
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic studies of isomeric [2]rotaxanes consisting of two different tetrathiafulvalene units reveal that the movement of cyclobis(paraquat-p-phenylene) can be controlled†
Sofie K.
Jensen
 ,
Mathias S.
Neumann
,
Rikke
Frederiksen
,
Mathias L.
Skavenborg
,
Mads C.
Larsen
,
Stinne E.
Wessel
and
Jan O.
Jeppesen
*
,
Mathias S.
Neumann
,
Rikke
Frederiksen
,
Mathias L.
Skavenborg
,
Mads C.
Larsen
,
Stinne E.
Wessel
and
Jan O.
Jeppesen
*
Department of Physics, Chemistry and Pharmacy, University of Southern Denmark, Campusvej 55, 5230 Odense, Denmark. E-mail: joj@sdu.dk
First published on 23rd October 2023
Abstract
Controlling the movement in artificial molecular machines is a key challenge that needs to be solved before their full potential can be harnessed. In this study, two isomeric tri-stable [2]rotaxanes 1·4PF6 and 2·4PF6 incorporating both a tetrathiafulvalene (TTF) and a monopyrrolotetrathiafulvalene (MPTTF) unit in the dumbbell component have been synthesised to measure the energy barriers when the tetracationic cyclobis(paraquat-p-phenylene) (CBPQT4+) ring moves across either a TTF2+ or an MPTTF2+ dication. By strategically exchanging one of the thiomethyl barriers on either the TTF unit or the MPTTF unit with the bulkier thioethyl group, the movement of the CBPQT4+ ring in 14+ and 24+ can be controlled to take place in only one direction upon tetra-oxidation. Cyclic voltammetry and 1H NMR spectroscopy were used to investigate the switching mechanism and it was found that upon tetra-oxidation of 14+ and 24+, the CBPQT4+ ring moves first to a position where it is located between the TTF2+ and MPTTF2+ dications producing high-energy co-conformations which slowly interconvert into thermodynamically more stable co-conformations. The kinetics of the movement occurring in the tetra-oxidised [2]rotaxanes 18+ and 28+ were studied at different temperatures allowing the free energy of the transition state, when CBPQT4+ moves across TTF2+ (21.5 kcal mol−1) and MPTTF2+ (20.3 kcal mol−1) at 298 K, to be determined. These results demonstrate for the first time that the combination of a TTF and an MPTTF unit can be used to induce directional movement of the CBPQT4+ ring in molecular machines with a 90% efficiency.
Introduction
Life relies on motion induced by a variety of molecular machines. Nature uses these machines to regulate chemical processes, ensuring that a specific sequence of actions is followed, and that work performed in one process is not cancelled in the next. Inspired by naturally occurring molecular machines, such as the ATP synthase1 or the kinesin motor protein,2 there is a great current interest in designing and synthesising artificial molecular machines (AMMs)3 that mimic biomachines/biomotors and use their underlying mechanistic principles to create systems capable of performing rotary and linear movements at the molecular level.2b,3,4,5 Large amplitude motion in AMMs, similar to what can be observed in biomachines, can be achieved by conformational or configurational changes in covalently bonded molecules5g,6 or in supramolecular architectures.5h,7 In both cases, control of directionality is of paramount importance if rectified motion, such as unidirectional linear motion or unidirectional circumrotational motion in AMMs should be realised. In the latter case, significant progress toward this end has been made possible as a consequence of the formation/discovery of the mechanical bond.8 Directional movements in mechanically interlocked molecules (MIMs), such as catenanes9 and rotaxanes,9 can be obtained either through systems that rely on an energy ratchet mechanism3,10 or on an information ratchet mechanism.3,10b The information ratchet operates continuously in a non-equilibrium steady state, while the energy ratchet operates in a stepwise manner. In both cases, the key requirement is the ability to change both the thermodynamical distribution between two states and the kinetical interconversion between them, in order to obtain kinetic asymmetry.3,10b,11 Exemplary studies have shown (i) how simultaneous modification of recognition sites and covalently linked steric barriers can be used to create unidirectional movement in catenanes,7d (ii) how an asymmetric set of steric and electrostatic barriers can be used to pump macrocycles across a linear axle12 and latest (iii) how the introduction of asymmetry in a macrocycle can be used to control the directionality of the movement of the macrocycle on a symmetric dumbbell component in a [2]rotaxane.13 Mechanistic studies on AMMs, therefore, are essential if the next generation of AMMs with enhanced control of movement should be developed.MIMs based on the tetracationic electron-accepting macrocycle cyclobis(paraquat-p-phenylene) (CBPQT4+) and the electron-donating tetrathiafulvalene (TTF) unit7c,14 have been used extensively for the construction of AMMs. TTF and many of its derivatives show a high binding with CBPQT4+ as a result of the formation of charge-transfer (CT) interactions between the electron-deficient bipyridinium moieties present in CBPQT4+ and the electron-rich TTF unit.15 By oxidising the TTF unit to the TTF radical cation (TTF˙+) or the TTF dication (TTF2+), the favourable CT interactions between TTF and CBPQT4+ are replaced by coulombic repulsion between TTF˙+/TTF2+ and the tetracationic CBPQT4+ ring, forcing CBPQT4+ to move away from the oxidised TTF unit.7c,15b This motif has successfully been used as the switching gear in numerous redox-switchable [2]rotaxanes and [2]catenanes based on CBPQT4+ and TTF7a,c,14,16 and illustrates that the electroactive TTF unit can be used as a stimuli-controlled station for the CBPQT4+ ring. However, the symmetric nature of most TTF derivatives result in non-directional movement of CBPQT4+ away from the oxidised TTF unit, i.e. the encircling CBPQT4+ ring can leave the oxidised TTF unit in both directions with equal probability (Scheme S1a, ESI†). This problem can be circumvented if the monopyrroloTTF (MPTTF) unit is used as the electroactive component. On account of its non-symmetrical nature, the MPTTF unit can be used to induce directional movement of CBPQT4+ following its oxidation, since the combination of the thiomethyl (SMe) and the thioalkyl (SR) groups acts as a steric barrier that forces the CBPQT4+ ring to escape from the oxidised MPTTF unit in only one direction (Scheme S1b, ESI†), i.e. across the pyrrole moiety present in MPTTF.‡
Most recently, some of us have discovered that TTF/MPTTF also can act as stimuli-induced electrostatic barriers for the movement of CBPQT4+. It was found that both the di-oxidised TTF2+ and MPTTF2+ dications can act as electrostatic barriers for the CBPQT4+ ring when both units are present in a tetra-stable [2]rotaxane.14d Although it was not possible to quantify the size of the barriers when the CBPQT4+ ring moves across the di-oxidised units, it was found that the barrier for CBPQT4+ to move across the MPTTF2+ dication is about 0.5 kcal mol−1 smaller than over the TTF2+ dication.14d Later, we used a simple bistable [2]rotaxane as the platform to make a complete profiling of the energy landscape when the CBPQT4+ ring moves across an MPTTF2+ dication in both the forward and the backward directions and it was estimated that the Gibbs free energy of activations (ΔG‡) for these processes were 21.8 (forward) and 23.3 (backward) kcal mol−1, respectively, in CD3CN at 298 K.7e However, no studies have so far been directed toward quantifying the barrier when CBPQT4+ moves across a TTF2+ dication, and more importantly, to quantify to which extent the combination of an MPTTF and a TTF unit following their oxidations can be used to induce directional movement of the CBPQT4+ ring in MIMs. Toward this end, we have designed (Scheme 1) a set of isomeric [2]rotaxanes 1·MPTTF4+ and 2·MPTTF4+, incorporating both a TTF (light green) and an MPTTF (dark green) unit in the dumbbell component, that allow us to quantify the size of the energy barriers when the CBPQT4+ ring (blue) moves across either a TTF2+ or an MPTTF2+ dication. By strategically exchanging one of the SMe barriers on either the TTF unit or the MPTTF unit with the much bulkier thioethyl (SEt) group, the movement of the CBPQT4+ ring can be isolated to take place in only one direction, on account of the fact17 that the combination of the SEt and the SR groups acts as a stopper for CBPQT4+. Following tetra-oxidation of 1·MPTTF4+ and 2·MPTTF4+, it was found that the CBPQT4+ ring moves in between the TTF2+ and MPTTF2+ dications producing 1·TEG8+ and 2·TEG8+, respectively. These two co-conformations represent metastable states (out-of-equilibrium) of the oxidised [2]rotaxanes 18+ and 28+, which slowly interconvert into the thermodynamically more stable co-conformations 1·OP8+ and 2·OP8+, respectively, when CBPQT4+ moves across either the TTF2+ dication in 18+ or the MPTTF2+ dication in 28+. This observation allowed us to determine the rate constants (k1) and associated ΔG‡ values when CBPQT4+ moves across a TTF2+ or an MPTTF2+ dication and to elucidate the impact of the increased electrostatic repulsion present when the CBPQT4+ ring is located between two doubly oxidised units (i.e. TTF2+ and MPTTF2+).
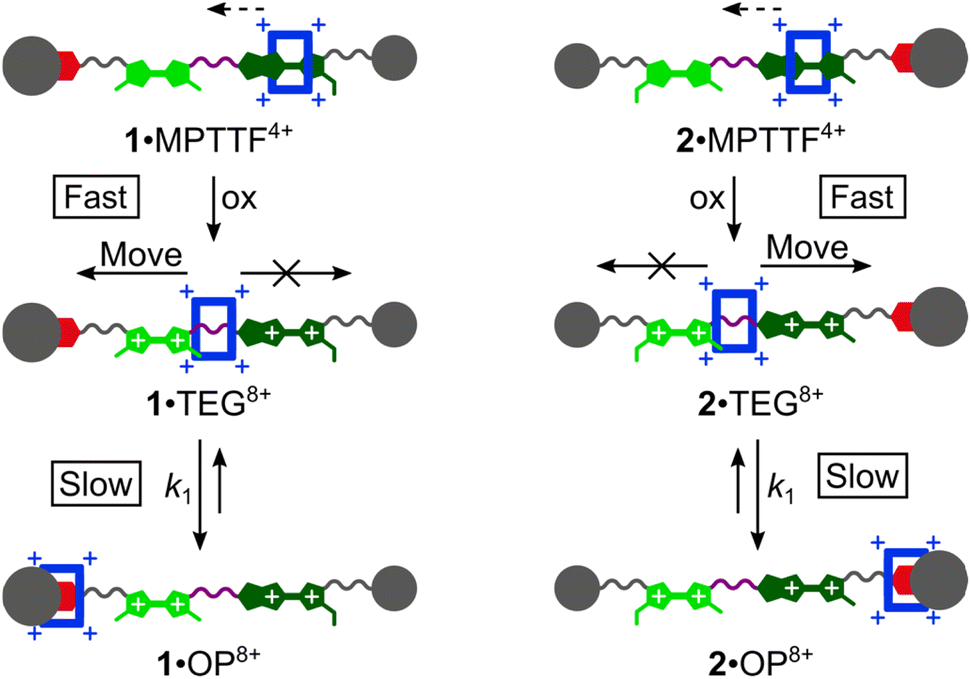 | ||
| Scheme 1 Oxidation of the two isomeric [2]rotaxanes 1·MPTTF4+ and 2·MPTTF4+ producing initially 1·TEG8+ and 2·TEG8+, respectively, which subsequently in a slow and rate-limiting step converts into 1·OP8+ and 2·OP8+, respectively allowing the rate constants k1 for the movement of CBPQT4+ (blue) from the TEG linker across either a TTF2+ (light green) or an MPTTF2+ (dark green) to the OP (red) unit to be determined. In 1·TEG8+ and 2·TEG8+, the movement of CBPQT4+ to the right and left, respectively, is rendered impossible by attachment of a large and bulky thioethyl (SEt) group to either the MPTTF2+ (1·TEG8+) or the TTF2+ (2·TEG8+) unit. | ||
Here, we describe (i) the template-directed synthesis and characterisation of two isomeric tri-stable [2]rotaxanes 1·4PF6 and 2·4PF6 (Fig. 1) in which the ring component is CBPQT4+ (blue) and the dumbbell component contains three potential stations for CBPQT4+, namely an MPTTF15c,18 unit (dark green), a TTF15b,d,19 unit (light green), and an oxyphenylene7c,e,14d,17 (OP) unit (red). In each case, the dumbbell component is terminated by a diisopropylphenyl (DIPP) stopper7e at one end and a triarylmethyl stopper,7c,e,14d,17 which is directly connected to the OP unit, at the other end. The TTF and MPTTF units are positioned in the middle of the dumbbell components and are connected by a triethylene glycol (TEG) chain in such a way that the pyrrole moiety of the MPTTF unit points toward the TTF unit in both 1·4PF6 and 2·4PF6. Based on the three stations' relative affinities toward CBPQT4+, it is expected that the majority of 1·4PF6 and 2·4PF6 exist as the translational isomers 1·MPTTF·4PF6 and 2·MPTTF·4PF6, respectively, in which the CBPQT4+ ring is located around the MPTTF station (Fig. 1). Subsequently, (ii) the thermodynamic and electrochemical properties of 14+ and 24+ are described, followed by describing (iii) thorough kinetic investigations of the tetra-oxidised [2]rotaxanes 18+ and 28+ using time-resolved 1H NMR spectroscopy at variable temperatures. Finally, (iv) it is shown that the difference between the size of the TTF2+ and the MPTTF2+ electrostatic barriers are heavily temperature dependent.
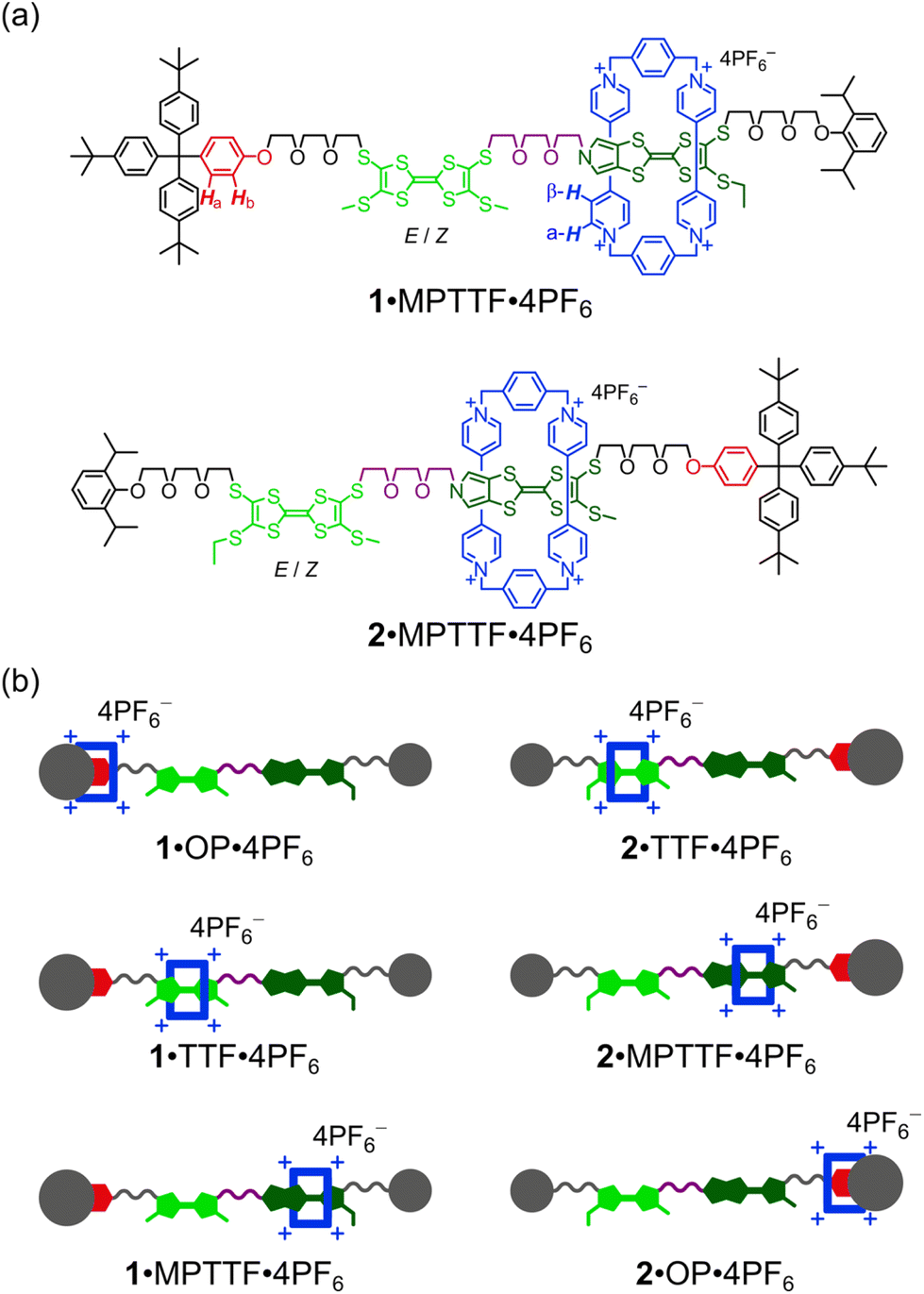 | ||
| Fig. 1 (a) Molecular formulas of the tri-stable [2]rotaxanes 1·4PF6 and 2·4PF6 (only one translational isomer for each is shown). (b) Cartoon representations of the three possible translational isomers of 1·4PF6 and 2·4PF6, respectively (all translational isomers also exist as a mixture of E- and Z-isomers). | ||
Results and discussion
Synthesis
The [2]rotaxanes 1·4PF6 and 2·4PF6 were prepared as shown in Schemes S2 and S3 (ESI†). A modular approach was used in which the various parts of the target molecules were prepared individually and joined together in the latter steps of the synthetic pathways to produce the dumbbells 14 and 24. Formation of the [2]rotaxanes 1·4PF6 and 2·4PF6 was achieved by using the dumbbells 14 and 24, respectively, as templates for the formation of the CBPQT4+ tetracation under high pressure conditions20 from the dicationic precursor2115·2PF6 and the dibromide 16 (see ESI†).Mass spectrometric investigations
All new compounds reported in this paper were characterised by electrospray ionisation (ESI) mass spectrometry. The ESI mass spectra (Fig. S1 and S2, ESI†) of the [2]rotaxanes 1·4PF6 and 2·4PF6 revealed peaks corresponding to the doubly positively charged [M − 2PF6]2+ and triply positively charged [M − 3PF6]3+ ions.Photophysical investigations
The photophysical properties of the dumbbells 14 and 24 and the [2]rotaxanes of 14+ and 24+ were studied in solution at 298 K. The UV/Vis/NIR absorption spectra recorded of 14 (Fig. S3a, ESI†) and 24 (Fig. S3b, ESI†) in MeCN/CH2Cl2 only exhibit weak tails in the visible region at λ ≤ 500 nm and both solutions appear yellow. In contrast, solutions of 14+ and 24+ both appear green, and the absorption spectra recorded of 14+ (Fig. S3c, ESI†) and 24+ (Fig. S3d, ESI†) in MeCN showed a broad CT absorption band centered at 807 and 809 nm, respectively, which is characteristic7a,15b–d,22 of structures containing a TTF/MPTTF unit positioned inside the cavity of CBPQT4+. These observations clearly suggest that CBPQT4+ encircles the rod section of the dumbbell component in the [2]rotaxanes 14+ and 24+ and that either the TTF or the MPTTF unit is located inside the cavity of CBPQT4+.1H NMR spectroscopic investigations
While both mass spectrometry and absorption spectroscopy indicate qualitatively that the CBPQT4+ ring is present on the rod section of the dumbbell component of 14+ and 24+, 1H NMR spectroscopy was used as a quantitative tool for monitoring the precise position of CBPQT4+ on the rod section and hence allowing the relative amounts of the different translational isomers present in 14+ and 24+ to be determined. All of the compounds containing the TTF unit were isolated as mixtures of E/Z isomers on account of the inherent E/Z isomerism of the TTF unit and 1H NMR spectroscopy confirmed that the ratio between the isomers was approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in all cases.
:1 in all cases.
A comparison of the 1H NMR spectra (400 MHz, CD3CN, 298 K) of the [2]rotaxane 14+ (Fig. 2) and the dumbbell 14 (Fig. S4, ESI†) revealed new signals resonating at δ = 8.75–9.13 ppm, δ = 7.65–8.13 ppm, δ = 7.65–7.84 ppm and δ = 5.63–5.82 ppm that can be associated with the α-H, β-H, xylyl-H and CH2N+ protons, respectively, present in the CBPQT4+ ring; this suggests that CBPQT4+ is present in 14+ in agreement with the findings obtained from the mass spectrometric and photophysical investigations (vide supra).
 | ||
| Fig. 2 Partial 1H NMR spectrum of an isomeric mixture of the [2]rotaxane 14+ (400 MHz, CD3CN, 298 K, 1.5 mM), where the assignments in light green are associated with 1·TTF4+ and the dark green signals are associated with 1·MPTTF4+, while the assignments in grey are associated with a mixture of the two translational isomers. The C(CH3)3 signal is not shown in its full height. | ||
A careful examination of the 1H NMR spectrum (400 MHz) of the [2]rotaxane 14+ shows that it exists as a mixture of two stable translational isomers, where CBPQT4+ encircles either the TTF (i.e.1·TTF4+) or the MPTTF (i.e.1·MPTTF4+) station, respectively, and that their exchange is slow on the 1H NMR timescale (400 MHz) in CD3CN at 298 K. Thus, several protons in both the dumbbell component and in the CBPQT4+ ring gave rise to two sets of signals, one for 1·MPTTF4+ and one for 1·TTF4+, allowing the population of each of the two translational isomers at equilibrium to be determined. The most diagnostic evidence, which shows that 1·MPTTF4+ is present in the isolated mixture is the appearance of an AB system (J = 1.8 Hz) at δ = 6.11 and 6.14 ppm, which can be assigned7a to the resonances associated with the two pyrrole-H protons in the MPTTF unit when it is located inside the cavity of CBPQT4+. The resonances for the two pyrrole-H protons in 1·MPTTF4+ are upfield shifted as compared to the dumbbell 14, where they appear at δ = 6.62–6.67 ppm, a finding that is fully consistent with CBPQT4+ encircling the MPTTF unit.7a Furthermore, the two methylene protons of the SEt (SCH2CH3) group attached to the MPTTF unit in 1·MPTTF4+ are downfield shifted relative to their position in the dumbbell 14 on account of the deshielding effect of CBPQT4+ when it encircles the MPTTF unit.7c,17 The SCH2CH3 protons resonate as a quartet (J = 7.4 Hz) at δ = 3.11 ppm in 1·MPTTF4+, compared with a quartet (J = 7.3 Hz) at δ = 2.85 ppm in the dumbbell 14. Consistently, the SCH2 protons associated with the thioethoxy (SCH2CH2O) group attached directly to the MPTTF unit in 1·MPTTF4+ are similarly shielded and were found (Fig. 2) to resonate as a triplet (J = 6.3 Hz) at δ = 3.30 ppm, while the same protons in the dumbbell 14 resonate as a triplet (J = 6.4 Hz) at δ = 3.01 ppm. The protons associated with the two thiomethyl (SCH3) groups, attached to the TTF unit in 1·MPTTF4+, gave rise to a total of four singlets resonating at δ = 2.31, 2.32, 2.37 and 2.38 ppm, respectively, and are located at almost identical chemical shift values as in the dumbbell 14 (δ = 2.35, 2.36 and 2.38 ppm).§ The existence of two sets of SCH3 signals in 1·MPTTF4+ at δ = 2.31 and 2.32 ppm and at δ = 2.37 and 2.38 ppm, therefore, indicate that the SCH3 protons experience two slightly different chemical environments which only can be accounted for by the presence of both an E- and a Z-isomer in 1·MPTTF4+.
The existence of 1·TTF4+ is evidenced (Fig. 2) by the presence of three singlets resonating at δ = 2.52, 2.59 and 2.59 ppm,¶ which can be associated with the SCH3 protons in the E/Z isomeric mixture of 1·TTF4+. They have experienced a downfield shift relative to the position at which they were found to resonate in the dumbbell 14 (δ = 2.35, 2.36 and 2.38 ppm) and previous investigations7c have shown that such behaviour is entirely consistent with CBPQT4+ encircling a TTF unit. The small multiplet centered at δ = 6.46 ppm can be assigned14d to the pyrrole-H protons in the MPTTF unit when the TTF unit is encircled by CBPQT4+. Compared to 1·MPTTF4+ (δ = 3.11 ppm), the signal for the SCH2CH3 protons in 1·TTF4+ (δ = 2.85 ppm) has not been shifted significantly downfield and resonates at almost the same position as the SCH2CH3 protons in the dumbbell 14 (δ = 2.85 ppm).
Although the [2]rotaxane 14+, contains three potential stations for CBPQT4+ to reside around, a careful inspection of the 1H NMR spectrum recorded of 14+ in CD3CN at 298 K did not show any indication of the presence of the translational isomer where CBPQT4+ encircles the OP unit (i.e.1·OP4+). Since the [2]rotaxane 14+, is constructed in such a way that the TTF unit is separated from the OP unit by a combination of an SMe group and a thiotriethylene glycol (TTEG) substituent which is known7c,18b to act as a “speed bump” for CBPQT4+, the shuttling of CBPQT4+ between the MPTTF and OP units is expected to be slow on the 1H NMR timescale. Consequently, if 1·OP4+ is present in the isolated mixture of the [2]rotaxane 14+ a third set of signals should appear in the 1H NMR spectrum. The absence of a signal around 6.25 ppm demonstrates that 1·OP4+ is not present in measurable amounts in the isolated mixture of 14+, since it is known from previous studies7c that the OP-Ha protons resonate as a doublet at δ = 6.25 ppm when the CBPQT4+ ring encircles an OP unit in TTF based [2]rotaxanes. Using the integrals of appropriate signals as probes (Table S1, ESI†), the distribution between the two different translational isomers in the [2]rotaxane 14+ can be obtained and it was found that 1·MPTTF4+ is the predominant translational isomer, accounting for 82% of the mixture, whereas 1·TTF4+ only account for 18% as illustrated in Scheme S2 (ESI†).
As for the [2]rotaxane 14+, the [2]rotaxane 24+ exists (Fig. S7, ESI†) as a mixture of two interconverting translational isomers (i.e.2·MPTTF4+ and 2·TTF4+) where their exchange is slow on the 1H NMR timescale (400 MHz) in CD3CN at 298 K. Using a similar analysis (Table S2, ESI†) as carried out on the [2]rotaxane 14+, it can be estimated that the [2]rotaxane 24+ almost exclusively (95%) exists as the translational isomer 2·MPTTF4+ in CD3CN at 298 K, while 2·TTF4+ only is present in tiny amounts (5%) as illustrated in Scheme S3 (ESI†). The presence of 2·MPTTF4+ is evidenced (Fig. S7, ESI†) by the characteristic AB system (J = 2.0 Hz) resonating at δ = 6.17 and 6.20 ppm assigned|| to the pyrrole-H protons in the MPTTF unit, when the MPTTF unit is positioned inside CBPQT4+. Furthermore, the existence of 2·MPTTF4+ is confirmed by a singlet resonating at δ = 2.68 ppm in the 1H NMR spectrum of 24+, which is diagnostic17 for SCH3 protons attached to an MPTTF unit when it is encircled by CBPQT4+, while the two small singlets appearing at δ = 2.51 and 2.52 ppm are associated with 2·TTF4+ and can be assigned to the SCH3 protons attached to the TTF unit when it is located inside CBPQT4+.**
To support the experimentally found population of translational isomers in the two [2]rotaxanes, the theoretical distributions of translational isomers were calculated using the binding energies between CBPQT4+ and the three different stations, i.e. ΔG°(MPTTF)14d = −6.0 kcal mol−1, ΔG°(TTF)7c,14d = −4.4 kcal mol−1 and ΔG°(OP)7c = −1.7 kcal mol−1 in MeCN at 298 K. Assuming that the relative distribution between the translational isomers is related to the above listed binding energies of CBPQT4+ with the separate MPTTF, TTF and OP units, it was calculated (see ESI†) that the theoretical distribution between the three translational isomers is 94:6:0 (MPTTF:TTF:OP) in MeCN at 298 K. This is in perfect agreement with the 1H NMR spectroscopic findings (vide supra) which suggest that the proportions of the three translational isomers is 95:5:0 (2·MPTTF4+:2·TTF4+:2·OP4+) for 24+, while the amount of 1·MPTTF4+ compared to the amount of 1·TTF4+ in 14+ (82:18:0 (1·MPTTF4+:1·TTF4+:1·OP4+)) is lower than expected from the theoretical calculations. The observed difference in the population of translations isomers in 14+ and 24+ can most likely be accounted for by the fact that the relative position of the stopper groups and the SEt/SMe groups along the dumbbell component affects the stability of the different tertiary (3°) structures23 the two isomeric [2]rotaxanes 14+ and 24+ can adopt in solution.
Electrochemical investigations
The electrochemical properties of the dumbbells 14 and 24 and their corresponding [2]rotaxanes 14+ and 24+ were investigated using cyclic voltammetry (CV). The cyclic voltammograms (CVs) were all measured in nitrogen-purged MeCN solutions at 298 K, and we have mainly focused our studies on the oxidation processes of the TTF and MPTTF units. The obtained half-wave potentials (E1/2) are listed in Table 1, together with the corresponding anodic (Eox) and cathodic (Ered) peak potentials.| Compound | E ox 1 [V] | E red 1 [V] | E 1/2 1 [V] | E ox 2 [V] | E red 2 [V] | E 1/2 2 [V] | E ox 3 [V] | E red 3 [V] | E 1/2 3 [V] | E ox 4 [V] | E red 4 [V] | E 1/2 4 [V] |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a CV measurements of 14, 24, 14+ and 24+ in nitrogen-purged solutions (0.5 mM in MeCN (14+ and 24+) or <0.5 mM in MeCN (14 and 24)) were conducted with 0.1 M n-Bu4NPF6 as the electrolyte, a glassy carbon electrode as the working electrode, a Pt counter electrode and a Ag/AgNO3 reference electrode with a scan rate of 0.1 V s−1; Eox, Ered and E1/2 values vs. Fc/Fc+ and the estimated errors on the E values are ± 0.01 V. b Anodic peak potential. c Cathodic peak potential. d Half-wave potential, E1/2 = (Eox + Ered)/2. | ||||||||||||
| 14 | +0.02 | −0.03 | 0.00 | +0.09 | +0.05 | +0.07 | +0.41 | +0.36 | +0.39 | +0.41 | +0.36 | +0.39 |
| 24 | +0.02 | −0.03 | 0.00 | +0.10 | +0.05 | +0.08 | +0.41 | +0.36 | +0.39 | +0.41 | +0.36 | +0.39 |
| 1 4+ | +0.15 | +0.08 | +0.12 | +0.33 | +0.28 | +0.30 | +0.40 | +0.35 | +0.38 | +0.66 | +0.54 | +0.60 |
| 2 4+ | +0.17 | +0.09 | +0.13 | +0.34 | +0.28 | +0.31 | +0.40 | +0.35 | +0.38 | +0.66 | +0.53 | +0.60 |
The CVs of the two isomeric dumbbells 14 and 24 are as expected almost identical (Fig. 3a and b) and show four redox processes. The first redox process appears at E1/21 = 0.00 V vs. Fc/Fc+ and can be assigned to the first oxidation of the MPTTF unit (MPTTF → MPTTF˙+) to produce 141˙/1+/241˙/1+, since the MPTTF unit is easier to oxidise than the TTF unit.14d,23 Consequently, the second redox process appearing at E1/22 = +0.07/+0.08 V vs. Fc/Fc+ can be associated with the first oxidation of the TTF unit (TTF → TTF˙+), which will produce 142˙/2+/242˙/2+. The third and fourth redox processes are observed as two overlapping processes at E1/23 = E1/24 = +0.39 V vs. Fc/Fc+ and can be attributed to the second oxidations of the MPTTF and TTF units to produce the corresponding dications (i.e. MPTTF˙+ → MPTTF2+ and TTF˙+ → TTF2+, respectively). As observed previously in similar systems, it seems likely that MPTTF˙+ in 142˙/2+/242˙/2+ will be oxidised at a slightly lower potential relative to TTF˙+ to produce initially 141˙/3+/241˙/3+ followed by the formation of 144+/244+ (Schemes S4 and S5, ESI†).††
 | ||
| Fig. 3 Cyclic voltammograms of the dumbbells (a) 14 and (b) 24 and the [2]rotaxanes (c) 14+ and (d) 24+. The measurements were carried out at 298 K in nitrogen-purged MeCN solutions (0.5 mM for 14+ and 24+ or <0.5 mM for 14 and 24) with a glassy carbon electrode as the working electrode, a Pt counter electrode, a Ag/AgNO3 reference electrode, a scan rate of 0.1 V s−1 and n-Bu4NPF6 as the electrolyte (0.1 M). | ||
In the case of the [2]rotaxanes 14+ and 24+, a more complex situation occurs, since they both exist as a mixture of two different translational isomers (Schemes S1 and S2, ESI†). However, 1H NMR spectroscopy revealed (vide supra) that the predominant isomer in both cases is the translational isomer in which the CBPQT4+ ring encircles the MPTTF unit, i.e.1·MPTTF4+ (82%) and 2·MPTTF4+ (95%), respectively, and that 1·TTF4+ (18%) and 2·TTF4+ (5%) are only present in small amounts in 14+ and 24+, respectively. Thus, the CVs recorded (Fig. 3c and d) of the [2]rotaxanes 14+ and 24+ will provide us with the electrochemical profile of 1·MPTTF4+ and 2·MPTTF4+, respectively, since it seems reasonable to expect that the two minor isomers (i.e.1·TTF4+ and 2·TTF4+) only will give rise to small shoulders in the CVs and therefore not interfere to any significant degree with the electrochemical characterisation of 1·MPTTF4+ and 2·MPTTF4+.
The CVs recorded (Fig. 3c and d) of the [2]rotaxanes 14+ and 24+ are almost identical and the results show that 1·MPTTF4+ and 2·MPTTF4+ are characterised by four redox processes appearing at the same potentials (Table 1) when errors are taken into account, i.e. E1/2 = +0.12, +0.30, +0.38 and +0.60 vs. Fc/Fc+ for 1·MPTTF4+ and E1/2 = +0.13, +0.31, +0.38 and +0.60 vs. Fc/Fc+ for 2·MPTTF4+. A suggested mechanism for the four-step oxidation of 1·MPTTF4+ is shown in Scheme 2. While the MPTTF unit in the dumbbell 14 (Scheme S4, ESI†) was oxidised before the TTF unit, the opposite is observed in the case of 1·MPTTF4+. Since the MPTTF unit is located inside the CBPQT4+ ring in 1·MPTTF4+, the first oxidation of the MPTTF unit is expected to occur at a more positive potential (>+0.30 V)‡‡ compared to the same process in the dumbbell 14 as a result of the strong CT interactions taking place between the MPTTF unit and the bipyridinium moieties in CBPQT4+. Therefore, the first redox process (E1/21 = +0.12 V vs. Fc/Fc+) of 1·MPTTF4+ can be associated with the first oxidation of the TTF unit to TTF˙+ to give 1·MPTTF1˙/5+ (process I, Scheme 2), while the second redox process (process II, Scheme 2) appearing at E1/22 = +0.30 V vs. Fc/Fc+ can be assigned to the first oxidation of the MPTTF unit encircled by CBPQT4+ producing 1·MPTTF2˙/6+. A comparison between the third redox process (E1/23) of the dumbbell 14 (+0.39 V vs. Fc/Fc+) and the [2]rotaxane 1·MPTTF4+ (+0.38 V vs. Fc/Fc+) reveals that they appear at similar potentials when errors are taken into account. This observation indicates that upon oxidation of MPTTF to MPTTF˙+ creating 1·MPTTF2˙/6+, the CBPQT4+ ring will leave the mono-oxidised MPTTF unit on account of coulombic repulsion between the MPTTF˙+ radical cation and the CBPQT4+ ring. Since the planar pyrrole moiety (<15 kcal mol−1)18b of the MPTTF unit is a significantly smaller steric barrier than the bulky SEt group (>29 kcal mol−1),22c it is evident that the CBPQT4+ ring is forced to leave the MPTTF˙+ radical cation via the pyrrole-end producing 1·TEG2˙/6+ in which the CBPQT4+ ring is located on the TEG linker in between the TTF˙+ and MPTTF˙+ radical cations. As for the dumbbell 14, it seems likely that the second oxidation of the MPTTF unit in the [2]rotaxane 1·MPTTF4+ also will take place before the second oxidation of the TTF unit. Therefore, the third redox process (process III, Scheme 2) appearing at E1/23 = +0.38 V vs. Fc/Fc+ can be assigned to the second oxidation of the MPTTF unit to give 1·TEG1˙/7+. Finally, the fourth redox process observed at E1/24 = +0.60 V vs. Fc/Fc+ can be assigned to the oxidation of the TTF˙+ radical cation to the TTF2+ dication to produce 1·TEG8+ (process IV, Scheme 2). Compared to the dumbbell 14, the E1/24 value in the [2]rotaxane 1·MPTTF4+ is anodically shifted by 0.21 V, a finding which is fully consistent with previous results reported for a tetra-stable [2]rotaxane,14d indicating that the CBPQT4+ ring upon formation of 1·TEG1˙/7+, is forced to move closer to the TTF˙+ unit (Scheme 2), which causes the oxidation of TTF˙+ to TTF2+ (i.e. process IV) to be more difficult compared to the same process in the dumbbell 14. These results also show that the CBPQT4+ ring remains around the TEG linker connecting the TTF and MPTTF units after they have been oxidised on the ∼20 s timescale of the CV experiment.
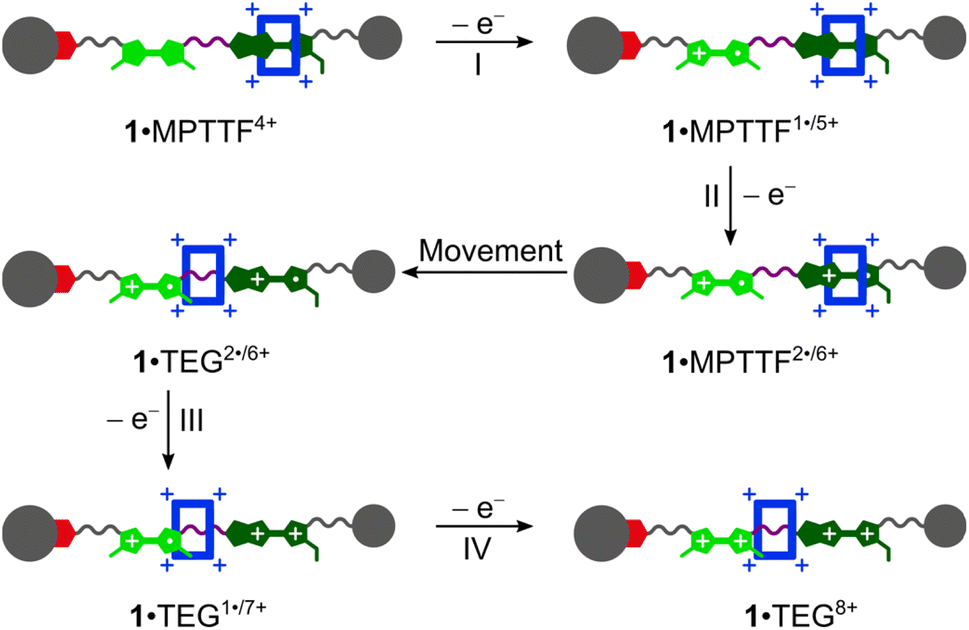 | ||
| Scheme 2 Suggested mechanism for tetra-oxidation of 1·MPTTF4+ to produce 1·TEG8+ in which the CBPQT4+ ring is located on the TEG linker connecting the TTF2+ and MPTTF2+ dications. | ||
A similar analysis can be carried out on the [2]rotaxane 2·MPTTF4+ and a suggested mechanism for the four-step oxidation of 2·MPTTF4+ is shown in Scheme S6 (ESI†).
Chemical oxidation of 14+ and 24+
The outcome of the electrochemical investigations clearly shows that 1·TEG8+ and 2·TEG8+ were produced upon tetra-oxidation of 1·MPTTF4+ and 2·MPTTF4+, respectively. Both 1·TEG8+ and 2·TEG8+ represent out-of-equilibrium24 states of 18+ and 28+, respectively, since the tetracationic CBPQT4+ ring is pushed into a high energy (metastable) state where it is in an unfavourable position between, and in close proximity to, both a TTF2+ and an MPTTF2+ dication. Although 1·TEG8+ and 2·TEG8+ were found to be stable on the short timescale (∼20 s) of the electrochemical experiments, it is expected that they will relax into more stable translational isomeric forms of 18+ and 28+, respectively, on a longer timescale. For this purpose, 1H NMR spectroscopy was used to probe the stability of 1·TEG8+ and 2·TEG8+.Initially, solutions of the [2]rotaxanes 14+ and 24+ in CD3CN at 298 K were oxidised with an excess (ten equiv.) of the chemical oxidant tris(4-bromophenyl)ammoniumyl hexachloroantimonate (TBPASbCl6). Subsequently, 1H NMR spectra (400 MHz) were recorded as fast as possible (∼5 min) after adding TBPASbCl6 followed by the recording of 1H correlation spectroscopy (COSY) spectra. Comparing the 1H NMR spectra recorded of 14+ at 298 K before (Fig. 2 and 4a) and after oxidation (Fig. 4b) reveals significant shifts to almost all of the signals in both the dumbbell and the ring components, which is consistent with the formation of both the MPTTF2+ and TTF2+ dications and, hence, the formation of the tetra-oxidised [2]rotaxane 18+. For instance, the signals for the pyrrole-H protons attached to the MPTTF unit shift (Fig. 4a) from δ = 6.11 and 6.14 ppm (AB system) for 1·MPTTF4+ to δ = 8.02 ppm (singlet) for 18+ (Fig. 4b). Likewise, the protons associated with the two different SCH3 groups attached to the TTF unit in 18+ are downfield shifted (δ = 2.99 and 3.01 ppm, Fig. 4b) compared to their position in which they are found resonating in the un-oxidised [2]rotaxane 1·MPTTF4+ (δ = 2.31, 2.32, 2.37 and 2.38 ppm, Fig. 2 and 4a). Previous studies have shown that such behaviour is entirely consistent with the formation of un-encircled MPTTF2+ and TTF2+ dications.7c,14d,17,22b,25 Furthermore, the decrease in the number of singlets associated with the SCH3 protons from four to two, when 1·MPTTF4+ is oxidised to 18+, is also consistent with the formation of TTF2+ because E/Z isomerism cannot exist once the central double bond of the TTF unit is broken upon oxidation to TTF2+. Hence only two SCH3 signals will be observed in the 1H NMR spectrum recorded of 18+ at 298 K on account of the non-symmetric structure of the tetra-oxidised dumbbell component.
 | ||
| Fig. 4 Partial 1H NMR spectra (CD3CN, 1.5 mM) recorded of (a) the [2]rotaxane 14+ (400 MHz) at 298 K, (b) the oxidised [2]rotaxane 18+ (400 MHz) at 298 K and (c) the oxidised [2]rotaxane 18+ (600 MHz) at 258 K. The oxidised [2]rotaxane 18+ was generated by adding an excess of the chemical oxidant tris(4-bromophenyl)ammoniumyl hexachloroantimonate (TBPASbCl6) to 14+ and the spectra were recorded ca. 5 min after adding TBPASbCl6 at the indicated temperature. The assignments in red are associated with 1·OP8+ (i.e. the OP station is located inside the CBPQT4+ ring) and the assignments in purple are associated with 1·TEG8+ (i.e. the CBPQT4+ ring is located at the TEG linker connecting the TTF2+ and MPTTF2+ units). The C(CH3)3 and CD2HCN signals are not shown in their full height. | ||
Secondly, it is also evident from the 1H NMR spectrum recorded of 18+ at 298 K that 18+ almost exclusively exists as a single translational isomer, namely 1·OP8+ in which the CBPQT4+ ring encircles the OP moiety. The new location of the CBPQT4+ ring along the dumbbell component of the oxidised [2]rotaxane 18+ is evident from the very large upfield shift (Fig. 4) of the resonances associated with the OP-Ha and OP-Hb protons (Fig. 1) in the oxidised [2]rotaxane 18+ relative to their positions in 1·MPTTF4+. In 1·MPTTF4+, the resonances appear (Fig. 2 and 4a) at δ = 7.12 ppm (OP-Ha) and δ = 6.80 ppm (OP-Hb), while they in 18+ appear (Fig. 4b) at δ = 6.27 ppm (OP-Ha) and δ = 2.54 ppm (OP-Hb) as a result of the anisotropic shielding effect that occurs when the CBPQT4+ ring encircles the OP moiety. Consistently, the OCH2 protons attached directly to the encircled OP moiety in 18+ are also significantly shielded and resonate at δ = 1.42 ppm compared to δ = 4.06 ppm in 1·MPTTF4+. Examination of the COSY spectrum for 18+ recorded in CD3CN at 298 K (Fig. S10, ESI†) clearly shows through-bond scalar couplings between the two shielded OP-Ha (δ = 6.80 ppm) and OP-Hb (δ = 2.54 ppm) protons and between the OCH2 (δ = 1.42 ppm) protons attached directly to the encircled OP moiety and the protons in the neighbouring CH2 (δ = 3.46 ppm) group present in the glycol chain attached to the OP moiety.
Overall, these observations unambiguously confirm that CBPQT4+ has moved to the OP moiety 5 min after 14+ was oxidised with TBPASbCl6 at 298 K in CD3CN and that 1·TEG8+ seems to be too short-lived to be observed at 298 K.§§ However, a careful inspection of the 1H NMR spectrum (Fig. 4b) recorded of 18+ at 298 K, 5 min after being oxidised, reveals signals that cannot be associated with 1·OP8+. For example, there is a singlet resonating at δ = 1.28 ppm and a small doublet (J = 7.8 Hz) resonating at δ = 6.78 ppm, but in the 1H NMR spectrum recorded 13 min later (Fig. S9b, ESI†) these signals were almost absent. These results clearly suggest that a dynamic process takes place in the oxidised solution of 18+ and that the decreasing signals appearing at δ = 1.28 ppm and δ = 6.78 ppm most likely can be assigned to the C(CH3)3 and OP-Hb protons, respectively, in 1·TEG8+.
To reduce the speed in which 1·TEG8+ was converted into 1·OP8+, a solution of 14+ in CD3CN was oxidised at a lower temperature (258 K) with an excess of TBPASbCl6 and once again a 1H NMR spectrum (400 MHz) was recorded as fast as possible (∼5 min) after mixing the two components. In this case, it transpires (Fig. 4c) that both 1·TEG8+ and 1·OP8+ are present in the oxidised mixture in almost equal proportions, since two sets of distinct signals with equal intensities can be observed in the 1H NMR spectrum for numerous of the protons present in 18+. For instance, in the aromatic region of the spectrum, two broad signals resonating at δ = 8.83 ppm and δ = 9.02 ppm appear which can be assigned to the bipyridinium α-H protons (Fig. 1) in 1·OP8+ and 1·TEG8+, respectively. In addition, it is clearly evident from the 1H NMR spectra recorded of 18+ at 298 K (Fig. 4b) and 258 K (Fig. 4c) that the signal assigned to the OP-Hb protons (δ = 6.79 ppm) in 1·TEG8+ has increased in intensity upon lowering the temperature from 298 K to 258 K.
Similar oxidation studies (see ESI†) have been carried out on the [2]rotaxane 24+. In this case, it was found (Fig. S11 and S13b, ESI†) that 2·OP8+ was the only translational isomer present at 298 K immediately (∼5 min) after 2·MPTTF4+ was oxidised with TBPASbCl6 and that the metastable 2·TEG8+ translational isomer only could be observed in significant amounts in the 1H NMR spectrum (Fig. S13c, ESI†) at lower temperature (253 K).
For both 18+ and 28+, the SEt substituent bound to either the MPTTF or the TTF unit, respectively, successfully block the movement of CBPQT4+ towards the DIPP moiety since no signals in the 1H NMR spectra that can be associated with CBPQT4+ encircling the glycol linker attached to the DIPP stopper group were observed.7e
Kinetic investigations of 18+ and 28+
Once the switching mechanism for 1·MPTTF4+ and 2·MPTTF4+ was established and it qualitatively was found that the metastable translational isomers 1·TEG8+ and 2·TEG8+ were formed upon oxidation of 1·MPTTF4+ and 2·MPTTF4+, respectively, the next step was to monitor the stability of 1·TEG8+ and 2·TEG8+ by following (Scheme 1) their conversion into the thermodynamically more stable translational isomers 1·OP8+ and 2·OP8+, respectively. If 1·TEG8+ and 2·TEG8+ are to be converted into 1·OP8+ and 2·OP8+, respectively, it requires that the CBPQT4+ ring moves from its position on the TEG linker between the TTF2+ and MPTTF2+ dications and over either the TTF2+ dication in 18+ or the MPTTF2+ dication in 28+ allowing the size of the electrostatic barrier, when CBPQT4+ moves across one of these two dications, to be quantified.A series of oxidation experiments were carried out at different temperatures and the conversion of 1·TEG8+ and 2·TEG8+ into 1·OP8+ and 2·OP8+, respectively, was monitored using variable temperature (VT) 1H NMR spectroscopy. The experiments were carried out by preparing stock solutions of the [2]rotaxanes 14+ and 24+ in CD3CN (1.5 mM) and keeping them at 233 K overnight followed by the addition of an excess of the oxidant TBPASbCl6 to a sample of the stock solution. Subsequently, 1H NMR spectra (600 MHz) were recorded as fast as possible after oxidation of 14+ at 243, 253, 258, 263 and 268 K and of 24+ at 243, 253, 263 and 268 K and then approximately every 5 min. Representative examples for 14+ (258 K) and 24+ (253 K) are shown in Fig. S16 (ESI†) and Fig. S17 (ESI†), respectively. It is evident from the time-resolved 1H NMR spectra that there is a net movement of CBPQT4+ from the TEG linker (purple) towards the OP unit (red), i.e.1·TEG8+ and 2·TEG8+ are converted into 1·OP8+ and 2·OP8+, respectively. These processes are unimolecular reactions and can accordingly be expected to follow first-order kinetics.26 Since the energy of 1·TEG8+ and 2·TEG8+ are significantly higher than the energy of 1·OP8+ and 2·OP8+, respectively, it can be assumed that k1 ≫ k−1, where k1 is the rate constant for the movement of CBPQT4+ from the TEG linker to the OP unit and k−1 is the rate constant for the reverse process (Scheme 1). Hence, k1 can be determined if appropriate data points are collected in the early stage of the experiments where the reverse process is not yet occurring to any significant extent.
The movement of CBPQT4+ from the TEG linker to the OP unit was followed using several signals associated with protons in both translational isomers as probes (Tables S3 and S4, ESI†). An indicative signal that illustrates this process very well at 253 K is the dominant singlet resonating at 1.25 ppm (Fig. 5b). This signal can be associated with the C(CH3)3 protons in 1·TEG8+ and it is clearly evident that the intensity of this singlet decreases as a function of time. Concomitant with this change, the intensity of the singlet resonating at 1.34 ppm, which can be assigned to the C(CH3)3 protons in 1·OP8+, is found to increase over the same time frame illustrating that 1·TEG8+ is converted into 1·OP8+. In each case, the experimental data were subjected to a first-order analysis, and k1 values were obtained for the movement of CBPQT4+ from the TEG linker to the OP unit in CD3CN at different temperatures. The straight line obtained by plotting lnI as a function of t, where I is the integral of the signal in question and t is the time, confirmed¶¶ the first-order nature of the process. Representative examples for the conversion of 1·TEG8+ into 1·OP8+ and for 2·TEG8+ into 2·OP8+ are shown in Fig. 5c and S18c (ESI†), respectively. The k1 values and the corresponding free energies of activation (ΔG‡(k1)) were obtained directly from the slope of these straight lines and are recorded in Tables S3 and S4 (ESI†). Finally, the rate constant (kav1) and the derived energy of activation ΔG‡(kav1) for the movement of CBPQT4+ from the TEG linker to the OP unit at each temperature were obtained (Table 2) as an average of the k1 values obtained for each of the different probes collected in Tables S3 and S4 (ESI†).
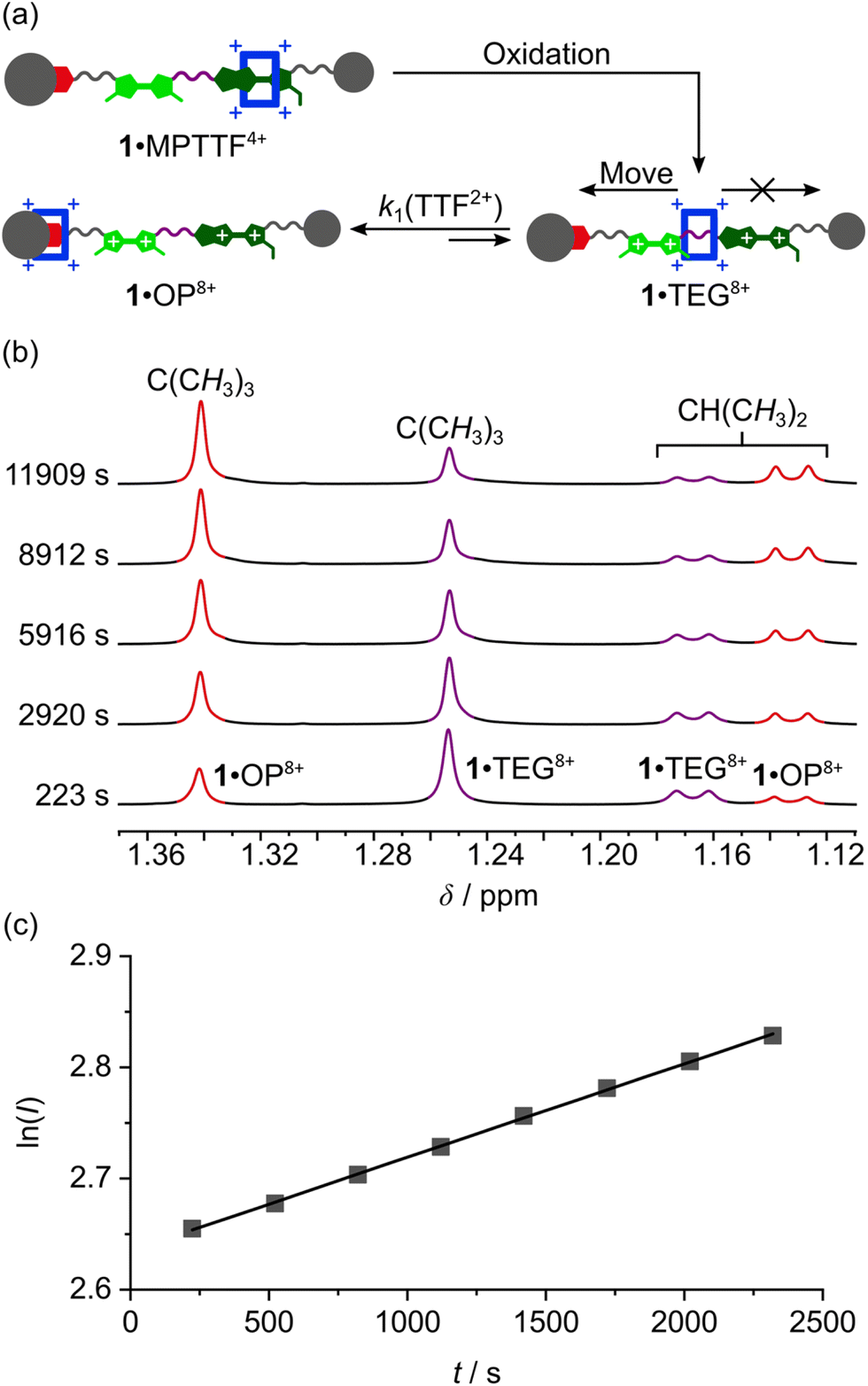 | ||
| Fig. 5 (a) A cartoon representation illustrating the oxidation of 1·MPTTF4+ leading initially to the formation of 1·TEG8+ followed by the movement of CBPQT4+ across the TTF dication (i.e. TTF2+) to produce 1·OP8+. (b) A series of partial 1H NMR spectra (600 MHz, 253 K, CD3CN) of the oxidised [2]rotaxane 18+ recorded at different delay times after addition of an excess (28–32 equiv.) of TBPASbCl6 to 14+ (c = 1.5 mM) showing the increasing signals for the resonances associated with the C(CH3)3 protons (δ = 1.34 ppm) and the CH(CH3)2 protons (δ = 1.13 ppm), respectively, in 1·OP8+, and the decreasing signals for the resonances associated with the C(CH3)3 protons (δ = 1.25 ppm) and the CH(CH3)2 protons (δ = 1.17 ppm), respectively, in 1·TEG8+. (c) Plot of lnI against t at 253 K for the movement of CBPQT4+ across the TTF2+ unit, where I is the integral of the signal at δ = 1.34 ppm. The eight data points have been fitted by the best straight line (black line), giving a correlation coefficient of 1.000, indicating that first-order kinetics are in operation. The slope of the line corresponds to the rate constant k1 for the movement of CBPQT4+ over the TTF2+ unit in 18+, according to the relationship lnI = k1t. | ||
| T [K] | # probesc | n | k av1 , [×10−4 s−1] | ΔG‡(kav1)b,e [kcal mol−1] | |
|---|---|---|---|---|---|
|
a The kav1 values were obtained by taking the average of the k1 values collected in Tables S3 and S4 (ESI).
b The ΔG‡(kav1) values were obtained using the relationship ΔG‡ = −RTln(kav1h/kBT) where R is the gas constant, T is the absolute temperature, kav1 is the average rate constant, h is Planck's constant and kB is the Boltzmann constant.
c # probes represent the number of probes used to calculate the average kav1.
d
n is the number of data points used to obtain the individual rate constant k1 as described in Tables S3 and S4 (ESI).
e The errors are calculated from Koumura et al.27 with Δt = 0.1 s, ΔT = 0.3 K and ΔI = 5%.
|
|||||
| 1 8+ | 243 | 6 | 6 | 0.53 ± 0.07 | 18.90 ± 0.07 |
| 253 | 10 | 8 | 0.81 ± 0.15 | 19.48 ± 0.10 | |
| 258 | 7 | 7 | 1.30 ± 0.10 | 19.63 ± 0.04 | |
| 263 | 12 | 5 | 1.87 ± 0.26 | 19.83 ± 0.08 | |
| 268 | 13 | 5 | 2.40 ± 0.29 | 20.09 ± 0.07 | |
| 2 8+ | 243 | 7 | 19 | 0.36 ± 0.03 | 19.08 ± 0.04 |
| 253 | 8 | 9 | 1.21 ± 0.04 | 19.28 ± 0.03 | |
| 263 | 15 | 5 | 3.43 ± 0.28 | 19.52 ± 0.05 | |
| 268 | 8 | 5 | 5.74 ± 0.43 | 19.62 ± 0.05 | |
Since the ΔG‡(kav1) values have been determined at different temperatures, the enthalpic (ΔH‡(kav1)) and entropic (ΔS‡(kav1)) contributions to the processes where 1·TEG8+ is converted into 1·OP8+ and 2·TEG8+ is converted into 2·OP8+ can be calculated from plots (Fig. 6) of ΔG‡(kav1) against T. Straight lines, each with a good fit, can be approximated to the experimental data, and the kinetic parameters obtained from these plots are summarised in Table 3, together with the extrapolated ΔG‡(kav1) values at 298 K. It is evident from inspection of the data in Table 3 that 1·TEG8+ is converted into 1·OP8+ more slowly than 2·TEG8+ is converted into 2·OP8+ at 298 K. In fact, the Gibbs free energy of activation is 1.2 kcal mol−1 higher in the former than in the latter, indicating that the MPTTF2+ dication constitutes a significantly smaller barrier for CBPQT4+ compared to the TTF2+ dication at 298 K. This difference in Gibbs free energy of activation (ΔΔG‡) can be used to describe how CBPQT4+ will move in the hypothetical [2]rotaxane R4+ (Fig. 7a) following its oxidation to R8+. Structurally, the [2]rotaxane R4+ is very similar to 14+ and 24+, where the only differences are that the DIPP stopper has been replaced by a triarylmethyl stopper connected to an OP unit and that the large bulky SEt group has been replaced by a much smaller SMe group. In principle, the [2]rotaxane R4+ is tetra-stable, because it is composed of two primary TTF-based stations (i.e. TTF and MPTTF) and two identical secondary OP stations. Consequently, it can theoretically exist, as a mixture of four different translational isomers (i.e.R·OP4+left, R·TTF4+, R·MPTTF4+ and R·OP4+right), but as in the case of 14+ and 24+, it can be expected that the majority of R4+ will exist as the R·MPTTF4+ translational isomer|||| and that R·TTF4+ only will be present in a small amount, while the population of the two OP translational isomers of R4+ will be very close to zero. Similar to what is observed for 1·MPTTF4+ and 2·MPTTF4+, oxidation of R·MPTTF4+ to R8+ is expected initially to lead to the formation of the metastable isomer R·TEG8+ (Fig. 7b), where the CBPQT4+ ring is located between the TTF2+ and MPTTF2+ dications. Since the TTF and MPTTF units in the [2]rotaxane R8+ only are substituted with SMe groups, the CBPQT4+ ring now has the option to cross either the TTF2+ dication or the MPTTF2+ dication to produce R·OP8+left or R·OP8+right, respectively. Using the ΔΔG‡ value of 1.2 kcal mol−1 (vide supra), it can be calculated (see ESI†) that approximately 90% of CBPQT4+ will move across the MPTTF2+ dication and only 10% over the TTF2+ dication when it has the possibility to choose between these two different dications at 298 K.
 | ||
| Fig. 6 Linear plots of ΔG‡(kav1) as functions of temperature (T) for the movement of CBPQT4+ across either TTF2+ (light green points, 18+) or MPTTF2+ (dark green points, 28+) in CD3CN. The ΔG‡(kav1) values were obtained as described in Table 2. The data points have been fitted to best straight lines, giving correlation coefficients of 0.982 (18+) and 0.999 (28+), respectively. The slope and intercept of each line of best fit give the values −ΔS‡(kav1) and ΔH‡(kav1) (see Table 3), respectively, from the equation ΔG‡(kav1) = ΔH‡(kav1) − T × ΔS‡(kav1). | ||
| k av1 298K [×10−3 s−1] | t av1/2 298K [ms] | ΔG‡(kav1)298K [kcal mol−1] | ΔH‡(kav1) [kcal mol−1] | ΔS‡(kav1) [cal mol−1 K−1] | |
|---|---|---|---|---|---|
|
a
k
av1 298K, tav1/2 298K, ΔG‡(kav1)298K, ΔH‡(kav1) and ΔS‡(kav1) are the values for the activation parameters.
b The ΔS‡(kav1) and ΔH‡(kav1) values were obtained from the intercept and slope of the straight line in the plot (Fig. 6) of ΔG‡(kav1) against T using the relationship ΔG‡(kav1) = ΔH‡(kav1) − T × ΔS‡(kav1), where T is the absolute temperature.
c The ΔG‡(kav1)298K values were extrapolated from the equation ΔG‡(kav1) = ΔH‡(kav1) − T × ΔS‡(kav1), while the kav1 298K values were calculated from the equation kav1 298K = exp(−ΔG‡(kav1)298K/RT) × kBT/h.
d Values for tav1/2 298K were determined using the equation tav1/2 298K = ln2/kav1 298K.
e The errors were calculated from Koumura et al.27 with Δt = 0.1 s, ΔT = 0.3 K and ΔI = 5%.
|
|||||
| 1 8+ | 1.08 | 0.64 | 21.5 ± 0.2 | 7.6 ± 1.4 | −46.6 ± 5.5 |
| 2 8+ | 8.27 | 0.08 | 20.3 ± 0.1 | 13.7 ± 0.7 | −22.0 ± 2.7 |
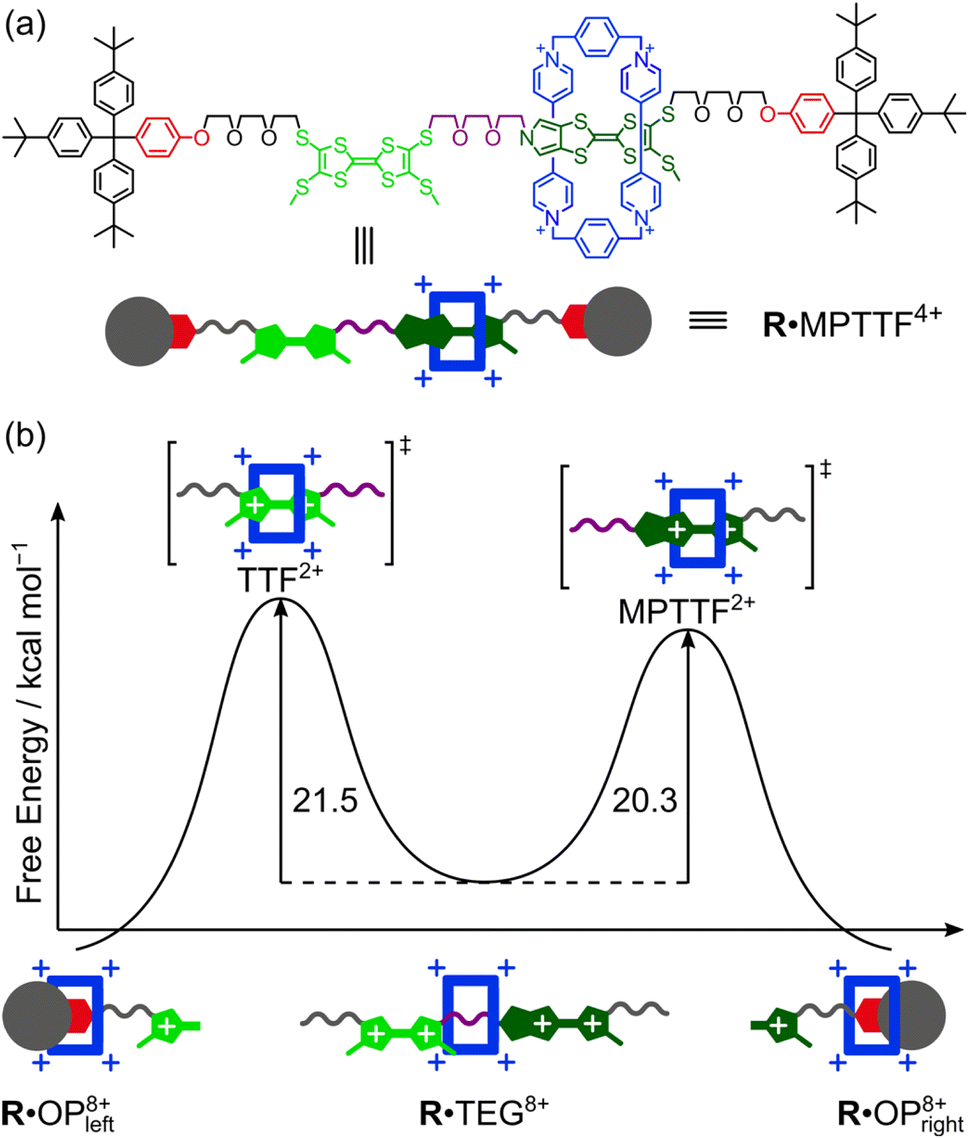 | ||
| Fig. 7 (a) Molecular formula of the hypothetical tetra-stable [2]rotaxane R4+ (only one translational isomer is shown which will exist as a mixture of E- and Z-isomers) and a cartoon representation of the expected dominating R·MPTTF4+ translational isomer. (b) Energy diagram showing the Gibbs free energies of activation (ΔG‡) for the movement of CBPQT4+ away from the TEG linker across either a TTF2+ dication or an MPTTF2+ dication to an OP unit in CD3CN at 298 K. | ||
The fact that the metastable states of the two oxidised [2]rotaxanes (i.e.1·TEG8+ and 2·TEG8+) show (Table 3) significantly different sets of activation parameters (ΔG‡(kav1), ΔH‡(kav1), ΔS‡(kav1)) indicates that the rate-limiting steps for the movement of the CBPQT4+ ring across the TTF2+ and MPTTF2+ dications have significantly different qualities. The negative activation entropies observed for both 18+ and 28+, suggest that there is an increase in the ordering of the transition state structures 18+‡ and 28+‡ relative to the initial structures of 1·TEG8+ and 2·TEG8+, respectively. In the [2]rotaxane 18+, the movement of the CBPQT4+ ring from the TEG chain across the TTF2+ dication to the OP station has a small enthalpy of activation, ΔH‡(kav1) = +7.6 ± 1.4 kcal mol−1, while the activation entropy is large and negative, ΔS‡(kav1) = −46.6 ± 5.5 cal mol−1 K−1. The magnitude of this entropic factor is consistent with the need for a significant increase in the ordering of both the SMe group and the TTEG linker to allow passage of the CBPQT4+ ring over the SMe/TTEG steric barrier.7c On its way across the SMe/TTEG steric barrier, the CBPQT4+ ring is repelled electrostatically by the two positive charges on the TTF2+ dication leading to a further decrease in conformational degrees of freedom of the transition state structure. Once the CBPQT4+ ring has managed to get over the first SMe/TTEG steric barrier and now encircles the TTF2+ dication, it seems likely that the energy for the movement of CBPQT4+ over the second SMe/TTEG steric barrier will be smaller, because of the large electrostatic repulsion between CBPQT4+ and the TTF2+ dication. By contrast, the [2]rotaxane 28+ shows an overall activation barrier that is enthalpic in nature (ΔH‡(kav1) = +13.7 ± 0.7 kcal mol−1) with a much reduced entropic contribution (ΔS‡(kav1) = −22.0 ± 2.7 cal mol−1 K−1). The large activation enthalpy observed in the [2]rotaxane 28+ can most likely be accounted for by the fact that the CBPQT4+ ring needs to move across the MPTTF2+ dication (electrostatic barrier) leading to an increase in enthalpy before it is forced to move over the SMe/TTEG steric barrier. Since the MPTTF unit only is attached to one SMe group and is more elongated compared to the TTF unit, the transition state structure of 28+ is most likely less ordered and consequently has lower entropic contribution relative to the transition state structure of 18+.
Finally, it is evident from Fig. 6 and Table 3 that the size of the TTF2+ and the MPTTF2+ electrostatic activation barriers are heavily temperature dependent. At high temperatures (T > 298 K), the MPTTF2+ dication constitutes a significantly smaller barrier for CBPQT4+ compared to the TTF2+ dication, while the reverse is true at low temperature (T < 200 K). This observation demonstrates that temperature potentially can be used to control in which direction the CBPQT4+ ring will move upon oxidation of MIMs incorporating both a TTF and an MPTTF unit.
Conclusions
In conclusion, the syntheses of two isomeric tri-stable [2]rotaxanes 1·4PF6 and 2·4PF6, with three different stations, a tetrathiafulvalene (TTF), a monopyrrolotetrathiafulvalene (MPTTF) and an oxyphenylene (OP) unit, for the tetracationic cyclobis(paraquat-p-phenylene) (CBPQT4+) ring have been reported. In both cases, 1H NMR spectroscopic analysis revealed that the [2]rotaxanes exist primarily as the translational isomer in which CBPQT4+ encircles the MPTTF station (i.e.1·MPTTF4+ and 2·MPTTF4+). Following tetra-oxidation of 1·MPTTF4+ and 2·MPTTF4+, either electrochemically or chemically, it was found that a metastable state was formed in which the CBPQT4+ ring was located on the triethylene (TEG) glycol linker connecting the TTF2+ and MPTTF2+ dications producing 1·TEG8+ and 2·TEG8+, respectively. The steric hindrance exhibited from the SEt group situated on either the MPTTF unit in 1·TEG8+ or the TTF unit in 2·TEG8+ made it possible to study the kinetics when these high-energy co-conformations slowly interconvert into thermodynamically more stable co-conformations (i.e.1·OP8+ and 2·OP8+). The conversion could be followed by 1H NMR spectroscopy at different temperatures allowing us to calculate the rate constants and associated energies of activation. From our kinetic studies, we were able to determine the free energy of the transition state when CBPQT4+ moves across either a TTF2+ (21.5 kcal mol−1) or an MPTTF2+ (20.3 kcal mol−1) electrostatic barrier at 298 K. Consequently, it can be concluded that the TTF2+ dication constitutes a significantly higher electrostatic barrier for CBPQT4+ compared to the MPTTF2+ dication at 298 K. In fact, the Gibbs free energy of activation is 1.2 kcal mol−1 higher in the former than in the latter showing that approximately 90% of CBPQT4+ will move across the MPTTF2+ dication and only 10% over the TTF2+ dication when it has the possibility to choose between these two different dications. Furthermore, the electrostatic barrier for the movement of CBPQT4+ across the MPTTF2+ (20.3 kcal mol−1) dication in the tetra-oxidised [2]rotaxane 28+ is 1.5 kcal mol−1 smaller compared to the movement of CBPQT4+ across a similar MPTTF2+ (21.8 kcal mol−1) dication in a bistable [2]rotaxane7e containing only an MPTTF unit in the dumbbell component. This observation can be accounted for by the increased electrostatic repulsion present when the CBPQT4+ ring is located between two doubly oxidised units (i.e. TTF2+ and MPTTF2+) in 28+. Our results demonstrate for the first time that the combination of a TTF and an MPTTF unit following their oxidations can be used to induce directional movement of the CBPQT4+ ring in molecular machines with a 90% efficiency – a feature that might be used to construct an electroactive pumping unit that works under oxidative conditions allowing rotaxanes and catenanes incorporating TTF and MPTTF to be designed which show electrically driven unidirectional motion, i.e. rotary and linear molecular motors.Data availability
Synthetic procedures and characterisation data along with the kinetic data are available in the ESI.†Author contributions
Conceptualisation and methodology: RF, MLS and JOJ. SKJ and MLS performed the synthesis with contributions from MCL and SEW. Investigation: SKJ performed the described investigations of the two [2]rotaxanes with supervision by RF and JOJ. Writing: SKJ, MSN, RF and JOJ prepared the original manuscript with revisions made by SKJ, MSN and JOJ.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Independent Research Fund Denmark | Natural Sciences (FNU, project no. 9040-00169B) and the Villum Foundation.Notes and references
- (a) Ó. Gutiérrez-Sanz, P. Natale, I. Márquez, M. C. Marques, S. Zacarias, M. Pita, I. A. C. Pereira, I. López-Montero, A. L. De Lacey and M. Vélez, Angew. Chem., Int. Ed., 2016, 55, 6216–6220 CrossRef PubMed; (b) S. Mukherjee and A. Warshel, Photosynth. Res., 2017, 134, 1–15 CrossRef CAS PubMed.
- (a) S. Liepelt and R. Lipowsky, Phys. Rev. Lett., 2007, 98, 258102 CrossRef PubMed; (b) M. Von Delius, E. M. Geertsema and D. A. Leigh, Nat. Chem., 2010, 2, 96–101 CrossRef CAS PubMed; (c) M. L. Mugnai, C. Hyeon, M. Hinczewski and D. Thirumalai, Rev. Mod. Phys., 2020, 92, 025001 CrossRef CAS.
- S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS PubMed.
- E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2007, 46, 72–191 CrossRef CAS PubMed.
- (a) S. Kassem, T. Van Leeuwen, A. S. Lubbe, M. R. Wilson, B. L. Feringa and D. A. Leigh, Chem. Soc. Rev., 2017, 46, 2592–2621 RSC; (b) S. Erbas-Cakmak, S. D. P. Fielden, U. Karaca, D. A. Leigh, C. T. McTernan, D. J. Tetlow and M. R. Wilson, Science, 2017, 358, 340–343 CrossRef CAS PubMed; (c) L. Zhang, V. Marcos and D. A. Leigh, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9397–9404 CrossRef CAS PubMed; (d) Y. Qiu, Y. Feng, Q.-H. Guo, R. D. Astumian and J. F. Stoddart, Chem, 2020, 6, 1952–1977 CrossRef CAS; (e) M. N. Tasbas, E. Sahin and S. Erbas-Cakmak, Coord. Chem. Rev., 2021, 443, 214039 CrossRef CAS; (f) A. Mondal, R. Toyoda, R. Costil and B. L. Feringa, Angew. Chem., Int. Ed., 2022, 61, e202206631 CrossRef CAS PubMed; (g) S. Borsley, E. Kreidt, D. A. Leigh and B. M. W. Roberts, Nature, 2022, 604, 80–85 CrossRef CAS PubMed; (h) L. Zhang, Y. Qiu, W.-G. Liu, H. Chen, D. Shen, B. Song, K. Cai, H. Wu, Y. Jiao, Y. Feng, J. S. W. Seale, C. Pezzato, J. Tian, Y. Tan, X.-Y. Chen, Q.-H. Guo, C. L. Stern, D. Philp, R. D. Astumian, W. A. Goddard III and J. F. Stoddart, Nature, 2023, 613, 280–286 CrossRef CAS PubMed.
- N. Koumura, R. W. J. Zijlstra, R. A. Van Delden, N. Harada and B. L. Feringa, Nature, 1999, 401, 152–155 CrossRef CAS PubMed.
- (a) J. O. Jeppesen, K. A. Nielsen, J. Perkins, S. A. Vignon, A. Di Fabio, R. Ballardini, M. T. Gandolfi, M. Venturi, V. Balzani, J. Becher and J. F. Stoddart, Chem.–Eur. J., 2003, 9, 2982–3007 CrossRef CAS; (b) J. D. Badjic, V. Balzani, A. Credi, S. Silvi and J. F. Stoddart, Science, 2004, 303, 1845–1849 CrossRef CAS PubMed; (c) S. S. Andersen, A. I. Share, B. L. C. Poulsen, M. Kørner, T. Duedal, C. R. Benson, S. W. Hansen, J. O. Jeppesen and A. H. Flood, J. Am. Chem. Soc., 2014, 136, 6373–6384 CrossRef CAS PubMed; (d) M. R. Wilson, J. Solà, A. Carlone, S. M. Goldup, N. Lebrasseur and D. A. Leigh, Nature, 2016, 534, 235–240 CrossRef CAS PubMed; (e) M. S. Neumann, A. F. Smith, S. K. Jensen, R. Frederiksen, M. L. Skavenborg and J. O. Jeppesen, Chem. Commun., 2023, 59, 6335–6338 RSC.
- C. J. Bruns and J. F. Stoddart, The Nature of the Mechanical Bond:From Molecules to Machines, John Wiley & Sons, Inc., Hoboken, New Jersey, 2016 Search PubMed.
- J. W. Steed and J. L. Atwood, Supramolecular Chemistry, John Wiley & Sons Ltd, Chippenham, Wiltshire, 2nd edn, 2009 Search PubMed.
- (a) M. N. Chatterjee, E. R. Kay and D. A. Leigh, J. Am. Chem. Soc., 2006, 128, 4058–4073 CrossRef CAS PubMed; (b) J. S. W. Seale, Y. Feng, L. Feng, R. D. Astumian and J. F. Stoddart, Chem. Soc. Rev., 2022, 51, 8450–8475 RSC.
- R. D. Astumian, Nat. Commun., 2019, 10, 3837 CrossRef PubMed.
- (a) C. Cheng, P. R. McGonigal, S. T. Schneebeli, H. Li, N. A. Vermeulen, C. Ke and J. F. Stoddart, Nat. Nanotechnol., 2015, 10, 547–553 CrossRef CAS PubMed; (b) C. Pezzato, M. T. Nguyen, C. Cheng, D. J. Kim, M. T. Otley and J. F. Stoddart, Tetrahedron, 2017, 73, 4849–4857 CrossRef CAS.
- L. M. Zhao, L. S. Zheng, X. Wang and W. Jiang, Angew. Chem., Int. Ed., 2022, 61, e202214296 CrossRef CAS PubMed.
- (a) A. C. Fahrenbach, Z. Zhu, D. Cao, W.-G. Liu, H. Li, S. K. Dey, S. Basu, A. Trabolsi, Y. Y. Botros, W. A. Goddard III and J. F. Stoddart, J. Am. Chem. Soc., 2012, 134, 16275–16288 CrossRef CAS PubMed; (b) H. V. Schröder and C. A. Schalley, Beilstein J. Org. Chem., 2018, 14, 2163–2185 CrossRef PubMed; (c) S. S. Andersen, A. W. Saad, R. Kristensen, T. S. Pedersen, L. J. O'Driscoll, A. H. Flood and J. O. Jeppesen, Org. Biomol. Chem., 2019, 17, 2432–2441 RSC; (d) M. Jensen, R. Kristensen, S. S. Andersen, D. Bendixen and J. O. Jeppesen, Chem.–Eur. J., 2020, 26, 6165–6175 CrossRef CAS PubMed; (e) X. Y. Chen, H. Chen and J. Fraser Stoddart, Angew. Chem., Int. Ed., 2022, 62, e202211387 CrossRef PubMed.
- (a) D. Philp, A. M. Z. Slawin, N. Spencer, J. F. Stoddart and D. J. Williams, J. Chem. Soc., Chem. Commun., 1991, 1584–1586 RSC; (b) W. Devonport, M. A. Blower, M. R. Bryce and L. M. Goldenberg, J. Org. Chem., 1997, 62, 885–887 CrossRef CAS; (c) J. O. Jeppesen, J. Perkins, J. Becher and J. F. Stoddart, Org. Lett., 2000, 2, 3547–3550 CrossRef CAS PubMed; (d) M. B. Nielsen, J. O. Jeppesen, J. Lau, C. Lomholt, D. Damgaard, J. P. Jacobsen, J. Becher and J. F. Stoddart, J. Org. Chem., 2001, 66, 3559–3563 CrossRef CAS PubMed.
- M. Asakawa, P. R. Ashton, V. Balzani, A. Credi, C. Hamers, G. Mattersteig, M. Montalti, A. N. Shipway, N. Spencer, J. F. Stoddart, M. S. Tolley, M. Venturi, A. J. P. White and D. J. Williams, Angew. Chem., Int. Ed., 1998, 37, 333–337 CrossRef CAS PubMed.
- R. Kristensen, M. S. Neumann, S. S. Andersen, P. C. Stein, A. H. Flood and J. O. Jeppesen, Org. Biomol. Chem., 2022, 20, 2233–2248 RSC.
- (a) J. O. Jeppesen, J. Becher and J. F. Stoddart, Org. Lett., 2002, 4, 557–560 CrossRef CAS PubMed; (b) J. O. Jeppesen, S. A. Vignon and J. F. Stoddart, Chem.–Eur. J., 2003, 9, 4611–4625 CrossRef CAS PubMed; (c) R. Kristensen, S. S. Andersen, G. Olsen and J. O. Jeppesen, J. Org. Chem., 2017, 82, 1371–1379 CrossRef CAS PubMed.
- (a) Z.-T. Li, P. C. Stein, N. Svenstrup, K. H. Lund and J. Becher, Angew. Chem., Int. Ed. Engl., 1995, 34, 2524–2528 CrossRef CAS; (b) P. R. Ashton, V. Balzani, J. Becher, A. Credi, M. C. T. Fyfe, G. Mattersteig, S. Menzer, M. B. Nielsen, F. M. Raymo, J. F. Stoddart, M. Venturi and D. J. Williams, J. Am. Chem. Soc., 1999, 121, 3951–3957 CrossRef CAS.
- F.-G. Klärner and F. Wurche, J. Prakt. Chem., 2000, 342, 609–636 CrossRef.
- P. L. Anelli, P. R. Ashton, R. Ballardini, V. Balzani, M. Delgado, M. T. Gandolfi, T. T. Goodnow, A. E. Kaifer, D. Philp, M. Pietraszkiewicz, L. Prodi, M. V. Reddington, A. M. Z. Slawin, N. Spencer, J. F. Stoddart, C. Vicent and D. J. Williams, J. Am. Chem. Soc., 1992, 114, 193–218 CrossRef CAS.
- (a) J. O. Jeppesen, J. Perkins, J. Becher and J. F. Stoddart, Angew. Chem., Int. Ed., 2001, 40, 1216–1221 CrossRef CAS; (b) J. O. Jeppesen, S. Nygaard, S. A. Vignon and J. F. Stoddart, Eur. J. Org. Chem., 2005, 196–220 CrossRef; (c) S. Nygaard, B. W. Laursen, A. H. Flood, C. N. Hansen, J. O. Jeppesen and J. F. Stoddart, Chem. Commun., 2006, 144–146 RSC; (d) S. Nygaard, K. C. F. Leung, I. Aprahamian, T. Ikeda, S. Saha, B. W. Laursen, S.-Y. Kim, S. W. Hansen, P. C. Stein, A. H. Flood, J. F. Stoddart and J. O. Jeppesen, J. Am. Chem. Soc., 2007, 129, 960–970 CrossRef CAS PubMed; (e) S. S. Andersen, M. Jensen, A. Sørensen, E. Miyazaki, K. Takimiya, B. W. Laursen, A. H. Flood and J. O. Jeppesen, Chem. Commun., 2012, 48, 5157–5159 RSC.
- S. W. Hansen, P. C. Stein, A. Sørensen, A. I. Share, E. H. Witlicki, J. Kongsted, A. H. Flood and J. O. Jeppesen, J. Am. Chem. Soc., 2012, 134, 3857–3863 CrossRef CAS PubMed.
- (a) J. H. Van Esch, R. Klajn and S. Otto, Chem. Soc. Rev., 2017, 46, 5474–5475 RSC; (b) R. Merindol and A. Walther, Chem. Soc. Rev., 2017, 46, 5588–5619 RSC; (c) S. A. P. Van Rossum, M. Tena-Solsona, J. H. Van Esch, R. Eelkema and J. Boekhoven, Chem. Soc. Rev., 2017, 46, 5519–5535 RSC; (d) J. Matern, Y. Dorca, L. Sánchez and G. Fernández, Angew. Chem., Int. Ed., 2019, 58, 16730–16740 CrossRef CAS PubMed.
- S. Nygaard, Y. Liu, P. C. Stein, A. H. Flood and J. O. Jeppesen, Adv. Funct. Mater., 2007, 17, 751–762 CrossRef CAS.
- K. J. Laidler, Chemical Kinetics, Harper Collins, New York, 1987 Search PubMed.
- N. Koumura, E. M. Geertsema, M. B. Van Gelder, A. Meetsma and B. L. Feringa, J. Am. Chem. Soc., 2002, 124, 5037–5051 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic details and procedures with full characterisation, 1D and 2D NMR spectra and full description of kinetic experiments. See DOI: https://doi.org/10.1039/d3sc04408d |
| ‡ The combination of an SMe and an SR group constitutes a larger barrier for the CBPQT4+ ring than the pyrrole moiety which results in CBPQT4+ initially moving across the pyrrole moiety upon oxidation of the MPTTF unit.7e |
| § In the dumbbell 14, the SCH3 protons only give rise to three singlets, most likely because of overlap between two of the four singlets, which is supported by the fact that one of the observed singlets resonating at δ = 2.36 ppm is twice as intense as the other two singlets resonating at δ = 2.35 and 2.38 ppm. |
| ¶ As for the dumbbell 14, the SCH3 protons in 1·TTF4+ only give rise to three singlets, because of overlap between two of the four singlets, supported by the fact that one of the observed singlets resonating at δ = 2.52 ppm is twice as intense as the other two singlets. |
| || A comparison of the 1H NMR spectra (400 MHz) recorded of the dumbbell 24 (Fig. S6†) and the [2]rotaxane 24+ (Fig. S7†) in CD3CN at 298 K, reveals that the pyrrole-H protons in 2·MPTTF4+ have been upfield shifted compared to the dumbbell 24, where they resonate as a singlet at δ = 6.63 ppm. |
| ** The existence of two SCH3 signals for 2·TTF4+ indicates that it exists as a mixture of E- and Z-isomers. |
| †† It has previously been shown by the use of appropriate MPTTF and TTF model compounds that both the first (ΔE1/21 = −0.08 V) and the second (ΔE1/22 = −0.02 V) oxidation process associated with the MPTTF unit take place at potentials that are less positive than those observed for the TTF unit.14d |
| ‡‡ In a single-station [2]rotaxane, containing an MPTTF station in the dumbbell component and CBPQT4+ as the ring component, the first redox-wave was shifted 0.33 V toward a more positive potential, compared to the same process in the corresponding dumbbell.7a |
| §§ Upon oxidation of 14+, the small amount (18%) of 1·TTF4+, present in the isomeric mixture of 14+ will also be oxidised and converted either into 1·TEG8+ or directly into 1·OP8+. |
| ¶¶ At each temperature and for each probe, first-order kinetics are observed to be in operation, i.e. good straight lines are observed (see Tables S3 and S4, ESI†) when lnI is plotted against t, where I is the integral of the signal in question and t is the time. This outcome is a consequence of the fact that the data points were collected in the early stage of the experiments where the reverse process is not yet occurring to any significant extent (see ESI†). |
| |||| It should be emphasised that R·MPTTF4+ has not yet been synthesised, but only represents a model compound used to illustrate how much CBPQT4+ will move across an MPTTF2+ dication compared to a TTF2+ dication when they both are substituted with SMe groups. |
| This journal is © The Royal Society of Chemistry 2023 |