
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Regioselective ortho halogenation of N-aryl amides and ureas via oxidative halodeboronation: harnessing boron reactivity for efficient C–halogen bond installation†
Ganesh H.
Shinde‡
 a,
Ganesh S.
Ghotekar‡
a,
Francoise M.
Amombo Noa
b,
Lars
Öhrström
b,
Per-Ola
Norrby
c and
Henrik
Sundén
*ab
a,
Ganesh S.
Ghotekar‡
a,
Francoise M.
Amombo Noa
b,
Lars
Öhrström
b,
Per-Ola
Norrby
c and
Henrik
Sundén
*ab
aDepartment of Chemistry and Molecular Biology, University of Gothenburg, SE-41296 Gothenburg, Sweden. E-mail: henrik.sunden@chem.gu.se
bDepartment of Chemistry and Chemical Engineering, Chalmers University of Technology, SE-41296 Gothenburg, Sweden
cData Science and Modelling, Pharmaceutical Sciences, R&D, AstraZeneca Gothenburg, Pepparedsleden 1, Mölndal SE-43183, Sweden
First published on 23rd October 2023
Abstract
The installation of the C–halogen bond at the ortho position of N-aryl amides and ureas represents a tool to prepare motifs that are ubiquitous in biologically active compounds. To construct such prevalent bonds, most methods require the use of precious metals and a multistep process. Here we report a novel protocol for the long-standing challenge of regioselective ortho halogenation of N-aryl amides and ureas using an oxidative halodeboronation. By harnessing the reactivity of boron over nitrogen, we merge carbonyl-directed borylation with consecutive halodeboronation, enabling the precise introduction of the C–X bond at the desired ortho position of N-aryl amides and ureas. This method offers an efficient, practical, and scalable solution for synthesizing halogenated N-heteroarenes under mild conditions, highlighting the superiority of boron reactivity in directing the regioselectivity of the reaction.
Introduction
The amide functional group is an important structural motif found in a variety of natural products and biologically active molecules. For example, 25% of known drugs exhibit an amide motif in their complex structure and several of these substances contain the N-aryl amide functional group, for example, atorvastatin, aubagio, and prilocaine.1 Thus, transformations that are designed to cause structural modifications in the proximity of the amide functional group are of utmost importance as structural modifications, often drastically change the physiological and biological properties of pharmaceutically active molecules.2 In this regard, regioselective ortho-halogenation of N-aryl amides is of high interest as the halogen serves as one of the most versatile synthetic handles for functional group interconversions.3However, traditional electrophilic aromatic halogenation of N-aryl amides typically results in mixtures of para and ortho/para substitution (Fig. 1Aa).4b,d,e A higher selectivity can be obtained with transition metal-catalyzed C–H functionalization (Fig. 1Ab).4 However, these halogenations are limited by the need for tailored activating groups and inert atmosphere conditions, which can impede synthetic efficiency.5 In addition, these transformations are generally restricted to meta and para-substituted aryl amides due to the formation of di-substituted halogenated side products.4b–f
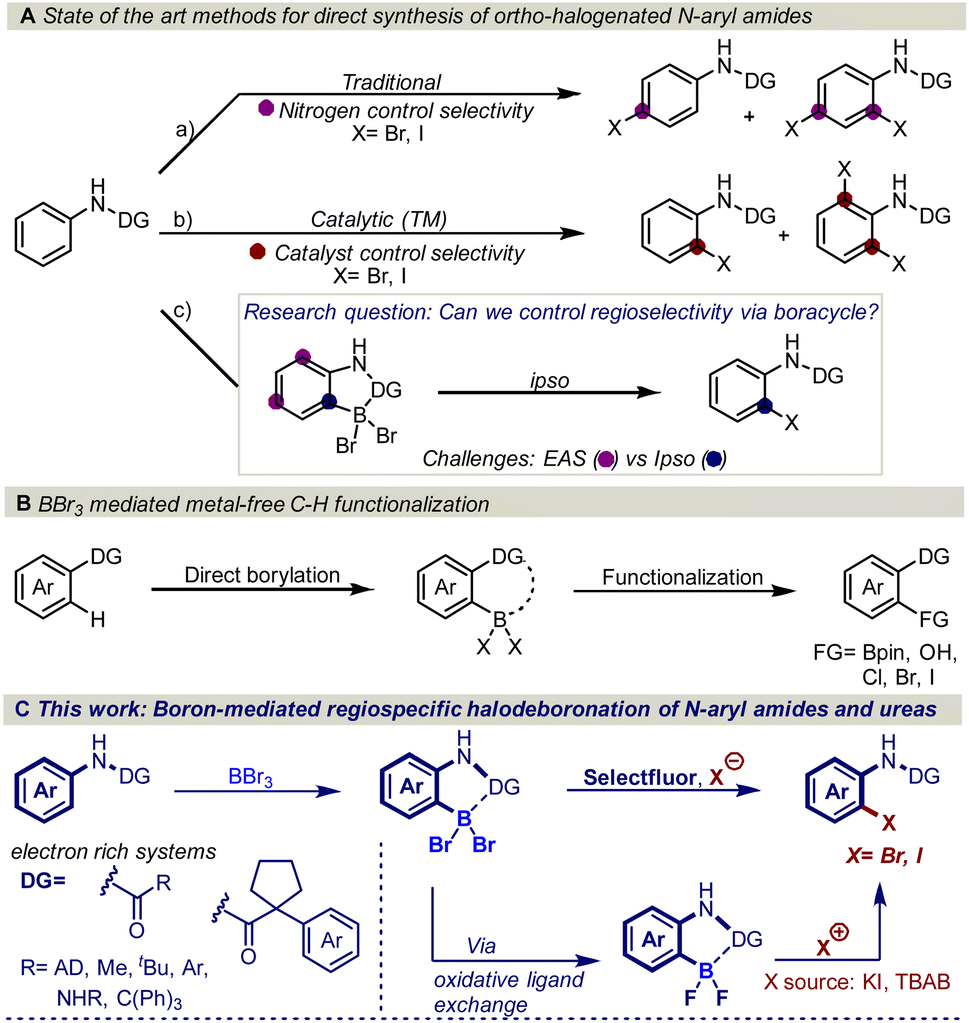 | ||
| Fig. 1 (A) Literature reports on ortho halogenation of N-aryl amides and our research question. (B) Recent reports on BBr3-mediated C–H borylation and functionalization. (C) This work: boron-mediated regioselective halodeboronation of N-aryl amides and ureas. | ||
Despite the maturity of the field ortho functionalization remain elusive. To address the selectivity issue, an alternative strategy would be to install boron on the aromatic ring to alter the aromatic reactivity (Fig. 1Ac) and achieve ipso-substitution via an oxidative halodeboronation at the C–B aromatic carbon instead of electrophilic aromatic substitution (EAS), providing boron control over nitrogen control. An attractive approach to installing the boron is to use the anilide amide as a handle to direct the aromatic borylation, as first developed by Murakami in 2010 (Fig. 1B). Murakami disclosed a BBr3-mediated synthesis of pyridine-based boracycles, which has proven to be an exceptionally attractive approach in functionalizing N-heteroarenes under metal-free conditions.6 Recently, BBr3-mediated borylative functionalization via stable boracycles has developed into a reliable synthetic route for installing hydroxyl and Bpin functionality in the ortho-position to a variety of directing groups (Fig. 1B).7 More recently, we showed that the chemistry is not limited to only hydroxyl and Bpin, but it can also be used for installing halogens to phenyl pyridines and aryl aldehydes (Fig. 1B).8 One of the key findings in this study is that the reaction proceeds through a difluoroboracycle, which plays a critical role in controlling the reaction's regioselectivity and preventing side reactions. However, the reaction has a limitation in that it requires an electronically deficient system. This is because electronically rich systems can lead to challenges in achieving regioselective halogenations, as competing electrophilic aromatic substitution (EAS) reactions may occur. The reactivity of electronically rich systems is highly dependent on the directing groups, making it difficult to control regioselectivity, especially with electron-rich directing groups. Inspired by the synthetic opportunities that would arise from an oxidative halodeboronation on an electronically rich system, we set out to test the capabilities of a new difluoroborane system on electronically rich N-aryl amides (Fig. 1C). Herein we report a convenient protocol for the unexplored regioselective ortho-halogenation of N-aryl amides and ureas. Our developed protocol shows that by using a boron handle, it is possible to control the regioselectivity of the halogenation through ipso-addition at the C–B carbon.
Results and discussion
Our study was initiated by examining the sequential addition of BBr3 and Selectfluor to N-phenylpivalamide (1a) in the expectation of forming BBr2-complex 2a, which would undergo oxidative ligand exchange with Selectfluor to yield BF2-complex 4a and cationic bromine. Ultimately, this reaction pathway was expected to generate ortho-brominated anilide 5avia ipso-substitution. However, a mixture of regioisomers (5a and 5b) was obtained, which suggests that the electronically rich anilide substrate was challenging to react selectively and led to side reactions involving the radical and cationic forms of bromine promoted by Selectfluor. It is known that the combination of bromide and Selectfluor generates the cationic form of bromine, and we hypothesize that over time, the cationic form of bromine will generate bromide radicals,9 bromine, or HBr, which could explain the formation of the side product 5b. A potential solution to address this issue would be to avoid adding Selectfluor directly to boracycle 2a and instead prepare the electrophilic halogenating reagent by mixing Selectfluor with an anionic halogen source prior to its addition to 2a. This would create a mixture of a cationic halogen source and fluoride, both of which are necessary for the formation of aryl-BF2 species (4a). Our experiments showed that by premixing 1 equivalent of Selectfluor with 1.2 equivalents of potassium iodide (KI) and subsequently adding this mixture to a solution of 2a in an acetonitrile-water system at 60 °C, we were able to obtain ortho-iodinated product 3a exclusively with a yield of 41% (Table 1, entry 2). We attempted to further enhance the yield by increasing the loading of Selectfluor and KI, which led to the successful generation of 3a in 93% yield (Table 1, entry 4 vs. entries 2–3). Further, we investigated other inorganic and organic iodine sources and the result showed that KI delivered the best result (ESI, optimization Table S1,† entries 5–8). To be noted, without BBr3 no product formation was observed, which shows the importance of the six-member ring boron handle in this transformation (Table 1, entry 5). Next, subjecting boracycle 2a with direct electrophilic iodinating reagents provided 3a in a poor yield (Table 1, entries 6–8) and no reaction, respectively (Table 1, entry 9).|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Halogen source (equiv.) | Oxidant (equiv.) | Solvent | Time (h) | Yield 3ac | NMR ratio 5a:5b |
| a Reaction conditions iodination: step (i) 1a (0.22 mmol), BBr3 (0.26 mmol), in 0.5 mL anhydrous CH2Cl2 at 22 °C, 2 h; step (ii) Selectfluor (SF) (0.44 mmol), potassium iodide (KI) (0.48 mmol) in 1.5 mL CH3CN and 1 mL water at 22 °C, 2 h then 60 °C for 5 h. b Without BBr3. c Isolated yields. Barluenga's reagent = bis(pyridine)iodonium(I) tetrafluoroborate. | ||||||
| 1 | — | Selectfluor (2) | CH3CN:Water | 2 | — | 0.26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
| 2 | KI (1.2) | Selectfluor (1) | CH3CN:Water | 5 | 41% | — |
| 3 | KI (1.7) | Selectfluor (1.5) | CH3CN:Water | 5 | 66% | — |
| 4a | KI (2.2) | Selectfluor (2) | CH3CN:Water | 5 | 93% | — |
| 5b | KI (2.2) | Selectfluor (2) | CH3CN:Water | 5 | 0% | — |
| 6 | NIS (1) | — | CH3CN:Water | 5 | 14% | — |
| 7 | ICl (1) | — | CH3CN:Water | 5 | 12% | — |
| 8 | Barluenga's reagent (1) | — | CH3CN:Water | 5 | 17% | — |
| 9 | I2 (2) | — | CH3CN:Water | 5 | 0% | — |

With our optimized reaction conditions in hand, the reaction scope was investigated. Several para-substituted N-aryl amides including donating groups, halogenated, and withdrawing groups, were subjected to the protocol, yielding ortho-iodinated products 3a–3h in moderate to excellent yields (42–93%, Scheme 1). Similarly, substitution at meta-position including methyl, strongly donating methoxy, halogens and withdrawing trifluoromethyl (–CF3) were also well tolerated providing exclusively the desired iodinated product 3i–3m in good to excellent yields (51–96%). Suggesting that the boracycle formation on the less hindered carbon is much faster than the hindered one. A sluggish reaction was observed in the case of substitution at ortho-position for substrates 1n and 1o. This could be explained by the steric hindrance at the ortho-position which potentially hinders the necessary planar conformation of the amide in the boracycle intermediate. However, by modifying the first step time, this issue could be addressed and 3n could be obtained in 65% and 3o 48%, respectively. Disubstitution on the N-aryl ring provided 3p–3r in good to excellent yields (57–92%). Furthermore, extended aromatics 3s–3u and substrates with a higher degree of complexity 3v–3w are also compatible with the reaction, provided the desired products in moderate to excellent yields without any side reactions on the phenyl rings. Notably, under similar conditions diiodination was performed on oxydibenzene substrate 1x giving desired product 3x in 62% yield. The reaction is scalable and can be performed on gram-scale (5.64 mmol) without much loss of efficiency and compound 3a can be obtained in 82% yield (Scheme 1). Compound 1y exhibiting an unsymmetrical N-aryl ring delivered the single isomer exclusively in 62% yield (3y). The biologically active procain derivative 1z with an electron-deficient system also provided desired product albeit in lower yield (3z). Next, we investigated the generality of the protocol with respect to the directing group. It was found that with slight alterations in the reaction conditions, including heating at 40 °C, the reaction was compatible with a range of directing groups, and the ortho-iodination exhibited excellent regioselectivity, yielding products in good to excellent yields (Scheme 2). For instance, electron-donating groups at both para and meta positions (7a–7c, Scheme 2) were well tolerated, providing the desired iodinated products in yields ranging from 77–87%. Halogen-bearing substrates on para and ortho positions also delivered iodinated products in moderate to good yields (7d–7g, 42–75%, Scheme 2). It is noteworthy to mention that these di-halogenated substrates possess high importance due to their facile cyclization for the synthesis of biologically active compounds.12 Electron withdrawing groups on the directing group, such as –CF3, –NO2, do not impede the reaction (7h 55%, 7i 85%, Scheme 2). Electron-rich heteroaromatics worked exceptionally well without any side reaction (7j 81%, 7k 86%, Scheme 2). Substitution on the aniline part was also well tolerated under similar conditions (7l 71%). Notably, alkyl-based directing groups such as adamantane-1-carboxamide and acetamide also provided desired products (7m 73%, 7n 31%).
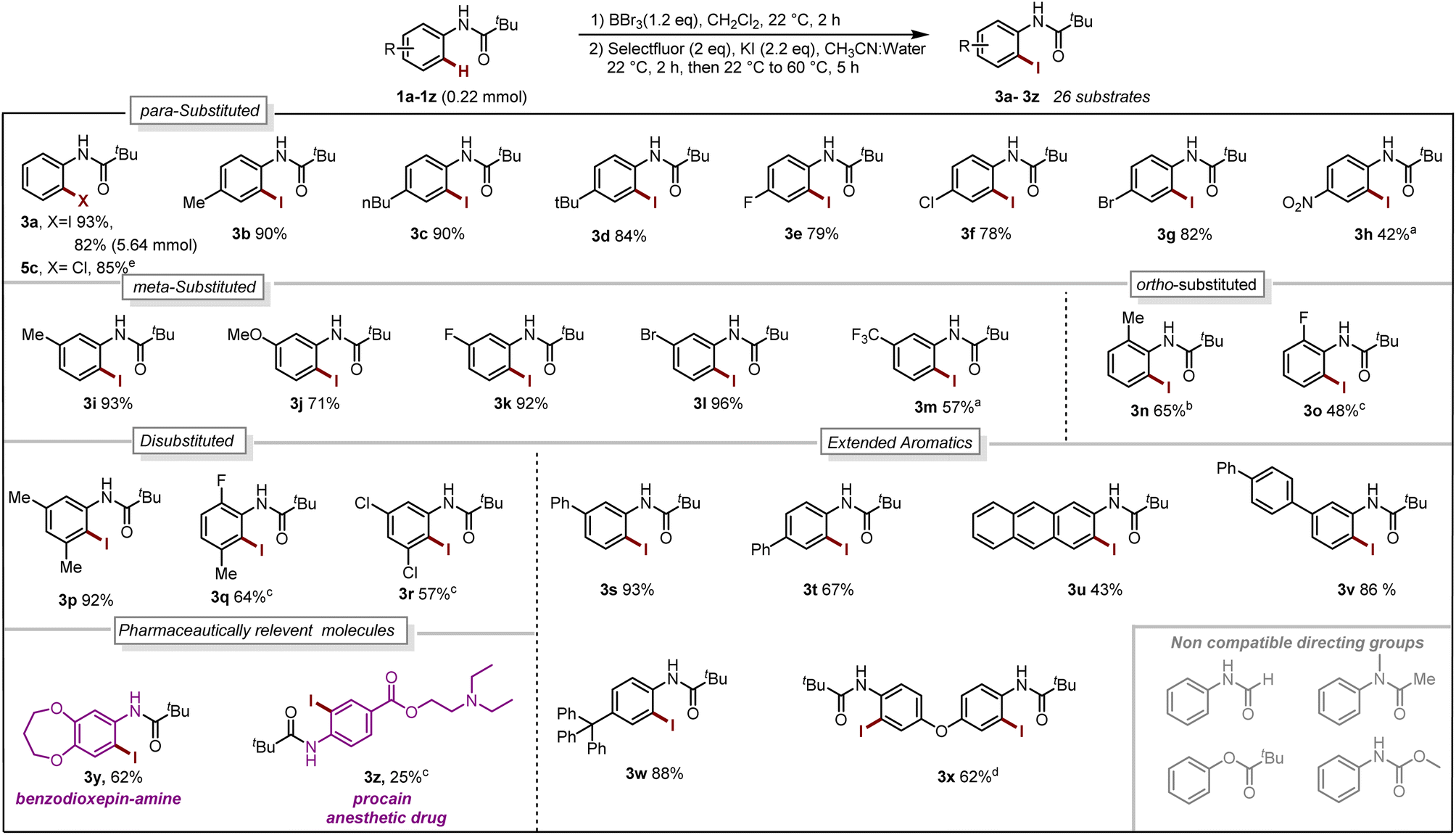 | ||
| Scheme 1 Reaction Scope. Iodination (1a–1z 0.22 mmol):a 2.2 mmol of BBr3.bstep-1 for 16 h. cStep-1 at 40 °C, 16 h. d0.48 mmol of BBr3, 0.88 mmol of Selectfluor, 0.92 mmol of KI. Chlorination:e 0.1 mmol of 4a, 0.1 mmol of chloramine T trihydrate, CH3CN:Water, 70 °C, 4 h (for reaction condition see Section 4.4 in ESI†). | ||
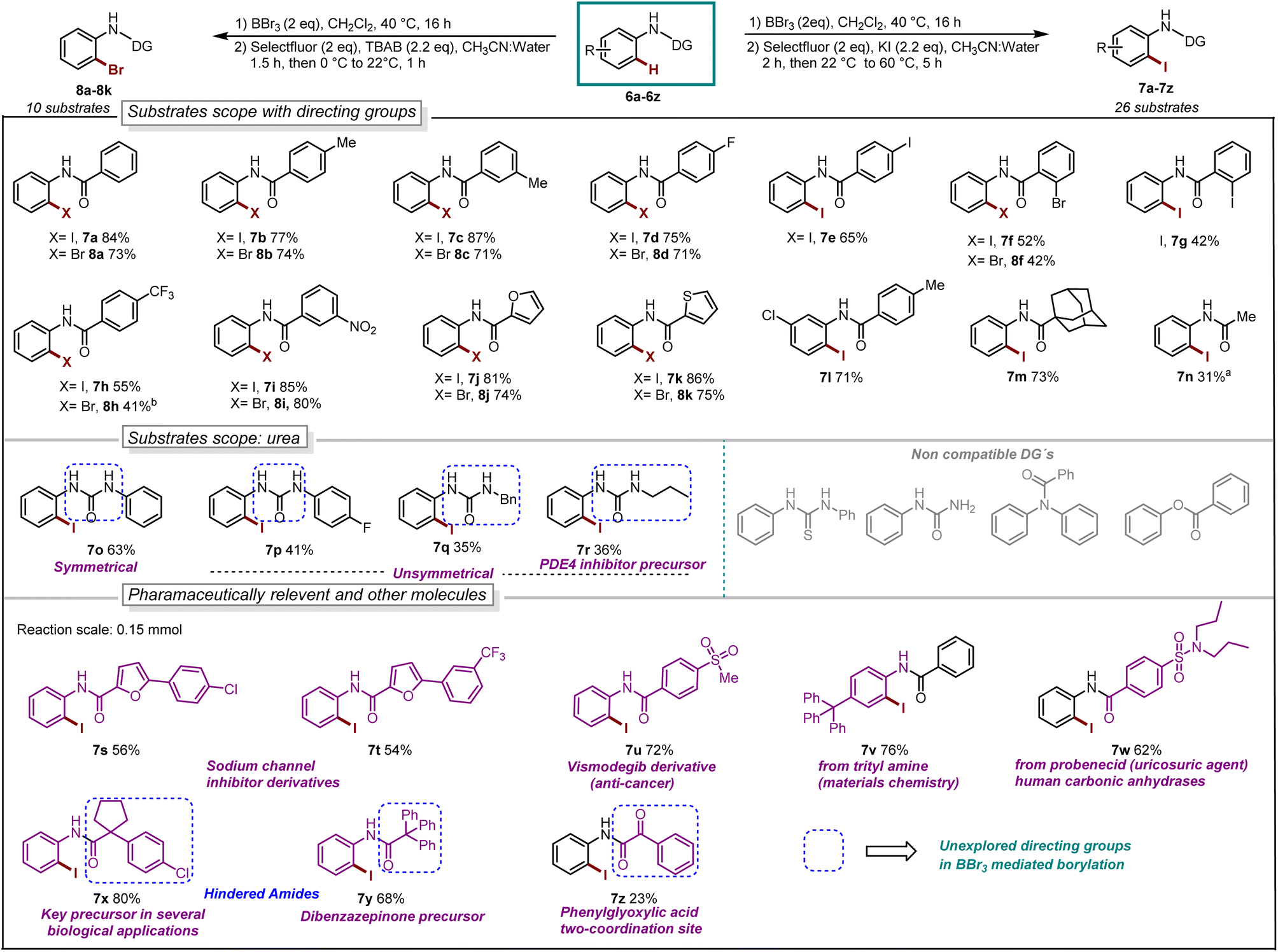 | ||
| Scheme 2 Reaction scope, iodination: aBBr3 (0.66 mmol), 60 °C, 24 h. Bromination: bstep-2 at 40 °C, 1.5 h. | ||
It is also possible to install bromine to the ortho-position of anilides by using TBAB as the bromine source. The bromination worked exceptionally well for a series of substrates such as donating groups (8a–8c, 71–74%), halogens (8d 71% and 8f 42%), withdrawing groups (8h 41% and 8i 80%), heteroaromatics (8j 74% and 8k 75%).
Next, we explored urea substrates for our deborylative iodination. Urea represents a more complex substrate class in BBr3 chemistry due to its polarity, which gives rise to difficulties in purification. The reaction was found to proceed efficiently with a range of ureas, affording low to moderate yields of the corresponding iodinated ureas with tolerance for phenyl (7o 63%), p-fluorophenyl- (7p 41%), benzyl- (7q 35%) and propyl- (7r 36%) substituents. It is noteworthy to mention that this is the first approach for the halogenation as well as the borylation of N-aryl urea using BBr3. Next, we examined late-stage skeletal editing on a series of bioactive molecules and other applicative substrates.
The deborylative iodination worked exceptionally well for sodium channel inhibitors10a with tolerance for furan heterocycle and –CF3 functionality (7s 56%, 7t 54%, Scheme 2), and vismodegib derivative precursor (7u 72%), hindered p-trityl anilide10b (7v 76%), probenecid derivative10c (7w 62%) respectively. Notably, hindered amide substrates such as 6x10d,e and 6y undergo smooth transformation under our condition providing valuable and challenging substrates in good to excellent yields (7x 80%, 7y 68%). Substrate with two coordinating sites such as 6z also provided desired iodination product albeit in lower yield (7z 23%). To explore the utility of deborylative halogenation we carried out a few transformations on halogenated products (Scheme 3A and B). The biologically active 3,4-dihydroquinolinone derivative 9a was synthesized from substrate 5a using a known palladium-catalyzed approach in 43% yield.11 Next, the benzimidazo-isoquinlinone12a9b and azepino-fused isoindolinone core12b9c were synthesized from 8f in 51% and 76% yield respectively (Scheme 3A). Further, the biologically active benzoyl carbazole13a9d and benzoxazole core13b9e–9f were synthesized using known palladium-aryne and copper chemistry from substrates 7a and 7l in 50%, 65% and 70% yield, respectively (Scheme 3B). In a copper-catalyzed reaction, iodo urea 7o was converted into 2-benzimidazolones149g in 80% yield (Scheme 3B).
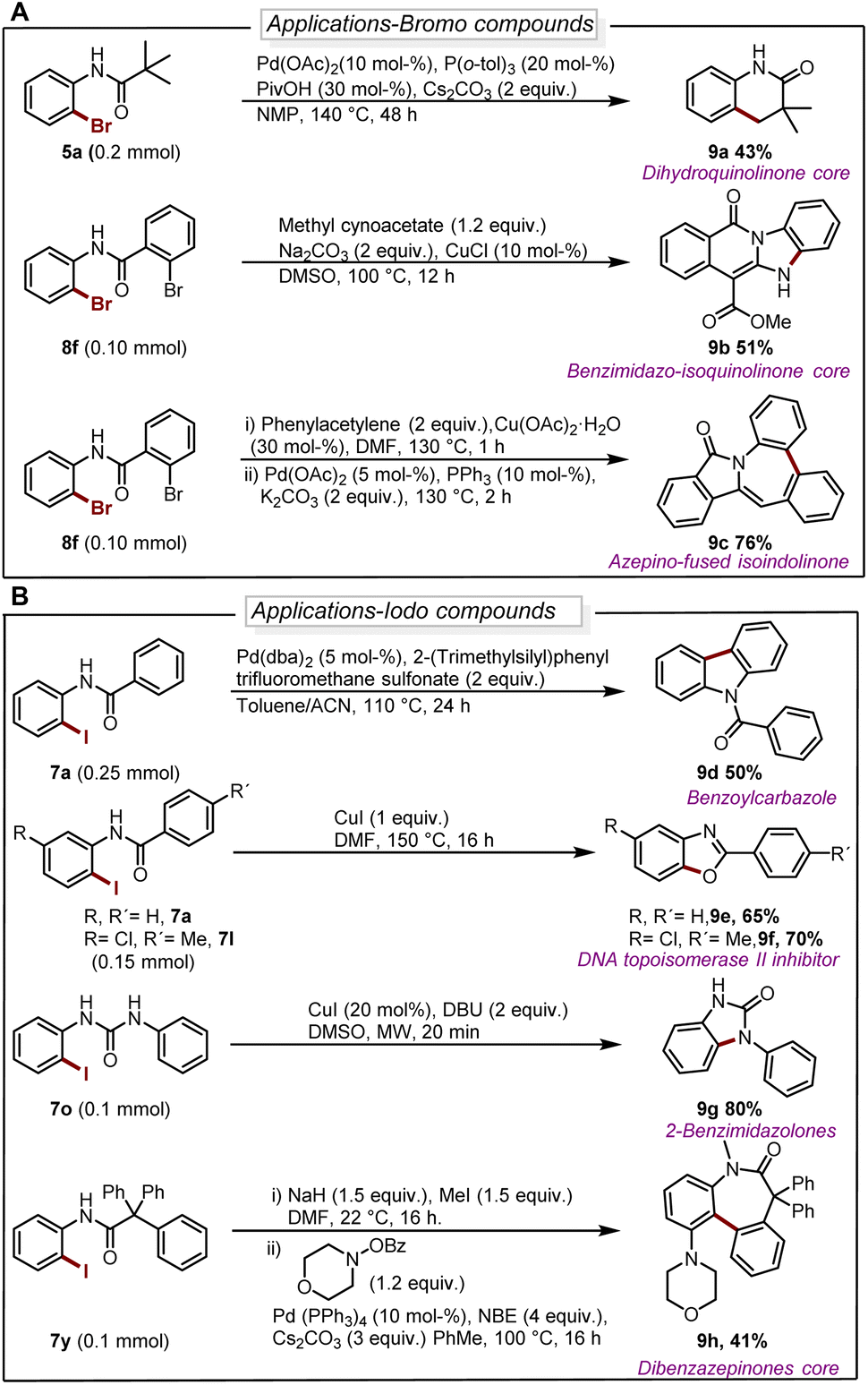 | ||
| Scheme 3 (A) Diversification of brominated substrates. For reaction details see ESI.† (B) Diversification of iodinated substrates. For reaction details see ESI.† | ||
Finally, we demonstrated the utility of this strategy in the synthesis of the dibenzazepinones core15 in a reaction where 9h can be obtained from 7y in a yield of 41% (Scheme 3B).
Next, we analyzed the selectivity of our protocol, we conducted experiments with the oxidative halogenation conditions and the results are collected in Scheme 4a and b. In the presence of Selectfluor and bromine source tetra butyl ammonium bromide (TBAB), we observed electrophilic aromatic substitution (EAS) of 1a providing a mixture of brominated products 5b and 5c with no selectivity for the ortho-bromination (Scheme 4a). Further, with similar conditions in the presence of a boron handle, we observed exclusively the ortho bromination and 5a could be isolated in 78% yield (Scheme 4b). This result reveals that, using the boron complex the reactivity completely shifted to ipso rather than EAS. This experiment supports the fact that our protocol is regioselective where the reactivity of the system is governed by boron rather than nitrogen.
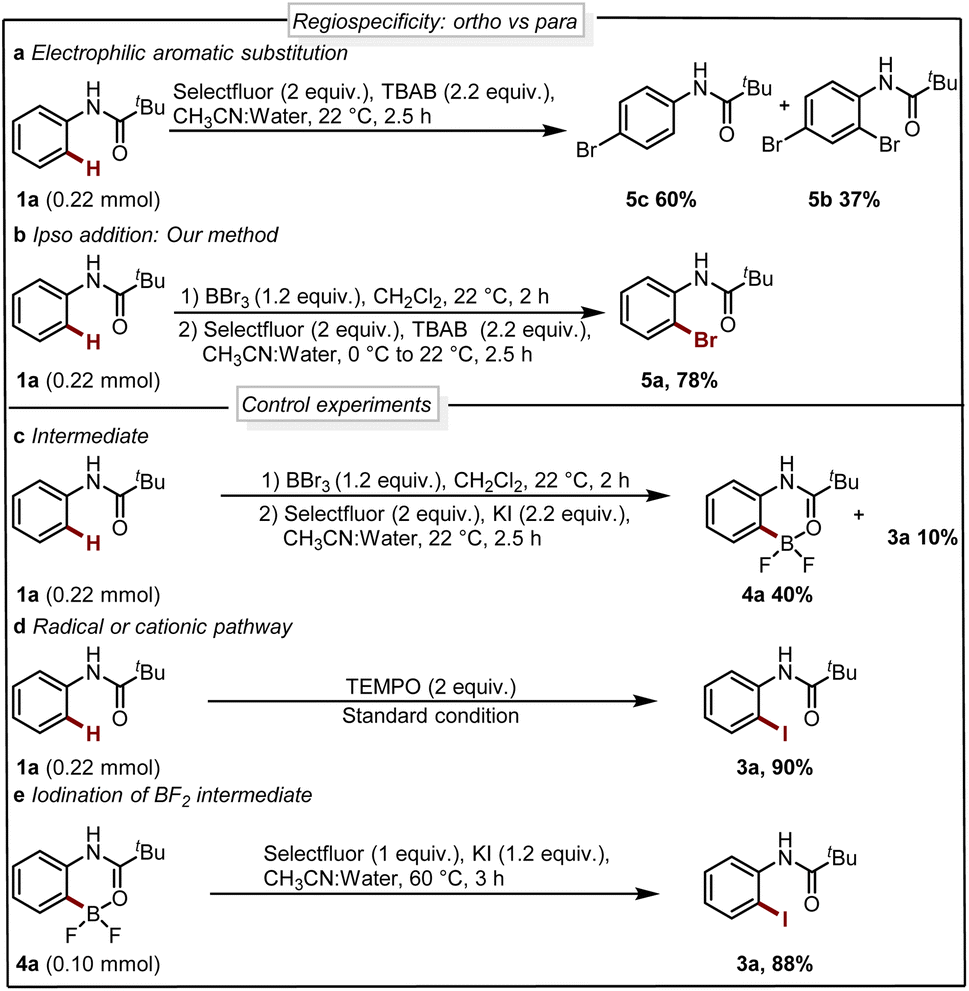 | ||
| Scheme 4 (a) Nitrogen control reaction (EAS). (b) Boron control reaction (ipso). (c–e) Control experiments. | ||
Seeking to gain mechanistic insight into the bromide-to-fluoride ion exchange behavior and C–B bond cleavage, we performed a series of experimental and computational studies. We identified two crucial steps to be investigated, i.e. (i) the generation of the difluoroborane species and (ii) the occurrence of ionic ipso-addition. To confirm the difluoroborane species 4a we quenched the reaction prematurely and isolated the 4a in 40% yield along with 10% of 3a (Scheme 4c). This result shows the formation of intermediate 4a in the reaction. The compound 4a is also confirmed by a single crystal X-ray structure (Fig. 2, ESI Section 9†). In order to get insights into the radical or ionic ipso addition mechanism, we conducted the reaction in the presence of radical scavenger TEMPO (Scheme 4d). However, the excess TEMPO did not affect the reaction outcome providing desired iodinated product in 90% yield. Furthermore, to reveal the reactivity and support the mechanism we subjected BF2 intermediate 4a to the reaction conditions of the oxidative halodeboronation (Scheme 4e) and obtained 88% yield of desired product 3a.
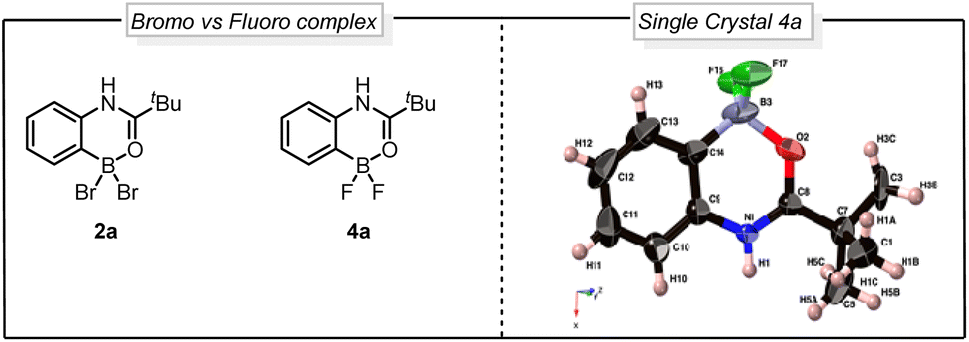 | ||
| Fig. 2 Comparison between bromo vs. fluoro complex and single crystal X-ray structure of 4a. | ||
We performed a computational investigation intended to model the ipso addition using trimethyl amine-iodo as a model source for iodination (Scheme 5). Thus, we adopted DFT at the ωB97xD/LACVP* level with a PBF continuum model for water solvation, to evaluate the effect of dibromo-boracycle vs. difluoro-boracycle on ipso addition. The water model may slightly overestimate the stabilization provided by solvent compared to the actual solvent mix but is expected to give results superior to any other type of solvent model available to us.
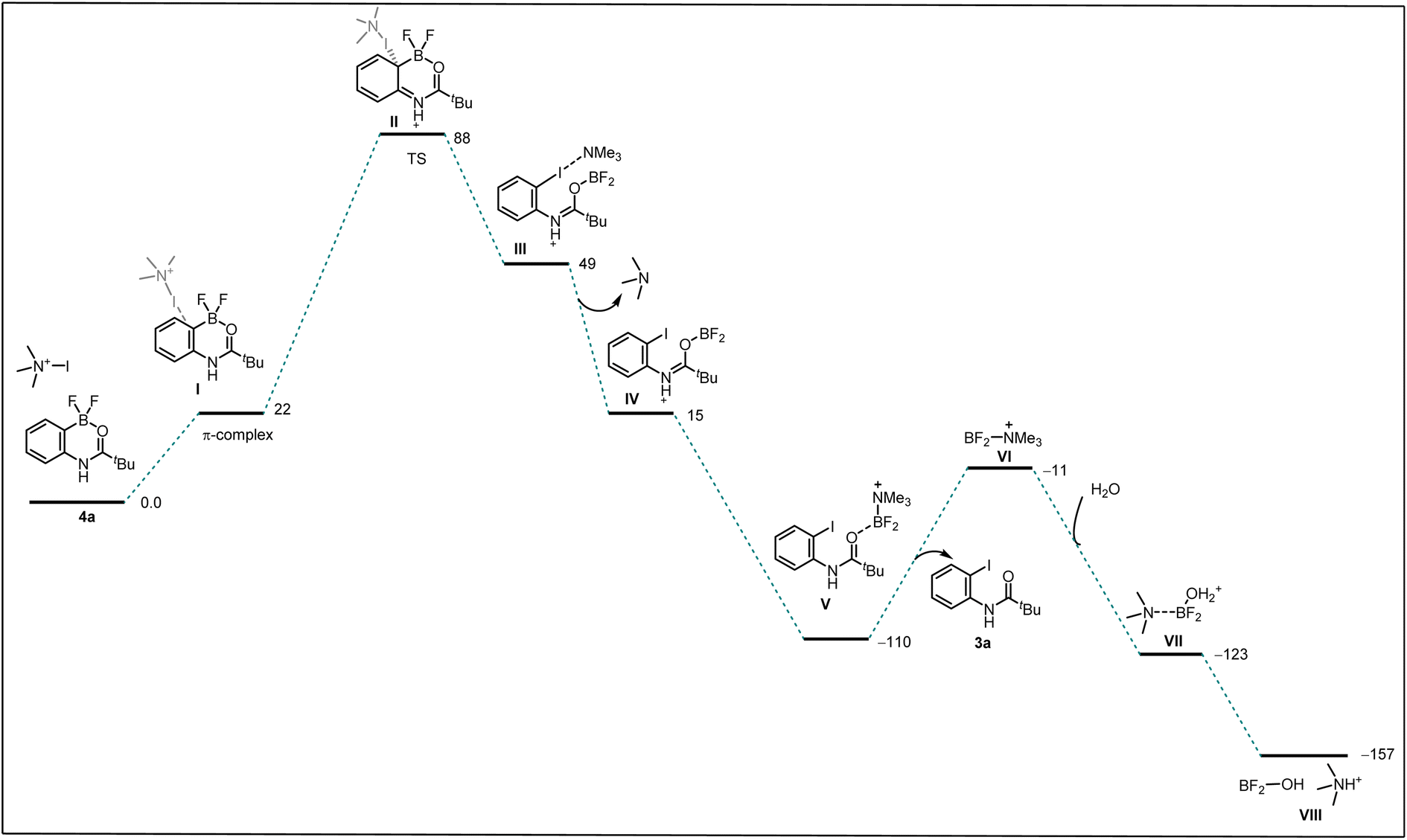 | ||
| Scheme 5 Free energy surface of ipso-substitution. We use a simplified model for the base, which is hypothesized to act as a carrier of electrophilic I+, to drive the final dissociation of boron, and to deprotonate the water adding to the boron product. | ||
We started by considering tetrahedral difluoro-boracycle 4a reacting with iodo reagent to π-complex (I) which is endergonic by 22 kJ mol−1. This reacts through TS(II) to complex (III), with a barrier of 88 kJ mol−1 compared to separated starting materials. In complex (III) boron is trigonal and has a weak interaction with π electrons of the aromatic ring. The iodine carrier, here modelled by NMe3, is weakly associated to the iodine by a halogen bond.
Dissociation of NMe3 is monotonous on the potential energy surface, but exergonic. Reassociation to boron is very strongly exergonic and drives the overall reaction. Dissociation of final product 3a is slightly endergonic, but boron can in turn associate to water, and through dissociation and final proton transfer arrive at the endpoint of the computational investigation, (VIII). Overall, formation of 3a and byproducts from 4a is exergonic by 157 kJ mol−1, with a barrier of 88 kJ mol−1. The limiting TS, (II), was located by coordinate driving of the iodine-carbon distance to shorter values. At a C–I distance of 2.2 Å, the structure is similar to the final structure of the TS, with similar energy, and could be used as a starting point for the subsequent TS search. Similar coordinate drives were performed for all positions of the aromatic ring, as well as for the ipso-position of the bromo-boracycle substrate 2a. In all cases, the energy rose to >150 kJ mol−1 without snapping into a product-like geometry, clearly showing that iodination is very strongly preferred at the ipso position of 4a compared to 2a.
To understand the reactivity difference between bromo boracycle 2a and fluoro-boracycle 4a (Fig. 2, single crystal X-ray structure), we analyzed the structures of both compounds, as well as the immediate product (III) from ipso-substitution of both complexes, where the C–B bond has been broken. In the bromo complex, the boron is electron deficient as compared to the fluoro complex. Despite the higher electronegativity of fluorine, the shorter B–F bonds, and the potential for π-donation from fluorine to the formally empty orbital of boron in (III) stabilize the boron. The BBr2 complex 2a has significantly shorter bonds to oxygen and carbon.
In a tetrahedral environment, the boron can compensate for the low electron density by stronger bonds to the other atoms, but in the trigonal planar complex (III), the BBr2 moiety lacks the stabilization which can be found from p-donation in the BF2 moiety. In other words, RBBr2 is a stronger Lewis acid than RBF2. In the tetrahedral complex, there is not much π-donation from fluorine to boron; the boron p-orbital is not empty as it is occupied with lone pair of carbonyl. Thus, the empty p-orbital can only be stabilized by π-donation from the three attached atoms, oxygen and two halides. The oxygen contribution is the same in both complexes, but the halides are very different. For instance, Br basically cannot do π-stabilization, and the orbitals of Br lone pairs are of the wrong size and distance to give efficient π overlap. On the other hand, F has a perfectly matched, filled p-orbital, which gives a very good π-stabilization.
To rationalize the observed reactivity, based on control experiments and DFT studies we propose a mechanism (Scheme 6) that starts with the formation of boracycle 2avia an electrophilic aromatic substitution in the presence of boron tribromide. Next, the bromo-boracycle 2a converts into fluoro boracycle 4a in the presence of an oxidative system (A). In the classical oxidative process,9 Selectfluor oxidizes halogen anion X− to halogen cation X+. However, in our case, the oxidative condition plays a dual role which is the generation of electrophile and nucleophilic fluorines for the ligand exchange on boron. In the presence of this oxidative condition, the fluoro boracycle undergoes ipso addition on cationic iodine to form intermediate B which upon deboryalation forms desired iodinated product 3a.
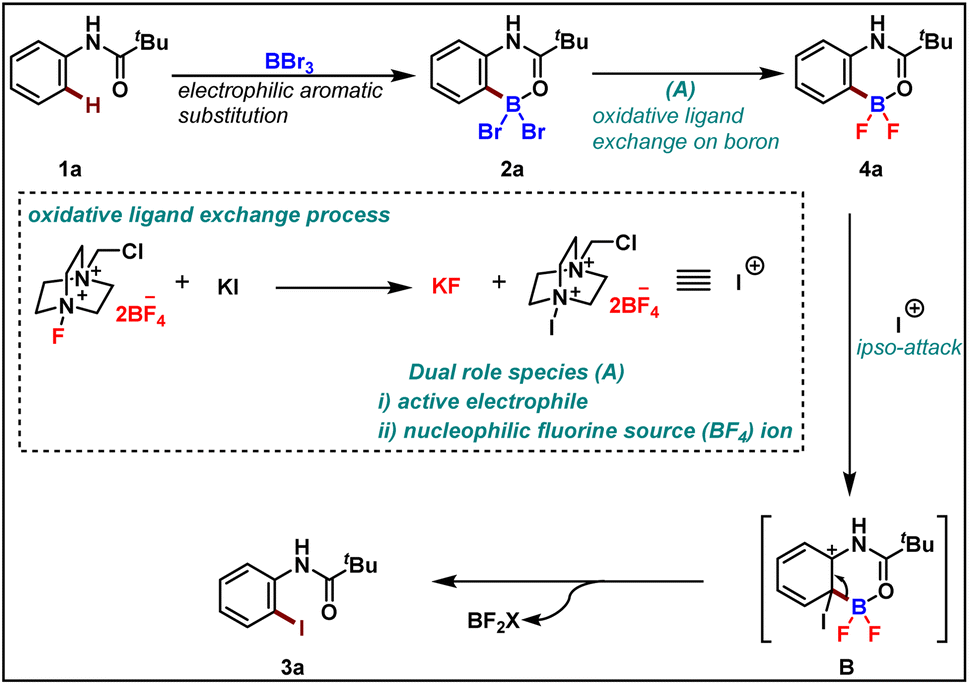 | ||
| Scheme 6 Proposed mechanism. | ||
Conclusion
In summary, we have demonstrated a regioselective, efficient, and practical ortho halogenation of N-aryl amides and ureas promoted by a boron handle. The power of this method is showcased by halogenation on a set of different carbonyls as directing groups. Our strategy leverages the dual role of Selectfluor as a fluoride source and oxidant, enabling regioselective deborylative ortho-halogenation of N-aryl amides and ureas. The proposed mechanism involves oxidative ligand exchange on boron followed by ipso-addition with concomitant release of the boron species. This newfound reactivity of the C–B bond and B–X bond holds promise as an alternative for the functionalization of a broad range of N-heterocycles in the coming years. Overall, our protocol demonstrates its applicability in the synthesis of urea substrates, pharmaceutically active molecules, and hindered amides, showcasing its strength in material science, medicinal chemistry, and synthetic organic chemistry.Data availability
All detailed procedures, characterization data, and spectra are available in the ESI.†Author contributions
H. S. supervised the overall project. G. H. S. conceived the idea and designed the study with G. S. G. F. M. A. N. and L. Ö. contributed to the crystal structure. P. O. N. contributed to the computational study. G. S. G., F. M. A. N., L. Ö. and P. O. N. provided valuable suggestions on the manuscript. H. S. and G. H. S. co-wrote the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by grants from the Adlerbertska Research Foundation and Carl Tryggers Stiftelse. We also thank the Olle Engkvist Foundation, Chalmers areas of Advance Energy, Materials, Health and Nano, and the Chalmers Materials Analysis Laboratory.Notes and references
- (a) N. A. McGrath, M. Brichacek and J. T. Njardarson, J. Chem. Educ., 2010, 87, 1348–1349 CrossRef CAS; (b) X. D. Yang, X. H. Zeng, Y. H. Zhao, X. Q. Wang, Z. Q. Pan, L. Li and H. Bin Zhang, J. Comb. Chem., 2010, 12, 307–310 CrossRef CAS PubMed; (c) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed; (d) M. M. Heravi and V. Zadsirjan, RSC Adv., 2020, 10, 44247–44311 RSC.
- S. Kumari, A. V. Carmona, A. K. Tiwari and P. C. Trippier, J. Med. Chem., 2020, 63, 12290–12358 CrossRef CAS PubMed.
- (a) U. Ladziata, A. Y. Koposov, K. Y. Lo, J. Willging, V. N. Nemykin and V. V. Zhdankin, Angew. Chemie-Int. Ed., 2005, 44, 7127–7131 ( Angew. Chem. , 2005 , 117 , 7289–7293 ) CrossRef CAS PubMed; (b) N. Zheng, K. W. Anderson, X. Huang, H. N. Nguyen and S. L. Buchwald, Angew. Chemie-Int. Ed., 2007, 46, 7509–7512 ( Angew.Chem. , 2007 , 119 , 7653 ) CrossRef CAS PubMed; (c) E. A. Merritt and B. Olofsson, Angew. Chemie-Int. Ed., 2009, 48, 9052–9070 ( Angew. Chem. , 2009 , 121 , 9214–9234 ) CrossRef CAS PubMed; (d) J.-X. Yan, H. Li, X.-W. Liu, J.-L. Shi, X. Wang and Z.-J. Shi, Angew. Chemie-Int. Ed., 2014, 53, 4945–4949 ( Angew. Chem. , 2014 , 126 , 5045–5049 ) CrossRef CAS PubMed; (e) S. Z. Tasker and T. F. Jamison, J. Am. Chem. Soc., 2015, 137, 9531–9534 CrossRef CAS PubMed; (f) Shaifali, P. Mehara, A. Kumar and P. Das, ChemCatChem , 2021, 13, 2459–2464 CrossRef CAS.
- (a) J. M. Fourquez, A. Godard, F. Marsais and G. B. Quéguiner, J. Heterocycl. Chem., 1995, 32, 1165–1170 CrossRef CAS; (b) R. B. Bedford, M. F. Haddow, C. J. Mitchell and R. L. Webster, Angew. Chemie-Int. Ed., 2011, 50, 5524–5527 ( Angew. Chem. , 2011 , 123 , 5638 ) CrossRef CAS PubMed; (c) B. Urones, Á. M. Martínez, N. Rodríguez, R. Gómez Arrayás and J. C. Carretero, Chem. Commun., 2013, 49, 11044–11046 RSC; (d) J. Chen, X. Xiong, Z. Chen and J. Huang, Synlett, 2015, 26, 2831–2834 CrossRef CAS; (e) D. Liang, X. Li, C. Wang, Q. Dong, B. Wang and H. Wang, Tetrahedron Lett., 2016, 57, 5390–5394 CrossRef CAS; (f) S. Kathiravan and I. A. Nicholls, Chem. - Eur. J., 2017, 23, 7031–7036 CrossRef CAS PubMed; (g) R. Das and M. Kapur, J. Org. Chem., 2017, 82, 1114–1126 CrossRef CAS PubMed; (h) Z. lin Li, K. kang Sun and C. Cai, Org. Biomol. Chem., 2018, 16, 5433–5440 RSC; (i) Y. Du, Z. Xi, L. Guo, H. Lu, L. Feng and H. Gao, Tetrahedron Lett., 2021, 72, 153074 CrossRef CAS; (j) E. Kianmehr and H. Afaridoun, Synthesis, 2021, 53, 1513–1523 CrossRef CAS.
- (a) D. A. Petrone, J. Ye and M. Lautens, Chem. Rev., 2016, 116, 8003–8104 CrossRef CAS PubMed; (b) R. Das and M. Kapur, Asian J. Org. Chem., 2018, 7, 1524–1541 CrossRef CAS.
- N. Ishida, T. Moriya, T. Goya and M. Murakami, J. Org. Chem., 2010, 75, 8709–8712 CrossRef CAS PubMed.
- (a) L. Niu, H. Yang, R. Wang and H. Fu, Org. Lett., 2012, 14, 2618–2621 CrossRef CAS PubMed; (b) S. A. Iqbal, J. Cid, R. J. Procter, M. Uzelac, K. Yuan and M. J. Ingleson, Angew. Chem., Int. Ed., 2019, 58, 15381–15385 ( Angew. Chem. , 2019 , 131 , 15525–15529 ) CrossRef CAS PubMed; (c) J. Lv, X. Chen, X. S. Xue, B. Zhao, Y. Liang, M. Wang, L. Jin, Y. Yuan, Y. Han, Y. Zhao, Y. Lu, J. Zhao, W. Y. Sun, K. N. Houk and Z. Shi, Nature, 2019, 575, 336–340 CrossRef CAS PubMed; (d) J. Lv, B. Zhao, Y. Yuan, Y. Han and Z. Shi, Nat. Commun., 2020, 11, 1316 CrossRef CAS PubMed; (e) G. Wu, X. Fu, Y. Wang, K. Deng, L. Zhang, T. Ma and Y. Ji, Org. Lett., 2020, 22, 7003–7007 CrossRef CAS PubMed; (f) G. Wu, B. Pang, Y. Wang, L. Yan, L. Chen, T. Ma and Y. Ji, J. Org. Chem., 2021, 86, 5933–5942 CrossRef CAS PubMed; (g) S. Li, C. Hu, X. Cui, J. Zhang, L. L. Liu and L. Wu, Angew. Chem., Int. Ed., 2021, 60, 26238–26245 ( Angew.Chem. , 2021 , 133 , 26442–26449 ) CrossRef CAS PubMed; (h) Z. J. Wang, X. Chen, L. Wu, J. J. Wong, Y. Liang, Y. Zhao, K. N. Houk and Z. Shi, Angew. Chem., Int. Ed., 2021, 60, 8500–8504 ( Angew. Chem. , 2021 , 133 , 8581–8585 ) CrossRef CAS PubMed; (i) S. Rej and N. Chatani, J. Am. Chem. Soc., 2021, 143, 2920–2929 CrossRef CAS PubMed; (j) S. Rej, A. Das and N. Chatani, Chem. Sci., 2021, 12, 11447–11454 RSC; (k) J. Lv, X.-J. Zhang, M. Wang, Y. Zhao and Z. Shi, Chem. - Eur. J., 2022, 28, e202104100 CrossRef CAS PubMed; (l) O. Sadek, A. L. Gac, N. Hidalgo, S. Mallet-Ladeira, K. Miqueu, G. Bouhadir and D. Bourissou, Angew. Chem., Int. Ed. , 2022, 61, e202110102 ( Angew. Chem. , 2022 , 134 , e202110102 ) CrossRef CAS PubMed; (m) G. Wu, Z. Yang, X. Xu, L. Hao, L. Chen, Y. Wang and Y. Ji, Org. Lett., 2022, 24, 3570–3575 CrossRef CAS PubMed; (n) X. Xu, G. Wu, Z. Yang, X. Liu, L. Hao, Y. Wang, Z. Ma and Y. Ji, Org. Lett., 2022, 24, 7163–7167 CrossRef CAS PubMed; (o) Review: S. Rej and N. Chatani, Angew. Chem., Int. Ed., 2022, 61, e202209539 ( Angew. Chem. , 2022 , 134 , e202209539 ) CrossRef CAS PubMed; (p) G. Berionni, Angew. Chem., Int. Ed., 2022, 61, e202210284 ( Angew. Chem. , 2022 , 134 , e202210284 ) CrossRef CAS PubMed; (q) S. A. Iqbal, M. Uzelac, I. Nawaz, Z. Wang, H. Jones, G. S. Nichol, K. Yuan, C. Millet, G. A. Chotana and M. J. Ingleson, Chem. Sci., 2023, 1, 3865–3872 RSC; (r) D. Zhao and G. Yang, Synfacts, 2023, 19, 0554 CrossRef; (s) Z. Yang, L. Hao, X. Xu, Y. Wang, G. Wu and Y. Ji, Org. Lett., 2023, 25, 5875–5879 CrossRef CAS PubMed; (t) P. Kumar Someswara Ashwathappa, T. Higashi, V. Desrosiers, A. A. Omaña and F. G. Fontaine, Angew. Chemie-Int. Ed., 2023, 62, 1–7 CrossRef PubMed; (u) T. Wang, Z.-J. Wang, M. Wang, L. Wu, X. Fang, Y. Liang, J. Lv and Z. Shi, Angew. Chem., Int. Ed., 2023, e202313205 CAS.
- G. H. Shinde and H. Sundén, Chem. - Eur. J., 2023, 29, e202203505 CrossRef CAS PubMed.
- (a) R. G. Syvret, K. M. Butt, T. P. Nguyen, V. L. Bulleck and R. D. Rieth, J. Org. Chem., 2002, 67, 4487–4493 CrossRef CAS PubMed; (b) C. Ye and J. M. Shreeve, J. Org. Chem., 2004, 69, 8561–8563 CrossRef CAS PubMed; (c) Z. Daǧalan, R. Koçak, A. Daştan and B. Nişancl, Org. Lett., 2022, 24, 8261–8264 CrossRef PubMed.
- (a) M. E. Kort, I. Drizin, R. J. Gregg, M. J. C. Scanio, L. Shi, M. F. Gross, R. N. Atkinson, M. S. Johnson, G. J. Pacofsky, J. B. Thomas, W. A. Carroll, M. J. Krambis, D. Liu, C. C. Shieh, X. F. Zhang, G. Hernandez, J. P. Mikusa, C. Zhong, S. Joshi, P. Honore, R. Roeloffs, K. C. Marsh, B. P. Murray, J. Liu, S. Werness, C. R. Faltynek, D. S. Krafte, M. F. Jarvis, M. L. Chapman and B. E. Marron, J. Med. Chem., 2008, 51, 407–416 CrossRef CAS PubMed; (b) F. Dumur and F. Goubard, New J. Chem., 2014, 38, 2204–2224 RSC; (c) M. D'Ascenzio, S. Carradori, D. Secci, D. Vullo, M. Ceruso, A. Akdemir and C. T. Supuran, Bioorg. Med. Chem. , 2014, 22, 3982–3988 CrossRef PubMed; (d) G. Tang, Z. Qiu, X. Lin, W. Li, L. Zhu, S. Li, H. Li, L. Wang, L. Chen, J. Z. Wu and W. Yang, Bioorg. Med. Chem. Lett., 2010, 20, 3507–3510 CrossRef CAS PubMed; (e) J. Tng, J. Lim, K. C. Wu, A. J. Lucke, W. Xu, R. C. Reid and D. P. Fairlie, J. Med. Chem., 2020, 63, 5956–5971 CrossRef CAS PubMed.
- J. Yan, H. Li, X. Liu, J. Shi, X. Wang and Z. Shi, Angew. Chemie - Int. Ed, 2014, 53, 4945–4949 ( Angew Chem , 2014 , 126 , 5045–5049 ) CrossRef CAS PubMed.
- (a) J. Lu, X. Gong, H. Yang and H. Fu, Chem. Commun., 2010, 46, 4172–4174 RSC; (b) H. K. Saini, N. K. Nandwana, S. Dhiman, K. Rangan and A. Kumar, Eur. J. Org Chem., 2017, 2017, 7277–7282 CrossRef CAS.
- (a) R. W. Bowman, H. Heaney and P. H. G. Smith, Tetrahedron Lett., 1982, 23, 5093–5096 CrossRef; (b) C. Lu, N. A. Markina and R. C. Larock, J. Org. Chem., 2012, 77, 11153–11160 CrossRef CAS PubMed.
- Z. Li, H. Sun, H. Jiang and H. Liu, Org. Lett., 2008, 10, 3263–3266 CrossRef CAS PubMed.
- X. Abel-Snape, A. Whyte and M. Lautens, Org. Lett., 2020, 22, 7920–7925 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2279239. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc04628a |
| ‡ G. H. S. and G. S. G. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |