
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSkeletal rearrangement through photocatalytic denitrogenation: access to C-3 aminoquinolin-2(1H)-ones†
Swati
Singh
 ,
Gopal
Chakrabortty
and
Sudipta
Raha Roy
*
,
Gopal
Chakrabortty
and
Sudipta
Raha Roy
*
Department of Chemistry, Indian Institute of Technology Delhi, Hauz Khas, New Delhi, 110016, India. E-mail: srr@chemistry.iitd.ac.in
First published on 17th October 2023
Abstract
The addition of an amine group to a heteroaromatic system is a challenging synthetic process, yet it is an essential one in the development of many bioactive molecules. Here, we report an alternative method for the synthesis of 3-amino quinolin-2(1H)-one that overcomes the limitations of traditional methods by editing the molecular skeleton via a cascade C–N bond formation and denitrogenation process. We used TMSN3 as an aminating agent and a wide variety of 3-ylideneoxindoles as synthetic precursors for the quinolin-2(1H)-one backbone, which demonstrates remarkable tolerance of sensitive functional groups. The control experiments showed that the triazoline intermediate plays a significant role in the formation of the product. The spectroscopic investigation further defined the potential reaction pathways.
Introduction
Selective inclusion of amine functionality on the N-heteroaromatic ring is always a fascinating tool for pharmaceutical industries as it is present in one-third of small-molecule marketed drugs.1 In this regard, as a prominent heterocycle, 3-aminated quinolin-2(1H)-one, exhibits impressive pharmacological effects, including anticancer activity,2 KCNQ2 channel opener,3 neuronal maxi-K channel activator,4 antitubercular agent and inhibitor of the Hsp90 protein (Scheme 1a).5 Moreover, selective N-heteroaromatic amination can also be a building block of diverse synthetic scaffolds.6 Conventionally, introducing an amine group on the quinolin-2(1H)-one ring is conducted by electrophilic aromatic nitration and subsequent reductive hydrogenation (Scheme 1b).7 The regioselectivity in this procedure must be precisely controlled, and it demands extremely acidic conditions.8 Other approaches that are frequently utilized to synthesize 3-aminated quinolin-2(1H)-ones are the Hofmann rearrangement of quinolinone-3-carboxylic acid and the cyclization process of N-chloroacetyl-ortho-aminobenzophenone derivatives.9 Later, Messaoudi and group disclosed the copper-catalyzed approach for 3-aminoquinolinones from 3-bromoquinolinones at elevated temperatures (Scheme 1b).10 As a result, it continues to motivate the synthetic community to develop new methodologies to overcome the associated challenges, such as harsh conditions and site selectivity, and forgo prefunctionalized precursors.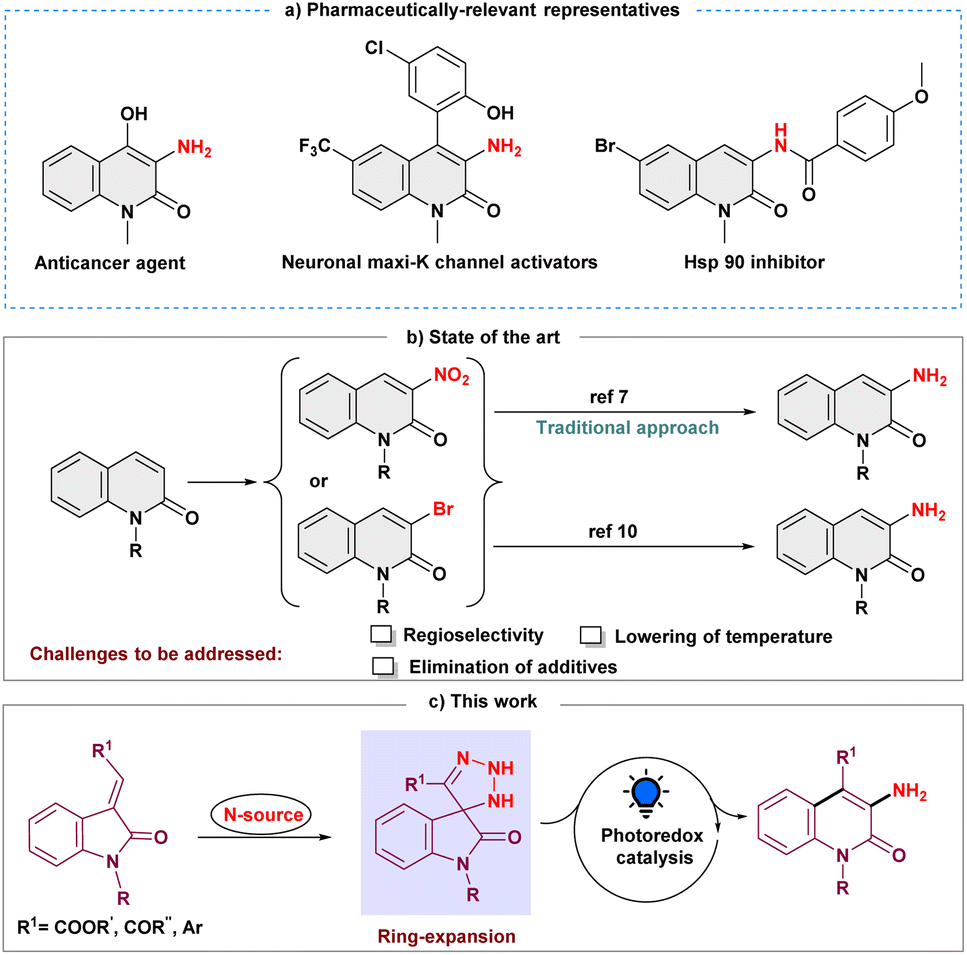 | ||
| Scheme 1 Photoinduced C-3 amination of quinolin-2-ones and the reported approaches. | ||
In recent years, skeletal editing has drawn attention in the synthetic community to address inaccessible reactions and maintain the selectivity that is difficult to achieve with peripheral C–H bond editing.11 To meet these demands, several efforts have been made to synthesize pyridine scaffolds from pyrrole's backbone, including the well-known Ciamician–Dennstedt rearrangement, which provides 3-halopyridines through the haloform-derived carbene. The Dimroth rearrangement is yet another skeletal editing reaction in which the endocyclic and exocyclic nitrogen substituents switch their place to form a six-membered N-heterocycle.12 Similarly, synthetic procedures using an isatin moiety as a skeletal editing precursor have been reported for the synthesis and subsequent functionalization of quinolin-2-ones.13 Encouraged by these skeletal editing strategies, we envisioned that easily accessible 3-ylideneoxindoles could also serve as a synthetic precursor for the quinolin-2-one backbone, and thus, the development of a method for obtaining selective 3-aminated quinolin-2(1H)-ones from 3-ylideneoxindoles under milder conditions remains an intriguing objective.
Moreover, with the ability to act as electrophilic, nucleophilic, and radical acceptors, azides are a potent synthon for introducing amines into the organic framework through the irreversible expulsion of dinitrogen.14 Additionally, it has been demonstrated that azides efficiently undergo a 1,3-dipolar cycloaddition reaction with alkenes and alkynes constituting triazoline and triazole rings, respectively.15 However, the feasibility of using azide as an amine precursor in skeletal editing has not been extensively investigated. Thus, while the synergistic effect of the cycloaddition and ring expansion process for amination is appealing, converting the intermediate triazoline ring into an amine moiety at ambient temperature is still challenging.16 Our perception was that the inclusion of a photocatalyst would turn the nucleophilic skeletal expansion process into a radical one, making the endeavor possible. The discovery by Li and Yang of visible-light mediated denitrogenative coupling using benztriazoles for radical amination provided strong support for our hypothesis.17 Here, we present a novel photoinduced skeletal expansion strategy for regioselective denitrogenative amination to access C-3 aminoquinolin-2(1H)-ones (Scheme 1c).
Results and discussion
To evaluate the viability of the photoinduced amination process along with the search for suitable reaction conditions, we initiated our investigations employing ethyl (E)-2-(1-methyl-2-oxoindolin-3-ylidene)acetate (1a) as a model substrate and azidotrimethylsilane (TMSN3) (2) as an aminating agent (Table 1, see the ESI† for more details). After a thorough investigation of commercially available photocatalysts (Table 1, entries 1–4), we found that Ir(dFppy)3 (PC4), using 4-dimethylaminopyridine (DMAP) as the base in MeCN/H2O mixed solvent and irradiated with a 3 W blue LED, gave us the desired product 3a in an 82% yield. With [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 (PC1), however, the desired product was obtained in a yield of 40%. The desired product could not be obtained using alternative photocatalysts, including Ru(dtbbpy)3(PF6)2 (PC2) or MesAcr+BF4− (PC3). We found no significant improvement when we replaced DMAP with other common bases, such as the organic bases DABCO and DIPEA or the inorganic base K2CO3 (Table 1, entry 5). Subsequently, evaluation of different solvents, such as DCE and tBuOH demonstrated that MeCN consistently produced a higher yield (Table 1, entry 6). The addition of water was notable, which had a substantial beneficial effect on the formation of 3a (Table 1, entry 7). In addition, when we used sodium azide (NaN3) as an alternate aminating agent, it did not provide the expected product (Table 1, entry 8). The significance of a base, a photocatalyst, and light on our model reaction (Table 1, entry 9–11) to generate the desired aminated product 3a was implied by control experiments.|
|
||
|---|---|---|
| Entry | Variations from above | Yieldb (%) |
| a Reaction conditions: unless otherwise specified, 3-ylideneoxindoles (1a) (0.2 mmol), 2 (0.4 mmol), DMAP (0.24 mmol), and PC (2 mol%) in 40 μL of water and 1.6 mL solvent irradiated with a blue LED (455 nm) for 24 h under an argon atmosphere. b yields determined by NMR using trichloroethylene as a standard. c Under dark conditions. | ||
| 1 | None | 82 |
| 2 | PC1 instead of PC4 | 40 |
| 3 | PC2 instead of PC4 | n.r. |
| 4 | PC3 instead of PC4 | n.r. |
| 5 | DABCO/DIPEA/K2CO3 instead of DMAP | 33/63/34 |
| 6 | DCE/tBuOH instead of CH3CN | 41/42 |
| 7 | MeCN without the addition of 40 μL H2O | 53 |
| 8 | NaN3 instead of TMSN3 | n.r. |
| 9 | No DMAP | n.r. |
| 10 | No PC4 | Trace |
| 11c | No light | n.r. |
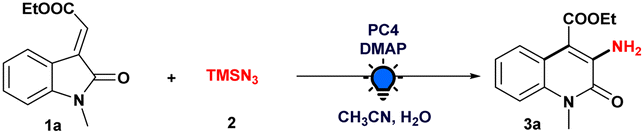
Furthermore, X-ray crystallography and NMR analysis confirmed the formation of 3a and also helped us to pinpoint the amine moiety on the quinolin-2(1H)-one ring. Though Croix et al. revealed the cycloaddition-ring expansion of the 3-substituted 1H-pyrrolo[2,3-b]pyridin-2(3H)-one derivative with an azide source at elevated temperature under microwave irradiation, they produce mixtures of the two regioisomeric aminated product 3- and 4-amino-naphthyridin-2(1H)-one derivatives with less preference towards the 3-aminated one.18
Therefore, after optimizing the reaction conditions, we explored the scope of ethyl (E)-2-oxoindolin-3-ylidene acetate derivatives to produce a 3-aminated quinolin-2(1H)-one derivative in order to establish the generality of the photoinduced heteroaryl amination process (Scheme 2). To begin, we evaluated our standard reaction conditions on a series of N-alkylated 3-ylideneoxindole derivatives in the presence of TMSN3. N-Isopropyl, N-benzyl 3-ylideneoxindoles, and N-cyclopropyl methyl 3-ylideneoxindoles, in addition to our model substrate 1a, which afforded the corresponding aminated heteroarenes (3a–d). Intriguingly, the cyclopropyl ring was preserved in the final product when ethyl (E)-2-(1-(cyclopropylmethyl)-2-oxoindolin-3-ylidene)acetate was used. Furthermore, even an unprotected 3-ylideneoxindole derivative was tolerated under optimized reaction conditions and provided the desired product 3e in 85% yield. In addition, N-alkyl groups containing double and triple-bond functional moieties were preserved during the course of the photoinduced reaction, and moderate to good yields of the desired products (3f–g) were obtained.
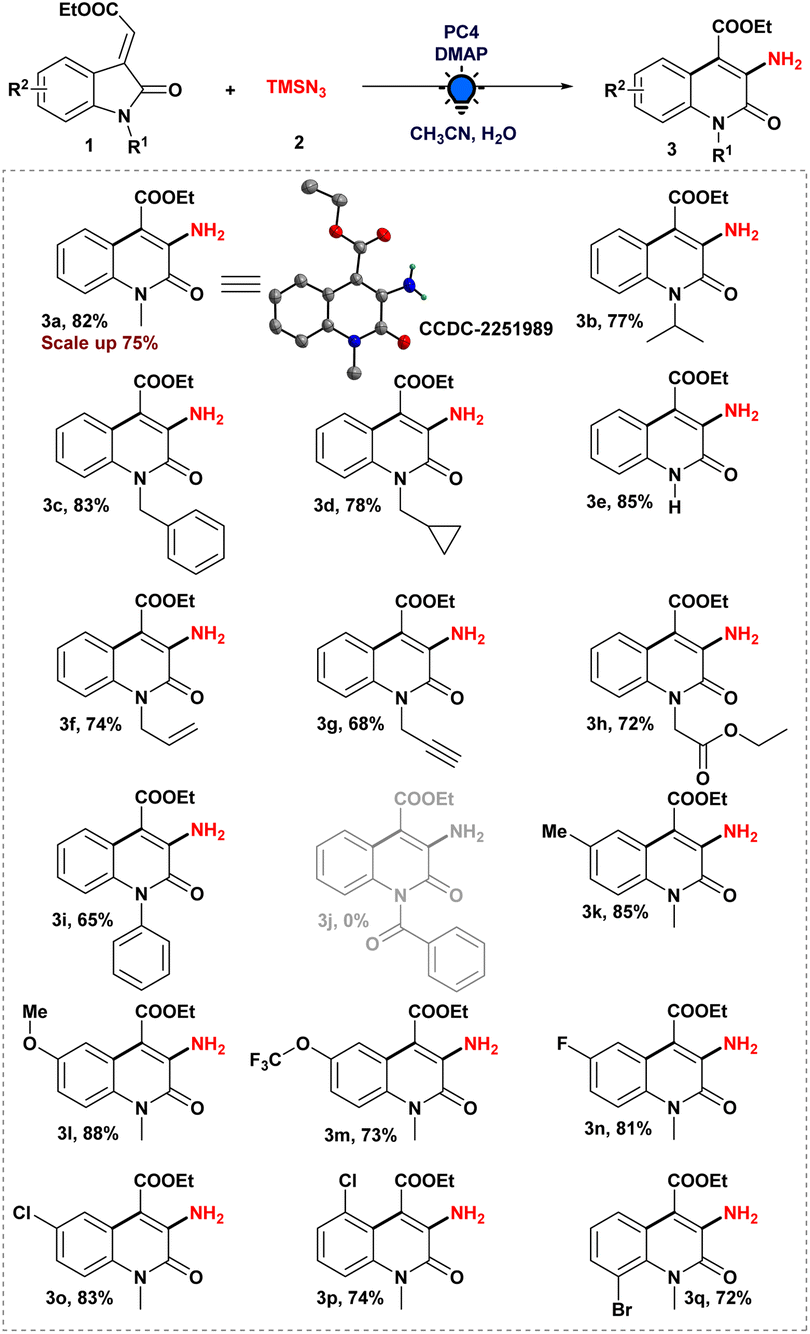 | ||
| Scheme 2 Scope of the 3-ylidene oxindoles. Reaction conditions: unless otherwise specified, 3-ylideneoxindoles (1) (0.2 mmol), 2 (0.4 mmol), DMAP (0.24 mmol), and PC4 (2 mol%) in 40 μL of water and 1.6 mL acetonitrile irradiated with a blue LED (455 nm) for 24 h under an argon atmosphere. | ||
Also, 3-ylideneoxindoles containing N-alkyl group having an electron-withdrawing substituent (–CH2CO2Et) were capable of producing the aminated product (3h) with a yield of 72%. In our optimized reaction system, N-phenyl 3-ylideneoxindoles furnished the expected product (3i) with excellent yield. Unfortunately, N-benzoyl 3-ylideneoxindoles failed to provide the desired product (3j) in our developed strategy.
Then, we tried to understand the impact of the electronic effect on our reaction strategy by anchoring electron-rich to electron-poor systems with 3-ylideneoxindole derivatives. We were gratified to find that our developed protocol was successful in producing good to excellent yields of the corresponding product (3k–l) from 3-ylideneoxindoles bearing –Me and –OMe groups. Moreover, 3-ylideneoxindoles substituted with –OCF3, –F, –Cl, and –Br groups were well tolerated, affording the desired 3-amino quinolone derivatives (3m–q) in decent yield. It was worth mentioning that the position of the halo groups on the aromatic ring did not restrict the ring expansion process, which was the crucial step in the synthesis of 3-amino quinolone derivatives. To investigate the synthetic potential of our amination methodology in a more efficient manner, a tenfold scale-up reaction was conducted under optimized reaction conditions, and to our delight, we obtained a 75% yield of the aminated product 3a, demonstrating the practical applications of our reaction system.
Subsequently, we studied the scope of various functionalities on the olefinic bond of oxindoles. As shown in Scheme 3, ester-substituted 3-ylideneoxindoles with alkyl as well as aryl groups were well tolerated in the system giving the desired product (3r–u) with 71 to 87% yield.
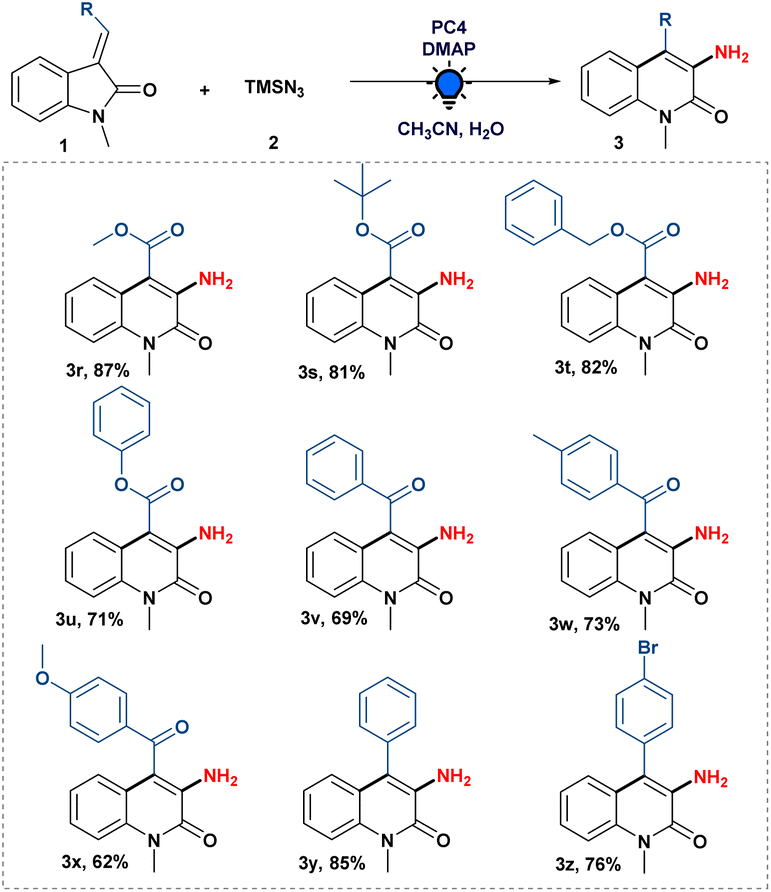 | ||
| Scheme 3 Scope of the 3-ylidene oxindoles. Reaction conditions: unless otherwise specified, 3-ylideneoxindoles (1) (0.2 mmol), 2 (0.4 mmol), DMAP (0.24 mmol) and PC4 (2 mol%) in 40 μL of water and 1.6 mL acetonitrile irradiated with a blue LED (455 nm) for 24 h under an argon atmosphere. | ||
Next, we examined the compatibility of benzoyl substitution on the olefinic bond of oxindoles to produce 3-amino-4-benzoyl-1-methylquinolin-2(1H)-one (3v). It is crucial to recognize that such moieties have never been synthesized. Subsequently, we looked into the amination reaction using (E)-1-methyl-3-(2-oxo-2-phenylethylidene)indolin-2-one attached with an electron-donating (–OMe and –Me) group, which gave us good to excellent yields of the desired compound (3w–x). Moreover, 3-amino-4-phenyl-1-methylquinolin-2(1H)-one (3y) was also obtained by the developed amination strategy with (E)-3-benzylidene-1-methylindolin-2-one. When (Z)- and (E)-3-(4-bromobenzylidene)-1-methylindolin-2-one were treated separately with our optimized conditions, we were pleased to know that a halogen (–Br) group substituted on the phenyl ring of the olefinic bond of oxindoles was well tolerated to give the expected product (3z) in good yield, regardless of the stereochemistry of the 3-ylideneoxindoles. It is evident from the literature that 3-benzylidene-1-methylindolin-2-one derivatives could function as a photo-switch when exposed to light;19 therefore, photoisomerization of (Z)- and (E)-3-(4-bromobenzylidene)-1-methylindolin-2-one during the optimised reaction conditions cannot be ruled out to get the desired product 3z (see the ESI†).
The significance of continuous visible-light irradiation was also demonstrated by means of subsequent light on/off studies, and the graph illustrates the requirement for light radiation for this skeletal editing approach (see the ESI† for details). The reaction process and the purpose of the photocatalyst or other additives were then investigated through a series of control experiments (Scheme 4). When the model reaction was performed using the radical scavenger BHT, the reaction was completely suppressed, and the involvement of radical intermediacy during this transformation was ascertained. Moreover, the BHT-adduct of radical intermediate (4) was detected by HR-MS (Scheme 4a).
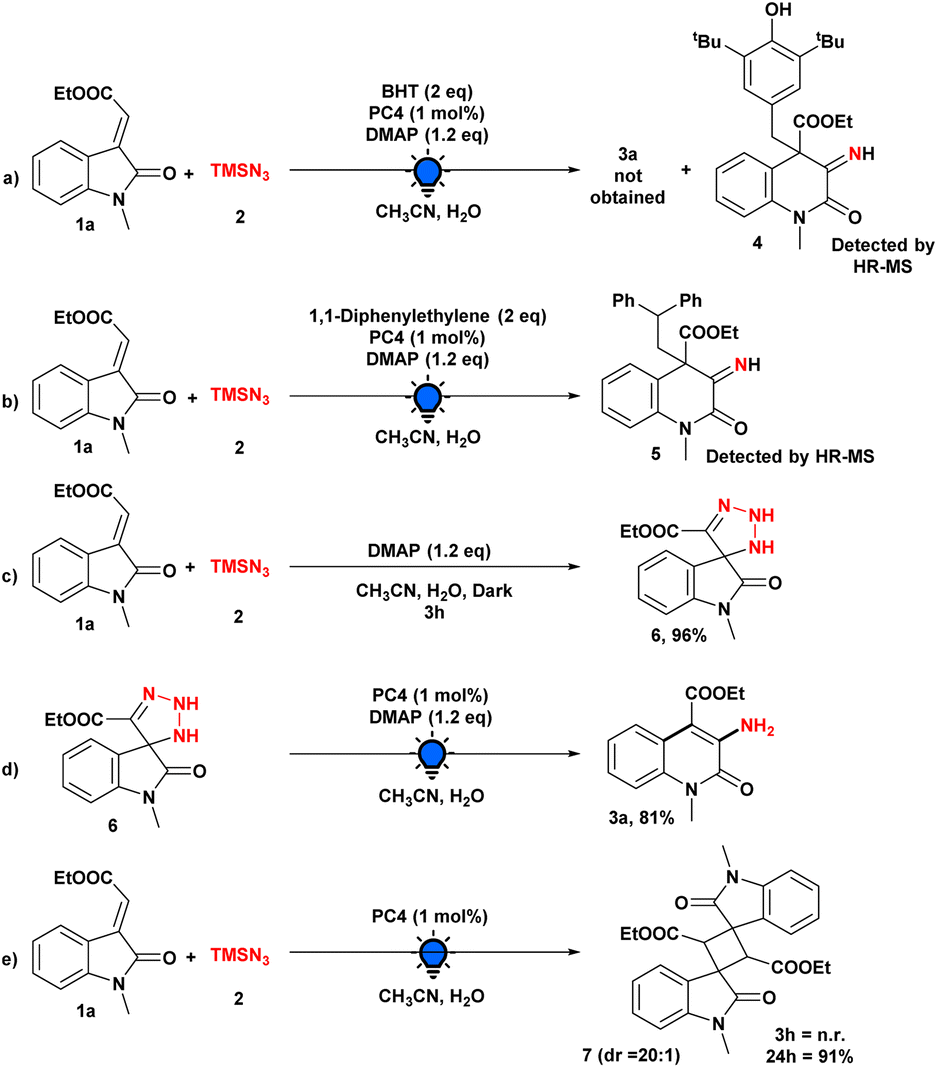 | ||
| Scheme 4 Control experiments. | ||
In addition, we were able to detect the radical intermediate adduct (5) by introducing 1,1-diphenylethylene into the model reaction and analyzing an aliquot of the reaction mixture with HR-MS (Scheme 4b). We then performed our model reaction with and without PC4 in the dark for 3 h; in both cases, we obtained a triazoline intermediate (6), which was isolated and characterized by NMR spectroscopy, demonstrating that the photocatalyst does not play a substantial role in this step (Scheme 4c). In addition, when isolated triazoline intermediate (6) was used under optimized conditions, the desired product was produced in 96% yield, providing additional evidence that the reaction pathway included a triazoline intermediate (Scheme 4d). Moreover, when we performed the model reaction in the absence of the base, there was no formation of any product after 3 h (Scheme 4e). However, after 24 h, we observed the 2 + 2 cycloaddition product 7 rather than the desired product 3a, demonstrating the essential role of the base.20
To substantiate the results obtained from control experiments, various spectroscopic studies were performed. UV-vis studies exclude the possibility of complex formation between PC4, 1a, TMSN3, and DMAP at the ground state (see the ESI† for details). To ascertain the exact role of a photocatalyst during this photoinduced skeletal rearrangement amination process, we studied the steady-state emission spectra of the photocatalyst (PC4) in the presence of TMSN3 (2), DMAP, and the 6 separately and conceived the Stern–Volmer plot (Fig. 1A and B). The sequential addition of 6 to a solution of PC4 (0.02 M) resulted in a steady decrease in the fluorescence intensity of PC4. In contrast, the sequential addition of 2 or DMAP showed only a minor decrease in the emission intensity of PC4. In addition, a higher level of quenching of PC4 emission intensity was observed with 1a (see the ESI†), which further supported the possibility of a [2 + 2] cycloaddition product as a side product obtained during control experiments. However, we can anticipate that the faster rate of triazoline intermediate formation perhaps completely suppressed the [2 + 2] cycloaddition reaction as we observed from control experiments (Scheme 4c and e). Furthermore, the Stern–Volmer plots of PC4 with increasing concentrations of triazoline intermediate (6), DMAP, and TMSN3 revealed that 6 contributes significantly to the steady-state fluorescence quenching of PC4 in the excited state.
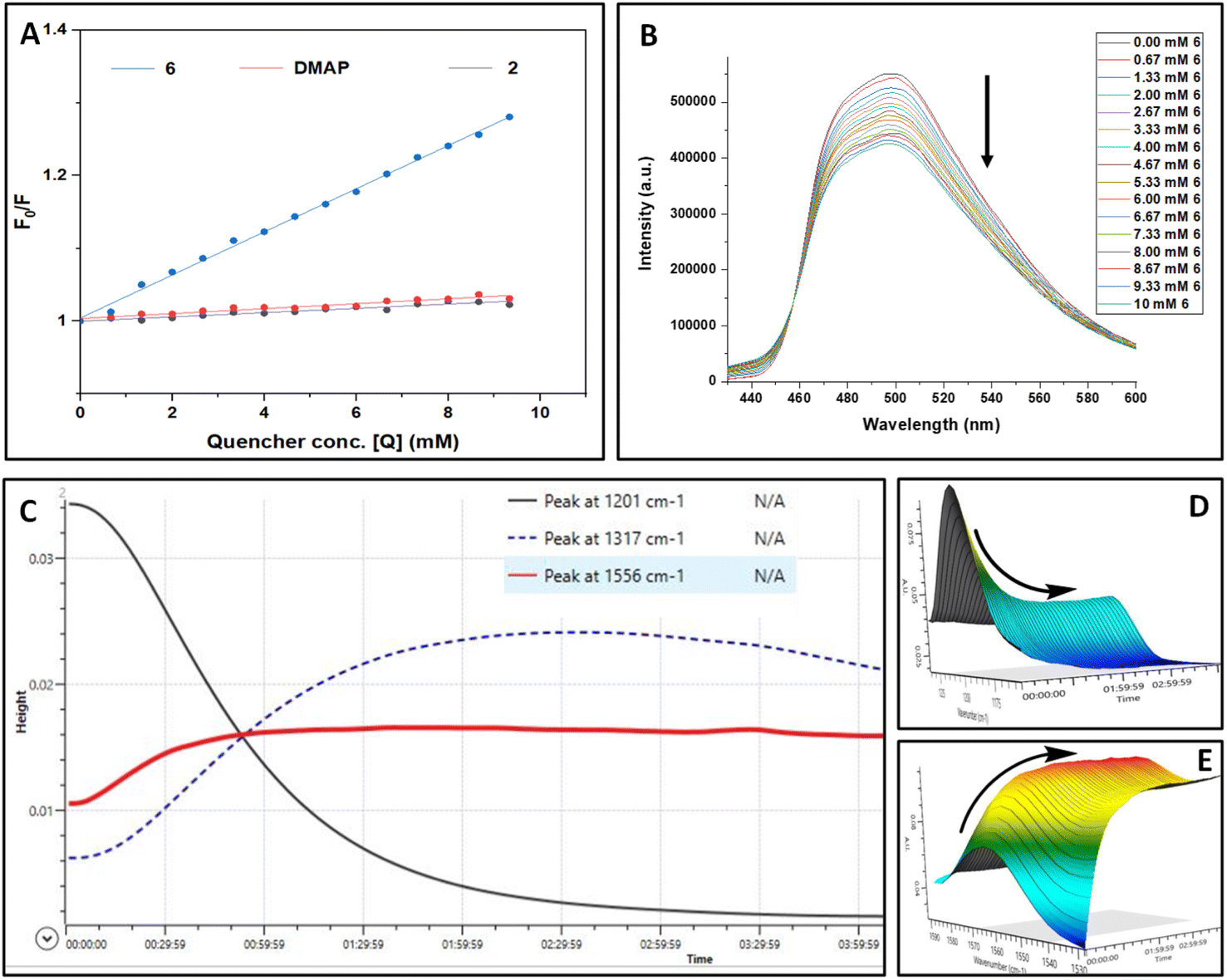 | ||
Fig. 1 (A) Combined Stern–Volmer plot of a solution of PC4 (0.02 M) in acetonitrile with 6 (blue line), DMAP (red line), and 2 (black line) as the quencher. (B) Fluorescence quenching spectra of a solution of PC4 (0.02 M) in acetonitrile with 6 (0.2 M) as the quencher. (C) ReactIR reaction progress data. (D–E) Surface spectral changes in the ReactIR reaction profile for 1a decreasing (black line, ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) 1201 cm−1, D), 3a increasing (red line, 1556 cm−1, E), and 6 ( 1317 cm−1, blue dotted line). 1201 cm−1, D), 3a increasing (red line, 1556 cm−1, E), and 6 ( 1317 cm−1, blue dotted line). | ||
In addition, in situ FTIR analysis revealed that as the reaction progresses, there is rapid consumption of 1a to form triazoline intermediates 6 (Fig. 1C–E). The reaction profile depicted in Fig. 1C shows that the peak for 1a (black line, : 1201 cm−1) gradually decreases, whereas the peak for 6 (blue dotted line, : 1317 cm−1) first increased gradually and then decreased, demonstrating the involvement of intermediate 6 which transformed into the product 3a (red line, : 1556 cm−1).
Based on the aforementioned mechanistic studies and literature precedents, a plausible mechanism for the amination strategy is depicted in Scheme 5. Initially, we reasoned that TMSN3 reacts with H2O and DMAP generates an azide anion (N3−), which further participates in [3 + 2] cycloaddition with the exocyclic olefinic bond of 1a to generate the triazoline intermediate 6. Then, upon irradiation with light, Ir(III) transformed into Ir(III)* which was then involved in SET with the 6 to form the triazoline radical cation A. This procedure conforms to the results of the fluorescence quenching experiment. Then, the deprotonation of A with the assistance of DMAP leads to the formation of the corresponding radical intermediate B. Furthermore, the ring-expansion process facilitates the formation of another radical intermediate C, and, subsequently, the elimination of nitrogen (N2) to generate an intermediate D, which further rearranges and is involved in SET with a reduced Ir(II) to produce anionic intermediate F. Finally, the protonation process gave intermediate G followed by the enolization process, which delivers the desired aminated product 3a.
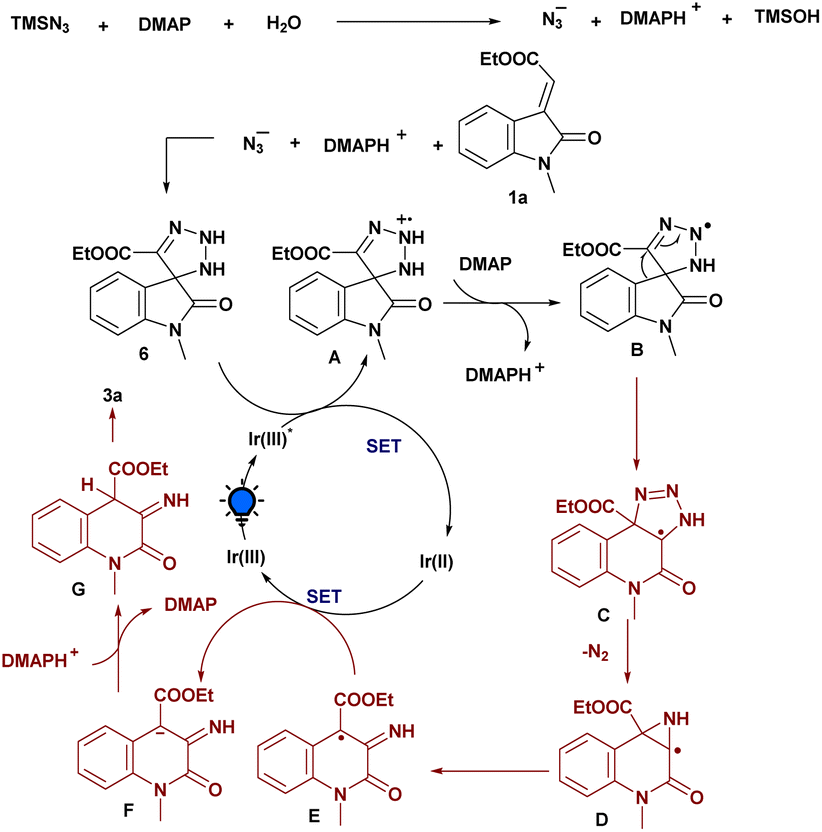 | ||
| Scheme 5 Plausible reaction mechanism for the photoinduced amination of 3-ylideneoxindoles. | ||
Finally, the applicability of this skeletal rearrangement reaction to synthesize C-3 aminoquinolin-2(1H)-one derivatives was showcased by the post-transforming reactions (Scheme 6). The decarboxylation of 3a produces 3-amino quinolin-2(1H)-one (8), while benzoylation of the amine group yields the corresponding amide product (9).21 In addition, the pharmaceutical utility of our protocol was demonstrated by the synthesis of an HSP90 protein inhibitor (10) in moderate yield.5b
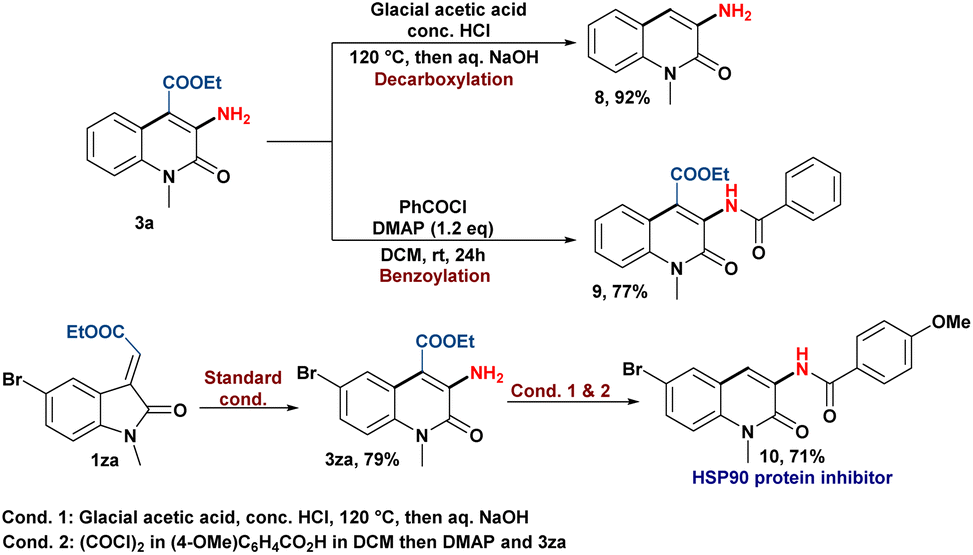 | ||
| Scheme 6 Synthetic applications of the amination reaction. | ||
Conclusion
In summary, an intriguing approach was disclosed for peripheral editing of 3-ylideneoxindoles with TMSN3 driven by visible light. This methodology utilized a step-economic and milder approach for regioselective C-3 amination of quinolin-2(1H)-one compared to the literature precedents, which require elevated temperature or extreme acidic conditions. Along with broad functionality on 3-ylideneoxindoles, the benzoyl group and aryl group on the olefinic bond of 3-ylideneoxindoles were also compatible providing the C-4 benzoyl/aryl substituted 3-aminoquinolin-2(1H)-one. Mechanistic investigation was carried out to comprehensively view the reaction path and photocatalytic involvement in the skeletal editing of the triazoline intermediate to deliver the 3-aminated quinolin-2(1H)-one.Data availability
All experimental data, detailed procedures, NMR spectra and characterization data for all compounds can be found in the ESI.†Author contributions
SRR and SS conceived the research concept. SS performed majority of the synthetic work and the analytical components of this project. GC also performed the synthesis and characterisation of several compounds reported in this manuscript. SS and GC prepared the ESI.† All authors discussed the results and contributed to the conceptualization of the project and editing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SRR thanks SERB (CRG/2022/002306) for financial support. SS and GC thank the CSIR and UGC, New Delhi, for research fellowships. The authors acknowledge CRF, IIT Delhi, for instrument facilities.References
- (a) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed; (b) T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546–576 RSC; (c) D. C. Blakemore, L. Castro, I. Churcher, D. C. Rees, A. W. Thomas, D. M. Wilson and A. Wood, Nat. Chem., 2018, 10, 383–394 CrossRef CAS PubMed; (d) W. Z. Bi, K. Sun, C. Qu, X. L. Chen, L. B. Qu, S. H. Zhu, X. Li, H. T. Wu, L. K. Duan and Y. F. Zhao, Org. Chem. Front., 2017, 4, 1595–1600 RSC; (e) P. S. Fier, S. Kim and R. D. Cohen, J. Am. Chem. Soc., 2020, 142, 8614–8618 CrossRef CAS PubMed; (f) W. S. Ham, H. Choi, J. Zhang, D. Kim and S. Chang, J. Am. Chem. Soc., 2022, 144, 2885–2892 CrossRef CAS PubMed.
- N. Talaat, M. Abass, H. Mohamed Hassanin and D. Abdel-Kader, Synth. Commun., 2022, 52, 1756–1767 CrossRef CAS.
- P. Hewawasam, N. Chen, M. Ding, J. T. Natale, C. G. Boissard, S. Yeola, V. K. Gribkoff, J. Starrett and S. I. Dworetzky, Bioorg. Med. Chem. Lett., 2004, 14, 1615–1618 CrossRef CAS PubMed.
- P. Hewawasam, W. Fan, J. Knipe, S. L. Moon, C. G. Boissard, V. K. Gribkoff and J. E. Starrett, Bioorg. Med. Chem. Lett., 2002, 12, 1779–1783 CrossRef CAS PubMed.
- (a) J. Escribano, C. Rivero-Hernández, H. Rivera, D. Barros, J. Castro-Pichel, E. Pérez-Herrán, A. Mendoza-Losana, Í. Angulo-Barturen, S. Ferrer-Bazaga, E. Jiménez-Navarro and L. Ballell, ChemMedChem, 2011, 6, 2252–2263 CrossRef CAS PubMed; (b) D. Audisio, S. Messaoudi, L. Cegielkowski, J.-F. Peyrat, J.-D. Brion, D. Methy-Gonnot, C. Radanyi, J.-M. Renoir and M. Alami, ChemMedChem, 2011, 6, 804–815 CrossRef CAS PubMed.
- (a) D. Moser, Y. Duan, F. Wang, Y. Ma, M. J. O'Neill and J. Cornella, Angew. Chem., 2018, 130, 11201–11205 CrossRef; (b) F. J. R. Klauck, H. Yoon, M. J. James, M. Lautens and F. Glorius, ACS Catal., 2019, 9, 236–241 CrossRef CAS; (c) F. Zhao, C. L. Li and X. F. Wu, Chem. Commun., 2020, 56, 9182–9185 RSC; (d) C. Ghiazza, T. Faber, A. Gómez-Palomino and J. Cornella, Nat. Chem., 2022, 14, 78–84 CrossRef CAS PubMed; (e) S. Z. Sun, Y. M. Cai, D. L. Zhang, J. B. Wang, H. Q. Yao, X. Y. Rui, R. Martin and M. Shang, J. Am. Chem. Soc., 2022, 144, 1130–1137 CrossRef CAS PubMed; (f) M. Jakubczyk, S. Mkrtchyan, M. Shkoor, S. Lanka, Š. Budzák, M. Iliaš, M. Skoršepa and V. O. Iaroshenko, J. Am. Chem. Soc., 2022, 144, 10438–10445 CrossRef CAS PubMed; (g) C. Ghiazza, L. Wagner, S. Fernández, M. Leutzsch and J. Cornella, Angew. Chem., Int. Ed., 2023, 62 DOI:10.1002/anie.202212219.
- (a) P. Roschger, W. Fiala and W. Stadlbauer, J. Heterocycl. Chem., 1992, 29, 225–231 CrossRef CAS; (b) A. Katritzky, E. Scriven, S. Majumder, R. Akhmedova, N. Akhmedov and A. Vakulenko, Arkivoc, 2005, 2005, 179–191 Search PubMed; (c) M. Orlandi, D. Brenna, R. Harms, S. Jost and M. Benaglia, Org. Process Res. Dev., 2018, 22, 430–445 CrossRef CAS.
- N. Nishiwaki, M. Sakashita, M. Azuma, C. Tanaka, M. Tamura, N. Asaka, K. Hori, Y. Tohda and M. Ariga, Tetrahedron, 2002, 58, 473–478 CrossRef CAS.
- (a) K. H. Raitio, J. R. Savinainen, J. Vepsäläinen, J. T. Laitinen, A. Poso, T. Järvinen and T. Nevalainen, J. Med. Chem., 2006, 49, 2022–2027 CrossRef CAS PubMed; (b) P. Cheng, Q. Zhang, Y.-B. Ma, Z.-Y. Jiang, X.-M. Zhang, F.-X. Zhang and J.-J. Chen, Bioorg. Med. Chem. Lett., 2008, 18, 3787–3789 CrossRef CAS PubMed.
- S. Messaoudi, J.-D. Brion and M. Alami, Adv. Synth. Catal., 2010, 352, 1677–1687 CrossRef CAS.
- (a) J. C. Reisenbauer, O. Green, A. Franchino, P. Finkelstein and B. Morandi, Science, 2022, 377, 1104–1109 CrossRef CAS PubMed; (b) J. Wang, H. Lu, Y. He, C. Jing and H. Wei, J. Am. Chem. Soc., 2022, 144, 22433–22439 CrossRef CAS PubMed; (c) E. E. Hyland, P. Q. Kelly, A. M. McKillop, B. D. Dherange and M. D. Levin, J. Am. Chem. Soc., 2022, 144, 19258–19264 CrossRef CAS PubMed; (d) D.-W. Ji, Y.-C. Hu, X.-T. Min, H. Liu, W.-S. Zhang, Y. Li, Y. J. Zhou and Q.-A. Chen, Angew. Chem., Int. Ed., 2023, 62, e202213074 CrossRef CAS PubMed; (e) D. M. Soro, J. B. Roque, J. W. Rackl, B. Park, S. Payer, Y. Shi, J. C. Ruble, A. L. Kaledin, M.-H. Baik, D. G. Musaev and R. Sarpong, J. Am. Chem. Soc., 2023, 145, 11245–11257 CrossRef CAS PubMed.
- (a) B. D. Dherange, P. Q. Kelly, J. P. Liles, M. S. Sigman and M. D. Levin, J. Am. Chem. Soc., 2021, 143, 11337–11344 CrossRef CAS PubMed; (b) C. Wentrup, M. S. Mirzaei, D. Kvaskoff and A. A. Taherpour, J. Org. Chem., 2021, 86, 8286–8294 CrossRef CAS PubMed; (c) P. Finkelstein, J. C. Reisenbauer, B. B. Botlik, O. Green, A. Florin and B. Morandi, Chem. Sci., 2023, 14, 2954–2959 RSC.
- (a) Y. Tangella, K. L. Manasa, N. H. Krishna, B. Sridhar, A. Kamal and B. N. Babu, Org. Lett., 2018, 20, 3639–3642 CrossRef CAS PubMed; (b) A. Ali, H. K. Harit, M. Devi, D. Ghosh and R. P. Singh, J. Org. Chem., 2022, 87, 16313–16327 CrossRef CAS PubMed.
- (a) D. Huang and G. Yan, Adv. Synth. Catal., 2017, 359, 1600–1619 CrossRef CAS; (b) D. Ichinari, Y. Ashikari, K. Mandai, Y. Aizawa, J. Yoshida and A. Nagaki, Angew. Chem., Int. Ed., 2020, 59, 1567–1571 CrossRef CAS PubMed; (c) J. P. Soni, M. Kadagathur and N. Shankaraiah, Asian J. Org. Chem., 2021, 10, 3186–3200 CrossRef CAS; (d) A. Nayl, A. A. Aly, W. A. A. Arafa, I. M. Ahmed, A. I. Abd-Elhamid, E. M. El-Fakharany, M. A. Abdelgawad, H. N. Tawfeek and S. Bräse, Molecules, 2022, 27, 3716 CrossRef CAS PubMed.
- (a) E. Lallana, R. Riguera and E. Fernandez-Megia, Angew. Chem., Int. Ed., 2011, 50, 8794–8804 CrossRef CAS PubMed; (b) F. Sebest, L. Casarrubios, H. S. Rzepa, A. J. P. White and S. Díez-González, Green Chem., 2018, 20, 4023–4035 RSC; (c) S. Santra, R. Bean, B. Heckert, Z. Shaw, V. Jain, L. Shrestha, R. Narayanam and Q. Austin, Polym. Chem., 2020, 11, 3723–3731 RSC; (d) F. Sebest, K. Lachhani, C. Pimpasri, L. Casarrubios, A. J. P. White, H. S. Rzepa and S. Díez-González, Adv. Synth. Catal., 2020, 362, 1877–1886 CrossRef CAS; (e) H. M. Pineda-Castañeda, Z. J. Rivera-Monroy and M. Maldonado, ACS Omega, 2023, 8, 3650–3666 CrossRef PubMed; (f) D. Bauer, S. M. Sarrett, J. S. Lewis and B. M. Zeglis, Nat. Protoc., 2023, 18, 1659–1668 CrossRef CAS PubMed.
- (a) Y. Zhang, X. Dong, Y. Wu, G. Li and H. Lu, Org. Lett., 2018, 20, 4838–4842 CrossRef CAS PubMed; (b) K. S. Stankevich, A. K. Lavrinenko and V. D. Filimonov, J. Mol. Model., 2021, 27, 305 CrossRef CAS PubMed.
- (a) Y. Jian, M. Chen, B. Huang, W. Jia, C. Yang and W. Xia, Org. Lett., 2018, 20, 5370–5374 CrossRef CAS PubMed; (b) J. Li, S. Wang, J. Zhao and P. Li, Org. Lett., 2022, 24, 5977–5981 CrossRef CAS PubMed.
- C. Croix, S. Massip and M.-C. Viaud-Massuard, Chem. Commun., 2018, 54, 5538–5541 RSC.
- (a) C. Raji Reddy, V. Ganesh and A. K. Singh, RSC Adv., 2020, 10, 28630–28634 RSC; (b) D. Doellerer, D. R. S. Pooler, A. Guinart, S. Crespi and B. L. Feringa, Chem. –Eur. J., 2023, e202301634 CrossRef CAS PubMed.
- (a) M. Milanesio, D. Viterbo, A. Albini, E. Fasani, R. Bianchi and M. Barzaghi, J. Org. Chem., 2000, 65, 3416–3425 CrossRef CAS PubMed; (b) S. K. Pagire, A. Hossain, L. Traub, S. Kerres and O. Reiser, Chem. Commun., 2017, 53, 12072–12075 RSC.
- Y. Cao, X. Liu, J. Zhang, Z. Liu, Y. Fu, D. Zhang, M. Zheng, H. Zhang and M.-H. Xu, ACS Chem. Neurosci., 2023, 14, 829–838 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2251989. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc04447e |
| This journal is © The Royal Society of Chemistry 2023 |