig. 4 CRISPR-ACC for detection of endogenous BMAL1 mRNA in cells. (A) Workflow comparison of CRISPR-ACC and RT-PCR for mRNA detection in cells. (B) Fluorescence signals of CRSPR-ACC system for detecting B16F10 cell total RNA, quantified using the LightCycler 96 real-time PCR system (Roche). (C) Comparison of mBMAL1 expression levels in different cells detected using CRISPR-ACC and RT-PCR. Statistical significance: **P < 0. 01, ns (p > 0.05), no significant differences. | ||
Conclusions
In summary, we have developed a CRISPR/Cas13a-powered autocatalytic cleavage circuit (CRISPR-ACC) for rapid and accurate detection of a circadian clock gene. Using BMAL1 mRNA as a model, we designed a three-stranded RNA reporter for Cas13a-mediated collateral cleavage, allowing the rapid target-triggered autocatalysis cleavage with minimal background signal. Taking advantage of both sequence-specific recognition of CRISPR–Cas13a and autocatalysis-driven signal amplification, the CRISPR-ACC system offers significant improvements in both analytical sensitivity and specificity. In addition, the CRISPR-ACC system possessed high specificity with single base resolution in a position-dependent manner. As a proof-of-concept, we also demonstrated its utility in detecting BMAL1 from cell extracts, showing strong positive correlation with the RT-PCR method. In addition, it is still important to effectively improve Cas13a's ability to detect single nucleotide mismatches. Our future work will focus on functionalizing the spacer region of crRNA by adding stem-loop structures or engineering Cas protein, which may help improve Cas13a's ability to recognize single nucleotide mutations, and further improve the detection accuracy and precision of CRISPR-ACC system. Overall, the proposed CRISPR-ACC system provides a simple, rapid, sensitive and selective method for circadian clock gene evaluation, and thus paves a new avenue to study the circadian-related diseases.Author contributions
Zhiyuan Feng: validation, data curation, investigation, writing. Yi Xue: validation, data curation, writing. Yangfang Yun: computational modelling analysis, data curation, writing. Zheng Liu: writing, investigation, data curation. Jingjing Zhang: manuscript editing, conceptualization, methodology.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 22004063; 22274072), Natural Science Foundation of Jiangsu Province (no. 20200303).Notes and references
- A. Sancar, L. A. Lindsey-Boltz, S. Gaddameedhi, C. P. Selby, R. Ye, Y.-Y. Chiou, M. G. Kemp, J. Hu, J. H. Lee and N. Ozturk, Biochemistry, 2015, 54, 110–123 CrossRef CAS PubMed.
- C. Savvidis and M. Koutsilieris, Mol. Med., 2012, 18, 1249–1260 CAS.
- A. Sancar and R. N. Van Gelder, Science, 2021, 371, eabb0738 CrossRef CAS PubMed.
- R. Braun, W. L. Kath, M. Iwanaszko, E. Kula-Eversole, S. M. Abbott, K. J. Reid, P. C. Zee and R. Allada, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E9247–E9256 CAS.
- S. S. El-Naidany, A. E. Eltorgman, I. Elmadbouh, A. M. Naguib and M. F. A. Assar, Gene Rep., 2023, 30, 101711 CrossRef CAS.
- S. Chatterjee, D. Nam, B. Guo, J. M. Kim, G. E. Winnier, J. Lee, R. Berdeaux, V. K. Yechoor and K. Ma, J. Cell Sci., 2013, 126, 2213–2224 CAS.
- Z.-L. Zeng, M.-W. Wu, J. Sun, Y.-L. Sun, Y.-C. Cai, Y.-J. Huang and L.-J. Xian, J. Biochem., 2010, 148, 319–326 CrossRef CAS PubMed.
- Y. Yang, L. A. Lindsey-Boltz, C. M. Vaughn, C. P. Selby, X. Cao, Z. Liu, D. S. Hsu and A. Sancar, J. Biol. Chem., 2021, 297, 101068 CrossRef CAS PubMed.
- L. Shan, W. Zheng, B. Bai, J. Hu, Y. Lv, K. Chen, X. Wang, Y. Pan, X. Huang, H. Zhu and S. Dai, Cancer Med., 2023, 12, 4472–4485 CrossRef CAS PubMed.
- C. Sun, C. Li, W. Liu and H. B. Schiöth, Biomedicines, 2022, 10, 3108 CrossRef CAS PubMed.
- M. Y. Kim, S. Jung, J. Kim, H. J. Lee, S. Jeong, S. J. Sim and S. K. Kim, Sci. Rep., 2021, 11, 6463 CrossRef CAS PubMed.
- C. Paris, V. Moreau, G. Deglane, E. Voirin, P. Erbacher and N. Lenne-Samuel, Nucleic Acids Res., 2010, 38, e95–e95 CrossRef PubMed.
- K. Oishi, K. Sakamoto, T. Okada, T. Nagase and N. Ishida, Biochem. Biophys. Res. Commun., 1998, 253, 199–203 CrossRef CAS PubMed.
- M. J. McDonald and M. Rosbash, Cell, 2001, 107, 567–578 CrossRef CAS PubMed.
- Y. Umemura, N. Koike, Y. Tsuchiya, H. Watanabe, G. Kondoh, R. Kageyama and K. Yagita, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2114083119 CrossRef CAS PubMed.
- C. Niu, X. Xing and C. Zhang, Sens. Diagn., 2023, 2, 155–162 RSC.
- S.-Y. Wang, Y.-C. Du, D.-X. Wang, J.-Y. Ma, A.-N. Tang and D.-M. Kong, Anal. Chim. Acta, 2021, 1185, 338882 CrossRef CAS PubMed.
- Y. Sha, R. Huang, M. Huang, H. Yue, Y. Shan, J. Hu and D. Xing, Chem. Commun., 2021, 57, 247–250 RSC.
- X. Guo, T. Tian, X. Deng, Y. Song, X. Zhou and E. Song, Anal. Chim. Acta, 2022, 1209, 339853 CrossRef CAS PubMed.
- Y. Ma, Q. Mou, P. Yan, Z. Yang, Y. Xiong, D. Yan, C. Zhang, X. Zhu and Y. Lu, Chem. Sci., 2021, 12, 11740–11747 RSC.
- P. Song, P. Zhang, K. Qin, F. Su, K. Gao, X. Liu and Z. Li, Talanta, 2022, 246, 123521 CrossRef CAS PubMed.
- H. Zeng, P. Zhang, X. Jiang, C. Duan, Y. Yu, Q. Wu and X. Yang, Anal. Chim. Acta, 2022, 1217, 340009 CrossRef CAS PubMed.
- Y. Yu, H. Zeng, Q. Wu, X. Jiang, C. Duan, J. Long, M. Chen and X. Yang, Anal. Chim. Acta, 2022, 1227, 340266 CrossRef CAS PubMed.
- M. Huang, R. Huang, H. Yue, Y. Shan and D. Xing, Sens. Actuators, B, 2020, 325, 128799 CrossRef CAS.
- Y. Cui, S. Fan, Z. Yuan, M. Song, J. Hu, D. Qian, D. Zhen, J. Li and B. Zhu, Talanta, 2021, 224, 121878 CrossRef CAS PubMed.
- Y. V. Gerasimova and D. M. Kolpashchikov, Chem. Soc. Rev., 2014, 43, 6405–6438 RSC.
- R. Li, Y. Zhu, X. Gong, Y. Zhang, C. Hong, Y. Wan, X. Liu and F. Wang, J. Am. Chem. Soc., 2023, 145, 2999–3007 CrossRef CAS PubMed.
- K. Shi, S. Xie, R. Tian, S. Wang, Q. Lu, D. Gao, C. Lei, H. Zhu and Z. Nie, Sci. Adv., 2021, 7, eabc7802 CrossRef CAS PubMed.
- Y. Shan, X. Zhou, R. Huang and D. Xing, Anal. Chem., 2019, 91, 5278–5285 CrossRef CAS PubMed.
- J. Wang, Q. Xia, J. Wu, Y. Lin and H. Ju, Anal. Chim. Acta, 2021, 1187, 339131 CrossRef CAS PubMed.
- P. D. Hsu, D. A. Scott, J. A. Weinstein, F. A. Ran, S. Konermann, V. Agarwala, Y. Li, E. J. Fine, X. Wu, O. Shalem, T. J. Cradick, L. A. Marraffini, G. Bao and F. Zhang, Nat. Biotechnol., 2013, 31, 827–832 CrossRef CAS PubMed.
- L. Gao, D. B. T. Cox, W. X. Yan, J. C. Manteiga, M. W. Schneider, T. Yamano, H. Nishimasu, O. Nureki, N. Crosetto and F. Zhang, Nat. Biotechnol., 2017, 35, 789–792 CrossRef CAS PubMed.
- L. Liu, X. Li, J. Ma, Z. Li, L. You, J. Wang, M. Wang, X. Zhang and Y. Wang, Cell, 2017, 170, 714–726 CrossRef CAS PubMed.
- Y. Ke, S. Huang, B. Ghalandari, S. Li, A. R. Warden, J. Dang, L. Kang, Y. Zhang, Y. Wang, Y. Sun, J. Wang, D. Cui, X. Zhi and X. Ding, Adv. Sci., 2021, 8, 2003611 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sd00069a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |