DOI:
10.1039/D3SC04660E
(Edge Article)
Chem. Sci., 2023,
14, 11907-11913
Unlocking mild-condition benzene ring contraction using nonheme diiron N-oxygenase†
Received
4th September 2023
, Accepted 9th October 2023
First published on 11th October 2023
Abstract
Benzene ring contractions are useful yet rare reactions that offer a convenient synthetic route to various valuable chemicals. However, the traditional methods of benzene contraction rely on noble-metal catalysts under extreme conditions with poor efficiency and uncontrollable selectivity. Mild-condition contractions of the benzene ring are rarely reported. This study presents a one-step, one-pot benzene ring contraction reaction mediated by an engineered nonheme diiron N-oxygenase. Using various aniline substrates as amine sources, the enzyme causes the phloroglucinol-benzene-ring contraction to afford a series of 4-cyclopentene-1,3-dione structures. A reaction detail study reveals that the nonheme diiron N-oxygenase first oxidizes the aromatic amine to a nitroso intermediate, which then attacks the phloroglucinol anion and causes benzene ring contraction. Besides, we have identified two potent antitumor compounds from the ring-contracted products.
Introduction
Ring contractions (RCTs) are powerful tools for synthesizing atom-number-reduced structures from existing ring skeletons. Carbon-ring contractions provide elegant and efficient synthetic routes to many drugs, natural products, and industrial materials.1–5 Among all carbon-ring structures, benzene is notably the most widely distributed one in nature, rendering it a promising reservoir for RCTs. Representative benzene ring contracted structures, such as cyclopentene and saturated cyclic hydrocarbons, carry substantial significance within the pharmaceutical and industrial domains (Fig. 1A).6,7 However, owing to the aromaticity, non-polarity, and high stability, contracting or cleaving the benzene ring gives rise to inherent thermodynamic barriers. Current strategies for opening benzene rings require the involvement of specific functional groups, heavy metals, high temperature, electrochemical methods, or photochemical techniques, in order to overcome the inherent thermal barrier (representative examples in Fig. 1B).6–15 Therefore, mild-condition enzymatic contraction of the benzene ring is of great importance and yet challenging. In this study, we report an aqueous-phase phloroglucinol benzene RCT facilitated by an engineered non-heme diiron N-oxygenase, AzoC-III (Fig. 1C).
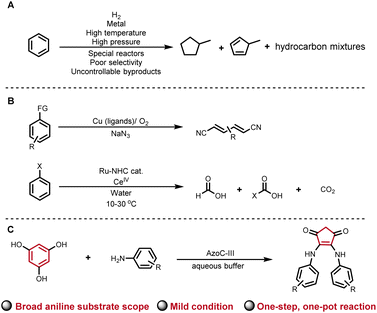 |
| | Fig. 1 Benzene ring contractions and cleavages. (A) Traditional benzene ring contraction in the petroleum industry. (B) Examples of current benzene ring cleavage strategies. (C) Non-heme diiron N-oxygenase mediated benzene ring contraction in this study. | |
The typical prerequisites for RCT encompass four key elements: firstly, the imperative incorporation of activating factors like heat, nucleophilic reagents, Lewis acid, light, oxidation, or heavy metals;16–22 secondly, the generation of active intermediates, be they ions, carbenoids, or radicals, in facilitating the contraction process;23–25 thirdly, the strategic positioning of functional groups, such as sulfones, halogen atoms, diazole, azide, organic boron, organic silicon, thioether, enol ethers, vicinal diols, or enolized ketones, adjacent the contracting site;26 fourthly, the requirement of a spontaneous or environmentally triggered chemical rearrangement, such as the pinacol rearrangement, Favorskii rearrangement, Wolff rearrangement, cyclic silyl-enol ether rearrangement, etc., in stabilizing the active intermediates and ensuring the fulfilment of the contractions.27–31 With these fundamental elements, traditional RCTs often transpire under rigorous conditions, yielding intricate byproducts.
Non-heme diiron N-oxygenase AzoC natively catalyzes four-electron oxidation of an amine to a nitroso group during azoxymycins biosynthesis in Streptomyces chattanoogensis.32,33 Recently, this enzyme's catalytic capability has been extended for the chemoenzymatic synthesis of phenol diarylamines employing its intrinsic nitroso-producing ability. Mechanistically, AzoC-II, the substrate-scope expanded AzoC mutant, catalyzes the four-electron oxidation of various para- and meta-substituted aniline substrates to afford the corresponding nitroso intermediates, which couple to the phenoxide anion (the enolyzed form of phenol) and culminate in the formation of the final diarylamine products.34 In organic chemistry, the nitroso group is widely recognized as a versatile and captivating building block for forging C–N and N–N bonds.35–37 Consequently, the nitroso group fulfils the role of the initial activating factor in the RCT process, while diarylhydroxylamine, an intermediate formed after nitroso addition to the phenoxide anion during phenol diarylamine biosynthesis, might contribute as the second element of the active intermediate; furthermore, the alkaline solution (pH 8 buffer) could potentially serve as the fourth element, inducing environmental phenol rearrangement.38 By intertwining these principles, we investigated whether mild-condition RCT could be realized through this chemoenzymatic phenol sp2 C–H amination strategy.
Results and discussion
Enzymatic RCT design
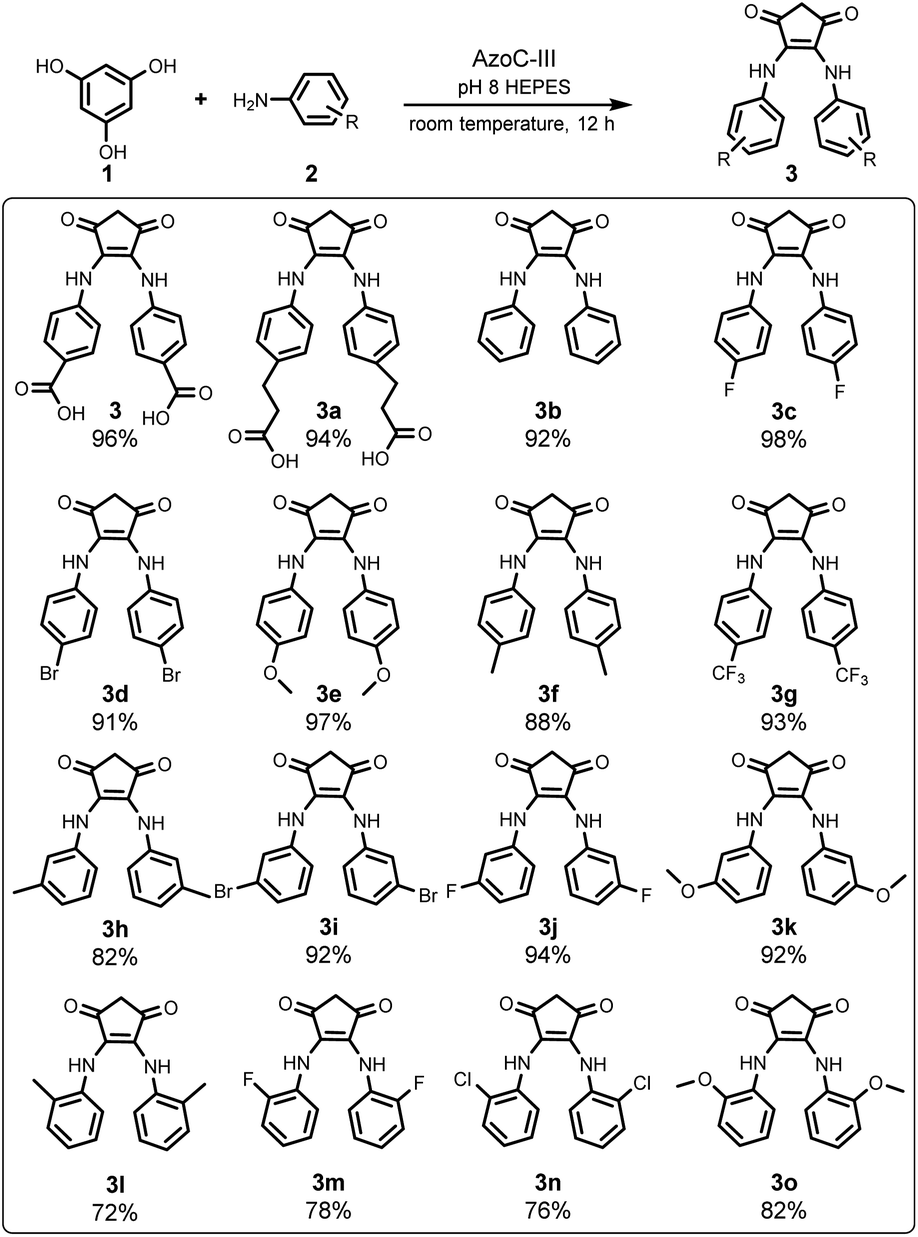
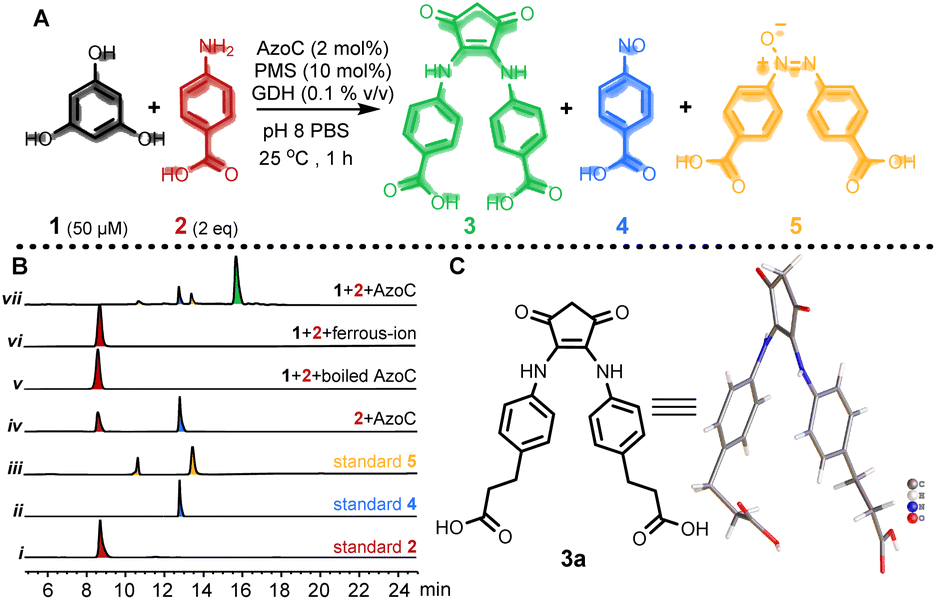
Our investigation commenced by seeking an appropriate starting benzene ring substrate. In the context of diarylamine synthesis, the reactivity of phenol experiences a substantial elevation as the number of hydroxyl groups increases. For example, while a phenol with a solitary hydroxy group fails to yield diarylamine, introducing two hydroxy groups leads to a 78% diarylamine yield.34 This observation led us to hypothesize that a benzene ring with three hydroxy groups might exhibit heightened reactivity in benzene ring activation. Among benzene tri-phenols, phloroglucinol stands out because it possesses three chemically equivalent sp2 C–H bonds. This advantage could avoid the formation of undesired imbalanced chemical products. Hence, we selected phloroglucinol (1) as the model phenol substrate, and employed p-aminobenzoic acid (2) as the amine partner. We then conducted the enzymatic phenol amination reaction in pH 8 PBS buffer using phenazine methosulfate (PMS) as the electron mediator and glucose dehydrogenase (GDH) as the NADH regenerating system (Fig. 2A).34 A series of control reactions were performed as well. According to HPLC analysis, the enzyme predominantly transformed 2 to p-nitroso benzoic acid (4), as expected, when 1 was not added (Fig. 2B, trace iv); however, the incubation of 1 and 2 with AzoC resulted in the formation of a new peak 3 (Fig. 2B, trace vii) alongside the azoxy product (5, a non-enzymatic dimerization product from 4, as reported previously).32,33 Meanwhile, the incubation of 1 and 2 with boiled enzyme or ferrous ions showed no reaction (Fig. 2B, traces v and vi). These results indicated that both the nitroso-producing activity of AzoC and the presence of 1 are crucial for generating 3. We then performed a scale-up reaction and purified 3. 1H NMR analysis of 3 revealed a distinctive non-benzene chemical shift signal at 3.14 (Fig. S4†), which was absent in any substrates or characterized shunt products, suggesting a possible ring cleavage. To corroborate this finding, we further used 3-(4-aminophenyl)propionic acid and aniline as amine sources, and isolated 3a and 3b, respectively. 1H NMR analysis of 3a and 3b also unveiled non-benzene chemical shift signals at 3.06 and 3.08, respectively (Fig. S8 and S12†). We then referred 3, 3a and 3b to single crystal cultivation and obtained the crystals of 3a and 3b in methanol solution (Fig. 2C, S7 and S11,†3a CCDC ID: 2193171, 3b CCDC ID: 2209966).39 The X-ray diffraction analysis indicated that both 3a and 3b comprised a 1,3-cyclopentanedione moiety, distinctly indicating a noticeable ring contraction of 1. These results enabled the assignment of the 1H NMR signal around 3.1 as the hallmark chemical shift of the RCT products.
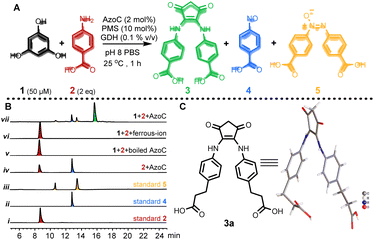 |
| | Fig. 2 Enzyme triggered ring contraction of phloroglucinol. (A) AzoC mediated RCT. (B) HPLC analysis of the enzyme reaction mixtures at 305 nm (note: 1 is undetectable with our HPLC method). (i) Standard 2; (ii) standard 4; (iii) standard 5; (iv) 100 μM 2 with 1 μM AzoC; (v) 50 μM 1 and 100 μM 2, with 1 μM boiled AzoC; (vi) 50 μM 1 and 100 μM 2, with 2 μM Mohr's salt (ferrous iron source); (vii) 50 μM 1 and 100 μM 2, with 1 μM AzoC. (C) X-ray structure of 3a (CCDC ID: 2193171). Note: Standard 5 showed two peaks due to the spontaneous N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N E/Z isomerization of the azoxy bond. N E/Z isomerization of the azoxy bond. | |
Reaction conditions optimization
We then optimized the reaction conditions. With 1 and 2 as substrates, we analyzed the influences of buffer type, substrates ratio, and GDH stock addition on the yield of 3 (Table 1). As a result, optimal reaction conditions with a 96% yield of 3 were obtained when the reaction was performed by incubating 1 eq. of 1, 3 eq. of 2, 10% eq. of PMS, 10% eq. of TEMPO (nitroso polymerization inhibitor), 0.1% v/v GDH stock solution (electron source), and 3% mol enzyme in pH 8 HEPES buffer for 1 h (entry 8 in Table 1). Interestingly, the increase of the 2/1 ratio above 3 has not improved the yield of 3 and meanwhile has not caused the production of side-products other than 4 and 5 (entries 5–7 in Table 1 and Fig. S56†), which indicates 3 is the chemically stable ring contracting product rather than an active intermediate.
Table 1 Reaction conditions optimization
Enzyme engineering
With the optimized reaction conditions in hand, we then set out to further expand the amine substrate scope of AzoC. AurF (GenBank accession number: CAE02601), a non-heme diiron N-oxygenase bearing 39% sequence identity to AzoC, can effectively oxidize a variety of para-, meta- and ortho-carboxy substituted aniline substrates to the corresponding nitro products.40,41 In our phenol diarylamine biosynthetic study,34 a mutant enzyme, AzoC-II, which recognizes various para- and meta-substituted anilines, was readily obtained through a simple two-substrate-binding-residues switch of AzoC to the counterpart-residues of AurF. Since AzoC-II does not accept ortho-substituted anilines, we further engineered this enzyme using AurF as the structural model. Bioinformatic analysis of AurF and AzoC revealed a residue difference in the substrate binding pocket (the red and blue marked amino acid residues in Fig. 3A). The co-crystal structure of AurF with para-nitrobenzoic acid (PDB:3-CHT, Fig. 3B) indicated the involvement of Y93, V97, T100, N200 and R302 in substrate-binding (corresponding to residues Y94, T98, L101, N202, and Q304 in AzoC).42 Among these, Y93 and N200 in AurF are identical to their counterparts in AzoC. The RAurF304QAzoC mutation, which replaced the basic amino acid R with the polar neutral amino acid Q in AzoC, potentially explained the recognition of various para-substituted anilines by AzoC (AurF predominantly favors acid-substituted anilines). Thus, we narrowed our focus to residues T98 and L101. Considering that T98 and L101 in AzoC and V97 and T100 in AurF are all uncharged amino acids, we constructed a mutant library by replacing these two residues with smaller, uncharged amino acids. Following two rounds of screening, we successfully isolated the AzoC-T98G-L101G mutant (AzoC-III), which exhibited the broadest substrate scope encompassing diverse electronic substitution on para-, meta- and ortho-sites. The structural-mimic model of AzoC-III (Fig. 3C) indicates an enlarged substrate binding pocket compared to that of AurF (Fig. 3B). This potentially explains the extended substrate scope of AzoC-III.
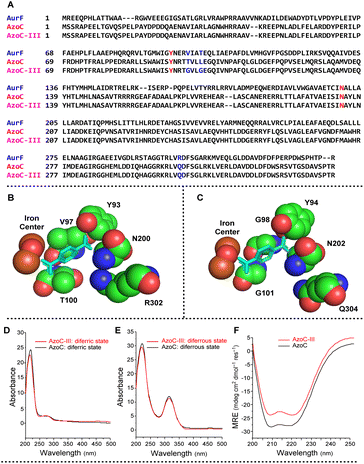 |
| | Fig. 3 Engineering and characterization of AzoC. (A) Sequence alignment of AurF, AzoC and AzoC-III. (B) Substrate binding pocket of AurF (PDB:3-CHT). (C) Substrate binding pocket of AzoC-III, the structure was built using SWISS-MODEL based on PDB:3-CHT. (D) UV/visible spectra of AzoC and AzoC-III at the diferric state: 10 μM enzyme in pH 8 HEPES buffer was bubbled with oxygen for 5 min, then subjected to UV/vis analysis. (E) UV/vis spectra of AzoC and AzoC-III at the diferrous state: 10 μM enzyme in pH 8 HEPES buffer was mixed with 5 fold sodium dithionite for 5 min, then subjected to UV/vis analysis. (F) CD spectra of AzoC and AzoC-III: 10 μM enzyme in pH 8 HEPES was analyzed. | |
Identification of the mutant enzymes
The successful execution of the RCT reaction heavily relies on the enzymatic nitroso-producing activity. However, AzoC-III was engineered based on the nitro-producing enzyme AurF. In non-heme diiron oxygenases, the oxygen–iron coordination geometry is a critical determinant of the enzymatic reactivity.43,44 UV/vis spectra analysis is an effective method to characterize the oxygen–iron coordination. Consequently, our assessment delved into the UV/visible spectra analysis of both AzoC and AzoC-III, spanning their diferric (oxygen-coordinated to the diiron center, in the oxidizing form) and diferrous (no oxygen-coordinated to the diiron center, in the reducing form) states. As depicted in Fig. 3D and E, the UV/vis spectra of AzoC-III and AzoC closely aligned in both the diferric and diferrous states. This congruence underscored the substantial resemblance between the oxygen–iron coordination geometries of AzoC-III and AzoC. Essentially, the UV/visible data provided microstructural confirmation of the likeness at the active iron center between AzoC-III and the native enzyme AzoC. Considering that the activity of a metalloenzyme is influenced not only by the active metal center but also by the protein scaffold,45 our exploration next extended to the macrostructural analysis of AzoC-III and AzoC by using circular dichroism (CD) spectroscopy, which is a powerful protein-secondary-structure investigation tool. As depicted in Fig. 3F, the negative bands at 208 nm and 222 nm in the spectra of both AzoC and AzoC-III signified the prevailing α-helical structural characteristic. This insight indicated that the engineering of AzoC to AzoC-III did not disrupt the overall α-helical protein scaffold at the macrostructural level. By examining these findings, it's clear that AzoC-III underwent changes neither in its protein scaffold nor in its active center. Instead, the changes should be centered around the substrate binding pocket.
Substrate scope expansion
Subsequently, we studied the substrate scope of this RCT, utilizing AzoC-III as the catalyst under the optimized reaction conditions. As depicted in Fig. 4, our reaction converted a diverse array of aniline substrates, including varying electronic substitutions at the para-, meta-, and ortho-sites, to the RCT products in good to excellent yields. Upon delving into the regioselectivities, the para- and meta-substituted anilines gave excellent yields (3c–3g and 3h–3k in Fig. 4, respectively), while ortho-substituted anilines (3l–3o in Fig. 4) only showed good yields, indicating that even with the enzyme engineering, AzoC-III displays a regioselectivity bias against ortho-substitutions. Interestingly, in all para-, meta- and ortho-substitutions, anilines with higher water solubilities exhibited remarkably better yields (such as 3c, 3e, 3g, 3j, 3k, 3m and 3o) compared to those with lower water solubilities (such as 3d, 3f, 3h, 3i, 3l and 3n, the water solubilities were collected from the PubChem database46). This observation suggests that the water solubilities of anilines exert a more pronounced influence on this enzymatic RCT compared to their electron-inductive effects. In essence, the substrate scope examination revealed a broad tolerance for electronic substitution and a general capacity for regioselectivity of the aniline substrates, towards both the engineered enzyme AzoC-III and the RCT reaction itself.
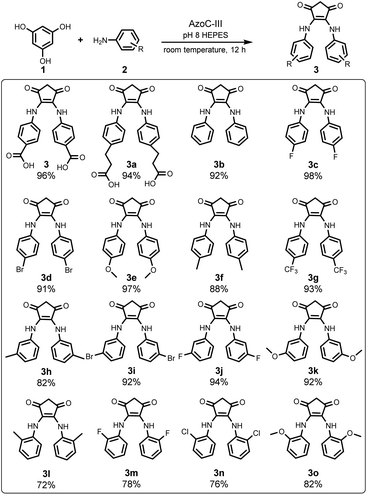 |
| | Fig. 4 Substrate scope of the RCT. Reaction conditions: 30 μM AzoC-III, 100 μM PMS, 100 μM TEMPO, 1 mM phloroglucinol and 3 mM amines were incubated with 0.1% v/v GDH stock solution in pH 8 HEPES buffer at 25 °C and 200 rpm for 12 h. | |
Reaction detail study
We then proceeded to study the reaction details in order to probe the potential mechanism. Aromatic nitroso groups exhibit both radical and ionic addition reactivities.47 Strikingly, during our optimization efforts and the prior diarylamine biosynthesis studies, introducing a radical scavenger (TEMPO) into the reaction system resulted in enhanced but not decreased yields (Table 1). Hence, the possibility of a radical addition route was excluded, leaving the ionic addition pathway more credible. We then studied the authentic state of phloroglucinol (1) during the reaction. The structure of 1 indicated that it has various enol and keto tautomerizations. In pH 8 PBS buffer, the UV/vis spectrum of 1 exhibited absorbance peaks at 278 nm and 350 nm, corresponding to the mono-deprotonated and double-deprotonated phenoxide anion states of phloroglucinol respectively (Fig. 5A and B).48 With all of this evidence, an ionic addition of the nitroso N atom (positively charged) onto the negatively charged phloroglucinol phenoxide anions was proposed. We then delved into unravelling the departing entity of the contracted carbon atom from the phloroglucinol benzene ring. The strategy started by incubating 3 mmol of nitrosobenzene and 1 mmol of 1 in 50 mL HEPES buffer (pH 8) within a pre-degassed and sealed flask for an hour, followed by the adjustment of the solution pH to 4 and the collection of the expelled gas. Remarkably, the Gas Chromatography (GC) analysis of the expelled gas revealed a peak exhibiting the same retention time as CO2 (Fig. S53†). Moreover, when the collected gas was bubbled through saturated limewater, a milky sediment was observed, the characteristic of CO2 formation. These results firmly confirmed CO2 as the departing species of the contracted carbon atom. We further explored the hydrogen source of the secondary amine of the ring-contracted product. To elucidate this, we subjected nitrosobenzene and 1 to non-enzymatic reaction in pH 8 D2O water (pH adjusted using NaOD). A control reaction using H2O and NaOH was conducted as well. High-resolution mass spectrometry (HRMS) analysis revealed a signal of +2 Da compared to the control sample (Fig. S54 and S55†), unequivocally establishing that the amine moieties' hydrogen atoms were originated from water. More importantly, the incubation of nitrosobenzene with 1 produced the RCT product 3b at the equivalent yield (91%) to the enzymatic reaction, revealing a non-enzymatic chemical synthetic route of the RCT products. Based on these findings, a probable reaction pathway was proposed (Fig. 5C).
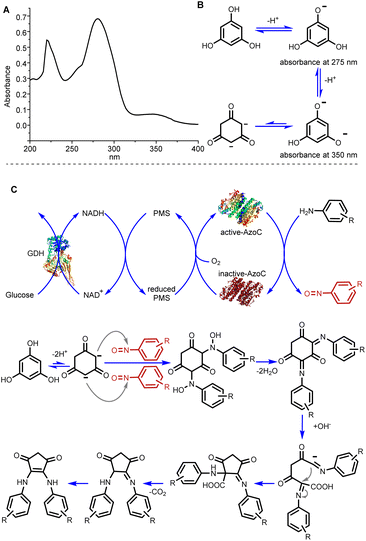 |
| | Fig. 5 Reaction detail study. (A) UV/vis spectrum of phloroglucinol (1) in pH 8 HEPES buffer. (B) Keto–enol tautomerization of phloroglucinol in pH 8 HEPES buffer. (C) Proposed pathway of the RCT. | |
Bioactivity study of RCT products
We proceeded to assay the bioactivities of the RCT products. As depicted in Fig. 4, the ring-contracted products 3b–3o all share a common cyclopentenedione moiety, a versatile structure frequently encountered in natural products and drugs, particularly those exhibiting antimicrobial and anti-tumor activity.49–54 Consequently, we evaluated their potent bioactivities against Staphylococcus aureus ATCC 25923, E. coli K12, Bacillus subtilis ATCC 67736, Saccharomyces cerevisiae YJM789, breast cancer cell line MDA-MB-648, and lymphoma cell lines Rec-1 and RAMOS. Although the majority of the RCT products displayed negligible antimicrobial activity, we did observe mild activities of compound 3 against S. aureus ATCC 25923 (MIC ≈ 125 μM, approximately 20 times that of chloramphenicol) and E. coli K12 (MIC ≈ 25 μM, roughly 10 times that of chloramphenicol). We also observed moderate anti-tumor activities. Specifically, 3a demonstrated moderate potency against RAMOS (IC50 ≈ 3.150 μM, Fig. S57†), and 3g displayed moderate effects against MDA-MB-648 (IC50 ≈ 7.840 μM, Fig. S58†).
Beyond these bioactivities, cyclic dione structures hold significance as versatile building blocks within various domains of chemistry, including the synthesis of heterocycles, photoelectric materials, polymeric materials, organic catalysts, etc.55,56 Moreover, diamine-substituted cyclopentane frameworks are pivotal moieties within asymmetric synthetic ligands and molecule recognition.57 Conventional synthetic methodologies for cyclic diones and diamine-substituted cyclopentane frameworks necessitate multi-step metallic reactions in organic solvents.58–60 In stark contrast, our RCT strategy offers an elegant and cutting-edge approach to these structures.
Conclusions
In summary, we have demonstrated a benzene ring contraction (RCT) facilitated by an engineered non-heme diiron N-oxygenase. By utilizing phloroglucinol as the RCT starting material, the engineered enzyme AzoC-III showcases remarkable versatility in accommodating a wide array of aniline substrates. This encompasses an extensive range of electron-inductive substitutions on the para-, meta-, and ortho-sites of anilines, culminating in the generation of diverse RCT products. Remarkably, this process has led to the discovery of two promising anti-tumor compounds. Additionally, our findings have disclosed an efficient non-enzymatic method for the one-pot chemical synthesis of diamine-substituted cyclopentanediones in an aqueous environment, utilizing nitroso compounds as nitrogen sources.
Data availability
All data are provided in the ESI.†
Author contributions
Y.-Y. G. conceived the idea and prepared the manuscript. Y.-Y. G., Z.-H. T. and Y.-C. H. performed the biosynthetic and mechanism experiments. C. H. M. and Y.-Y. G. performed the bioactivity study. D. C. B., Z. Y. J. and Y.-Y. G conducted the mechanism experiments design.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (22377024, 31901014, 21925103) and the Natural Science Foundation of Henan Province (232300421229).
References
- K. M. J. de Mattos-Shipley, C. E. Spencer, C. Greco, D. M. Heard, D. E. O'Flynn, T. T. Dao, Z. Song, N. P. Mulholland, J. L. Vincent, T. J. Simpson, R. J. Cox, A. M. Bailey and C. L. Willis, Chem. Sci., 2020, 11, 11570–11578 RSC.
- D. Heldt, A. D. Lawrence, M. Lindenmeyer, E. Deery, P. Heathcote, S. E. Rigby and M. J. Warren, Biochem. Soc. Trans., 2005, 33, 815–819 CrossRef CAS PubMed.
-
L. F. Silva Jr, Ring Contraction Reactions in the Total Synthesis of Biologically Active Natural Products, Stereoselective Synthesis of Drugs and Natural Products, 2013, pp. 1–20, DOI:10.1002/9781118596784.ssd018.
- X. Kang, G. Luo, L. Luo, S. Hu, Y. Luo and Z. Hou, J. Am. Chem. Soc., 2016, 138, 11550–11559 CrossRef CAS PubMed.
- L. F. Silva, Jr., Molecules, 2006, 11, 421–434 CrossRef PubMed.
- S. Hu, T. Shima and Z. Hou, Nature, 2014, 512, 413–415 CrossRef CAS PubMed.
-
D. S. Jones and P. P. Pujadó, Handbook of petroleum processing, Springer Science & Business Media, 2006 Search PubMed.
- X. Qiu, Y. Sang, H. Wu, X.-S. Xue, Z. Yan, Y. Wang, Z. Cheng, X. Wang, H. Tan, S. Song, G. Zhang, X. Zhang, K. N. Houk and N. Jiao, Nature, 2021, 597, 64–69 CrossRef CAS PubMed.
- Y. Shimoyama, T. Ishizuka, H. Kotani and T. Kojima, ACS Catal., 2019, 9, 671–678 CrossRef CAS.
- L. T. Scott and N. H. Roelofs, J. Am. Chem. Soc., 1987, 109, 5461–5465 CrossRef CAS.
- L. T. Scott, N. H. Roelofs and T. H. Tsang, J. Am. Chem. Soc., 1987, 109, 5456–5461 CrossRef CAS.
- M. Jakoobi, N. Halcovitch, G. F. S. Whitehead and A. G. Sergeev, Angew. Chem., Int. Ed., 2017, 56, 3266–3269 CrossRef CAS PubMed.
- W. Guo, W. Ding, Y. Yao, S. Rajca, Q. Li, H. Jiang, A. Rajca and Y. Wang, Org. Lett., 2023, 25, 3972–3977 CrossRef CAS PubMed.
- Z.-C. Yin, M. Li, C. Niu, W.-F. Wang, W.-R. Liu, Q.-W. Zhang and G.-W. Wang, Angew. Chem., Int. Ed., 2023, 62, e202304321 CrossRef CAS PubMed.
- J. Jian, X. Wu, M. Chen and M. Zhou, J. Am. Chem. Soc., 2020, 142, 10079–10086 CrossRef CAS PubMed.
- N. E. Behnke, J. H. Siitonen, S. A. Chamness and L. Kürti, Org. Lett., 2020, 22, 5715–5720 CrossRef CAS PubMed.
- Z. Huang, W.-X. Zhang and Z. Xi, Org. Lett., 2018, 20, 485–488 CrossRef CAS PubMed.
- K. Hurej, M. Pawlicki, L. Szterenberg and L. Latos-Grażyński, Angew. Chem., Int. Ed., 2016, 55, 1427–1431 CrossRef CAS PubMed.
- J. Jurczyk, M. C. Lux, D. Adpressa, S. F. Kim, Y.-h. Lam, C. S. Yeung and R. Sarpong, Science, 2021, 373, 1004–1012 CrossRef CAS PubMed.
- B. Patra, S. Sobottka, S. Mondal, B. Sarkar and S. Kar, Chem. Commun., 2018, 54, 9945–9948 RSC.
- E. Tayama, Ring-Substitution, Enlargement, and Contraction by Base-Induced Rearrangements of N-Heterocyclic Ammonium Salts, Heterocycles, 2016, 92, 793–828 CrossRef CAS.
- X. Yu, J. Hu, Z. Shen, H. Zhang, J.-M. Gao and W. Xie, Angew. Chem., Int. Ed., 2017, 56, 350–353 CrossRef CAS PubMed.
- D. Crich, A. L. J. Beckwith, G. F. Filzen and R. W. Longmore, J. Am. Chem. Soc., 1996, 118, 7422–7423 CrossRef CAS.
- M. Piesch, S. Reichl, M. Seidl, G. Balázs and M. Scheer, Angew. Chem., Int. Ed., 2019, 58, 16563–16568 CrossRef CAS PubMed.
- K. Takasu, Y. Nagamoto and Y. Takemoto, Chem.–Eur. J., 2010, 16, 8427–8432 CrossRef CAS PubMed.
- Z.-L. Song, C.-A. Fan and Y.-Q. Tu, Chem. Rev., 2011, 111, 7523–7556 CrossRef CAS PubMed.
- D. Bégué, H. Santos-Silva, A. Dargelos and C. Wentrup, J. Phys. Chem. A, 2017, 121, 5998–6003 CrossRef PubMed.
- C. Chapuis, F. Robvieux, C. Cantatore, C. Saint-Léger and L. Maggi, Helv. Chim. ACTA, 2012, 95, 428–447 CrossRef CAS.
- J. Limanto, E. R. Ashley, J. Yin, G. L. Beutner, B. T. Grau, A. M. Kassim, M. M. Kim, A. Klapars, Z. Liu, H. R. Strotman and M. D. Truppo, Org. Lett., 2014, 16, 2716–2719 CrossRef CAS PubMed.
- M. J. Mitcheltree, Z. A. Konst and S. B. Herzon, Tetrahedron, 2013, 69, 5634–5639 CrossRef CAS PubMed.
- J. Oliver-Meseguer, M. Boronat, A. Vidal-Moya, P. Concepción, M. Á. Rivero-Crespo, A. Leyva-Pérez and A. Corma, J. Am. Chem. Soc., 2018, 140, 3215–3218 CrossRef CAS PubMed.
- Y.-Y. Guo, H. Li, Z.-X. Zhou, X.-M. Mao, Y. Tang, X. Chen, X.-H. Jiang, Y. Liu, H. Jiang and Y.-Q. Li, Org. Lett., 2015, 17, 6114–6117 CrossRef CAS PubMed.
- Y.-Y. Guo, Z.-H. Li, T.-Y. Xia, Y.-L. Du, X.-M. Mao and Y.-Q. Li, Nat. Commun., 2019, 10, 4420 CrossRef PubMed.
- Y.-Y. Guo, Z.-H. Tian, L. Wang, Z.-D. Lai, L. Li and Y.-Q. Li, ACS Catal., 2023, 13, 1412–1417 CrossRef CAS.
- H.-Y. He, H. Niikura, Y.-L. Du and K. S. Ryan, Chem. Soc. Rev., 2022, 51, 2991–3046 RSC.
- A. J. Waldman, T. L. Ng, P. Wang and E. P. Balskus, Chem. Rev., 2017, 117, 5784–5863 CrossRef CAS PubMed.
- Y. Katsuyama and K. Matsuda, Curr. Opin. Chem. Biol., 2020, 59, 62–68 CrossRef CAS PubMed.
- M. Murakata and M. Kimura, Tetrahedron Lett., 2010, 51, 4950–4952 CrossRef CAS.
- Deposition numbers 2193171 and 2209966 contain the supplementary crystallographic data for this paper.†.
- E. Chanco, Y. S. Choi, N. Sun, M. Vu and H. Zhao, Helv. Chim. Acta, 2014, 22, 5569–5577 CAS.
- R. Winkler, M. E. A. Richter, U. Knuepfer, D. Merten and C. Hertweck, Angew. Chem., Int. Ed., 2006, 45, 8016–8018 CrossRef CAS PubMed.
- Y. S. Choi, H. Zhang, J. S. Brunzelle, S. K. Nair and H. Zhao, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6858–6863 CrossRef CAS PubMed.
- A. J. Jasniewski and L. Que, Chem. Rev., 2018, 118, 2554–2592 CrossRef CAS PubMed.
- T. M. Makris, V. V. Vu, K. K. Meier, A. J. Komor, B. S. Rivard, E. Muenck, L. Que, Jr. and J. D. Lipscomb, J. Am. Chem. Soc., 2015, 137, 1608–1617 CrossRef CAS PubMed.
- A. J. Reig, M. M. Pires, R. A. Snyder, Y. Wu, H. Jo, D. W. Kulp, S. E. Butch, J. R. Calhoun, T. Szyperski, E. I. Solomon and W. F. DeGrado, Nat. Chem., 2012, 4, 900–906 CrossRef CAS PubMed.
- S. Kim, P. A. Thiessen, E. E. Bolton, J. Chen, G. Fu, A. Gindulyte, L. Han, J. He, S. He, B. A. Shoemaker, J. Wang, B. Yu, J. Zhang and S. H. Bryant, Nucleic Acids Res., 2016, 44, D1202–D1213 CrossRef CAS PubMed.
- Y. Gao, S. Yang, W. Xiao, J. Nie and X.-Q. Hu, Chem. Commun., 2020, 56, 13719–13730 RSC.
- M. Lohrie and W. Knoche, J. Am. Chem. Soc., 1993, 115, 919–924 CrossRef CAS.
- L. Chen, B. Liu, J.-J. Deng, J.-S. Zhang, W. Li, A. Ahmed, S. Yin and G.-H. Tang, RSC Adv., 2018, 8, 17898–17904 RSC.
- H. Tan, C. Zheng, Z. Liu and D. Z. Wang, Org. Lett., 2011, 13, 2192–2195 CrossRef CAS PubMed.
- S.-H. Gao, Y.-X. Jia, X.-Z. Zhao and Y.-Q. Tu, Chin. J. Chem., 2006, 24, 595–597 CrossRef CAS.
- S. Das, S. Chandrasekhar, J. S. Yadav and R. Grée, Chem. Rev., 2007, 107, 3286–3337 CrossRef CAS PubMed.
- T. Das, P. Saha and V. K. Singh, Org. Lett., 2015, 17, 5088–5091 CrossRef CAS PubMed.
- H. Pellissier, Chem. Rev., 2016, 116, 14868–14917 CrossRef CAS PubMed.
- K. De Bruycker, S. Billiet, H. A. Houck, S. Chattopadhyay, J. M. Winne and F. E. Du Prez, Chem. Rev., 2016, 116, 3919–3974 CrossRef CAS PubMed.
- D. B. Rubinov, I. L. Rubinova and A. A. Akhrem, Chem. Rev., 1999, 99, 1047–1066 CrossRef CAS PubMed.
- J. González-Sabín, F. Rebolledo and V. Gotor, Chem. Soc. Rev., 2009, 38, 1916–1925 RSC.
- A. Jeppesen, B. E. Nielsen, D. Larsen, O. M. Akselsen, T. I. Sølling, T. Brock-Nannestad and M. Pittelkow, Org. Biomol. Chem., 2017, 15, 2784–2790 RSC.
- V. E. Zwicker, K. K. Y. Yuen, D. G. Smith, J. Ho, L. Qin, P. Turner and K. A. Jolliffe, Chem.–Eur. J., 2018, 24, 1140–1150 CrossRef CAS PubMed.
- H. Junek, G. Zuschnig, R. Thierrichter, G. Gfrerer and H. Sterk, Monatsh. Chem., 1982, 113, 1045–1058 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Ze-Hua
Tian
,
ChunHua
Ma
*,
Ze-Hua
Tian
,
ChunHua
Ma

![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :
: