
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA label-free silicon-based spherical nucleic acid enzyme (SNAzyme) for ultrasensitive chemiluminescence detection of acute myocardial infarction-related nucleic acids†
Shiyang
Qu
 ,
Lin
Shi
,
Huan
Li
and
Tao
Li
*
,
Lin
Shi
,
Huan
Li
and
Tao
Li
*
Department of Chemistry, University of Science and Technology of China, 96 Jinzhai Road, Hefei, Anhui 230026, China. E-mail: tlitao@ustc.edu.cn
First published on 29th November 2022
Abstract
As a nano-form of G-quadruplex (G4) based DNAzymes, spherical nucleic acid enzymes (SNAzymes) with gold nanoparticles (AuNPs) as cores have shown great promise for applications in nucleic acid detection. SNAzymes can be constructed by modifying thiolated G4 DNAs onto AuNPs through various methods. However, those constructed with label-free G4 DNAs were found to have a weak enzyme activity due to the G4 destruction on the surface of AuNPs. To address this, here we constructed label-free silicon-based SNAzymes using silica nanoparticles (SiO2) as the core for the first time. We took advantage of the versatility of SiO2 and loaded N-(4-aminobutyl)-N-ethylisoluminol (ABEI) on the surface of silicon spheres (SiO2@ABEI). Then, we found that SiO2@ABEI can achieve high-efficiency adsorption of arbitrary G4 sequences within a few minutes without high requirements for modification methods. In this way, a highly sensitive CL nanoprobe integrated with a catalyst and a chemiluminescence (CL) reagent was successfully constructed. Finally, we use the nanoprobe for the ultrasensitive chemiluminescence detection of acute myocardial infarction (AMI)-related nucleic acids, achieving a detection range of 10 fM–100 pM and an excellent detection limit of 0.8 fM.
1 Introduction
Spherical nucleic acids (SNAs), first proposed by Mirkin's team, are now used in a wide range of fields including biological analysis, materials science and disease treatment.1–4 In a previous study of our group, G-quadruplex (G4) was modified onto the surface of gold nanoparticles (AuNPs) and showed extremely high peroxidase activity after binding cofactor hemin.5 This SNA is actually a nanoform of DNAzymes, i.e. SNAzymes, and has been used to achieve high-sensitivity detection of microRNAs in serum by chemiluminescence (CL) and electrochemiluminescence (ECL).6,7In the construction of SNAzymes, thiol-labelled G4 is often required to form stable Au–S bonds with AuNPs. Considering the cost of thiol-labelled DNAs,8 constructing SNAzymes with label-free G4 DNAs is here taken into account. To date, there have been many methods for modifying label-free DNA onto AuNPs. The salt aging method has achieved the labelling of SH-free DNA on the surface of AuNPs based on the affinity between adenine (A) and AuNPs.9,10 However, this method usually takes at least two days and is prone to AuNP aggregation during salt aging. In subsequent studies, label-free DNAs were modified by a low-pH method,11,12 freeze–thaw method8,13 and microwave heating drying method,14 in which the modification time was greatly shortened. However, the above methods have relatively high requirements for DNA sequences, requiring poly-A or poly-T sequences at the end of the DNA. The G4 required for SNAzyme construction will easily form secondary structures in the modification process, which is also not conducive to modification.8,14 Meanwhile, due to the affinity of the G base for AuNPs, G4 cannot be formed on AuNPs, which affects the catalytic activity of SNAzymes. (Fig. S1†). Therefore, a fast and efficient modification method is urgently needed, which can complete the modification of label-free G4.
Silica nanoparticles (SiO2) have a uniform size, simple synthesis, good biocompatibility, and a large number of hydroxyl groups on the surface. As a result, various organic functional groups can be further modified on the surface by organosilanes and crosslinking agents, such as amino,15 carboxyl,16 thiol,17 aldehyde,18 and epoxy groups,19etc. After the modification of SiO2, biological molecules such as nucleic acids,20 proteases21 and antibodies22 as well as functional molecules such as fluorescent23 and quantum dots24 can be loaded by electrostatic adsorption or covalent reaction. Luo et al. have simultaneously labeled ABEI and DNA onto the SiO2 surface to achieve the construction of a CL probe.25 At present, there are many methods for modifying DNA on the surface of SiO2.26–29 Either way, DNA needs to be labelled with functional groups such as amino, carboxyl or sulfhydryl groups. However, there are a few reports on the modification of label-free DNA on the surface of SiO2.
In this work, we constructed a novel label-free SNAzyme with SiO2 as the core. Firstly, carboxylated SiO2 (SiO2–COOH) was prepared by the multi-functionalization of SiO2, and then N-(4-aminobutyl)-N-ethylisoluminol (ABEI) was immobilized on the surface of SiO2 by amide reaction. Next, we found that unlabelled G4 could be well adsorbed on the surface of SiO2@ABEI, and then under the action of K+, G4 folded and bound hemin to form SNAzymes. In this way, a silicon-based SNAzyme integrating a CL substrate and catalyst was successfully constructed, hereinafter referred to as SiO2–SNAzyme@ABEI. Compared with SNAzymes constructed by AuNPs and unlabelled G4 (AuNPs–SNAzyme), SiO2@ABEI does not have high requirements for the sequence of DNA. Even G4, which easily forms secondary structures, can be successfully modified to the surface of silicon spheres. Moreover, the modification method of G4 is very simple, which just needs to be left standing at room temperature for a few minutes without heating, freezing or adding other reagents. Since AuNPs will affect the sequence arrangement when modifying unlabelled G4, this may affect the catalytic effect of AuNPs–SNAzyme, but label-free SiO2–SNAzyme@ABEI doesn't have such a problem and still has good peroxidase activity (Fig. S1†). At the same time, we applied this novel CL probe to the ultrasensitive detection of microDNA-499 (miDNA-499, which is the DNA analogue of miRNA-499), which is a marker of acute myocardial infarction (AMI).30
2 Experimental
2.1 Materials and reagents
Tetraethoxysilane (TEOS), mercaptopropyl trimethoxysilane (MPTMS) and succinic anhydride (SA) were purchased from Aladdin Biochemical Technology Co., Ltd. (Shanghai, China). (3-Aminopropyl)trimethoxysilane (APTES) was purchased from Energy Chemical Co., Ltd. (Shanghai, China). Cyclohexane, glutaraldehyde and propyl trimethoxysilane (GPTMS) were purchased from Macklin Biochemical Co., Ltd (Shanghai, China). N-(4-Aminobutyl)-N-ethylisoluminol (ABEI), 2,2′-azinobis(3-ethylbenzothiazoline-6-sulfonic acid) ammonium salt (ABTS), 4-maleimide N-hydroxysuccinimide butyrate ester (GMBS), N-hydroxysuccinimide (NHS) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) were purchased from Bide Medical Technology Co., Ltd. Hexanol, ammonium hydroxide (NH3·H2O), toluene, N,N-dimethylformamide (DMF), anhydrous ethanol and chloroauric acid (HAuCl4) were provided by Sinopharm Chemical Reagent Co., Ltd. (China). Magnesium acetate (MgAc2), potassium acetate (KAc), Triton X-100, 4-morpholineethanesulfonic acid (MES), hemin and all DNA sequences were provided by Sangon Biological Company (Shanghai, China), and all DNA sequences are displayed in Table S1.† The serum used in the experiment was provided by the First Affiliated Hospital of Nanjing Medical University. All reagents used in this experiment are of analytical grade.2.2 Instruments
A LUMIstar Omega Plate Reader (BMG LABTECH, Germany) was used for CL signal detection. A Millipore Milli-Q was used to prepare ultrapure water. A Cary-60 was used to collect UV-vis spectra. Gel electrophoresis images were collected on a Bio-Rad Gel Doc EZ imager system. Transmission electron microscopy (TEM) characterization of nanoparticles was completed using a Hitachi 7700. A NanoFCM was used to analyse the number of nanoparticles. A Thermo Fisher Fourier spectrometer was used to pick up infrared signals. A NanoBrook 90Plus PALS was used to determine the zeta potential of nanoparticles.2.3 Preparation of SiO2, SiO2–NH2, and SiO2–COOH
The synthesis of SiO2 and aminated SiO2 (SiO2–NH2) is referred to previous studies with some changes.23,31 15 mL cyclohexane, 3.2 mL hexanol, and 3.54 mL Triton X-100 were mixed in a flask. Then, 960 μL water and 200 μL TEOS were added and stirred at room temperature for 20 min, and then 120 μL NH3·H2O were added and stirred at room temperature for 24 h. 15 mL acetone was added for demulsification, and repeated washing with acetone, ethanol and water was conducted at a speed of 8000 RPM. The synthesis of SiO2–NH2 was basically the same as that of SiO2. After the reaction, 100 μL TEOS and 100 μL APTES were added and reacted again for 24 h. Finally, the solid powder of the two silicon spheres can be obtained by drying the precipitate in a vacuum drying oven.50 mg SiO2–NH2 and 10 g SA were dissolved in 30 mL DMF, stirred at room temperature in a nitrogen atmosphere for 8 h, and then centrifuged and washed in DMF, ethanol and water in turn. After drying, carboxylated SiO2 (SiO2–COOH) was obtained.
For modification of other functional groups of SiO2, please refer to the ESI.†
2.4 Preparation of AuNPs–DNA
2.5 Preparation of SiO2–DNA@ABEI
2.6 Comparison of catalytic activities of SiO2–SNAzyme@ABEI and AuNPs–SNAzyme by colorimetry
First, 25 μL of 2 mg mL−1 SiO2–DNA@ABEI or 25 μL of 8 nM AuNPs–DNA prepared by the above five methods were dissolved in 49 μL of 10 mM Tris-HCl EDTA (TE) buffer containing 50 mM KAc. The mixture was reacted at 37 °C for 2 h. Then, 10 μL of 5 μM hemin was pipetted into the system and reacted at 37 °C for 1 h. Then, 10 μL of 36 mM ABTS was added. After adding 6 μL of 10 mM H2O2, it was left for 3 min. Finally, a Cary-60 was used to collect UV-vis spectra.2.7 Polyacrylamide gel electrophoresis (PAGE)
In the construction of the detection platform for miDNA-499, the assembly of the three-arm structure was optimized with 12% polyacrylamide gel. First, 0.2 μM miDNA-499, 0.2 μM SiO2-capture and 0.2 μM capture-n-G4 (n = 6–10) were mixed in 1× Tris-acetate EDTA (TAE) buffer and reacted at 37 °C for 2 h. Then, 2 μL of 500 mM KAc was added to form G4 for 1 h. The total volume of each sample was 20 μL. The gel was run with 8 V cm−1 for 3 h at 4 °C. Then, the gel was dyed with GelRed for 40 min and imaged using a Gel Doc EZ Imager system.2.8 Procedures for miDNA-499 detection
3 Results and discussion
3.1 Synthesis of label-free SiO2–SNAzyme@ABEI
Fig. 1 depicts the construction of SiO2–SNAzyme@ABEI. First, SiO2 nanoparticles are synthesized by the water-in-oil microemulsion method, and then APTES is added to functionalize SiO2 with amino groups. Next, SA is introduced to convert the amino groups into carboxyl groups, which are further activated by NHS and EDC. And then, ABEI with amino groups can be modified on the SiO2 surface. Then, unlabelled DNA with a G-rich sequence is loaded on the surface of SiO2, and under the action of K+, G4 forms and binds hemin to form SiO2–SNAzyme@ABEI.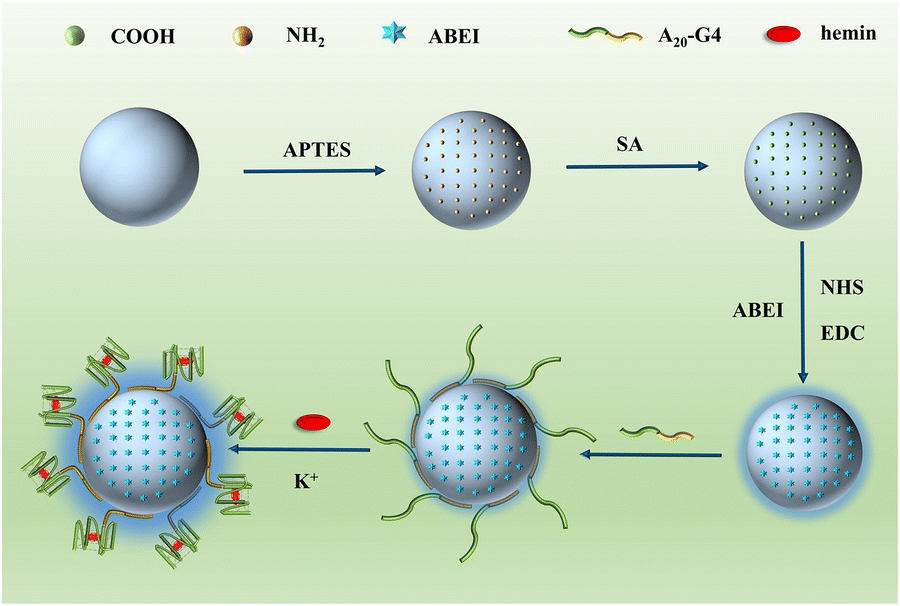 | ||
| Fig. 1 Schematic for the construction of SiO2–SNAzyme@ABEI. | ||
The characterization of the above synthesis process is shown in Fig. 2. The TEM image in Fig. 2A shows that the morphology of SiO2 synthesized by the microemulsion method is uniform, and the average diameter is 56 ± 2 nM, which is accordance with a previous study.32 The addition of ABEI and DNA doesn't affect the morphology of SiO2, which is still in a monodispersed state (Fig. 2B and C). The characterization of the zeta potential further illustrates the completion of each step of the modification process (Fig. 2D). The potential values of SiO2, SiO2–NH2, and SiO2–COOH are −19.1 mV, +18.23 mV, and −14.41 mV, respectively, and the change trend of the potential is in line with previous literature reports.21 The successful modification of amino and carboxyl groups was also well demonstrated by ninhydrin staining and Fourier transform infrared spectroscopy (Fig. S2†). The potential value was positively shifted after the addition of ABEI, which may be related to the formation of amide bonds consuming the negatively charged carboxyl groups.33 After the addition of G4, the potential value decreases due to the negative charge of the DNA phosphate backbone, which also indicates the successful modification of DNA. In addition, the modification of ABEI and DNA was also monitored using UV-vis spectra (Fig. 2E). Before adding functional molecules, none of the three SiO2 samples has obvious absorption peaks. After ABEI is loaded, SiO2@ABEI shows obvious absorption peaks at wavelengths of 290 nm and 325 nm, which are consistent with the absorption peaks of free ABEI. After the DNA is added, a new absorption peak appears at 260 nm, and the absorption peaks of ABEI and DNA appear at the same time, indicating that the two were successfully modified.34
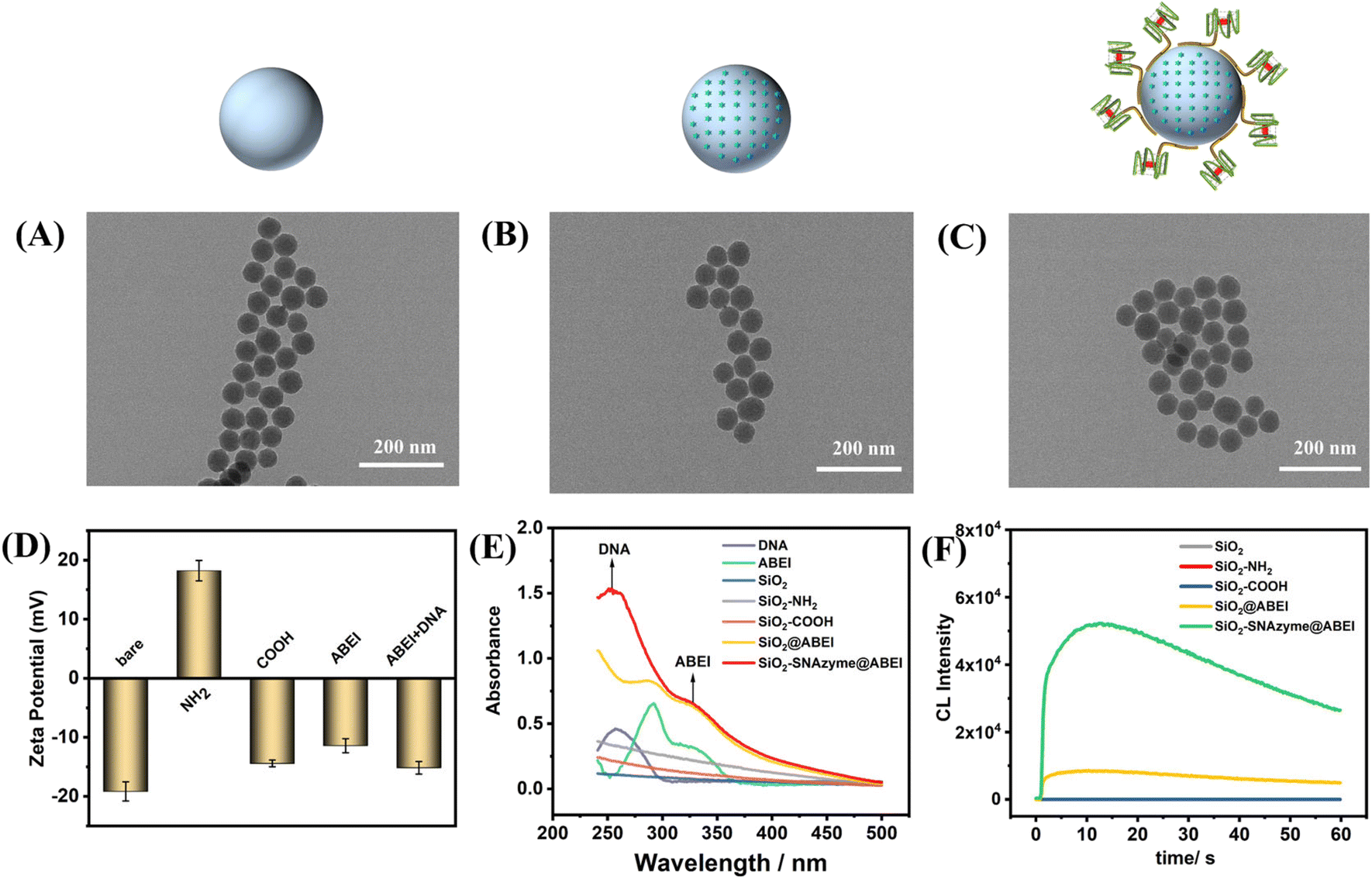 | ||
| Fig. 2 Characterization of the formation process of SiO2–SNAzyme@ABEI. (A–C) TEM characterization of (A) SiO2, (B) SiO2@ABEI and (C) SiO2–SNAzyme@ABEI. (D) Zeta potential of SiO2, SiO2–NH2, SiO2–COOH, SiO2@ABEI and SiO2–SNAzyme@ABEI. (E) UV-vis spectra of DNA, ABEI, SiO2, SiO2–NH2, SiO2–COOH, SiO2@ABEI and SiO2–SNAzyme@ABEI. (F) CL kinetic curves of SiO2, SiO2–NH2, SiO2–COOH, SiO2@ABEI and SiO2–SNAzyme@ABEI. | ||
In order to obtain the most excellent CL performance of SiO2–SNAzyme@ABEI, we optimized the modification method of ABEI. After being modified by different organosilanes or cross-linking agents, SiO2 is successfully functionalized with various organic functional groups, and then further interacts with the amino group of ABEI. The specific synthesis process is detailed in the ESI.† The loading capacity of different functional groups on ABEI was detected by CL (Fig. S3A†). The result shows that carboxylated SiO2 has the strongest loading capacity for ABEI with the assistance of NHS and EDC, which is consistent with reported results.33,35 We also found that the modification density of carboxyl groups also affected the loading capacity of ABEI. The CL signal of the material gradually rises with the increase of the feeding ratio of SiO2–NH2 and SA, and reaches the highest at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :200 (Fig. S3B†). We next monitored the CL properties of SiO2–SNAzyme@ABEI (Fig. 2F). Under the conditions of pH 12, 2 mM H2O2 and 100 nM hemin, SiO2, SiO2–NH2 and SiO2–COOH have almost no CL signal. After the material is loaded with ABEI, the CL signal is increased by nearly 200 times. By further modification of G4, the formation of SNAzymes makes the CL signal again boosted by a factor of nearly 6.
:200 (Fig. S3B†). We next monitored the CL properties of SiO2–SNAzyme@ABEI (Fig. 2F). Under the conditions of pH 12, 2 mM H2O2 and 100 nM hemin, SiO2, SiO2–NH2 and SiO2–COOH have almost no CL signal. After the material is loaded with ABEI, the CL signal is increased by nearly 200 times. By further modification of G4, the formation of SNAzymes makes the CL signal again boosted by a factor of nearly 6.
3.2 Comparison of label-free SiO2–SNAzyme@ABEI and AuNPs–SNAzyme
Next, in order to illustrate the advantages of using SiO2 as the core of SNAzymes, we compare it with AuNPs, which is currently the most widely used in the construction of SNAzymes. The rapid modification methods of DNA on AuNPs include the heating–drying method,14 butanol extraction method,36 freezing method8 and low-pH method.11 The modification of DNA in SiO2 is based on the magnetic stirring method.32,35,37,38 Here, we design three types of G4 with poly(A20), poly(T20), and poly(C20) tags at the 5′ terminal and a random sequence of DNA with poly(A20) tags at the 5′ terminal as ctrl. The four kinds of DNAs were modified on the surface of AuNPs and SiO2 by using the above-mentioned five methods of modifying gold or silicon. After the modification is completed, the success of DNA modification could be judged according to gold aggregation or no gold aggregation. While the surface of SiO2–DNA@ABEI is rich in CL reagent, in order to avoid the influence of ABEI on the results, the modification of G4 was determined by colorimetry instead of CL.Fig. 3(A–E) show the experimental results of the above scheme. The results show that the modification of non-labeled G4 on AuNPs is difficult. After adding Mg2+, the aggregation of gold nanoparticles occurs in almost all modification methods. Only T20-G4 using the microwave heating–drying method, and A20-ctrl using the freezing or low pH method are able to be successfully modified on AuNPs, which is basically consistent with results reported in the literature.8,11,14 We speculate that the reason for the failure of DNA modification may be the formation of the secondary structure of G4 during the modification process which greatly affects the adsorption of DNA and gold. However, with SiO2 as the core, no matter what modification method or G4 sequence is used, SiO2–SNAzymes@ABEI can well catalyze the colorimetric reaction of ABTS, resulting in an obvious absorption peak at 420 nm. Fig. S4† confirms the efficient adsorption capacity of SiO2@ABEI for G4 by measuring the UV-vis spectrum of the supernatant solution. Fig. S5† shows that G4 functionalization of SiO2@ABEI can be completed in a few minutes, and the reaction time of the two hardly affects the modification efficiency.
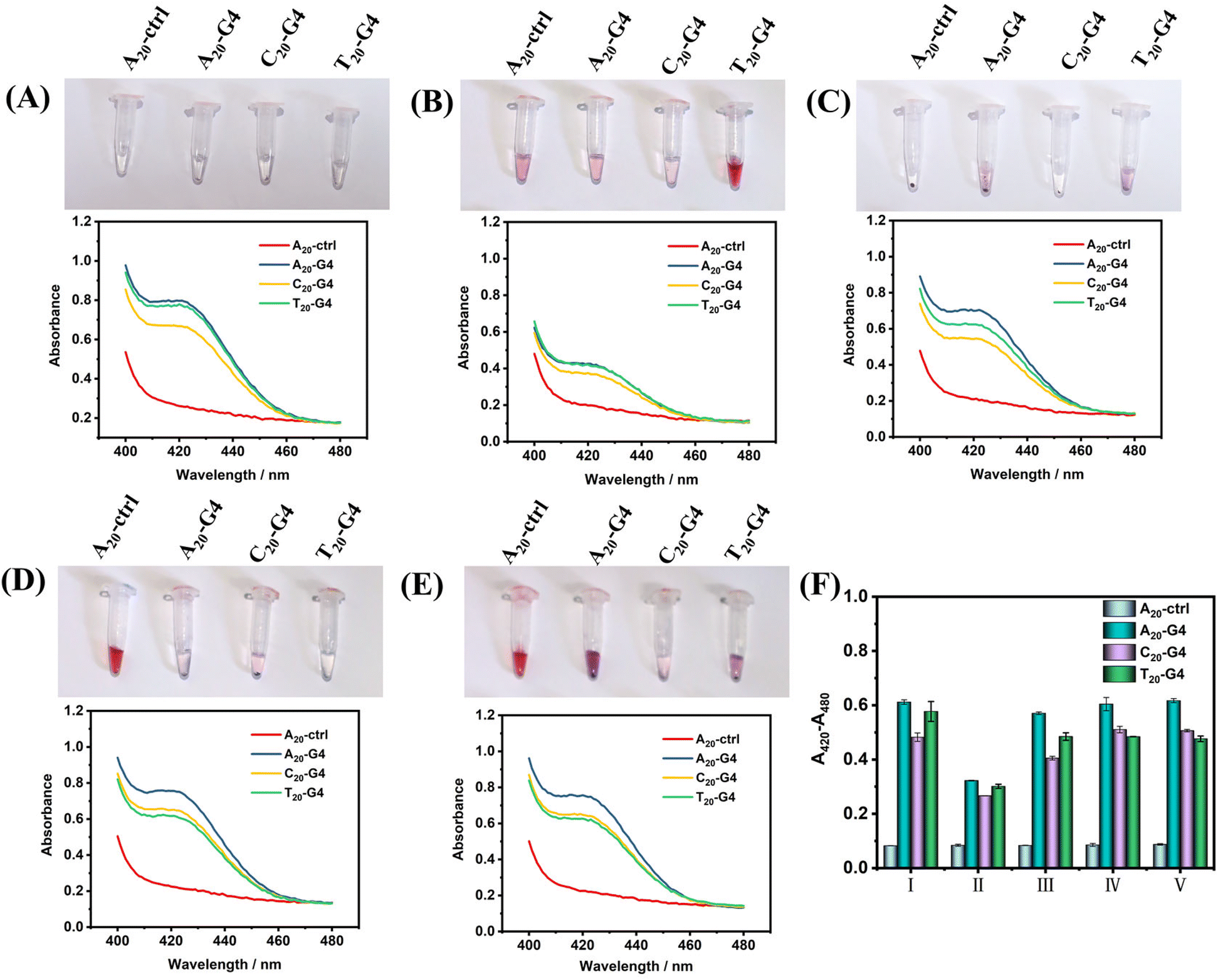 | ||
| Fig. 3 Comparison of label-free SiO2–SNAzyme@ABEI and AuNPs–SNAzyme. (A–E) The photographs show the results of AuNPs modified with four kinds of DNA and the UV-vis spectra show the catalytic activity of ABTS after SiO2@ABEI modification with four kinds of DNA by (A) the magnetic stirring method, (B) microwave heating–drying method, (C) butanol extraction method, (D) freezing method and (E) low-pH method. (F) Quantitative results of catalytic activity of SiO2–SNAzyme@ABEI by the (I) magnetic stirring method, (II) microwave heating–drying method, (III) butanol extraction method, (IV) freezing method and (V) low-pH method. | ||
Fig. 3F summarizes the quantification results of the catalytic activity of SiO2–SNAzyme@ABEI constructed by the five methods. The SNAzymes prepared by the methods have similar catalytic activities except for that by microwave heating–drying, which may be due to the loss of some G4 and SiO2 during microwave heating. Meanwhile, comparing the modification effects of the three types of G4, the A20-G4 modified SNAzyme consistently shows the highest catalytic activity. Therefore, the poly(A20) tag will be used as the linker for adsorption between DNA and SiO2 in subsequent experiments. Finally, we compared the catalytic activities of AuNPs–SNAzyme and SiO2–SNAzyme@ABEI, both prepared by the microwave heating–drying method (Fig. S1†). The results show that SiO2–SNAzyme@ABEI has good catalytic activity, but AuNPs–SNAzyme does not. The principle of the microwave heating–drying method for DNA modification is that the affinity of thymine (T) to gold is low. When heating and drying, the low-affinity poly(T) is distributed in the outer layer due to repulsion effects, so the DNA can “stand-up” and gold aggregation can be avoided,14 but this will also lead to the G-rich sequence being adsorbed on the gold surface, which immensely affects the binding of G4 to hemin, so the catalysis can't be completed, but SiO2 does not have such a problem (Fig. S1A†). Finally, we compared two types of SiO2–SNAzyme@ABEI: one was modified with amino labeled G4 (SiO2–NH2G4@ABEI) and the other was modified with unlabelled G4 (SiO2–G4@ABEI) (Fig. S6†). First, we counted the number of SiO2 nanoparticles using a Flow NanoAnalyzer, and then divided by Avogadro's constant to calculate the molar concentration of 0.5 mg mL−1 SiO2 probe to be approximately 20 pM. We compared the CL intensities of SiO2–SNAzyme@ABEI and G4–DNAzyme. The results show that the CL intensity produced by 20 pM SiO2–SNAzyme@ABEI is consistent with that of 20 nM G4 DNAzymes, suggesting an about 1000-fold signal enhancement. Meanwhile, the enhancement effect of SiO2–NH2G4@ABEI is almost the same as that of SiO2–G4@ABEI. As a result, the modification of DNA on the SiO2 surface by the adsorption method is similar to that by the organic crosslinking method.
In summary, compared with AuNPs, using SiO2 as the core of SNAzymes can produce an excellent adsorption effect on unlabelled G4, and there are no strict requirements for the sequence of DNA and modification method.
3.3 Construction of label free SiO2-capture@ABEI probe for CL detection of AMI-related miDNA-499
Based on the above results, we constructed a SiO2 nanoprobe for the ultrasensitive CL detection of AMI-related miDNA-499. Fig. 4A describes the construction process of this probe. First, SiO2@ABEI is synthesized. Next, label-free SiO2-capture (Sc) acts as a probe to rapidly adsorb on the surface of the SiO2. After washing, capture-G4 and miDNA-499 are added. miDNA-499 is used as a trigger chain to induce the formation of the DNA three-arm structure on the surface of SiO2. After centrifugation to remove excess capture-G4, hemin and K+ were added. After centrifugation again to remove unbound hemin, the target-controlled label-free SiO2-capture@ABEI is constructed.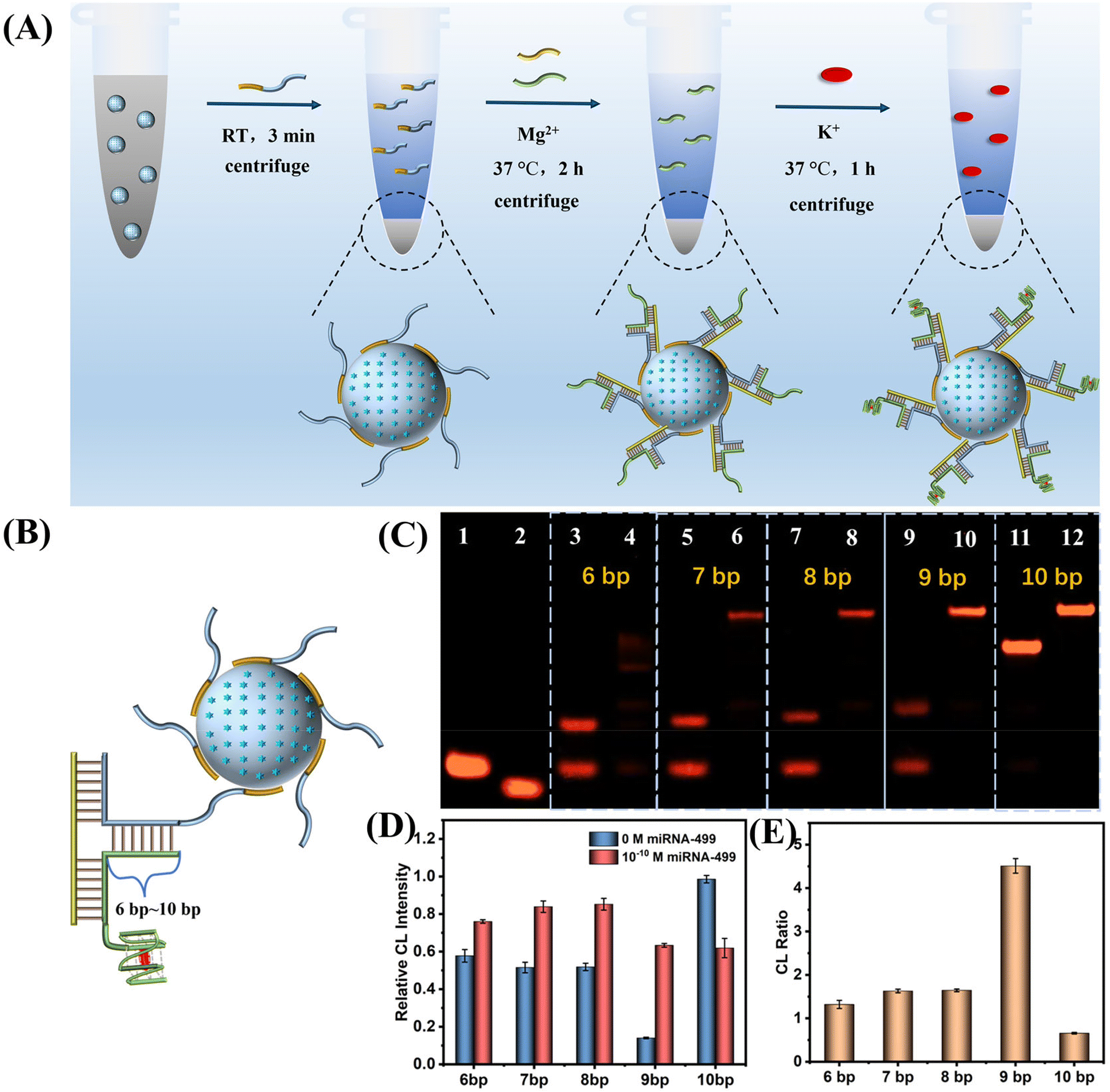 | ||
| Fig. 4 (A) Schematic for construction of the SiO2-capture@ABEI nanoprobe. (B) Schematic diagram of the three-arm structure of DNA from 6 bp to 10 bp. (C) Native PAGE characterization of optimization of three-arm DNA structure. Lane 1, SiO2-capture (Sc); lane 2, miDNA-499; lane 3, capture-6-G4 + Sc; lane 4, capture-6-G4 + Sc + miDNA-499; lane 5, capture-7-G4 + Sc; lane 6, capture-7-G4 + Sc + miDNA-499; lane 7, capture-8-G4 + Sc; lane 8, capture-8-G4 + Sc + miDNA-499; lane 9, capture-9-G4 + Sc; lane 10, capture-9-G4 + Sc + miDNA-499; lane 11, capture-10-G4 + Sc; lane 12, capture-12-G4 + Sc + miDNA-499. (D and E) The DNA three-arm structure was optimized by CL using 100 pM miRNA-499 as the target. (D) CL intensity and (E) corresponding CL ratio. | ||
In order to obtain the best miDNA-499 detection performance, we optimized the sequence of the three-arm structure. First, we designed multiple DNA sequences, called capture-n-G4 (n = 6–10), which can form a 6 bp to 10 bp complementarity with Sc (Fig. 4B). Native polyacrylamide gel electrophoresis (PAGE) images for the hybridization products of capture-n-G4, Sc and target miDNA-499 are shown in Fig. 4C. When the complementarity number reaches 9 bp, the best stability of the three-arm structure is obtained with an extremely low background (Fig. 4C, lane 9 and lane 10). We also optimized the three-arm structure by CL, which was consistent with the results of PAGE, with the highest signal-to-noise ratio at 9 bp (Fig. 4D and E). At the same time, in order to reduce the non-specific adsorption of SiO2 to DNA, we optimized the modification concentration of SiO2-capture to saturate the adsorption on silica spheres (Fig. S7†). The results show that when the initial modification concentration of A20-G4 is 12 μM, the catalytic activity reaches the highest, which also indicates that the adsorption of SiO2 to DNA is saturated.
Next, under the optimal conditions, the above probes were used to perform ultrasensitive CL detection for miDNA-499. Fig. 5A shows the change trend of CL kinetic curves with the various concentrations of miDNA-499. In the concentration range of 10 fM to 100 pM, the maximum value of the kinetic curve gradually increases, and the CL intensity shows a linear relationship with the logarithm of miDNA-499 concentration (Fig. 5B). The correction curve shows that the system has a limit of detection (LOD) of 0.8 fM (S/N = 3), which is even better compared to other miRNA detection methods (Table S2†). Meanwhile, we also used the SiO2 nanoprobe constructed from amino-labeled DNA for CL detection of miDNA-499 (Fig. S8†). The results show that the linear detection range of miDNA is still 10 fM–100 pM, which is consistent with the results of using label-free DNA to construct SiO2 nanoprobes.
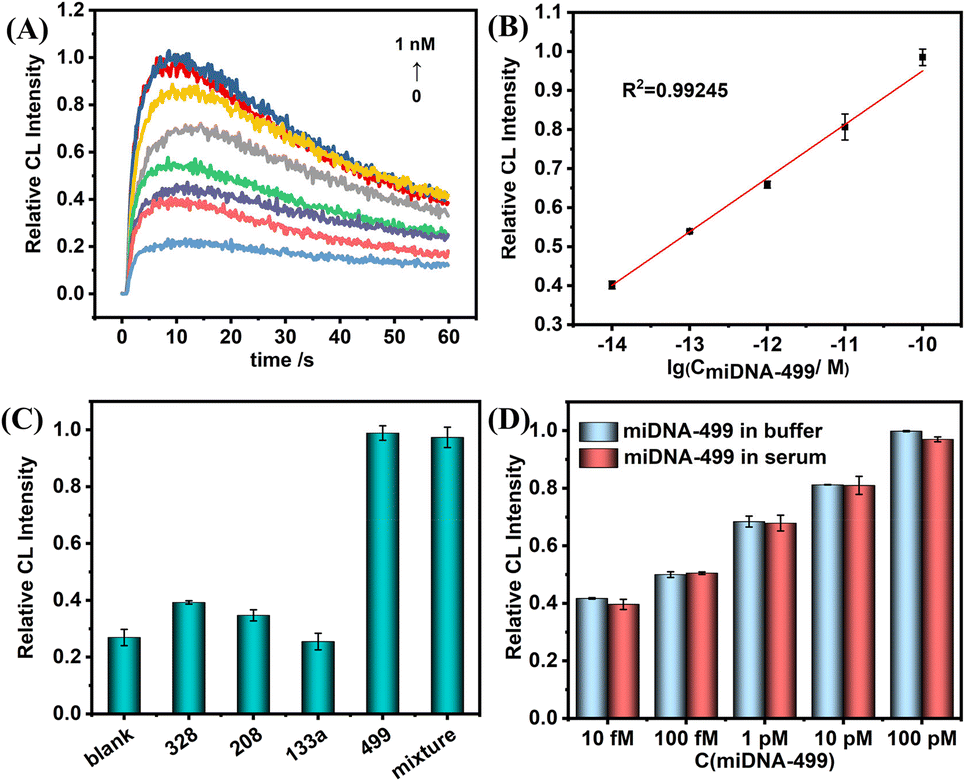 | ||
| Fig. 5 (A) Representative kinetic curves of CL responses corresponding to different concentrations of miDNA-499. (B) Calibration curve of CL responses for miDNA-499 in the range of 10 fM–100 pM. (C) CL responses of specific analysis for non-target miDNAs. The concentration of non-target miDNAs is 1 nM and that of miDNA-499 is 100 pM (D) CL responses of detecting miDNA-499 in buffer and in 1% serum. | ||
To test the selectivity of this nanoprobe, we selected three AMI-related nucleic acids (miDNA-133a, miDNA-328, and miDNA-208, DNA analogues of microRNA-133a, microRNA-328, and microRNA-208, respectively) as an interfering substance. The results show that even though the concentration of non-target miDNAs is 10 times that of miDNA-499, there is still no obvious CL signal enhancement. The CL signal intensity was almost the same as that with miDNA-499 alone after mixing the four miDNAs (Fig. 5C). At the same time, the nanoprobe has the ability to differentiate a single base mismatch (Fig. S9†). Meanwhile, we found that the label-free nanoprobe has good anti-interference ability. In 1% serum, the SiO2-capture@ABEI nanoprobe has excellent anti-degradation ability (Fig. S10†). In addition, the CL intensity corresponding to each concentration of miDNA-499 is basically the same as that in the buffer (Fig. 5D). Otherwise, as shown in Table 1, the detection platform has satisfactory recovery (90–106%) in 1% serum and the relative standard deviation (RSD) is below 4.7%.
| Sample | Add | Found | Recovery (%) | RSD (%) |
|---|---|---|---|---|
| 1 | 100 pM | 95.4 pM | 95.4 | 1.1 |
| 2 | 10 pM | 10.6 pM | 106.0 | 2.4 |
| 3 | 1 pM | 0.9 pM | 90.0 | 2.8 |
| 4 | 100 fM | 102 fM | 102.0 | 4.7 |
| 5 | 10 fM | 10.6 fM | 106.0 | 2.3 |
In summary, the label-free SiO2@capture-ABEI probe has good sensitivity, specificity and anti-interference ability for miDNA-499, so we think it has the potential to identify AMI.
4 Conclusions
In summary, the label-free SiO2–SNAzyme@ABEI overcomes the problem that AuNPs–SNAzyme cannot modify G4 of any sequence, and does not have high requirements for the modification method, and can be prepared by mixing at room temperature for a few minutes without any treatment. The label-free SiO2-capture@ABEI nanoprobe prepared by the same method for the CL detection of AMI-related miDNA-499 also achieves great detection sensitivity, with the lowest detection concentration being as low as 10 fM. Meanwhile, it has excellent selectivity and anti-interference ability. The label-free silicon-based SNAzymes we constructed greatly reduce the preparation cost and lay a good foundation for further application in life analysis, disease treatment and other fields.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank Prof. Liansheng Wang at the First Affiliated Hospital of Nanjing Medical University for providing the serum specimens. This work was supported by the National Natural Science Foundation of China [No. 22074133], and the Anhui Provincial Natural Science Foundation (No. 2008085MB42).Notes and references
- C. A. Mirkin, R. L. Letsinger, R. C. Mucic and J. J. Storhoff, Nature, 1996, 382, 607–609 CrossRef CAS PubMed.
- M. Liu, X. Mao, L. Huang, C. Fan, Y. Tian and Q. Li, Anal. Chem., 2020, 92, 1333–1339 CrossRef CAS PubMed.
- J.-W. Oh, D.-K. Lim, G.-H. Kim, Y. D. Suh and J.-M. Nam, J. Am. Chem. Soc., 2014, 136, 14052–14059 CrossRef CAS PubMed.
- M. N. Wang, Y. Chen, W. J. Cai, H. Feng, T. Y. Du, W. W. Liu, H. Jiang, A. Pasquarelli, Y. Weizmann and X. M. Wang, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 308–316 CrossRef CAS PubMed.
- Y. Sun, L. Shi, Q. Wang, L. Mi and T. Li, Anal. Chem., 2019, 91, 3652–3658 CrossRef CAS PubMed.
- L. Shi, Y. Sun, L. Mi and T. Li, ACS Sens., 2019, 4, 3219–3226 CrossRef CAS PubMed.
- Y. Sun, Q. Wang, L. Mi, L. Shi and T. Li, Anal. Chem., 2019, 91, 12948–12953 CrossRef CAS PubMed.
- M. Hu, C. Yuan, T. Tian, X. Wang, J. Sun, E. Xiong and X. Zhou, J. Am. Chem. Soc., 2020, 142, 7506–7513 CrossRef CAS PubMed.
- A. Opdahl, D. Y. Petrovykh, H. Kimura-Suda, M. J. Tarlov and L. J. Whitman, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 9–14 CrossRef PubMed.
- H. Pei, F. Li, Y. Wan, M. Wei, H. Liu, Y. Su, N. Chen, Q. Huang and C. Fan, J. Am. Chem. Soc., 2012, 134, 11876–11879 CrossRef CAS PubMed.
- X. Zhang, B. Liu, N. Dave, M. R. Servos and J. Liu, Langmuir, 2012, 28, 17053–17060 CrossRef CAS PubMed.
- Z. Huang, B. Liu and J. Liu, Langmuir, 2016, 32, 11986–11992 CrossRef CAS PubMed.
- Y. Ye, S. Hou, X. Wu, X. Cheng and S. He, Langmuir, 2022, 38, 4625–4632 CrossRef CAS PubMed.
- M. Huang, E. Xiong, Y. Wang, M. Hu, H. Yue, T. Tian, D. Zhu, H. Liu and X. Zhou, Nat. Commun., 2022, 13, 968 CrossRef CAS PubMed.
- X. Shen, Y. Wang, Y. Zhang, J. Ouyang and N. Na, Adv. Funct. Mater., 2018, 28, 1803286 CrossRef.
- P. Alonso-Cristobal, P. Vilela, A. El-Sagheer, E. Lopez-Cabarcos, T. Brown, O. L. Muskens, J. Rubio-Retama and A. G. Kanaras, ACS Appl. Mater. Interfaces, 2015, 7, 12422–12429 CrossRef CAS PubMed.
- X. Wang, S. Li, G. Yang, C. Jin and S. Huang, Langmuir, 2020, 36, 2654–2662 CrossRef CAS PubMed.
- N. Duan, C. Li, M. Song, Z. Wang, C. Zhu and S. Wu, Spectrochim. Acta, Part A, 2022, 265, 120342 CrossRef CAS PubMed.
- Y. Li, J. Li, X. Gao, S. Qi, J. Ma and J. Zhu, Appl. Surf. Sci., 2018, 462, 362–372 CrossRef CAS.
- Y. An, R. Li, F. Zhang and P. He, Talanta, 2021, 235, 122790 CrossRef CAS PubMed.
- D. Li, W. Y. Teoh, J. J. Gooding, C. Selomulya and R. Amal, Adv. Funct. Mater., 2010, 20, 1767–1777 CrossRef CAS.
- W. Linyu, Y. Manwen, F. Chengzhi and Y. Xi, Luminescence, 2017, 32, 1039–1044 CrossRef PubMed.
- J. E. Smith, C. D. Medley, Z. Tang, D. Shangguan, C. Lofton and W. Tan, Anal. Chem., 2007, 79, 3075–3082 CrossRef CAS PubMed.
- Q. Jia, J. Ge, W. Liu, S. Liu, G. Niu, L. Guo, H. Zhang and P. Wang, Nanoscale, 2016, 8, 13067–13077 RSC.
- X. Hun, F. Liu, Z. Mei, L. Ma, Z. Wang and X. Luo, Biosens. Bioelectron., 2013, 39, 145–151 CrossRef PubMed.
- H. Li, Y. Cao, T. Wu, Y. Zhang, Z. Zheng, J. Lv, A. Mao, Y. Zhang, Q. Tang and J. Li, Anal. Chem., 2021, 93, 11043–11051 CrossRef CAS PubMed.
- T. Tanaka, K. Hatakeyama, M. Sawaguchi, A. Iwadate, Y. Mizutani, K. Sasaki, N. Tateishi, H. Takeyama and T. Matsunaga, Biotechnol. Bioeng., 2006, 95, 22–28 CrossRef CAS PubMed.
- K. Srinivasan, K. Subramanian, A. Rajasekar, K. Murugan, G. Benelli and K. Dinakaran, Bull. Mater. Sci., 2017, 40, 1455–1462 CrossRef CAS.
- D. Long, Y. Tu, Y. Chai and R. Yuan, Anal. Chem., 2021, 93, 12995–13000 CrossRef CAS PubMed.
- Y. Devaux, M. Vausort, E. Goretti, P. V. Nazarov, F. Azuaje, G. Gilson, M. F. Corsten, B. Schroen, M.-L. Lair, S. Heymans and D. R. Wagner, Clin. Chem., 2012, 58, 559–567 CrossRef CAS PubMed.
- L. Wang, C. Yang and W. Tan, Nano Lett., 2005, 5, 37–43 CrossRef CAS PubMed.
- H. Fan, X. Wang, F. Jiao, F. Zhang, Q. Wang, P. He and Y. Fang, Anal. Chem., 2013, 85, 6511–6517 CrossRef CAS PubMed.
- W. Kong, X. Zhao, Q. Zhu, L. Gao and H. Cui, Anal. Chem., 2017, 89, 7145–7151 CrossRef CAS PubMed.
- M. You, S. Yang, W. Tang, F. Zhang and P. He, Biosens. Bioelectron., 2018, 112, 72–78 CrossRef CAS PubMed.
- J. Shu, Z. Han, T. Zheng, D. Du, G. Zou and H. Cui, Anal. Chem., 2017, 89, 12636–12640 CrossRef CAS PubMed.
- Y. Hao, Y. Li, L. Song and Z. Deng, J. Am. Chem. Soc., 2021, 143, 3065–3069 CrossRef CAS PubMed.
- C. Chen, F. Pu, Z. Huang, Z. Liu, J. Ren and X. Qu, Nucleic Acids Res., 2011, 39, 1638–1644 CrossRef CAS PubMed.
- H. Li, Y. Li, J. Li, F. Yang, L. Xu, W. Wang, X. Yao and Y. Yin, Anal. Chem., 2020, 92, 4094–4100 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sd00170e |
| This journal is © The Royal Society of Chemistry 2023 |