
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent developments for intermolecular enantioselective amination of non-acidic C(sp3)–H bonds
Heng-Hui
Li
 ,
Xuemeng
Chen
and
Søren
Kramer
*
,
Xuemeng
Chen
and
Søren
Kramer
*
Department of Chemistry, Technical University of Denmark, 2800 Kgs. Lyngby, Denmark. E-mail: sokr@kemi.dtu.dk
First published on 8th November 2023
Abstract
Enantioenriched chiral amines are of exceptional importance in the pharmaceutical industry. Recently, several new methods for the installation of these functional groups directly from non-acidic C(sp3)–H bonds by catalytic intermolecular enantioselective amination have been reported. These methods represent significant advances of the field and most of them display high levels of enantioselectivity, utilize the C(sp3)–H substrate as the limiting reagent, feature good functional group tolerance, and show compatibility with late-stage C(sp3)–H amination of advanced substrates. This perspective provides an overview of the recent developments in this rapidly advancing field and outlines possibilities and limitations, which will help identify unsolved challenges and guide future research efforts.
1. Introduction
Chiral amines are frequent substructures in natural products, agrochemicals, and pharmaceuticals (Scheme 1).1 Easy access to enantioenriched chiral amines is pivotal for the pharmaceutical industry due to the privileged nature of this motif which plays an important role for bioactivity and pharmacokinetics. Many different strategies for enantioselective C–N bond formation have been developed such as reductive amination of ketones,2 allylic and propargylic substitution,3 amination of alkyl halides,4 hydroamination of alkenes,5 and carbene insertion to N–H bonds.6 These methods represent remarkable advances for accessing enantioenriched chiral amines.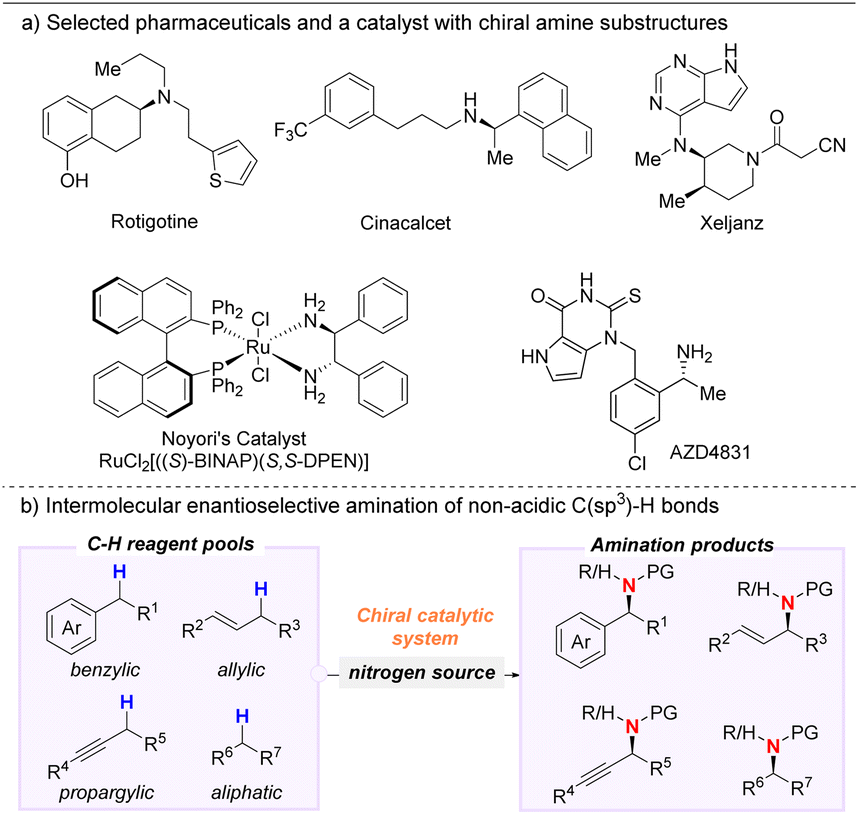 | ||
| Scheme 1 Perspective context and overview. (a) Selected examples of pharmaceuticals and a catalyst containing chiral amine motifs. (b) Overview of reaction types covered in this perspective. | ||
As a complementary strategy, C–H functionalization relies on the presence of an activated C–H bond rather than the displacement of a traditional functional group. This could be advantageous in scenarios where the functional group to be displaced is incompatible with the shortest synthesis route to a target molecule, or if late-stage diversification of advanced drug candidates is desired but they do not contain the necessary functional group, or if the goal is late-stage diversification by installation of different functionalities but the installation of each different functionality requires displacement of different functional groups. In such cases, site-selective C–H functionalization can offer a complementary strategy potentially solving these issues and thereby shorten synthesis routes to target molecules.
Over the last two decades, the field of C–H functionalization has witnessed tremendous advances.7 The ability to selectively transform relatively inert bonds, C–H bonds, into a diverse range of functional groups opens new avenues for the construction of molecules as well as new opportunities in drug development.8 Especially, late-stage introduction of small functional groups is highly important in a medicinal setting.8b In this perspective, we will focus on reactions transforming non-acidic C(sp3)–H bonds into enantioenriched amines by C–N bond formation. In this perspective, we define non-acidic C(sp3)–H bonds as C(sp3)–H bonds that are not readily functionalized by enolate/enamine chemistry, such as electrophilic α-amination of aldehydes, ketones, 1,3-ketoesters, etc., by well-established methods involving enantioselective organocatalysis or metal enolate methodologies.9
The majority of early work on enantioselective amination of non-acidic C(sp3)–H dealt with intramolecular amination reactions;10 however, seminal work on intermolecular enantioselective C(sp3)–H amination still dates back more than 20 years11 with continued development since then.12,13 Notably, during the last five years, the field of catalytic enantioselective intermolecular amination of non-acidic C(sp3)–H bonds has witnessed rapid advances. These recent developments will be covered in this perspective.
Intramolecular C(sp3)–H amination methods can lead to valuable changes in site- and chemo-selectivity compared to intermolecular variants.12–14 Nonetheless, intermolecular reactions often place less restrictions on the substrates than intramolecular reactions and thus have more potential for generality and late-stage functionalization. Hence, we will focus on intermolecular enantioselective C(sp3)–H amination reactions rather than intramolecular reactions.
We will only discuss methods aimed at enantioselective amination with achiral amination reagents rather than methods based on chiral amination reagents for diastereoselective C–N bond formation.15 Furthermore, a few elegant examples of amination by enantioselective desymmetrization have been reported within the timeframe of this perspective,16 but we will focus on reactions where the new C–N bond is installed directly on the stereogenic carbon atom.
The transformations in this perspective are categorized according to the type of non-acidic C(sp3)–H bond being functionalized: benzylic, allylic, propargylic, or aliphatic. Further subdivisions are made based on the metal catalyst. We end the perspective by discussing unsolved challenges as well as future opportunities in the field.
2. Benzylic C(sp3)–H amination
2.1 Rhodium catalysis
One of the most explored strategies for C–H amination relies on metal nitrenes, particularly involving rhodium catalysts.11,13 Recently, further advances with for this strategy were achieved. In 2019, Dauban et al. reported a general methodology for enantioselective intermolecular benzylic C(sp3)–H sulfamation of ethylarenes using the chiral rhodium(II) catalyst, (S)-Rh-1 and pentafluorobenzyl sulfamate, PfbsNH2, as nitrogen source (Scheme 2a).17 The reaction proceeds via a nitrene pathway with PhI(OPiv)2 as the stoichiometric oxidant. Importantly, the method utilizes the C–H substrate as the limiting reagent. Fluorination of various reaction components (PfbsNH2 as nitrene precursor, PhCF3 as solvent, and perfluorinated phthaloyl moiety on the catalyst) led to significant improvement in reaction yield and enantioselectivity. Ultimately, low catalyst loading (0.1 mol%) could be used for the C–H substrate scope, which for the most part provided the desired product in high yields and enantioselectivities. The enantioselectivity is unaffected by the presence of electron-withdrawing or electron-donating substituents on the aryl moiety of the C–H substrate. Nonetheless, the yields tend to be lower for electron-withdrawing groups. While the reaction works very well for ethylarenes, extension of the alkyl chain from ethyl to propyl led to a significant decrease in enantioselectivity. The methodology was successfully applied to late-stage modification of complex molecules, such as an analog of methyl dehydroabietate and derivatives of sulbactam and cinnamic acid. Deprotection to liberate the free chiral primary amines was performed by treatment with pyridine at 75 °C.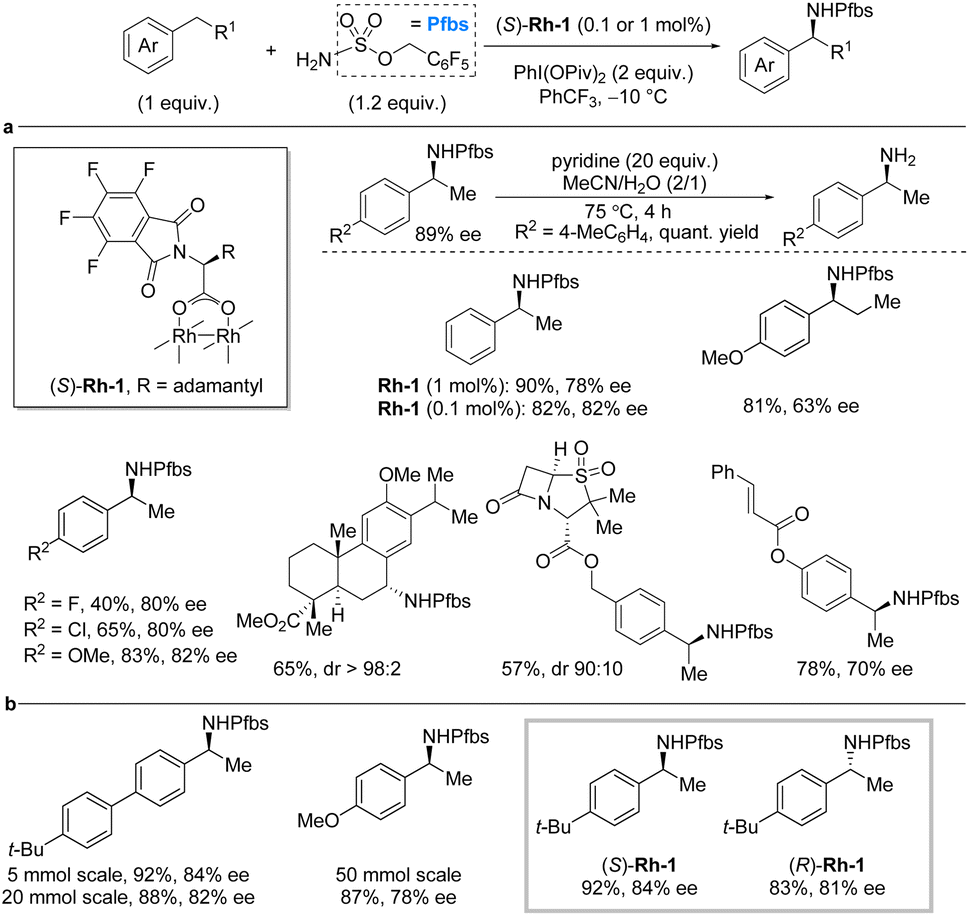 | ||
| Scheme 2 Rhodium-catalyzed enantioselective benzylic sulfamation. (a) Deprotection protocol and selected examples from scope investigation. (b) Scale-up of the developed method. | ||
The following year, Dauban and co-workers investigated potential of the (S)-Rh-1-catalyzed benzylic amination with PfbsNH2 on larger scale (Scheme 2b).18 Under the previously reported reaction conditions and with a catalyst loading of 0.1 mol%, the reaction was tested on 5 mmol, 20 mmol, and 50 mmol scale. It was found that the scale only had a minor influence on the reaction outcome. Finally, it was also demonstrated that the use of (R)-Rh-1 provides access to the R-enantiomer of the chiral benzylic amine in comparable yield and enantiocontrol to (S)-Rh-1 as catalyst.
In 2019, Yoshino, Matsunaga et al. described a directed enantioselective benzylic C(sp3)–H amidation of 8-alkylquinoline under hybrid catalytic system consisting of [Cp*RhCl2]2, two different silver salts, and a chiral carboxylic acid (CCA) with dioxazolones as nitrogen source (Scheme 3).19 The binaphthyl-based CCA, synthesized from BINOL in five steps, was hypothesized to assist in the methylene C–H bond cleavage. While the reaction is limited to 8-alkylquinolines, the functional group tolerance is good, and the yields and enantioselectivities are generally high. The reaction uses the C–H substrate as the limiting reagent. Both electron-withdrawing and -donating groups are tolerated as well as different substituents on the dioxazolones. With minor modifications longer alkyl chains than ethyl were tolerated as demonstrated by both a propyl and a pentyl group. Deprotection of the amine was not demonstrated. H/D exchange experiments indicated that the C–H functionalization step was essentially irreversible.
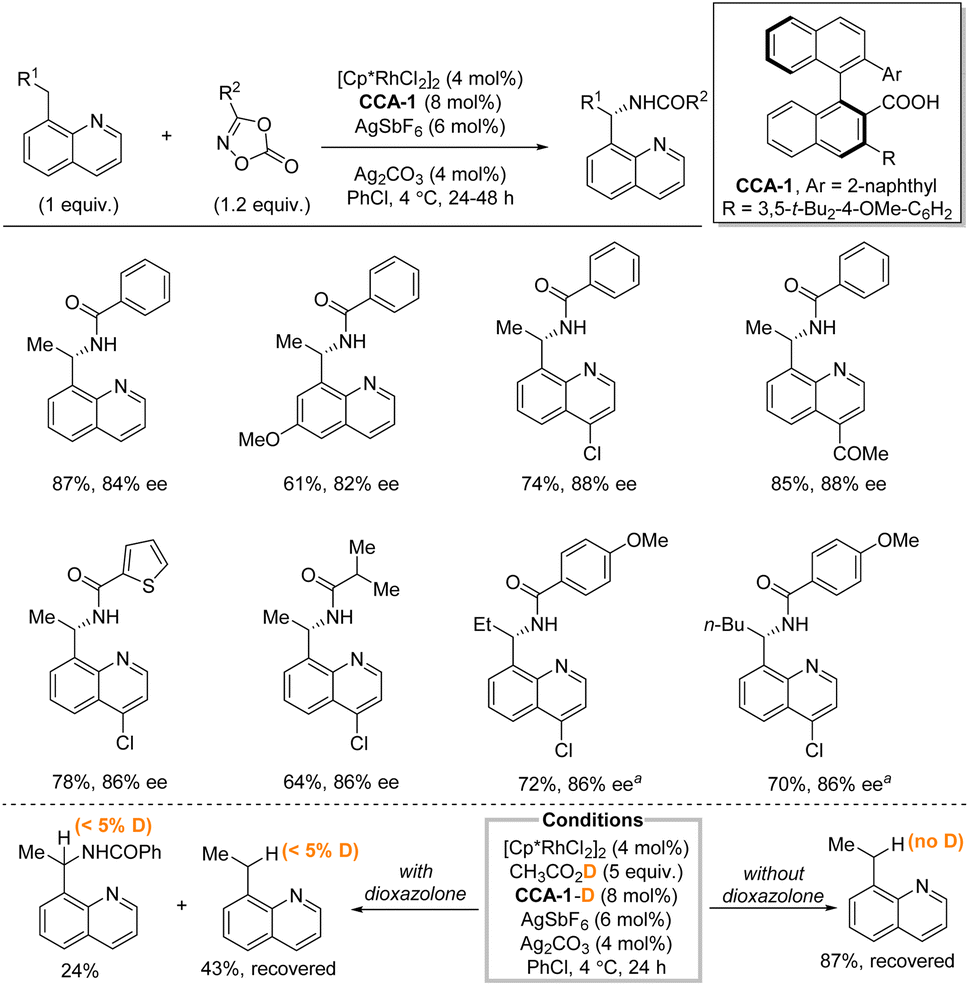 | ||
| Scheme 3 Enantioselective benzylic amidation using a rhodium/chiral carboxylic acid catalytic system. a AgSbF6 (16 mol%), Ag2CO3 (30 mol%), 72 h. | ||
In 2021, Phipps and co-workers reported enantioselective intermolecular C(sp3)–H sulfamation of 4-arylbutanols with sulfamates as nitrogen source, PhIO as oxidant, and a catalytic combination of rhodium-complex Rh-2 and chiral cation L1 (Scheme 4).20 The chiral cation is derived from quaternized cinchona alkaloids. For hydroxyl-bearing substrates, the reaction works well leading to good yields and high enantioselectivities for substrates bearing a range of common functional groups. The methodology utilizes the C–H substrate as the limiting reagent. Deprotection was only demonstrated for a cyclized amination product, which required PhSNa followed by HCl/EtOH, leading to the free amine in 32% yield, 89% ee. The hydroxyl group on the C–H substrate is essential for the successful reaction outcome as demonstrated by comparing reactions with phenylbutane and 4-phenylbutanol as C–H substrates. While 4-phenylbutanol affords excellent yield and enantioselectivity, phenylbutane only gave 3% yield and 28% ee. With Rh2(esp)2 as catalyst, both substrates gave poor yields in racemic form. The authors suggested that the hydroxyl group potentially interacts with the methylenesulfonate groups on the rhodium complex through a hydrogen bonding network. The linker length between the phenyl and hydroxyl moieties also influences the reaction outcome; however, by fine-tuning the catalyst, substrates with different linker-lengths can be aminated in 76–78% ee.
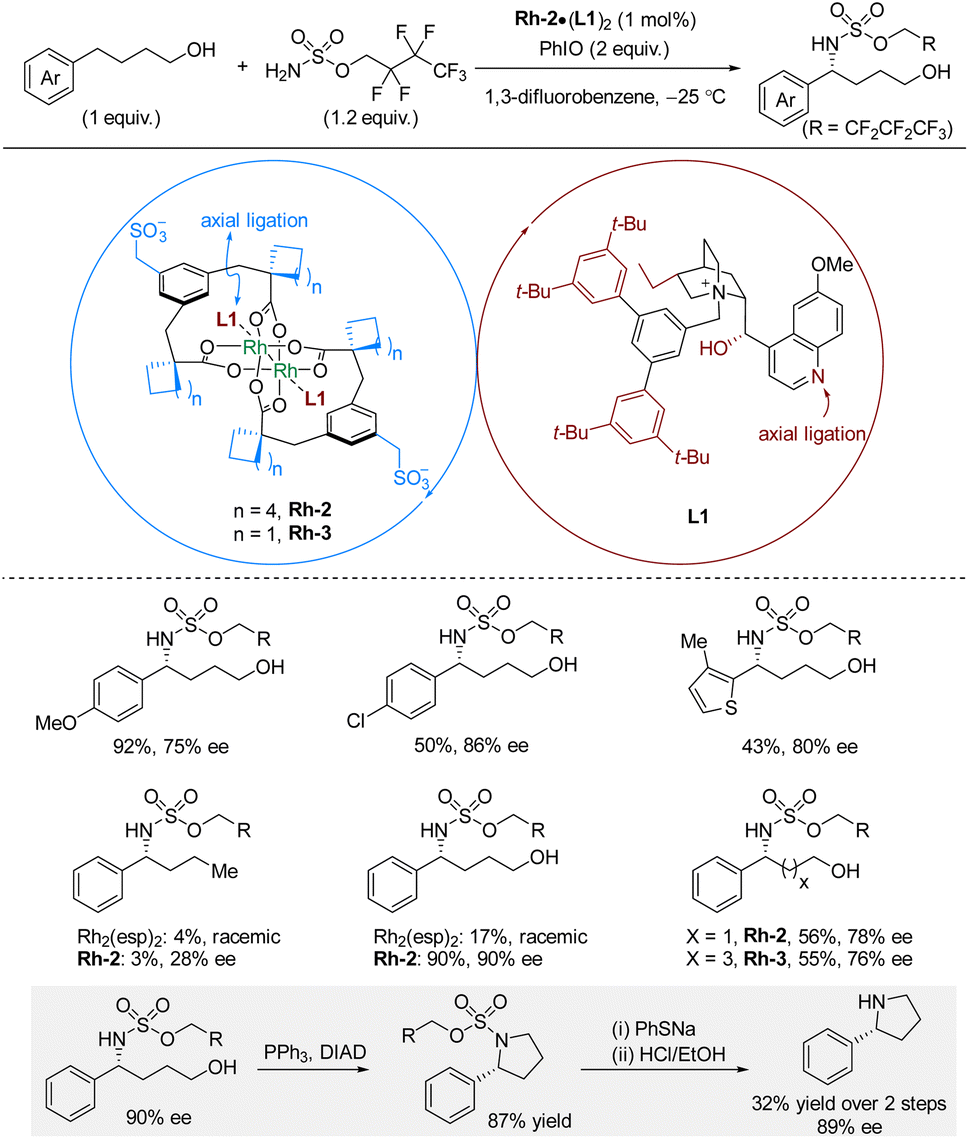 | ||
| Scheme 4 Rhodium-catalyzed enantioselective benzylic C–H sulfamation of alcohol-containing substrates. Esp = α,α,α′,α′-tetramethyl-1,3-benzenedipropanoate. | ||
In 2023, Miller and co-workers developed a peptide-based dirhodium(II) catalytic system that enabled enantioselective benzylic C(sp3)–H sulfamation using 2,2,2-trifluoroethoxylsulfonamide (TfesNH2) as nitrene precursor in the presence of PhI(OPiv)2 as oxidant (Scheme 5).21 This class of dirhodium(II) carboxylate system, based on aspartyl β-turn-biased tetrapeptides, was modified through the peptidyl ligand sequence to achieve high enantioselectivities. The crystal structure of the optimized catalyst unveiled the β-turn conformation of the peptidyl ligand and a well-defined hydrogen-bonding framework surrounding the dirhodium core. This amination reaction works well for a range of substrates containing different functional groups, and it typically affords high yields and enantioselectivities. For the most part, the methodology uses the C–H substrate as limiting reagent; however, in challenging cases, such as a para-CF3 and a primary alkyl bromide, the C–H substrate is used in excess (2 equiv.). Notably, substrates bearing longer alkyl chains, including functionalized ones, underwent the transformation smoothly. Interestingly, in contrast to the report by Phipps et al., the use of 4-phenylbutanol did not lead to any reaction. Deprotection of an isolated amination product was achieved by simply heating it in H2O/1,4-dioxane which, after benzoylation, afforded the benzoylated amine in 38% yield (90% ee). Finally, it was discovered that the catalyst aminates itself, but this self-amination phenomenon was not deleterious to the reaction outcome under the standard reaction conditions.
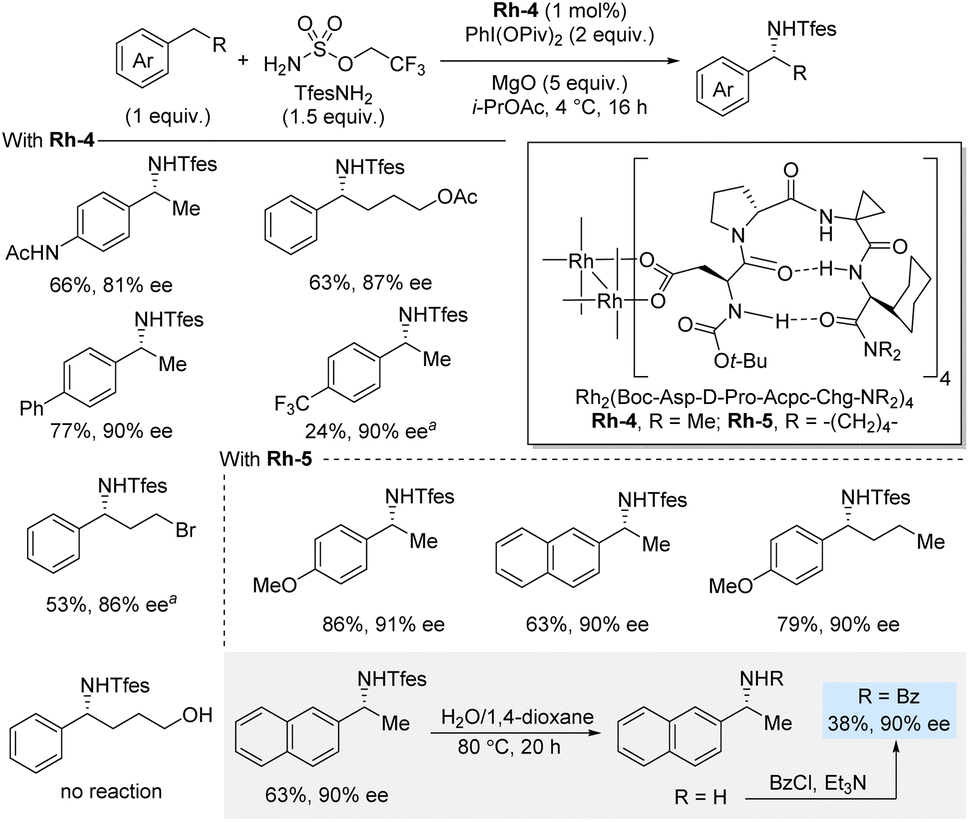 | ||
| Scheme 5 Enantioselective benzylic sulfamation catalyzed by a rhodium complex with chiral peptide-based ligands. a C–H substrate (2 equiv.). | ||
2.2 Iron/enzyme catalysis
For many years, C(sp3)–H amination using nitrene chemistry relied on the use of precious metal catalysts (vide supra).13 However, in 2017, Arnold et al. reported an enantioselective intermolecular benzylic C(sp3)–H sulfonamidation using an engineered iron-haem enzyme catalyst (Scheme 6).22 Specifically, directed evolution led to an efficient iron-enzyme catalyst (cytochrome P411CHA) producing the sulfonamidation products in excellent enantioselectivities and good turnover numbers (TON). As for most nitrene reactions, significantly better yields were observed for substrates bearing electron-donating groups compared to substrates bearing electron-withdrawing groups. Some cyclic alkyl substituents were well-tolerated affording products in up to 80% yield and 96% ee. In contrast, extension of the linear alkyl chain from the standard ethyl to a propyl group led to a significant drop in TON and yield, even with a p-OMe group. For all substrates, the products were formed in excellent enantioselectivities. Deprotection of the tosyl group was achieved by treatment with SmI2 and pyrrolidine leading to the free amine in 61% yield with no erosion of ee. A kinetic isotope effect (KIE) of kH/kD = 1.6 was observed by absolute rate measurements suggesting that the C–H bond insertion is partly rate-determining.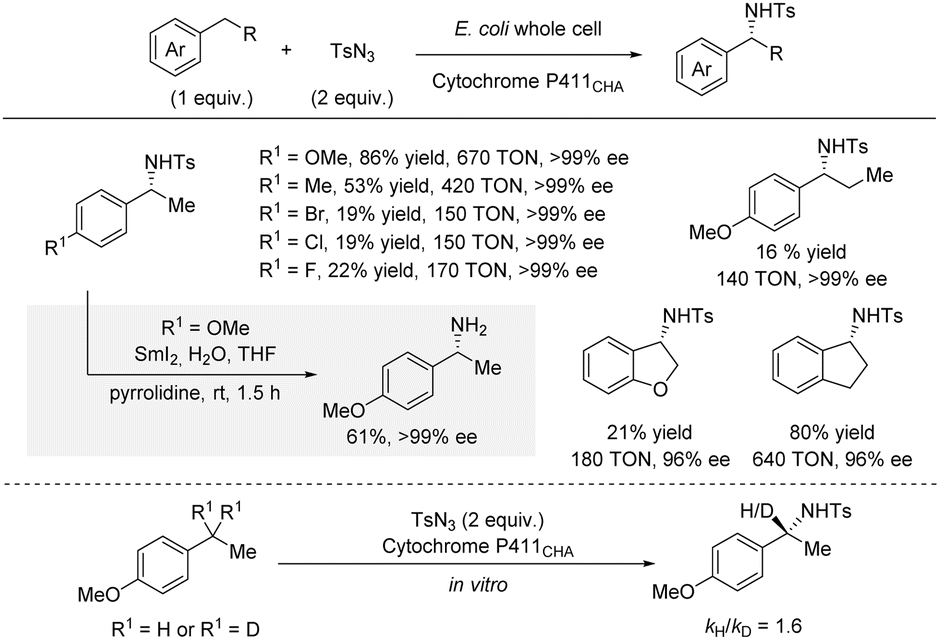 | ||
| Scheme 6 Enantioselective benzylic C–H sulfonamidation catalyzed by an engineered iron-haem enzyme. | ||
As observed throughout this perspective, most methods for intermolecular enantioselective C(sp3)–H amination installs the amine in protected form. Remarkably, in 2020, Arnold and co-workers reported a biocatalytic system which directly affords the free primary amines without any need for deprotection (Scheme 7).23 Directed evolution led to the iron-enzyme P411BPA which can utilize a readily available hydroxylamine ester as nitrene precursor. The C–H substrate was used as limiting reagent. This method works particularly well for arenes with cyclic alkyl substituents, affording up to 85% yield and 93% ee. For ethylarenes, the yields were more moderate with ethylbenzene providing 30% yield; nonetheless, the enantioselectivity and total turnover number (TTN) were still high. No examples of longer linear alkyl substituents than ethyl were reported.
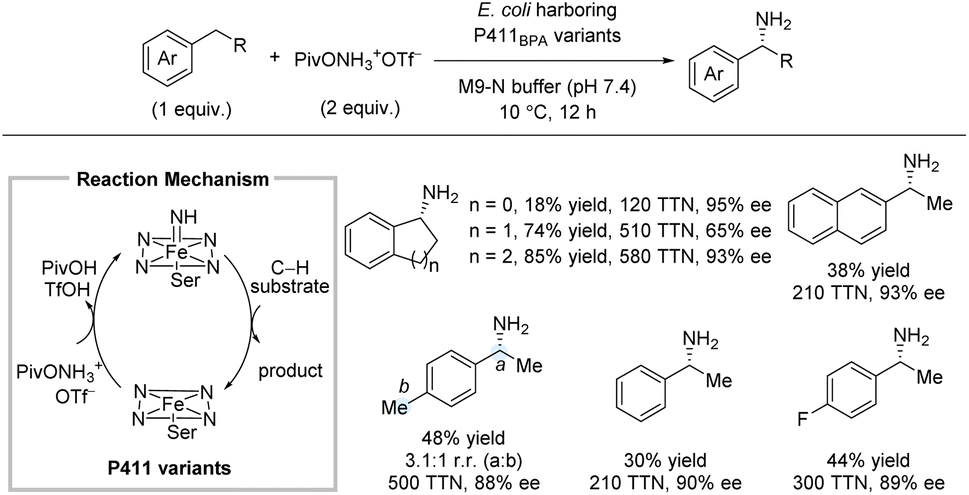 | ||
| Scheme 7 Enzyme-catalyzed enantioselective direct amination of benzylic C–H bonds. | ||
The following year, the same group reported an enzymatic enantioselective benzylic C(sp3)–H amidation using hydroxamate esters as nitrene precursors (Scheme 8).24 Directed evolution led to identification of whole cells containing an iron-enzyme (cytochrome P411) that can serve as a highly efficient catalyst for the amidation reaction. For the C–H substrate scope, high TTN was observed accompanied by excellent enantioselectivities. By using different cell strains both substrates containing electron-donating and electron-withdrawing groups were successfully amidated. Furthermore, both ethyl- and propylarenes could successfully be amidated in excellent enantioselectivities. Most examples evaluated TTN and enantioselectivity, but a few cases of isolated yields were also included which demonstrated that the desired products can be isolated in high yields and excellent enantioselectivities. No example of amine deprotection was demonstrated. To gain insight into the reaction mechanism, a combination of KIE and density functional theory (DFT) calculations was carried out. For substrates with H/D in the benzylic position, independent rate experiments showed kH/kD = 1.06 ± 0.2 while an intermolecular competition experiment showed kH/kD = 8.2 ± 0.4. These results indicate that C–H bond cleavage is not turnover-limiting. In combination with an observed first order dependence on the concentration of the hydroxamate ester, the turnover-limiting step is likely the formation of the metal-nitrenoid.
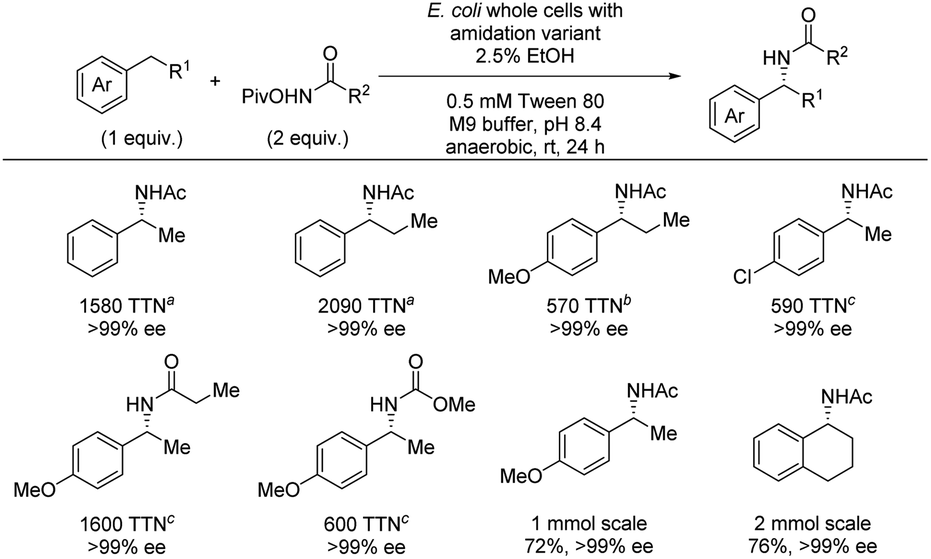 | ||
| Scheme 8 Enantioselective benzylic C–H amidation by enzyme catalysis. Different cell strains were used for different substrate classes: a With i-AMD9. b With iAMD8. c With iAMD8b-A74T. | ||
In 2023, Meggers and co-workers reported carboxylate-directed enantioselective intermolecular C(sp3)–H carbamation (Scheme 9).25 The reaction proceeds via a metal-nitrenoid pathway, and the reaction is selective for carbamation in the α-position of the carboxylate. Most of the included substrates led to carbamate-protected benzylamines in high yields and enantioselectivites. The reaction is not sensitive to the electronic effect of the substituents on the aromatic ring, as both p-MeO and p-CO2Me leads to comparable outcome as the unsubstituted phenyl substrate. The carboxylate is the limiting reagent. Tertiary stereocenters can also be accessed in high yields and enantioselectivities using this protocol. Notably, this protocol is not limited to benzylic carbamation as aliphatic carboxylates are also suitable substrates. However, the carboxylate moiety is essential for the reaction as it directs the C–N bond formation to the α-position. A reaction mechanism was proposed based on the experiments and DFT calculations. The reaction is initiated by formation of an iron-nitrene intermediate which is trapped by the carboxylate forming Fe-2. Intramolecular 1,5-HAT leads to intermediate Fe-3, which can rebound to form the C–N bond. This step is believed to be the enantioselectivity-determining step. Release of the product from Fe-4 closes the catalytic cycle.
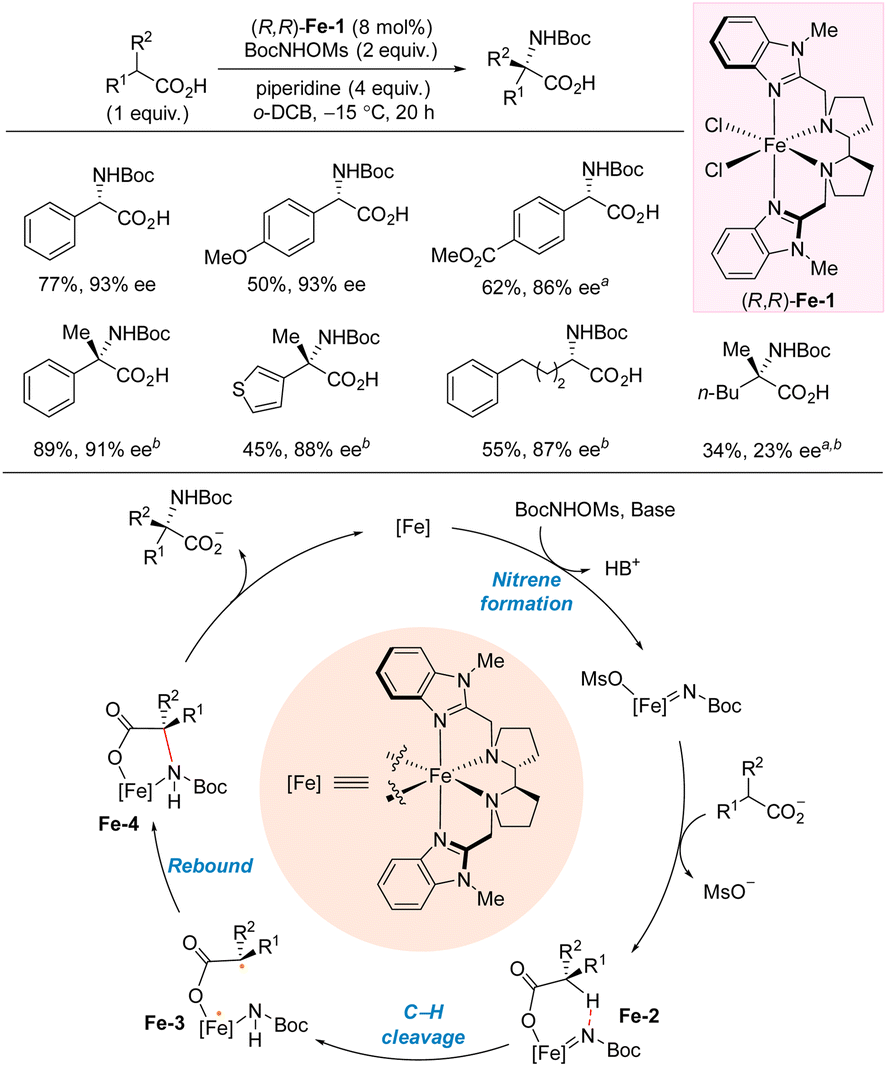 | ||
| Scheme 9 Intermolecular α-C(sp3)–H carbamation of carboxylic acids. a CH2Cl2 as solvent. b (R,R)-Fe-1 (15 mol%), BocNHOMs (5 equiv.), piperidine (7 equiv.) o-DCB = 1,2-dichlorobenzene. | ||
2.3 Silver catalysis
In 2020, Bach and co-workers disclosed silver-catalyzed enantioselective C(sp3)–H sulfonamidation at the allylic position of quinolones and pyridones using iminoiodinane PhI = NNs as nitrene precursor (Scheme 10).26 The heteroleptic silver catalyst is bound to one achiral 1,10-phenanthroline ligand and one chiral phenanthroline ligand bearing a octahydro-1H-4,7-methanoisoindol-1-one moiety which controls the orientation of the substrate by hydrogen bonding and thereby controls the enantioinduction. In general, for the different quinolones and pyridones reported, good yields and high enantioselectivities were obtained. Deprotection of the nosyl group by PhSH and K2CO3 was demonstrated in 81% yield (isolated after boc-protection). A KIE of 5.5 was observed in a competitive experiment; however, measurements of the independent rates revealed a KIE of 1.2 indicating that C–H bond cleavage is not involved in the rate-determining step. Instead, the authors propose that delivery of the nitrene fragment from PhI = NNs to the silver catalyst is rate-determining.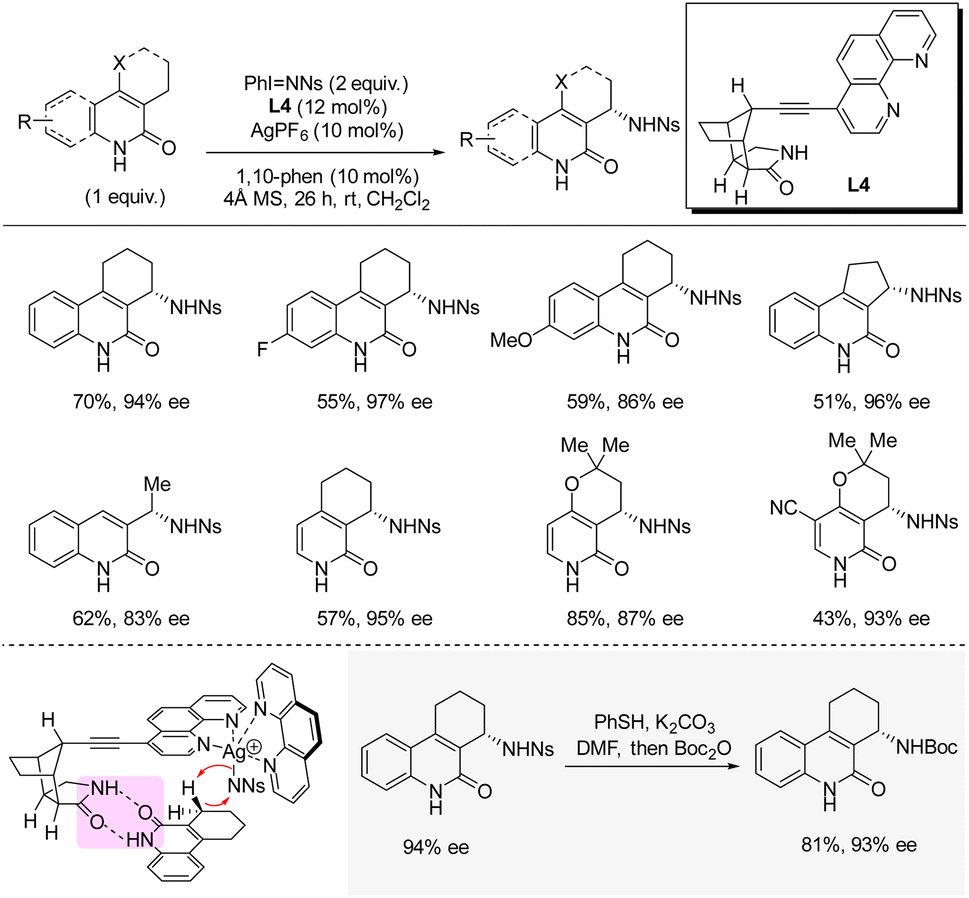 | ||
| Scheme 10 Silver-catalyzed enantioselective C–H sulfonamidation of quinolones and pyridones. | ||
2.4 Cobalt catalysis
In 2020, Zhang and co-workers developed an enantioselective intermolecular C(sp3)–H amination of benzylic esters with perfluorinated aryl azides via Co(II)-based metalloradical catalysis (Scheme 11).27,28 The catalyst was fine-tuned by modifying the remote substituent of the D2-symmetric chiral amidoporphyrin ligand to maximize noncovalent interactions between the catalyst and substrates. This led to catalyst Co-1, which provides chiral amino acid derivatives in up to 97% yield and 99% ee with the C–H substrate as the limiting reagent. To demonstrate the importance of the noncovalent interactions between catalyst and substrate, a comparison of substrates without and with the α-carbonyl group was performed leading to 47% yield, 17% ee and 91% yield, 97% ee, respectively. The developed reaction works well for both electron-withdrawing and -donating substituents on the aromatic ring, albeit the yields are higher with electron-donating groups. Heterobenzylic esters are tolerated, and a single example of a benzylic amide was included. Amination of C–H bonds that are both benzylic and allylic/propargylic is possible, but no examples of non-benzylic functionalization were demonstrated. Mechanistic studies were undertaken and indicated the existence of both an alkyl radical intermediate and a Co(III)-aminyl radical intermediate, which likely combine to form the product via a radical substitution pathway.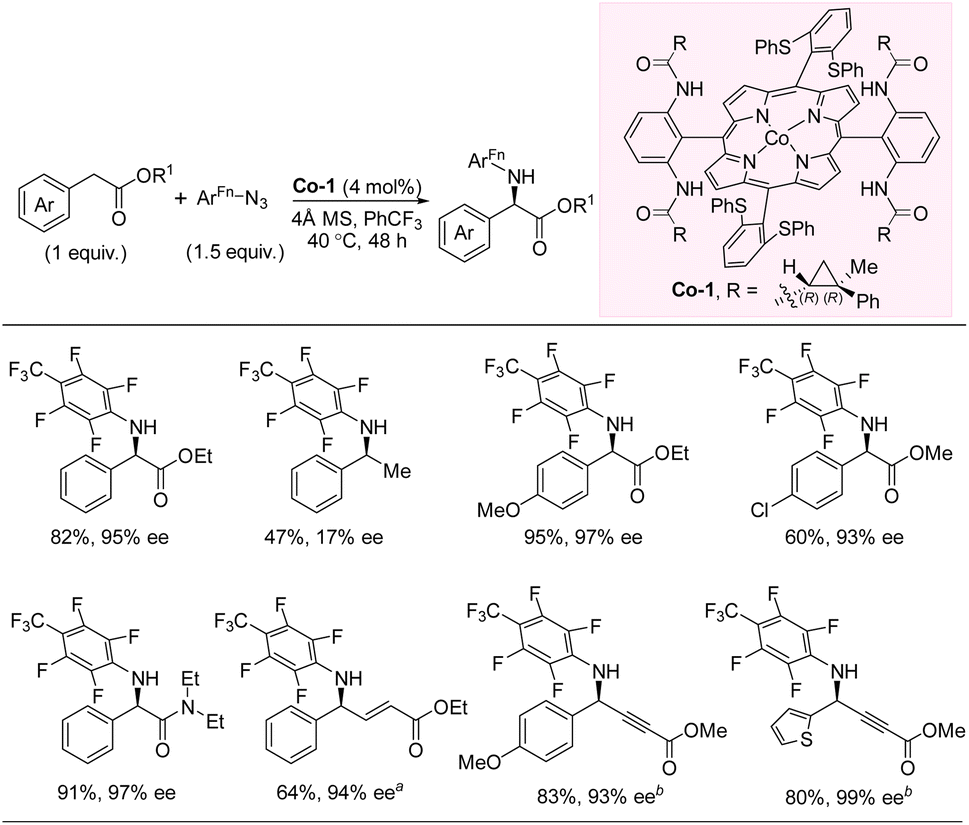 | ||
| Scheme 11 Intermolecular C–H amination with aryl azides via cobalt catalysis. (a) Benzene as solvent. (b) ArFn–N3 (1.2 equiv.). | ||
2.5 Manganese catalysis
In 2022, Liu and co-workers published a highly site- and enantioselective benzylic C(sp3)–H azidation of indolines catalyzed by a chiral manganese complex (Scheme 12).29,30 The authors identified the very bulky Mn(salen) complex Mn-1 as the best catalyst for the azidation using NaN3 as nitrogen source and PhIO as oxidant. The C–H substrate was used as limiting reagent. The azidation afforded high yields and enantioselectivities for a range of 3-substituted indolines, leading to quaternary stereocenters, as well as indolines lacking a 3-substituent. In general, <10% of the competing oxidation was observed. It was demonstrated that hydrogenation over Pd/C liberated the free primary amine in 95% yield without erosion of enantioselectivity (90% ee). A tetrahydroquinoline also underwent the enantioselective azidation albeit with decreased enantioselectivity. The reaction mechanism was studied by a combination of independent rate and competitive KIE experiments, Hammett study, radical probe experiment, and ESI-MS analysis. In combination, the mechanistic evidence supported a reaction pathway involving a benzylic radical pathway. Initially the reaction between the chiral Mn(salen) complex Mn-1 and NaN3 leads to formation of a Mn–N3 species. This species is then oxidized by PhIO generating Mn-2. Then, Mn-2 abstracts a hydrogen atom from the C–H substrate leading to a benzylic radical intermediate, which was supported by the radical probe experiment. Finally, capture of the benzylic radical intermediate by Mn(IV)–N3 affords the azidation product and simultaneously releases Mn(III)–OH which can regenerate the starting Mn(III)–N3 complex by reaction with NaN3.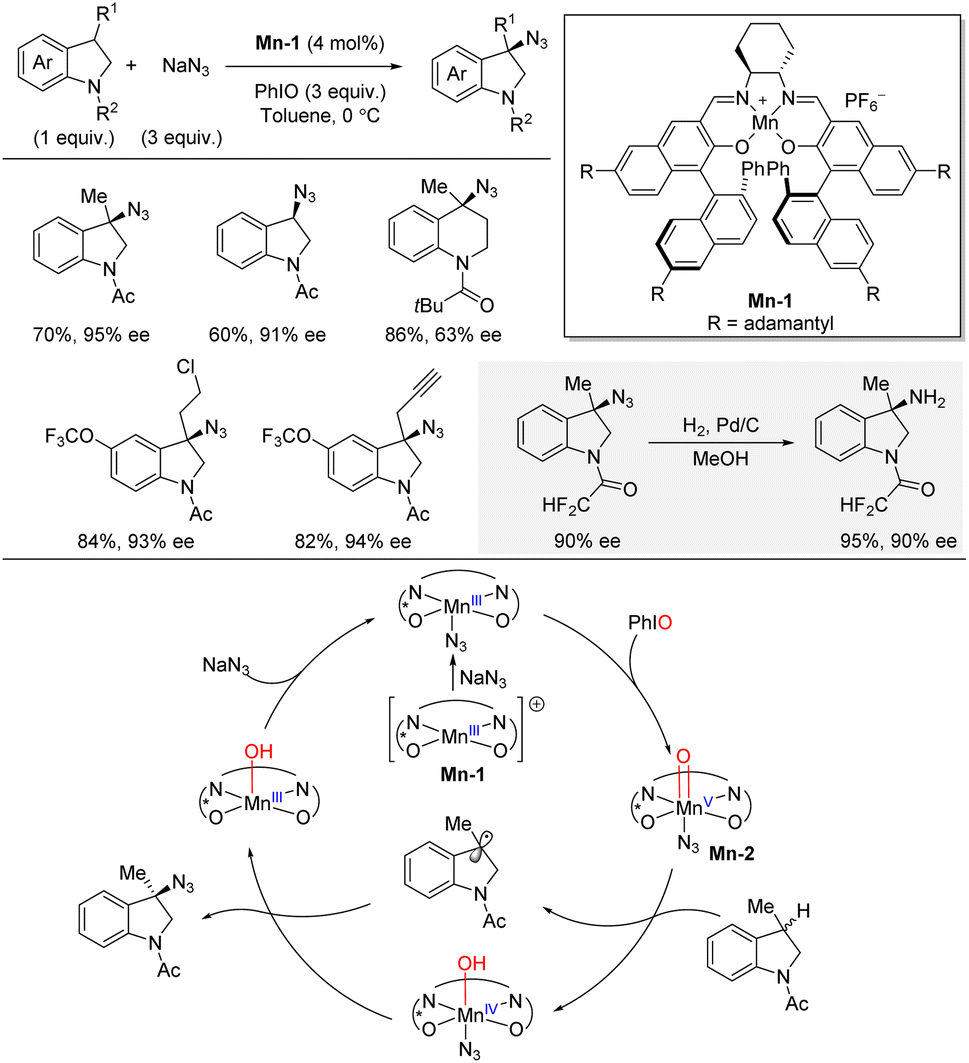 | ||
| Scheme 12 Manganese-catalyzed enantioselective benzylic C–H azidation of indolines. | ||
2.6 Copper catalysis
In 2023, Kramer and co-workers reported enantioselective benzylic C(sp3)–H amidation directly from the unfunctionalized primary amides by using copper catalysis in combination with photocatalysis and di-tert-butyl peroxide (DTBP) as oxidant (Scheme 13).31,32 The C–H substrate is the limiting reagent in this cross-dehydrogenative method which is compatible with a variety of different aromatic and aliphatic amides. For the C–H substrate scope, a broad range of functional groups were tolerated, except for strongly electron-donating and electron-withdrawing groups in the para-position. Notably, the enantioselective amidation in the presence of pyridines, pyrazole, and other heterocycles was also successful. Arenes with longer linear alkyl chains than ethyl were suitable substrates including substrates with alkyl chains bearing functional groups. Furthermore, late-stage amidation of medicinal molecules such as sulbactam and triclosan derivatives was demonstrated. Boc-protected ammonia could also be used as nitrogen precursor albeit in slightly lower yields and enantioselectivities. The use of 15N-labelled amides and carbamate, originating from 15NH4Cl as cheap 15N-source, provided facile access to 15N-labelled amides and primary and secondary amines. Deprotection of an amide was performed with Schwartz's reagent in 72% yield, while Boc-deprotection took place under acidic conditions in 97% yield. Reactions performed in the presence of radical traps as well as a crossover experiment supported a radical pathway rather than a radical-polar crossover mechanism. A KIE of 1.8 was observed by independent rate experiments indicating that the C–H bond cleavage is involved in the rate-determining steps. In this dual copper and photocatalysis method, the excited photocatalyst produces tert-butoxyl radicals from DTBP even at low temperature. The tert-butoxyl radical then abstracts a benzylic hydrogen atom forming a benzylic radical that is trapped by the chiral copper catalyst which installs the C–N bond in an enantioselective fashion.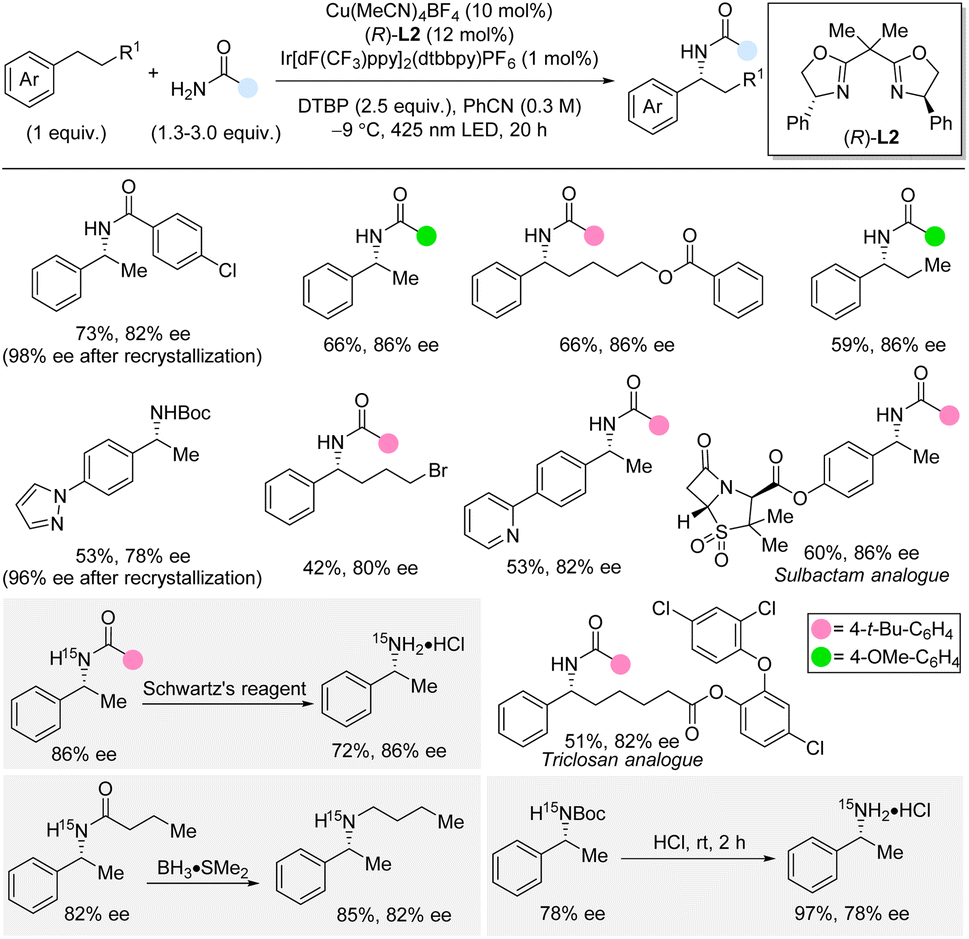 | ||
| Scheme 13 Enantioselective benzylic C–H amidation and carbamation by dual copper and photocatalysis. Schwartz's reagent = Cp2ZrHCl. | ||
Shortly afterwards, Zhou and co-workers reported a related intermolecular enantioselective benzylic C–H amidation by cationic copper catalysis using DTBP as an oxidant under thermal conditions (Scheme 14).32,33 In this method, excess of the C–H substrate (4 equiv.) is required, and the amide is used as limiting reagent. Nonetheless, under these conditions, a simple copper catalyst provides high yields and enantioselectivities for a range of amides and C–H substrates. In general, the method exhibited broad functional group tolerance, except for strongly electron-donating groups in the para-position (strongly electron-withdrawing group in the para-position was not reported). Arenes bearing cyclic or longer linear alkyl chains were suitable substrates. Interestingly, for 1-bromo-2-ethylbenzene, the electronic nature of the amide did not influence the enantioselectivity, while for ethylbenzene a strong influence of the amide was observed in a Hammett plot. Heterocycle compatibility was demonstrated for the amide coupling partner, but not for the C–H substrate, which is used in excess. Deprotection of the amide with Schwartz's reagent went smoothly affording the free primary amine in 94% yield. Mechanistic experiments, including TEMPO-trapping and a radical clock experiment, indicated the involvement of a benzylic radical intermediate. A KIE of 2.1 was obtained from independent rate measurements suggesting that the hydrogen atom abstraction is part of the rate-determining steps. The use of the large non-coordinating counterion BArF was essential for this amidation reaction, and the authors proposed a reaction mechanism involving cationic copper catalysis, where the cationic copper(I) catalyst is oxidized by DTBP, producing a tert-butoxyl radical. After benzylic hydrogen atom abstraction, the generated benzylic radical is trapped by an amide-bound copper(II) intermediate. Subsequent reductive elimination from copper(III) furnishes the new C–N bond.
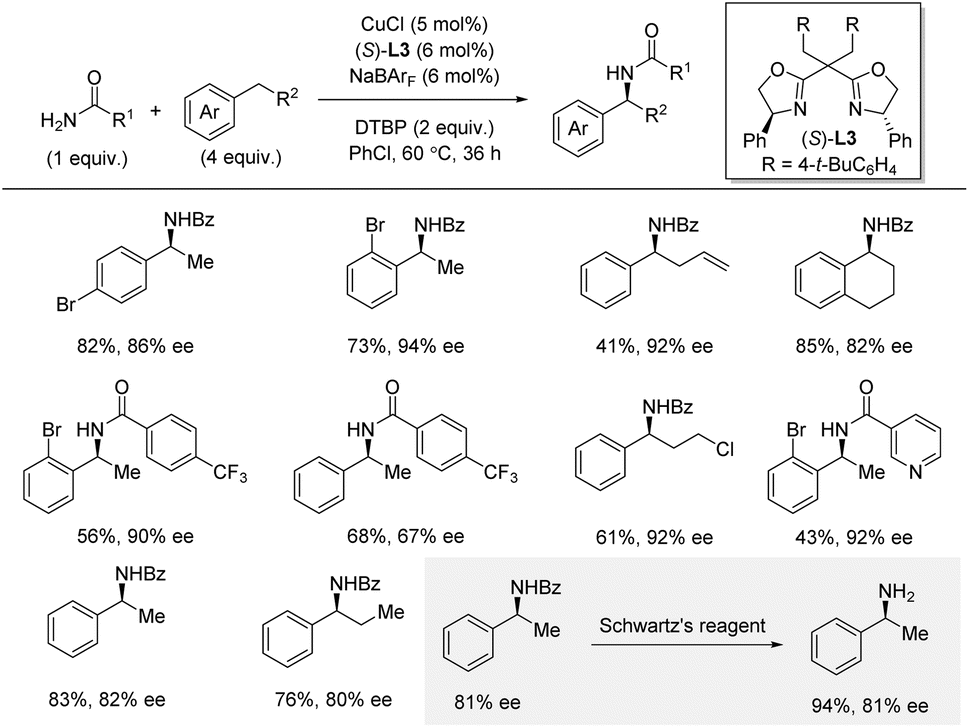 | ||
| Scheme 14 Cationic copper-catalyzed enantioselective benzylic C–H amidation. Schwartz's reagent = Cp2ZrHCl. | ||
3. Allylic C(sp3)–H amination
3.1 Rhodium catalysis
Similar to benzylic C–H amination, the metal nitrene strategy is also the most explored strategies for allylic C–H amination.11–13 In terms of recent advances, Blakey and co-workers disclosed the development of a rhodium-catalyzed enantioselective allylic C(sp3)–H amidation reaction with dioxazolones as nitrene precursors in 2020 (Scheme 15).34 The key to success is the use of a simple planar chiral indenyl ligand on rhodium. The C–H substrate is the limiting reagent in this amidation reaction. A range of different allylic C–H substrate classes can be used including aliphatic terminal alkenes, allylbenzenes, homoallylbenzenes, and styrenes. Internal aliphatic alkenes also worked but in low yields. In general, the enantioselectivities are high for all the included C–H substrates and dioxazolones while the yields span a broad range and are highly dependent on the nature of both the C–H and the dioxazolone. As this study focused on accessing amides, no examples of deprotection of the amide to access the corresponding primary amine were demonstrated. Based on DFT calculations and previous stoichiometric studies, a catalytic cycle was proposed. First, activation of the precatalyst Rh-6 by AgNTf2 and LiOAc forms the coordinatively unsaturated cationic indenyl rhodium complex. After alkene coordination, the C–H bond cleavage takes place in the rate- and enantio-determining step, as indicated by DFT calculations. Then, the dioxazolone coordinates and forms the nitrenoid complex by CO2 expulsion. The ensuing regioselectivity-determining C–N bond formation occurs at the site of the weakest rhodium–carbon bond.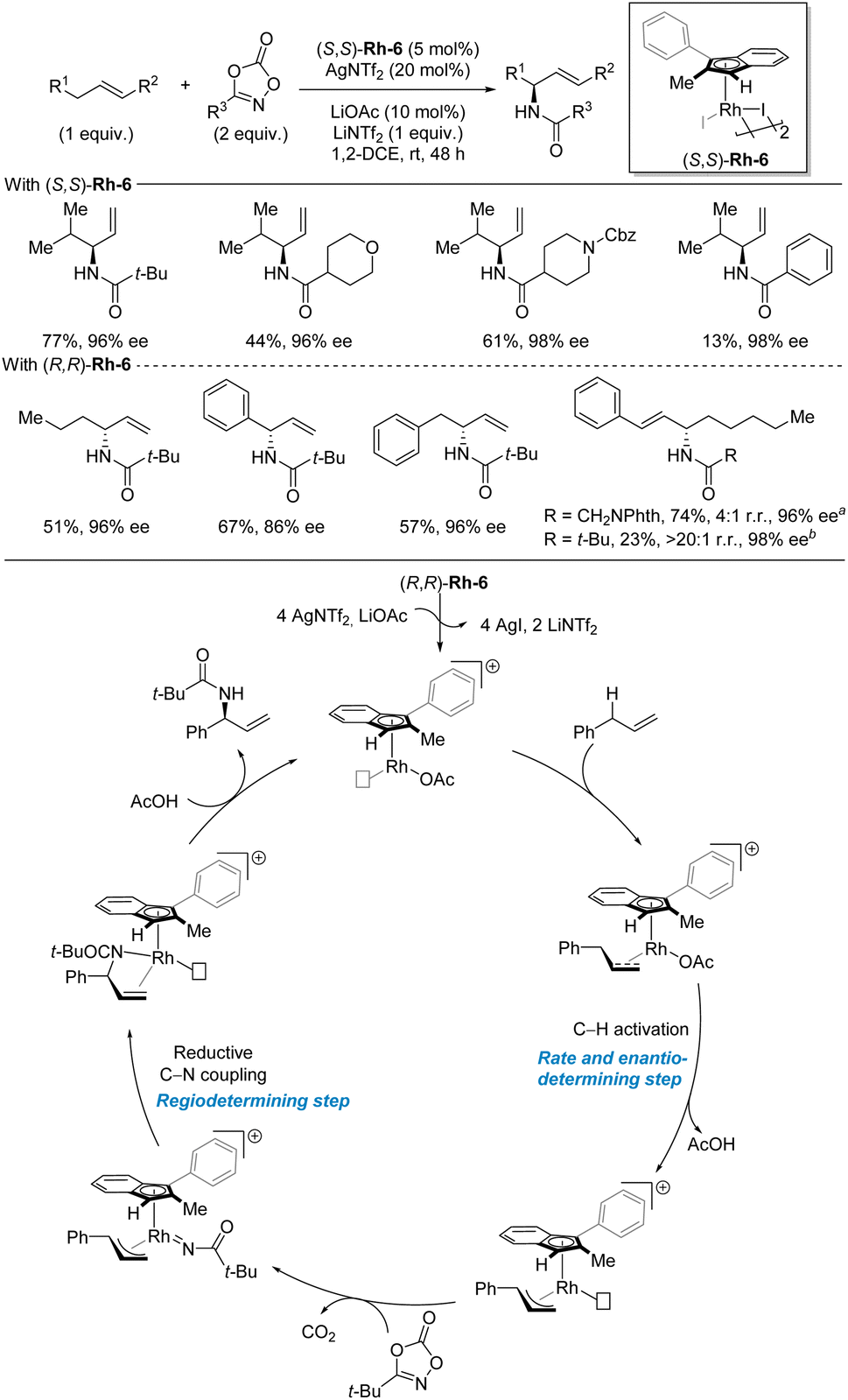 | ||
| Scheme 15 Rhodium-catalyzed enantioselective allylic C–H amidation. a Reaction temperature 60 °C. b Reaction temperature 40 °C. | ||
3.2 Palladium catalysis
In 2022, Gevorgyan and co-workers reported a palladium-catalyzed intermolecular allylic C(sp3)–H amination directly with primary and secondary amines under visible-light irradiation (Scheme 16).35 This protocol use an aryl bromide as an unusual HAT reagent and oxidant. In contrast to the classical Pd(0/II) catalytic mode, this strategy proceeds via a Pd(0/I/II) catalytic cycle. The light-induced bromide abstraction by palladium(0) leads to palladium(I) and an aryl radical. This aryl radical then undertakes HAT from the allylic C(sp3)–H. Subsequently, the allyl radical is trapped by the catalyst forming a palladium(II) intermediate, which after reductive elimination of the C–N bond regenerates palladium(0). The sterical hindrance from the ortho-substituents on the aryl bromide prevents undesired side reactions, including solvent interruption and Heck reaction. For the racemic reaction with Xantphos as ligand, an exceptional scope was demonstrated with a very broad variety of allylic substrates and a vast span of functional groups including numerous heterocycles. For the enantioselective version of the reaction with (R)-BINAP as ligand, a narrower substrate scope was reported. Nonetheless, the use of both primary and secondary amines as well as different allylic substrates afforded the aminated products in good to high enantioselectivities and moderate to high yields. Importantly, the C–H substrate is used as limiting reagent.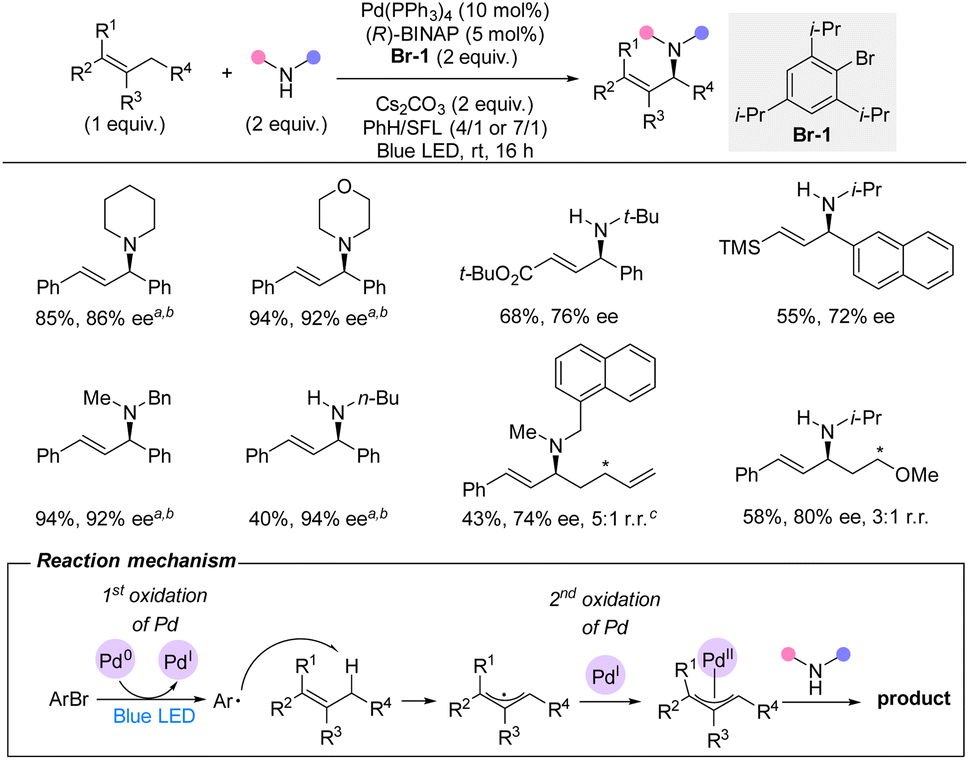 | ||
| Scheme 16 Palladium-catalyzed enantioselective allylic C–H direct amination with aliphatic amines. * Regioisomeric site. a Pd(OAc)2/PPh3 instead of Pd(PPh3)4. b TBAB (1.0 equiv.). c Alkene (2.5 equiv.). SFL = sulfolane. | ||
The methodologies in Sections 3.1 and 3.2 represent substantial advances for enantioselective allylic C(sp3)–H amination using precious metal catalysts. Nonetheless, considering the efficiency and low catalyst loadings used for allylic amination by substitution via allyl-metal intermediates, there is still a need for improving the efficiency and decreasing catalyst loadings.36
3.3 Iron/enzyme catalysis
Based on the findings for enantioselective benzylic amination catalyzed by the iron-enzyme P411BPA (Section 2.2), further directed evolution also led to identification of the iron-enzyme P411APA as an efficient catalyst for allylic C(sp3)–H amination with a hydroxylamine ester as nitrogen source (Scheme 17).23 Using the C–H substrate as limiting reagent, a range of styrenes directly produced the allylic amines in good yields and high enantioselectivities. A substrate with an ethyl group in the allylic position, instead of a methyl group, was shown to lower the yield, but the enantioselectivity was unaffected. The same trend was observed for an electron-donating group. In addition to styrene substrates, 2-hexene also afforded the chiral allylic amine albeit in more moderate enantioselectivity.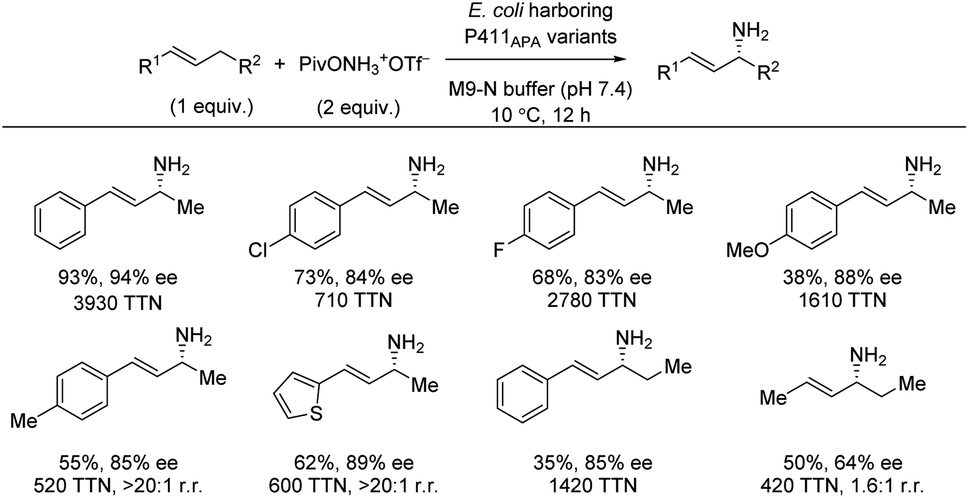 | ||
| Scheme 17 Enzyme-catalyzed enantioselective direct amination of allylic C–H bonds. | ||
3.4 Cobalt catalysis
In 2023, Zhang and co-workers reported cobalt-catalyzed intermolecular allylic amination using perfluoroaryl azide as amination reagent (Scheme 18).28,37 The aniline products were obtained in high yields, regioselectivities, E/Z-selectivities, and enantioselectivities. Notably, a mixture of regio- and stereoisomers were used for the allylic substrates which all converge into the same products. The reaction requires the presence of an ester/carbonyl on the allylic position of the products, and in most cases, the aniline products are both allylic and benzylic. Nonetheless, successful examples of non-benzylic, allylic amination were also demonstrated. Access to an allylic primary amine was achieved through oxidation and in situ acidic hydrolysis of the p-quinonimide intermediate. Combined experimental and computational studies indicated very rapid trapping of an allyl radical intermediate by a Co(III)-amido complex in a regio- and enantioselectivity-determining radical substitution step.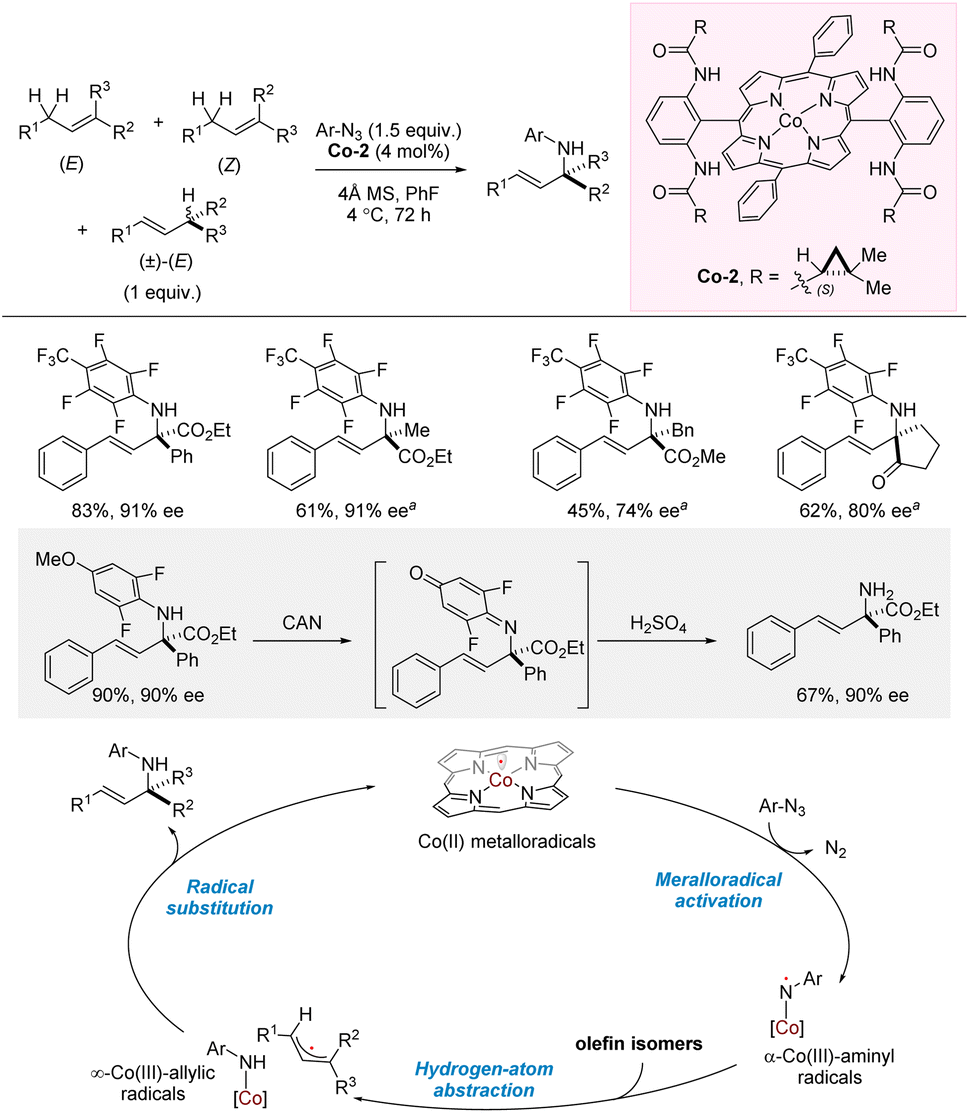 | ||
| Scheme 18 Cobalt-catalyzed allylic C–H amination from isomeric mixture of alkenes. aCo-1 (2 mol%), hexane, 40 °C, 72 h. CAN = NH4Ce(NO3)6. | ||
4. Propargylic C(sp3)–H amination
4.1 Iron/enzyme catalysis
In 2022, Arnold et al. demonstrated that enantioenriched propargylic primary amines can be accessed directly by biocatalyzed amination of propargylic C(sp3)–H bonds (Scheme 19).38 The catalyst is an iron-porphyrin P411 enzyme developed by directed evolution, and a simple hydroxylamine ester serves as nitrene precursor. The C–H substrate is used as limiting reagent. For the standard substrate, but-1-yn-1yl-benzene, high yield and enantioselectivity was obtained. Extension of the propargylic alkyl substituent from methyl to ethyl to propyl was accompanied by decreasing yields (79%, 40%, and 6% yield, respectively) but slightly increasing enantioselectivities. The introduction of different substituents on the aryl moiety of the C–H substrates tended to lower the yield; however, the C–H substrates tended to lower the yield; however, the enantioselectivity only varied a little. Notably, pyridine- and thiophene-containing substrates were successful. In general, high TTNs were obtained. DFT calculations showed that the nitrene insertion is a stepwise process where initial HAT leads to a propargylic radical intermediate which is rapidly trapped in an ensuing radical rebound step forming the C–N bond. The low energy barrier for the rebound process (TS2) suggests that the C–N bond formation step can outcompete radical reorientation and alkyl group rotation in the active site. Hence, the initial orientation of the propargylic radical determines the enantioselectivity of this amination process.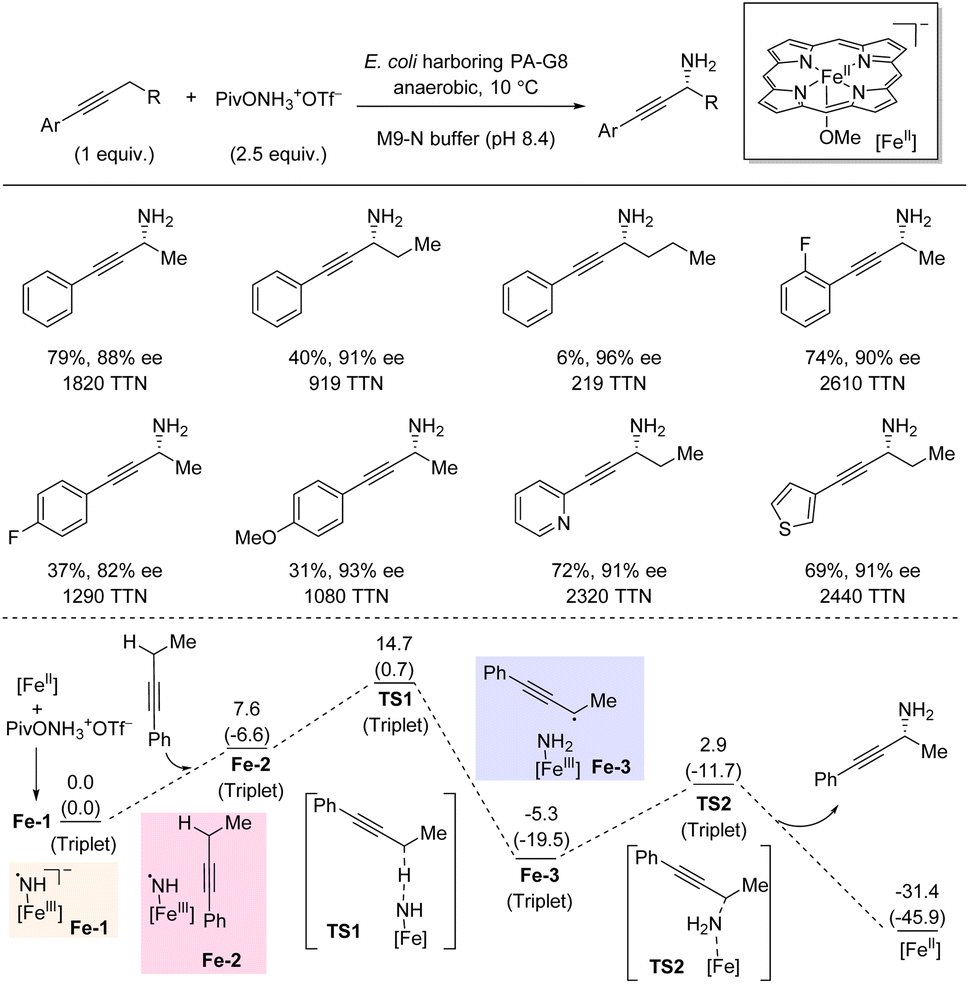 | ||
| Scheme 19 Enantioselective direct propargylic amination by enzyme catalysis. | ||
5. Aliphatic C(sp3)–H amination
5.1 Iron/enzyme catalysis
In 2022, Arnold and co-workers reported examples of enantioselective amination and amidation of aliphatic C(sp3)–H bonds by enzyme catalysis (Scheme 20).39 A hydroxylamine ester was used as nitrene precursor for amination, and the analogous hydroxamate ester was used for amidation. Directed evolution of an iron-containing P450 nitrene transferase led to an enzyme variant capable of amination and amidation of methyl- and ethyl-cyclohexane with excellent enantioselectivity and site-selectivity. For acyclic alkanes, amidation also proved possible albeit with moderate to low enantioselectivity. Although this method is far from universal, the transformation is exceptionally challenging due to the near-identical C–H bonds in the substrate. DFT calculations and KIE experiments suggested a stepwise radical pathway similar to the one described for propargylic amination (Section 4.1).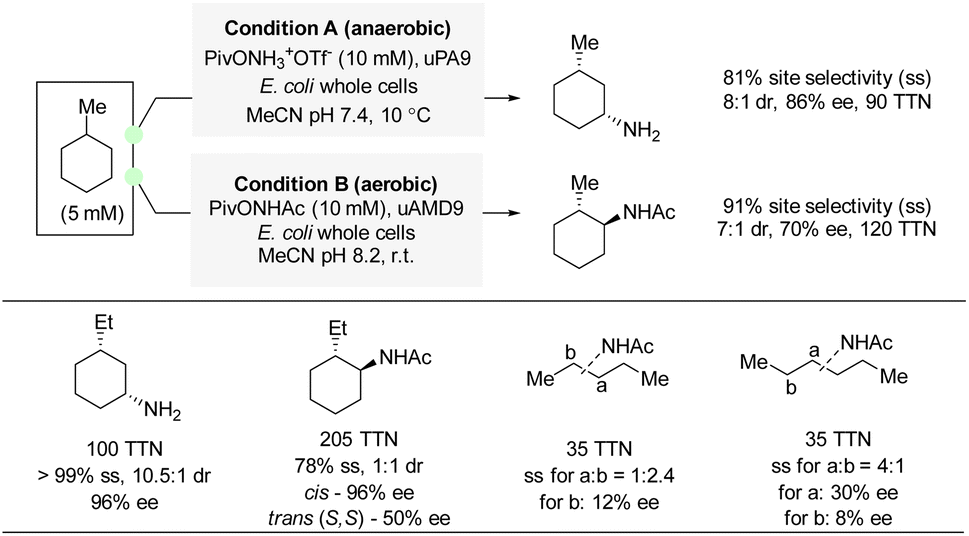 | ||
| Scheme 20 Enantioselective amination and amidation of aliphatic substrates by enzyme catalysis. | ||
Meggers et al. also included examples of aliphatic substrates in their α-carboxylate C(sp3)–H carbamation, albeit the presence of a carboxylate directing group is required (see Scheme 9).25
6. Conclusion and outlook
In this perspective, we have provided an overview of the last five years' developments for intermolecular enantioselective amination of non-acidic C(sp3)–H bonds. These methods represent great advances for this type of transformation. For each method, we have discussed advantages and limitations which will aid the reader in identifying both possibilities and restrictions to consider when applying the specific methods. Below, we have outlined some more general issues to address for future research in this field.Several methods have been developed which provide access to a broad range of enantioenriched amines from different types of C(sp3)–H bonds. The vast majority of methods targets fairly weak C(sp3)–H bonds. This feature enables good site-selectivity for complex molecules with many C(sp3)–H bonds, and it allows for easy prediction of the amination site. However, methods for enantioselective amination of stronger aliphatic C(sp3)–H bonds are exceptionally scarce, and this is clearly an important challenge to address in the future. While significant progress for benzylic, allylic, and propargylic C(sp3)–H bonds has been achieved recently, thus offering access to many different enantioenriched amines, each method also has distinct limitations. Accordingly, there is still a need for further development for robust and broadly applicable methods for these substrate classes as well.
The aim of the methods included in this perspective is the introduction of an amine. If the amination is the last step in a synthetic route, for example, for late-stage functionalization, then direct access to the free amine is ideal. However, if subsequent steps are required to access the target molecules, the installation of a protected amine can be advantageous. In such a scenario, it is important that the protecting group can be easily removed. This is indeed the case for many of the methods described above. Nonetheless, future methods should continue to consider the overall amination transformation including the protecting group removal. Access to orthogonal protecting groups will also be useful for applications in organic synthesis.
Finally, most examples presented in this perspective lead to primary amines. A few exceptions include the palladium-catalyzed allylic amination and borane reduction of amidation products. Nonetheless, methods for enantioselective synthesis of secondary and tertiary amines from non-acidic C(sp3)–H bonds are still rare and limited to a few substrate classes.
Although there are still many challenges to address, the recent advances demonstrate that intermolecular enantioselective amination is now possible for a range of different C(sp3)–H substrate classes. The continued development of complementary methods using different strategies (nitrenes, radical formation via HAT, enzymes, etc.) will hopefully ensure that suitable methods for the enantioselective amination of virtually any type of C(sp3)–H bond can be available in the future. We hope the overview provided here will help accelerate the progress toward this goal.
Author contributions
All authors contributed to the conceptualization and literature search. H.-H. Li wrote the first draft and made all schemes. All authors were involved in revising, editing, and proofreading.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are deeply appreciative of financial support from the Independent Research Fund Denmark (grant no. 0171-00018B), the Villum Foundation (grant no. 35812), and the Novo Nordisk Foundation (grant no. NNF21OC0071458).References
- (a) Methodologies in Amine Synthesis: Challenges and Applications, ed. A. Ricci and L. Bernardi, Wiley, 2021 Search PubMed; (b) Chiral Amine Synthesis: Methods, Developments and Applications, ed. T. C. Nugent, Wiley, 2021 Search PubMed.
- Y. Tian, L. Hu, Y.-Z. Wang, X. Zhang and Q. Yin, Org. Chem. Front., 2021, 8, 2328–2342 RSC.
- (a) J. F. Hartwig and L. M. Stanley, Acc. Chem. Res., 2010, 43, 1461–1475 CrossRef CAS; (b) S. L. Rössler, D. A. Petrone and E. M. Carreira, Acc. Chem. Res., 2019, 52, 2657–2672 CrossRef PubMed.
- (a) Q. M. Kainz, C. D. Matier, A. Bartoszewicz, S. L. Zultanski, J. C. Peters and G. C. Fu, Science, 2016, 351, 681–684 CrossRef CAS PubMed; (b) A. Bartoszewicz, C. D. Matier and G. C. Fu, J. Am. Chem. Soc., 2019, 141, 14864–14869 CrossRef CAS PubMed; (c) C. Chen, J. C. Peters and G. C. Fu, Nature, 2021, 596, 250–256 CrossRef CAS; (d) C. Chen and G. C. Fu, Nature, 2023, 618, 301–307 CrossRef CAS PubMed; (e) J.-J. Chen, J.-H. Fang, X.-Y. Du, J.-Y. Zhang, J.-Q. Bian, F.-L. Wang, C. Luan, W.-L. Liu, J.-R. Liu, X.-Y. Dong, Z.-L. Li, Q.-S. Gu, Z. Dong and X.-Y. Liu, Nature, 2023, 618, 294–300 CrossRef CAS PubMed.
- (a) S. Zhu, N. Niljianskul and S. L. Buchwald, J. Am. Chem. Soc., 2013, 135, 15746–15749 CrossRef CAS PubMed; (b) S.-L. Shi, Z. L. Wong and S. L. Buchwald, Nature, 2016, 532, 353–356 CrossRef CAS PubMed; (c) Y. Xi, S. Ma and J. F. Hartwig, Nature, 2020, 588, 254–260 CrossRef CAS PubMed.
- M.-L. Li, J.-H. Yu, Y.-H. Li, S.-F. Zhu and Q.-L. Zhou, Science, 2019, 366, 990–994 CrossRef CAS.
- For reviews, see: (a) H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417–424 CrossRef CAS PubMed; (b) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS; (c) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960–9009 CrossRef CAS; (d) C. Liu, J. Yuan, M. Gao, S. Tang, W. Li, R. Shi and A. Lei, Chem. Rev., 2015, 115, 12138–12204 CrossRef CAS PubMed; (e) J. F. Hartwig, J. Am. Chem. Soc., 2016, 138, 2–24 CrossRef CAS; (f) P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192–2452 CrossRef CAS PubMed.
- (a) T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546–576 RSC; (b) L. Guillemard, N. Kaplaneris, L. Ackermann and M. J. Johansson, Nat. Rev. Chem., 2021, 5, 522–545 CrossRef CAS PubMed.
- (a) J. M. Janey, Angew. Chem., Int. Ed., 2005, 44, 4292–4300 CrossRef CAS; (b) A. M. R. Smith and K. K. Hii, Chem. Rev., 2011, 111, 1637–1656 CrossRef CAS PubMed; (c) F. Zhou, F.-M. Liao, J.-S. Yu and J. Zhou, Synthesis, 2014, 46, 2983–3003 CrossRef CAS.
- For a review on intramolecular C–H amination, see: J. L. Jeffrey and R. Sarpong, Chem. Sci., 2013, 4, 4092–4106 RSC.
- For seminal work on intermolecular enantioselective C(sp3)–H amination, see: (a) X.-G. Zhou, X.-Q. Yu, J.-S. Huang and C.-M. Che, Chem. Commun., 1999, 2377–2378 RSC; (b) Y. Kohmura and T. Katsuki, Tetrahedron Lett., 2001, 42, 3339–3342 CrossRef CAS; (c) M. Yamawaki, H. Tsutsui, S. Kitagaki, M. Anada and S. Hashimoto, Tetrahedron Lett., 2002, 43, 9561–9564 CrossRef CAS; (d) R. P. Reddy and H. M. L. Davies, Org. Lett., 2006, 8, 5013–5016 CrossRef CAS PubMed; (e) Y. Nishioka, T. Uchida and T. Katsuki, Angew. Chem., Int. Ed., 2013, 52, 1739–1742 CrossRef CAS PubMed; (f) T. Höke, E. Herdtweck and T. Bach, Chem. Commun., 2013, 49, 8009–8011 RSC.
- For comprehensive reviews on amine synthesis, see: (a) Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247–9301 CrossRef CAS PubMed; (b) A. Trowbridge, S. M. Walton and M. J. Gaunt, Chem. Rev., 2020, 120, 2613–2692 CrossRef CAS PubMed.
- For reviews on C–H amination involving metal nitrenoids, see: (a) F. Collet, C. Lescot and P. Dauban, Chem. Soc. Rev., 2011, 40, 1926–1936 RSC; (b) D. Hazeland, P.-A. Nocquet and P. Compain, Org. Chem. Front., 2017, 4, 2500–2521 RSC; (c) H. Hayashi and T. Uchida, Eur. J. Org Chem., 2020, 8, 909–916 CrossRef; (d) M. Ju and J. M. Schomaker, Nat. Rev. Chem., 2021, 5, 580–594 CrossRef CAS PubMed.
- For selected examples, see: (a) H. Wang, Y. Park, Z. Bai, S. Chang, G. He and G. Chen, J. Am. Chem. Soc., 2019, 141, 7194–7201 CrossRef CAS PubMed; (b) Y. Park and S. Chang, Nat. Catal., 2019, 2, 219–227 CrossRef CAS; (c) K. M. Nakafuku, Z. Zhang, E. A. Wappes, L. M. Stateman, A. D. Chen and D. A. Nagib, Nat. Chem., 2020, 12, 697–704 CrossRef PubMed; (d) J. Jeong, H. Jung, D. Kim and S. Chang, ACS Catal., 2022, 12, 8127–8138 CrossRef CAS.
- C. Liang, F. Robert-Peillard, C. Fruit, P. Müller, R. H. Dodd and P. Dauban, Angew. Chem., Int. Ed., 2006, 45, 4641–4644 CrossRef CAS PubMed.
- (a) S. Fukagawa, Y. Kato, R. Tanaka, M. Kojima, T. Yoshino and S. Matsunaga, Angew. Chem., Int. Ed., 2019, 58, 1153–1157 CrossRef CAS; (b) B. Liu, P. Xie, J. Zhao, J. Wang, M. Wang, Y. Jiang, J. Chang and X. Li, Angew. Chem., Int. Ed., 2021, 60, 8396–8400 CrossRef CAS PubMed.
- A. Nasrallah, V. Boquet, A. Hecker, P. Retailleau, B. Darses and P. Dauban, Angew. Chem., Int. Ed., 2019, 58, 8192–8196 CrossRef CAS.
- A. Nasrallah, Y. Lazib, V. Boquet, B. Darses and P. Dauban, Org. Process Res. Dev., 2020, 24, 724–728 CrossRef CAS.
- S. Fukagawa, M. Kojima, T. Yoshino and S. Matsunaga, Angew. Chem., Int. Ed., 2019, 58, 18154–18158 CrossRef CAS PubMed.
- A. Fanourakis, B. D. Williams, K. J. Paterson and R. J. Phipps, J. Am. Chem. Soc., 2021, 143, 10070–10076 CrossRef CAS.
- N. van den Heuvel, S. M. Mason, B. Q. Mercado and S. J. Miller, J. Am. Chem. Soc., 2023, 145, 12377–12385 CrossRef CAS.
- C. K. Prier, R. K. Zhang, A. R. Buller, S. Brinkmann-Chen and F. H. Arnold, Nat. Chem., 2017, 9, 629–634 CrossRef CAS PubMed.
- Z.-J. Jia, S. Gao and F. H. Arnold, J. Am. Chem. Soc., 2020, 142, 10279–10283 CrossRef CAS PubMed.
- S. V. Athavale, S. Gao, Z. Liu, S. C. Mallojjala, J. S. Hirschi and F. H. Arnold, Angew. Chem., Int. Ed., 2021, 60, 24864–24869 CrossRef CAS.
- C.-X. Ye, D. R. Dansby, S. Chen and E. Meggers, Nat. Synth., 2023, 2, 645–652 CrossRef.
- R. R. Annapureddy, C. Jandl and T. Bach, J. Am. Chem. Soc., 2020, 142, 7374–7378 CrossRef CAS PubMed.
- L.-M. Jin, P. Xu, J. Xie and X. P. Zhang, J. Am. Chem. Soc., 2020, 142, 20828–20836 CrossRef CAS PubMed.
- For a review on metalloporphyrin-catalyzed C–H amination, see: R. Singh and A. Mukherjee, ACS Catal., 2019, 9, 3604–3617 CrossRef CAS.
- M. Cao, H. Wang, Y. Ma, C.-H. Tung and L. Liu, J. Am. Chem. Soc., 2022, 144, 15383–15390 CrossRef CAS PubMed.
- For a review on radical C(sp3)–H functionalization, see: D. L. Golden, S.-E. Suh and S. S. Stahl, Nat. Rev. Chem., 2022, 6, 405–427 CrossRef CAS PubMed.
- X. Chen, Z. Lian and S. Kramer, Angew. Chem., Int. Ed., 2023, 62, e202217638 CrossRef CAS PubMed.
- For a review on copper-catalyzed radical C(sp3)–H functionalization, see: Z. Zhang, P. Chen and G. Liu, Chem. Soc. Rev., 2022, 51, 1640–1658 RSC.
- L. Dai, Y.-Y. Chen, L.-J. Xiao and Q.-L. Zhou, Angew. Chem., Int. Ed., 2023, 62, e202304427 CrossRef CAS.
- C. M. B. Farr, A. M. Kazerouni, B. Park, C. D. Poff, J. Won, K. R. Sharp, M.-H. Baik and S. B. Blakey, J. Am. Chem. Soc., 2020, 142, 13996–14004 CrossRef CAS PubMed.
- K. P. S. Cheung, J. Fang, K. Mukherjee, A. Mihranyan and V. Gevorgyan, Science, 2022, 378, 1207–1213 CrossRef PubMed.
- (a) U. Kazmaier, Transition Metal Catalyzed Enantioselective Allylic Substitution in Organic Synthesis, Springer Berlin Heidelberg, Berlin, 2012 CrossRef; (b) R. L. Grange, E. A. Clizbe and P. A. Evans, Synthesis, 2016, 48, 2911–2968 CrossRef CAS; (c) O. Pàmies, J. Margalef, S. Cañellas, J. James, P. J. Guiry, C. Moberg, J.-E. Bäckvall, A. Pfaltz, M. A. Pericàs and M. Diéguez, Chem. Rev., 2021, 121, 4373–4505 CrossRef PubMed.
- P. Xu, J. Xie, D.-S. Wang and X. P. Zhang, Nat. Chem., 2023, 15, 498–507 CrossRef CAS.
- Z. Liu, Z.-Y. Qin, L. Zhu, S. V. Athavale, A. Sengupta, Z.-J. Jia, M. Garcia-Borràs, K. N. Houk and F. H. Arnold, J. Am. Chem. Soc., 2022, 144, 80–85 CrossRef CAS PubMed.
- S. V. Athavale, S. Gao, A. Das, S. C. Mallojjala, E. Alfonzo, Y. Long, J. S. Hirschi and F. H. Arnold, J. Am. Chem. Soc., 2022, 144, 19097–19105 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |