DOI:
10.1039/D3SC04390H
(Edge Article)
Chem. Sci., 2023,
14, 14262-14270
An orbitally adapted push–pull template for N2 activation and reduction to diazene-diide†
Received
21st August 2023
, Accepted 19th November 2023
First published on 21st November 2023
Abstract
A Lewis superacidic bis(borane) C6F4{B(C6F5)2}2 was reacted with tungsten N2-complexes [W(N2)2(R2PCH2CH2PR2)2] (R = Ph or Et), affording zwitterionic boryldiazenido W(II) complexes trans-[W(L)(R2PCH2CH2PR2)2(N2{B(C6F5)2(C6F4B(C6F5)3})] (L = ø, N2 or THF). These compounds feature only one N–B linkage of the covalent type, as a result of intramolecular boron-to-boron C6F5 transfer. Complex trans-[W(THF)(Et2PCH2CH2PEt2)2(N2{B(C6F5)2C6F4B(C6F5)3})] (5) was shown to split H2, leading to a seven-coordinate complex [W(H)2(Et2PCH2CH2PEt2)2(N2{B(C6F5)2}2C6F4)] (7). Interestingly, hydride storage at the metal triggers backward C6F5 transfer. This reverts the bis(boron) moiety to its bis(borane) state, now doubly binding the distal N, with structural parameters and DFT computations pointing to dative N→B bonding. By comparison with an N2 complex [W(H)2(Et2PCH2CH2PEt2)2(N2{B(C6F5)3}] (10) differing only in the Lewis acid (LA), namely B(C6F5)3, coordinated to the distal N, we demonstrate that two-fold LA coordination imparts strong N2 activation up to the diazene-diide (N22−) state. To the best of our knowledge, this is the first example of a neutral LA coordination that induces reduction of N2.
Introduction
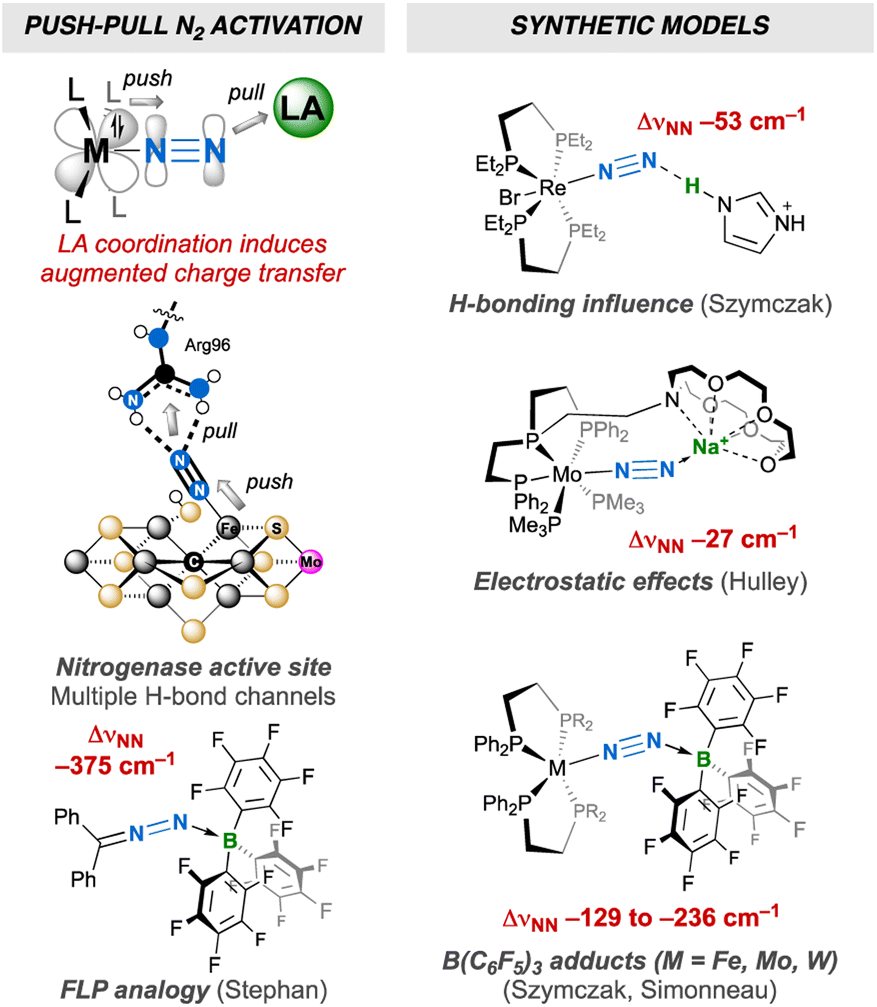
Dinitrogen activation by transition-metals as a means to transform this inert molecule under mild conditions has attracted the interest of chemists for almost 60 years.1 Over the last five decades, dinitrogen complexes of the d-,1 f-2 and recently s-3 and p-4 block elements have been described in the literature, exhibiting a wealth of coordination modes, oxidation states and reactivity for the N2 ligand. Some have shown potential as greener alternatives5 to the energy- and fossil fuel-intensive Haber–Bosch process,6 or for the synthesis of value-added (organo)nitrogen compounds.7 They can serve as models of the nitrogen-fixing enzymes nitrogenases, that feature an organometallic FeM (M = Mo, V or Fe) cofactor that can bind the diatomic molecule.8 Interaction of dinitrogen with a transition metal is well described by the Chatt–Dewar–Duncanson model:9 the metal centre “pushes” its d-electrons into the antibonding orbitals of N2. High activation may ultimately be reached in dinuclear edifices through “push–push” activation.1b,10 “Push–pull” activation of dinitrogen11 remains less explored. It consists of enhancing the polarization of dinitrogen when coordinated to a metal centre through non-covalent interaction with an electron-deficient species,12 mimicking to a certain extent the effect of secondary-sphere H-bonding between N2 and the multiple acidic functions of nitrogenases' active site.12f,i,13,14 Several groups have evidenced that a single non-covalent interaction of a neutral transition-metal12a or main group12b–g,j Lewis acid (LA), an alkali12f,h or Au(I)+ cation12j or a weak acid12i (neutral or charged) leads to enhanced activation of N2 thanks to augmented charge transfer (Fig. 1). A parallel between push–pull activation and the chemistry of frustrated Lewis pairs (FLPs)15 can be drawn, since the reactivity thereof resides in the synergistic action of a strong LA pulling electron density and a bulky Lewis base pushing its electron pair into antibonding orbitals of a substrate to activate. The chemistry of FLP still lacks a genuine N2 capture example,16 but the Stephan group has proposed the B(C6F5)3 adduct of diphenyldiazomethane as a model thereof (Fig. 1).17,18 The above-mentioned body of work thus provides clues of what to expect from push–pull or FLP-related N2 activation in terms of structure, whereas the higher polarization of the N2 unit in these compounds may ultimately lead to the discovery of new reactivity.12f,19 Besides, the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N→LA linkage may show FLP-type reactivity, such as Si–H bond heterolytic splitting, resulting in N2 silylation.12g,20 Of note, the analogous reaction employing H2 for N–H bond formation21 is yet to be achieved and is challenged by the propensity of H2 to easily substitute N2.22 Alternatively, [R3XH]+[R′3BH]– (X = N or P) ion pairs delivered by FLP H2 splitting23 have been employed to protonate N2.24,25
N→LA linkage may show FLP-type reactivity, such as Si–H bond heterolytic splitting, resulting in N2 silylation.12g,20 Of note, the analogous reaction employing H2 for N–H bond formation21 is yet to be achieved and is challenged by the propensity of H2 to easily substitute N2.22 Alternatively, [R3XH]+[R′3BH]– (X = N or P) ion pairs delivered by FLP H2 splitting23 have been employed to protonate N2.24,25
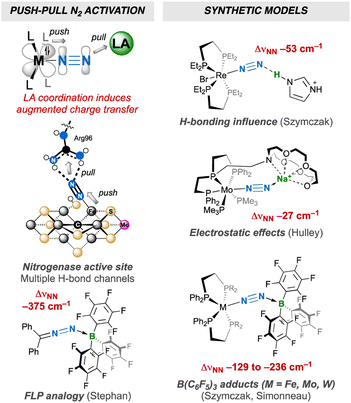 |
| | Fig. 1 N2 push–pull activation: bioinorganic relevance, FLP analogy and synthetic models. | |
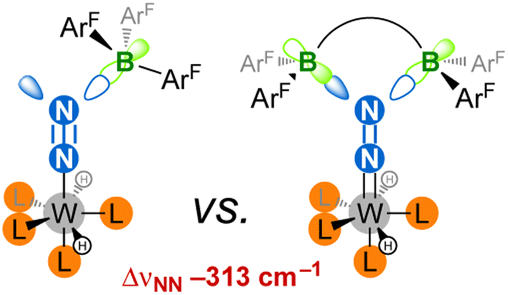
Main-group Lewis acids interact with a molecular orbital of an N2 complex that mixes a filled metal's d orbital and a π* orbital of N2.12f,j,26 To the best of our knowledge, the two-fold coordination of a neutral LA to the distal N was never reported,27,28 although it would be interesting to judge how far the resulting push–pull effect affects the N2 unit when compared to two-fold protonation24a,29 or functionalisation,30 without being altered by the association of the conjugated base or leaving group with the metal that generally takes place in the two former cases. Such an achievement would also complement previous studies aimed at measuring the push–pull influence of monotopic LAs,12 showing how multiple electron-deficient centres (as found in nitrogenases' active sites) may synergistically enhance N2 activation. To this end, we have explored the coordination of a strongly electrophilic bis(borane) to a terminal N2 complex (Fig. 1) and endeavour to compare the push–pull effect with the one induced by B(C6F5)3. We hoped that the two empty p orbitals of the tethered B atoms would spatially match with the two lobes of the distal N's p orbital. Such an “orbitally adapted” push–pull template should result in high N2 activation and polarisation as a result of greater stabilization of the d+π* frontier orbital (Fig. 2). In this article, we show that particular conditions should be met to observe two-fold coordination of the bis(borane). Indeed, hydride storage through H2 oxidative addition at the metal–N2 complex was key to achieve this goal, allowing us to discover that LA adducts of group 6 N2 complexes may react with H2 without N2 loss (but not through FLP-type splitting). It is shown that two-fold LA coordination exerts a very strong influence on the diatomic molecule, and comparison of structural, spectroscopic and computational data supports its activation up to the 2-e– reduced state.
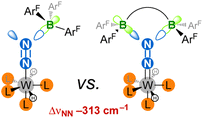 |
| | Fig. 2 This work: achievement of two-fold LA coordination on an N2 ligand results in an extreme push–pull effect. ArF = C6F5. | |
Results and discussion
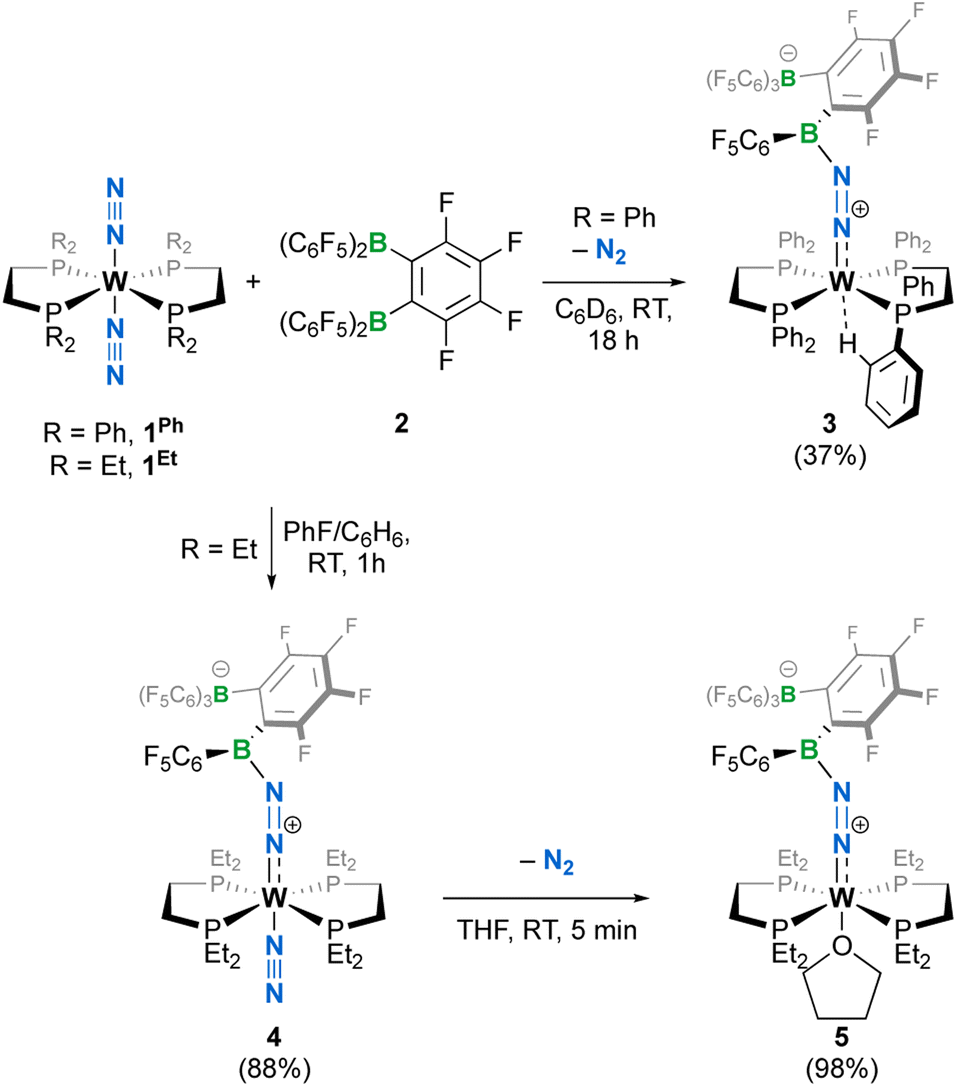
When we reacted equimolar amounts of the dinitrogen complexes [W{R2P(CH2)2PR2}2(N2)2] (1R with R = Ph or Et, Ph2P(CH2)2PPh2 = dppe and Et2P(CH2)2PEt2 = depe) with the Lewis superacidic31 bis(borane) C6F4-1,2-{B(C6F5)2}2 (2) reported by the Piers group,32 the rapid crystallization at room temperature of a new species [W (dppe)2(N2{B(C6F5)2C6F4B(C6F5)3})] 3 in moderate yield was observed (Scheme 1), whereas a depe analogue trans-[W(N2) (depe)2(N2{B(C6F5)2C6F4B(C6F5)3})] 4 was obtained in high yield from a PhF/C6H6 mixture. Complexes 3 and 4 were found to be poorly soluble in toluene-d8 or C6D6 and decomposed readily in CD2Cl2 or PhCl-d5. When dissolved in THF-d8, complex 3 converted immediately to a mixture of compounds whose identification was made difficult due to rapid polymerization of the medium. In the same solvent, 4 cleanly turns to a THF complex trans-[W(THF)(depe)2(N2{B(C6F5)2C6F4B(C6F5)3})] (5) after N2 substitution, as evidenced by visible gas evolution. The latter could be isolated in high yield after removal of solvent under vacuum. Generation of 5 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 reaction of 2 and 1Et in THF was not optimal as THF polymerization occurred upon solvation of the reactants. Complex 5 has been characterized in solution by 1H, 31P, 19F and 11B NMR spectroscopy. The 1H and 31P NMR were diagnostic of two pairs of inequivalent phosphine ligands with two pseudo-singlets in 31P{1H} NMR at 38.6 and 33.8 ppm, with 2JPP inferred by 2D experiments but too weak (<1 Hz) to be resolved (Fig. S5†). In 11B NMR (Fig. S6†), only a well resolved borate resonance was observed as a singlet at −15.05 ppm, indicating boron quaternarization. The other resonance could not be detected even at high concentrations, presumably as a consequence of quadrupolar coupling in an unfavourable electric field gradient situation that leads to fast relaxation of the nuclei.33 In 19F NMR, all fluorine atoms were found to be magnetically inequivalent (Fig. S8†). We surmised that steric crowding of the bis(borane) moiety might result in hindered rotation of C6F5 substituents at the NMR time scale, thus differentiating the fluorine nuclei.
:1 reaction of 2 and 1Et in THF was not optimal as THF polymerization occurred upon solvation of the reactants. Complex 5 has been characterized in solution by 1H, 31P, 19F and 11B NMR spectroscopy. The 1H and 31P NMR were diagnostic of two pairs of inequivalent phosphine ligands with two pseudo-singlets in 31P{1H} NMR at 38.6 and 33.8 ppm, with 2JPP inferred by 2D experiments but too weak (<1 Hz) to be resolved (Fig. S5†). In 11B NMR (Fig. S6†), only a well resolved borate resonance was observed as a singlet at −15.05 ppm, indicating boron quaternarization. The other resonance could not be detected even at high concentrations, presumably as a consequence of quadrupolar coupling in an unfavourable electric field gradient situation that leads to fast relaxation of the nuclei.33 In 19F NMR, all fluorine atoms were found to be magnetically inequivalent (Fig. S8†). We surmised that steric crowding of the bis(borane) moiety might result in hindered rotation of C6F5 substituents at the NMR time scale, thus differentiating the fluorine nuclei.
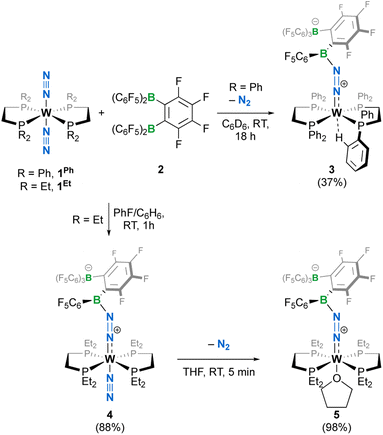 |
| | Scheme 1 Syntheses of complexes 3–5; isolated yields in parentheses. | |
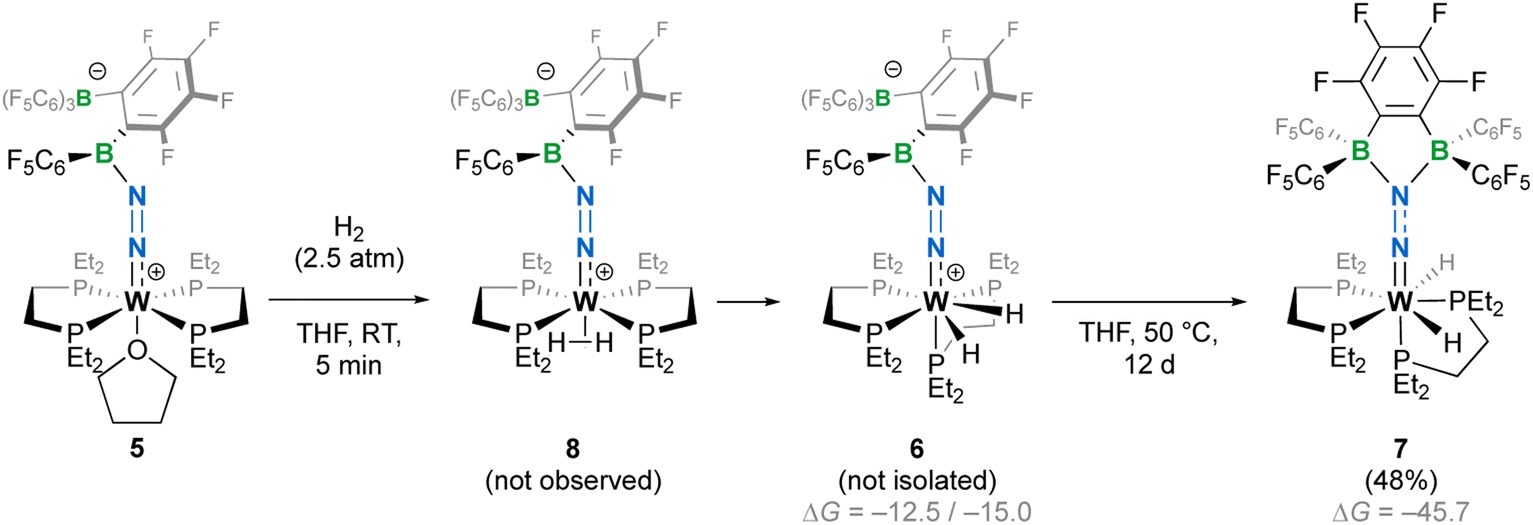
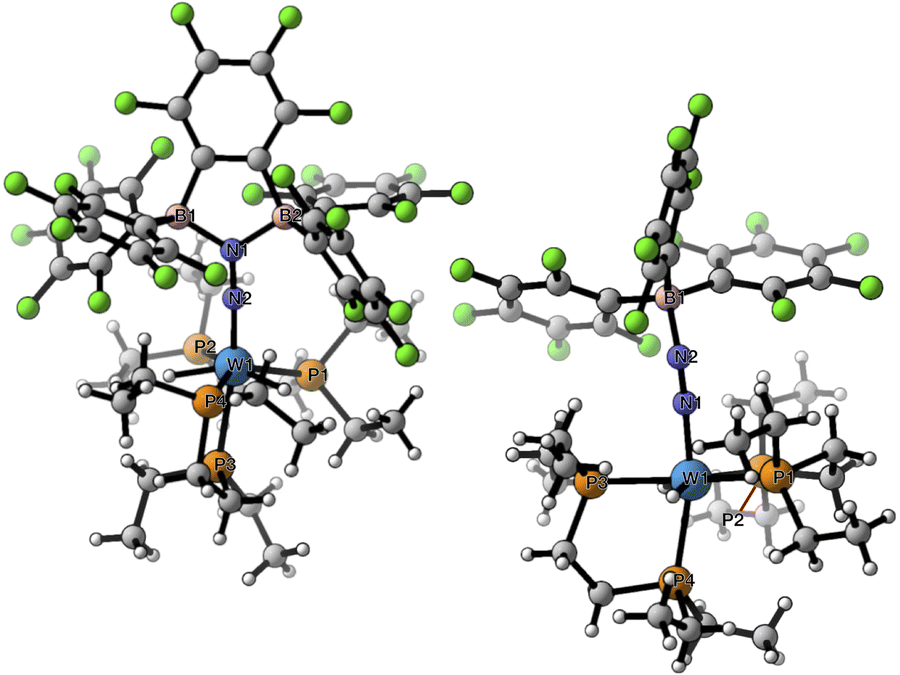
The crystalline materials afforded in the above-mentioned reactions could be analysed by single crystal X-ray diffraction (sc-XRD), but the obtained structures for 5 were of too poor quality to be discussed. The solid-state structures of the spectroscopically similar complexes 3 and 4 reveal that the bis(borane) moiety interacts with the terminal nitrogen via a single boryl centre through covalent, double B–N bonding (Fig. 3). These are the first examples of dinitrogen borylation where the BR2 group is sourced from a triarylborane.34,35 The second boron atom is found as a tetraaryl borate centre, in agreement with boron NMR analysis. This κ1B arrangement of the bis(boron) moiety results from a C6F5 transfer, similar to what was observed upon methyl abstraction on [Cp2Zr(CH3)2] by 2 to form the anion [B(Me)(C6F5)(C6F4{B(C6F5)3}],32c and likely occurs as a means to mitigate steric repulsion between the C5F6 groups and the phosphines' substituents. The boron atoms are essentially coplanar with the bridging C6F4 group, but steric tension in these edifices is revealed by the two boron centres being pushed away from each other, with C–C–B angles in the phenylene junctions approaching 130°. Meanwhile, the borate moiety significantly deviates from the ideal tetrahedral geometry. Short W–N distances (3: 1.788(3); 4: 1.860(6) Å) and long N–N ones (3: 1.248(5); 4: 1.249(8) Å) suggest a formal W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NN motif, and the trans effect from the second N2 ligand in 4 is likely to be responsible for the elongated W–N distance compared to that of 3. Whereas infrared spectroscopy did not allow us to clearly identify the boryldiazenido N–N bond stretches (see Fig. S1–S3 and S9, S10†), which was not surprising given the elongated N2 unit found in 3 and 4, resonance Raman spectroscopy of 5 shows an intense line in the spectrum at 1364 cm−1 (Fig. S11†). 15N isotopic enrichment shifts this line to 1343 cm−1 (Fig. S12†). These frequencies agree with a NN linkage as they are close to those of the νNN of azo-dyes (ca. 1400 cm−1).37 A weak band observed at 2220 cm−1 in the IR spectrum of 4 was attributed to the terminal N2 ligand. The bent N–N–B angles (ca 140°) are typically found in other examples of boryldiazenido–tungsten complexes.12g,20a,b,28,33a,b,38 The B–N bonds are ca. 1.40 Å long, indicative of significant double B–N bonding. Collectively, these parameters point to a formal description of these compounds as W(II)-boryldiazenido complexes. Interestingly, the main compositional difference between 3 and 4 is also found when comparing the adducts of B(C6F5)3 with 1Ph12g or 1Et12j: in complex 3 the trans coordination site features an agostic interaction involving the Cortho–H of a phenyl substituent of the dppe ligand, whereas the depe analogue 4 retains its second N2 ligand. We believe that the relative instability of 3 originates from the labile agostic bond that may expose the Lewis acidic, 16-electron tungsten centre to further reactivity. Because we failed to observe double coordination of the bis(borane) to the terminal N2 ligand of complexes 1R, we briefly explored the reactivity of 5 (Scheme 2). The instability of 3 in common solvents precluded such study. Among the various tests we have carried out, the reaction with dihydrogen was particularly rewarding in view of our initial goal. Treating 5 with H2 produced almost immediately a new complex [W(H)2(depe)2(N2{B(C6F5)2C6F4B(C6F5)3})] (6) that readily decomposed upon attempting its isolation by concentration under vacuum. The 31P NMR points to the occurrence of two isomers for 6, possibly due to a hindered free rotation induced by the bulkiness of the bis(boron) moiety (Fig. S14†). Each shows four similar signals, indicative of a dissymmetry of the first coordination sphere. The 11B NMR spectrum is basically unchanged. Over the course of several days, 6 evolves upon heating into a new species [W(H)2(depe)2(N2{B(C6F5)2}2C6F4)] (7) for which the 11B NMR spectrum shows significant change in the bis(boron) moiety (disappearance of the borate signal at δ = −15.1 ppm and a new, deshielded resonance at −3.4 ppm). Yellow crystals of 7 could be recovered and analysed by sc-XRD (Fig. 4), revealing oxidative addition of H2 and the seven-coordinate nature of 7 with one isomerized depe ligand.39 Remarkably, the bis(borane) pairs with the basic terminal N through two-fold single, dative-type bonding (B–N distances are ca. 1.61 vs. ca. 1.40 Å for 4) of its two boron atoms after a backward C6F5 transfer. This results in augmented, push–pull type charge transfer to the N2 ligand, as shown by the remarkably long N–N bond of 1.291(3) Å, going beyond that of a N–N double bond.40 As a specific comparison, N–N bond lengths of two-fold 1,2-B(C6F5)3 adducts of diazene (N2H2, 1.230(3) Å) and diazenide (N2H−, 1.2227(18) Å))41 or LA adducts of diazenes (1.227(2)–1.263(4) Å) are much shorter.42 To the best of our knowledge, this is the first example of a neutral adduct of an end-on N2 ligand with a ditopic Lewis acid.
NN motif, and the trans effect from the second N2 ligand in 4 is likely to be responsible for the elongated W–N distance compared to that of 3. Whereas infrared spectroscopy did not allow us to clearly identify the boryldiazenido N–N bond stretches (see Fig. S1–S3 and S9, S10†), which was not surprising given the elongated N2 unit found in 3 and 4, resonance Raman spectroscopy of 5 shows an intense line in the spectrum at 1364 cm−1 (Fig. S11†). 15N isotopic enrichment shifts this line to 1343 cm−1 (Fig. S12†). These frequencies agree with a NN linkage as they are close to those of the νNN of azo-dyes (ca. 1400 cm−1).37 A weak band observed at 2220 cm−1 in the IR spectrum of 4 was attributed to the terminal N2 ligand. The bent N–N–B angles (ca 140°) are typically found in other examples of boryldiazenido–tungsten complexes.12g,20a,b,28,33a,b,38 The B–N bonds are ca. 1.40 Å long, indicative of significant double B–N bonding. Collectively, these parameters point to a formal description of these compounds as W(II)-boryldiazenido complexes. Interestingly, the main compositional difference between 3 and 4 is also found when comparing the adducts of B(C6F5)3 with 1Ph12g or 1Et12j: in complex 3 the trans coordination site features an agostic interaction involving the Cortho–H of a phenyl substituent of the dppe ligand, whereas the depe analogue 4 retains its second N2 ligand. We believe that the relative instability of 3 originates from the labile agostic bond that may expose the Lewis acidic, 16-electron tungsten centre to further reactivity. Because we failed to observe double coordination of the bis(borane) to the terminal N2 ligand of complexes 1R, we briefly explored the reactivity of 5 (Scheme 2). The instability of 3 in common solvents precluded such study. Among the various tests we have carried out, the reaction with dihydrogen was particularly rewarding in view of our initial goal. Treating 5 with H2 produced almost immediately a new complex [W(H)2(depe)2(N2{B(C6F5)2C6F4B(C6F5)3})] (6) that readily decomposed upon attempting its isolation by concentration under vacuum. The 31P NMR points to the occurrence of two isomers for 6, possibly due to a hindered free rotation induced by the bulkiness of the bis(boron) moiety (Fig. S14†). Each shows four similar signals, indicative of a dissymmetry of the first coordination sphere. The 11B NMR spectrum is basically unchanged. Over the course of several days, 6 evolves upon heating into a new species [W(H)2(depe)2(N2{B(C6F5)2}2C6F4)] (7) for which the 11B NMR spectrum shows significant change in the bis(boron) moiety (disappearance of the borate signal at δ = −15.1 ppm and a new, deshielded resonance at −3.4 ppm). Yellow crystals of 7 could be recovered and analysed by sc-XRD (Fig. 4), revealing oxidative addition of H2 and the seven-coordinate nature of 7 with one isomerized depe ligand.39 Remarkably, the bis(borane) pairs with the basic terminal N through two-fold single, dative-type bonding (B–N distances are ca. 1.61 vs. ca. 1.40 Å for 4) of its two boron atoms after a backward C6F5 transfer. This results in augmented, push–pull type charge transfer to the N2 ligand, as shown by the remarkably long N–N bond of 1.291(3) Å, going beyond that of a N–N double bond.40 As a specific comparison, N–N bond lengths of two-fold 1,2-B(C6F5)3 adducts of diazene (N2H2, 1.230(3) Å) and diazenide (N2H−, 1.2227(18) Å))41 or LA adducts of diazenes (1.227(2)–1.263(4) Å) are much shorter.42 To the best of our knowledge, this is the first example of a neutral adduct of an end-on N2 ligand with a ditopic Lewis acid.
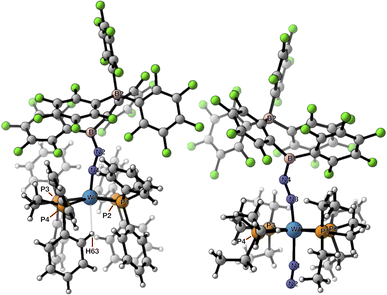 |
| | Fig. 3 Molecular structures of complexes 3 (left) and 4 (right) in the solid state rendered with CYLview20.36 Selected distances (Å) and angles (°): 3: W1–H63 2.437, W1–N1 1.788(3), N1–N2 1.248(5), N2–B1 1.400(5), W1–N1–N2 168.9(3), N1–N2–B1 141.7(4); 4: W1a–N1 2.155(7), N1–N2 1.117(11), W1a–N3 1.860(6), N3–N4 1.249(8), N4–B1 1.368(10), W1a–N1–N2 171.5(8), W1a–N3–N4 165.3(5), N3–N4–B1 143.9(6). | |
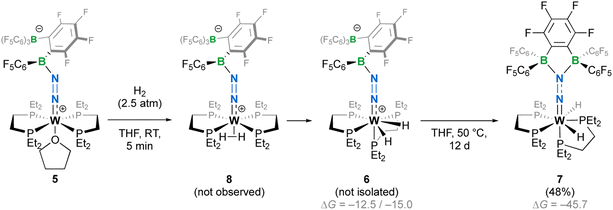 |
| | Scheme 2 Reaction of 5 with dihydrogen leading to seven-coordinate complexes 7. Isolated yields in parentheses. ΔG (DFT, B3PW91-6-31G(d,p)) are given in kJ mol−1. | |
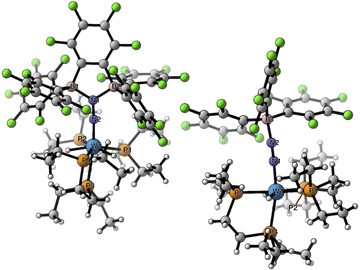 |
| | Fig. 4 Molecular structures of complexes 7 (left) and 10 (right) in the solid state. Selected distances (Å) and angles (°): 7: W1–P1 2.559(1), W1–P2 2.516(1), W1–P3 2.651(1), W1–P4 2.475(1), W1–N2 1.825(2), N1–N2 1.291(3), N1–B1 1.615(3), N1–B2 1.614(3), W1–H100 1.66(3), W1–H200 1.62(2), W1–N2–N1 177.0(2), N2–N1–B1 125.0(2), N2–N1–B2 121.8(2), B1–N1–B2 113.3(2). 10: W1–P1 2.4813(5), W1–P2 2.4792(5), W1–P3 2.4300(5), W1–P4 2.5214(5), W1–N1 1.8826(17), N1–N2 1.193(2), N1–B1 1.562(3), W1–H1 1.71(3), W1–H2 1.70(3), W1–N1–N2 167.21(15), N1–N2–B1 139.13(18). | |
DFT calculations were conducted to shed light on this transformation (see Fig. S40†). Structure optimization allowed us to locate a trans-σ-H2 complex 8 and a more stable cis-dihydride that could well correspond to 6. Two rotamers (6 and 6′) were found for the latter species, whose energy difference matches well with the experimentally observed ratio. Isomer 7 was found to be more stable than 6 by 31 kJ mol−1. Estimates of 1H chemical shifts for the W-bound protons that we were not able to locate in the 1H NMR spectra (see Fig. S13 and S18†) were also computed. They fall within the alkyl protons' region and probably overlap with the resonances linked to the phosphine's substituents (see Plot S1†), supporting the absence of resonances at higher fields.
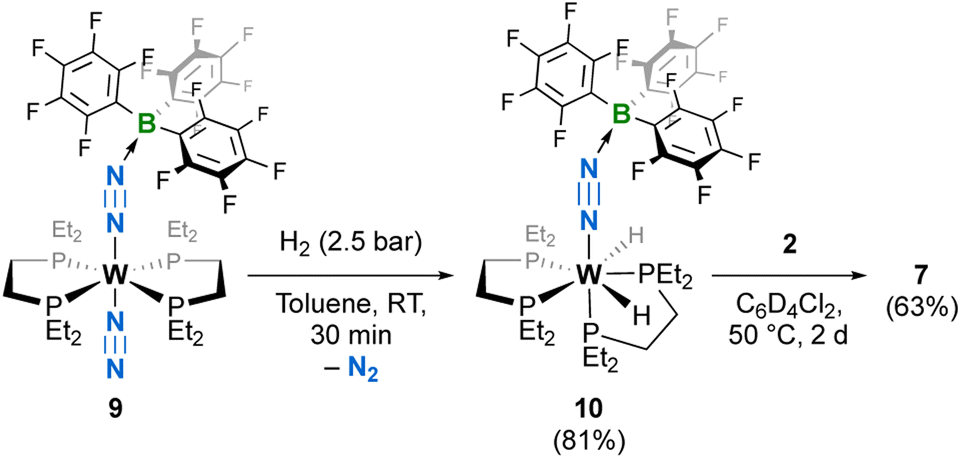
The activation of dihydrogen by 5 pushed us to verify whether a simpler system, that is the adduct of B(C6F5)3 with 1Et, trans-[W(N2)(depe)2(N2{B(C6F5)3}] (9), would react similarly—a positive result would also provide a comparison of the influence of a mono- vs. di-topic LA to N2 activation. Gratifyingly, under an H2 atmosphere, 9 evolved into the bis-hydride complex [W(H)2(depe)2(N2{B(C6F5)3}] (10) without N2 loss. We were able to isolate it in high yields and carried out its full characterization. We could not evidence FLP-type heterolytic splitting of H2 at the B–N linkage, similar to what we observed when we reacted an adduct of 1Ph and B(C6F5)3 with hydro-boranes and -silanes.12g Here, dihydrogen favoured reactivity at the reduced metal centre. sc-XRD analysis of 10 revealed a similar first coordination sphere to that of 7 (Fig. 4). The influence of dihydrogen oxidative addition on N2 activation may be measured by comparing 10 with the adduct of B(C6F5)3 with 1Et (9).12j The two structures are rather similar (Table 1), with a slightly higher activation level for N2 in 10 (1.196(2) vs. 1.179(9) Å for 9). This is also reflected by the IR stretching frequencies (1767 cm−1 for 9vs. 1712 cm−1 for 10). The N–N–B angle is also more acute in 10, which may be due to reduced steric hindrance of the isomerized first coordination sphere (Table 1). Interestingly, 7 could also be obtained from 10 in high yields by simply reacting it with one equivalent of bis(borane) 2 (Scheme 3), with B(C6F5)3 being displaced upon slight thermal activation.
Table 1 Comparison of pertinent structural parameters, NN stretching frequencies. Values in parentheses were calculated by DFT (B3PW91, 6-31G(d,p), and D3BJ). Computed infrared frequencies were corrected with a scaling factor = 0.958. n.a. = not applicable. n.d. = not determined. as = asymmetric. s = symmetric
| Bonds (Å)/angles (°)/ν (cm−1) |
1Et
|
9
|
10
|
7
|
|
Taken from ref. 12j.
Averaged data; standard deviation calculated from the measurement set are not the average of the standard deviation of individual measurements.
FT-ATR infrared spectroscopy.
Resonance Raman spectroscopy.
Taken from ref. 43.
|
| W–N (theor.) |
2.016 |
1.908(6) |
1.883(2) (1.873) |
1.825(2) (1.831) |
| N–N (theor.) |
1.123(2) |
1.179(9); 1.08(1) |
1.193(2) (1.184) |
1.291(3) (1.243) |
| N–B (theor.) |
n.a. |
1.55(1) |
1.562(3) (1.556) |
1.615(1) (1.626[0])b |
| W–Ptrans (theor.) |
n.a. |
n.a. |
2.5214(5) (2.512) |
2.651(1) (2.609) |
| W–N–N (theor.) |
178.6(1) |
169.0(6) |
167.2(2) (166.8) |
177.0(2) (175.9) |
| N–N–B (theor.) |
n.a. |
148.4(7) |
139.1(2) (135.3) |
123.4(23) (123.6[18])b |
| av. N–W–Peqb |
90.0(9) |
91(4) |
89(4) |
99(6) |
|
ν
NN
14N (theor.)/15Nc |
1890/1830 (as) |
1767 |
1712 (1827)/1659 |
1399 (1548)/1338 |
|
ν
NN
14N/15Nd |
1968/1903 (s) |
n.d. |
1714/1664 |
Not observed |
 |
| | Scheme 3 Synthesis of bis-hydride 10 by the reaction of H2 with the B(C6F5)3 adduct of 1Et, 9, and displacement of the borane by bis(borane) 2 to give 7. Isolated yields in parentheses. | |
Comparison of the structures of the N2 complexes 7 and 10 supports the depiction of the N2 unit in 7 as a diazene-diide (N22−) ligand (Table 1): as mentioned above, the N–N bond length (1.291(3) Å) goes beyond that of standard N–N double bonds. The linearisation of the W–N–N array in 7 (177 vs. 167° for 10) and the significant shortening of the W–N bond (1.82 vs. 1.88 Å for 10) are also in line with the diatomic molecule being an LX-type ligand. In addition, the smaller N–N–B angles point to a fully sp2-hybridized distal nitrogen, as expected for the N22− dianion. 15N labelling of 7 and 10 allowed us to unambiguously locate N–N stretches in their IR spectra (Table 1, Fig. S22, S23 and S31, S32†). Surprisingly, no shifting lines could be observed when comparing the Raman spectra of 14N- and 15N-7 (Fig. S24 and S25†), while in the case of 10, a low-intensity line at 1714 cm−1 shifted to 1664 cm−1 for the 15N isotopologue (Fig. S33 and S34†). The strong bathochromic shift recorded for the stretching frequency of 7 (1399 vs. 1712 cm−1 for 10) and the structural data indeed support the NN2− depiction.37,40–42 The reaction of 10 with 2 points to diborane coordination-triggered 2-e– reduction of N2, and somewhat recalls the evolution of the N–N bond upon the two-fold protonation of related group 6 N2 complexes affording hydrazido complexes.29 The resulting positively charged compounds show N–N bond lengths ranging from 1.30–1.37 Å depending on the trans ligand and H-bonding in the lattice. Interestingly, the Schrock group was, to the best of our knowledge, the only group to report structural data for both Mo-diazenido and Mo-hydrazido complexes, the latter being obtained by protonation of the former. Surprisingly, the N–N bond lengths are equal for these two compounds (1.302(13) vs. 1.304(6), respectively). Although this single point of comparison prevents a general conclusion to be drawn, it is remarkable that in our case, the effect of incremental LA coordination is much more pronounced. Unfortunately, comparison of 15N NMR chemical shifts could not be performed since despite several hours of acquisition, we were not able to detect resonances for 7 (see Fig. S7, S17 and S29† for the 15N NMR spectra of 5, 6 and 10, respectively).
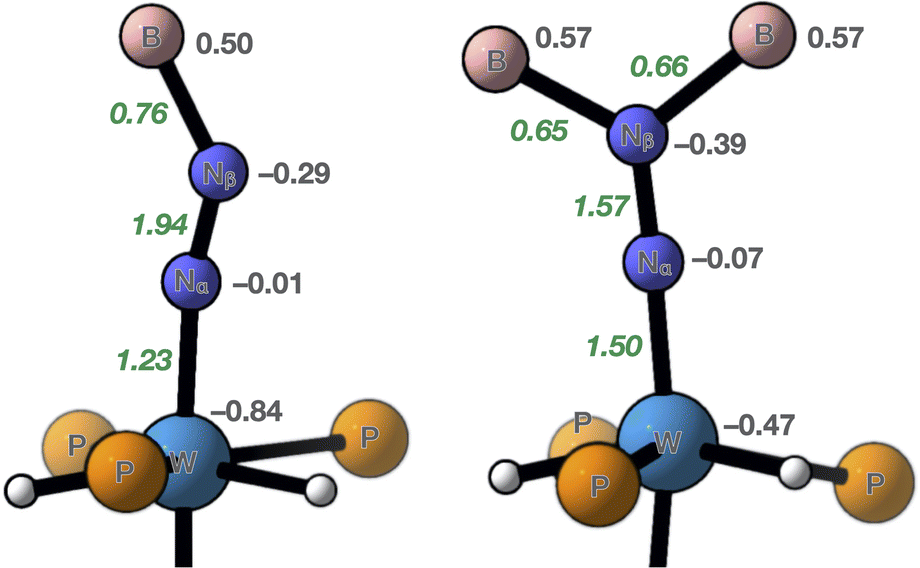
DFT calculations including dispersion (D3BJ) were run on 7 and 10 (see the ESI for details and atomic coordinates of the computed structures). Key metrical parameters were found to be close to those experimentally determined or within the margin of error of DFT (Table 1). Comparison of the atomic charges obtained by a Natural Population Analysis (NPA) evidences that the negative charge carried by the N2 unit in 7 (−0.46) is 1.5 times higher than that found in 10 (−0.30). In both cases, it is almost totally localized on the distal nitrogen, in agreement with previous observations,12f and the N–N bond in 7 is significantly more polarised. A much higher negative charge is carried by the metal atom in 10, in line with a diminished metal-to-N2 charge transfer and a lower formal oxidation state attributed to its W centre. Wiberg bond indices (WBI) also reflect the trends revealed by the structures of 7 and 10: a higher N–N WBI is found in 10 (1.94 vs. 1.57 in 7, Fig. 5), but a lower W–N one (1.23 vs. 1.50 in 7). As far as the B–N bonds are concerned, a smaller N–B WBI in 7 parallels the longer N–B distances found in its crystal structure (Table 1). Analysis of the Natural Bonding Orbitals (NBOs) further differentiates 7 and 10. In 10, the W–N bonding involves mainly atomic orbitals of σ symmetry, while for 7 it is computed to rest on π-symmetric orbitals exclusively (see Fig. S51–S53† for visualization of key pNBOs and Natural Localized Molecular Orbitals [NLMOs] of 7 and 10). In both cases, the W–N NBO is polarized towards the nitrogen atom. Interestingly, a lone pair is found on the proximal nitrogen of 7, which undergoes significant stabilizing donor–acceptor interactions with the W–Ptrans antibond. This is where the W–N σ-symmetric bonding lies, and this is what explains why in 7 the W–Ptrans bond is significantly elongated compared to that of 10 (Table 1). In other terms, it translates the stronger trans influence of the diazene-diide ligand. Conversely, 10 carries a lone pair of pure d character on W for which second-order perturbation theory estimates an important stabilization by a donor–acceptor interaction with a π* of N2, thus embodying π back-donation. Moving to the N–N linkage, a single π bond is found for 7 along the σ component, whereas 10 has two, indicating an intact triple N–N linkage, unlike 7. N–B NBOs are polarised towards nitrogen to the same extent in both compounds (ca. 75%), yet the natural atomic hybrids on nitrogen differ significantly from 7 to 10, reflecting sp2 and sp hybridization, respectively (see Tables S7 and S8† for a summary of pertinent values issued by NBO analysis). Collectively, these computational observations also support a 2-e– reduced N2 unit, stabilised by a W(IV) centre through σ and π donation from the nitrogen ligand, as well as by two-fold dative N→B bonding in 7.
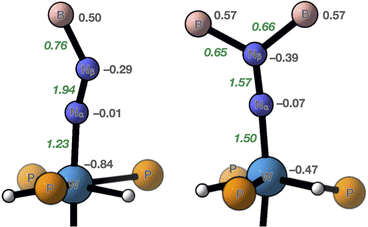 |
| | Fig. 5 Summary of key parameters from theoretical calculations run on 7 (right) and 10 (left). Grey figures are NPA charges and green italic ones, Wiberg bond indices. | |
Conclusions
We have explored the coordination of the strongly electrophilic bis(borane) 2 to terminal dinitrogen complexes [W{R2P(CH2)2PR2}2(N2)2] (1R) with the hope to observe κ2B coordination to the distal nitrogen, thus building an orbitally adapted push–pull template for N2 activation. The reaction with either 1Ph or 1Et did not furnish the expected outcome with formation of covalent N–B bonds in complexes trans-[W(L)(R2PCH2CH2PR2)2(N2{B(C6F5)2(C6F4B(C6F5)3})] (L = ø, N2 or THF) (3–5) after boron-to-boron C6F5 transfer. The reactivity of trans-[W(THF)(depe)2(N2{B(C6F5)2C6F4B(C6F5)3})] (5) with dihydrogen was then explored. It triggered backward C6F5 transfer upon oxidative addition of H2, without N2 loss, and afforded complex [W(H)2(depe)2(N2{B(C6F5)2}2C6F4)] (7) having the bis(borane) coordinating the distal nitrogen via two-fold dative bonding. Its especially long N–N bond (1.29 Å) goes beyond reference NN distances (1.21–1.25 Å). The B(C6F5)3 analogue of 7, complex [W(H)2(depe)2(N2{B(C6F5)3}] (10), could be prepared by oxidative addition of H2 on adduct [W(N2)(depe)2(N2{B(C6F5)3}] 9. In 10, B(C6F5)3 can be displaced by bis(borane) 2 to furnish 7. Comparison of 7 with 10 led us to propose that the N2 ligand underwent 2e– reduction upon coordination of the bis(borane), due to an extreme push–pull effect. To the best of our knowledge, this is the first example of LA-coordination-induced reduction of an N2 ligand, a transformation that is commonly observed upon protonation or functionalisation.29,30 This points to a possible strong influence of the multiple H-bonding channel when N2 is coordinated to the FeMo cofactor of the nitrogenases. Guidelines for future N2 fixation FLP systems may also be drawn from our study as we demonstrate that bis(borane) can trigger reduction of the diatomic molecule when combined to an electron-rich species, a reactivity commonly observed in small molecule activation by FLPs. Future work will focus on the reactivity of 7 and 10, new complexes that have the rare but remarkable ability to activate N2 and H2 at a single metal centre;22d,44 promoting N–H bond formation in such species should be facilitated by the proximity of the two hydrides with the N2 ligand.45
Data availability
All the relevant curated data can be found in the ESI† as stated in the footnote. Raw data were not uploaded into repositories.
Author contributions
D. S., M.-C. B., A. C. and L. E. run the experiments and analyzed the data. L. V. recorded single-crystal X-ray diffraction data and solved the structures. M. G. performed DFT calculations and assisted in the writing of the manuscript. A. S. obtained funding for the project and managed it, conceived the original idea, designed the experiments and took part in the analysis of the data. D. S. wrote the preliminary manuscript draft and A. S. took charge of writing the submitted and revised versions. D. S., M.-C. B., M. G. and A. S. were involved in the writing of the ESI.† All authors were involved in the proof-reading process.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors would like to thank Pr. François P. Gabbaï, Dr Mitsukimi Tsunoda and Dr Kirill I. Tugashov for providing expert advice on the synthesis of [HgC6F4]3 necessary to prepare bis(borane) 2. D. S., M.-C. B. and A. S. are indebted to the European Research Council for funding (ERC Starting Grant no 757501). A. C. is grateful to the French Ministry of National and Superior Education and Research (MENESR) for a PhD fellowship. This work was partly supported by the ANR PUNCh project (post-doc fellowship for L. E.), grant ANR-21-CE07-0003 of the French Agence Nationale de la Recherche.
References
-
(a) F. Masero, M. A. Perrin, S. Dey and V. Mougel, Chem.–Eur. J., 2021, 27, 3892–3928 CrossRef PubMed;
(b) D. Singh, W. R. Buratto, J. F. Torres and L. J. Murray, Chem. Rev., 2020, 120, 5517–5581 CrossRef CAS PubMed;
(c)
Transition Metal-Dinitrogen Complexes: Preparation and Reactivity, ed. Y. Nishibayashi, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2019 Search PubMed;
(d) R. J. Burford and M. D. Fryzuk, Nat. Rev. Chem, 2017, 1, 1–13 CrossRef;
(e)
M. D. Walter, in Advances in Organometallic Chemistry, ed. P. J. Pérez, Elsevier, 2016, vol. 65, pp. 261–377 Search PubMed.
-
(a)
Y. Tanabe, in Transition Metal-Dinitrogen Complexes, 2019 Search PubMed;
(b) Z. R. Turner, Inorganics, 2015, 3, 597–635 CrossRef CAS;
(c) M. G. Gardiner and D. N. Stringer, Materials, 2010, 3, 841–862 CrossRef CAS;
(d) W. J. Evans and D. S. Lee, Can. J. Chem., 2005, 83, 375–384 CrossRef CAS.
-
(a) B. Rösch, T. X. Gentner, J. Langer, C. Färber, J. Eyselein, L. Zhao, C. Ding, G. Frenking and S. Harder, Science, 2021, 371, 1125–1128 CrossRef PubMed;
(b) R. Mondal, K. Yuvaraj, T. Rajeshkumar, L. Maron and C. Jones, Chem. Commun., 2022, 58, 12665–12668 RSC.
-
(a) M.-A. Légaré, G. Bélanger-Chabot, R. D. Dewhurst, E. Welz, I. Krummenacher, B. Engels and H. Braunschweig, Science, 2018, 359, 896–900 CrossRef PubMed;
(b) M.-A. Légaré, M. Rang, G. Bélanger-Chabot, J. I. Schweizer, I. Krummenacher, R. Bertermann, M. Arrowsmith, M. C. Holthausen and H. Braunschweig, Science, 2019, 363, 1329–1332 CrossRef PubMed;
(c) M.-A. Légaré, G. Bélanger-Chabot, M. Rang, R. D. Dewhurst, I. Krummenacher, R. Bertermann and H. Braunschweig, Nat. Chem., 2020, 12, 1076–1080 CrossRef PubMed;
(d) S. Bennaamane, B. Rialland, L. Khrouz, M. Fustier-Boutignon, C. Bucher, E. Clot and N. Mézailles, Angew. Chem., Int. Ed., 2023, 62, e202209102 CrossRef CAS PubMed.
-
(a) Y. Tanabe and Y. Nishibayashi, Chem. Soc. Rev., 2021, 50, 5201–5242 RSC;
(b) Q. J. Bruch, G. P. Connor, N. D. McMillion, A. S. Goldman, F. Hasanayn, P. L. Holland and A. J. M. Miller, ACS Catal., 2020, 10, 10826–10846 CrossRef CAS;
(c) M. J. Chalkley, M. W. Drover and J. C. Peters, Chem. Rev., 2020, 120, 5582–5636 CrossRef CAS PubMed;
(d) S. L. Foster, S. I. P. Bakovic, R. D. Duda, S. Maheshwari, R. D. Milton, S. D. Minteer, M. J. Janik, J. N. Renner and L. F. Greenlee, Nat. Catal., 2018, 1, 490–500 CrossRef;
(e) N. Stucke, B. M. Flöser, T. Weyrich and F. Tuczek, Eur. J. Inorg. Chem., 2018, 2018, 1337–1355 CrossRef CAS;
(f)
Nitrogen Fixation, ed. Y. Nishibayashi, Springer International Publishing AG, Cham, Switzerland, 2017 Search PubMed;
(g) Y. Roux, C. Duboc and M. Gennari, ChemPhysChem, 2017, 18, 2606–2617 CrossRef CAS PubMed.
-
(a)
M. Appl, Ammonia, 2. Production Processes, 2011 Search PubMed;
(b) G. Ertl, Angew. Chem., Int. Ed., 2008, 47, 3524–3535 CrossRef CAS PubMed;
(c) J. W. Erisman, M. A. Sutton, J. Galloway, Z. Klimont and W. Winiwarter, Nat. Geosci., 2008, 1, 636 CrossRef CAS;
(d) R. Schlögl, Angew. Chem., Int. Ed., 2003, 42, 2004–2008 CrossRef PubMed;
(e)
V. Smil, Enriching the Earth: Fritz Haber, Carl Bosch, and the Transformation of World Food Production, Mit Press, 2001 Search PubMed;
(f) V. Smil, Nature, 1999, 400, 415 CrossRef CAS;
(g)
J. R. Jennings, Catalytic Ammonia Synthesis: Fundamentals and Practice, Springer, New York, 1991 CrossRef.
-
(a) S. Kim, F. Loose and P. J. Chirik, Chem. Rev., 2020, 120, 5637–5681 CrossRef CAS PubMed;
(b) Z.-J. Lv, J. Wei, W.-X. Zhang, P. Chen, D. Deng, Z.-J. Shi and Z. Xi, Natl. Sci. Rev., 2020, 7, 1564–1583 CrossRef CAS PubMed.
-
(a) O. Einsle and D. C. Rees, Chem. Rev., 2020, 120, 4969–5004 CrossRef CAS PubMed;
(b) L. C. Seefeldt, Z.-Y. Yang, D. A. Lukoyanov, D. F. Harris, D. R. Dean, S. Raugei and B. M. Hoffman, Chem. Rev., 2020, 120, 5082–5106 CrossRef CAS PubMed;
(c) B. M. Hoffman, D. Lukoyanov, Z.-Y. Yang, D. R. Dean and L. C. Seefeldt, Chem. Rev., 2014, 114, 4041–4062 CrossRef CAS PubMed;
(d) K. Tanifuji and Y. Ohki, Chem. Rev., 2020, 120, 5194–5251 CrossRef CAS PubMed.
- D. Dubois and R. Hoffmann, Nouv. J. Chim., 1977, 1, 479–492 CAS.
- S. Gambarotta and J. Scott, Angew. Chem., Int. Ed., 2004, 43, 5298–5308 CrossRef CAS PubMed.
-
(a)
A. Coffinet, A. Simonneau and D. Specklin, in Encyclopedia of Inorganic and Bioinorganic Chemistry, ed. R. A. Scott, Wiley-VCH Verlag GmbH & Co. KGaA, 2020, pp. 1–25 Search PubMed;
(b) A. J. Ruddy, D. M. C. Ould, P. D. Newman and R. L. Melen, Dalton Trans., 2018, 47, 10377–10381 RSC;
(c) A. Simonneau and M. Etienne, Chem.–Eur. J., 2018, 24, 12458–12463 CrossRef CAS PubMed.
-
(a) J. Chatt, J. R. Dilworth, R. L. Richards and J. R. Sanders, Nature, 1969, 224, 1201–1202 CrossRef CAS PubMed;
(b) J. Chatt, R. H. Crabtree and R. L. Richards, J. Chem. Soc. Chem. Commun., 1972, 534 RSC;
(c) J. Chatt, R. H. Crabtree, E. A. Jeffery and R. L. Richards, J. Chem. Soc. Dalton Trans., 1973, 1167–1172 RSC;
(d) F. Studt, B. A. MacKay, S. A. Johnson, B. O. Patrick, M. D. Fryzuk and F. Tuczek, Chem.–Eur. J., 2005, 11, 604–618 CrossRef CAS PubMed;
(e) H. Broda, S. Hinrichsen, J. Krahmer, C. Nather and F. Tuczek, Dalton Trans., 2014, 43, 2007–2012 RSC;
(f) J. B. Geri, J. P. Shanahan and N. K. Szymczak, J. Am. Chem. Soc., 2017, 139, 5952–5956 CrossRef CAS PubMed;
(g) A. Simonneau, R. Turrel, L. Vendier and M. Etienne, Angew. Chem., Int. Ed., 2017, 56, 12268–12272 CrossRef CAS PubMed;
(h) L. G. Pap, A. Couldridge, N. Arulsamy and E. Hulley, Dalton Trans., 2019, 48, 11004–11017 RSC;
(i) J. P. Shanahan and N. K. Szymczak, J. Am. Chem. Soc., 2019, 141, 8550–8556 CrossRef CAS PubMed;
(j) D. Specklin, A. Coffinet, L. Vendier, I. del Rosal, C. Dinoi and A. Simonneau, Inorg. Chem., 2021, 60, 5545–5562 CrossRef CAS PubMed.
-
(a) P. C. Dos Santos, R. Y. Igarashi, H.-I. Lee, B. M. Hoffman, L. C. Seefeldt and D. R. Dean, Acc. Chem. Res., 2005, 38, 208–214 CrossRef CAS PubMed;
(b) T. Spatzal, K. A. Perez, O. Einsle, J. B. Howard and D. C. Rees, Science, 2014, 345, 1620–1623 CrossRef CAS PubMed;
(c) S. M. Keable, J. Vertemara, O. A. Zadvornyy, B. J. Eilers, K. Danyal, A. J. Rasmussen, L. De Gioia, G. Zampella, L. C. Seefeldt and J. W. Peters, J. Inorg. Biochem., 2018, 180, 129–134 CrossRef CAS PubMed;
(d) D. Sippel, M. Rohde, J. Netzer, C. Trncik, J. Gies, K. Grunau, I. Djurdjevic, L. Decamps, S. L. A. Andrade and O. Einsle, Science, 2018, 359, 1484–1489 CrossRef CAS PubMed;
(e) W. Kang, C. C. Lee, A. J. Jasniewski, M. W. Ribbe and Y. Hu, Science, 2020, 368, 1381–1385 CrossRef CAS PubMed. Although the structural data reported in this latter reference have been the matter of a discussion, the role of acidic residues in the nitrogenases' active site for N2 activation remains a solid hypothesis, see ref. 8a–c and 13a.
(f) J. W. Peters, O. Einsle, D. R. Dean, S. DeBeer, B. M. Hoffman, P. L. Holland and L. C. Seefeldt, Science, 2021, 371, eabe5481 CrossRef CAS PubMed;
(g) K. Wonchull, C. C. Lee, A. J. Jasniewski, M. W. Ribbe and Y. Hu, Science, 2021, 371, eabe5856 CrossRef PubMed.
- This has pushed several groups to explore the influence of inter- or intramolecular acidic or basic functions to mimic secondary-sphere interactions:
(a) P. Bhattacharya, D. E. Prokopchuk and M. T. Mock, Coord. Chem. Rev., 2017, 334, 67–83 CrossRef CAS;
(b) S. E. Creutz and J. C. Peters, Chem. Sci., 2017, 8, 2321–2328 RSC.
-
(a)
J. C. Slootweg and A. R. Jupp, Frustrated Lewis Pairs, Springer Nature, 2020 Search PubMed;
(b) D. W. Stephan, Science, 2016, 354, aaf7229 CrossRef PubMed;
(c) D. W. Stephan, Acc. Chem. Res., 2015, 48, 306–316 CrossRef CAS PubMed;
(d) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS PubMed;
(e)
Frustrated Lewis Pairs I: Uncovering and Understanding, ed. G. Erker and D. W. Stephan, Springer Berlin Heidelberg, 2013 Search PubMed;
(f)
Frustrated Lewis Pairs II: Expanding the Scope, ed. G. Erker and D. W. Stephan, Springer Berlin Heidelberg, 2013 Search PubMed;
(g) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46–76 CrossRef CAS PubMed.
- Zhu and colleagues proposed a series of in silico models, see:
(a) J. Zhu, Chem.–Asian J., 2019, 14, 1413–1417 CrossRef CAS PubMed;
(b) A. M. Rouf, Y. Huang, S. Dong and J. Zhu, Inorg. Chem., 2021, 60, 5598–5606 CrossRef CAS PubMed;
(c) C. Dai, Y. Huang and J. Zhu, Organometallics, 2022, 41, 1480–1487 CrossRef CAS;
(d) J. Zeng, R. Qiu and J. Zhu, Chem.–Asian J., 2023, 18, e202201236 CrossRef CAS PubMed.
- R. L. Melen, Angew. Chem., Int. Ed., 2018, 57, 880–882 CrossRef CAS PubMed.
- C. Tang, Q. Liang, A. R. Jupp, T. C. Johnstone, R. C. Neu, D. Song, S. Grimme and D. W. Stephan, Angew. Chem., Int. Ed., 2017, 56, 16588–16592 CrossRef CAS PubMed.
- L. L. Cao, J. Zhou, Z.-W. Qu and D. W. Stephan, Angew. Chem., Int. Ed., 2019, 58, 18487–18491 CrossRef CAS PubMed.
-
(a) A. Coffinet, D. Specklin, L. Vendier, M. Etienne and A. Simonneau, Chem.–Eur. J., 2019, 25, 14300–14303 CrossRef CAS PubMed;
(b) A. Coffinet, D. Zhang, L. Vendier, S. Bontemps and A. Simonneau, Dalton Trans., 2021, 50, 5582–5589 RSC;
(c) A. Coffinet, D. Specklin, Q. Le Dé, S. Bennaamane, L. Muñoz, L. Vendier, E. Clot, N. Mézailles and A. Simonneau, Chem.–Eur. J., 2023, 29, e202203774 CrossRef CAS PubMed.
-
(a) C. Sivasankar, P. K. Madarasi and M. Tamizmani, Eur. J. Inorg. Chem., 2020, 1383–1395 CrossRef CAS;
(b) M. Hölscher and W. Leitner, Chem.–Eur. J., 2017, 23, 11992–12003 CrossRef PubMed.
- Selected examples:
(a) A. Sacco and M. Rossi, Inorg. Chim. Acta, 1968, 2, 127–132 CrossRef CAS;
(b) L. J. Archer and T. A. George, Inorg. Chem., 1979, 18, 2079–2082 CrossRef CAS;
(c) D. G. H. Hetterscheid, B. S. Hanna and R. R. Schrock, Inorg. Chem., 2009, 48, 8569–8577 CrossRef CAS PubMed;
(d) H. Fong, M.-E. Moret, Y. Lee and J. C. Peters, Organometallics, 2013, 32, 3053–3062 CrossRef CAS PubMed.
- G. C. Welch and D. W. Stephan, J. Am. Chem. Soc., 2007, 129, 1880–1881 CrossRef CAS PubMed.
-
(a) M. Tamizmani and C. Sivasankar, Eur. J. Inorg. Chem., 2017, 2017, 4239–4245 CrossRef CAS;
(b) L. R. Doyle, A. J. Wooles and S. T. Liddle, Angew. Chem., Int. Ed., 2019, 58, 6674–6677 CrossRef CAS PubMed.
- Uranium nitride was shown to be hydrogenated in the presence of borane via an FLP-related mechanism: L. Chatelain, E. Louyriac, I. Douair, E. Lu, F. Tuna, A. J. Wooles, B. M. Gardner, L. Maron and S. T. Liddle, Nat. Commun., 2020, 11, 337 CrossRef CAS PubMed.
- F. F. Martins and V. Krewald, Eur. J. Inorg. Chem., 2023, e202300268 CrossRef.
- A bis(borane) adduct of a side-on Sm–N2 complex was reported recently. However, it is not clear whether the reduction of dinitrogen in this complex is the result of bis(borane) coordination via a push–pull mechanism, or occurs through capture by Sm prior to interaction with bis(borane). S. Xu, L. A. Essex, J. Q. Nguyen, P. Farias, J. W. Ziller, W. H. Harman and W. J. Evans, Dalton Trans., 2021, 50, 15000–15002 RSC.
- Diborane(4) compound B2Br4(SMe2)2 has also been explored for N2 functionalisation and furnishes boranylboryldiazenido complexes: L. C. Haufe, M. Arrowsmith, M. Dietz, A. Gärtner, R. Bertermann and H. Braunschweig, Dalton Trans., 2022, 51, 12786–12790 RSC.
- Selected examples of structurally characterized group 6 hydrazido complexes:
(a) G. A. Heath, R. Mason and K. M. Thomas, J. Am. Chem. Soc., 1974, 96, 259–260 CrossRef CAS;
(b) M. Hidai, T. Kodama, M. Sato, M. Harakawa and Y. Uchida, Inorg. Chem., 1976, 15, 2694–2697 CrossRef CAS;
(c) J. E. Barclay, A. Hills, D. L. Hughes, G. J. Leigh, C. J. Macdonald, M. A. Bakar and H. Mohd.-Ali, J. Chem. Soc., Dalton Trans., 1990, 2503–2507 RSC;
(d) D. V. Yandulov and R. R. Schrock, Inorg. Chem., 2005, 44, 1103–1117 CrossRef CAS PubMed;
(e) K. Arashiba, Y. Miyake and Y. Nishibayashi, Nat. Chem., 2011, 3, 120–125 CrossRef CAS PubMed.
- For two-fold Nβ covalent functionalisation with main group electrophiles, see:
(a) H. Oshita, Y. Mizobe and M. Hidai, Chem. Lett., 1990, 1303–1306 CrossRef CAS;
(b) H. Oshita, Y. Mizobe and M. Hidai, Organometallics, 1992, 11, 4116–4123 CrossRef CAS;
(c) H. Oshita, Y. Mizobe and M. Hidai, J. Organomet. Chem., 1993, 456, 213–220 CrossRef CAS;
(d) M.-E. Moret and J. C. Peters, J. Am. Chem. Soc., 2011, 133, 18118–18121 CrossRef CAS PubMed;
(e) P. A. Rudd, N. Planas, E. Bill, L. Gagliardi and C. C. Lu, Eur. J. Inorg. Chem., 2013, 2013, 3898–3906 CrossRef CAS;
(f) D. L. M. Suess and J. C. Peters, J. Am. Chem. Soc., 2013, 135, 4938–4941 CrossRef CAS PubMed;
(g) Q. Liao, N. Saffon-Merceron and N. Mézailles, Angew. Chem., Int. Ed., 2014, 53, 14206–14210 CrossRef CAS PubMed;
(h) Q. Liao, N. Saffon-Merceron and N. Mézailles, ACS Catal., 2015, 5, 6902–6906 CrossRef CAS.
- L. Greb, Chem.–Eur. J., 2018, 24, 17881–17896 CrossRef CAS PubMed.
-
(a) V. C. Williams, W. E. Piers, W. Clegg, M. R. J. Elsegood, S. Collins and T. B. Marder, J. Am. Chem. Soc., 1999, 121, 3244–3245 CrossRef CAS;
(b) P. A. Chase, L. D. Henderson, W. E. Piers, M. Parvez, W. Clegg and M. R. J. Elsegood, Organometallics, 2006, 25, 349–357 CrossRef CAS;
(c) V. C. Williams, C. Dai, Z. Li, S. Collins, W. E. Piers, W. Clegg, M. R. J. Elsegood and T. B. Marder, Angew. Chem., Int. Ed., 1999, 38, 3695–3698 CrossRef CAS;
(d) S. P. Lewis, N. J. Taylor, W. E. Piers and S. Collins, J. Am. Chem. Soc., 2003, 125, 14686–14687 CrossRef CAS PubMed;
(e) S. P. Lewis, L. D. Henderson, B. D. Chandler, M. Parvez, W. E. Piers and S. Collins, J. Am. Chem. Soc., 2005, 127, 46–47 CrossRef CAS PubMed;
(f) S. P. Lewis, J. Chai, S. Collins, T. J. J. Sciarone, L. D. Henderson, C. Fan, M. Parvez and W. E. Piers, Organometallics, 2009, 28, 249–263 CrossRef CAS.
- For related compounds whose 11B resonances could not be detected, see ref. 12g, 20a, 20b, 28 and
(a) A. Rempel, S. K. Mellerup, F. Fantuzzi, A. Herzog, A. Deißenberger, R. Bertermann, B. Engels and H. Braunschweig, Chem.–Eur. J., 2020, 26, 16019–16027 CrossRef CAS PubMed;
(b) A. Bouammali, A. Coffinet, L. Vendier and A. Simonneau, Dalton Trans., 2022, 51, 10697–10701 RSC. Other unrelated diamagnetic boron species that are NMR silent
can be found in:
(c) J. M. Farrell and D. W. Stephan, Angew. Chem., Int. Ed., 2015, 54, 5214–5217 CrossRef CAS PubMed;
(d) L. Zhu and R. Kinjo, Angew. Chem., Int. Ed., 2023, 62, e202306519 CrossRef CAS PubMed.
- A N2-derived titanium nitride was shown to undergo similar N-borylation through C6F5 group transfer: Z. Mo, T. Shima and Z. Hou, Angew. Chem., Int. Ed., 2020, 59, 8635–8644 CrossRef CAS PubMed.
- For a review on N2 borylation, see: A. Simonneau, New J. Chem., 2021, 45, 9294–9301 RSC.
-
C. Y. Legault, CYLview20, http://www.cylview.org 2020 Search PubMed.
- A. V. Zemskov, G. N. Rodionova, Yu. G. Tuchin and V. V. Karpov, J. Appl. Spectrosc., 1988, 49, 1020–1024 CrossRef.
- H. Ishino, Y. Ishii and M. Hidai, Chem. Lett., 1998, 27, 677–678 CrossRef.
- Related bis-diphosphine group 6 NO complexes were shown to activate H2 with the same first sphere isomerisation: A. Dybov, O. Blacque and H. Berke, Eur. J. Inorg. Chem., 2010, 3328–3337 CrossRef CAS.
- P. Pyykkö and M. Atsumi, Chem.–Eur. J., 2009, 15, 12770–12779 CrossRef PubMed.
- Z. Hussain, Y. Luo, Y. Wu, Z. Qu, S. Grimme and D. W. Stephan, J. Am. Chem. Soc., 2023, 145, 7101–7106 CrossRef CAS PubMed.
- F. Reiß, A. Schulz and A. Villinger, Chem.–Eur. J., 2014, 20, 11800–11811 CrossRef PubMed.
- F. Tuczek, K. H. Horn and N. Lehnert, Coord. Chem. Rev., 2003, 245, 107–120 CrossRef CAS.
-
(a) W. H. Bernskoetter, E. Lobkovsky and P. J. Chirik, Organometallics, 2005, 24, 6250–6259 CrossRef CAS;
(b) R. P. Yu, J. M. Darmon, S. P. Semproni, Z. R. Turner and P. J. Chirik, Organometallics, 2017, 36, 4341–4343 CrossRef CAS;
(c) S. M. Rummelt, H. Zhong, N. G. Léonard, S. P. Semproni and P. J. Chirik, Organometallics, 2019, 38, 1081–1090 CrossRef CAS PubMed.
- M. M. Deegan and J. C. Peters, Chem. Sci., 2018, 9, 6264–6270 RSC.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*