
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA corresponding V-target lattice structure to enhance selection pressure for aptamer selection†
Yong-Tao
Wang
,
Meng
Wang
,
Ke-Zhu
Yang
and
Zhi-Ling
Zhang
 *
*
College of Chemistry and Molecular Sciences, Wuhan University, Wuhan 430072, China. E-mail: zlzhang@whu.edu.cn
First published on 26th January 2023
Abstract
Aptamers, synthetic single-stranded oligonucleotide molecules that specifically bind to targets, are obtained through systematic evolution of ligands by exponential enrichment (SELEX). Microfluidic-SELEX (M-SELEX) technology has attracted wide attention due to its advantages of integration and high separation efficiency. However, most of the current M-SELEX technologies mainly exhibit characteristics of high separation efficiency to separate oligonucleotide molecules with strong affinity to targets to improve selection pressure, without considering the binding process of targets and oligonucleotide libraries inside microfluidic chips. In this paper, a corresponding V-target lattice structure was constructed in a microfluidic chip, which could enhance the binding process between targets and oligonucleotide libraries. Computational simulations were used to demonstrate that the structure could enhance the mass transfer process in the channel. And then the hemagglutinin protein of influenza A H5N1 virus (HA-H5N1) was used as the target protein for aptamer selection. After three rounds of positive and negative selection, an aptamer with high affinity and high specificity for HA-H5N1 protein was obtained, and the dissociation constant (Kd) was 6.7 ± 2.8 nM. This work provides a new research idea on how to improve selection pressure in SELEX to obtain aptamers with high affinity and high specificity. The microfluidic chip with the corresponding V-lattice structure, which can enhance mass transfer, can be used to select high affinity and high specificity aptamers and also has the potential to be used in detection, sensing and other fields.
Introduction
Nucleic acid-based aptamers are artificial single-stranded DNAs or RNAs, which can specifically bind to almost all target substances, including ions,1 proteins,2,3 viruses,4 bacteria,5etc. Aptamers are very attractive because they possess affinity and specificity close to other affinity reagents such as antibodies,6 and also have the characteristics of easy chemical modification, a wide range of targets, low cost, low batch to batch variation, good stability, etc.7 Owing to these advantages, aptamers have been widely applied in viral inhibition,8,9 therapy,10 diagnosis11,12 and bio-sensing13,14 over the past few decades.In general, aptamers are developed through an in vitro iterative process called systematic evolution of ligands by exponential enrichment (SELEX),15 which mainly involves the binding of targets to random oligonucleotide libraries, the isolation of candidate aptamers, and the preparation of evolved libraries.15,16 Multiple rounds of iteration of this process ultimately result in aptamers which can specifically bind to targets. A key point in the SELEX process is improvement of selection pressure, that is, how to improve the differentiation between oligonucleotides strongly bound to targets and oligonucleotides relatively weakly bound to targets, resulting in oligonucleotides with only strong affinity for targets. In order to solve this problem, a variety of technologies have been introduced in the SELEX process, including capillary electrophoresis technology,17,18 magnetic separation technology,19 microfluidic technology,20,21 sol–gel technology,22,23etc. Among many technologies, microfluidic technology has attracted much attention due to its characteristics of integration, small sample consumption, miniaturization and high separation efficiency.24 Currently, most microfluidic-SELEX (M-SELEX) technologies mainly take advantage of high separation efficiency of microfluidic chips to efficiently separate oligonucleotide molecules with strong affinity to targets, so as to improve selection pressure and reduce selection rounds.24 However, the binding process of targets and oligonucleotide libraries in microfluidic chips is not considered. In microfluidic chips, fluid flow is mainly laminar flow,25 and the proximity between oligonucleotide libraries and targets depends only on molecular diffusion. A low diffusion rate will reduce the binding process between oligonucleotides and targets, thus reducing selection pressure. Therefore, how to enhance selection pressure in M-SELEX is a problem that needs to be solved.
There are two kinds of enhanced mass transfer techniques in microfluidic chips: active mixing and passive mixing. Active mixing relies on external energy fields, such as pressure fields,26 electric fields27 and acoustic vibrations28 to cause forced mixing of fluid in chips, whose mixing efficiency is extremely high, but owing to the characteristics of complex equipment, high cost and poor stability, its development is limited. Another class of micromixers uses static structures in the channel to induce fluid mixing, called passive mixing. This method has high mixing efficiency, and has been widely developed because of its low cost and good stability. The most typical passive micromixer is Stroock's herringbone (HB) micromixer,25 which adds a series of herringbone structures at the bottom of a channel. These structures will generate two opposite swirling vortices in a channel, changing the flow of fluid to achieve the mixing of fluid.29 Passive mixing technology has been widely used to increase mass transfer between two and three liquid streams,30 promote cell separation and enrichment,31–33 and improve surface reaction efficiency.34,35 Most of the existing research studies are limited to increasing mixing efficiency by the structure at the bottom or top of a channel, which cannot achieve efficient mixing in three-dimension. Therefore, we hope to solve this problem by adding structures to the top and bottom of the channel at the same time.
In our previous work, a magnetic nanosphere (MN) array structure was formed by a magnetic control method at the bottom of a channel,36 which was used for aptamer selection.37,38 The target array structure at the bottom of the channel could achieve lateral mass transfer, but lacks vertical mass transfer. Therefore, in order to enhance the binding process between an oligonucleotide solution and the target-modified MN array, a corresponding V-target lattice structure was constructed in a magnetism-controlled microfluidic chip. The V-shaped structure at the top of the channel corresponds to the staggered target lattice structure at the bottom of channel one to one. This configuration not only increased the lateral disturbance, but also allowed for the generation of vertical vortices, which cannot be generated by a single-layer structure alone. The software COMSOL Multiphysics was used to simulate the flow in a microfluidic chip and evaluate whether the structure could enhance the mass transfer between the target array and oligonucleotide solution. At the same time, streptavidin and biotin were used as models to explore the binding efficiency and reaction process of the target array in chips with different structures. Experimental results showed that the V-shaped structure corresponding to the staggered lattice structure enables the target array to interact more effectively with the molecules in solution. Finally, the magnetism-controlled microfluidic chip with the corresponding V-target lattice structure was used to select aptamers, taking the hemagglutinin protein of influenza A H5N1 virus (HA-H5N1) as the target substance. The selection pressure can be improved mainly through the following aspects: firstly, the V-shaped structure at the top of the channel forcibly transferred oligonucleotides in the solution body to the surface and interior of the target array through recycling, which increased the chance of interaction between targets and oligonucleotides. Secondly, the interaction between the V-shaped structure at the top and the target array at the bottom increased the velocity and vorticity of the target array surface, which increased binding competitiveness between targets and oligonucleotides. Thirdly, the hemagglutinin protein of influenza B virus (HA-B) with a similar structure to the target protein was used as a negative protein to remove non-specific adsorbed oligonucleotides. In addition, the selection process was monitored in real time with the help of a fiber optic spectrometer, which made the selection process more visual. After three rounds of positive and negative selection, an aptamer with high affinity and high specificity for HA-H5N1 protein was obtained, and the dissociation constant (Kd) was 6.7 ± 2.8 nM.
Experimental section
Materials and chemicals
The initial ssDNA library contained a randomized sequence of 60 nucleotides (nts) flanked by 20-nt primer binding sites (5′-Alexa Fluor 488-AGCAGCACAGAGGTCAGATG(N60)CCTATGCGTGCTA-CCGTGAA-3′) and was labeled with fluorescein amide (Alexa Fluor 488) (excitation/emission: 488 nm/519 nm) at the 5′-end. The forward primer (5′-Alexa Fluor 488-AGCAGCACAGAGGTCA-GATG-3′) also was labeled with Alexa Fluor 488 at the 5′-end, whereas the reverse primer (5′-biotin-TTCACGGTAGCACG-CATAGG-3′) was labeled with biotin at the 5′-end. All these oligonucleotides were synthesized and purified by Sangon Biotechnology (Shanghai, China). Recombinant HA protein from influenza A H5N1 (90%, A/Hubei/1/2010), recombinant HA protein from influenza B (95%, B/Malaysia/2506/2004) and influenza A H5N1 hemagglutinin/HA antibody (mouse monoclonal antibody) were offered by Sino Biological (Beijing, China). Goat anti-mouse IgG-Alexa Fluor 488 was purchased from Bioss (Beijing, China). Bovine serum albumin (BSA), N-hydroxysuccinimide (NHS) and 1-ethyl-(3-dimethylaminopropyl) carbodiimine hydrochloride (EDC) were purchased from Sigma-Aldrich (Shanghai, China). Streptavidin (SA) was purchased from VWR Life Science. A polymerase chain reaction (PCR) kit was purchased from TransGen Biotech (Beijing, China). The 50 bp ssDNA ladder was purchased from Bioruler (Jiangsu, China). SU8 2015 and AZ9260 photoresists (PRs) were obtained from AZ Electronic Materials (USA). Poly(dimethylsiloxane) (PDMS, RTV615A + RTV615B) used for chips was bought from GE Toshiba Silicones (Japan). The following buffers were used in the experiment: 1× binding buffer (20 mM Hepes, 150 mM NaCl, 2 mM KCl, 2 mM MgCl2, 2 mM CaCl2 (pH 7.4)); 1× washing buffer (1× binding buffer + 0.05% Tween-20).To prepare target-coated MNs, the target protein was incubated with 400 μL of EDC/NHS-activated MNs for 12 h with mild rotation at room temperature. And then unbound protein was removed followed by washing three times with 400 μL of 1× PBS (pH 7.4). Unoccupied sites of MNs were blocked by incubation with 1% BSA for 30 min at room temperature with mild rotation. Finally, the target-functionalized MNs were dissolved in 400 μL of 1× PBS (pH 7.4) and stored at 4 °C until use.
Microfluidic device fabrication
The fabrication process of the magnetism-controlled microfluidic chip with the corresponding V-lattice structure was as follows: a layer of negative photoresist SU8 2015 was spin-coated on a clean silicon wafer, and the rectangular base of the bottom microchannel was obtained after exposure with the first mask. Then, another layer of photoresist was spin-coated again and the microstructure at the top in the channel was obtained after exposure with the second mask. After the mold was made, PDMS components (RTV615A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :RTV615B = 10:1 (w:w)) were poured into it, cured at 75 °C for 4 h, and cut with a scalpel to obtain the fluidic channel with the microstructure. A layer of positive photoresist AZ9260 was spin-coated on clean indium tin oxide (ITO) conductive glass. After exposure with a mask, ITO glass with the staggered lattice structure on the surface was obtained. Then, staggered nickel patterns were electroplated on the lattices that were not covered by the photoresist. After the photoresist was washed with ethanol, an ITO substrate with the staggered nickel pattern structure was obtained. Staggered nickel patterns would generate a gradient magnetic field under the action of a permanent magnet. PDMS (RTV615A:RTV615B = 15:1 (w:w)) was used to encapsulate the ITO substrate. Finally, the encapsulated ITO substrate and the fluidic channel were plasma cleaned for 2 min and then bonded together to make a magnetic-controlled microfluidic chip with different structures.
:RTV615B = 10:1 (w:w)) were poured into it, cured at 75 °C for 4 h, and cut with a scalpel to obtain the fluidic channel with the microstructure. A layer of positive photoresist AZ9260 was spin-coated on clean indium tin oxide (ITO) conductive glass. After exposure with a mask, ITO glass with the staggered lattice structure on the surface was obtained. Then, staggered nickel patterns were electroplated on the lattices that were not covered by the photoresist. After the photoresist was washed with ethanol, an ITO substrate with the staggered nickel pattern structure was obtained. Staggered nickel patterns would generate a gradient magnetic field under the action of a permanent magnet. PDMS (RTV615A:RTV615B = 15:1 (w:w)) was used to encapsulate the ITO substrate. Finally, the encapsulated ITO substrate and the fluidic channel were plasma cleaned for 2 min and then bonded together to make a magnetic-controlled microfluidic chip with different structures.
Computational simulations
The model was built with AutoCAD, and all simulations were performed with the finite-element method software, COMSOL Multiphysics. The inlet fluid was set as a fully developed flow, the average velocity was set as 0.833 mm s−1, the pressure at the outlet was set as ambient pressure, and the anti-slip boundary condition was set at a fixed wall surface. The velocity and vorticity profiles in the microchannel with different structures were obtained by solving the Navier–Stokes equations in steady state. The particle trajectories in the microchannel at different times were obtained by solving the Navier–Stokes equations in transient state.Binding profiles between MNs-SA array and biotin solution
To investigate whether the top structure could enhance the binding process between the target array and oligonucleotides in solution, a model of MNs-SA and 5′-biotin-TTCACGGTAGCAC-GCATAGG-Alexa Fluor 488-3′ was built. MNs-SA solution was pumped into the magnetic-controlled microfluidic chip with different structures at a flow rate of 10 μL min−1 to form the target array, and biotin solutions with different concentrations were pumped at a flow rate of 2 μL min−1 for the reaction for 1 h. Then, the fluorescence intensities of the biotin solutions before and after reaction were measured, and the binding efficiencies under different structures and concentrations were calculated. To explore the reaction process of the target array, 100 nM of biotin solution was selected to react with the target array. Fluorescence images were recorded at different times using an inverted fluorescence microscope (TiU, Nikon, Japan), and fluorescence intensity was monitored at different times with a fiber optic spectrometer (QE65000, Ocean Optics, China).Selection of aptamers in the microfluidic chip with the corresponding V-lattice structure
The selection process of aptamers in the microfluidic chip with the corresponding V-lattice structure was as follows: firstly, a microfluidic chip was placed between two permanent magnets to induce a gradient magnetic field between the nickel lattices, and then the target array was formed when MNs-HA-H5N1 solution and MNs-HA-B solution were pumped into the positive and negative selection regions at a flow rate of 10 μL min−1, respectively. Secondly, 200 pmol of initial DNA libraries were dispersed in 150 μL 1× binding buffer, and then they were heated at 95 °C for 10 min, cooled to 0 °C for 10 min and incubated at room temperature for another 5 min to achieve folding into diverse spatial structures. Finally, the pre-treated libraries were pumped into the negative selection regions at a flow rate of 2 μL min−1 to remove the nonspecific adsorbed oligonucleotides, and oligonucleotides excluded from nonspecific adsorption were incubated with the positive selection regions. After incubation, oligonucleotides that were not bound to target proteins or weakly bound were removed with 1× washing buffer at a rate of 2 μL min−1, and 20 μL MNs-HA-H5N1-DNA complex was collected.Preparation of the evolved ssDNA libraries
The collected products were amplified by PCR apparatus (Biometra, Germany). The 50 μL PCR mixture consisted of 5 μL template, 28 μL DEPC water, 1 μL Alexa Fluor 488-FP (10 μM), 1 μL biotin-RP (10 μM), 5 μL 10× GC Enhancer, 5 μL 10× Taq buffer, 4 μL dNTPs and 1 μL Taq DNA polymerase. The thermal cycling conditions were as follows: 95 °C for 10 min, 25 cycles at 95 °C for 30 s, 56 °C for 30 s, and 72 °C for 30 s, followed by an extra extension at 72 °C for 10 min. In order to isolate dsDNA modified with biotin, the amplified products were incubated with MNs-SA at room temperature for 45 min. And then, they were denatured with 0.1 M NaOH solution to release the ssDNA. Finally, evolved ssDNA libraries were obtained by purification with a 10 K ultrafiltration tube (Amicon Ultra-0.5, Merck Millipore, China) for the next round of selection.Cloning and sequencing
In each round, the fluorescence intensity of the MNs-HA-ssDNA array after washing was monitored with a fiber optic spectrometer in situ. When the fluorescence intensity of the positive screen unit did not change much, the selection was complete. Unlabeled primers were used for PCR amplification, and 20 clones were randomly selected for cloning and sequencing (Sangon Biotechnology, Shanghai, China). The obtained sequences were analysed by the software Clustalx, and sequences with high homology were selected for the affinity and specificity testing.Determination of binding affinity and specificity
The dissociation constant (Kd) of the aptamers were determined by a fluorescence binding method. Aptamers labeled with FAM were diluted to different concentrations (0, 25, 50, 100, 150, 200 nM) with 1× binding buffer. The pre-treated aptamers reacted with MNs-HA-H5N1 for 1 h. The fluorescence intensity of the composite solution was determined after unbound oligonucleotides were removed with 1× PBS buffer. The formula for calculating the Kd value of the aptamer is: Y = BmaxX/(Kd + X), where Y is the fluorescence intensity, X is the concentration of the aptamer, and Bmax is the saturation fluorescence intensity. The aptamer with high affinity was reacted with MNs modified with different proteins (HA-H5N1, HA-B, HA-H7N9, HA-H9N2, BSA) to evaluate the specificity of the aptamer. Mfold software was used to simulate the secondary structure of the aptamer.Results and discussion
Design of the structure in the microfluidic chip for aptamer selection
The selection process for aptamers in the microfluidic chip with the corresponding V-lattice structure is shown in Scheme 1. MNs modified with proteins would be captured by the gradient magnetic field to form the target array at the bottom of the channels in the positive and negative selection units, respectively, when they were pumped into the chip. And then, they were incubated with random ssDNA libraries under flow conditions. After incubation, oligonucleotides weakly bound and unbound to the targets were removed by continuous flow, and the MNs-HA-H5N1-DNA complex was collected for PCR amplification. The amplified products were incubated with MNs-SA to isolate biotin-modified dsDNA which was denatured by NaOH solution to release ssDNA, subsequently. Evolved ssDNA libraries were obtained by purification with the ultrafiltration tube for the next round of selection. The whole selection process was monitored in situ with an inverted fluorescence microscope, and selection rounds were evaluated using a fiber optic spectrometer. When the selection was complete, sequencing and affinity testing were performed.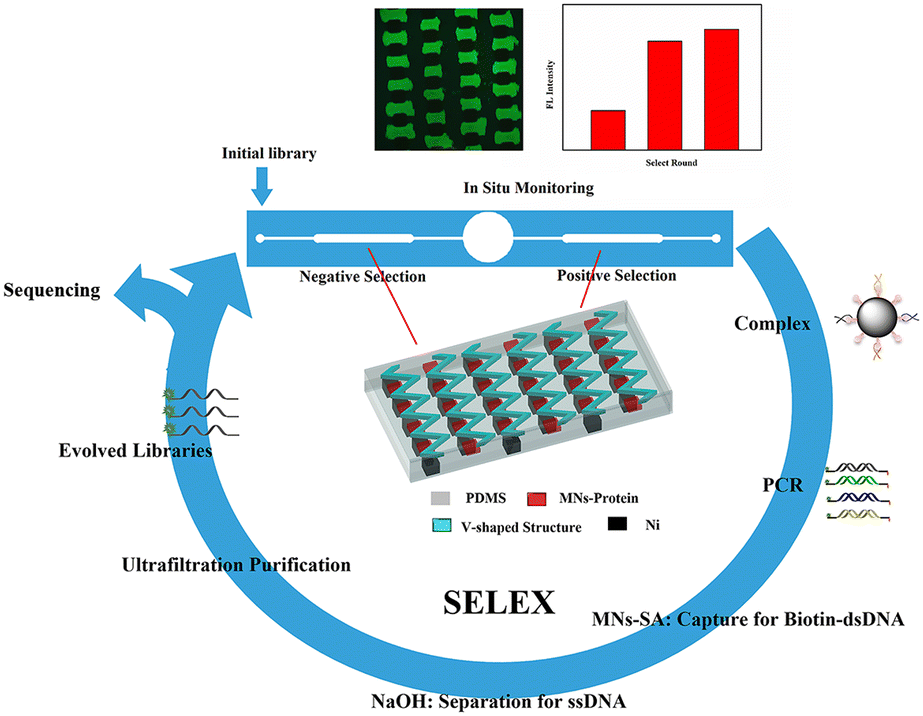 | ||
| Scheme 1 Principle of the magnetism-controlled microfluidic chip with the corresponding V-lattice structure for aptamer selection. | ||
The binding process of targets and random libraries was very significant in M-SELEX technology. Therefore, based on the staggered target lattice structure at the bottom of the channel, a static structure was established at the top of the channel to enhance the binding process of the targets and random libraries through the interaction of the top and bottom structures. When there was no superstructure (Fig. 1A), a staggered array structure at the bottom could only realize lateral mass transfer and cannot transfer the solution from the top of the channel to the array.37 Therefore, based on the staggered array structure at the bottom of the channel, a corresponding V-target lattice structure (Fig. 1B) was designed with the hope of realizing efficient mass transfer in three dimensions in the channel. Meanwhile, a forward herringbone structure was designed (Fig. 1C) at the top of the channel for contrast. A forward herringbone structure could generate two opposite rotating vortices in the channel, which could transfer the solution to the bottom of the channel for reaction. The detailed parameters of the three structures are shown in Fig. S1.†
 | ||
| Fig. 1 Three channel models with no superstructure (A), with the corresponding V-shaped structure (B), and with the forward herringbone structure (C). | ||
Enhancement of the mass transfer with the corresponding V-lattice structure
Computational simulations were performed to explore the velocity and vorticity field distributions of chips with different channel configurations. The velocity field and vorticity field distributions of the different cross-sections with different channel structures are shown in Fig. S2.† When there was no superstructure, the velocity and vorticity in the channel were low compared with the velocity and vorticity in the channel with the addition of a superstructure, indicating that the superstructure could enhance the mass transfer in the channel. Then, the cross-section passing through the target array was selected. Fig. 2A–C show the velocity field simulation results of three channel configurations. Without the superstructure, the velocity of the target lattice surface was very low (Fig. 2A). And most of the solution in the channel flowed directly away and would not be transferred to the surface of the target lattices for reaction (Movie S1 and S2†). When the V-shaped structure corresponding to the staggered lattice structure at the bottom was added at the top of the channel, the velocity of the target surface has been increased compared with no superstructure (Fig. 2B), indicating that mass transfer was enhanced. Due to the one-to-one correspondence between the V-shaped structure and the lattice structure, the solution could be uniformly transferred to the surface of each target lattice (Movie S3 and S4), thus greatly enhancing the reaction efficiency. Meanwhile, the V-shaped structure could also increase the surface velocity of each target lattice, thus enhancing the interaction competitiveness between targets and oligonucleotides. This was beneficial to obtain aptamers with strong affinity in the selection. And the added forward herringbone structure could also transfer the solution to the surface of the target lattices (Fig. 2C), but when the arm of the forward herringbone structure was directly above the target lattices, the velocity near these target lattices was very low. The reaction efficiency was limited by mass transfer because few solutions flow through these regions. | ||
| Fig. 2 Cross-sectional velocity field distribution in the channel with no superstructure (A), with the corresponding V-shaped structure (B) and with the forward herringbone structure (C). The scale unit of the velocity field is m s−1. Cross-sectional vorticity field distribution in the channel with no superstructure (D), with the corresponding V-shaped structure (E) and with the forward herringbone structure (F). The scale unit of the vorticity field is s−1. | ||
The cross-section simulation results of the vorticity field in three channel configurations are shown in Fig. 2D–F. The vorticity of the target lattice surface was low when there was no superstructure (Fig. 2D), indicating that mass transfer in solution would mainly rely on molecular diffusion. And when the V-shaped structure at the top of the channel corresponded to the target lattice structure at the bottom one to one (Fig. 2E), the vorticity of the target lattice surface could be uniformly increased. The V-shaped structure at the top interacted with the lattice structure at the bottom, allowing both lateral and vertical flows to be generated to achieve efficient mass transfer in three dimensions. Thus, the solution could be forcibly transferred to the target lattice surface to enhance the reaction efficiency. And after the addition of the forward herringbone structure, the vorticity of the target lattice surface was also increased (Fig. 2F). However, when the arm of the forward herringbone structure was directly above the target lattices, the vorticity near these target lattices was very low. In these areas, the reaction efficiency was limited because the molecules in the solution could mainly reach the surface of the target lattices through diffusion. The results of the velocity field and vorticity field showed that the corresponding V-lattice structure could realize efficient mass transfer in three dimensions in the chip.
It can be seen from Fig. S2† that the distributions of the velocity field and vorticity field on the target lattice surface presented periodic changes when the V-shaped structure corresponded to the lattice structure one to one. Therefore, in order to further understand the interaction between the V-shaped structure and the target lattice structure, we made the target lattice into a microsphere stacking model, and selected one unit to simulate the velocity field and vorticity field. The results showed that the corresponding V-lattice structure could increase the velocity of the lattice surface (Fig. 3A and D). When there was no superstructure, the surface velocity of the lattice formed by microsphere stacking was about 8 × 10−6 m s−1, and the vorticity was about 1 × 103 s−1 (Fig. 3B and C). When the superstructure was added, the surface velocity of the lattice formed by microsphere stacking was about 8 × 10−5 m s−1, and the vorticity was about 2 × 103 s−1 (Fig. 3E and F). The V-shaped structure could increase the surface velocity of the target lattice by 10 times (Fig. 3B and E) and the vorticity by 2 times (Fig. 3C and F), approximately, which led to the enhancement of the binding competitiveness between molecules and targets. The velocity of the target lattice surface represented the volume of the solution flowing through the surface per unit time. Obviously, within a certain range, the higher the velocity, the more solution flowing through the surface of the lattice, and the easier the reaction would occur. Therefore, the volume integral of the internal velocity of the target lattice could indicate how much volume of the solution flowed into the target lattice to react per unit time. The results showed that the volume integral was 8.62 × 10−19 m4 s−1 with no superstructure and 14.53 × 10−19 m4 s−1 with the corresponding V-shaped structure, and the mass transfer effect was improved by 68.6%.
 | ||
| Fig. 3 Velocity field distribution of one unit in the channel with no superstructure (A) and with the corresponding V-shaped structure (D). Velocity field distribution of the target lattice with no superstructure (B) and with the corresponding V-shaped structure (E). The scale unit of the velocity field is m s−1. Vorticity field distribution of one unit in the channel with no superstructure (C) and with the corresponding V-shaped structure (F). The scale unit of the vorticity field is s−1. | ||
Binding experiments between the MNs-SA array and biotin solution verified the computational simulation results that the corresponding V-lattice structure can enhance mass transfer in the channel. The results are shown in Fig. 4A. Under different concentration conditions, the binding efficiency was improved after the addition of the structure at the top, and when the V-shaped structure and the target lattice structure were corresponded one to one, the reaction efficiency was the highest. When the concentration of the biotin solution was 25 nM, the binding efficiency was 23.3% without a superstructure, increased to 30.5% when the forward herringbone structure was added, and further increased to 38.5% when the V-shaped structure was corresponding to the target lattice structure. Compared with no superstructure, the mass transfer effect was improved by 65.2% with the corresponding V-lattice structure in the chip, which was basically consistent with the result of the volume integral of the target lattice in one unit in the simulation results.
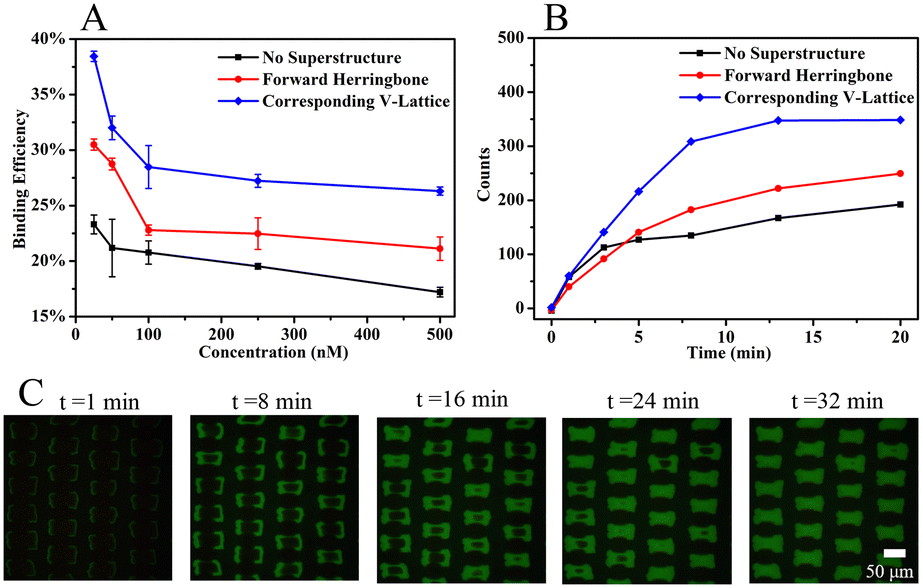 | ||
| Fig. 4 (A) Binding efficiency between different structures at different concentrations. Error bars correspond to standard deviation (n = 3). (B) Fluorescence intensity within 20 min between different structures. (C) Fluorescence images of the target array at different times with the corresponding V-lattice structure. | ||
100 nM of biotin solution was selected to react with the target array, and the fluorescence intensity of the target array within 20 min was monitored with a fiber optic spectrometer. The results are shown in Fig. 4B. When the reaction was carried out for 13 min, the fluorescence signal was basically unchanged in the chip with the corresponding V-lattice structure, indicating that most of the target substances had reacted completely, while the fluorescence signal was still slowly enhanced when there was no structure or forward herringbone structure at the top. After comparison, it could be concluded that the reaction efficiency was the highest when the V-shaped structure was corresponding to the lattice structure. An inverted fluorescence microscope was used to record the fluorescence images at different times, and the results are shown in Fig. 4C. The solution was folded and stretched on the front and back surfaces of the lattices, resulting in the reaction of the front and back surfaces of the target lattices first, while the reaction of two sides was slower, mainly because the flow rate between the two sides was too fast due to the folding of the fluid between two lattices, and biotin in the solution had already flowed away before reacting with MNs-SA on the side. At the same time, the V-shaped structure at the top of the channel forced the molecules in the solution into the target lattices to react. The computational simulations and experimental results showed that the corresponding V-lattice structure could achieve efficient mass transfer in three dimensions in the microfluidic chip.
Improving the binding process of SELEX
Hemagglutinin protein of influenza B virus (HA-B) was used as the negative selection protein to obtain highly specific aptamers. On the one hand, oligonucleotide molecules that could bind to other types of HA proteins were removed. On the other hand, oligonucleotide molecules adsorbent on the substrate of the chip would be removed. And to determine whether the top structure could enhance the binding process of targets and random libraries in M-SELEX, an oligonucleotide solution was introduced into the microfluidic chips with different structures and incubated with the target array, and then the fluorescence intensity of the target array in different structures was determined after the non-specifically bound oligonucleotides were removed by continuous flow. As shown in Fig. 5A, the fluorescence intensity of the target array was low without the superstructure, indicating that the binding process was weak, while the fluorescence intensity was the highest with the corresponding V-lattice structure, indicating that the binding process was the strongest. The binding process of the targets and random ssDNA libraries was enhanced because the structure at the top of the channel could forcibly transfer oligonucleotide molecules in the solution body to the surface and interior of the target array through recycling. The addition of the forward herringbone structure did not take into account the staggered lattice structure of the bottom. When the arm of the herringbone structure was directly above the lattices at the bottom, these target lattices would react more slowly. The reaction efficiency of each target lattice would be uniformly increased when the V-shaped structure at the top corresponded to the target lattice structure at the bottom one to one. The V-shaped structure interacted with the target lattice structure, which uniformly increased the velocity and vorticity of the target lattice surface. This would increase the binding competitiveness between the targets and the oligonucleotides, which was conducive to obtain an aptamer with high affinity. | ||
| Fig. 5 (A) Fluorescence intensity of the target array with different structures after incubation with random ssDNA libraries. (B) Fluorescence intensity of different washing times after incubation. Error bars correspond to standard deviation (n = 3). | ||
After the targets were bound to the random libraries, weakly bound and unbound oligonucleotide molecules were removed by continuous flow. The fluorescence intensity of the target array was monitored in situ at different times. As shown in Fig. 5B, the fluorescence intensity decreased rapidly within the first 2 min, indicating that oligonucleotide molecules that did not bind to the targets were mainly removed. The fluorescence intensity decreased slowly from 2 min to 6 min, demonstrating that oligonucleotide molecules with relatively weak binding to the targets were mainly removed. Then, the fluorescence intensity changed a little, indicating that the weakly bound and unbound oligonucleotide molecules could be effectively removed by continuous flow for 6 min in the microfluidic chip with the corresponding V-lattice structure.
In situ monitoring of the selection process
Owing to the advantage that it can enhance the binding process between the random libraries and targets, the microfluidic chip with the corresponding V-lattice structure was used to select aptamers for HA-H5N1 protein. The selection chip is shown in Fig. S3A.† Under the action of a permanent magnet, a gradient magnetic field would be formed between the staggered nickel lattices. MNs would be trapped between two nickel lattices to form the target array (Fig. S3B†). The target protein was modified onto the MN surface by EDC/NHS activation. The changes of particle size and zeta (ζ) potential before and after the protein modification onto the MN surface are shown in Fig. S4A and B.† Subsequently, MNs modified with HA-H5N1 protein were incubated with influenza A H5N1 HA antibody and goat anti-mouse IgG-Alexa Fluor 488 (Fig. S4C–E†). These results indicated that HA-H5N1 protein was successfully modified onto the MN surface without damaging the active site of the protein.Fluorescence images during the selection process were monitored in situ, as shown in Fig. 6A–C. After the weakly bound and unbound oligonucleotides were removed by continuous flow, the positive selection unit showed an obvious fluorescence signal, indicating that the random ssDNA libraries were clearly bound to the targets. According to the result of 12% denatured polyacrylamide gel electrophoresis (Fig. 6D), the complex was successfully amplified. After incubation with MNs-SA, the dsDNA modified with biotin was successfully separated from the amplified products (Fig. S5A–C†). Optimization of the MNs-SA amount is shown in Fig. S5G.† And then, they were denatured with NaOH solution to release ssDNA (Fig. S5D–F†). At last, the evolved ssDNA libraries were obtained by purification with a 10 K ultrafiltration tube (Fig. S5H†). The fluorescence signal was monitored in situ, which evaluated the selection process of different rounds. As shown in Fig. 6E, the fluorescence intensity did not increase significantly, indicating that the selection was finished. After the first round of selection, the fluorescence intensity increased rapidly, demonstrating that the chip with the corresponding V-lattice structure could enrich aptamers that bind specifically to the target, rapidly. The whole selection process was controlled and visual, which saved selection time and reagents.
 | ||
| Fig. 6 Microscope images of the positive selection unit. (A): Bright field, (B): fluorescence field, and (C): merged. (D) PAGE results: PCR product of the complex (lane 1); 50 bp marker (lane 2). (E) Fluorescence intensity of each round of selection for HA protein. | ||
Affinity and specificity of selected aptamers
After the selection was complete, 20 clones were randomly selected for cloning and sequencing. Homology of the obtained sequences was analysed by Clustalx software and the phylogenetic tree of the obtained sequences was analysed by TreeView X software (Fig. S6†). Among the 20 sequences obtained, sequence 6 was repeated 3 times and sequence 11 was repeated 2 times. The repeated sequence was preferentially selected for affinity measurement. At last, four sequences (4-aptamer, 6-aptamer, 7-aptamer, 11-aptamer) were selected for affinity test. As shown in Fig. 7A–D, the Kd values of aptamers 4, 6, 7, and 11 were 18.9 ± 11.4 nM, 20.3 ± 5.7 nM, 6.7 ± 2.8 nM, and 45.3 ± 3.9 nM, respectively. 7-Aptamer was used for specificity measurement thanks to the lowest Kd value. As shown in Fig. S7A,† 7-aptamer could specifically bind to the HA-H5N1 protein compared to other proteins (HA-H5N1, HA-B, HA-H7N9, HA-H9N2, BSA), indicating that 7-aptamer has high specificity. Mfold was used to simulate the secondary structure of the aptamers (Fig. S7B–E†). To conclude, the corresponding V-lattice structure in microfluidic chips can enhance the selection pressure of the SELEX process, resulting in high affinity and high specificity of aptamers.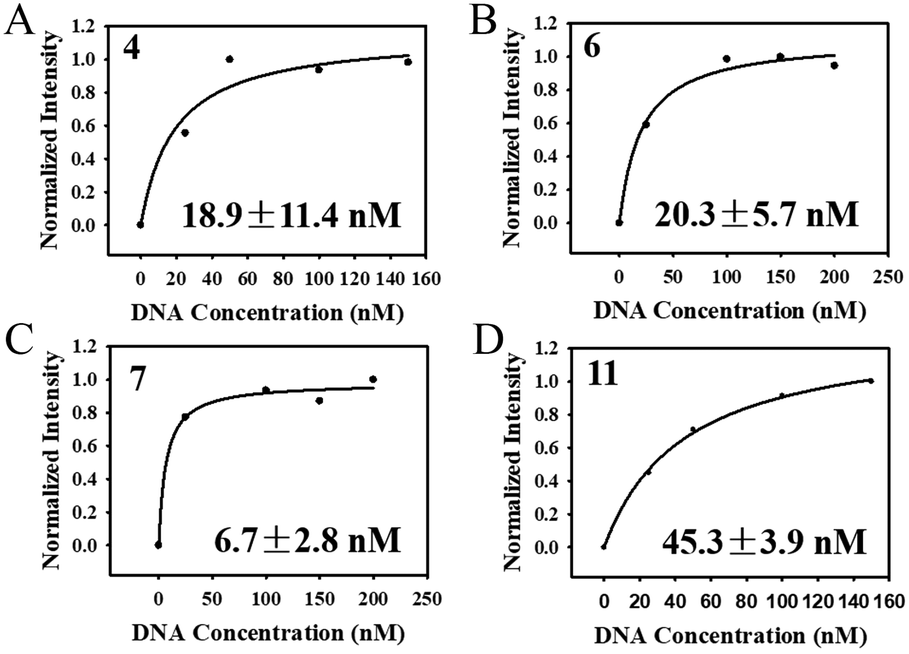 | ||
| Fig. 7 (A–D) Determination of the dissociation constants for HA-H5N1 protein with 4-aptamer, 6-aptamer, 7-aptamer and 11-aptamer. | ||
Conclusions
To sum up, the corresponding V-target lattice structure in the microfluidic chip was constructed to increase the selection pressure in the M-SELEX process. The V-shaped structure at the top corresponded to the staggered target lattice structure at the bottom, which could increase the chance and competitiveness of reaction between targets and oligonucleotides. Then, the microfluidic chip with the corresponding V-target lattice structure was used to select aptamers. After three rounds of positive selection and negative selection, an aptamer with high affinity and high specificity for HA-H5N1 protein was obtained, and the Kd value was 6.7 ± 2.8 nM. This work provides a new research idea to obtain aptamers with high affinity and high specificity. This configuration, in which two passive mixing structures are integrated at both the top and bottom of the channel, is suitable for selecting aptamers of any target, and also has the potential to be used in detection, sensing and other fields owing to its efficient mass transfer.Author contributions
Yong-Tao Wang: conceptualization, data curation, formal analysis, investigation, methodology, writing – original draft, visualization, and writing – review & editing. Meng Wang: writing – review & editing and data curation. Ke-Zhu Yang: data curation. Zhi-Ling Zhang: conceptualization, writing – original draft, writing – review & editing, funding acquisition, and supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22074107 and 22274118).Notes and references
- H. Sun, W. Li, Z. Z. Dong, C. Hu, C. H. Leung, D. L. Ma and K. Ren, Biosens. Bioelectron., 2018, 99, 361–367 CrossRef CAS PubMed.
- Y. Song, Z. Zhu, Y. An, W. Zhang, H. Zhang, D. Liu, C. Yu, W. Duan and C. J. Yang, Anal. Chem., 2013, 85, 4141–4149 CrossRef CAS PubMed.
- C. C. Wu, H. I. Lin, C. H. Yang, K. W. Chang, G. B. Lee and S. C. Shiesh, Lab Chip, 2015, 15, 486–494 RSC.
- R. Wang, J. Zhao, T. Jiang, Y. M. Kwon, H. Lu, P. Jiao, M. Liao and Y. Li, J. Virol. Methods, 2013, 189, 362–369 CrossRef CAS PubMed.
- D. Yılmaz, T. Muslu, A. Parlar, H. Kurt and M. Yüce, J. Biotechnol., 2022, 354, 10–20 CrossRef PubMed.
- S. D. Jayasena, Clin. Chem., 1999, 45, 1628–1650 CrossRef CAS.
- H. Lin, W. Zhang, S. Jia, Z. Guan, C. J. Yang and Z. Zhu, Biomicrofluidics, 2014, 8, 041501–041515 CrossRef PubMed.
- M. Sun, S. Liu, T. Song, F. Chen, J. Zhang, J. A. Huang, S. Wan, Y. Lu, H. Chen, W. Tan, Y. Song and C. Yang, J. Am. Chem. Soc., 2021, 143(51), 21541–21548 CrossRef CAS PubMed.
- X. Y. Zhang, L. Q. Wang, J. X. Liu, Z. M. Zhang, L. L. Zhou, X. H. Huang, J. G. Wei, M. Yang, Q. W. Qin and S. W. Wang, Aquaculture, 2022, 547, 737478–737487 CrossRef CAS.
- K. X. Tan, S. Pan, J. Jeevanandam and M. K. Danquah, Int. J. Pharm., 2019, 558, 413–425 CrossRef CAS PubMed.
- K. Wang, D. Fan, Y. Liu and E. Wang, Biosens. Bioelectron., 2015, 73, 1–6 CrossRef CAS PubMed.
- X. Yang, Y. Zhuo, S. Zhu, Y. Luo, Y. Feng and Y. Xu, Biosens. Bioelectron., 2015, 64, 345–351 CrossRef CAS PubMed.
- J. A. Phillips, Y. Xu, Z. Xia, Z. H. Fan and W. H. Tan, Anal. Chem., 2009, 81, 1033–1039 CrossRef CAS PubMed.
- W. Zhang, Z. He, L. Yi, S. Mao, H. Li and J. M. Lin, Biosens. Bioelectron., 2018, 102, 652–660 CrossRef CAS PubMed.
- C. Tuerk and L. Gold, Science, 1990, 249, 505–510 CrossRef CAS PubMed.
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818–822 CrossRef CAS PubMed.
- C. Zhu, L. Li, G. Yang, S. Fang, M. Liu, M. Ghulam, C. X. Hao, Y. B. Chen and F. Qu, Anal. Chim. Acta, 2019, 1070, 112–122 CrossRef CAS PubMed.
- C. Zhu, G. Yang, M. Ghulam, L. Li and F. Qu, Biotechnol. Adv., 2019, 37, 107432–107447 CrossRef CAS PubMed.
- R. Stoltenburg, C. Reinemann and B. Strehlitz, Anal. Bioanal. Chem., 2005, 383, 83–91 CrossRef CAS PubMed.
- X. H. Lou, J. R. Qian, Y. Xiao, L. Viel, A. E. Gerdon, E. T. Lagally, P. Atzberger, T. M. Tarasow, A. J. Heeger and H. T. Soh, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 2989–2994 CrossRef CAS PubMed.
- X. Liu, H. Li, W. Jia, Z. Chen and D. Xu, Lab Chip, 2017, 17, 178–185 RSC.
- J. Y. Ahn, S. Lee, M. Jo, J. Kang, E. Kim, O. C. Jeong, T. Laurell and S. Kim, Anal. Chem., 2012, 84, 2647–2653 CrossRef CAS PubMed.
- M. C. Lim, E. S. Lim, J. A. Lim, S. W. Choi and H. J. Chang, Appl. Biochem. Biotechnol., 2022, 194, 3901–3913 CrossRef CAS PubMed.
- Y. Liu, N. Wang, C. W. Chan, A. Lu, Y. Yu, G. Zhang and K. Ren, Front. Cell Dev. Biol., 2021, 9, 730035–730045 CrossRef PubMed.
- A. D. Stroock, S. K. W. Dertinger, A. Ajdari, I. Mezi, H. A. Stone and G. M. Whitesides, Science, 2002, 295, 647–651 CrossRef CAS PubMed.
- X. Niu and Y. K. Lee, J. Micromech. Microeng., 2003, 13, 454–462 CrossRef.
- J. L. Lin, K. H. Lee and G. B. Lee, Electrophoresis, 2005, 26, 4605–4615 CrossRef CAS PubMed.
- T. D. Luong, V. N. Phan and N. T. Nguyen, Microfluid. Nanofluid., 2010, 10, 619–625 CrossRef.
- N. S. Lynn and D. S. Dandy, Lab Chip, 2007, 7, 580–587 RSC.
- X. Feng, Y. Ren, L. Hou, Y. Tao, T. Jiang, W. Li and H. Jiang, Lab Chip, 2019, 19, 2936–2946 RSC.
- B. L. K. Coles, M. Labib, M. Poudineh, B. T. Innes, J. Belair-Hickey, S. Gomis, Z. Wang, G. D. Bader, E. H. Sargent, S. O. Kelley and D. van der Kooy, Lab Chip, 2021, 21, 4464–4476 RSC.
- S. L. Stott, C. H. Hsu, D. I. Tsukrov, M. Yu, D. T. Miyamoto, B. A. Waltman, S. M. Rothenberg, A. M. Shah, M. E. Smas, G. K. Korir, F. P. Floyd, Jr., A. J. Gilman, J. B. Lord, D. Winokur, S. Springer, D. Irimia, S. Nagrath, L. V. Sequist, R. J. Lee, K. J. Isselbacher, S. Maheswaran, D. A. Haber and M. Toner, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 18392–18397 CrossRef CAS PubMed.
- C. G. Wang, Y. Xu, S. N. Li, Y. Zhou, Q. L. Qian, Y. F. Liu and X. Q. Mi, Mater. Today Bio, 2022, 16, 100346–100356 CrossRef CAS PubMed.
- J. O. Foley, A. Mashadi-Hossein, E. Fu, B. A. Finlayson and P. Yager, Lab Chip, 2008, 8, 557–564 RSC.
- P. Zhao, J. L. Zheng, Y. Liang, F. C. Tian, L. Peng, D. Q. Huo and C. J. Hou, ACS Sustainable Chem. Eng., 2021, 9, 15386–15393 CrossRef CAS.
- X. Yu, X. Feng, J. Hu, Z. L. Zhang and D. W. Pang, Langmuir, 2011, 27, 5147–5156 CrossRef CAS PubMed.
- S. L. Hong, Y. T. Wan, M. Tang, D. W. Pang and Z. L. Zhang, Anal. Chem., 2017, 89, 6535–6542 CrossRef CAS PubMed.
- H. Y. Dong, Q. H. Xie, D. W. Pang, G. Chen and Z. L. Zhang, Chem. Commun., 2021, 57, 3555–3558 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sd00219a |
| This journal is © The Royal Society of Chemistry 2023 |