DOI:
10.1039/D3SD00046J
(Paper)
Sens. Diagn., 2023,
2, 726-735
Braided copper cobaltite/MWCNT composites enable acetylcholine detection at sub-nanomolar levels in vitro†
Received
21st February 2023
, Accepted 18th April 2023
First published on 18th April 2023
Abstract
Developing a sensor for neurotransmitter detection is of paramount medical importance in the early diagnosis of neurological and psychiatric disorders and monitoring the treatment efficacy of drugs targeting the cholinergic system. To address this need, we present a non-enzymatic electrochemical sensor based on a copper cobaltite/MWCNT composite for detecting acetylcholine, a neurotransmitter found in human body fluids. The sensor utilises a bisolvent interface-assisted synthesis technique to produce highly crystalline braided networks of copper cobaltite, which were subsequently characterised using high-resolution transmission electron microscopy (HRTEM) to study their evolution and crystallinity. The composite-modified sensor exhibited outstanding detection capabilities with a limit of detection (LOD) of 0.8 nM, superior selectivity in the presence of interfering molecules, and exceptional stability. To miniaturise the bulky three-electrode system, an all-integrated flexible sensor was fabricated and was successfully employed to detect acetylcholine released by sub-cultured human neural cells, demonstrating its potential for real-time diagnostic applications.
Introduction
Acetylcholine (ACh) is a neurotransmitter essential for various physiological processes, including memory, learning, attention, and muscle movement. ACh detection in biological samples is of utmost clinical significance for the early diagnosis and surveillance of various neurological and psychiatric disorders, such as Alzheimer's disease, Parkinson's disease, and schizophrenia. ACh detection can also aid in understanding the mechanism of action of cholinergic system-targeting drugs such as cholinesterase inhibitors and muscarinic receptor agonists.1–4 However, the lower concentration and short lifetime of ACh and its lack of electro, fluorescent, and UV activity have made its in vivo detection difficult.2 The detection of ACh in the human body has traditionally been performed using labour-intensive and expensive methods. However, electrochemical sensing has emerged as a preferred method due to its cost-effectiveness, compactness, and ability to provide timely results.3,4 In recent years, significant progress has been made in the electrochemical sensing of ACh. A wide range of sensors has been developed, primarily based on enzymatic processes.3,5,6 Despite the success of enzymatic electrochemical sensors for detecting ACh, there is a growing need for alternative non-enzymatic sensors. This is due to factors such as the high cost associated with enzymatic sensors, the labour-intensive and complex process of immobilising and purifying enzymes, and the reduced stability of enzymatic sensors.4,7 The development of non-enzymatic sensors for the sensitive and selective detection of ACh in real-time poses a significant challenge for the scientific community. The feasibility of non-enzymatic reactions is limited to highly electroactive materials, such as transition metal oxides.1 Thus, the design and fabrication of non-enzymatic sensors for ACh detection utilising these materials represent a significant area of ongoing research.
The use of transition metal compounds and their composites has been a prominent approach in the development of non-enzymatic electrochemical sensors for the detection of ACh.1 Previous studies have demonstrated the potential of a variety of transition metal composites, including NiO nanostructures, Fe2O3 nanoparticles, MnO2-integrated metal–organic frameworks, Cu@Cu2O nanostructures, and CuCo2O4 nanostructures, for non-enzymatic ACh detection.8–12 Among metal oxides, spinel compounds have gained attention as intelligent materials due to their unique combination of bimetallic properties that surpass their monometallic analogues.13 Copper-substituted cobaltites, in particular, are widely studied for their improved electrical conductivity, high redox activity, tunable morphology and properties, good chemical stability, environmental benignity, and cost-effectiveness.8,13–15 In this study, copper cobaltite (CuCo2O4) was chosen as a material for modifying the electrode to detect ACh effectively. The literature shows various methods for synthesising copper cobaltites with tailored morphology and dimension. Our previous work has established bisolvent interface-assisted synthesis as an efficient technique for preparing physicochemically tuned nanomaterials.16,17 The addition of highly conductive fillers such as multi-walled carbon nanotubes (MWCNT) or graphene oxide (GO) to the synthesised copper cobaltite enhances electrical conductivity and can be used as modifying electrodes.18–20 Among these conductive fillers, MWCNT offer faster electron transfer by virtue of their enhanced electrical conductivity, specific surface area, and improved stability.21 In light of these studies, it can be concluded that copper cobaltite composites can be effectively utilised as non-enzymatic electrochemical sensors for ACh detection.
This study presents a morphologically-tuned CuCo2O4/MWCNT composite synthesised via an interface-assisted technique for real-time detection of ACh. To the best of our knowledge, this is the first report on the morphological tuning of braided copper cobaltite using the interface-assisted technique for better electrochemical activity. Although recent research has focused on developing cobaltite-based non-enzymatic sensors for ACh detection, fewer studies have been conducted on the real-time detection of ACh released by human neural cells.4,8 This work aims to broaden the current studies on non-enzymatic electrochemical sensors for real-time detection of ACh. The CuCo2O4/MWCNT composite was synthesised through an n-butanol/water interface-assisted technique, which allows for control of the morphology of the resulting nanostructures. The electrochemical activity of the resulting nanostructures was then characterised and found to be suitable for the non-enzymatic detection of ACh. The composite was then modified with a small amount of MWCNT to enhance conductivity. The CuCo2O4/MWCNT composite-modified sensor showed an enhanced redox current response to the redox reaction of ACh, with a better detection limit, even in the presence of interfering groups. To miniaturise the bulky three-electrode system, an all-integrated flexible sensor was fabricated. The fabricated sensor's utility for real-time applications was demonstrated by detecting ACh released by sub-cultured human neural cells. In conclusion, this study presents a potential, scalable, and cost-effective non-enzymatic sensor for the sensitive and selective real-time detection of ACh.
Materials and methods
Material synthesis and characterisation
In this study, copper cobaltite (CC) nanostructures were synthesised via a bisolvent interface-assisted technique, as reported in our previous works.16,22 Specifically, 28 mmol of urea was dissolved in 20 mL of n-butanol, and an aqueous solution of varying ratios of copper and cobalt nitrate precursors (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, 1:10, and 1:20) were added. The reaction mixture was stirred at 100 rpm at 95 °C for 6 hours. The resulting products, denoted as CC 1, CC 2, and CC 3, corresponding to the precursor ratios of 1:2, 1:10, and 1:20, respectively, were then filtered, washed with an ethanol–water mixture, and dried under ambient conditions. Higher precursor ratios above 1:20 were not considered due to practical difficulties in dissolving the precursors in water. The optimised sample, CC 3, was then combined with different weight percentages of multi-walled carbon nanotubes (MWCNTs) (4–6 nm diameter and 5–20 μm length) to form CC/MWCNT composite.17 The MWCNTs were first dispersed in water using probe sonication with a 5% HPMC solution. The required amount of CC was added to this dispersion and subjected to bath sonication for 10 minutes to obtain a printable composite ink. De-ionized water (ELGA Purelab Quest UV, 18.2 MΩ) was used throughout the synthesis.
:2, 1:10, and 1:20) were added. The reaction mixture was stirred at 100 rpm at 95 °C for 6 hours. The resulting products, denoted as CC 1, CC 2, and CC 3, corresponding to the precursor ratios of 1:2, 1:10, and 1:20, respectively, were then filtered, washed with an ethanol–water mixture, and dried under ambient conditions. Higher precursor ratios above 1:20 were not considered due to practical difficulties in dissolving the precursors in water. The optimised sample, CC 3, was then combined with different weight percentages of multi-walled carbon nanotubes (MWCNTs) (4–6 nm diameter and 5–20 μm length) to form CC/MWCNT composite.17 The MWCNTs were first dispersed in water using probe sonication with a 5% HPMC solution. The required amount of CC was added to this dispersion and subjected to bath sonication for 10 minutes to obtain a printable composite ink. De-ionized water (ELGA Purelab Quest UV, 18.2 MΩ) was used throughout the synthesis.
The morphologies of the as-synthesized products were characterised using field emission scanning electron microscopy (FESEM, FEI Quanta FEG 200F) and high-resolution transmission electron microscopy (HRTEM, JEOL/JEM 2100 and Thermo Fisher Talos F200 S). The elemental composition of the products was confirmed using energy dispersive spectroscopy (EDS, Octane Plus, Gemini 300/EDS) coupled with SEM, and the uniform distribution of individual elements was analysed through elemental mapping (Thermo Fisher Talos F200 S). The elemental composition was further verified using inductively coupled plasma mass spectroscopy (ICP-MS, Thermo Fisher iCAP RQ ICP-MS). The chemical and structural properties of the compounds were studied using attenuated total reflectance-infrared spectroscopy (ATR-IR, PerkinElmer Frontier MIR spectrometer) and Raman spectroscopy (Horiba Labram HR Evolution Confocal Raman Spectrometer) with a 532 nm laser. The structural features and crystallinity were analysed using X-ray diffraction (XRD) studies with PANALYTICAL Aeris Research (source: Cu Kα).
Electrochemical studies
A conventional three-electrode system was used for the electrochemical characterisation of the prepared compounds, consisting of a material-modified glassy carbon working electrode (GCE), Ag/AgCl(aq) reference electrode, and platinum counter electrode. The active surface of the GCE was cleaned and polished with ethanol, DI water and 0.05 μM alumina slurry before modification. The GCE was then electrochemically activated using a continuous cyclic voltammetric scan in 0.1 M H2SO4. For electrode modification, 6 μL of the compound dispersed in ethanol was drop cast onto the GCE, followed by 2 μL of a 5% Nafion solution. In the present study, 0.1 M NaOH was employed as the electrolyte for electrochemical analysis. Electrochemical impedance spectroscopy (EIS) and cyclic voltammetry (CV) were performed using the Origalys OGFEIS and OGF500 electrochemical workstation, respectively. The scan rate was fixed at 50 mV s−1 unless otherwise stated. The limit of detection (LOD) was determined from the calibration plot of anodic peak current vs. concentration using the formula 3Sb/m, where Sb represents the standard deviation of the blank, and m represents the slope of the calibration curve. Interference studies were conducted by comparing the anodic peak currents of the modified electrode in the presence of 500 nM ACh and 5 μM concentration of various interfering molecules such as ascorbic acid, uric acid, histamine, glutamic acid, dopamine, leucine, glycine, arginine, and cysteine. To assess the shelf life of the sensor, the modified electrode was stored at 4 °C, and the anodic current towards ACh was periodically analysed.
To detect the presence of ACh in serum, samples were collected from two healthy individuals and diluted ten-fold with 0.1 M NaOH. The resulting solution was subsequently analysed for the presence of ACh via the cyclic voltammetry (CV) technique. To fabricate the flexible sensor, adhesive polyimide applied flexible substrate was used for assembling electrodes. CC/MWCNT composite, conductive silver paste, and pyrolytic graphite sheet were used as the working, reference, and counter electrodes, respectively. The copper tape was used for providing electrical connections. For the detection of ACh released by neural cells, human SH-SY5Y neural cells were cultured at 37 °C in Dulbecco's modified Eagle's medium (DMEM, Thermo Fisher Scientific, MA, USA) containing 10% foetal bovine serum (FBS; Thermo Fisher Scientific, MA, USA) and 1% penicillin–streptomycin (Thermo Fisher Scientific, MA, USA) and incubated in a humidified environment containing 5% CO2 and 95% air. 100 μM choline chloride, the precursor of ACh, was added to the media, and the release of ACh was triggered by adding 200 μL 500 mM KCl solution after 2 hours. The released ACh was detected using the cyclic voltammetry technique with a composite-modified electrode. The specific details of the chemicals utilized for synthesis and electrochemical detection can be found in the ESI† section of the study.
Results and discussion
The morphologically tuned copper cobaltite (CC) nanostructures were synthesised at the quasi-two-dimensional interface of two immiscible solvents, n-butanol and water. The controlled release of urea and metal nitrate precursors to the interface with high interfacial tension was achieved by dissolving them in different immiscible solvents. In the n-butanol phase, urea underwent a series of decomposition reactions that resulted in the release of OH− and CO32−, serving as an alkaline source for the CC synthesis. The OH− and CO32− ions triggered the generation of Cu/Co carbonate hydroxide species, which, when oxidised in the presence of oxygen in a hot n-butanol/water bisolvent, resulted in the formation of CC nanostructures. Hence the following mechanism was proposed for the formation of CC:14,16,23| | | 6CO(NH2)2 → C3H6N6 + 6NH3↑ + 3CO2↑ | (1.1) |
| | | NH3 + ROH → NH4+ + OR− | (1.2) |
| | OR− + H2O ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) ROH + OH− ROH + OH− | (1.3) |
| | | CO2 + 2OH− → CO32− + H2O | (1.4) |
| | | (Cu2+, Co2+) + 2OH− + CO32− → (Cu, Co)2(CO3)(OH)2 | (1.5) |
| | | Cu2(CO3)(OH)2 + 2Co2(CO3)(OH)2 + O2 → 2CuCo2O4 + 3CO2↑ + 3H2O | (1.6) |
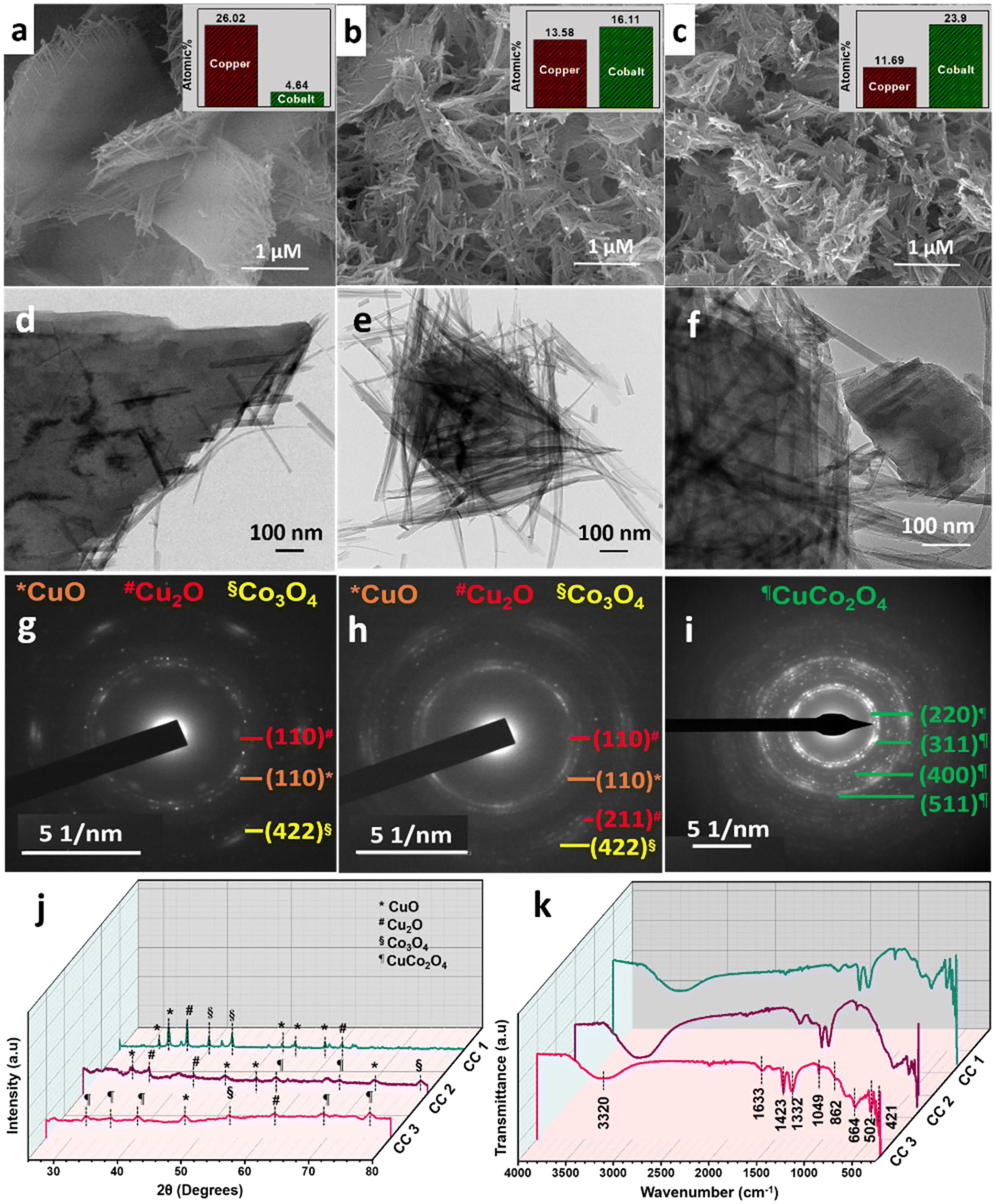
In particular, a 1:2 ratio of Cu:Co nitrates were used in the synthesis of CuCo2O4, which resulted in the formation of 2D sheet-like structures with tangled ends (CC 1), as confirmed by the FESEM image (Fig. 1a). However, the elemental composition determined by EDS analysis (ESI† Fig. S1) revealed that the product contained a higher concentration of Cu. To achieve the CuCo2O4 ratio, different Cu:Co precursor ratios (1:10 and 1:20) were used, and the corresponding CC nanostructures were termed CC 2 and CC 3, respectively. The FESEM images in Fig. 1a–c revealed a gradual transition in morphology from sheet-like structures with tangled ends to completely braided structures from CC 1 to CC 3. The EDS analysis reflected the successful formation of CuCo2O4 with a Cu:Co ratio of 1:2 in CC 3 structures (ESI† Fig. S1d), supported by the ICP-MS results in Table 1.
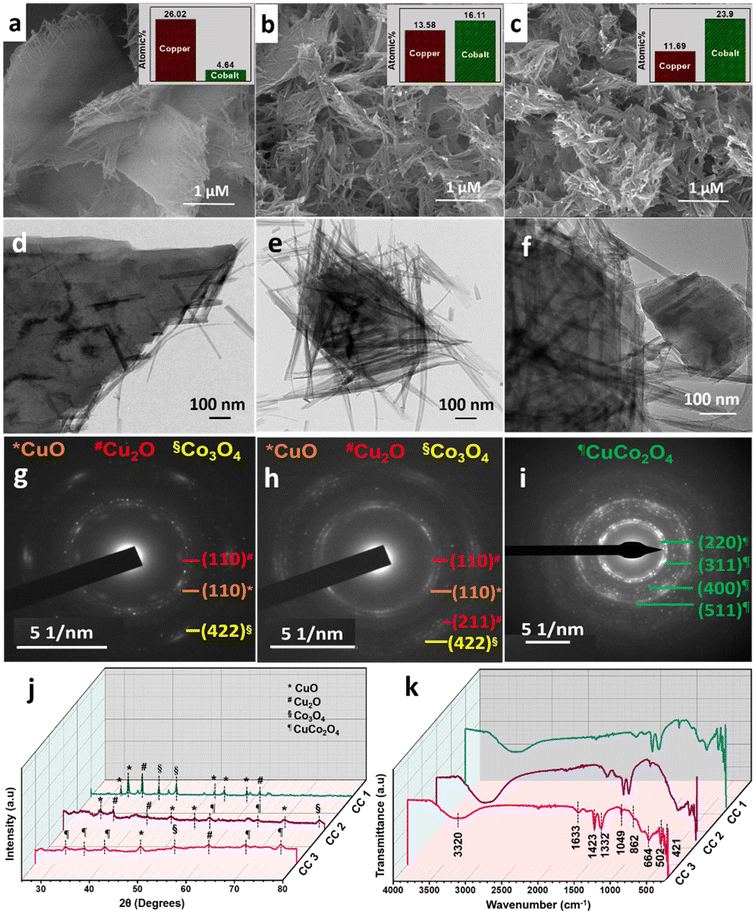 |
| | Fig. 1 FESEM images of (a) CC 1, (b) CC 2, and (c) CC 3. The corresponding elemental compositions obtained from EDS analysis are given in the inset. HRTEM images and the related SAED patterns of (d and g) CC 1, (e and h) CC 2, and (f and i) CC 3. (j) XRD patterns and (k) ATR-IR spectra of CC nanostructures. | |
Table 1 ICP-MS results of CC nanostructures
| Sample |
63Cu (%) |
59Co (%) |
| CC 1 |
19.37 |
4.47 |
| CC 2 |
10.38 |
10.10 |
| CC 3 |
6.52 |
11.24 |
The HRTEM images (Fig. 1d–f) confirmed the transition in morphology from sheet-like structures to braided structures as the cobalt wt% increased. The corresponding SAED patterns (Fig. 1g–i) exhibited a gradual increase in the Bragg spot intensity of the samples from CC 1 to CC 3. Moreover, as observed in the SAED patterns, samples CC 1 and CC 2 exhibited planes corresponding to copper and cobalt oxides. In comparison, the SAED pattern of CC 3 indicated the planes corresponding to CuCo2O4 with high crystallinity. The powder XRD patterns given in Fig. 1j further confirmed the dominance of CC 3 over other compositions in forming CuCo2O4. The significant set of planes (![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 10), (110), (112), (020), (202), and (110), (
10), (110), (112), (020), (202), and (110), (![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 02), (020), (311) exhibited by samples CC 1 and CC 2 correspond to the CuO (JCPDS 00-045-0937 and 00-041-0254) present in the samples. Similarly, the planes (111) and (220) exhibited by both CC 1 and CC 2 correspond to Cu2O (JCPDS 00-005-0667). Further, the planes (222), (400), and plane (622) exhibited by CC 1 and CC 2, respectively, correspond to Co3O4 (JCPDS 00-042-1467). Interestingly, CC 1 did not exhibit planes of CuCo2O4, while CC 2 samples contain planes (422) and (440) corresponding to CuCo2O4 (JCPDS 00-001-1155). On the other hand, the significant planes (220), (311), (222), (531), and (533) exhibited by CC 3 correspond to CuCo2O4 (JCPDS 00-001-1155). Few other planes, (12) corresponding to CuO, (220) corresponding to Cu2O, and (422) corresponding to Co3O4, might be originated from the impurities present in the sample. The standard JCPDS cards of the corresponding oxides are provided in ESI† Fig. S2. Further, the chemical nature of the synthesised samples was analysed using the ATR-IR technique, and the corresponding spectrum is given in Fig. 1k. The absorption bands below 700 cm−1 confirmed the metal–oxygen interactions in the samples.14 Bands present between 700–1500 cm−1 could be due to the vibrations of carbonate ions.24 The specific bands at 1430, 1054, and 744 cm−1 correspond to the ν3(E′),
02), (020), (311) exhibited by samples CC 1 and CC 2 correspond to the CuO (JCPDS 00-045-0937 and 00-041-0254) present in the samples. Similarly, the planes (111) and (220) exhibited by both CC 1 and CC 2 correspond to Cu2O (JCPDS 00-005-0667). Further, the planes (222), (400), and plane (622) exhibited by CC 1 and CC 2, respectively, correspond to Co3O4 (JCPDS 00-042-1467). Interestingly, CC 1 did not exhibit planes of CuCo2O4, while CC 2 samples contain planes (422) and (440) corresponding to CuCo2O4 (JCPDS 00-001-1155). On the other hand, the significant planes (220), (311), (222), (531), and (533) exhibited by CC 3 correspond to CuCo2O4 (JCPDS 00-001-1155). Few other planes, (12) corresponding to CuO, (220) corresponding to Cu2O, and (422) corresponding to Co3O4, might be originated from the impurities present in the sample. The standard JCPDS cards of the corresponding oxides are provided in ESI† Fig. S2. Further, the chemical nature of the synthesised samples was analysed using the ATR-IR technique, and the corresponding spectrum is given in Fig. 1k. The absorption bands below 700 cm−1 confirmed the metal–oxygen interactions in the samples.14 Bands present between 700–1500 cm−1 could be due to the vibrations of carbonate ions.24 The specific bands at 1430, 1054, and 744 cm−1 correspond to the ν3(E′), 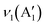 , and ν4(E′′) vibrations of the fingerprint peaks of carbonate ions, and the peak at 1342 cm−1 corresponds to the C–N stretching of unreacted urea.24 The bands at 1633 and 3320 cm−1 correspond to the bending and stretching bands of –OH in the adsorbed water molecules.14 Hence, the physicochemical investigations assert the formation of highly crystalline CuCo2O4 braided networks at the n-butanol/water interface.
, and ν4(E′′) vibrations of the fingerprint peaks of carbonate ions, and the peak at 1342 cm−1 corresponds to the C–N stretching of unreacted urea.24 The bands at 1633 and 3320 cm−1 correspond to the bending and stretching bands of –OH in the adsorbed water molecules.14 Hence, the physicochemical investigations assert the formation of highly crystalline CuCo2O4 braided networks at the n-butanol/water interface.
The appropriateness of the prepared CC nanostructures for electrochemical applications was explored using the cyclic voltammetric technique. For this, the CV scans of CC-modified GCE electrodes were recorded in the presence of 0.1 M NaOH, and the results are presented in ESI† Fig. S3a. The results showed an enhancement in the electrochemical activity of the samples from CC 1 to CC 3, CC 3 being the most electroactive material. The enhancement in the electrochemical activity in CC 3 might be due to the formation of pure CuCo2O4 and the combined effect of its morphology and crystallinity. The morphology tuning of highly crystalline CuCo2O4 into braided networks enhanced the diffusion of reactive species to the electrode surface, facilitating faster electron transport. The excellent crystallinity exhibited by CC 3, as seen in the SAED patterns, also acts as a synergetic factor for the increased electrochemical activity of CC 3. The prepared CC samples were then analysed to detect one of the essential neurotransmitters, ACh. The result presented in ESI† Fig. S3b showed a more significant redox current for CC 3-modified electrode, which is by ESI† Fig. S3a. Thus, considering the desired morphology, crystallinity, and enhanced electrochemical activity towards ACh, CC 3 sample was optimised for further studies.
The temporal evolution of nanostructures and the crystallinity of braided networks at the bisolvent interface are crucial to their electrochemical performances. To look into these factors, the growth mechanism of CC nanostructures at the n-butanol/water interface was investigated using the HRTEM studies. In detail, the reaction mixture of the optimised sample CC 3 was collected at different time intervals (30 minutes, 1 hour, 2 hours, and 3 hours), and the morphology and crystallinity of the intermediates were compared with the final product as given in Fig. 2a–j. The HRTEM images of Fig. 2a–e displayed an agglomerated flake-like structure at the initial phase of the reaction, which eventually turned into a braided form during the reaction. The visible change from a flake-like structure to a braided one was observed at around 3 hours. The SAED patterns provided in Fig. 2f–j also confirmed a gradual change from diffusive Bragg rings to bright Bragg spots corresponding to an increase in the crystallinity of the observed planes during the reaction. The SAED patterns in the initial period of the reaction exhibited diffusive Bragg rings corresponding to the planes of the oxides of both copper and cobalt. However, after 2 hours of reaction, all the rings in the SAED analysis of the reaction mixture corresponded to CuCo2O4.
 |
| | Fig. 2 HRTEM images and their corresponding SAED patterns of CC 3 at varying reaction time intervals. (a and f) 30 minutes, (b and g) 1 hour, (c and h) 2 hours, (d and i) 3 hours, and (e and j) after the completion of the reaction. Fig. 1f and i are used for comparison in Fig. 2 as e and j. (k) High-angle annular dark-field image and the corresponding elemental mapping images of (l) copper, (m) cobalt, (n) oxygen, and (o) mix. | |
Further, the elemental mapping analysis by the HRTEM technique (Fig. 2k–o) confirmed the uniform distribution of copper, cobalt, and oxygen throughout the sample. A comparison of the elemental mapping of CC 1 and CC 2 is provided in ESI† Fig. S4, which reveals the dominance of cobalt in the braided region. This can be correlated to the structure of copper and cobalt oxides synthesised in a similar bisolvent-assisted synthesis. The FESEM images of copper and cobalt oxides in ESI† Fig. S5 displayed a flake-like structure for copper oxide and a wire-like structure for cobalt oxide. The HRTEM investigations affirm the transformation in the morphology of CC nanostructures from sheet-like to braided networks with increasing cobalt content.
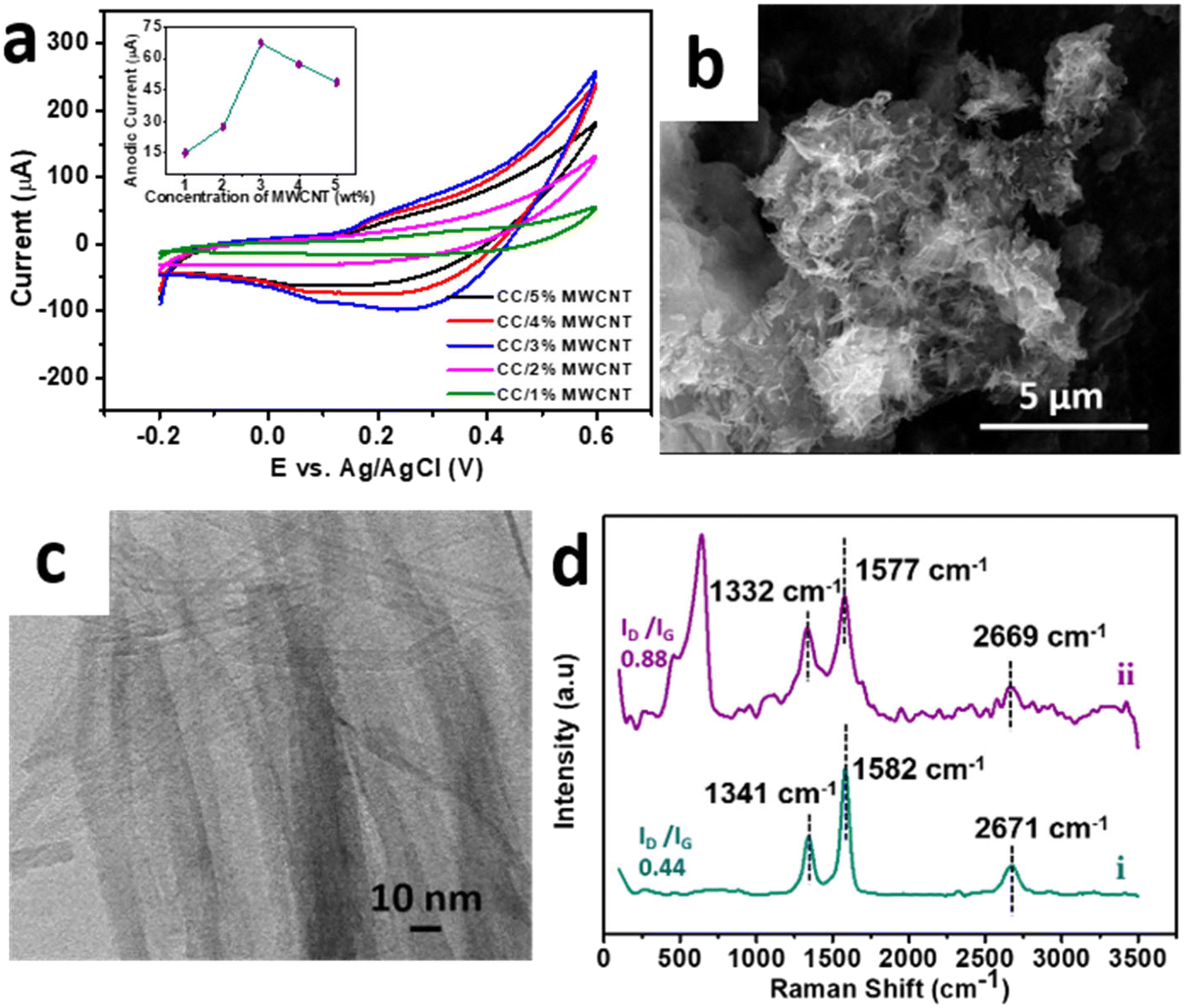
To enhance the performance in electrochemical applications, the optimised sample CC 3 was composited with a minimal amount of high aspect ratio MWCNT to form a CC/MWCNT composite. To optimize the amount of MWCNT to be incorporated in the composite, different CC/MWCNT composites were prepared by incorporating 1–5% MWCNT. CV scans of these composite-modified electrodes were recorded in 0.1 M NaOH. The corresponding CV plots in Fig. 3a demonstrated a drastic increase in the anodic peak current in the 3% MWCNT incorporated composite. Thus, it was presumed that 3 wt% MWCNT is the critical concentration for the composite to cross the percolation threshold, and hence, 3% MWCNT was optimised for the preparation of the CC/MWCNT composite.17
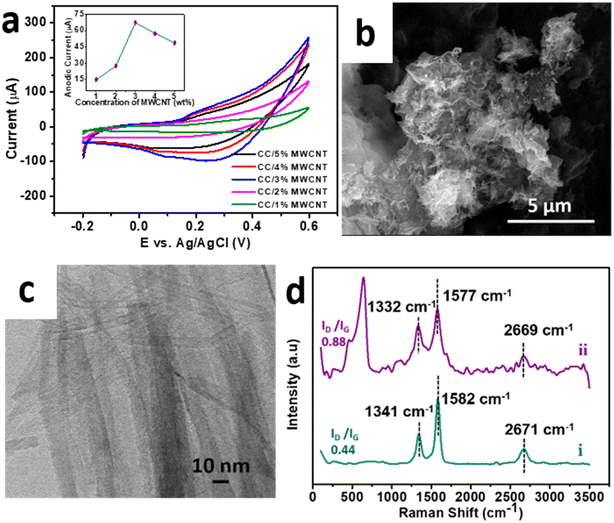 |
| | Fig. 3 (a) Cyclic voltammograms of CC/MWCNT composites of different weight percentages MWCNT in the presence of 0.1 M NaOH at a scan rate of 50 mV s−1. The corresponding anodic peak currents with respect to the concentrations of MWCNT are provided in the inset. (b) FESEM, and (c) HRTEM images of CC/MWCNT composite. (d) Raman spectra of (i) pristine MWCNT and (ii) CC/MWCNT composite. | |
The optimised CC/MWCNT composite morphology was examined using FESEM and HRTEM studies (Fig. 3b and c), which confirmed the formation of an interconnected network between CC and MWCNT. MWCNT was found to act as channels intertwining with the braided CC, thus facilitating a faster electron transfer. The corresponding EDS spectrum shown in ESI† Fig. S6 proved the presence of copper, cobalt, oxygen, and carbon in the sample, further confirming the formation of the CC/MWCNT composite. The structural environment of the composite was examined using Raman spectroscopy, and the results are presented in Fig. 3d. Pristine MWCNT exhibited two prominent peaks at ∼1341 cm−1 (D band) and ∼1582 cm−1 (G band), corresponding to the defects in the carbon system and the in-plane vibration of the carbon–carbon backbones, respectively.25 The observed peaks of the pristine MWCNT were then compared with the composite. As inferred from our previous work, the downshift in both bands indicates an electron transfer from CC to MWCNT.17 An increased ID/IG ratio of the composite compared to pristine MWCNT showed a possible interaction between the CC and MWCNT.26 The Raman spectrum of pure CC 3 is provided in ESI† Fig. S7 for comparison.
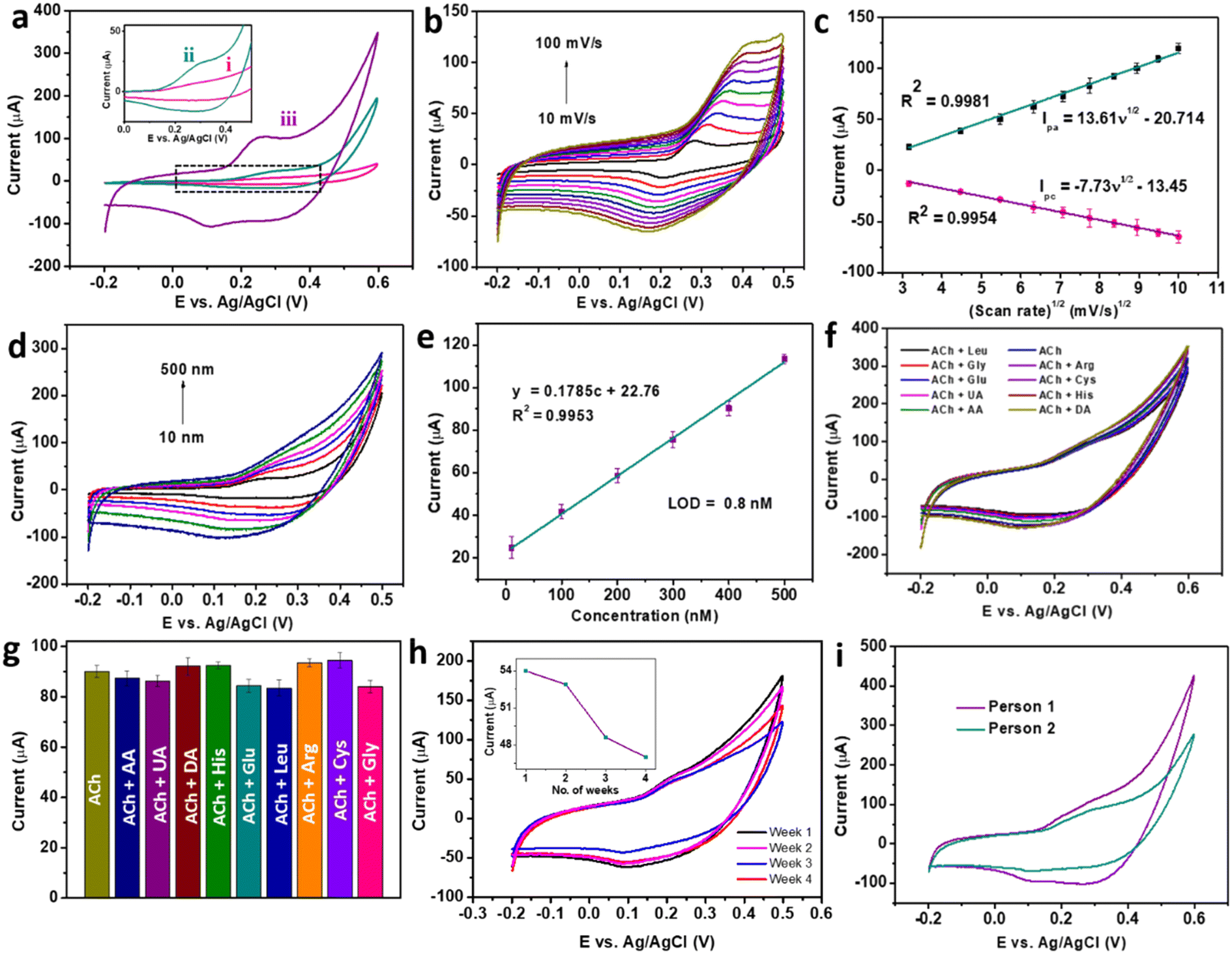
The electrochemical activity of braided structured CC for the detection of ACh was further enhanced by replacing the CC-modified electrode with CC/MWCNT-modified GCE. Consequently, the optimised CC/MWCNT composite-modified GCE was used to detect 500 nM ACh, as presented in Fig. 4a. Both CC-modified and CC/MWCNT-modified electrodes exhibited a clear redox peak in the presence of 500 nM ACh, which corresponds to the redox reactions of ACh to betaine. The mechanism of redox reactions at the electrode–electrolyte interface for the electrochemical detection of ACh is proposed here. In the given basic environment, CuCo2O4 undergoes hydrolysis reaction as given below:8
| | | CuCo2O4 + OH− + H2O → CuOOH + 2CoOOH + e− | (2.1) |
The oxidation of ACh further proceeds
via a two-step reaction as given below:
4,8| | | CH3COOCH2CH2N+(CH3)3 + H2O → CH3COOH + HO(CH2)2N+(CH3)3 | (2.2) |
| | | MOOH + HO(CH2)2N+(CH3)3 + OH− HOOCCH2N+(CH3)3 + H2O + M(OH)2 (M = Cu/Co) | (2.3) |
In the presence of an alkaline solution, ACh is hydrolysed into choline and acetic acid. The alcoholic group of choline is then oxidised to the carboxylic group at the composite-modified electrode to form betaine.
8 The synergistic redox effects of copper and cobalt catalysed the oxidation of ACh to betaine.
4 The higher electrochemical response of CC/MWCNT-modified electrode could be attributed to the increased current flow resulting from an enhanced surface area.
27 The electrochemically active surface area can be deduced using Randles–Sevcik equation,
28,29 where the active surface area of CC/MWCNT electrode was calculated as 0.119 cm
2. Additionally, the conductive networks of MWCNTs offered better electron transfer between the electrode–electrolyte interface and enhanced electrochemical activity of CC/MWCNT towards ACh. The electron transfer from CC to MWCNT, as deduced from the Raman spectra, explains the role of MWCNT in the faster transport of these electrons, thereby increasing the overall reaction rate. The importance of braided networks in controlling the electronics at the interface was investigated from the effect of scan rate on the anodic/cathodic peak currents. For this, successive CV scans of the composite-modified electrode were recorded in the presence of 500 nM ACh in a range of scan rates from 10 mV s
−1 to 100 mV s
−1. The results in
Fig. 4b showed increased peak currents with scan rate. The slight shift in both the anodic and cathodic peak currents with respect to the increase in the scan rate might be due to the quasi-reversible nature of the reaction.
30 Plot of peak currents and scan rates resulted in a linear regression with regression equations
Ipa = 13.61
ν1/2 − 20.71 (
R2 = 0.9981) and
Ipc = −7.73
ν1/2 − 13.45 (
R2 = 0.9954) (
Fig. 4c). The linear relationship between the anodic and cathodic peak currents with the square root of the scan rate, pointing to the nature of the reaction as diffusion-controlled.
31 This observation affirmed the braided morphology of the material, which can facilitate faster diffusion, thereby enhancing the electrochemical activity.
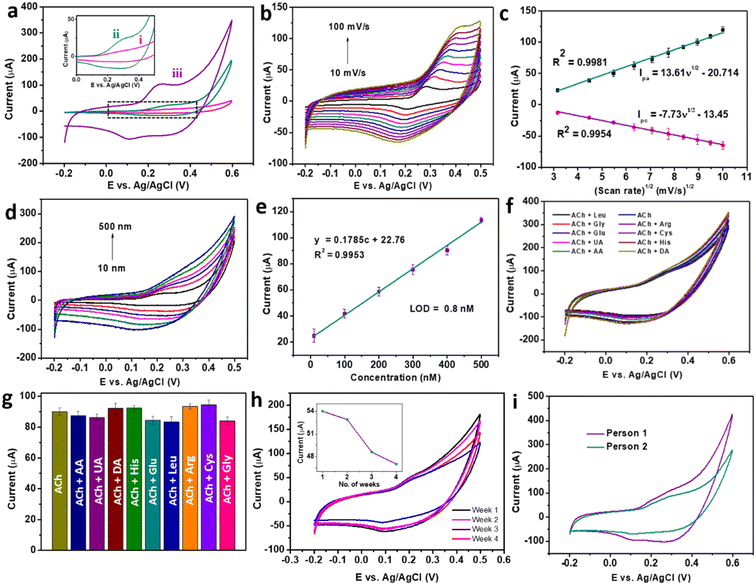 |
| | Fig. 4 (a) Cyclic voltammograms of (i) bare GCE (ii) CC 3-modified GCE and (iii) CC/MWCNT composite-modified GCE in the presence of 500 nM ACh in 0.1 M NaOH. (b) Cyclic voltammograms of CC/MWCNT composite-modified GCE in the presence of 500 nM ACh solution at varying scan rates, and (c) the corresponding calibration plot of redox peak current vs. scan rate1/2. (d) Cyclic voltammograms of CC/MWCNT composite-modified GCE in the presence of varying concentrations of ACh in the range of 10 nM to 500 nM at a scan rate of 50 mV s−1 and (e) the corresponding calibration curve. (f) Cyclic voltammograms of 500 nM ACh were recorded using CC/MWCNT-modified electrodes in the presence of 5 μM concentrations of interfering groups and (g) comparison of the anodic current responses. (h) Cyclic voltammograms of CC/MWCNT-modified electrode towards 200 nM ACh were taken periodically from week 1 to week 4 for storage stability studies. The inset shows the anodic current response with the number of weeks. (i) Cyclic voltammograms for detecting ACh in the serum of two individuals using a CC/MWCNT-modified sensor. | |
The adeptness of a sensor for practical applications depends on its high sensitivity, selectivity, and stability. CV studies were used to ensure the sensor satisfies these qualities. Initially, the composite-modified electrode was analysed to detect ACh over a range of concentrations. CV scans recorded in a concentration range of 10 nM to 500 nM produced a set of cyclic voltammograms with a gradual increase in the peak currents with concentration (Fig. 4d). Further quantification of the detection ability was done from the calibration plot of anodic peak current vs. concentration, as shown in Fig. 4e. The calibration plot produced a regression equation y = 0.1785c + 22.76, with an R2 = 0.9953, from which the LOD was calculated as 0.8 nM using the equation LOD = 3Sb/m, where Sb is the standard deviation of the blank, and m is the slope of the calibration plot of anodic peak current vs. concentration.17 The comparison of the obtained detection with the existing ACh sensors (ESI† Table S1) proved the dominance of the CC/MWCNT modified sensor over the reported works. As the detection of ACh in the biofluids involves the co-existence of other electroactive interfering biomolecules, the selective detection of ACh was ensured using the modified sensor. For this, CV scans were recorded in the presence of the solutions containing 500 nM ACh and 5 μM of the interfering species in 0.1 M NaOH. The results in Fig. 4f showed no additional signals in the presence of higher concentrations of interfering groups. The corresponding anodic peaks were compared with that of 500 nM ACh (Fig. 4g). No considerable change in the anodic peak currents in the presence of higher concentrations of co-existing interfering molecules substantiates the selectivity of the sensor. To analyse the stability of the modified sensor, ten successive CV scans of the composite-modified electrode were recorded in the presence of 500 nM ACh, and the results are presented in ESI† Fig. S8a. The meagre relative standard deviation (RSD) of 2.67% validated the stability of the sensor. The reproducibility of the sensor was validated using five different CC/MWCNT-modified electrodes to detect 500 nM ACh. The resulting CV plots given in ESI† Fig. S8b showed a RSD of only 2.82%, validating the reproducibility of the sensor. The storage stability of the sensor was then analysed by storing the modified electrode in 0.1 M NaOH at 4 °C. The periodic monitoring of the anodic peak currents toward 500 nM ACh using this modified electrode showed that the sensor retained 48.7 μA current (90.2% w.r.t the original anodic current) after three weeks, thus validating its storage stability (Fig. 4h).
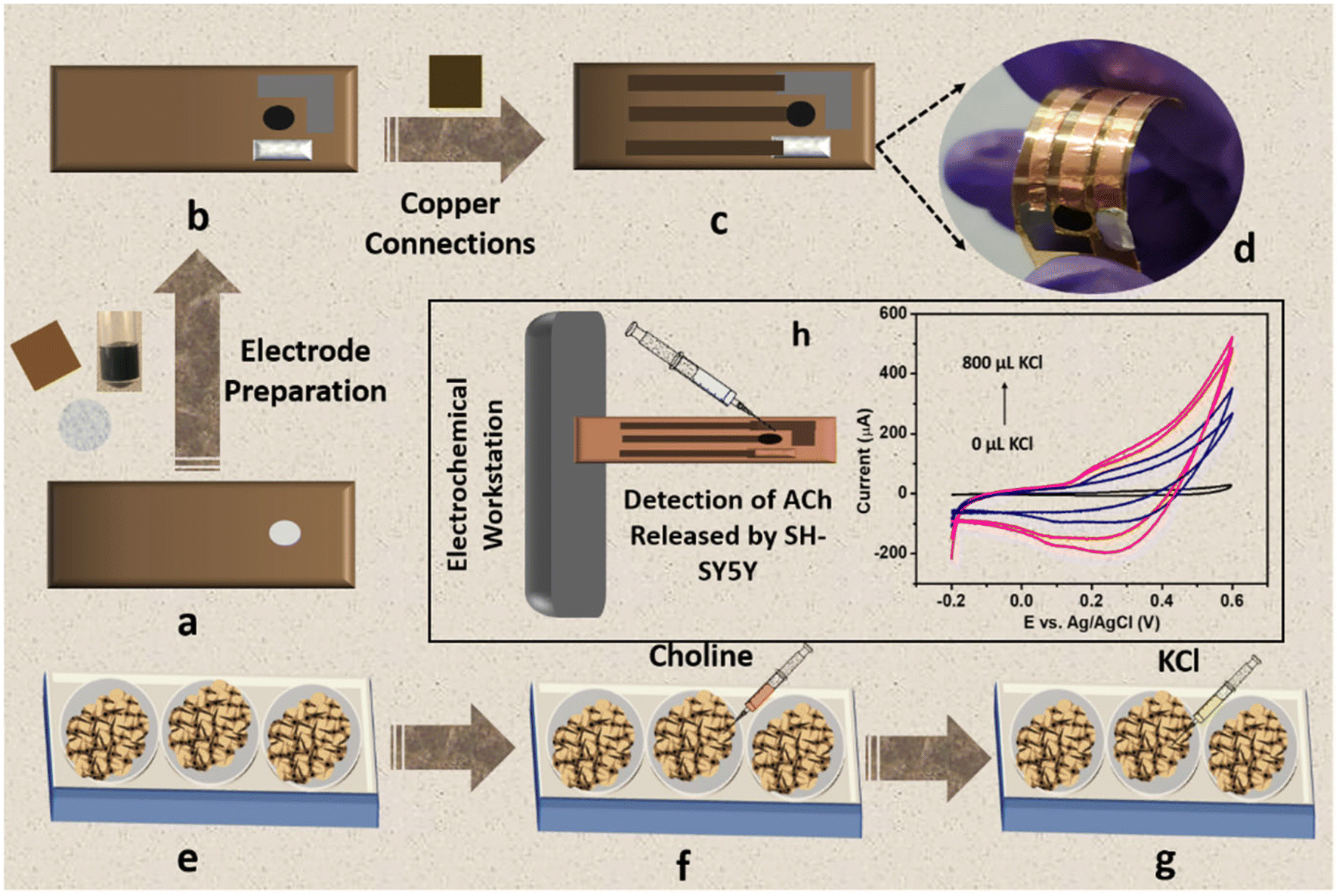
Owing to the excellent sensitivity, stability, and selectivity exhibited by the CC/MWCNT-modified electrodes, the sensor was analysed to detect real-life samples. Human serum was collected from two healthy individuals and diluted ten times using 0.1 M NaOH. The CV scans recorded using the composite-modified sensor in the presence of diluted serum showed a good redox peak (Fig. 4i), which proved the ability of the sensor to detect ACh present in human serum. Further, the measured peak current is analogous to the optimum level of ACh present in the human body.32 As a preliminary step toward developing a portable device for point-of-care applications, the bulky three-electrode system was miniaturised to an all-integrated flexible sensor. To attain this goal, a transparent polyester film was chosen as the flexible substrate, on which an adhesive polyimide tape was applied to provide electrical isolation of the electrodes. Conductive silver paste cured at 130 °C for 30 minutes as the reference electrode and pyrolytic graphite sheet as the counter electrode were assembled on the flexible substrate with proper electrical connections using copper tape. CC/MWCNT composite was used as the working electrode. Fig. 5a–d depicts the schematic representation of the flexible sensor fabrication and the prepared sensor's photographic image. Finally, the fabricated sensor was used for the real-time monitoring of ACh released by human neural cells. The human neuronal cells SH-SY5Y were selected as the in vitro model and were sub-cultured in Dulbecco's modified Eagle's medium containing 10% foetal bovine serum and 1% penicillin–streptomycin and incubated at 37 °C in a humidified environment containing 5% CO2 and 95% air. After incubation, 100 μM choline, the precursor of ACh, was added to the culturing media to ensure the release of ACh. Two hours later, 200 μL 500 mM KCl solution was added successively to trigger the release of ACh by the neural cells (Fig. 5e–g). The flexible sensor detected the thus released ACh and the results are shown in Fig. 5h. The results showed the presence of distinct redox peaks with increasing peak currents on the successive addition of KCl, which ensured the drastic release of ACh on the addition of KCl. In addition, the sensor exhibited a faster response time of 6 seconds, which could be attributed to the braided structure of the electroactive CC and the presence of highly conductive MWCNT. Thus, the modified sensor was proven to detect ACh released by human neural cells, demonstrating its ability for real-time applications.
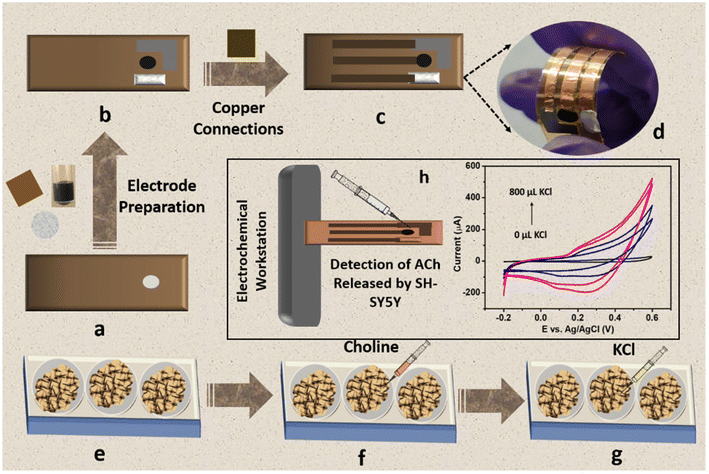 |
| | Fig. 5 Schematic representation of the fabrication of flexible sensor and its application in real-time detection of ACh released by human neural cells. (a) Flexible transparent polyester substrate applied with adhesive polyimide tape. (b) Electrode preparation using CC/MWCNT composite as the working electrode, conductive silver paste as the reference electrode, and pyrolytic graphite sheet as the counter electrode. (c) Schematic representation of the prepared flexible sensor and (d) its photographic image. (e) Sub-culturing of human neural cells SH-SY5Y in DMEM medium, (f) addition of the precursor choline to the medium, and (g) addition of KCl to trigger the release of ACh. (h) Electrochemical detection of ACh released by SH-SY5Y neural cells using the fabricated flexible sensor. | |
Conclusions
In summary, a braided-structured composite of copper cobaltite and multi-walled carbon nanotubes was used to develop a non-enzymatic electrochemical sensor for ACh detection. The nanostructure of the copper cobaltite was tailored to a braided network through an interface-assisted technique, and the temporal evolution of the nanostructures and crystallinity of the braided networks were studied using high-resolution transmission electron microscopy. The braided CC/MWCNT composite showed superior performance in the non-enzymatic detection of the neurotransmitter acetylcholine with a limit of detection of 0.8 nM, excellent stability for three weeks, and high selectivity even in the presence of high concentrations of interferents. Additionally, the ability of the sensor to detect ACh from human serum demonstrates its potential for use in health monitoring. An all-integrated flexible sensor was fabricated for the real-time monitoring of ACh. Real-time monitoring of ACh was performed using sub-cultured human neural cells, highlighting the cost-effectiveness, compactness, and user-friendliness of the CC/MWCNT sensors for detecting ACh in the human body. Thus, the proposed sensor would be a potential candidate for the non-enzymatic electrochemical detection of ACh, which contributes to the early detection of many deadly diseases. The tuned morphology and electrochemical activity of the material also promise the use of the material for the electrochemical detection of other biomolecules in the human body. The proposed flexible sensor is an initial approach for developing a miniaturised device that produces a timely response, helping the medical community quickly diagnose and treat diseases early. However, an extensive investigation is required to study the mechanism of the electrochemical reaction happening at the electrode–electrolyte interface to design further novel materials for detecting other biomolecules, which is planned as the future aspect of this work.
Author contributions
The manuscript was written through the contributions of all authors. All authors have given approval for the final version of the manuscript.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
MMM greatly acknowledges the funding from INSPIRE faculty award (DST/INSPIRE/04/2015/002050) by the Department of Science and Technology (DST). We acknowledge SAIF IIT Madras for HRSEM analysis, SAIF-STIC CUSAT for HRTEM analysis, CeNS Bangalore for HRTEM mapping, St. Thomas College, Thrissur for XRD Analysis, and CMC NIT Calicut for Raman analysis.
References
- R. R. Poolakkandy and M. M. Menamparambath, Electrochem. Sci. Adv., 2021, 1, 1–24 CrossRef.
- M. P. S. Mousavi, M. K. Abd El-Rahman, A. M. Mahmoud, R. M. Abdelsalam and P. Bühlmann, ACS Sens., 2018, 3, 2581–2589 CrossRef CAS PubMed.
- F. T. C. Moreira, M. G. F. Sale and M. Di Lorenzo, Biosens. Bioelectron., 2017, 87, 607–614 CrossRef CAS PubMed.
- G. Ashraf, M. Asif, A. Aziz, T. Iftikhar, Z. T. Zhong, S. Zhang, B. Liu, W. Chen and Y. Di Zhao, J. Hazard. Mater., 2022, 426, 128133 CrossRef CAS PubMed.
- N. Chauhan, S. Chawla, C. S. Pundir and U. Jain, Biosens. Bioelectron., 2017, 89, 377–383 CrossRef CAS PubMed.
- S. Hou, Z. Ou, Q. Chen and B. Wu, Biosens. Bioelectron., 2012, 33, 44–49 CrossRef CAS PubMed.
- A. Shadlaghani, M. Farzaneh, D. Kinser and R. C. Reid, Sensors, 2019, 19, 1–15 CrossRef PubMed.
- P. Balasubramanian, T. S. T. Balamurugan, S. M. Chen and T. W. Chen, ACS Sustainable Chem. Eng., 2019, 7, 7642–7651 CrossRef CAS.
- W. da Silva and C. M. A. Brett, J. Electroanal. Chem., 2020, 872, 114050 CrossRef CAS.
- P. Balasubramanian, T. S. T. Balamurugan, S. M. Chen, T. W. Chen and T. Sathesh, ACS Sustainable Chem. Eng., 2019, 7, 5669–5680 CrossRef CAS.
- N. Sattarahmady, H. Heli and R. D. Vais, Biosens. Bioelectron., 2013, 48, 197–202 CrossRef CAS PubMed.
- S. M. A. Shibli, K. S. Beenakumari and N. D. Suma, Biosens. Bioelectron., 2006, 22, 633–638 CrossRef CAS PubMed.
- V. Sudha, K. Annadurai, S. M. S. Kumar and R. Thangamuthu, Ionics, 2019, 25, 5023–5034 CrossRef CAS.
- S. Liu, K. S. Hui and K. N. Hui, ACS Appl. Mater. Interfaces, 2016, 8, 3258–3267 CrossRef CAS PubMed.
- R. Nakhowong and R. Chueachot, J. Alloys Compd., 2017, 715, 390–396 CrossRef CAS.
- R. Rahman Poolakkandy, S. Kaladi Chondath, N. Puthiyottil, D. Davis and M. M. Menamparambath, Langmuir, 2020, 36, 872–879 CrossRef CAS PubMed.
- R. R. Poolakkandy, A. R. Neelakandan, M. F. Puthiyaparambath, R. G. Krishnamurthy, R. Chatanathodi and M. M. Menamparambath, J. Mater. Chem. C, 2022, 10, 3048–3060 RSC.
- S. Ramesh, A. Kathalingam, K. Karuppasamy, H. S. Kim and H. S. Kim, Composites, Part B, 2019, 166, 74–85 CrossRef CAS.
- S. Ramesh, K. Karuppasamy, D. Vikraman, P. Santhoshkumar, C. Bathula, R. R. Palem, A. Kathalingam, H. S. Kim, J. H. Kim and H. S. Kim, J. Alloys Compd., 2022, 892, 162182 CrossRef CAS.
- F. S. Omar, A. Numan, N. Duraisamy, M. M. Ramly, R. Ramesh and R. Ramesh, Electrochim. Acta, 2017, 227, 41–48 CrossRef CAS.
- X. Xing, R. Liu, K. Cao, U. Kaiser and C. Streb, Chem. – Eur. J., 2019, 25, 11098–11104 CrossRef CAS PubMed.
- R. R. Poolakkandy, N. Ar, K. A. Padmalayam, G. K. Rajanikant and M. M. Menamparambath, ACS Appl. Nano Mater., 2023, 6, 1347–1359 CrossRef CAS.
- S. Samanta and R. Srivastava, J. Electroanal. Chem., 2016, 777, 48–57 CrossRef CAS.
- A. Karmakar and S. K. Srivastava, ACS Appl. Mater. Interfaces, 2017, 9, 22378–22387 CrossRef CAS PubMed.
- A. Yadav, A. Upadhyaya, J. Gope, S. K. Gupta and C. M. S. Negi, J. Mater. Sci.: Mater. Electron., 2020, 31, 1451–1460 CrossRef CAS.
- J. Zhao, J. Chen, S. Xu, M. Shao, Q. Zhang, F. Wei, J. Ma, M. Wei, D. G. Evans and X. Duan, Adv. Funct. Mater., 2014, 24, 2938–2946 CrossRef CAS.
- N. Demir, K. Atacan, M. Ozmen and S. Z. Bas, New J. Chem., 2020, 44, 11759–11767 RSC.
- A. Jirjees Dhulkefl, K. Atacan, S. Z. Bas and M. Ozmen, Anal. Methods, 2020, 12, 499–506 RSC.
- K. Atacan, J. Alloys Compd., 2019, 791, 391–401 CrossRef CAS.
- A. Babaei and A. Reza Taheria, Sens. Actuators, B, 2013, 176, 543–551 CrossRef CAS.
- X. Chen, Q. Liu, M. Liu, X. Zhang, S. Lin, Y. Chen, J. Zhuang and D. P. Yang, Microchim. Acta, 2018, 185, 340 CrossRef PubMed.
- M. Watanabe, A. Kimura, K. Akasaka and S. Hayashi, Biochem. Med. Metab. Biol., 1986, 36, 355–362 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a
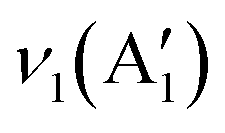 , and ν4(E′′) vibrations of the fingerprint peaks of carbonate ions, and the peak at 1342 cm−1 corresponds to the C–N stretching of unreacted urea.24 The bands at 1633 and 3320 cm−1 correspond to the bending and stretching bands of –OH in the adsorbed water molecules.14 Hence, the physicochemical investigations assert the formation of highly crystalline CuCo2O4 braided networks at the n-butanol/water interface.
, and ν4(E′′) vibrations of the fingerprint peaks of carbonate ions, and the peak at 1342 cm−1 corresponds to the C–N stretching of unreacted urea.24 The bands at 1633 and 3320 cm−1 correspond to the bending and stretching bands of –OH in the adsorbed water molecules.14 Hence, the physicochemical investigations assert the formation of highly crystalline CuCo2O4 braided networks at the n-butanol/water interface.