
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReversible Cl/Cl− redox in a spinel Mn3O4 electrode†
Sean K.
Sandstrom‡
a,
Qiuyao
Li‡
b,
Yiming
Sui‡
a,
Mason
Lyons
c,
Chun-Wai
Chang
c,
Rui
Zhang
d,
Heng
Jiang
a,
Mingliang
Yu
a,
David
Hoang
a,
William F.
Stickle
e,
Huolin L.
Xin
*d,
Zhenxing
Feng
 *c,
De-en
Jiang
*bf and
Xiulei
Ji
*a
*c,
De-en
Jiang
*bf and
Xiulei
Ji
*a
aDepartment of Chemistry, Oregon State University, Corvallis, OR 97331, USA. E-mail: david.ji@oregonstate.edu
bInterdisciplinary Materials Science Program, Vanderbilt University, Nashville, TN 37235, USA. E-mail: de-en.jiang@vandderbilt.edu
cSchool of Chemical, Biological, and Environmental Engineering, Corvallis, OR 97331, USA. E-mail: zhenxing.feng@oregonstate.edu
dDepartment of Physics and Astronomy, University of California, Irvine, CA 92697, USA. E-mail: huolin.xin@uci.edu
eHewlett-Packard Co., Corvallis, OR 97330, USA
fDepartment of Chemical and Biomolecular Engineering, Vanderbilt University, Nashville, TN 37235, USA
First published on 24th October 2023
Abstract
A unique prospect of using halides as charge carriers is the possibility of the halides undergoing anodic redox behaviors when serving as charge carriers for the charge-neutrality compensation of electrodes. However, the anodic conversion of halides to neutral halogen species has often been irreversible at room temperature due to the emergence of diatomic halogen gaseous products. Here, we report that chloride ions can be reversibly converted to near-neutral atomic chlorine species in the Mn3O4 electrode at room temperature in a highly concentrated chloride-based aqueous electrolyte. Notably, the Zn2+ cations inserted in the first discharge and trapped in the Mn3O4 structure create an environment to stabilize the converted chlorine atoms within the structure. Characterization results suggest that the Cl/Cl− redox is responsible for the observed large capacity, as the oxidation state of Mn barely changes upon charging. Computation results corroborate that the converted chlorine species exist as polychloride monoanions, e.g., [Cl3]− and [Cl5]−, inside the Zn2+-trapped Mn3O4, and the presence of polychloride species is confirmed experimentally. Our results point to the halogen plating inside electrode lattices as a new charge-storage mechanism.
Introduction
As the world continues to shift away from fossil fuels toward renewable energy sources, there is an ever-growing demand for energy-storage solutions that are safe, low-cost, and use abundant and environmentally benign material resources.1–3 Unfortunately, the current Li-ion batteries are lacking in each of these considerations.4,5 Thus, immense efforts have been devoted to other more sustainable battery systems, e.g., Na-ion,6,7 K-ion,8,9 and Zn-ion batteries.10–12 Beyond these systems that operate via the reversible (de)insertion of cation charge carriers in electrode hosts, more recently, systems that use anions as the ion charge carriers have begun to attract significant interest.13,14 These so-called anion-shuttle batteries, including dual-ion batteries15 and anion rocking-chair batteries,16,17 are attractive due to their potentially high energy densities and low costs, as well as resource availability.A unique feature of using anionic charge carriers is the possibility of the anions undergoing anodic reactions to form neutral species, which can be deemed as plating of non-metals. Redox of Cl−, for example, has been studied and utilized in batteries since the 19th century.18 However, the Cl2/Cl− redox process generally suffers from poor reversibility due to loss of gaseous Cl2.19–25 Recently, Dai et al. demonstrated Cl2‖alkali metal batteries that operate via the reversible Cl2/Cl− redox at carbon-based electrodes.26,27 The effective trapping of Cl2 as the charge product at the electrode hosts was deemed essential in affording the reversibility of these systems. Wang et al. showed high reversibility of the Cl2/Cl− redox couple in an aqueous NaCl electrolyte by storing the as-produced Cl2 in nonaqueous, water-immiscible phases such as CCl4.28 Other approaches for overcoming the loss of gaseous Cl2 include the formation of interhalogen species with higher boiling points, e.g., BrCl and ICl, or operating the electrode below the boiling point of Cl2 liquid.29–33
Another approach for avoiding the escape of gaseous Cl2, which has yet to be explored, would be to “plate” neutral Cl atoms inside a host material. Such a mechanism would require a host material to effectively anchor the plated Cl atoms, where the strong binding between the inserted Cl and the host inhibits the formation of the Cl–Cl diatomic bond. Our group recently reported on the reversible anodic charge-storage behaviors of a spinel Mn3O4 electrode after trapping Zn2+ cations in the first discharge in a concentrated ZnCl2-based electrolyte.34 After Zn2+ is trapped, the as-formed Zn0.25Mn3O4 electrode exhibited a specific charge capacity of over 200 mA h g−1 with an average operating potential above 1.6 V vs. Zn2+/Zn. Herein, we report that the large capacity observed in the charging process is not due to the oxidation of Mn-ions of Mn3O4, where the oxidation state of Mn remains nearly unchanged during cycling, but from a conversion from chloride to partially charged polychloride species stored in the distorted lattice of the Mn3O4 cathode.
Results and discussion
Electrochemical performance of the Mn3O4 electrode
The electrochemical performance of the Mn3O4 (space group, I41/amd, PDF # 24-0734, Fig. S1†)34,35 electrode was first evaluated using a concentrated aqueous water-in-salt electrolyte (WiSE) of 20 M ZnCl2 + 5 M NH4Cl. As reported before, when cycled in a two-electrode cell with Zn metal as the counter and reference electrode, the Mn3O4 electrode demonstrates a relatively small first discharge capacity of 128 mA h g−1 at a potential of ∼0.6 V (vs. Zn2+/Zn and hereafter) and a current rate of 50 mA g−1 (Fig. 1a).34 The subsequent charging process results in a substantially larger capacity of 218 mA h g−1 at an average potential of 1.66 V (the potential at half the capacity). This exceedingly large hysteresis in the first cycle suggests that the initial insertion of Zn2+ cations is largely irreversible and that the oxidative insertion of anions was deemed responsible for most of the charging capacity. The large potential hysteresis disappears after the first cycle, and the second discharge delivers a capacity of 226 mA h g−1 at a slightly smaller potential than the charging process, thereby indicating the excellent reversibility of the anion-hosting reaction. A self-discharge test was performed after the first cycle to assess the stability and reversibility of the charged electrode. After idling the charged electrode for 12 hours, the electrode delivered a Coulombic efficiency of 94.3%, suggesting the good stability and reversibility of the charge products in the electrode (Fig. S2†). Note that the first charge capacity with Zn2+ trapped is much larger than the case when we charged the Mn3O4 cathode directly with a capacity of 84 mA h g−1 (Fig. 1b and S3†). To further evaluate the effect of the initial Zn2+-trapping process, we cycled the Mn3O4 electrode against an activated carbon counter electrode with another high-concentration chloride-based aqueous electrolyte of 15 M tetraethylammonium chloride (TEACl) (Fig. S4†). The initial discharge capacity was only ∼13 mA h g−1, indicating that the bulky tetraethylammonium cation was not inserted into Mn3O4 to any meaningful extent. The subsequent charge capacity was ∼64 mA h g−1, which is closer to when we charged the pristine Mn3O4 electrode first in the 20 M ZnCl2 + 5 M NH4Cl WiSE. This significant capacity difference suggests that the initial trapping of Zn2+ cations transforms the Mn3O4 structure in such a way that it facilitates the anodic process of anion storage. The question is: Can the trapped Zn2+ ions promote the storage of other anions?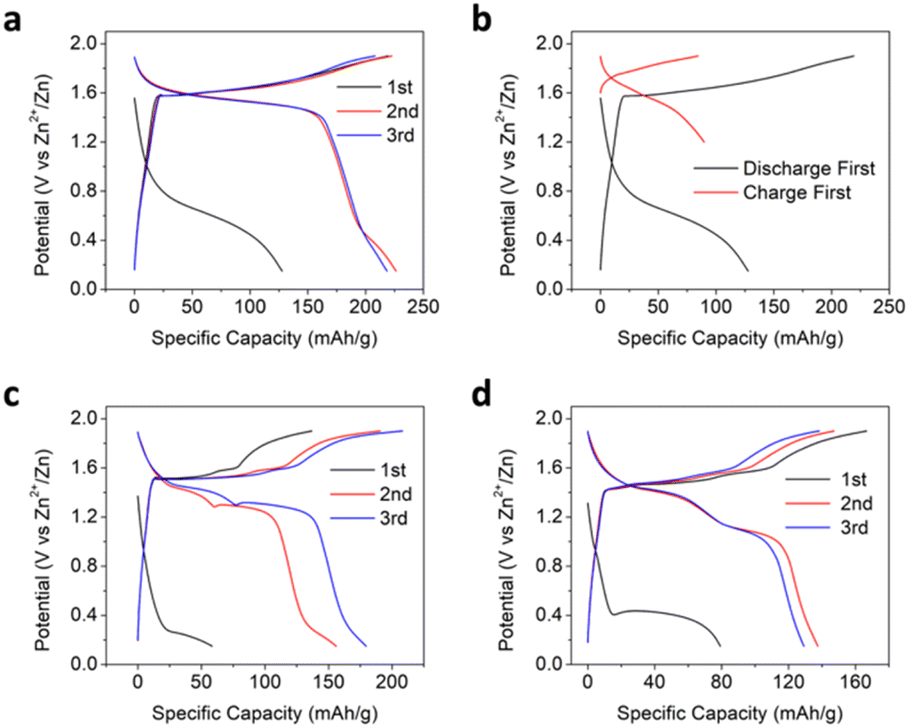 | ||
| Fig. 1 (a) Galvanostatic charge–discharge (GCD) potential profiles of the Mn3O4 electrode in the 20 M ZnCl2 + 5 M NH4Cl WiSE. (b) Comparison of the first cycle GCD profiles of the Mn3O4 electrode in the WiSE when it is discharged first to a lower cutoff potential of 0.2 V and when it is charged first with the lower cutoff potential raised to 1.2 V to eliminate Zn2+ insertion. GCD profiles of the Mn3O4 electrode in common aqueous Zn-ion electrolytes of (c), 2 M ZnSO4 and (d), 2 M Zn(ClO4)2. | ||
We also investigated the redox behaviors of the Mn3O4 electrode in common aqueous Zn-ion electrolytes of 2 M ZnSO4 and 2 M Zn(ClO4)2. In both electrolytes, the Mn3O4 electrode also exhibits a low-potential plateau around 0.4 V in the first discharge. However, the Mn3O4 electrode showed two apparent charge plateaus in these electrolytes, with the first plateau at potentials well below 1.66 V, the potential in the chloride-based WiSE (Fig. 1c and d). This two-plateau behavior is commonly observed for manganese oxide cathodes in mildly acidic aqueous electrolytes and is often attributed to H+ and Zn2+ coinsertion36,37 or a combination of Zn2+ (de)insertion and MnOx/Mn2+ dissolution/deposition reactions.38 The distinctly different redox behaviors of the Mn3O4 electrode in the chloride-based electrolyte and the non-chloride electrolytes are also displayed in the cyclic voltammetry (CV) curves, where Mn3O4 exhibits two distinct pairs of redox peaks in the 2 M ZnSO4 in contrast to one pair in the chloride WiSE (Fig. S5†). We conducted inductively coupled plasma optical emission spectroscopy (ICP-OES) after the second discharge, where the dissolved concentration of Mn in the ZnSO4 electrolyte corresponds to 3.6 times the observed discharge capacity. However, in the ZnCl2 WiSE electrolyte, the dissolution of Mn could only account for 4.3% of the observed discharge capacity. The results suggest that the operation of Mn3O4 in dilute electrolytes such as 2 M ZnSO4 occurs by the dissolution of Mn-ions and deposition of manganese oxides, where the Zn-trapping and the associated promoted anion storage are irrelevant.
Characterization of the operation of the Mn3O4 electrode in the ZnCl2-based WiSE electrode
Ex situ X-ray diffraction (XRD) results indicate significant amorphization of the Mn3O4 structure after the initial Zn2+ insertion (Fig. S6†). This loss in the long-range order could explain how the relatively large Cl−/Cl anions/atoms are able to be lodged within the compact structure after the following charge. The energy dispersive X-ray spectroscopy (EDS) elemental mapping associated with TEM confirms the Zn2+ insertion inside the structure of Mn3O4 after the initial discharge (Fig. 2a). Interestingly, the Zn content appears to be particularly enriched towards the surface of the Mn3O4 particles. Indeed, the high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) images indicate that the surface of the Mn3O4 particle is furnished with Zn2+ cations rather than O2− anions after discharging (Fig. 2b). Elemental mapping obtained after the subsequent charging process shows a uniform increase in Cl content, thereby indicating the insertion of Cl−/Cl anions/atoms (Fig. 2c). Of note, the Zn content is significantly more concentrated at the Mn3O4 surface after the charging process, as shown by the EDS mapping and the associated line scanning profile, which indicates that the inserted Zn2+ cations become trapped towards the surface of the particle (Fig. 2c and S7†) and the trapped Zn2+ cations are not static during cycling. | ||
| Fig. 2 (a) An HAADF-STEM image of the Mn3O4 electrode and associated EDS elemental mappings of the Mn, O, Cl, and Zn contents after the first discharge. (b) HAADF-STEM images of the near-surface region of the Mn3O4 before and after the first discharge, where the surface of the Mn3O4 is furnished with Zn-ions after the first discharge. (c) HAADF-STEM image of the Mn3O4 electrode and associated elemental mappings of the Mn, O, Cl, and Zn contents after the first charge. | ||
To reveal the charge storage mechanism of the Mn3O4 electrode in the chloride-based WiSE, we examined the Mn oxidation state at different state of charge (SoC) using ex situ synchrotron-based X-ray absorption spectroscopy (XAS).39–41 The Mn K-edge X-ray absorption near-edge structure (XANES) studies showed slight changes in the edge positions from the pristine, to the discharged, and charged samples (Fig. 3a and S8†). From fully discharged to fully charged in the first cycle, the valence state of Mn was calculated to increase by +0.09 (from Mn2.83+ to Mn2.92+). In theory, such an oxidation state change would result in a small specific capacity of only 31.6 mA h g−1. However, we obtained a much larger specific capacity of 218 mA h g−1 for the first charge of the Mn3O4 electrode (Fig. 1a). These very subtle changes in the oxidation state of Mn were corroborated by X-ray photoelectron spectroscopy (XPS), where the calculated oxidation states of Mn in the spectra of the pristine and charged electrodes were +2.63 and +2.75, respectively (Fig. 3b). Moreover, there were no obvious differences in the soft XAS (sXAS) spectra of Mn between the pristine, discharged, and charged samples (Fig. 3c), further indicating the lack of significant Mn redox involved in the charge storage mechanism of the Mn3O4 electrode in the chloride WiSE. This absence of the Mn redox in the (dis)charge processes of the Mn3O4 electrode supports the possibility of Cl/Cl− redox as being the major contributor to the observed capacity because only Cl− and water can be oxidized in the charging process other than Mn, where Cl− should be oxidized before water due to its lower redox potential at such a high concentration.
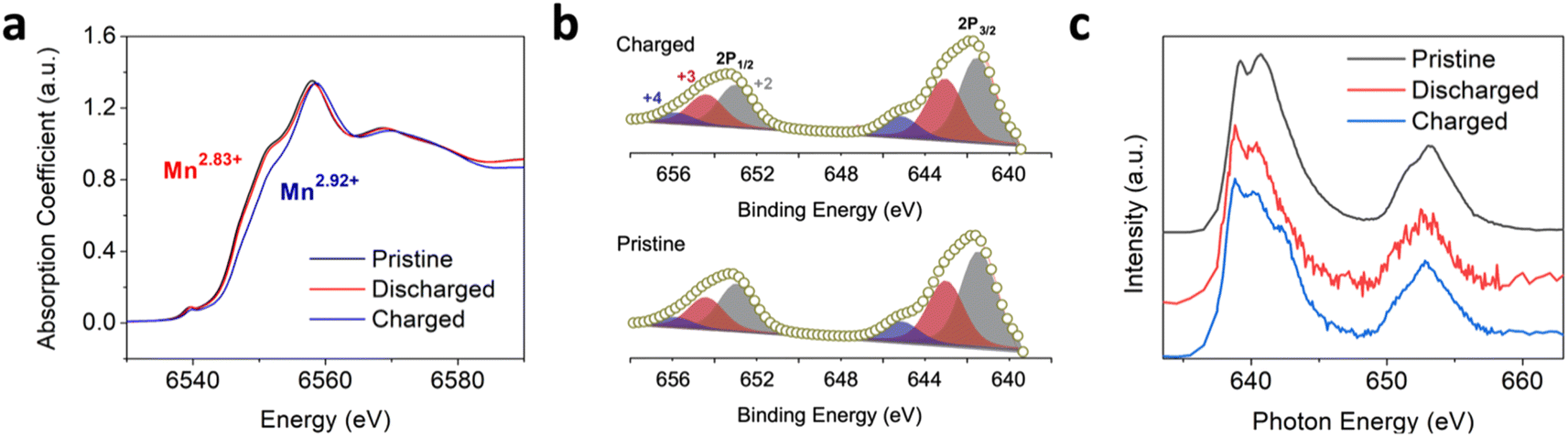 | ||
| Fig. 3 (a) Normalized XANES of the Mn K-edge spectra of the pristine Mn3O4 electrode and the Mn3O4 electrode after the initial discharge and charge. The calculated oxidation state changes from +2.83 to +2.92 from the fully discharged to the fully charged state of the Mn3O4 electrode in the first cycle. (b) XPS spectra of the charged (upper) and pristine (lower) Mn3O4 electrode with Mn 2p3/2 and Mn 2p1/2 profiles. (c) Mn L-edge sXAS spectra of the pristine, discharged, and charged Mn3O4 electrodes. | ||
Effect of Zn2+-trapping on Cl/Cl− plating/stripping and the generation of polychloride anions
To gain a theoretical understanding of how the initial Zn2+-trapping process facilitates the subsequent Cl−/Cl hosting and redox reaction, we conducted density functional theory (DFT) calculations to investigate the behavior of Cl insertion in Mn3O4 before and after Zn trapping. Fig. 4a and b exhibit the configurations with Zn inserted into the octahedral (Oh) site (denoted as ZnOh) and the tetrahedral (Td) site (denoted as ZnTd) of Mn3O4, respectively. We found that the Td site is slightly more energetically favorable by 0.12 eV than the Oh site for Zn. During geometry optimization, the Mn atoms adjacent to Zn spontaneously move from the Td sites to Oh sites, leading to the formation of a layer of Td-site vacancies, resulting in a characteristic of spinel-to-layered structure transformation. This structural transformation results in a deformation of the crystal structure, accompanied by a volume expansion of approximately 4%. The calculated insertion energy of Zn into Mn3O4 is −1.0 eV (Table S1†), suggesting the stability of the structure after the initial Zn-trapping process.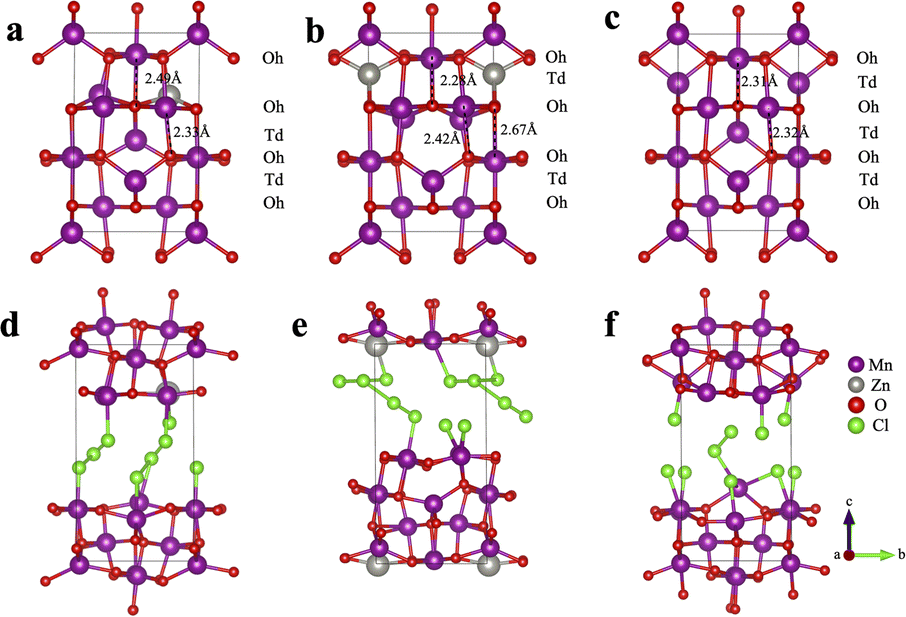 | ||
| Fig. 4 (a) The optimized structure of Zn trapped in the octahedral site in Mn3O4 (denoted as ZnOh). (b) The optimized structure of Zn trapped in the tetrahedral site in Mn3O4 (denoted as ZnTd). (c) The structure of pristine Mn3O4. (d–f) Optimized structures upon Cl insertion into ZnOh, ZnTd, and pristine Mn3O4, respectively. The stoichiometry is Zn0.25Mn3O4Cl1.75 for the Zn-trapped models and Mn3O4Cl1.75 for the pristine Mn3O4 model. | ||
Subsequently, we studied the behavior of Cl insertion in Mn3O4 following the initial Zn-trapping process. Interestingly, upon comparing the energetics of various potential Cl intercalation sites, the computation results suggest the formation of polychloride monoanions within the structures. Specifically, within the ZnOh model, the inserted Cl atoms form trichloride [Cl3]− anions, while within the ZnTd model, they formed T-shaped pentachloride [Cl5]− anions (Fig. 4d, e, and S9†).42–47 This unique finding sheds light on a new mechanism of Cl storage. The presence of these polychloride species helps explain the stability of “plated” chlorine species within the crystal structure at room temperature. Notably, the oxidation of chloride into polychlorides has been demonstrated to be thermodynamically favorable in highly concentrated acidic chloride.47 In our work, the WiSE electrolyte used is both highly acidic and features a high chloride concentration, making the oxidation of chloride into polychloride anions feasible. In contrast, for the pristine Mn3O4 structure without Zn-trapping, only a small amount of dichlorides (Cl2) was formed after the insertion of Cl atoms (Fig. S9c†). This finding provides an explanation for the considerably lower capacity observed in the pristine Mn3O4 electrode (Fig. 1b) in comparison to the Zn-trapped electrode. Furthermore, all three crystal structures experience significant volume expansion exceeding 40% upon Cl insertion. This substantial volume expansion may contribute to the observed amorphization suggested by the ex situ XRD patterns after cycling the electrode (Fig. S6†).
In order to assess the impact of the initial Zn-trapping on the Cl insertion process, we calculated the Cl insertion energy. Remarkably, the Cl insertion energies for the Zn-trapped models were found to be much lower compared to the pristine Mn3O4 model (1.9, 2.0, and 4.0 eV for the ZnOh, ZnTd, and Mn3O4 models, respectively). This observation indicates that the initial Zn-trapping process plays a crucial role in facilitating the oxidation of Cl− ions and the formation/insertion of polychloride ions, thus resulting in an enhancement in Cl storage capacity.
In addition, the hosting of these polychloride anions may explain the slight oxidation of Mn observed in the XAS and XPS spectra of the charged electrode. While the majority of the observed capacity can be attributed to the redox of the inserted Cl− species, a slight oxidation of Mn occurs to accommodate the negatively charged polychloride monoanions (Fig. 3a–c). This observation of slight oxidation in Mn aligns with the findings from the Bader charge analysis,48 where a slight increase in the Bader charge of Mn is observed after Cl insertion (Table S2†). Moreover, the COHP (crystal orbital Hamilton population) analysis49 reveals the contribution of antibonding states near the Fermi level to the Mn–Cl bonds in the ZnOh and ZnTd models in contrast to the pristine Mn3O4 model. This demonstrates that the chemical bonding between Mn and Cl is weaker in the Zn-trapped models, suggesting a facile extraction of Cl− species after Zn2+-trapping (Fig. S11†).
To confirm the formation and hosting of the polychloride monoanions predicted by the calculations, we conducted ultraviolet-visible (UV-vis) spectroscopy on the electrolyte after the charging process. Surprisingly, there was no noticeable difference between the spectra of the 20 M ZnCl2 + 5 M NH4Cl WiSE before and after charging the Mn3O4 electrode. This may be due to the strong binding between the polychloride monoanions and the host structure, considering the excellent self-discharge performance. Another explanation is that any diffused-out polychloride would have reacted with the Zn metal counter electrode before being detected by the UV-vis measurement of the electrolyte.50 To address the issue, we conducted the UV-vis on another highly concentrated chloride-based aqueous electrolyte of 15 M TEACl before and after charging the Mn3O4 electrode with an activated carbon free-standing film as the counter electrode (Fig. 5a). After charging, a broad peak appeared at ∼270 nm by the electrolyte, which is very likely indicative of the [Cl3]− anion.51,52 To further confirm the existence of the polychloride monoanions formed with the Mn3O4 structure after charging in the 20 M ZnCl2 + 5 M NH4Cl WiSE, we immersed the fully charged Mn3O4 electrode in an aqueous solution of 0.5 M KI. It was observed that the color of the KI solution quickly turned brownish, thereby indicating the presence of the polychloride monoanions as the charge products with the Mn3O4 host (Fig. 5b). In stark contrast, when the Mn3O4 electrode charged in the 2 M ZnSO4 electrolyte was immersed in the KI solution, the color of the solution did not change.
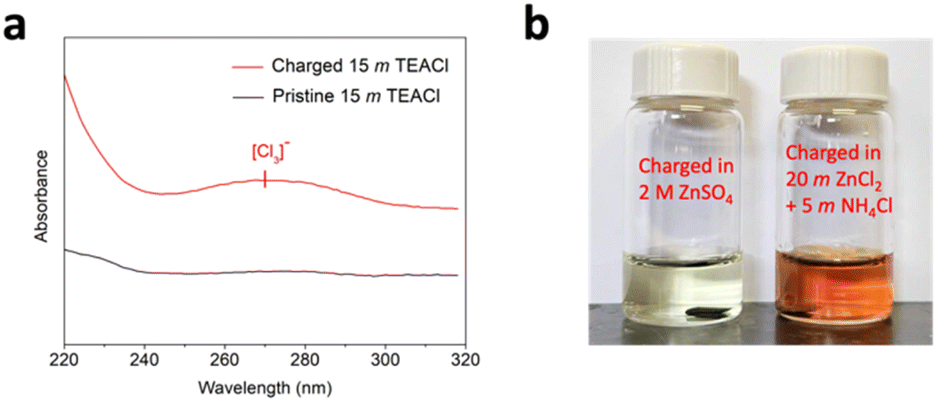 | ||
| Fig. 5 (a) UV-vis spectra of the pristine 15 M TEACl electrolyte and the 15 M TEACl electrolyte after the Mn3O4 electrode was charged in this electrolyte. (b) The 0.5 M KI solutions after immersing the Zn2+-trapped Mn3O4 electrode after charging in the 2 M ZnSO4 electrolyte (left) and after charging in the 20 M ZnCl2 + 5 M NH4Cl WiSE (right). | ||
Conclusions
In summary, we have demonstrated and characterized a new charge storage mechanism of reversible Cl/Cl− “plating/stripping” at a spinel Mn3O4 electrode in a high-concentration chloride-based aqueous electrolyte. The initial irreversible insertion of Zn2+ cations transforms the Mn3O4 structure in such a way that it helps facilitate the subsequent reversible Cl/Cl− redox reactions. The Mn3O4 host acts as a “semi-catalyst” that allows the oxidation of Cl− to Cl but not to Cl2 gas. Characterization by EDS, XAS, and XPS spectroscopies confirms that Mn redox is not responsible for most of the observed capacity. Instead, computation and experimental results suggest that the oxidized Cl− anions preferentially form polychloride monoanions when hosted within the Zn2+-trapped Mn3O4. Our results present a new strategy to use semi-catalysts as electrode materials that can effectively facilitate reversible Cl/Cl− “plating/stripping” reactions at room temperature.Experimental
Chemicals and materials
Nanoscale hausmannite Mn3O4 powder was synthesized by a room-temperature precipitation method.34,35 To begin, 1.126 g of MnSO4·H2O (Alfa Aesar) was dissolved into 150 mL of deionized (DI) water. The aqueous MnSO4 solution was then titrated with 29% NH4OH solution via dropwise additions under magnetic stirring until it reached a pH of 11. Once the solution was at pH 11, it was left to react under magnetic stirring for 1 h before being stored overnight at room temperature. The brown precipitate was then washed with DI water and centrifuged until a pH of 7 was obtained. Lastly, the precipitate was dried at 80 °C overnight to obtain the final Mn3O4 powder.For the preparation of the 20 M ZnCl2 + 5 M NH4Cl WiSE, anhydrous zinc chloride (metals basis, 99.95%) was purchased from Thermo Scientific. Ammonium chloride (ACS, 99.5%) and the water (HPLC grade) were purchased from Alfa Aesar.
Electrochemical measurements
Electrochemical tests were done using Swagelok cells. The Mn3O4 working electrodes were composed of 70 wt% active mass, 20 wt% KetjenBlack, and 10 wt% polyvinylidene fluoride binder coated on carbon fiber paper current collectors. The typical active mass loading was ∼2 mg cm−2. The Mn3O4 free-standing film electrodes used for obtaining the ex situ XRD patterns, self-discharge experiments, and the KI solution immersion tests were composed of 70 wt% active mass, 20 wt% KetjenBlack, and 10 wt% polytetrafluoroethylene (PTFE) binder. GCD measurements were obtained using a LANDT Battery Test System CT3002A, and CV tests were conducted with a VMP-3 multi-channel workstation.Materials characterization
XRD patterns were obtained using a Rigaku Ultima IV diffractometer with Cu Kα radiation (λ = 1.5406 Å) at a scan rate of 1° per minute. TEM data were recorded on an aberration-corrected (scanning) transmission electron microscope operated at 300 keV with a cold field-emission source (JEM-ARM300F Grand ARM). The energy dispersive X-ray spectroscopic (EDS) data were taken with dual 100 mm2 silicon drift detectors (SDD). Hard XAS was performed at the Advanced Photon Source 10-BM in transmission mode. XAS data was processed using Athena for pre-edge background subtraction and normalization. The Mn oxidation state was determined with XANES edge position at a normalized absorbance of 0.5 and linearly interpolated with Mn3O4 and Mn2O3 standards as +2.67 (6544.8 eV) and +3 (6548.7 eV) oxidation states, respectively. sXAS at Mn L-edge was performed using total electron yield (TEY) mode at beamline 7.3.1 of Advanced Light Source (ALS) of Lawrence Berkeley National Laboratory (LBNL).For XPS, a Physical Electronics Quantera II Hybrid was used for the analysis. The system energy scale was calibrated to Cu 2p3/2 at 932.6 eV and Au 4f at 84.0 eV. The base pressure of the system was 3 × 10−7 Pa. The XPS experiments were measured on as-loaded samples at room temperature. XPS was performed using monochromatized Al Kα radiation (hν = 1486.6 eV, at 50 watts and 100 μm beam diameter). The electron analyzer pass energy was set to 69 eV, with an emission angle of 45°. The specimens were neutralized using a combination of an electron flood gun set to 0.6 eV at 20 μA and an ion flood gun set to 0.1 kV. The XPS data were charge corrected to the C 1s aliphatic carbon binding energy at 284.8 eV. The XPS spectra were analyzed by fitting using CasaXPS software.
UV-vis spectroscopy analyses of the electrolytes were performed using a UV-vis-NIR spectrophotometer (PerkinElmer Lambda 750).
Theoretical calculations
DFT calculations were performed via the Vienna ab initio simulation package (VASP).53 The ion–electron interaction was depicted with the projector augmented wave (PAW) method. The electron exchange correlation was described by the Perdew, Burke, and Ernzerhof (PBE) functional with generalized gradient approximation (GGA).54 The cutoff energy of the plane-wave basis set was set to 450 eV. The Brillouin zone was sampled by (3 × 3 × 2) Γ-centered k-point mesh. The valence electrons of Mn, Zn, O, and Cl are 3d54s2, 3d104s2, 2s2sp4, and 3s23p5, respectively. The convergence threshold for structural optimization and electronic energy was set to be 0.01 eV Å−1 in force and 1 × 10−5 eV, respectively. The unit cell and atomic position were allowed to relax along with structure optimization. Spin polarization was considered in all computations. The DFT + U method was employed as the correction to simulate the strongly correlated materials, including transition metals.55 The values of U and J for Mn were set to be 4.5 and 0.5 eV, respectively.56,57In this work, the crystal structures were visualized using VESTA.58 The atomic net charge was generated with code developed by Henkelman's group via the Bader charge analysis.48 The crystal orbital Hamilton population (COHP) analysis was obtained using LOBSTER to analyze the chemical bond.49
The insertion energy of Zn (ΔEZI) and the insertion energy of Cl (ΔECI) were calculated by the following equations:
| ΔEZI = E(Bulk + Zn) − E(Bulk) − E(Zn) | (1) |
| ΔECI = E(Bulk + n × Cl) − E(Bulk) − n × 0.5E(Cl2) | (2) |
According to previous studies, the magnetic ordering of the Mn3O4 unit cell was set to be (↑↑↓↓↑↑).59
Data availability
Additional characterization and computation results are provided in the ESI.†Author contributions
S. K. Sandstrom: investigation, formal analysis, visualization, data curation, writing – original draft. Q. Li: investigation, formal analysis, visualization, data curation. M. Lyons: investigation, formal analysis, data curation. C.-W. Chang: investigation, formal analysis, data curation. R. Zhang: investigation, formal analysis, data curation. Y. Sui: investigation, formal analysis, data curation. H. Jiang: investigation. M. Yu: investigation. D. Hoang: investigation. W. F. Stickle: investigation, formal analysis, data curation. H. L. Xin: funding acquisition, writing – review and editing, supervision. Z. Feng: funding acquisition, writing – review and editing, supervision. D. Jiang: funding acquisition, writing – review and editing, supervision. X. Ji: conceptualization, funding acquisition, writing – review and editing, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
X. J. and D. J. thank the U.S. National Science Foundation (NSF) for the financial support with the Awards DMR 2221645 and DMR 2221646. M. L., C.-W. C. and Z. F. thank the U.S. NSF for financial support from CBET 2016192 and CBET 1949870. The hard XAS measurements were done at beamline 10-BM of The Materials Research Collaborative Access Team (MRCAT) at the Advanced Photon Source. MRCAT operations are supported by the Department of Energy (DOE) and the MRCAT member institutions. This research used resources of the Advanced Photon Source; a U.S. DOE Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. The soft X-ray absorption spectroscopy measurements were performed at beamline 6.3.1 of Advanced Light Source, which is an Office of Science User Facility operated for the U.S. DOE Office of Science by Lawrence Berkeley National Laboratory and supported by the DOE under Contract No. DEAC02-05CH11231. The UCI experiments were supported by the Office of Basic Energy Sciences of the U.S. Department of Energy, under award no. DE-SC0021204. R. Z.'s effort on this project was supported by HLX's startup funding. The authors thank Jesse M. Muratli and the Keck Collaboratory for Plasma Spectrometry at Oregon State University for ICP-OES measurements.Notes and references
- J. Song, K. Xu, N. Liu, D. Reed and X. Li, Mater. Today, 2021, 45, 191–212 CrossRef CAS.
- C. Bauer, S. Burkhardt, N. P. Dasgupta, L. A.-W. Ellingsen, L. L. Gaines, H. Hao, R. Hischier, L. Hu, Y. Huang, J. Janek, C. Liang, H. Li, J. Li, Y. Li, Y.-C. Lu, W. Luo, L. F. Nazar, E. A. Olivetti, J. F. Peters, J. L. M. Rupp, M. Weil, J. F. Whitacre and S. Xu, Nat. Sustain., 2022, 5(3), 176–178 CrossRef.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- K. Turcheniuk, D. Bondarev, V. Singhal and G. Yushin, Nature, 2018, 467–470 CrossRef CAS PubMed.
- W. Tahil, Implications of Future PHEV Production for Lithium Demand, Martainville: Meridian International Research, 2007 Search PubMed.
- S. Y. Hong, Y. Kim, Y. Park, A. Choi, N.-S. Choi and K. T. Lee, Energy Environ. Sci., 2013, 6(7), 2067–2081 RSC.
- Q. Li, Q. Wei, W. Zuo, L. Huang, W. Luo, Q. An, V. O. Pelenovich, L. Ma and Q. Zhang, Chem. Sci., 2017, 8(1), 160–164 RSC.
- W. Zhang, Y. Liu and Z. Guo, Sci. Adv., 2019, 5(5), eaav7412 CrossRef CAS PubMed.
- J. Mao, C. Wang, Y. Lyu, R. Zhang, Y. Wang, S. Liu, Z. Wang, S. Zhang and Z. Guo, J. Mater. Chem. A, 2022, 10(37), 19090–19106 RSC.
- L. Cao, D. Li, T. Pollard, T. Deng, B. Zhang, C. Yang, L. Chen, J. Vatamanu, E. Hu, M. J. Hourwitz, L. Ma, M. Ding, Q. Li, S. Hou, K. Gaskell, J. T. Fourkas, X.-Q. Yang, K. Xu, O. Borodin and C. Wang, Nat. Nanotechnol., 2021, 16(8), 902–910 CrossRef CAS PubMed.
- D. Dong, T. Wang, Y. Sun, J. Fan and Y.-C. Lu, Nat Sustainability, 2023, 1–11 Search PubMed.
- D. Zhao, X. Pu, S. Tang, M. Ding, Y. Zeng, Y. Cao and Z. Cheng, Chem. Sci., 2023, 14(30), 8206–8213 RSC.
- S. K. Sandstrom, X. Chen and X. Ji, Carbon Energy, 2021, 3(4), 627–653 CrossRef CAS.
- Q. Liu, Y. Wang, X. Yang, D. Zhou, X. Wang, P. Jaumaux, F. Kang, B. Li, X. Ji and G. Wang, Chem, 2021, 7(8), 1993–2021 CAS.
- F. P. McCullough, C. A. Levine and R. V. Snelgrove, US Pat., 4830938, 1989 Search PubMed.
- M. A. Reddy and M. Fichtner, J. Mater. Chem., 2011, 21(43), 17059–17062 RSC.
- X. Zhao, S. Ren, M. Bruns and M. Fichtner, J. Power Sources, 2014, 245, 706–711 CrossRef CAS.
- L. Winter and G. Degner, Minute Epics of Flight, 1933 Search PubMed.
- J. Jorné, J. T. Kim and D. Kralik, J. Appl. Electrochem., 1979, 9, 573–579 CrossRef.
- D. L. Thomas and D. N. Bennion, J. Electrochem. Soc., 1989, 136(12), 3553 CrossRef CAS.
- C. F. Holmes, Batteries for Implantable Biomedical Devices, 1986, pp. 133–180 Search PubMed.
- J. T. Kim and J. Jorné, J. Electrochem. Soc., 1977, 124(10), 1473 CrossRef CAS.
- J. T. Kim and J. Jorné, J. Electrochem. Soc., 1980, 127(1), 8 CrossRef CAS.
- K. A. Klinedinst and M. J. Domeniconi, J. Electrochem. Soc., 1980, 127(3), 539 CrossRef CAS.
- P. R. Gifford and J. B. Palmisano, J. Electrochem. Soc., 1988, 135(3), 650 CrossRef CAS.
- G. Zhu, X. Tian, H.-C. Tai, Y.-Y. Li, J. Li, H. Sun, P. Liang, M. Angell, C.-L. Huang, C.-S. Ku, W.-H. Hung, S.-K. Jiang, Y. Meng, H. Chen, M.-C. Lin, B.-J. Hwang and H. Dai, Nature, 2021, 596(7873), 525–530 CrossRef CAS PubMed.
- G. Zhu, P. Liang, C.-L. Huang, C.-C. Huang, Y.-Y. Li, S.-C. Wu, J. Li, F. Wang, X. Tian, W.-H. Huang, S.-K. Jiang, W.-H. Hung, H. Chen, M.-C. Lin, B.-J. Hwang and H. Dai, J. Am. Chem. Soc., 2022, 144(49), 22505–22513 CrossRef CAS PubMed.
- S. Hou, L. Chen, X. Fan, X. Fan, X. Ji, B. Wang, C. Cui, J. Chen, C. Yang, W. Wang, C. Li and C. Wang, Nat. Commun., 2022, 13(1), 1281 CrossRef CAS PubMed.
- J. Xu, T. P. Pollar, C. Yang, N. K. Dandu, S. Tan, J. Zhou, J. Wang, X. He, X. Zhang, A.-M. Li, E. Hu, X.-Q. Yang, A. Ngo, O. Borodin and C. Wang, Joule, 2023, 7(1), 83–94 CrossRef CAS.
- Q. Guo, K.-I. Kim, S. Li, A. M. Scida, P. Yu, S. K. Sandstrom, L. Zhang, S. Sun, H. Jiang, Q. Ni, D. Yu, M. M. Lerner, H. Xia and X. Ji, ACS Energy Lett., 2021, 6(2), 459–467 CrossRef CAS.
- Y. Sui, M. Lei, M. Yu, A. Scida, S. K. Sandstrom, W. Stickle, T. D. O'Larey, D.-e. Jiang and X. Ji, ACS Energy Lett., 2023, 8(2), 988–994 CrossRef CAS.
- C. Yang, J. Chen, X. Ji, T. P. Pollard, X. Lü, C.-J. Sun, S. Hou, Q. Liu, C. Liu, T. Qing, Y. Wang, O. Borodin, Y. Ren, K. Xu and C. Wang, Nature, 2019, 569(7755), 245–250 CrossRef CAS PubMed.
- G. Liang, B. Liang, A. Chen, J. Zhu, Q. Li, Z. Huang, X. Li, Y. Wang, X. Wang, B. Xiong, X. Jin, S. Bai, J. Fan and C. Zhi, Nat. Commun., 2023, 14(1), 1856 CrossRef CAS PubMed.
- H. Jiang and X. Ji, Carbon Energy, 2020, 2(3), 437–442 CrossRef CAS.
- H. Jiang, Z. Wei, L. Ma, Y. Yuan, J. J. Hong, X. Wu, D. P. Leonard, J. Holoubek, J. J. Razink, W. F. Stickle, F. Du, T. Wu, J. Lu and X. Ji, Angew. Chem., 2019, 131(16), 5340–5345 CrossRef.
- W. Sun, F. Wang, S. Hou, C. Yang, X. Fan, Z. Ma, T. Gao, F. Han, R. Hu, M. Zhu and C. Wang, J. Am. Chem. Soc., 2017, 139(29), 9775–9778 CrossRef CAS PubMed.
- Y. Jin, L. Zou, L. Liu, M. H. Engelhard, R. L. Patel, Z. Nie, K. S. Han, Y. Shao, C. Wang, J. Zhu, H. Pan and J. Liu, Adv. Mater., 2019, 31(29), 1900567 CrossRef PubMed.
- X. Guo, J. Zhou, C. Bai, X. Li, G. Fang and S. Liang, Mater. Today Energy, 2020, 16, 100396 CrossRef.
- M. Wang and Z. Feng, Chem. Commun., 2021, 57(81), 10453–10468 RSC.
- M. Wang and Z. Feng, Curr. Opin. Electrochem., 2021, 30, 100803 CrossRef CAS.
- M. Wang, L. Árnadóttir, Z. J. Xu and Z. Feng, Nano-Micro Lett., 2019, 11, 1–18 CrossRef PubMed.
- H. Keil, K. Sonnenberg, C. Müller, R. Herbst-Irmer, H. Beckers, S. Riedel and D. Stalke, Angew. Chem., Int. Ed., 2021, 60(5), 2569–2573 CrossRef CAS PubMed.
- F. A. Redeker, H. Beckers and S. Riedel, Chem. Commun., 2017, 53(96), 12958–12961 RSC.
- M. P. Bogaard, J. Peterson and A. D. Rae, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1981, 37(7), 1357–1359 CrossRef.
- R. Brückner, H. Haller, M. Ellwanger and S. Riedel, Chem.–Eur. J., 2012, 18(18), 5741–5747 CrossRef PubMed.
- J. Taraba and Z. Zak, Inorg. Chem., 2003, 42(11), 3591–3594 CrossRef CAS PubMed.
- D. Degoulange, G. Rousse and A. Grimaud, ACS Energy Lett., 2023, 8, 4397–4405 CrossRef CAS.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21(8), 084204 CrossRef CAS PubMed.
- V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, J. Phys. Chem. A, 2011, 115(21), 5461–5466 CrossRef CAS PubMed.
- A. Van den Bossche, E. De Witte, W. Dehaen and K. Binnemans, Green Chem., 2018, 20(14), 3327–3338 RSC.
- H. Sun, L. Yu, X. Jin, X. Hu, D. Wang and G. Z. Chen, Electrochem. Commun., 2005, 7(7), 685–691 CrossRef CAS.
- B. Wen, C. Yang, J. Wu, J. Liu, B. Li, J. Yang and Y. Liu, ACS Energy Lett., 2023, 8, 4204–4209 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54(16), 11169 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77(18), 3865 CrossRef CAS PubMed.
- V. I. Anisimov, J. Zaanen and O. K. Andersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44(3), 943 CrossRef CAS PubMed.
- Y.-F. Li, S.-C. Zhu and Z.-P. Liu, J. Am. Chem. Soc., 2016, 138(16), 5371–5379 CrossRef CAS PubMed.
- Y.-F. Li and Z.-P. Liu, J. Am. Chem. Soc., 2018, 140(5), 1783–1792 CrossRef CAS PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44(6), 1272–1276 CrossRef CAS.
- C. Franchini, R. Podloucky, J. Paier, M. Marsman and G. Kresse, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75(19), 195128 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc04545e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |